BioImpacts. 10(2):123-135.
doi: 10.34172/bi.2020.15
Original Research
Proteomic profiling of Serratia marcescens by high-resolution mass spectrometry
Bhavya Somalapura Gangadharappa 1, 2, Sharath Rajashekarappa 3, *  , Gajanan Sathe 4, 5
, Gajanan Sathe 4, 5
Author information:
1Department of Biotechnology, M.S. Ramaiah Institute of Technology, Bengaluru-560054, Karnataka, India
2Visvesvaraya Technological University, Belagavi-590018, Karnataka, India
3Department of Food Technology, Davangere University, Davangere-577007, Karnataka, India
4Institute of Bioinformatics, International Technology Park, Bangalore-560066, Karnataka, India
5Center for Molecular Medicine, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore-560029, Karnataka, India
Abstract
Introduction: Serratia marcescens,
an opportunistic human pathogen, is reported as an important cause of nosocomial infection and outbreaks. Although the genome of S. marcescens (ATCC 13880) was completely sequenced by 2014, there are no studies on the proteomic profile of the organism. The objective of the present study is to analyze the protein profile of S. marcescens (ATCC 13880) using a high resolution mass spectrometry (MS).
Methods: Serratia marcescens
ATCC 13880 strain was grown in Luria-Bertani broth and the protein extracted was subjected to trypsin digestion, followed by basic reverse phase liquid chromatography fractionation. The peptide fractions were then analysed using Orbitrap Fusion Mass Spectrometry and the raw MS data were processed in Proteome Discoverer software.
Results:
The proteomic analysis identified 15 009 unique peptides mapping to 2541 unique protein groups, which corresponds to approximately 54% of the computationally predicted protein-coding genes. Bioinformatic analysis of these identified proteins showed their involvement in biological processes such as cell wall organization, chaperone-mediated protein folding and ATP binding. Pathway analysis revealed that some of these proteins are associated with bacterial chemotaxis and beta-lactam resistance pathway.
Conclusion:
To the best of our knowledge, this is the first high-throughput proteomics study of S. marcescens (ATCC 13880). These novel observations provide a crucial baseline molecular profile of the S. marcescens proteome which will prove to be helpful for the future research in understanding the host-pathogen interactions during infection, elucidating the mechanism of multidrug resistance, and developing novel diagnostic markers or vaccine for the disease.
Keywords: Proteomics,
Serratia marcescens
, Proteome discoverer, Orbitrap fusion
Copyright and License Information
© 2020 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Serratia marcescens is a gram-negative bacillus, belonging to the family Enterobacteriaceae. This opportunistic human pathogen may cause various infections ranging from urinary and respiratory tract infection, wound infection, endocarditis, meningitis, to conjunctivitis pneumonia, and may even lead to septicemia.
1,2
This pathogen is naturally present in soil and water, and accounts for about 1%-2% of the nosocomial infections and outbreaks occurring in intensive care units (ICUs), surgery units, medical wards, and neonatal ICUs.
3
A recent study reported that S. marcescens is the third most common pathogen involved in neonatal ICU outbreaks.
4
The overall rates of mortality due to S. marcescens vary from 25%-58%.
5,6
The drug resistance of S. marcescens to a broad spectrum of antibiotics complicates treatment and infection control in hospitals.
7
Recently, the World Health Organization (WHO) listed S. marcescens as one among the top ten global priority pathogens with multiple drug-resistance.
8,9
The complete genome sequence of S. marcescens strain ATCC 13880 was sequenced in 2014 and the total genome assembly constitutes 5 131 448 bp long including 4724 protein coding sequences.
10
Advances in high throughput technologies, such as proteomics, is emerging as an alternative strategy to address the multidrug resistance by understanding the mechanisms involved in drug resistance.
11,12
Proteomics can offer multiple advantages by the identification and characterization of novel proteins which are directly or indirectly involved in drug resistance that could be used as potential targets in future.
13-15
In the last two decades, mass spectrometry (MS)-based proteomics has been effectively used to study proteome of microorganism, protein-protein interactions, differentially expressed proteins in order to better understand the molecular mechanism of microbial pathogenesis,
16
and antibiotic resistance mechanisms.
17
Compared to other important gram-negative bacteria, there are limited proteomic studies on S. marcescens.
18-22
Despite the previous efforts in S. marcescens proteome characterization, there are no studies involving a detailed proteomic profiling of S. marcescens ATCC 13880 strain.
In the present study, we have carried out a detailed proteome analysis of S. marcescens using high-resolution MS to provide the baseline data for future research. The study resulted in the identification of 2541 proteins, most of which were found to be associated with cell wall organization, chaperone-mediated protein folding, solute transport, signal transduction, ATP binding, metal ion binding, and protein transmembrane transporter activity. Proteins involved in important pathways including chemotaxis pathway, beta lactam resistance pathway, and bacterial secretion system pathway were also identified in the analysis. The information derived from this study could serve as a platform for future proteomic studies on the S. marcescens ATCC 13880.
Materials and Methods
Materials
Sodium lauryl sulfate (SDS), dithiothreitol (DTT), iodoacetamide (IAA), formic acid, acetonitrile (ACN), triethylammonium bicarbonate (TEABC) were supplied. All chemicals used were of analytical grade and were obtained from Sigma-Aldrich (St. Louis, MO, USA). Luria-Bertani (LB) (HiMedia Laboratories Pvt. Ltd, Mumbai, India), Sequencing grade modified trypsin (Promega, Madison, WI, USA), Bicinchoninic acid assay (BCA) (Thermo Fisher Scientific, Rockford, IL, USA). J. T. Baker MS grade water (Fischer Scientific, Waltham, MA, USA), 30 kDa Molecular weight cutoff (MWCO) spin filters (EMD Millipore, Billerica, MA, USA), Sep-Pak C18 columns (Waters Corporation, Milford, MA, USA), XBridge C18, 5 µm 250 x 4.6 mm column (Waters, Milford, MA, USA), Trap column (75 µm x 2 cm) packed in-house with Magic C18 AQ (Michrom Bioresources, Inc., Auburn, CA, USA) were also purchased.
Equipment
New Brunswick Scientific Incubator Shaker (IL Exella E24) (Edison, NJ, USA), Branson 450 Digital Sonifier w/ Probe (Marshall Scientific, Hampton, NH, USA), Speed VacTM SPD1010 system (Thermo Fisher Scientific, Bremen, Germany), Agilent 1100 series HPLC system (Agilent, Santa Clara, CA, USA), Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany), Eppendorf Centrifuge 5424 R (Eppendorf, Hamburg, Germany), Eppendorf ThermoMixer C (Eppendorf, Hamburg, Germany) were applied.
Preparation of Serratia marcescens culture
Serratia marcescens strain was purchased from American Type Culture Collection (ATCC number 13880) and was grown on LB agar plates (37°C overnight). The LB liquid medium was prepared in 500 mL Erlenmeyer flask. Then, colonies from the LB plate were picked and inoculated in 500 mL Erlenmeyer flask and incubated for 18-24 hours at 37°C with shaking (300 RPM). The final concentration of the bacterial suspension was adjusted to 0.5 McFarland standards (~1.5 x 108 CFU/mL at 600 nm wavelength) and then bacterial suspension was subjected to centrifugation for a period of 10 minutes at 1500 x g at 4°C. Further, these cell pellets were collected and washed four times with ice-cold PBS and stored at −80°C.
Sample preparation and fractionation
The cell pellets with buffer containing 4% SDS were subjected to five cycles of sonication for 30 seconds and 20% amplitude with an interval of 5 seconds each in Branson 450 Digital Sonifier (Marshall Scientific, Hampton, NH, USA). The lysates were centrifuged at 14 000 RPM for 10 minutes and the clear supernatant was collected and aliquoted. One aliquot was used for protein estimation using BCA assay and the other aliquot was stored at –80°C till further analysis. In-solution digestion and fractionation was performed as described previously.
23,24
In brief, 500 µg of protein was subjected to reduction using 100 mM DTT and alkylation using 10 mM IAA, followed by buffer exchange with 8 M urea to reduce the SDS concentration, using a 30 kDa MWCO spin filters (EMD Millipore, Billerica, MA). The buffer exchanged sample was diluted to bring down the concentration of urea to 2 M and proteins were then digested using sequencing grade modified trypsin (Promega, Madison, WI. Cat #: V5111) overnight at 37°C.
The peptide digest was cleaned using Sep-Pak C18 columns using two solvents; solvent A - 0.1% formic acid and solvent B - 40% ACN, 0.1% formic acid. Briefly, the Sep-Pack C18 column was activated by passing 5 mL of 100% ACN, and then equilibrated the column twice with 5 mL of 0.1% formic acid. The sample was reconstituted in 5 mL of 0.1% formic acid and loaded onto the Sep-Pack C18 column. The first flow through was collected and re-loaded onto the column and then, the column was washed twice with 5 mL of 0.1% formic acid. Peptides were eluted by passing 3 mL of 40% ACN, 0.1% formic acid and the eluate was collected in fresh Falcon 15 mL tube (This step was repeated). The eluate containing cleaned peptides collected from Sep-Pak C18 columns was vacuum-dried using SAVANT SPD 1010 Speed Vac Concentrator as reported previously.
25
The vacuum-dried samples were reconstituted in basic reverse phase liquid chromatography (bRPLC) solvent A containing 10 mM TEABC in water at pH 8.5 and loaded on XBridge C18, 5 µm 250 x 4.6 mm column connected to Agilent 1100 series HPLC system. The peptide digest was resolved using a gradient of 0 to 40% solvent B containing 10 mM TEABC in ACN at pH 8.5 in 50 minutes. The total fractionation time was 60 minutes. A total of 96 fractions were collected and concatenated to 6 fractions based on the hydrophobicity as reported earlier.
26
These fractions were then vacuum dried and stored at -20°C until further LC-MS/MS analysis was performed. All buffers and reagents used in the study are provided in Supplementary file 1.
LC-MS/MS analysis
MS analysis was performed using the Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) coupled to an Easy-nLC-1200 nanoflow LC system (Thermo Scientific). The peptide digests from each fraction were reconstituted in 20 µL of solvent A (0.1% formic acid in water) and around 10 µL of this sample having 2 µg of peptide was loaded onto trap column (75 µm x 2 cm) packed in-house with Magic C18 AQ (Michrom Bioresources, Inc., Auburn, CA, USA, 5 µm particle size, pore size 100 Å) at a flow rate of 5 µL/min with solvent A (0.1% formic acid in water). The separation of peptides was performed on an analytical column (75 µm x 15 cm) using a gradient of solvent A (0.1% formic acid) and solvent B (90% ACN with 0.1% formic acid) over 90 minutes at a flow rate of 300 nL/min. Six LC-MS/MS runs were performed. Data-dependent MS acquisition in positive mode was achieved at a resolution of 120 000 for the top ten precursor ions with a scan range of 350–1600 m/z. For MS/MS, top intense precursor ions from a 3-second duty cycle were preferred and subjected to higher-energy collision dissociation with 32% normalized collision energy. The fragment ions were detected at a mass resolution of 15 000 at m/z of 200 as explained previously.
23,27
Data analysis
The MS/MS searches were performed using SEQUEST (version 2.4.1) search algorithm through Proteome Discoverer (version 2.2) software (Thermo Scientific) against S. marcescens (ATCC 13880) NCBI protein list (https://www.ncbi.nlm.nih.gov/). The search database was created by downloading all the proteins using “Serratia marcescens subsp. marcescens ATCC 13880” as search key words from NCBI. The search workflow included SEQUEST (version 2.4.1), spectrum selector, search nodes, spectrum files, Sequest HT, and Percolator. Carbamidomethylation of cysteine was set as a fixed modification, acetylation of protein N-termini, and oxidation of methionine, and deamidation of asparagine and glutamine was set as a variable modification. MS and MS/MS mass tolerances were set to 10 ppm and 0.02 Da, respectively. The data was searched against the decoy database, and 1% false discovery rate (FDR) at both peptides and peptide spectral match levels was applied for the analysis.
28
The Bioinformatics analysis of the proteomic data obtained in this study was performed using PANTHER (http://www.pantherdb.org/),
29
DAVID (V. 6.8),
30,31
STRING (V. 11.0) (https://string-db.org/),
32
and KEGG Pathway (http://www. genome.ad.jp/kegg/).
33
Data availability
Raw MS files obtained in this experiment have been uploaded to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org),
34
through the PRIDE partner repository with the dataset identifier PXD011776.
Results
Proteomic analysis of S. marcescens (ATCC 13880)
The objective of the present study was to perform proteomic profiling to make a comprehensive proteome map of S. marcescens (ATCC 13880). Overview of the proteomic workflow to study the proteome of S. marcescens is shown in Fig. 1. Organism grown in LB medium post sonication extraction yielded ~8 mg/mL protein. Around 500 µg of the protein was subjected to in-solution digestion. Digested peptides were fractionated using bRPLC and these fractionated peptides were concatenated to 6 fractions and analyzed on LC-MS/MS. The analysis of these fractions resulted in the acquisition of 130448 MS/MS spectra, and the database searches against S. marcescens (ATCC 13880) NCBI protein database using SEQUEST resulted in the identification of 16 306 peptide-spectrum matches. A total of 15 009 unique peptides were identified (1% FDR), which mapped to 2541 protein groups accounting for 54% of the S. marcescens (ATCC 13880) proteome. The identified proteins and peptides are given in Tables S1 and S2 (see Supplementary file 2 and 3). The majority (78%) of the reported proteins were identified based on two or more unique peptides. On average, 6 unique peptides were found for each of the identified proteins. Representative mass spectra are shown in Supplementary file 4.
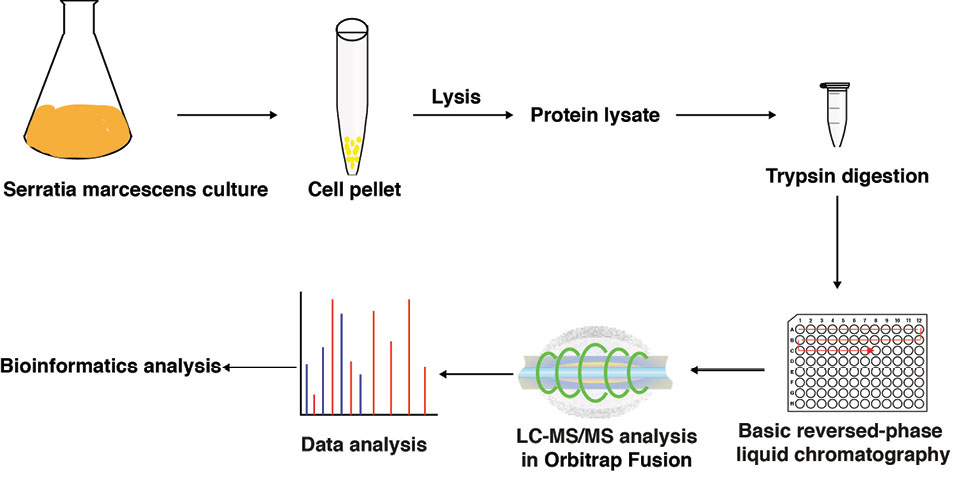
Fig. 1.
Overview of the proteomic workflow applied to study the proteome of S. marcescens: S. marcescens ATCC 13880 was cultured, protein was isolated from cell pellets and digested using trypsin enzyme. The digest was fractionated using bRPLC, acquired data on mass spectrometry and analyzed the data using Bioinformatics approach.
.
Overview of the proteomic workflow applied to study the proteome of S. marcescens: S. marcescens ATCC 13880 was cultured, protein was isolated from cell pellets and digested using trypsin enzyme. The digest was fractionated using bRPLC, acquired data on mass spectrometry and analyzed the data using Bioinformatics approach.
Gene Ontology analysis
The selected list of proteins was analysed for their gene ontology (GO) using PANTHER classification system. The identified proteins were classified as those involved in biological processes, molecular functions, and according to their cellular localization (Fig. 2 and Table 1). With reference to the biological process, majority of the proteins (31%) were involved in binding, 24% in catalytic activity, 16% molecular transduction activity, another 16% in transcription regulator activity, and 13% in transporter activity.
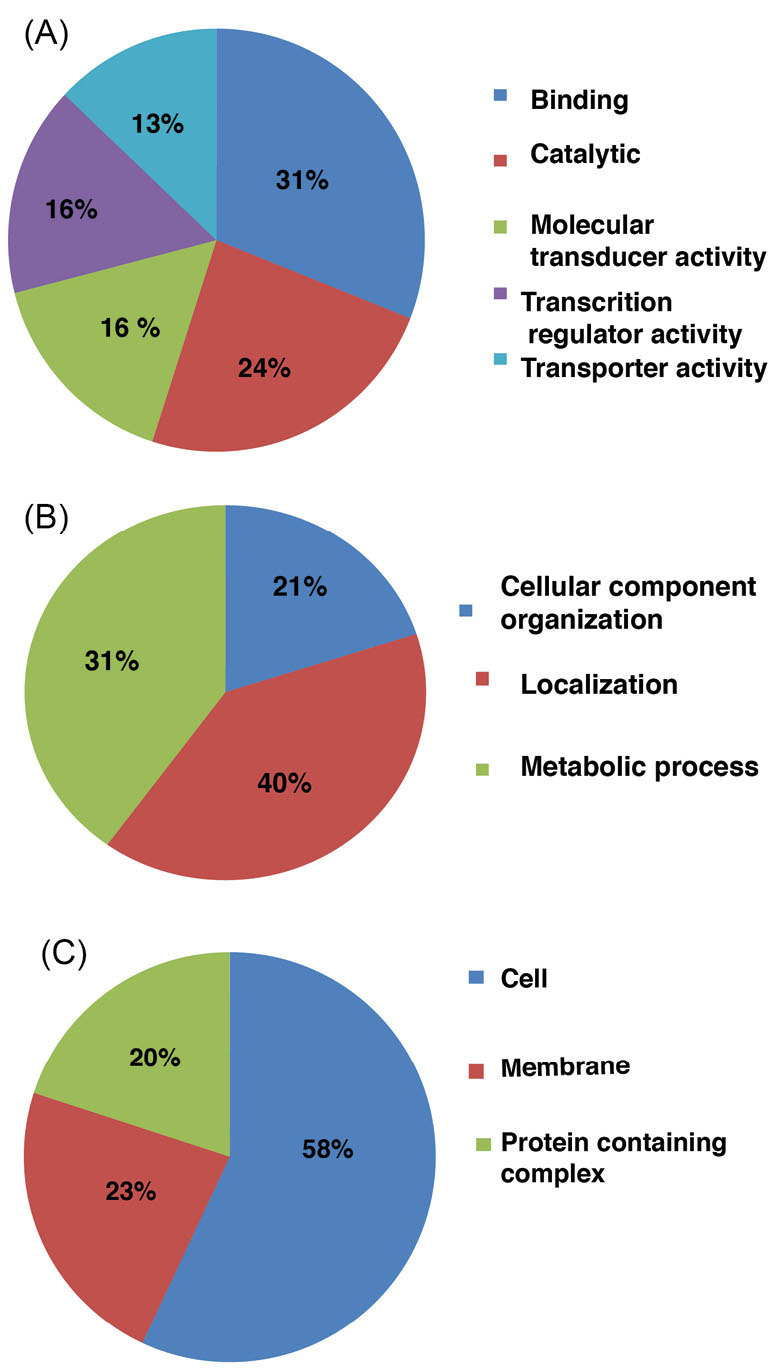
Fig. 2.
Gene Ontology analysis: A histogram depicting the distribution of Serratia marcescens proteins among various (A) biological processes and (B) molecular functions and (C) subcellular localizations.
.
Gene Ontology analysis: A histogram depicting the distribution of Serratia marcescens proteins among various (A) biological processes and (B) molecular functions and (C) subcellular localizations.
Table 1.
Functional analysis of the proteins identified in the study
|
Description
|
Molecular function
|
Biological processes
|
Cellular localization
|
Domain
|
|
Molecular chaperone proteins identified
|
| Hsp33 family molecular chaperone HslO |
Unfolded protein binding |
Protein folding |
Cytoplasm |
Redox-active center |
| Chaperone protein papD |
Chaperone |
Cell wall organization, chaperone-mediated protein folding |
Outer membrane-bounded periplasmic space |
Signal |
| Periplasmic fimbrial chaperone focC |
Chaperone |
Cell wall organization, chaperone-mediated protein folding |
Outer membrane-bounded periplasmic space |
Signal |
| Chaperone protein FimB |
Chaperone |
Cell wall organization, chaperone-mediated protein folding |
Outer membrane-bounded periplasmic space |
Signal |
| Chaperone protein YcdY |
Chaperone |
Bacterial-type flagellum-dependent swarming motility |
Cytosol |
Coiled coil |
| Chaperone protein fimC |
Chaperone |
Cell wall organization, chaperone-mediated protein folding |
Outer membrane-bounded periplasmic space |
Signal |
| Molecular chaperone DnaJ |
Unfolded protein binding |
Protein folding, response to heat |
Cytoplasm |
Repeat, Zinc-finger |
| Molecular chaperone DnaK |
Unfolded protein binding |
Protein folding, response to heat |
Cytoplasm |
Repeat, Zinc-finger |
| Molecular chaperone HtpG |
Unfolded protein binding |
Protein folding |
Cytoplasm |
Unknown |
| Molecular chaperone OsmY |
Unknown |
Unknown |
Unknown |
Unknown |
| Molecular chaperone Skp |
Unfolded protein binding |
Signaling |
Outer membrane-bounded periplasmic space |
Coiled coil |
| Chaperonin GroEL |
Unfolded protein binding |
Protein refolding |
Cytoplasm |
Coiled coil |
| ATP-dependent chaperone ClpB |
Unfolded protein binding |
Protein folding |
Cytoplasm |
Coiled coil |
| Molecular chaperone HtpG |
Unfolded protein binding |
Protein folding |
Cytoplasm |
Coiled coil |
| Co-chaperone YbbN |
Unknown |
Cell redox homeostasis |
Cell |
Thioredoxin |
| Fe-S protein assembly chaperone HscA |
Unknown |
Unknown |
Unknown |
Unknown |
| Heat shock chaperone IbpB |
Chaperone |
Protein stabilization |
Cytoplasm |
Unknown |
| Co-chaperone GroES |
Unknown |
Unknown |
Unknown |
Unknown |
| Co-chaperone DjlA |
Chaperone binding |
Signaling |
Integral component of membrane |
Transmembrane |
| Co-chaperone HscB |
ATPase activator activity |
Protein complex oligomerization, protein folding |
Unknown |
Coiled coil |
| Chaperone-modulator protein CbpM |
Unknown |
Unknown |
Unknown |
Coiled coil |
|
Outer membrane proteins
|
| Outer membrane protein A |
Porin activity |
Conjugation, ion transport |
Cell outer membrane |
Signal |
| Outer membrane assembly protein AsmA |
Unknown |
Regulation of protein targeting to membrane |
Periplasmic space |
Signal |
| Outer membrane autotransporter barrel domain-containing protein |
Serine-type endopeptidase activity |
Unknown |
Outer membrane |
Coiled coil |
| Outer membrane channel protein TolC |
Efflux transmembrane transporter activity |
Unknown |
Outer membrane |
Signal |
| Outer membrane lipid asymmetry maintenance protein MlaD |
Unknown |
Phospholipid transport |
Integral component of membrane |
Transmembrane, |
| Outer membrane lipoprotein chaperone LolA |
Lipoprotein transporter activity |
Protein transport |
Periplasmic space |
Signal |
| Outer membrane protein assembly factor |
Unknown |
Gram-negative-bacterium-type cell outer membrane assembly |
Cell outer membrane |
Signal |
| Outer membrane protein assembly factor BamA |
Unknown |
Gram-negative-bacterium-type cell outer membrane assembly |
Cell outer membrane |
Repeat |
| Outer membrane protein assembly factor BamB |
Unknown |
Gram-negative-bacterium-type cell outer membrane assembly |
Cell outer membrane |
Signal |
| Outer membrane protein assembly factor BamC |
Unknown |
Gram-negative-bacterium-type cell outer membrane assembly |
Cell outer membrane |
Signal |
| Outer membrane protein assembly factor BamD |
Unknown |
Gram-negative-bacterium-type cell outer membrane assembly |
Cell outer membrane |
Signal |
| Outer membrane protein assembly factor BamE |
Unknown |
Gram-negative-bacterium-type cell outer membrane assembly |
Cell outer membrane |
Signal |
| Outer membrane protein OmpW |
Unknown |
Transmembrane transport |
Cell outer membrane |
Signal |
| Outer membrane protein OmpX |
Identical protein binding |
Protein insertion into membrane from inner side |
Outer membrane-bounded periplasmic space |
Signal |
| Outer membrane usher protein |
Fimbrial usher porin activity |
Pilus assembly |
Cell outer membrane |
Transmembrane |
|
Two component systems proteins
|
| Two-component sensor histidine kinase BarA |
Phosphorelay sensor kinase activity |
Unknown |
Integral component of membrane |
Transmembrane |
| Two-component system QseEF-associated lipoprotein QseG |
Unknown |
Unknown |
Unknown |
Coiled coil |
| Two-component system response regulator ArcA |
DNA binding |
Phosphorelay signal transduction system |
Unknown |
Unknown |
| Two-component system response regulator BaeR |
DNA-binding transcription factor activity |
Regulation of response to drug |
Cytosol |
Coiled coil |
| Two-component system response regulator BasR |
DNA binding |
Phosphorelay signal transduction system |
Unknown |
Unknown |
| Two-component system response regulator CreB |
DNA binding |
Phosphorelay signal transduction system |
Unknown |
Unknown |
| Two-component system response regulator DcuR |
ATP binding |
Regulation of response to drug |
Integral component of membrane |
Transmembrane |
| Two-component system response regulator GlrR |
DNA binding |
Phosphorelay signal transduction system |
Unknown |
Unknown |
| Two-component system response regulator NarL |
DNA binding |
Phosphorelay signal transduction system |
Unknown |
Unknown |
| Two-component system response regulator OmpR |
DNA-binding transcription factor activity |
Phosphorelay signal transduction system |
Cytosol |
Unknown |
| Two-component system response regulator PhoP |
DNA-binding transcription factor activity |
Phosphorelay signal transduction system |
Cytosol |
Unknown |
| Two-component system response regulator RstA |
DNA-binding transcription factor activity |
Phosphorelay signal transduction system |
Cytosol |
Unknown |
| Two-component system response regulator UvrY |
DNA binding |
Phosphorelay signal transduction system |
Cytoplasm |
Unknown |
| Two-component system response regulator YehT |
DNA binding |
Phosphorelay signal transduction system |
Cell inner membrane |
Unknown |
| Two-component system sensor histidine kinase CpxA |
ATP binding |
Signal transduction by protein phosphorylation |
Integral component of membrane |
Unknown |
| Two-component system sensor histidine kinase DcuS |
ATP binding |
Phosphorelay signal transduction system |
Integral component of membrane |
Transmembrane |
| Two-component system sensor histidine kinase EnvZ |
ATP binding |
Phosphorelay signal transduction system |
Integral component of membrane |
Transmembrane |
| Two-component system sensor histidine kinase PhoQ |
ATP binding |
Phosphorelay signal transduction system |
Integral component of membrane |
Transmembrane |
| Two-component system sensor histidine kinase PhoR |
ATP binding |
Cellular response to phosphate starvation |
Integral component of membrane |
Transmembrane |
| Two-component system sensor histidine kinase QseC |
ATP binding |
Cellular response to phosphate starvation |
Integral component of membrane |
Transmembrane |
| Two-component system sensor histidine kinase RcsC |
ATP binding |
Cellular response to phosphate starvation |
Integral component of membrane |
Transmembrane |
Functional enrichment analysis of the identified proteins
The identified proteins were further categorized into different classes by sorting them based on their names manually. These classes include molecular chaperone proteins, outer membrane proteins, two-component systems proteins, and type VI secretion system proteins.
Protein network and pathway analysis
To understand the biological network complex involved, STRING analysis was performed.
32
The analysis showed protein–protein interaction networks that are associated with protein export, cellular localization, bacterial secretion, protein insertion into membrane, membrane organization, and signal transductions. The detailed interactions of these networks are given in Fig. 3. Pathway analysis of the S. marcescens proteomic data resulted in the identification of two component systems pathway, chemotaxis pathway (Fig. 4), beta-lactam resistance pathway (Fig. 5), and bacterial secretion system pathways (Table 2).
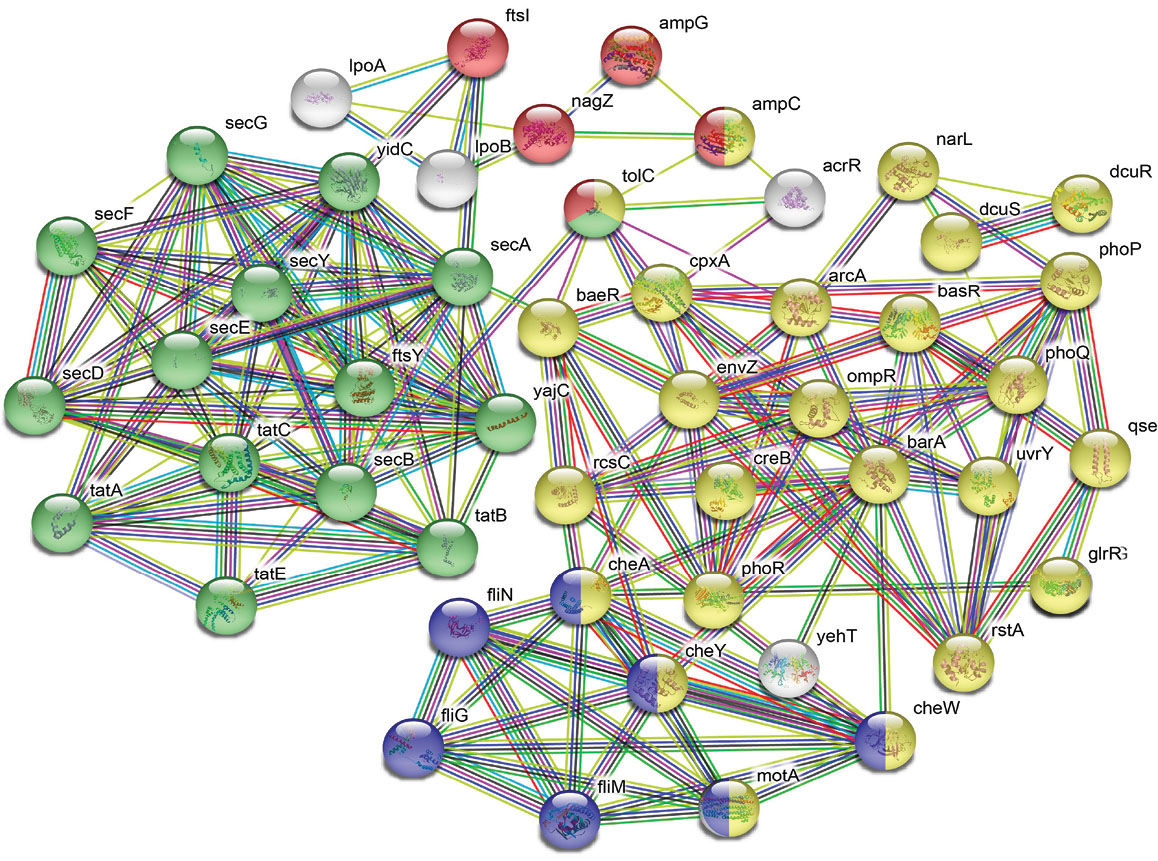
Fig. 3.
Interaction network analysis: STRING interaction network depicts the association among the proteins. Color balls are representing individual protein. Red: involved in beta-lactam resistance. Green: Bacterial secretion system, Blue: Bacterial chemotaxis and Yellow: Two-component systems.
.
Interaction network analysis: STRING interaction network depicts the association among the proteins. Color balls are representing individual protein. Red: involved in beta-lactam resistance. Green: Bacterial secretion system, Blue: Bacterial chemotaxis and Yellow: Two-component systems.
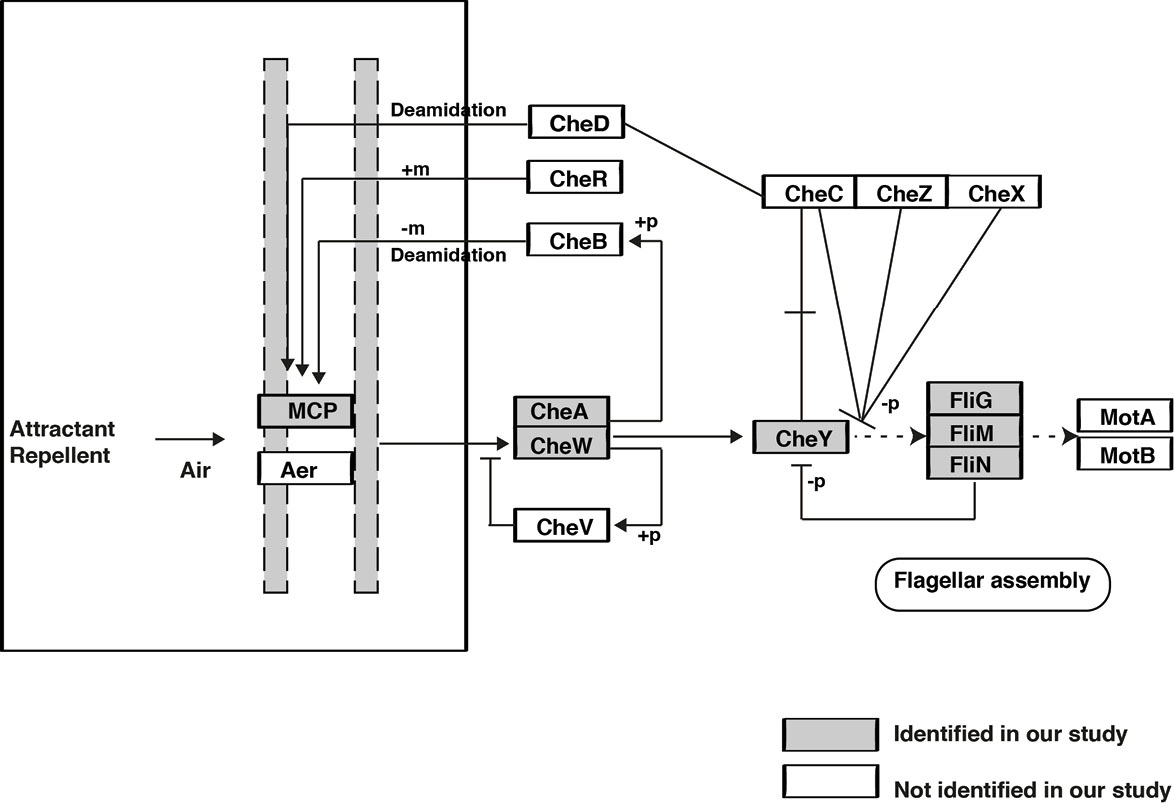
Fig. 4.
Pathway map representation of identified proteins involved in bacterial chemotaxis pathway.
.
Pathway map representation of identified proteins involved in bacterial chemotaxis pathway.
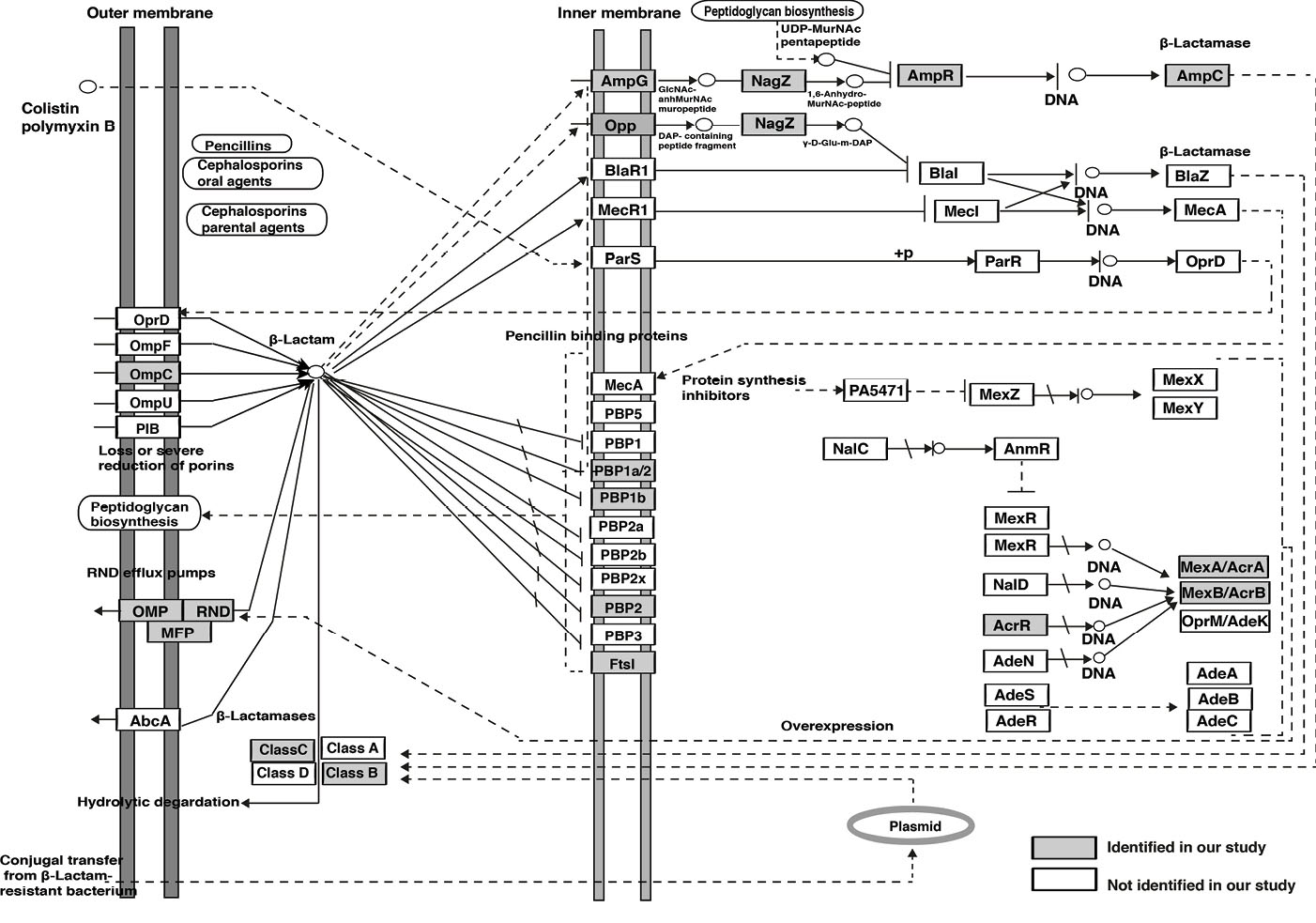
Fig. 5.
Pathway map representation of identified proteins involved in beta-lactam resistance pathway.
.
Pathway map representation of identified proteins involved in beta-lactam resistance pathway.
Table 2.
List of Type VI secretary proteins identified
|
Description
|
Gene symbol
|
T6SS subunit
|
Domain
|
| Hcp1 family Type VI secretion system effector |
hcp1
|
Hcp1 |
Unknown |
| Type VI secretion system ATPase TssH |
clpV
|
TssH |
Coiled coil |
| Type VI secretion system baseplate subunit TssF |
vasA
|
TssF |
Unknown |
| Type VI secretion system baseplate subunit TssG |
tssG
|
TssG |
Unknown |
| Type VI secretion system baseplate subunit TssK |
tssK
|
TssK |
Unknown |
| Type VI secretion system contractile sheath large subunit |
tssC
|
tssC |
Unknown |
| Type VI secretion system contractile sheath small subunit |
tssB
|
tssB |
Unknown |
| Type VI secretion system lipoprotein TssJ |
tssJ
|
TssJ |
Unknown |
| Type VI secretion system membrane subunit TssM |
icmF
|
TssM |
Transmembrane |
| Type VI secretion system protein TssA |
tssA
|
TssA |
Unknown |
| Type VI secretion system protein TssL |
tssL
|
TssL |
Transmembrane |
| Type VI secretion system tip protein VgrG |
tssI
|
VgrG |
Unknown |
| Type VI secretion system-associated FHA domain protein TagH |
tagH
|
TagH |
Unknown |
| Type VI secretion system-associated protein TagF |
tagF
|
TagF |
Unknown |
Discussion
The sequencing of bacterial genome has contributed a lot to elucidating various aspects of bacterial infection. However, a major obstruct in utilizing the bacterial genomic sequence for the study of pathogenesis is the fact that there are many parts of the genes with unknown function in each organism sequenced to date.
35
Hence, a detailed proteomic profiling of such organisms are important to draw conclusions on the host-pathogen interaction during an infection. Recent advances in MS instrumentation and the application of these sensitive high-throughput analytical instruments to study the proteins is opening new perspectives in microbiology.
36
In the last two decades much attention has been paid to study the proteome of different human pathogenic bacteria.
37,38
S. marcescens is one of the pathogenic bacteria and many strains of this bacterium have been sequenced at genome and proteome levels.
10,18,20,22
39-41
To the best of our knowledge, the genome of S. marcescens (ATCC 13880) sequence is available and the proteome of the same is not available till date.
10
Hence, this study was performed to understand the proteome of S. marcescens (ATCC 13880). This strain was initially isolated from pond water and later known to be pathogenic to humans especially children and the immunocompromised patients in the ICU units.
42,43
This is the first detailed proteomic analysis for S. marcescens (ATCC 13880) in which we were able to identify 2541 protein groups accounting for 54% of the S. marcescens (ATCC 13880) proteome. In this study, we reported the identification of important class of proteins which are essential for the growth and function of S. marcescens including molecular chaperone proteins, outer membrane proteins, two-component system proteins and type VI secretion system proteins. Different studies have shown that these classes of proteins play an important role in the physiology and virulence of S. marcescens.
44-49
Analysis of molecular chaperone proteins
In this study, we identified 21 molecular chaperone proteins and analysis of these proteins showed that they are mainly involved in cell wall organization, chaperone-mediated protein folding and response to heat. Further, we found that most of them are located in cytoplasm and outer membrane-bounded periplasmic space having coiled coil or signal domain. These classes of proteins include 17 KD protein Skp and molecular chaperone protein dnaK (DnaK). Molecular chaperone 17 KD protein Skp is a trimeric periplasmic 17 KD protein that helps outer membrane proteins in their folding and incorporation into membranes.
50
Skp is a significantly important protein in the chaperone system which has been distinguished for its function in outer membrane protein (OMP) biogenesis,
51
where it interacts with unfolded proteins.
52
Skp assists in releasing the translocated proteins from the plasma membrane and corrects folding of numerous OMPs.
53,54
However, the molecular mechanisms of how Skp performs its chaperone role have remained unclear,
55,56
and it has been also reported that it does not show sequence similarity to any other chaperone.
50
The protein DnaK, another important protein in the category, is a heat shock protein 70 (hsp70) which is a member of the molecular chaperones family. Studies show that hsp70s are essential part of the cell’s protein folding, degradation and repair machinery.
57
DnaK proteins coat polypeptide chains as they come out from membranes or ribosomes, averting accumulation reactions and premature folding.
58
DnaK helps cell to prevent denaturation of proteins and support the deprivation of the damaged proteins during stress conditions and play a vital role in chromosomal DNA replication (Table 1).
18
Analysis of outer membrane proteins
The outer membrane of gram-negative bacteria plays a role in membrane permeability and avoid the entry of toxic compounds and allow the influx of nutrient molecules.
59
OMPs perform multiple functions including solute transport, membrane structure, signal transduction, stability and catalysis.
60
In this study, we found 15 such outer membrane proteins. Functional analysis of the important proteins of this class includes outer membrane protein A (OmpA) and Outer membrane protein X (OmpX).
The OmpA belongs to the family of OMPs and it is conserved among gram-negative bacteria.
61
OmpA proteins have been reported to play important pathogenic roles such as adhesion to mucosal surfaces, intracellular survival or invasion, stimulators of proinflammatory cytokine production, and antimicrobial peptide resistance.
62
In S. marcescens, OmpA is an outer membrane protein with porin activity and conjugation or ion transport processes. The OmpA has been reported to play structural and ion-permeable porin roles which help in bacterial survival during osmotic stress conditions.
63
OmpX proteins are small molecular weight (15 to 18 KD) proteins and belong to a family of conserved bacterial proteins where they support adhesion to and entry into mammalian cells.
64,65
These proteins fold in an eight-beta-barrel structure through membrane-spanning domains that project from the cell surface.
66
OmpX are involved in physiological processes such as antibiotic resistance, invasion, signal transduction, internalization in epithelial cells, binding external proteins, contributing to channeling and virulence.
64,67-69
Analysis of two-component system proteins
The ability to sense and respond to its surroundings is fundamental to the survival of a bacterium. Two-component regulatory systems are reported as key mechanism through which bacteria perceive and respond to their environment.
70,71
Two component system proteins have been studied to be associated with physiological functions including competence, antibiotic resistance, cell-cell communication, adaptation to environments, and sporulation.
72
In this study we reported 22 such two-component system proteins. PhoP/PhoQ system is one among the 22 component system proteins reported in this study. It is a two-component system that controls the expression of genes associated with virulence, the adaptation to Mg
2+
restrictive environments, and controls various cellular activities in numerous gram-negative species.
73
Studies reported that in non-pathogenic bacteria PhoP/PhoQ system plays a role in the control of the magnesium homeostasis, whereas in pathogenic bacteria it plays an essential role in the expression of vital virulence phenotypes.
74
It is also reported that activity of the PhoP/PhoQ system permits Serratia to survive scarce environmental Mg
2+
availability, acidic pH, and the action of the cationic antimicrobial compound polymyxin B.
75
Analysis of type VI secretion system proteins
Gram-negative bacteria have evolved several secretion systems to deliver toxins and proteins into target cell, which are essential for virulence and survival within hosts. Type VI secretion system is widely used by gram-negative bacteria and it functions as nano-machine which expands from cytoplasm to the outer membrane to inject effector proteins into target cells.
76,77
T6SS is composed of at least 13 core-component proteins and is associated with auxiliary component proteins.
77
These core-component proteins are categorized into different subunits: the membrane complex (TssJLM), the baseplate complex (TssAEFGK), needle sheath complex (TssBC), sheath recycling ATPase (TssH), and the injection apparatus (Hcp, VgrG).
78
In the present study we found 12 such core-component proteins (TssABC, Hcp1, TssFGH, VgrG, TssJKLM) and two additional auxiliary component proteins (TagH and TagF) (Table 2). Interestingly, most of these proteins found to be in the cytoplasmic region which suggests that these cytoplasm proteins can be further investigated for enhanced understanding of secreted protein transport during the pathogenesis.
19
These proteins have been reported to play a role in various diseases.
2,79
Protein network and pathway analysis
Proteins associated with bacterial chemotaxis pathway and beta lactam resistance pathway were identified by the proteomic profiling of S. marcescens chemotaxis, the capacity of gram-negative bacteria to move in response to external stimuli has long been concerned in the bacterial colonization and pathogenicity.
80
In this study we identified 7 proteins which belong to chemotaxis pathway (Fig. 4). The prokaryotic chemotaxis pathway senses the external chemoeffector concentrations by the transmembrane receptor methyl-accepting chemotaxis protein (MCP).
81
The binding of chemoeffector ligand to a chemoreceptor is signaled across the membrane to regulate the autophosphorylation activity of the CheA kinase with the help of an adaptor protein CheW. Once CheA is phosphorylated, the phosphoryl group is transferred to response regulator CheY. Phosphorylated CheY diffuses away from the signaling cluster and binds with the flagella motor switch proteins FliG, FliM and FliN to cause a change in the rotation direction of the motor. Interruption of any component of the chemotaxis signal transduction pathway (MCP receptor-CheA/CheW-CheY-flagella cascade) reduces the ability of pathogens to colonize and cause disease.
82
Drug resistant bacterial infections have significant impact on the morbidity and mortality, as a consequence of this WHO has raised serious concern to address the growing multidrug resistance in gram-negative bacteria.
9
Bacteria have developed several strategies of defense against classes of antibiotics, including elimination of antibiotics through efflux pumps, enzymatic inactivation of antibiotics (production of beta-lactamase enzymes) and expression of low affinity targets (penicillin-binding proteins).
83
In this study we reported ten key proteins which belong to beta-lactam resistance pathway that may play an important role under these three strategies. TolC-outer membrane channel protein and AcrAB-multidrug efflux pump transcriptional activator found to play a role in efflux pumps (RND family),
84,85
AmpG- muropeptide transporter AmpG, nagZ-beta N-acetylhexosaminidase, AmpR- LysR family transcriptional regulator, SRT/SST family class C beta-lactamase, and class B metallo-beta-lactamases associated with enzymatic inactivation of antibiotics,
86
whereas, carboxypeptidase/penicillin-binding protein 1a, penicillin-binding protein 1b and penicillin-binding protein 2 play a role in the expression of low affinity targets.
87,88
Further studies are essential to understand the detailed role of these proteins in drug resistance mechanism.
Few studies have reported the proteomic analysis of this species, which helped in elucidating the molecular mechanisms underlying the resistance to, and removal of, some toxic elements such as manganese from the environment. Zakeri et al identified 130 proteins from the strain ZF03 of S. marcescens under radium stress condition using MS/MS analysis.
18
In another study Fritsch et al used LC-MS/MS to identify 842 proteins from the type VI secretion system (T6SS) dependent secretome of S. marcescens Db11 strain.
19
Yi et al used 2D MALDI-TOF MS analysis and reported 23 expressed proteins in S. marcescens ATCC 14041.
20
Cianfanelli et al identified 1025 proteins using LC-MS/MS based proteomic approach to measure the secretion of T6SS and VgrG dependent effector proteins in S. marcescens Db10 strain.
21
Recently, Tuveng et al reported the identification of 232 secreted proteins from S. marcescens BJL200 strain.
22
In one of the recent studies, Queiroz et al reported the alterations in the proteomic composition of S. marcescens in response to manganese (II).
89
They performed the comparative study with the presence and absence of Mn (II). They reported the identification of structural proteins and enzymes related to major cellular and metabolic processes required for growth and survival of gram-negative bacteria. Additionally, they have shown the differential expression of these proteins in the absence and presence of Mn II. Through this study, they report the application of S. marcescens LG1 strain in bioremediation applications, in particular removal of Mn (II) from the drinking water and industrial water.
The highlight of the present study is the use of high-sensitive mass-spectrometry to report a large number of proteins in S. marcescens (ATCC 13880) which was not available earlier. We believe that the proteins reported in this study may be used to compare the proteomic changes in the stress-induced/disease conditions and other situations, which may help to draw the inferences on the role of this organism in antibiotic resistance.
Conclusion
Serratia marcescens, one of the important human pathogens is ubiquitously distributed which causes nosocomial infections and outbreaks especially in children and immunocompromised patients. The situation is further worsened by the development of resistance to beta-lactam antibiotics of S. marcescens. Recent advances and successful microbial proteomic experiments with MS-based proteomics proves that MS can be used as a tool to understand the interaction between pathogens and their host such as that in infections and drug resistance. It involves the study of protein–protein interactions on a small and proteome-wide scale, defining proteome of several organelles, analysis of post-translational modifications, and quantitative estimation of change in the abundance of proteins. MS can be used to identify and quantify thousands of proteins in samples which have lots of applications in the field of clinical microbiology. LC-MS/MS-based proteomic studies will set the stage for the identification of proteins that are involved in virulence and pathogenesis of this organism and the host-pathogen interaction during infection. In the present study, we performed a detailed proteomic analysis of S. marcescens ATCC 13880, using MS-based advanced proteomic and bioinformatic approach to elucidate the proteome profile of S. marcescens ATCC 13880 which resulted in the identification of 2541 proteins. To the best of our knowledge, this study provides an in-depth coverage of the proteome and functional analysis of identified proteins. The study supplements the microbial proteome project and presents an essential knowledge of the S. marcescens ATCC 13880 proteome.
Acknowledgments
Authors thank the Management and Principal of MS Ramaiah Institute of Technology, Bangalore for their support and facility.
Funding sources
Nil.
Ethical statement
Not applicable.
Competing interests
The authors declare there is no conflict of interests.
Authors’ contribution
SR, GS, and BSG conceived and planned the project, the main conceptual ideas, and design of the experiments. BSG implemented the experiments. SR, GS, and BSG contributed to data analysis and interpretation of the results. BSG wrote the manuscript. SR and GS reviewed final version of the manuscript. All authors provided helpful feedback.
Supplementary Materials
Supplementary file 1, 2, 3 and 4 contain buffers and reagents used in the study, Table S1, Table S2 and representative mass spectra, respectively.
(pdf)
Research Highlights
What is the current knowledge?
simple
-
√ The complete genome sequence of S. marcescens strain
ATCC13880 is available and computationally predicted to
have 4724 protein coding sequences.
-
√ Proteome of other strains of S. marcescens is available where
as the whole proteome of S. marcescens ATCC 13880 remains
largely unexplored.
What is new here?
simple
-
√ The study provided experimental evidence at the level of
protein expression for the computationally predicted gene
annotations for the genome of S. marcescens strain ATCC
13880.
-
√ The study provided the list of 2541 unique protein groups,
which corresponds to ~54 % of the computationally predicted
protein-coding genes with peptide level evidence and their
associated Bioinformatics analysis.
-
√ The data and outcome of the study is valuable in addition to
the microbial proteome project.
References
- Khanna A, Khanna M, Aggarwal A. Serratia marcescens - a rare opportunistic nosocomial pathogen and measures to limit its spread in hospitalized patients. J Clin Diagn Res 2013; 7:243-6. doi: 10.7860/JCDR/2013/5010.2737 [Crossref] [ Google Scholar]
- Mahlen SD. Serratia infections: from military experiments to current practice. Clin Microbiol Rev 2011; 24:755-91. doi: 10.1128/CMR.00017-11 [Crossref] [ Google Scholar]
- Gastmeier P, Loui A, Stamm-Balderjahn S, Hansen S, Zuschneid I, Sohr D. Outbreaks in neonatal intensive care units - they are not like others. Am J Infect Control 2007; 35:172-6. doi: 10.1016/j.ajic.2006.07.007 [Crossref] [ Google Scholar]
- Raymond J, Aujard Y. Nosocomial infections in pediatric patients: a European, multicenter prospective study European Study Group. Infect Control Hosp Epidemiol 2000; 21:260-3. doi: 10.1086/501755 [Crossref] [ Google Scholar]
- Kim SB, Jeon YD, Kim JH, Kim JK, Ann HW, Choi H. Risk factors for mortality in patients with Serratia marcescens bacteremia. Yonsei Med J 2015; 56:348-54. doi: 10.3349/ymj.2015.56.2.348 [Crossref] [ Google Scholar]
- Shih HI, Lee HC, Lee NY, Chang CM, Wu CJ, Wang LR. Serratia marcescens bacteremia at a medical center in southern Taiwan: high prevalence of cefotaxime resistance. J Microbiol Immunol Infect 2005; 38:350-7. [ Google Scholar]
- Debast SB, Melchers WJ, Voss A, Hoogkamp-Korstanje JA, Meis JF. Epidemiological survey of an outbreak of multiresistant Serratia marcescens by PCR-fingerprinting. Infection 1995; 23:267-71. [ Google Scholar]
- Rello J, Kalwaje Eshwara V, Lagunes L, Alves J, Wunderink RG, Conway-Morris A. A global priority list of the TOp TEn resistant Microorganisms (TOTEM) study at intensive care: a prioritization exercise based on multi-criteria decision analysis. Eur J Clin Microbiol Infect Dis 2019; 38:319-23. doi: 10.1007/s10096-018-3428-y [Crossref] [ Google Scholar]
- Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 2018; 18:318-27. doi: 10.1016/S1473-3099(17)30753-3 [Crossref] [ Google Scholar]
- Daligault HE, Davenport KW, Minogue TD, Broomall SM, Bruce DC, Chain PS. Genome Assembly of Serratia marcescens Type Strain ATCC 13880. Genome Announc 2014; 2:e00967. doi: 10.1128/genomeA.00967-14 [Crossref] [ Google Scholar]
- Schirle M, Bantscheff M, Kuster B. Mass spectrometry-based proteomics in preclinical drug discovery. Chem Biol 2012; 19:72-84. doi: 10.1016/j.chembiol.2012.01.002 [Crossref] [ Google Scholar]
- Walgren JL, Thompson DC. Application of proteomic technologies in the drug development process. Toxicol Lett 2004; 149:377-85. doi: 10.1016/j.toxlet.2003.12.047 [Crossref] [ Google Scholar]
- Steen H, Mann M. The ABC's (and XYZ's) of peptide sequencing. Nat Rev Mol Cell Biol 2004; 5:699-711. doi: 10.1038/nrm1468 [Crossref] [ Google Scholar]
- Van Oudenhove L, Devreese B. A review on recent developments in mass spectrometry instrumentation and quantitative tools advancing bacterial proteomics. Appl Microbiol Biotechnol 2013; 97:4749-62. doi: 10.1007/s00253-013-4897-7 [Crossref] [ Google Scholar]
- Ozdemir V. Microbiome Special Issue: Food, Drugs, Diagnostics, and Built Environments. OMICS 2018; 22:89. doi: 10.1089/omi.2018.0001 [Crossref] [ Google Scholar]
- List E, Berryman D, Bower B, Sackmann-Sala L, Gosney E, Ding J. The use of proteomics to study infectious diseases. IInfect Disord Drug Targets 2008; 8:31-45. doi: 10.2174/187152608784139640 [Crossref] [ Google Scholar]
- Lee CR, Lee JH, Park KS, Jeong BC, Lee SH. Quantitative proteomic view associated with resistance to clinically important antibiotics in Gram-positive bacteria: a systematic review. Front Microbiol 2015; 6:828. doi: 10.3389/fmicb.2015.00828 [Crossref] [ Google Scholar]
- Zakeri F, Sadeghizadeh M, Kardan MR, Shahbani Zahiri H, Ahmadian G, Masoumi F. Differential proteome analysis of a selected bacterial strain isolated from a high background radiation area in response to radium stress. J Proteomics 2012; 75:4820-32. doi: 10.1016/j.jprot.2012.05.020 [Crossref] [ Google Scholar]
- Fritsch MJ, Trunk K, Diniz JA, Guo M, Trost M, Coulthurst SJ. Proteomic identification of novel secreted antibacterial toxins of the Serratia marcescens type VI secretion system. Mol Cell Proteomics 2013; 12:2735-49. doi: 10.1074/mcp.M113.030502 [Crossref] [ Google Scholar]
- Yi S, Wang W, Bai F, Zhu J, Li J, Li X. Antimicrobial effect and membrane-active mechanism of tea polyphenols against Serratia marcescens. World J Microbiol Biotechnol 2014; 30:451-60. doi: 10.1007/s11274-013-1464-4 [Crossref] [ Google Scholar]
- Cianfanelli FR, Alcoforado Diniz J, Guo M, De Cesare V, Trost M, Coulthurst SJ. VgrG and PAAR Proteins Define Distinct Versions of a Functional Type VI Secretion System. PLoS Pathog 2016; 12:e1005735. doi: 10.1371/journal.ppat.1005735 [Crossref] [ Google Scholar]
- Tuveng TR, Hagen LH, Mekasha S, Frank J, Arntzen MO, Vaaje-Kolstad G. Genomic, proteomic and biochemical analysis of the chitinolytic machinery of Serratia marcescens BJL200. Biochim Biophys Acta Proteins Proteom 2017; 1865:414-21. doi: 10.1016/j.bbapap.2017.01.007 [Crossref] [ Google Scholar]
- Advani J, Verma R, Chatterjee O, Devasahayam Arokia Balaya R, Najar MA, Ravishankara N. Rise of Clinical Microbial Proteogenomics: A Multiomics Approach to Nontuberculous Mycobacterium-The Case of Mycobacterium abscessus UC22. OMICS 2019; 23:1-16. doi: 10.1089/omi.2018.0116 [Crossref] [ Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009; 6:359-62. doi: 10.1038/nmeth.1322 [Crossref] [ Google Scholar]
- Nirujogi RS, Muthusamy B, Kim MS, Sathe GJ, Lakshmi PT, Kovbasnjuk ON. Secretome analysis of diarrhea-inducing strains of Escherichia coli. Proteomics 2017; 17(6). doi: 10.1002/pmic.201600299 [Crossref]
- Song C, Ye M, Han G, Jiang X, Wang F, Yu Z. Reversed-phase-reversed-phase liquid chromatography approach with high orthogonality for multidimensional separation of phosphopeptides. Anal Chem 2010; 82:53-6. doi: 10.1021/ac9023044 [Crossref] [ Google Scholar]
- Dammalli M, Dey G, Kumar M, Madugundu AK, Gopalakrishnan L, Gowrishankar BS. Proteomics of the Human Olfactory Tract. OMICS 2018; 22:77-87. doi: 10.1089/omi.2017.0155 [Crossref] [ Google Scholar]
- Dammalli M, Dey G, Madugundu AK, Kumar M, Rodrigues B, Gowda H. Proteomic Analysis of the Human Olfactory Bulb. OMICS 2017; 21:440-53. doi: 10.1089/omi.2017.0084 [Crossref] [ Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 2003; 13:2129-41. doi: 10.1101/gr.772403 [Crossref] [ Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57. doi: 10.1038/nprot.2008.211 [Crossref] [ Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1-13. doi: 10.1093/nar/gkn923 [Crossref] [ Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019; 47:D607-13. doi: 10.1093/nar/gky1131 [Crossref] [ Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28:27-30. doi: 10.1093/nar/28.1.27 [Crossref] [ Google Scholar]
- Jones P, Cote R. The PRIDE proteomics identifications database: data submission, query, and dataset comparison. Methods Mol Biol 2008; 484:287-303. doi: 10.1007/978-1-59745-398-1_19 [Crossref] [ Google Scholar]
- Raskin DM, Seshadri R, Pukatzki SU, Mekalanos JJ. Bacterial genomics and pathogen evolution. Cell 2006; 124:703-14. doi: 10.1016/j.cell.2006.02.002 [Crossref] [ Google Scholar]
- Lin H, He QY, Shi L, Sleeman M, Baker MS, Nice EC. Proteomics and the microbiome: pitfalls and potential. Expert Rev Proteomics 2019; 16:501-11. doi: 10.1080/14789450.2018.1523724 [Crossref] [ Google Scholar]
- Saleh S, Staes A, Deborggraeve S, Gevaert K. Targeted proteomics for studying pathogenic bacteria. Proteomics 2019; 19:e1800435. doi: 10.1002/pmic.201800435 [Crossref] [ Google Scholar]
- Soares NC, Bou G, Blackburn JM. Editorial: Proteomics of Microbial Human Pathogens. Front Microbiol 2016; 7:1742. doi: 10.3389/fmicb.2016.01742 [Crossref] [ Google Scholar]
- Chung WC, Chen LL, Lo WS, Kuo PA, Tu J, Kuo CH. Complete Genome Sequence of Serratia marcescens WW4. Genome Announc 2013; 1:e0012613. doi: 10.1128/genomeA.00126-13 [Crossref] [ Google Scholar]
- Wang Y, Yuan Y, Zhou L, Su Q, Fang X, Li T. Draft genome sequence of Serratia marcescens strain LCT-SM213. J Bacteriol 2012; 194:4477-8. doi: 10.1128/JB.00933-12 [Crossref] [ Google Scholar]
- Williams EA, Hopson H, Rodriguez A, Kongari R, Bonasera R, Hernandez-Morales AC. Complete Genome Sequence of Serratia marcescens Siphophage Serbin. Microbiol Resour Announc 2019; 8:e00422. doi: 10.1128/MRA.00422-19 [Crossref] [ Google Scholar]
- Mataseje LF, Boyd DA, Delport J, Hoang L, Imperial M, Lefebvre B. Serratia marcescens harbouring SME-type class A carbapenemases in Canada and the presence of blaSME on a novel genomic island, SmarGI1-1. J Antimicrob Chemother 2014; 69:1825-9. doi: 10.1093/jac/dku040 [Crossref] [ Google Scholar]
- Moya-Torres A, Mulvey MR, Kumar A, Oresnik IJ, Brassinga AK. The lack of OmpF, but not OmpC, contributes to increased antibiotic resistance in Serratia marcescens. Microbiology 2014; 160:1882-92. doi: 10.1099/mic.0.081166-0 [Crossref] [ Google Scholar]
- Castelli ME, Garcia Vescovi E, Soncini FC. The phosphatase activity is the target for Mg2+ regulation of the sensor protein PhoQ in Salmonella. J Biol Chem 2000; 275:22948-54. doi: 10.1074/jbc.M909335199 [Crossref] [ Google Scholar]
- English G, Trunk K, Rao VA, Srikannathasan V, Hunter WN, Coulthurst SJ. New secreted toxins and immunity proteins encoded within the Type VI secretion system gene cluster of Serratia marcescens. Mol Microbiol 2012; 86:921-36. doi: 10.1111/mmi.12028 [Crossref] [ Google Scholar]
- Lien YW, Lai EM. Type VI Secretion Effectors: Methodologies and Biology. Front Cell Infect Microbiol 2017; 7:254. doi: 10.3389/fcimb.2017.00254 [Crossref] [ Google Scholar]
- McMahon KJ, Castelli ME, Garcia Vescovi E, Feldman MF. Biogenesis of outer membrane vesicles in Serratia marcescens is thermoregulated and can be induced by activation of the Rcs phosphorelay system. J Bacteriol 2012; 194:3241-9. doi: 10.1128/JB.00016-12 [Crossref] [ Google Scholar]
- Sapriel G, Wandersman C, Delepelaire P. The SecB chaperone is bifunctional in Serratia marcescens: SecB is involved in the Sec pathway and required for HasA secretion by the ABC transporter. J Bacteriol 2003; 185:80-8. doi: 10.1128/jb.185.1.80-88.2003 [Crossref] [ Google Scholar]
- Wei JR, Tsai YH, Soo PC, Horng YT, Hsieh SC, Ho SW. Biochemical characterization of RssA-RssB, a two-component signal transduction system regulating swarming behavior in Serratia marcescens. J Bacteriol 2005; 187:5683-90. doi: 10.1128/JB.187.16.5683-5690.2005 [Crossref] [ Google Scholar]
- Walton TA, Sousa MC. Crystal structure of Skp, a prefoldin-like chaperone that protects soluble and membrane proteins from aggregation. Mol Cell 2004; 15:367-74. doi: 10.1016/j.molcel.2004.07.023 [Crossref] [ Google Scholar]
- Entzminger KC, Chang C, Myhre RO, McCallum KC, Maynard JA. The Skp chaperone helps fold soluble proteins in vitro by inhibiting aggregation. Biochemistry 2012; 51:4822-34. doi: 10.1021/bi300412y [Crossref] [ Google Scholar]
- Harms N, Koningstein G, Dontje W, Muller M, Oudega B, Luirink J. The early interaction of the outer membrane protein phoe with the periplasmic chaperone Skp occurs at the cytoplasmic membrane. J Biol Chem 2001; 276:18804-11. doi: 10.1074/jbc.M011194200 [Crossref] [ Google Scholar]
- Chen R, Henning U. A periplasmic protein (Skp) of Escherichia coli selectively binds a class of outer membrane proteins. Mol Microbiol 1996; 19:1287-94. doi: 10.1111/j.1365-2958.1996.tb02473.x [Crossref] [ Google Scholar]
- Schafer U, Beck K, Muller M. Skp, a molecular chaperone of gram-negative bacteria, is required for the formation of soluble periplasmic intermediates of outer membrane proteins. J Biol Chem 1999; 274:24567-74. [ Google Scholar]
- Kawe M, Pluckthun A. GroEL walks the fine line: the subtle balance of substrate and co-chaperonin binding by GroEL A combinatorial investigation by design, selection and screening. J Mol Biol 2006; 357:411-26. doi: 10.1016/j.jmb.2005.12.005 [Crossref] [ Google Scholar]
- Purdy GE, Fisher CR, Payne SM. IcsA surface presentation in Shigella flexneri requires the periplasmic chaperones DegP, Skp, and SurA. J Bacteriol 2007; 189:5566-73. doi: 10.1128/JB.00483-07 [Crossref] [ Google Scholar]
- Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol 2013; 14:630-42. doi: 10.1038/nrm3658 [Crossref] [ Google Scholar]
- Flynn GC, Pohl J, Flocco MT, Rothman JE. Peptide-binding specificity of the molecular chaperone BiP. Nature 1991; 353:726-30. doi: 10.1038/353726a0 [Crossref] [ Google Scholar]
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 2003; 67:593-656. doi: 10.1128/mmbr.67.4.593-656.2003 [Crossref] [ Google Scholar]
- Khalid S, Bond PJ, Carpenter T, Sansom MS. OmpA: gating and dynamics via molecular dynamics simulations. Biochim Biophys Acta 2008; 1778:1871-80. doi: 10.1016/j.bbamem.2007.05.024 [Crossref] [ Google Scholar]
- Krishnan S, Prasadarao NV. Outer membrane protein A and OprF: versatile roles in Gram-negative bacterial infections. FEBS J 2012; 279:919-31. doi: 10.1111/j.1742-4658.2012.08482.x [Crossref] [ Google Scholar]
- Smith SG, Mahon V, Lambert MA, Fagan RP. A molecular Swiss army knife: OmpA structure, function and expression. FEMS Microbiol Lett 2007; 273:1-11. doi: 10.1111/j.1574-6968.2007.00778.x [Crossref] [ Google Scholar]
- Hong H, Szabo G, Tamm LK. Electrostatic couplings in OmpA ion-channel gating suggest a mechanism for pore opening. Nat Chem Biol 2006; 2:627-35. doi: 10.1038/nchembio827 [Crossref] [ Google Scholar]
- Vogt J, Schulz GE. The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence. Structure 1999; 7:1301-9. doi: 10.1016/s0969-2126(00)80063-5 [Crossref] [ Google Scholar]
- Kolodziejek AM, Schnider DR, Rohde HN, Wojtowicz AJ, Bohach GA, Minnich SA. Outer membrane protein X (Ail) contributes to Yersinia pestis virulence in pneumonic plague and its activity is dependent on the lipopolysaccharide core length. Infect Immun 2010; 78:5233-43. doi: 10.1128/IAI.00783-10 [Crossref] [ Google Scholar]
- Li B, Huang Q, Cui A, Liu X, Hou B, Zhang L. Overexpression of Outer Membrane Protein X (OmpX) Compensates for the Effect of TolC Inactivation on Biofilm Formation and Curli Production in Extraintestinal Pathogenic Escherichia coli (ExPEC). Front Cell Infect Microbiol 2018; 8:208. doi: 10.3389/fcimb.2018.00208 [Crossref] [ Google Scholar]
- Miller VL, Beer KB, Heusipp G, Young BM, Wachtel MR. Identification of regions of Ail required for the invasion and serum resistance phenotypes. Mol Microbiol 2001; 41:1053-62. doi: 10.1046/j.1365-2958.2001.02575.x [Crossref] [ Google Scholar]
- Otto K, Hermansson M. Inactivation of ompX causes increased interactions of type 1 fimbriated Escherichia coli with abiotic surfaces. J Bacteriol 2004; 186:226-34. doi: 10.1128/jb.186.1.226-234.2004 [Crossref] [ Google Scholar]
- Stoorvogel J, van Bussel MJ, van de Klundert JA. Biological characterization of an Enterobacter cloacae outer membrane protein (OmpX). J Bacteriol 1991; 173:161-7. doi: 10.1128/jb.173.1.161-167.1991 [Crossref] [ Google Scholar]
- Casino P, Rubio V, Marina A. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 2009; 139:325-36. doi: 10.1016/j.cell.2009.08.032 [Crossref] [ Google Scholar]
- Mitrophanov AY, Groisman EA. Signal integration in bacterial two-component regulatory systems. Genes Dev 2008; 22:2601-11. doi: 10.1101/gad.1700308 [Crossref] [ Google Scholar]
- Paterson GK, Blue CE, Mitchell TJ. Role of two-component systems in the virulence of Streptococcus pneumoniae. J Med Microbiol 2006; 55:355-63. doi: 10.1099/jmm.0.46423-0 [Crossref] [ Google Scholar]
- Groisman EA. The pleiotropic two-component regulatory system PhoP-PhoQ. J Bacteriol 2001; 183:1835-42. doi: 10.1128/JB.183.6.1835-1842.2001 [Crossref] [ Google Scholar]
- Perez JC, Shin D, Zwir I, Latifi T, Hadley TJ, Groisman EA. Evolution of a bacterial regulon controlling virulence and Mg(2+) homeostasis. PLoS Genet 2009; 5:e1000428. doi: 10.1371/journal.pgen.1000428 [Crossref] [ Google Scholar]
- Lin QY, Tsai YL, Liu MC, Lin WC, Hsueh PR, Liaw SJ. Serratia marcescens arn, a PhoP-regulated locus necessary for polymyxin B resistance. Antimicrob Agents Chemother 2014; 58:5181-90. doi: 10.1128/AAC.00013-14 [Crossref] [ Google Scholar]
- Clemens DL, Lee BY, Horwitz MA. The Francisella Type VI Secretion System. Front Cell Infect Microbiol 2018; 8:121. doi: 10.3389/fcimb.2018.00121 [Crossref] [ Google Scholar]
- Cascales E, Cambillau C. Structural biology of type VI secretion systems. Philos Trans R Soc Lond B Biol Sci 2012; 367:1102-11. doi: 10.1098/rstb.2011.0209 [Crossref] [ Google Scholar]
- Yang X, Pan J, Wang Y, Shen X. Type VI Secretion Systems Present New Insights on Pathogenic Yersinia. Front Cell Infect Microbiol 2018; 8:260. doi: 10.3389/fcimb.2018.00260 [Crossref] [ Google Scholar]
- Murdoch SL, Trunk K, English G, Fritsch MJ, Pourkarimi E, Coulthurst SJ. The opportunistic pathogen Serratia marcescens utilizes type VI secretion to target bacterial competitors. J Bacteriol 2011; 193:6057-69. doi: 10.1128/JB.05671-11 [Crossref] [ Google Scholar]
- Matilla MA, Krell T. The effect of bacterial chemotaxis on host infection and pathogenicity. FEMS Microbiol Rev 2018; 42(1). doi: 10.1093/femsre/fux052 [Crossref]
- Blair DF. How bacteria sense and swim. Annu Rev Microbiol 1995; 49:489-522. doi: 10.1146/annurev.mi.49.100195.002421 [Crossref] [ Google Scholar]
- Korolik V. The role of chemotaxis during Campylobacter jejuni colonisation and pathogenesis. Curr Opin Microbiol 2018; 47:32-7. doi: 10.1016/j.mib.2018.11.001 [Crossref] [ Google Scholar]
- Munita JM, Arias CA. Mechanisms of Antibiotic Resistance. Microbiol Spectr 2016; 4. doi: 10.1128/microbiolspec.VMBF-0016-2015 [Crossref]
- Muller C, Plesiat P, Jeannot K. A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and beta-lactams in Pseudomonas aeruginosa. Antimicrob Agents Chemother 2011; 55:1211-21. doi: 10.1128/AAC.01252-10 [Crossref] [ Google Scholar]
- Nikaido H, Takatsuka Y. Mechanisms of RND multidrug efflux pumps. Biochim Biophys Acta 2009; 1794:769-81. doi: 10.1016/j.bbapap.2008.10.004 [Crossref] [ Google Scholar]
- Mark BL, Vocadlo DJ, Oliver A. Providing beta-lactams a helping hand: targeting the AmpC beta-lactamase induction pathway. Future Microbiol 2011; 6:1415-27. doi: 10.2217/fmb.11.128 [Crossref] [ Google Scholar]
- Fernandez L, Hancock RE. Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin Microbiol Rev 2012; 25:661-81. doi: 10.1128/CMR.00043-12 [Crossref] [ Google Scholar]
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361-85. doi: 10.1111/j.1574-6976.2007.00095.x [Crossref] [ Google Scholar]
- Queiroz PS, Ruas FAD, Barboza NR, de Castro Borges W, Guerra-Sa R. Alterations in the proteomic composition of Serratia marcescens in response to manganese (II). BMC Biotechnol 2018; 18:83. doi: 10.1186/s12896-018-0493-3 [Crossref] [ Google Scholar]