BioImpacts. 7(1):13-23.
doi: 10.15171/bi.2017.03
Original Research
Properties of macerated herbal oil
Fahsai Kantawong 1, *, Supawatchara Singhatong 1, Aomjai Srilamay 1, Kantarose Boonyuen 1, Niroot Mooti 1, Phenphichar Wanachantararak 2, Thasaneeya Kuboki 3
Author information:
1Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand
2The Dental Research Center, Faculty of Dentistry, Chiang Mai University, Chiang Mai 50200, Thailand
3Laboratory of Biomedical and Biophysical Chemistry, Institute for Materials Chemistry and Engineering, Kyushu University, Fukuoka, Japan
Abstract
Introduction:
The addition of herbs into hot sesame oil could increase the oil-pulling efficiency of sesame oil. The aim of present study was to modify the proportion of herbs and sesame oil with the addition of other ingredients including menthol, camphor, and borneol and improve the medicinal properties and the scent of the oil.
Methods:
Macerated herbal oil was prepared by heat extraction of five species of herbs (Zingiber cassumunar, Zingiber zerumbet, Plantago major Linn, Citrus hystrix, and Amomum biflorum) with hot sesame oil. The study was performed to evaluate the anti-oxidant, anti-inflammatory, and anti-bacterial properties of this macerated herbal oil.
Results:
Macerated herbal oil was evaluated for antioxidant activity using DPPH and ABTS assays. It was shown that at dilution 1:2 in DMSO, the macerated herbal oil had DPPH and ABTS radical scavenging activities equal to 63% and 22%, respectively. Macerated herbal oil dilution 1:8 in DMSO demonstrated ferric reducing capacity equivalent to ascorbic acid (0.208 µM) and had reducing power equivalent to butylated hydroxytoluene (BHT) 7.41 µg/mL. MTT assay was performed using immortalized human mesenchymal stem cells (HMSCs) as a cell culture model. The result indicated that the cytotoxic concentration of the macerated herbal oil was ≥ 2.5 µL/mL in complete DMEM. Anti-inflammatory effects were evaluated using the nitrite assay and RT-PCR. It was found that the macerated herbal oil could inhibit nitrite accumulation in culture media. Change in the expression of COX-2, Nrf2, and NF-kB in RT-PCR confirmed the anti-inflammatory activity of the macerated herbal oil.
Conclusion:
It could be concluded that the macerated herbal oil could inhibit nitrite accumulation in culture media, which might be the inhibitory effect of the macerated herbal oil on COX-2 or Nrf2, the downstream modulator of the COX-2 pathway. Further intensive studies are needed for the optimization before bringing this macerated herbal oil into clinical application.
Keywords: Antibacterial, Anti-inflammatory, Antioxidant, HMSCs, Macerated herbal oil
Copyright and License Information
© 2017 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Macerated herbal oil has been used as a remedy for joint, muscle, and skin inflammation in India and Southeast Asia since the ancient times and even in the present time. It is well known that sesame oil has anti-inflammatory properties and has always been used as an alternative medicine in many parts of Asia.
1
Currently, there are many scientific studies trying to prove that sesame oil has therapeutic effects, including: prevention of oxidative stress and lipid peroxidation in liver,
2,3
restoration of kidney function,
4
healing of intestinal and cardiac systems,
5,6
relieving pneumonia and bronchitis in rats,
7
and osteoprotection.
8
Sesame oil contains important ingredients such as sesamol,
9-12
linoleic acid, and oleic acid,
13
which might play important roles in rendering such therapeutic effects.
Sparingly water soluble ingredients in herbs are insoluble in water but can be dissolved in solvents like methanol, ethanol, or acetone. According to the polarity of phytoconstituents, the solvents for extraction are optional. Hexane is a solvent that can extract most of the fatty acids, lipids, waxes, steroids, triterpenoids, steroidal alkaloids and non-polar components. Methanol is suitable for the extraction of components like saponins, alkaloids, glycosides, carotenoids, flavonoids, and phenolics. Hot oil maceration is the method for extracting the phytoconstituents because maceration is a simple and effective method for extracting oil soluble components from herbal material. Sesame oil has medicinal properties of its own that can be utilized as an adjunct to the healing properties of the macerated herbs. Then Sesame oil was selected in this study. Moreover, the use of sesame oil did not need the hexane or methanol elimination process. For example, sabinene and terpinen-4-ol in Zingiber cassumunar
14,15
and zerumbone in Zingiber zerumbet
16
are water insoluble, so sesame oil can be employed to extract these substances from herbs. Moreover, the combination of sesame oil and herbal ingredients might provide benefits in terms of preserving the quality of active ingredients and preventing the lipid oxidation.
In the present study, sesame oil was heated with five species of herbs. These herbs have been widely studied and are readily available in local markets. The selected species of herbs were the ones traditionally used, including Z. cassumunar, Z. zerumbet, Plantago major Linn, Citrus hystrix, and Amomum biflorum. The active ingredients in the herbs have specific characteristics as follows:
Zingiber cassumunar Roxb.: This species has been widely researched and it was shown that terpinen-4-ol was the active compound which caused leukemic cells apoptosis.
14
It also has anti-inflammatory and anti-cancer properties.
17-21
Zingiber zerumbet (L.) Smith: Z. zerumbet has many research results corroborating its medicinal properties. Its active ingredient zerumbone has antitumor,
22
antinociceptive,
23
anti-inflammatory, and antimicrobial properties.
24,25
Moreover, the extract of this plant was proven to be nontoxic to cells, without any genetic mutation.
26
Plantago major Linn: P. major is recognized in the family Plantaginaceae. A variety of researches have demonstrated the antiviral, antileukemic, and wound-healing effects of this plant,
27,28
together with its antioxidant and anticancer activities.
29,30
The polysaccharide extract from the seeds of this plant have been used as a matrix for controlled release of drugs (drug-releasing matrix).
31
Citrus hystrix DC: C. hystrix is a common tropical herb in the family Rutaceae. The leaves and peels of C. hystrix have antimicrobial and antioxidant properties,
32,33
because this plant contains many phenolic compounds.
34
The essential oils from the leaves and peels of C. hystrix demonstrate anti-bacterial effects on pathogens in the respiratory tract.
35
Amomum biflorum Jack: A. biflorum is in the family Zingiberaceae. This plant has been proven to have high contents of total phenolic compounds with antioxidant and anti-tyrosinase properties.
36
To improve the properties of macerated herbal oil, the proportion of herbs and sesame oil are needed to be modified. In the present study the amount of herbs and sesame oil were modified and other ingredients such as menthol, camphor, and borneol were added to improve the scent of the oil. The bioactivity of this traditional medicine was tested with human mesenchymal stem cells (HMSCs) because HMSCs exhibited changes in the expression level of inflammatory mediator gene under various conditions.
37,38
Before bringing this alternative medicine into clinical application as a topical drug for local inflammation or infection, the quality of the macerated herbal oil should be proved and improved for better therapeutic outcome.
Materials and methods
Macerated herbal oil preparation and total phenolic content
All the herbs were washed and sliced. A volume of 1500 mL of cold pressed sesame oil (purchased from a local manufacturer) was heated with 300 g of crushed cockle shells on a charcoal stove for 10 minutes. The quantities including 400 g of Zingiber cassumunar, 200 g of Z zerumbet, 50 g of P. major Linn, 200 g of C. hystrix peels, 50 g of C. hystrix leaves, and 100 g of A. biflorum were added into the heated sesame oil. All the components were slowly stirred for 1.5 hours. Finally, 150 g of menthol, 50 g of camphor, and 50 g of borneol were added to improve the scent of the oil. After 1.5 hours of heat treatment, the oil was filtered through a delicate sieve and left for cooling down. The herb residual was separated using Whatman No. 1. Clear yellow oil with the soft aroma of the herbs was aliquoted in closed containers before storing at 4°C until use. Main components of macerated herbal oil are shown in Table 1.
Table 1.
Main components of macerated herbal oil
|
Components
|
Weight percent (wt%)
|
|
Sesame oil
|
47 |
| Crushed cockle shells |
10 |
|
Zingiber cassumunar Roxb.
|
13 |
|
Zingiber zerumbet
|
7 |
|
Plantago major Linn.
|
2 |
|
Peels of Citrus hystrix
|
7 |
|
Leaves of Citrus hystrix
|
2 |
| Amomum biflorum |
3 |
| Menthol |
5 |
| Camphor |
2 |
| Borneol |
2 |
| Total |
100 |
Total phenolic assay was performed by the following method. Briefly, the macerated herbal oil was diluted with dimethyl sulfoxide (DMSO) to the dilutions of 1:2, 1:4, 1:8, 1:16, and 1:32. Thereafter, 50 µL of each dilution was mixed with 2.5 mL Folin Ciocalteau reagent, and 2.0 mL Na2CO3 was added and mixed before incubating at 45°C for 15 minutes. A blue color was observed, and the absorbance was measured at a wavelength of 764 nm. The mixture of 2.5 mL Folin Ciocalteau reagent and 2.0 mL Na2CO3 was used as blank for the zero setting. A standard calibration curve was prepared using different concentrations of gallic acid in methanol. Three replicate assays were performed for each dilution, and the total phenolic concentrations were calculated from the standard curve and reported as mg/mL equivalent to standard gallic acid.
DPPH radical scavenging and ABTS assay
DPPH radical scavenging and ABTS assay were performed. The macerated herbal oil was diluted with DMSO to the dilutions of 1:2, 1:4, 1:8, and 1:16. Then 50 µL of each dilution was mixed with 2,950 µL of 2,2-diphenyl-1-picrylhydrazyl (DPPH) reagent and allowed to react in the dark at room temperature for 15 minutes. The absorbance was measured at a wavelength of 515 nm using distilled water to set zero. Standard trolox was used as the positive control. DMSO solution of volume 2,950 µL plus 50 µL of each dilution was used as a sample blank. DPPH solution of volume 2,950 µL plus 50 µL DMSO was used as a control for calculation. Three replicate assays were performed for each dilution, and the %DPPH scavenging activity was calculated by equation (Eq.) 1:
(Eq.1)
For the ABTS assay, 10 µL of each dilution was mixed with 990 µL of working 2,2'-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) reagent (14 mmol/L ABTS reagent plus 5 mmol/L potassium persulfate solution in the ratio 1:1). The mixture was allowed to stand for 1 minute before measuring at 734 nm using distilled water to set zero. Standard trolox was used as the positive control. Working ABTS reagent should have absorbance around 0.700 ± 0.02 (0.68–0.72) at 734 nm. Three replicate assays were performed for each dilution and the inhibition percentage was calculated by Eq. 2:
(Eq.2)
Ferric reducing capacity and reducing power
The macerated herbal oil was diluted with DMSO to the dilutions of 1:8, 1:16, 1:32, 1:64, and 1:128. Then 60 µL of each dilution was mixed with 1,800 µL of the cocktail solution. After that, 180 µL of deionized water was added to the reaction tube. The reaction was mixed and the absorbance was detected at 593 nm as a baseline. The reaction mixture was incubated again at 37°C for 4 minutes, following which the absorbance of the mixture was measured again at 593 nm within 4 minutes. Three replicate assays were performed for each dilution. Various concentrations of ascorbic acid were used to generate the standard curve. The ferric reducing capacity was calculated from the standard curve and reported as ferric reducing capacity equivalent to ascorbic acid.
For reducing power, 2.5 µL of each dilution was mixed with 2.5 mL PBS. Then 2.5 mL of 1% potassium ferricynide (K3Fe[CN]6) was added to the mixture and incubated at 50°C for 20 minutes. After the reaction cooled down, 2.5 mL of 10% trichloroacetic acid was added and mixed. The reaction was centrifuged at 3000 rpm for 10 minutes. A volume of 2.5 mL of the supernatant was separated and mixed with 2.5 mL of deionized water. A volume of 0.5 mL of 0.1% ferric chloride was added to the reaction and allowed to stand for 10 minutes. The absorbance was measured at 700 nm. Three replicate assays were performed for the statistical analysis. Various concentrations of butylated hydroxytoluene (BHT) were used to generate a standard curve. The reducing power was calculated from the standard curve and reported as reducing power equivalent to BHT.
MTT assay and nitrite assay
MTT assay was performed following the protocol manual of assay.
39
Immortalized HMSCs (passage 5) (Health Science Research Resources Bank, Osaka, Japan) were cultured at a seeding density of 1 × 105 cells/well into a 6-well plate. Cells were allowed to adhere for 24 hours in complete Dulbecco’s Modified Eagle Medium (DMEM) at 37°C and 5% CO2. The macerated herbal oil was diluted in DMSO to the dilutions of 1:4, 1:8, 1:16, 1:32, and 1:64. Then 100 µL of each dilution was added to 10 mL of complete DMEM. Thus, the final concentrations of the macerated herbal oil in 1 mL DMEM were 2.5 µL, 1.25 µL, 0.625 µL, 0.313 µL, and 0.156 µL, respectively. After the cells were allowed to settle for 24 hours, the cell viability was determined using the MTT assay. Briefly, the entire DMEM was discarded and replaced with macerated herbal oil supplemented DMEM (3 mL/well). The cell culture was maintained at a temperature of 37°C in a 5% CO2 incubator for 48 h. The macerated herbal oil supplemented DMEM was discarded and replaced with 1 mL of 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reagent (0.5 mg MTT in 1 mL complete DMEM). The reaction was maintained at 37°C in 5% CO2 for 2 hours. The MTT supplemented DMEM was discarded and 1 mL of DMSO was added. The absorbance of the formazan product was measured at 570 nm and the background was corrected at 630 nm. The percentage of the cell viability was compared to that of control which was cultured in DMEM plus DMSO (100 µL DMSO in 10 mL DMEM). Three replicate assays were performed for the statistical analysis.
For the nitrite assay, the HMSCs (passage 5) were cultivated in a 6-well plate at a seeding density of 1 × 105 cells/well using phenol red-free DMEM containing 10% FBS. After 24 hours, the cells were incubated with bacterial lipopolysaccharide (LPS; 2 μg/mL), macerated herbal oil (0.2 µL/mL), and LPS (2 μg/mL) plus macerated herbal oil (0.2 µL/mL) or DMSO for 24 hours, and then the culture media was subjected to the nitrite assay. Briefly, 800 μL of the culture medium was mixed with 800 μL of the Griess reagent and left at room temperature for 10 minutes. The absorbance was measured at 550 nm using water to set zero. A standard curve was plotted using serial concentrations of sodium nitrite. Three replicate assays were performed for the statistical analysis.
Relative quantitative real-time PCR
For the anti-inflammatory assay (pre-treatment), cells were incubated for 2 hours with the macerated herbal oil (0.2 μg/mL) before being treated with bacterial LPS (2 μg/mL), or DMSO (100 µL DMSO in 10 mL DMEM) for 24 hours. For the anti-inflammatory assay (co-treatment), cell culture was performed as described in the nitrite assay. In the anti-inflammatory assay (post-treatment), cells were incubated for 2 hours with bacterial LPS (2 μg/mL) before being treated with the macerated herbal oil (0.2 μg/mL), or DMSO (100 µL DMSO in 10 mL DMEM) for 24 hours. For the investigation of the redox gene expression upon long-term exposure, the HMSCs (passage 5) were seeded with complete DMEM containing 10% FBS into a 6-well plate at a seeding density of 1 × 105 cells/well. After 24 hours, the cells were incubated with the macerated herbal oil (0.1 µL/mL) or DMEM + DMSO for 12 days.
After the culture period, the total RNA was extracted from the HMSCs using NucleoSpin® RNA II, according to the protocol provided by the manufacturer. A volume of 10 µL total RNA was treated again with 1 µL rDNase (Macherey-Nagel) and incubated at 37°C for 10 minutes. A 40 μL cDNA reaction volume was prepared with the use of 5× iScript Reverse Transcription Supermix (Bio-Rad), as described by the manufacturer’s protocol. Reverse transcription was performed in Eppendorf Mastercycler® using the following program: priming at 25°C for 5 minutes, reverse transcription at 42°C for 30 minutes, and enzyme inactivation at 85°C for 5 minutes. cDNA was stored at −20°C until real-time PCR was performed. No-RT control, which contained all the reaction components except for the reverse transcriptase, was used to detect the contamination of the genomic DNA. The primer pairs for real-time PCR are shown in Table 2.
Table 2.
Human target genes for relative quantitative PCR
|
Target genes
|
Primer sequences (5'->3')
|
| Cyclooxygenase-2 (COX-2) |
F: CCCTTGGGTGTCAAAGGTAA |
|
|
R: GCCCTCGCTTATGATCTGTC |
|
Nuclear factor erythroid-derived 2-related factor 2 (Nrf2)
|
F: AGTGGATCTGCCAACTACTC |
|
|
R: CATCTACAAACGGGAATGTCTG |
|
NF-kappa B (NF-kB)
|
F: ATGGCTTCTATGAGGCTGAG |
|
|
R: GTTGTTGTTGGTCTGGATGC |
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) |
F: AAGGGCTCATGACCACAGTC |
|
|
R: GGATGACCTTGCCCACAG |
The cDNA was diluted 1:10 and RT-PCR was performed with a SYBR Green RT-PCR mastermix (Toyobo) in LightCycler480 (Roche). The 10-μL reaction was composed of 5 μL of SYBR Green RT-PCR master mix, 1 μL of cDNA template, 3.5 µL of distilled water, and 0.25 μM of target-specific primer. The polymerase chain reaction protocol consisted of 95°C pre-incubation for 30 seconds, followed by 45 cycles of 95°C for 5 seconds, 55°C for 10 seconds, and 74°C for 15 s. After the real-time PCR, a melting curve analysis was performed to demonstrate the specificity of the PCR product as a single peak. A control, which contained all the reaction components except for the template, and no-RT control were included in all the experiments. Three replicate assays were performed for the statistical analysis. The relative quantification for the gene expression was normalized to GAPDH using Pffal method (Light Cycler®480 software release 1.5.0 SP4).
Nitric oxide scavenging assay
Sodium nitroprusside generates nitric oxide (NO), which interacts with oxygen to produce nitrite ions, can be measured by using the Greiss reagent. The macerated herbal oil was used as a scavenger of nitric oxide, which led to a decrease in the nitric oxide level. A volume of 2 mL of sodium nitroprusside (10 mM) was mixed with 0.5 mL of PBS and 0.5 mL of the macerated herbal oil dilutions of 1:256 and 1:512, and then incubated at 25°C for 150 minutes. After that, 0.5 mL of the previous reaction was mixed with 1 mL of 1% sulfanilamide and incubated at room temperature for 5 minutes. Thereafter, 1 mL of 0.1% naphthyl ethylenediamine dihydrochloride was added and incubated at 25°C for 150 minutes. The absorbance was read at 537 nm using water to set zero. The reaction of the same reagents with 0.5 mL DMSO was used as a control. The %inhibition was calculated by Eq. 3:
(Eq.3)
Anti-bacterial activity
This experiment was performed in order to determine the lowest concentration of an antimicrobial agent that can kill bacteria in a given time. Briefly, the macerated herbal oil was diluted 10-fold in brain–heart infusion (BHI) broth (100–10-7) and Streptococcus mutans (DMST 187777) was diluted in brain–heart infusion (BHI) broth to a concentration of 0.5 McFarland standard using an OD measurement at 600 nm. Thereafter, 100 µL of each 10-fold macerated herbal oil dilution and 100 µL of the bacterial suspension were added into each well of a 96-well tissue culture plate. As a negative control, an equivalent volume of BHI was added to the bacterial cultures instead of the 10-fold macerated herbal oil dilution. As a positive control, 0.12% chlohexidine was added to the bacterial cultures instead of the 10-fold macerated herbal oil dilution. For the minimum bactericidal concentration measurement, 10 µL aliquots from a 96-well tissue culture plate that were previously treated with macerated herbal oil were spread on the BHI agar plates and incubated at 37°C for 24 hours under anaerobic conditions.
Statistical analysis
The student’s t test and one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc multiple comparison test were used to determine the significant difference between the two samples from the three independent assays. The data are presented as mean ± standard deviation (SD). The statistical significance was set at P < 0.05.
Results
Macerated herbal oil preparation
After 1.5 hours of heat treatment, the oil was filtered through a delicate sieve. The oil was then left for cooling down and the other residual was separated using Whatman No. 1. Clear yellow herb-scented oil was obtained, as shown in Fig. 1A, which was stored at 4°C until use. Macerated herbal oil was tested for the total phenolic content using the Folin Ciocalteau method. The result revealed that the macerated herbal oil dilution 1:2 had a total phenolic concentration equivalent to the concentration of gallic acid, 1.94 mg/mL. The concentration of the phenolic compound decreased unproportional to the dilution (Fig. 1B). This result indicated that phenolic compounds were present in the prepared macerated herbal oil.
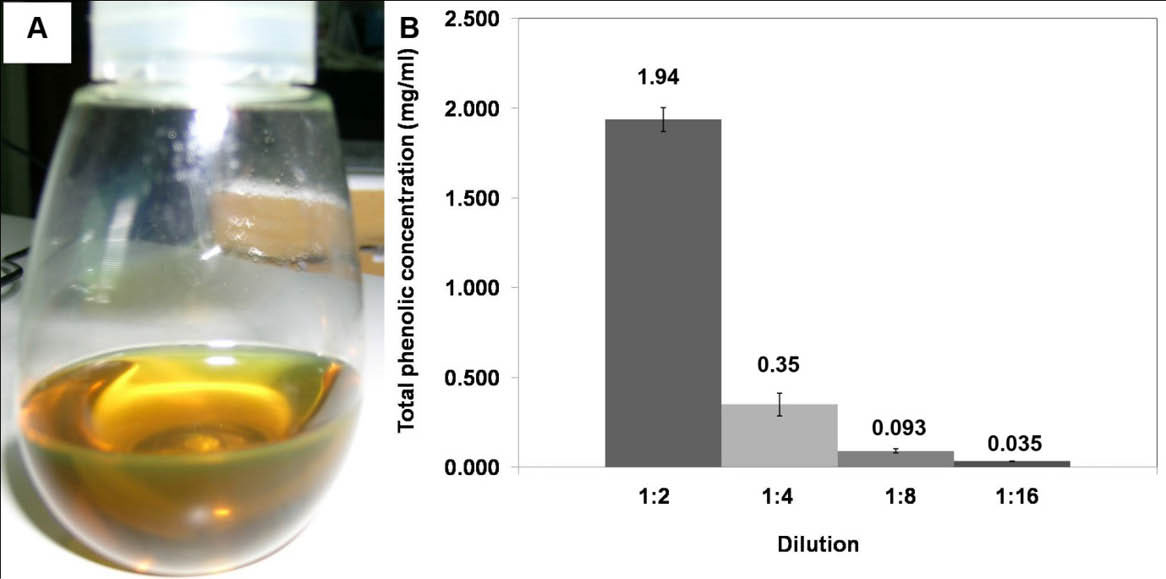
Fig. 1.
(A) Macerated herbal oil: clear yellow herb aroma oil was obtained in this experiment. (B) Total phenolic concentration: the
macerated herbal oil was diluted in DMSO to dilutions 1:2, 1:4, 1:8, and 1:16 which had total phenolic concentrations equivalent to gallic
acid 1.94 mg/mL, 0.35 mg/mL, 0.093 mg/mL, and 0.035 mg/mL, respectively.
.
(A) Macerated herbal oil: clear yellow herb aroma oil was obtained in this experiment. (B) Total phenolic concentration: the
macerated herbal oil was diluted in DMSO to dilutions 1:2, 1:4, 1:8, and 1:16 which had total phenolic concentrations equivalent to gallic
acid 1.94 mg/mL, 0.35 mg/mL, 0.093 mg/mL, and 0.035 mg/mL, respectively.
DPPH radical scavenging assay and ABTS radical scavenging assay
The antioxidant activity of the macerated herbal oil was tested using the DPPH assay and the ABTS assay. DPPH is a stable free radical with purple color. When the number of DPPH free radicals reduces, a yellow color is generated, and the reduction in their number results in the decrease of absorbance at 515 nm. The macerated herbal oil was diluted in DMSO to various dilutions. After the DPPH radicals had reacted with the macerated herbal oil, the absorbance was measured and compared to the absorbance of the DPPH reagent. The antioxidant activity of the macerated herbal oil was reported as %radical scavenging, as presented in Fig. 2A. The antioxidant activity of the macerated herbal oil was confirmed with the ABTS assay. ABTS reacted with potassium persulfate to generate ABTS free radicals which are blue in color. The reduction in the number of the ABTS radicals with several dilutions of the macerated herbal oil caused a decrease in the blue color, which could be measured at 734 nm. The %ABTS radical scavenging activity of the macerated herbal oil was calculated and is as presented in Fig. 2B.
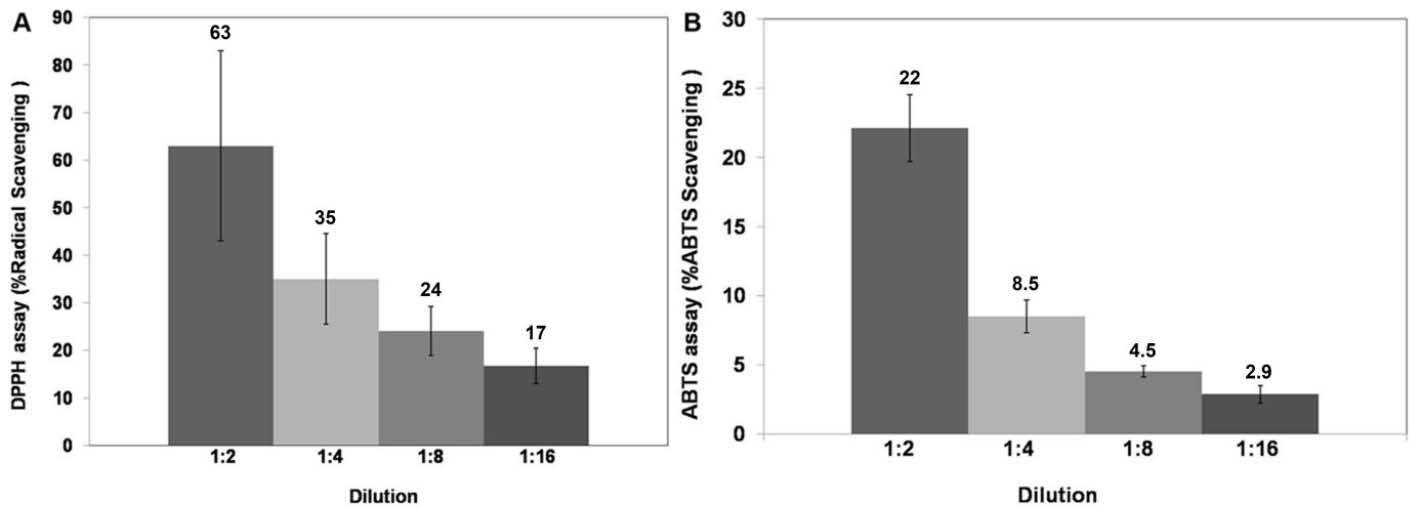
Fig. 2.
DPPH and ABTS assays: the macerated herbal oil was diluted in DMSO to dilutions of 1:2, 1:4, 1:8, and 1:16, and the antioxidant
activity was evaluated using the DPPH assay (A) and the ABTS assay (B). The results were presented as %radical scavenging (%inhibition).
.
DPPH and ABTS assays: the macerated herbal oil was diluted in DMSO to dilutions of 1:2, 1:4, 1:8, and 1:16, and the antioxidant
activity was evaluated using the DPPH assay (A) and the ABTS assay (B). The results were presented as %radical scavenging (%inhibition).
Ferric reducing capacity and reducing power
The ferric reducing capacity relies on the principle that the antioxidants can transfer electrons to the Fe3+ ions to become Fe2+ ions which have blue color. Absorbance can be measured at a wavelength of 593 nm. Fe2+ ions were formed after the reaction with the various concentrations of the macerated herbal oil, and the absorbance was compared to the standard curve. The antioxidant activity of the macerated herbal oil was reported as ferric reducing capacity equivalent to ascorbic acid (Fig. 3A).
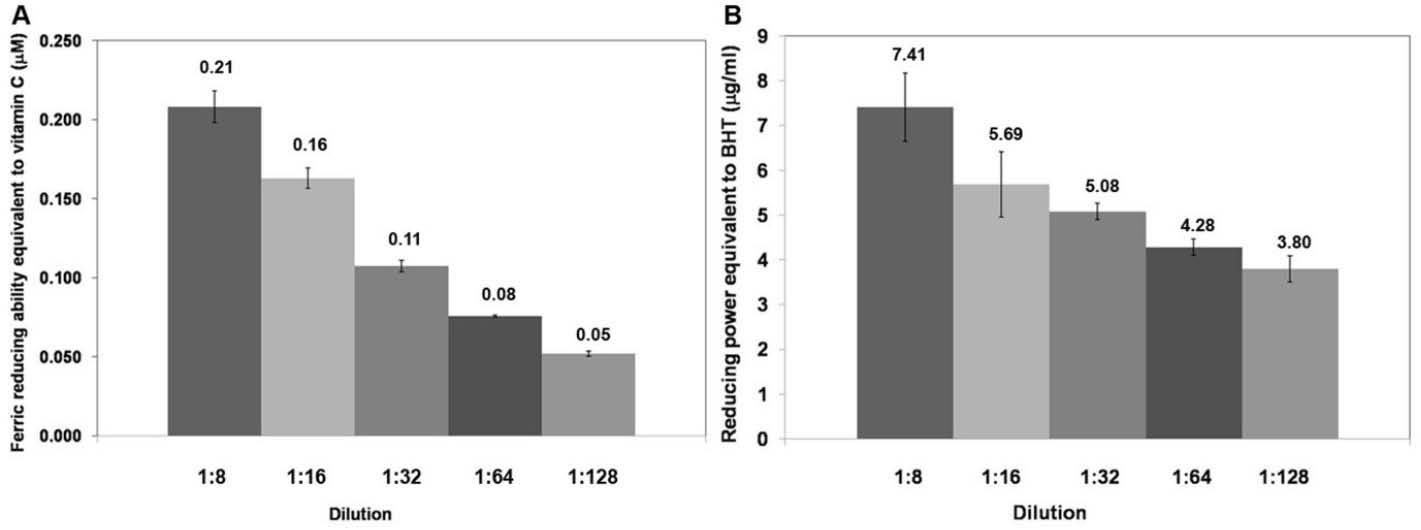
Fig. 3.
(A) Ferric reducing capacity: the macerated herbal oil was diluted in DMSO to dilutions 1:4, 1:8, 1:16, and 1:32 which had ferric
reducing capacities equivalent to ascorbic acid 0.351 μM, 0.093 μM, 0.035 μM, and 0.006 μM, respectively. (B) Reducing power: the
macerated herbal oil was diluted in DMSO to dilutions 1:4, 1:8, 1:16, and 1:32 which had reducing powers equivalent to BHT 0.351 μg/mL,
0.093 μg/mL, 0.035 μg/mL, and 0.006 μg/mL, respectively.
.
(A) Ferric reducing capacity: the macerated herbal oil was diluted in DMSO to dilutions 1:4, 1:8, 1:16, and 1:32 which had ferric
reducing capacities equivalent to ascorbic acid 0.351 μM, 0.093 μM, 0.035 μM, and 0.006 μM, respectively. (B) Reducing power: the
macerated herbal oil was diluted in DMSO to dilutions 1:4, 1:8, 1:16, and 1:32 which had reducing powers equivalent to BHT 0.351 μg/mL,
0.093 μg/mL, 0.035 μg/mL, and 0.006 μg/mL, respectively.
The experiment was confirmed with reducing power which relies on a similar principle. Ferric ions (Fe3+) in the FeSO4 solution possess the ability to obtain electrons from other substances in order to form Fe2+. Various concentrations of the macerated herbal oil were made to react with the FeSO4 solution, and then the absorbance was measured at 700 nm, which was proportional to the concentration of the ferrous ion (Fe2+). The increase in the absorbance indicated the considerable potential of the macerated herbal oil as a reducing agent. The absorbance was compared to the ability of BHT which was used to generate the standard curve. The result was reported as reducing power equivalent to BHT (Fig. 3B).
MTT assay and nitrite accumulation assay
Various concentrations of the macerated herbal oil were prepared using 100 µL of the macerated herbal oil dilutions at 1:4, 1:8, 1:16, 1:32, and 1:64 and were added to 10 mL of complete DMEM. Thus, the final concentrations of the macerated herbal oil in 1 mL DMEM were 2.5 µL, 1.25 µL, 0.625 µL, 0.313 µL, and 0.156 µL, respectively. The MTT assay indicated that only the 2.5 µL/mL concentration showed cytotoxic effect on the HMSCs (Fig. 4A). Taking into consideration the result of the MTT assay, the macerated herbal oil at the concentration of 0.2 µL/mL (100 µL of macerated herbal oil, dilution 1:50, in 10 mL of complete DMEM) was used for the nitrite assay. The accumulation of nitrite is an indicator of NO synthesis, which was measured in the culture medium using the Griess reagent. The HMSCs were seeded into a 6-well plate at a density of 1 × 105 cells/well and treated with the macerated herbal oil with or without LPS (2 µg/mL). It was demonstrated that LPS alone increased the concentration of nitrite in the culture medium. When LPS and the macerated herbal oil were presented together, the level of nitrite was not different from that of control. This result indicates that the macerated herbal oil had attenuated nitrite accumulation in the culture medium, which implies a reduction in the NO synthesis (Fig. 4B). The result obtained upon treating the HMSCs with the macerated herbal oil alone was not different from that of control.
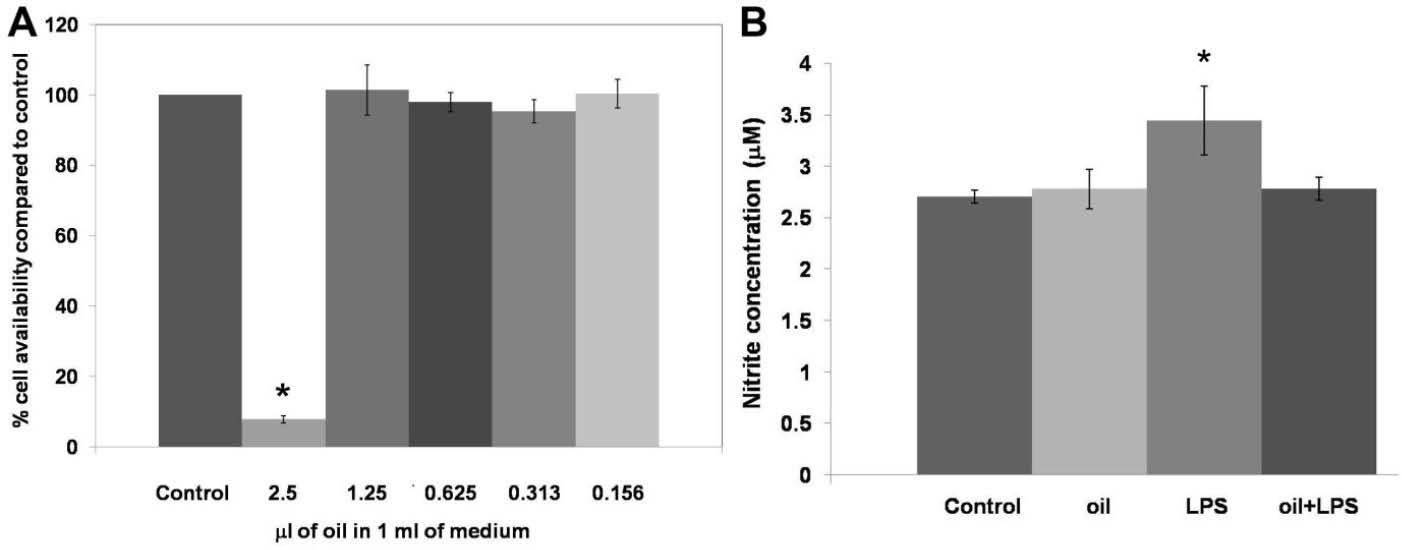
Fig. 4.
(A) Cell viability: various concentrations of the macerated herbal oil (2.5 μL/mL, 1.25 μL/mL, 0.625 μL/mL, 0.313 μL/mL, and
0.156 μL/mL) were incubated with HMSCs (1 × 105 cells/well) for 48 h. The cell viability was determined using the MTT assay and the
%viability was compared to the control group (cells cultured in DMEM + DMSO). Only the 2.5 μL/mL concentration showed cytotoxic effect
on the HMSCs (*P < 0.05).
(B) Nitrite assay: the macerated herbal oil concentration 0.2 μL/mL was used in this study. Three groups of
the cell cultures were designed to evaluate the NO production. The cells (1 × 105 cells/well) were incubated with oil, LPS, and oil + LPS
for 24 h. The nitrite assay was performed with the Griess reagent. The nitrite concentrations in the culture medium were compared to
the control group (cells cultured in DMEM + DMSO). The cells treated with LPS (2 μg/mL) showed increased nitrite accumulation in the
culture medium and nitrite accumulation was decreased in the presence of the macerated herbal oil (*P < 0.05, ANOVA, Tukey post hoc).
.
(A) Cell viability: various concentrations of the macerated herbal oil (2.5 μL/mL, 1.25 μL/mL, 0.625 μL/mL, 0.313 μL/mL, and
0.156 μL/mL) were incubated with HMSCs (1 × 105 cells/well) for 48 h. The cell viability was determined using the MTT assay and the
%viability was compared to the control group (cells cultured in DMEM + DMSO). Only the 2.5 μL/mL concentration showed cytotoxic effect
on the HMSCs (*P < 0.05).
(B) Nitrite assay: the macerated herbal oil concentration 0.2 μL/mL was used in this study. Three groups of
the cell cultures were designed to evaluate the NO production. The cells (1 × 105 cells/well) were incubated with oil, LPS, and oil + LPS
for 24 h. The nitrite assay was performed with the Griess reagent. The nitrite concentrations in the culture medium were compared to
the control group (cells cultured in DMEM + DMSO). The cells treated with LPS (2 μg/mL) showed increased nitrite accumulation in the
culture medium and nitrite accumulation was decreased in the presence of the macerated herbal oil (*P < 0.05, ANOVA, Tukey post hoc).
Gene expression
The results of pre-treatment are shown in Fig. 5A. Exposure of the cells to LPS for 24 hours caused a 12.3-fold up-regulation of the cox-2 gene, while exposure to the macerated herbal oil for 2 h followed by 24 hours of LPS treatment caused a 7.8-fold up-regulation of the cox-2 gene. The difference between these two groups was statistically significant (P < 0.05), which shows that pre-treatment of macerated herbal oil (0.2 µL/mL) could reduce cox-2 gene expression when it is applied before the treatment of LPS. No significant change in the Nrf2 and NF-kB expressions were observed at the 24-hour culture period compared to the control group.
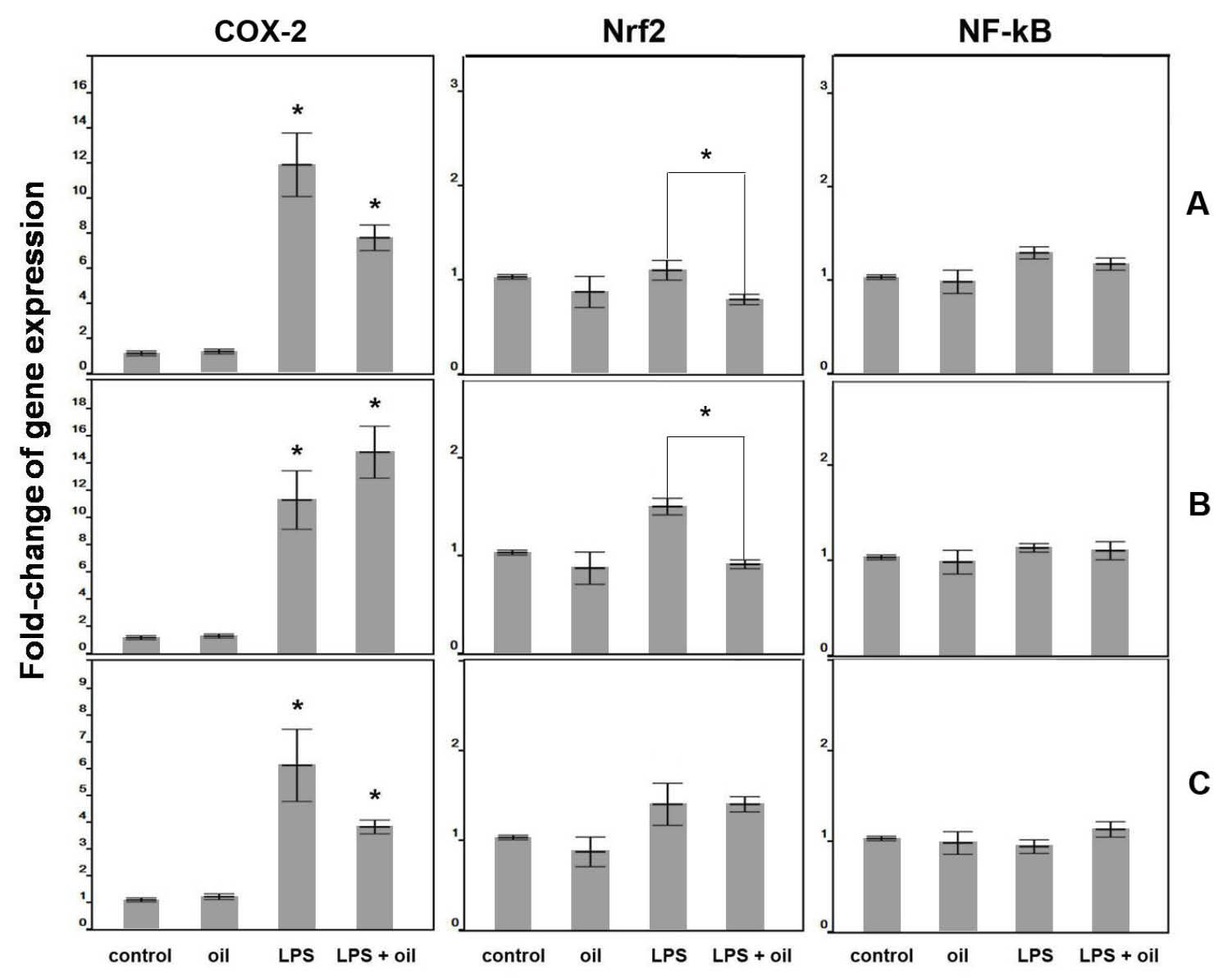
Fig. 5.
(A) Gene expression of pre-treatment: the macerated herbal oil concentration 0.2 μL/mL was used in this study. The cells (1 ×
105 cells/well) were treated using three different ways: (oil) cells were treated with oil for 24 h, (LPS) cells were treated with LPS for 24 h,
and (LPS + oil) cells were exposed to oil for 2 h before treating with LPS for 24 h. Total RNA extraction and RT-PCR were performed to
investigate the gene expression compared to control (cells cultured in DMEM + DMSO). The LPS and LPS + oil treated groups showed
significant up-regulation of the cox-2 gene compared to the control group (*P < 0.05). The LPS + oil treated group significantly lowered the
cox-2 gene expression when compared to the LPS group (*P < 0.05, ANOVA, Tukey post hoc). LPS + oil treated groups showed significant
down-regulation of the Nrf2 gene compared to LPS group (*P < 0.05, ANOVA, Tukey post hoc). (B) Gene expression of co-treatment:
the cells were incubated with the oil, LPS, and oil + LPS for 24 h. Both LPS and LPS + oil treated groups showed significant up-regulation
of the cox-2 gene compared to the control group (*P < 0.05, ANOVA). It was only the LPS-treated group that showed significant upregulation
of the Nrf2 gene which was decreased in the presence of oil (*P < 0.05, ANOVA, Tukey post hoc).
(C) Gene expression of
post-treatment:
the cells were treated using three different ways: (oil) cells were treated with oil for 24 h, (LPS) cells were treated with
LPS for 2 h before being cultured in DMEM + DMSO for 24 h, and (LPS + oil) cells were exposed to LPS for 2 h before treating with oil for
24 hours. The LPS and LPS + oil treated groups showed significant up-regulation of the cox-2 gene compared to the control group (*P <
0.05, ANOVA, Tukey post hoc). The LPS + oil treated group significantly lowered the cox-2 gene expression when compared to the LPS
group (*P < 0.05, ANOVA, Tukey post hoc).
.
(A) Gene expression of pre-treatment: the macerated herbal oil concentration 0.2 μL/mL was used in this study. The cells (1 ×
105 cells/well) were treated using three different ways: (oil) cells were treated with oil for 24 h, (LPS) cells were treated with LPS for 24 h,
and (LPS + oil) cells were exposed to oil for 2 h before treating with LPS for 24 h. Total RNA extraction and RT-PCR were performed to
investigate the gene expression compared to control (cells cultured in DMEM + DMSO). The LPS and LPS + oil treated groups showed
significant up-regulation of the cox-2 gene compared to the control group (*P < 0.05). The LPS + oil treated group significantly lowered the
cox-2 gene expression when compared to the LPS group (*P < 0.05, ANOVA, Tukey post hoc). LPS + oil treated groups showed significant
down-regulation of the Nrf2 gene compared to LPS group (*P < 0.05, ANOVA, Tukey post hoc). (B) Gene expression of co-treatment:
the cells were incubated with the oil, LPS, and oil + LPS for 24 h. Both LPS and LPS + oil treated groups showed significant up-regulation
of the cox-2 gene compared to the control group (*P < 0.05, ANOVA). It was only the LPS-treated group that showed significant upregulation
of the Nrf2 gene which was decreased in the presence of oil (*P < 0.05, ANOVA, Tukey post hoc).
(C) Gene expression of
post-treatment:
the cells were treated using three different ways: (oil) cells were treated with oil for 24 h, (LPS) cells were treated with
LPS for 2 h before being cultured in DMEM + DMSO for 24 h, and (LPS + oil) cells were exposed to LPS for 2 h before treating with oil for
24 hours. The LPS and LPS + oil treated groups showed significant up-regulation of the cox-2 gene compared to the control group (*P <
0.05, ANOVA, Tukey post hoc). The LPS + oil treated group significantly lowered the cox-2 gene expression when compared to the LPS
group (*P < 0.05, ANOVA, Tukey post hoc).
From the result of the co-treatment, as presented in Fig. 5B, it is evident that the LPS treatment and the LPS + oil treatment significantly induced the cox-2 gene expression, which indicates that the macerated herbal oil (0.2 µL/mL) could not inhibit the LPS-induced cox-2 gene expression in the HMSCs when they were presented together. However, the LPS + oil treatment resulted in the attenuation of the LPS-induced Nrf2 gene expression. No significant change in the NF-kB expression was observed at the 24-hour culture period compared to the control group.
The results of post-treatment are as shown in Fig. 5C. Exposure to LPS for 2 hours followed by 24 hours of normal culture caused a 6.2-fold up-regulation of the cox-2 gene, while exposure to LPS for 2 hours followed by 24 hours of macerated herbal oil treatment caused a 3.8-fold up-regulation of the cox-2 gene. The difference between these two groups was statistically significant (P < 0.05), which shows that post-treatment of macerated herbal oil (0.2 µL/mL) could reduce cox-2 gene expression when it is applied after the treatment of LPS. However, it was observed that the macerated herbal oil could not inhibit the LPS-induced Nrf2 gene expression in the post-treatment study. No significant change in the NF-kB expression was observed at the 24-hour culture period compared to the control group.
Nitric oxide scavenging assay
The macerated herbal oil showed NO scavenging activity up to the dilutions of 1: 512. The result, which is illustrated in Fig. 6, indicated that the herbal dilution 1: 256 had NO scavenging activity at 11%. The NO scavenging activity decreased proportionally to the dilution of the macerated herbal oil, demonstrating the activity to be at 5% at the dilution of 1:512.
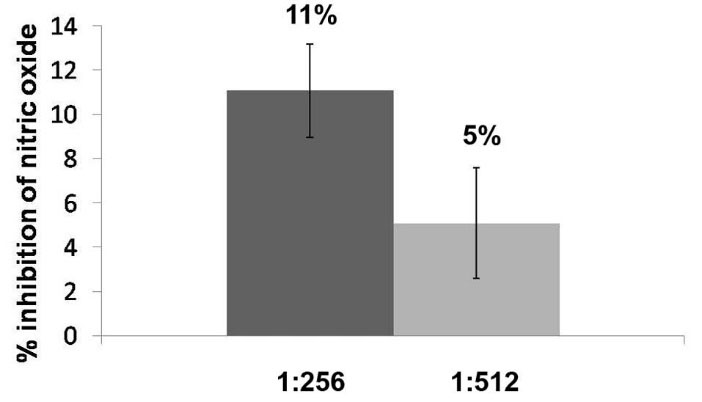
Fig. 6.
Nitric oxide scavenging activity: the nitric oxide
scavenging activity of the macerated herbal oil was reflected as
the %inhibition of nitric oxide. The macerated herbal oil dilutions
1:265 and 1:512 had %inhibition of nitric oxide of 11% and 5%,
respectively.
.
Nitric oxide scavenging activity: the nitric oxide
scavenging activity of the macerated herbal oil was reflected as
the %inhibition of nitric oxide. The macerated herbal oil dilutions
1:265 and 1:512 had %inhibition of nitric oxide of 11% and 5%,
respectively.
Anti-bacterial activity
Macerated herbal oil was diluted 10-fold in BHI broth and 100 µL of each dilution was added to 100 µL of bacterial suspension. As shown in Fig. 7, the macerated herbal oil presented antimicrobial activity against S. mutans. However, the antimicrobial activity was not very effective because only a 100 dilution is capable of inhibiting bacterial growth.
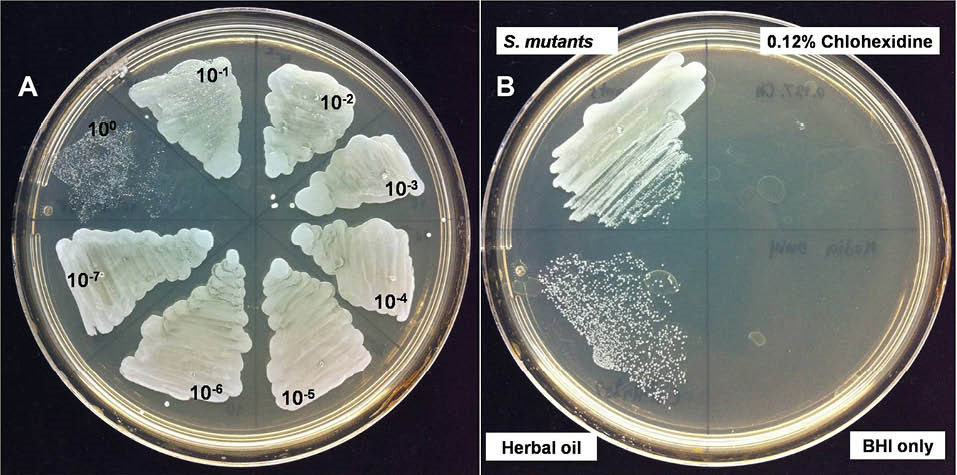
Fig. 7.
Anti-bacterial activity: (A) when 100 μL of each 10-
fold macerated herbal oil dilution was added to 100 μL bacterial
suspension, only the 100 dilution showed antimicrobial activity. (B)
The macerated herbal oil at dilution 100 was seen to inhibit bacterial
growth when compared to the negative control (S. mutants).
The macerated herbal oil at dilution 100 was less effective when
compared to the positive control (0.12% Chlohexidine). BHI was
used as the blank control.
.
Anti-bacterial activity: (A) when 100 μL of each 10-
fold macerated herbal oil dilution was added to 100 μL bacterial
suspension, only the 100 dilution showed antimicrobial activity. (B)
The macerated herbal oil at dilution 100 was seen to inhibit bacterial
growth when compared to the negative control (S. mutants).
The macerated herbal oil at dilution 100 was less effective when
compared to the positive control (0.12% Chlohexidine). BHI was
used as the blank control.
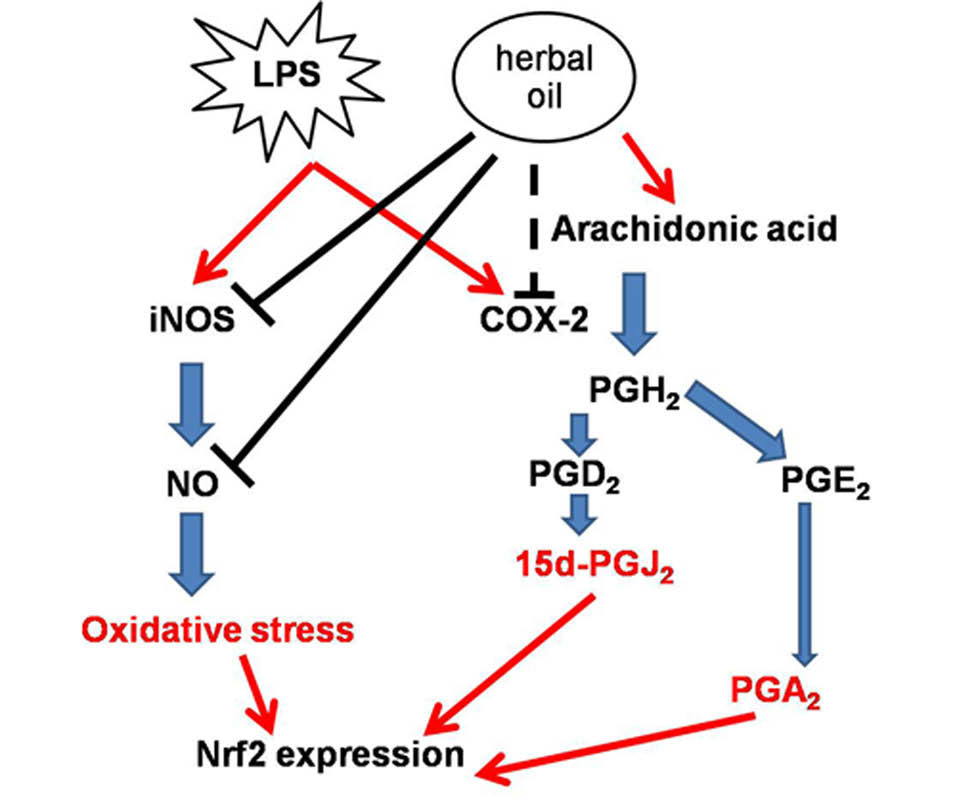
Fig. 8.
Postulated mechanism: LPS-induced inflammatory
responses by increasing expressions of iNOS and cox-2. iNOS
and cox-2 produced other inflammatory mediators which induce
Nrf2 expression. When macerated herbal oil and LPS were
presented together in co-treatment, linoleic acid in the macerated
herbal oil was used as a precursor of arachidonic acid which
might accelerate LPS- induced cox-2 expression. However, the
macerated herbal oil could inhibit iNOS expression together with
its NO scavenging activity leading to a reduction in the NO level,
which resulted in the down-regulation of the Nrf2 gene expression.
Nevertheless, the macerated herbal oil might be able to inhibit
LPS-induced cox-2 expression when it is applied before or after
the LPS. In the pre-treatment study, the HMSCs were exposed to
the macerated herbal oil only 2 h before replacing with LPS, so it
is possible that the oil might have employed its protective effects
on the cells before the addition of LPS, which led to the reduction
in the cox-2 gene expression. In the post-treatment study, after
the 2-h treatment with LPS, the HMSCs were exposed to the
macerated herbal oil for 24 h. When the macerated herbal oil was
added, it could express the anti-inflammatory effect by bringing
about the reduction in the cox-2 gene expression.
.
Postulated mechanism: LPS-induced inflammatory
responses by increasing expressions of iNOS and cox-2. iNOS
and cox-2 produced other inflammatory mediators which induce
Nrf2 expression. When macerated herbal oil and LPS were
presented together in co-treatment, linoleic acid in the macerated
herbal oil was used as a precursor of arachidonic acid which
might accelerate LPS- induced cox-2 expression. However, the
macerated herbal oil could inhibit iNOS expression together with
its NO scavenging activity leading to a reduction in the NO level,
which resulted in the down-regulation of the Nrf2 gene expression.
Nevertheless, the macerated herbal oil might be able to inhibit
LPS-induced cox-2 expression when it is applied before or after
the LPS. In the pre-treatment study, the HMSCs were exposed to
the macerated herbal oil only 2 h before replacing with LPS, so it
is possible that the oil might have employed its protective effects
on the cells before the addition of LPS, which led to the reduction
in the cox-2 gene expression. In the post-treatment study, after
the 2-h treatment with LPS, the HMSCs were exposed to the
macerated herbal oil for 24 h. When the macerated herbal oil was
added, it could express the anti-inflammatory effect by bringing
about the reduction in the cox-2 gene expression.
Discussion
The total phenolic contents and the antioxidant properties of macerated herbal oil were confirmed using different methods. The study was continued to find out how the addition of menthol, camphor, and borneol into this macerated herbal oil affected cells in the culture system. HMSCs were used because this macerated herbal oil was applied to relieve inflammation of mesenchymal tissues such as bones and muscles. Results from the MTT assay showed no cytotoxicity when the culture media contained oil concentration < 2.5 µL/mL (the result of Coomassie blue R staining was demonstrated in the supporting data). Nitrite assay was employed for the investigation of the anti-inflammatory effects; nitrite assay is a test based on the principle that inflammation raises the activity of the inducible nitric oxide synthase (iNOS). The increased nitric oxide synthase activity is evaluated by monitoring the change in the nitrite level in the cultured media. LPS (0.2 µg/mL) was used in this experiment to stimulate human mesenchymal stem cell inflammation. Obviously, the concentration of nitrite in the LPS-treated group was higher than that of the control group, but when the macerated herbal oil was presented with LPS (oil + LPS), it was found that the concentration of nitrite in the cultured medium was not different from that of the control group. The cells cultured with the macerated herbal oil alone (media + oil) showed no difference from the control group, thus indicating that the nitrite level was increased in the cultured media because of LPS and that it could be decreased by co-treatment with the macerated herbal oil. The study went on further to investigate gene expressions in the cultured cells. In the co-treatment (Fig. 5B), the cells cultured in the medium that contained both the macerated herbal oil and LPS expressed significantly higher levels of cox-2 gene when compared to control. However, there was no significant difference between the LPS-treated group and the oil + LPS treated group. The macerated herbal oil could not prevent the up-regulation of the cox-2 gene in the LPS-induced condition. Linoleic acid is an important ingredient in sesame oil, and it can be converted into arachidonic acid in mammalian cells.
40
COX-2 is an inducible enzyme abundant in the cells at the sites of inflammation, and it catalyzes the conversion of arachidonic acid to prostaglandin.
41
Thus, when the macerated herbal oil and LPS were presented together, the combination could not inhibit the cox-2 expression. The result indicates that the macerated herbal oil prevented the up-regulation of the Nrf2 gene in the LPS-induced condition. Recent research work has shown that Nrf2 has protective effect against oxidative stress. The study conducted by Itoh et al indicated that Nrf2 regulates the inflammation process downstream of 15d-PGJ2 by recruitment of inflammatory cells and by regulation of the gene expression within those cells.
41
Previous study by Wang et al showed that polyunsaturated fatty acids induce Nrf2 signaling pathway and that Nrf2 plays a role in the suppression of LPS-induced inflammation.
42
Thus it is possible that polyunsaturated fatty acids in the herbal oil could be able to do so. Pre- and post-treatment with the macerated herbal oil (0.2 µL/mL) could reduce the cox-2 gene expression when it was applied before or after the treatment of LPS. However, it was observed that the reduction in the cox-2 gene expression in the pre- and post-treatment groups could not reach the baseline because the up-regulation of the cox-2 gene in the pre- and post-treatment groups were significantly higher than that of the control group. Thus, evidently, post-treatment with the macerated herbal oil could not inhibit the LPS-induced Nrf2 gene expression. However, the Nrf2 gene expression in the case of pre-treatment with the macerated herbal oil could not be interpreted because the treatment with LPS alone did not show significant up-regulation of the Nrf2 gene (it could be noticed that the treatment with LPS alone significantly induced Nrf2 gene expression except that of the pre-treatment study). Thus, it could be postulated that the macerated herbal oil might play its anti-inflammatory role at the cox-2 or Nrf2 level, depending on the duration of the application. Furthermore, this study showed that the macerated herbal oil contained NO scavenging activity; NO is one of the mediators which regulates Nrf2 activity.
43
It could be postulated that the NO level decreased because of two reasons: Firstly, the macerated herbal oil attenuated iNOS activity and, secondly, the macerated herbal oil acted as the NO scavenger. These reasons explain the attenuation of the LPS-induced Nrf2 expression. The postulated mechanism has been presented in Fig. 8. No significant change in the NF-kB expression was found.
Various kinds of macerated herbal oil have been used in dental clinics with the aim of maintaining oral hygiene.
44
In the present study, the macerated herbal oil showed bactericidal activity but its efficiency as an antimicrobial agent is in need of improvement. The important components which might contribute to antimicrobial property and anti-inflammatory property were shown in the supporting data. Moreover, it was possible that the addition of menthol and borneol probably contributed to the antibacterial activity of the macerated herbal oil.
45
Because of the cytotoxic effect occurred in HMSCs when menthol, camphor, and borneol were added to the macerated herbal oil (Fig. 4A) so this macerated herbal oil should be applied for external use only. From this study, the use of macerated herbal oil could be applied as an alternative medical product.
Conclusion
The macerated herbal oil dilution 1:2 contained total phenolic concentration equivalent to gallic acid 1.94 mg/mL. The antioxidant activity of the macerated herbal oil was evaluated and confirmed with various antioxidant assays, namely, DPPH, ABTS, ferric reducing capacity, and reducing power. The MTT assay using HMSCs as the culture model pointed out that the cytotoxic concentration of the macerated herbal oil was ≥ 2.5 µL/mL. The anti-inflammatory effects were evaluated using nitrite assay and RT-PCR. It was found that the macerated herbal oil could inhibit nitrite accumulation in culture media. The results from RT-PCR demonstrated the inhibitory effect of the macerated herbal oil on cox-2 or Nrf2, the downstream modulator of the cox-2 pathway. Although the macerated herbal oil demonstrated bactericidal activity, the quality of the same needs to be improved.
Ethical approval
There is none to be declared.
Competing interests
No competing interests to be disclosed.
Acknowledgments
We are grateful to Prof. Kidoaki, Institute for Materials Chemistry and Engineering, Kyushu University, for HMSCs. We extend our gratitude to Miss Nawarat Viriyakhasem, Department of Biochemistry, Faculty of Medicine, Chiang Mai University, for her help regarding bacterial culture. We would also like to acknowledge the support from the Thailand Research Fund.
Research Highlights
What is current knowledge?
simple
-
√ This article proved that the macerated herbal oil contains
medicinal properties especially antioxidant and antiinflammatory
effects on HMSCs.
What is new here?
simple
-
√ The mechanism of anti-inflammatory effect of macerated
herbal oil on HMSCs was demonstrated.
-
√ The bactericidal activity of the macerated herbal oil was
observed in this study.
References
- Hsu DZ, Chen SJ, Chu PY, Liu MY. Therapeutic effects of sesame oil on monosodium urate crystal-induced acute inflammatory response in rats. Springerplus 2013; 2:659. doi: 10.1186/2193-1801-2-659 [Crossref] [ Google Scholar]
- Hsu DZ, Li YH, Chien SP, Liu MY. Effects of sesame oil on oxidative stress and hepatic injury after cecal ligation and puncture in rats. Shock 2004; 21:466-9. [ Google Scholar]
- Hsu DZ, Chien SP, Li YH, Chuang YC, Chang YC, Liu MY. Sesame oil attenuates hepatic lipid peroxidation by inhibiting nitric oxide and superoxide anion generation in septic rats. JPEN J Parenter Enteral Nutr 2008; 32:154-9. doi: 10.1177/0148607108314766 [Crossref] [ Google Scholar]
- Periasamy S, Liu CT, Hsu DZ, Liu MY. Sesame oil accelerates kidney healing following gentamicin-induced kidney injury in rats. Am J Nephrol 2010; 32:383-92. doi: 10.1159/000319853 [Crossref] [ Google Scholar]
- Periasamy S, Hsu DZ, Chandrasekaran VR, Liu MY. Sesame oil accelerates healing of 2,4,6-trinitrobenzenesulfonic acid-induced acute colitis by attenuating inflammation and fibrosis. JPEN J Parenter Enteral Nutr 2013; 37:674-82. doi: 10.1177/0148607112468768 [Crossref] [ Google Scholar]
- Liu CT, Periasamy S, Chang CC, Mo FE, Liu MY. Sesame oil therapeutically ameliorates cardiac hypertrophy by regulating hypokalemia in hypertensive rats. JPEN J Parenter Enteral Nutr 2014; 38:750-7. doi: 10.1177/0148607113491781 [Crossref] [ Google Scholar]
- Hsu DZ, Liu CT, Chu PY, Li YH, Periasamy S, Liu MY. Sesame oil attenuates ovalbumin-induced pulmonary edema and bronchial neutrophilic inflammation in mice. Biomed Res Int 2013; 2013:905670. doi: 10.1155/2013/905670 [Crossref] [ Google Scholar]
- El Wakf AM, Hassan HA, Gharib NS. Osteoprotective effect of soybean and sesame oils in ovariectomized rats via estrogen-like mechanism. Cytotechnology 2014; 66:335-43. doi: 10.1007/s10616-013-9580-4 [Crossref] [ Google Scholar]
- Jacklin A, Ratledge C, Welham K, Bilko D, Newton CJ. The sesame seed oil constituent, sesamol, induces growth arrest and apoptosis of cancer and cardiovascular cells. Ann N Y Acad Sci 2003; 1010:374-80. doi: 10.1196/annals.1299.068 [Crossref] [ Google Scholar]
- Alencar J, Gosset G, Robin M, Pique V, Culcasi M, Clément JL. Improving the stability and antioxidant properties of sesame oil: water-soluble spray-dried emulsions from new transesterified phenolic derivatives. J Agric Food Chem 2009; 57:7311-23. doi: 10.1021/jf9010627 [Crossref] [ Google Scholar]
- Fujimoto A, Shingai Y, Oyama TB, Kawanai T, Hashimoto E, Koizumi K. Apoptosis-inducing action of two products from oxidation of sesamol, an antioxidative constituent of sesame oil: a possible cytotoxicity of oxidized antioxidant. Toxicol In Vitro 2010; 24:1720-6. doi: 10.1016/j.tiv.2010.05.013 [Crossref] [ Google Scholar]
- Masuda T, Shingai Y, Fujimoto A, Nakamura M, Oyama Y, Maekawa T. Identification of cytotoxic dimers in oxidation product from sesamol, a potent antioxidant of sesame oil. J Agric Food Chem 2010; 58:10880-5. doi: 10.1021/jf103015j [Crossref] [ Google Scholar]
- Martinchik AN. [Nutritional value of sesame seeds]. Vopr Pitan 2011; 80:41-3. [ Google Scholar]
- Khaw-on P, Banjerdpongchai R. Induction of intrinsic and extrinsic apoptosis pathways in the human leukemic MOLT-4 cell line by terpinen-4-ol. Asian Pac J Cancer Prev 2012; 13:3073-6. doi: 10.7314/APJCP.2012.13.7.3073 [Crossref] [ Google Scholar]
- Pongprayoon U, Soontornsaratune P, Jarikasem S, Sematong T, Wasuwat S, Claeson P. Topical antiinflammatory activity of the major lipophilic constituents of the rhizome of Zingiber cassumunar Part I: The essential oil. Phytomedicine 1997; 3:319-22. doi: 10.1016/S0944-7113(97)80003-7 [Crossref] [ Google Scholar]
- Rana VS, Verdeguer M, Blazquez MA. Chemical composition of the essential oil of Zingiber zerumbet var darcyi. Nat Prod Commun 2012; 7:1369-70. [ Google Scholar]
- Jeenapongsa R, Yoovathaworn K, Sriwatanakul KM, Pongprayoon U, Sriwatanakul K. Anti-inflammatory activity of (E)-1-(3,4-dimethoxyphenyl) butadiene from Zingiber cassumunar Roxb. J Ethnopharmacol 2003; 87:143-8. doi: 10.1016/S0378-8741(03)00098-9 [Crossref] [ Google Scholar]
- Kaewchoothong A, Tewtrakul S, Panichayupakaranant P. Inhibitory effect of phenylbutanoid-rich Zingiber cassumunar extracts on nitric oxide production by murine macrophage-like RAW2647 cells. Phytother Res 2012; 26:1789-92. doi: 10.1002/ptr.4661 [Crossref] [ Google Scholar]
- Tangyuenyongwatana P, Kowapradit J, Opanasopit P, Gritsanapan W. Cellular transport of anti-inflammatory pro-drugs originated from a herbal formulation of Zingiber cassumunar and Nigella sativa. Chin Med 2009; 4:19. doi: 10.1186/1749-8546-4-19 [Crossref] [ Google Scholar]
- Nakamura S, Iwami J, Matsuda H, Wakayama H, Pongpiriyadacha Y, Yoshikawa M. Structures of new phenylbutanoids and nitric oxide production inhibitors from the rhizomes of Zingiber cassumunar. Chem Pharm Bull (Tokyo) 2009; 57:1267-72. doi: 10.1248/cpb.57.1267 [Crossref] [ Google Scholar]
- Matsuda H, Nakamura S, Iwami J, Li X, Pongpiriyadacha Y, Nakai M. Invasion inhibitors of human fibrosarcoma HT 1080 cells from the rhizomes of Zingiber cassumunar: structures of phenylbutanoids, cassumunols. Chem Pharm Bull (Tokyo) 2011; 59:365-70. doi: 10.1248/cpb.59.365 [Crossref] [ Google Scholar]
- Huang GC, Chien TY, Chen LG, Wang CC. Antitumor effects of zerumbone from Zingiber zerumbet in P-388D1 cells in vitro and in vivo. Planta Med 2005; 71:219-24. doi: 10.1055/s-2005-837820 [Crossref] [ Google Scholar]
- Khalid MH, Akhtar MN, Mohamad AS, Perimal EK, Akira A, Israf DA. Antinociceptive effect of the essential oil of Zingiber zerumbet in mice: possible mechanisms. J Ethnopharmacol 2011; 137:345-51. doi: 10.1016/j.jep.2011.05.043 [Crossref] [ Google Scholar]
- Zakaria ZA, Mohamad AS, Ahmad MS, Mokhtar AF, Israf DA, Lajis NH. Preliminary analysis of the anti-inflammatory activity of essential oils of Zingiber zerumbet. Biol Res Nurs 2011; 13:425-32. doi: 10.1177/1099800410386590 [Crossref] [ Google Scholar]
- Kader G, Nikkon F, Rashid MA, Yeasmin T. Antimicrobial activities of the rhizome extract of Zingiber zerumbet Linn. Asian Pac J Trop Biomed 2011; 1:409-12. doi: 10.1016/S2221-1691(11)60090-7 [Crossref] [ Google Scholar]
- Chang CJ, Tzeng TF, Liou SS, Chang YS, Liu IM. Absence of Genotoxic and Mutagenic Effects of Zingiber zerumbet (L) Smith (Zingiberaceae) Extract. Evid Based Complement Alternat Med 2012; 2012:406296. doi: 10.1155/2012/406296 [Crossref] [ Google Scholar]
- Chiang LC, Chiang W, Chang MY, Lin CC. In vitro cytotoxic, antiviral and immunomodulatory effects of Plantago major and Plantago asiatica. Am J Chin Med 2003; 31:225-34. doi: 10.1142/S0192415X03000874 [Crossref] [ Google Scholar]
- Thomé RG, dos Santos HB, dos Santos FV, da Silva Oliveira RJ, de Camargos LF, Pereira MN. Evaluation of healing wound and genotoxicity potentials from extracts hydroalcoholic of Plantago major and Siparuna guianensis. Exp Biol Med (Maywood) 2012; 237:1379-86. doi: 10.1258/ebm.2012.012139 [Crossref] [ Google Scholar]
- Mello JC, Guimarães NS, Gonzalez MV, Paiva JS, Prieto T, Nascimento OR. Hydroxyl scavenging activity accounts for differential antioxidant protection of Plantago major against oxidative toxicity in isolated rat liver mitochondria. J Pharm Pharmacol 2012; 64:1177-87. doi: 10.1111/j.2042-7158.2012.01496.x [Crossref] [ Google Scholar]
- Choi ES, Cho SD, Shin JA, Kwon KH, Cho NP, Shim JH. Althaea rosea Cavanil and Plantago major L suppress neoplastic cell transformation through the inhibition of epidermal growth factor receptor kinase. Mol Med Rep 2012; 6:843-7. doi: 10.3892/mmr.2012.977 [Crossref] [ Google Scholar]
- Saeedi M, Morteza-Semnani K, Sagheb-Doust M. Evaluation of Plantago major L seed mucilage as a rate controlling matrix for sustained release of propranolol hydrochloride. Acta Pharm 2013; 63:99-114. doi: 10.2478/acph-2013-0010 [Crossref] [ Google Scholar]
-
Chanthaphon S, Chanthachum S, Hongpattarakere T. Antimicrobial activities of essential oils and crude extracts from tropical Citrus spp. against food-related microorganisms. Songklanakarin J Sci Technol 2008; 30 (Suppl.1): 125-31.
- Chowdhury A. Antimicrobial, Antioxidant and Cytotoxic Activities of Citrus Hystrix DC. Fruits J Pharm Sci 2009; 8:177-80. doi: 10.3329/dujps.v8i2.6035 [Crossref] [ Google Scholar]
- Chan SW, Lee CY, Yap CF, Wan Aida WM, Wan Aida WM. Optimisation of extraction conditions for phenolic compounds from limau purut (Citrus hystrix) peels. International Food Research Journal 2009; 16:203-13. [ Google Scholar]
- Srisukh V, Tribuddharat C, Nukoolkarn V, Bunyapraphatsara N, Chokephaibulkit K, Phoomniyom S. Antibacterial activity of essential oils from Citrus hystrix (makrut lime) against respiratory tract pathogens. ScienceAsia 2012; 38:212-7. doi: 10.2306/scienceasia1513-1874.2012.38.212 [Crossref] [ Google Scholar]
- Srisook K, Salee P, Charoensuk Y, Srisook E. In vitro anti-oxidant and anti-tyrosinase activities of the rhizomal extractsfrom Amomum biflorum Jack. Thai Journal Of Botany 2010; 2:143-50. [ Google Scholar]
- Lee MW, Kim DS, Yoo KH, Kim HR, Jang IK, Lee JH. Human bone marrow-derived mesenchymal stem cell gene expression patterns vary with culture conditions. Blood Res 2013; 48:107-14. doi: 10.5045/br.2013.48.2.107 [Crossref] [ Google Scholar]
- Zhang R, Liu Y, Yan K, Chen L, Chen XR, Li P. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J Neuroinflammation 2013; 10:106. doi: 10.1186/1742-2094-10-106 [Crossref] [ Google Scholar]
- Eskandani M, Dadizadeh E, Hamishehkar H, Nazemiyeh H, Barar J. Geno/cytotoxicty and Apoptotic Properties of Phenolic Compounds from the Seeds of Dorema Glabrum Fisch CA. Bioimpacts 2014; 4:191-8. doi: 10.15171/bi.2014.019 [Crossref] [ Google Scholar]
- Salem N, Wegher B, Mena P, Uauy R. Arachidonic and docosahexaenoic acids are biosynthesized from their 18-carbon precursors in human infants. Proc Natl Acad Sci U S A 1996; 93:49-54. doi: 10.1073/pnas.93.1.49 [Crossref] [ Google Scholar]
- Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2). Mol Cell Biol 2004; 24:36-45. doi: 10.1128/MCB.24.1.36-45.2004 [Crossref] [ Google Scholar]
- Wang H, Khor TO, Saw CL, Lin W, Wu T, Huang Y. Role of Nrf2 in suppressing LPS-induced inflammation in mouse peritoneal macrophages by polyunsaturated fatty acids docosahexaenoic acid and eicosapentaenoic acid. Mol Pharm 2010; 7:2185-93. doi: 10.1021/mp100199m [Crossref] [ Google Scholar]
- Kobayashi E, Suzuki T, Yamamoto M. Roles nrf2 plays in myeloid cells and related disorders. Oxid Med Cell Longev 2013; 2013:529219. doi: 10.1155/2013/529219 [Crossref] [ Google Scholar]
- Gupta C, Kumari A, Garg AP, Catanzaro R, Marotta F. Comparative study of cinnamon oil and clove oil on some oral microbiota. Acta Biomed 2011; 82:197-9. [ Google Scholar]
- Kotan R, Kordali S, Cakir A. Screening of antibacterial activities of twenty-one oxygenated monoterpenes. Z Naturforsch C 2007; 62:507-13. doi: 10.1515/znc-2007-7-808 [Crossref] [ Google Scholar]