Bioimpacts. 10(4):227-234.
doi: 10.34172/bi.2020.29
Original Research
Flavones scaffold of Chromolaena odorata as a potential xanthine oxidase inhibitor: Induced Fit Docking and ADME studies
Babatomiwa Kikiowo 1, 2, *  , Adewale Joseph Ogunleye 1, Olumide Kayode Inyang 1, 2, Niyi Samuel Adelakun 1, 2, Olaposi Idowu Omotuyi 1, 2, Damilohun Samuel Metibemu 1, 2, Temitope Israel David 1, 2, Oluwatoyin Olajide Oludoyi 1, 2, Taiwo Tolulope Ijatuyi 2
, Adewale Joseph Ogunleye 1, Olumide Kayode Inyang 1, 2, Niyi Samuel Adelakun 1, 2, Olaposi Idowu Omotuyi 1, 2, Damilohun Samuel Metibemu 1, 2, Temitope Israel David 1, 2, Oluwatoyin Olajide Oludoyi 1, 2, Taiwo Tolulope Ijatuyi 2
Author information:
1Centre for Biocomputing and Drug Development, Adekunle Ajasin University, Akungba Akoko, Ondo State, Nigeria
2Department of Biochemistry, Adekunle Ajasin University, Akungba Akoko, Ondo State, Nigeria
Abstract
Introduction:
Gout is a type of painful inflammation initiated by the interactions between monosodium urate crystals and connective tissue. Xanthine oxidase (XO) catalyzes the oxidation of hypoxanthine to xanthine, then to uric acid. The primary treatments for gout include XO inhibitors. At present, allopurinol is the most used XO inhibitor for the treatment of gout. However, it can cause adverse effects commonly known as allopurinol hypersensitivity syndrome, thereby limiting its usage. Consequently, it is necessary to develop potent and less toxic inhibitors of XO. Chromolaena odorata is one of such plants under investigation for its diverse health benefits.
Methods:
Phytochemicals of C. odorata were screened against XO receptor, using molecular docking. The top five hit compounds of glide docking yield flavones scaffold which were subjected to induced fit docking (IFD) and absorption, distribution, metabolism, and excretion (ADME) studies.
Results:
The result showed that flavones scaffold of C. odorata can bind with higher affinity and lower free energy values when compared to that of the standard, allopurinol. The IFD scores of the flavones scaffold range from -1525.25 to -1527.99 kcal/mol.
Conclusion:
Our results have shown that flavones scaffold might have the potential to act as an effective drug candidate when compared to allopurinol in treating and/or preventing gout and some inflammatory condition.
Keywords: ADME, Flavones scaffold, Gout, Induced fit docking, Lipinski's rule of five, Xanthine oxidase
Copyright and License Information
© 2020 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Gout is a prevalent metabolic disease with high public distribution and continues to be a major health challenge. It affects circa 13% and 5% of male and female populations, respectively.
1
Gout is a type of painful inflammation initiated by the interactions between monosodium urate crystals and connective tissue
2
during purine catabolism by the enzyme of xanthine oxidase (XO).
3
XO is a very flexible enzyme that has a wide distribution among various species from bacteria to humans and within several tissues of mammals. It is a member of a class of enzymes called molybdenum iron-sulfur flavin hydroxylases.
4
XO plays an essential role in the breakdown of purines, it catalyzes the oxidation of hypoxanthine to xanthine, then to uric acid (UA), the last step in the catabolism of purine bases.
5
The build-up of UA in the body is accountable for the formation of several disorders, therefore, it plays a crucial role in the development of hyperuricemia and gout.
6
Inborn xanthine oxidase reductase (XOR) deficiency is a genetical dysfunction of XOR, and it is characterized by a very low or undetectable concentration of UA in the blood and urine, leading to a high level of urinary xanthine called xanthinuria.
7
Previous studies have reported that gout causes some complications, such as tophi, kidney stones and, joint deformities.
8
Drugs approved for the treatment of gout, include allopurinol, cyclooxygenase 2 inhibitors, steroids, and non-steroidal anti-inflammatory drugs. These drugs are effective but they are characterized by several side effects such as allergic skin conditions, skin rashes, fever, renal dysfunction, aseptic meningitis, renal and hepatic dysfunction.
9,10
Allopurinol is the most used XO inhibitor for the treatment of gout,
11
it inhibits XO and thereby increases re-utilization of hypoxanthine and xanthine for the formation of nucleotide and nucleic acid in a reaction catalyzed by hypoxanthine-guanine phosphoribosyltransferase, the consequent increase in nucleotide concentration results in feedback inhibition of de novo purine synthesis. Allopurinol thereby lowers UA concentrations in both urine and serum.
12
Allopurinol causes adverse effects commonly known as allopurinol hypersensitivity syndrome. Consequently, it is necessary to develop potent and less toxic inhibitors of XO. Recently, treating disease using medicinal plants is capturing new interest,
13
and research on medicinal plants has increased worldwide
14,15
due to fewer side effects and lower costs.
16
Ojieh et al.
17
reported that the main aim of sourcing for plants drug is to discover the active constituent in plants that can produce definite pharmacological effects since the results of such research would often serve as a lead compound for the treatment of diseases.
Chromolaena odorata is one of the common plants under investigation for its various health benefits. It is a tropical and subtropical species of flowering shrub in the family of sunflower, a scrambling and perennial plant belonging to Asteraceae , and has been classified as the weed. Previous studies showed that this plant possesses anti-inflammatory, anti-astringent, anti-diuretic and anti-hepatotropic activities,
18
and contains triterpenes, alkaloids and flavonoids.
19
The biological activities of flavonoids in the treatment of diseases have attracted high interest in drug research, they have been reported to possess numerous useful properties.
20
They interfere in carbohydrates metabolism and also have anti-allergic, anti-inflammatory, antiviral, and anticancer properties.
21
Flavonoids have a C6-C3-C6 parental skeleton, based on the structural difference on ring C, they are sub-classified into anthocyanidins, flavones, flavonols, flavanones, flavonols, and isoflavones. Thus, the present study uses in silico analysis via molecular docking, Induced fit docking (IFD), and pharmacological properties to evaluate the anti-gout phytoconstituents of C. odorata . Molecular docking is an in-silico approach that predicts the preferred orientation of small molecules in the active site of a receptor or target protein.
22
It predicts the best binding mode and bio affinities of ligands with their receptor,
23
at present it has been widely applied to virtual screening for the optimization of lead compounds. The basic tools of the molecular docking method are search algorithm and scoring functions for generating and analyzing different conformations of the ligand.
Materials and Methods
Protein preparation
Three-dimensional (3D) crystal structure of XO was retrieved from protein data bank with id: 1FIQ and prepared using the protein preparation wizard of Schrödinger Maestro 11.5.
24
Protein was prepared by assigning bond orders, adding hydrogens, filling missing loops and side-chain using prime, deleting water beyond 5.00 Å, generating tautomeric states of heteroatom groups at a pH of 7.0 ± 2 using Epik and heteroatom state was generated for ligand, then optimized at neutral pH. Using OPLS3 forcefield, restrained minimization was performed setting heavy atom root mean square deviation (RMSD) to 0.30Å.
Ligand preparation
Phytochemicals of C. odorata were obtained from reported literatures and their 2D structures were retrieved from the NCBI PubChem database. The 3D structures were built using LigPrep of Schrödinger Maestro 11.5 with an OPLS3 force field. Ligands ionization states were generated at a pH range of 7.0±2.0 (general pH of biological systems) using Epik.
25
Stereoisomers computation was left at retaining specified chiralities (vary other chiral centers), up to 4 possible stereochemical structures were generated per ligand.
Receptor grid generation
Receptor grid generation allows defining the position and size of the active site for ligand docking. The scoring grid was defined based on the co-crystalized ligand (Salicylic acid) using the receptor grid generation tool in Schrödinger Maestro 11.5. The van der Waals (vdW) radius scaling factor of nonpolar receptor atoms were scaled at 1.0, with a partial charge cut-off of 0.25.
Molecular docking procedure
On a defined receptor grid, the glide tool on Schrödinger Maestro 11.5 was used to carry out molecular docking studies. The prepared ligands were docked using standard precision (SP), setting ligand sampling to flexible, and then docked with extra precision (XP) with the ligand sampling set to none (refine only). The vdW radius scaling factor was scaled at 0.80 with a partial charge cut-off of 0.15 for ligand atoms.
Induced fit docking
IFD protocol was carried out on the top 5 hit compounds using Schrödinger Maestro 11.5. IFD
26
aims to introduce flexibility for both ligand and its target receptor during molecular docking. This is achieved by joining different iterations of docking of flexible ligands into a rigid receptor followed by the protein active site refinement to adopt the best conformation for a ligand. The scoring grid was defined based on the co-crystalized ligand (Salicylic acid) in 1FIQ and no constraints were defined. Sample ring conformation at an energy window of 2.5 kcal/mol were selected for ligand conformational sampling. In glide docking, the vdW radii of protein and ligand were at 0.5 and to generate a maximum of 20 poses per ligand. Refine residues within 5.0Å of ligand poses were refined with prime. Final structures within 30 kcal/mol of the best structure and the top 20 structures overall were re-docked with XP precision.
Molecular docking validation
The employed docking protocol was validated with the blasting of the FASTA sequences XO (PDB id:1FIQ), obtained from protein data bank into the CHEMBL database server (www.ebi.ac.uk/chembl/). From the search results, the bioactivity generated by the database with the IC50 value of 476, and an inhibition value of 121, was downloaded with a canonical smile of the compounds. The bioactivity was sorted out; missing or misplaced data were removed. Only 55 of drug-like compounds were recovered. The complied compounds were converted to 2D (in the sdf format) by data warrior software (version 2). The ligands were imported into Schrödinger Maestro 11.5 and the 3D structures were built by using LigPrep with an OPLS3 force field. The ligands were dock using glide into the target protein receptor using SP precision. A correlation coefficient graph was mapped between the docking scores of 55 compounds generated, and their corresponding PCHEMBL_VALUE was plotted as shown in Fig. 1. Spearman rank correlation coefficient graph was plotted to obtain the correlation (R2) between the PCHEMBL_VALUE and the docking scores of the compounds.
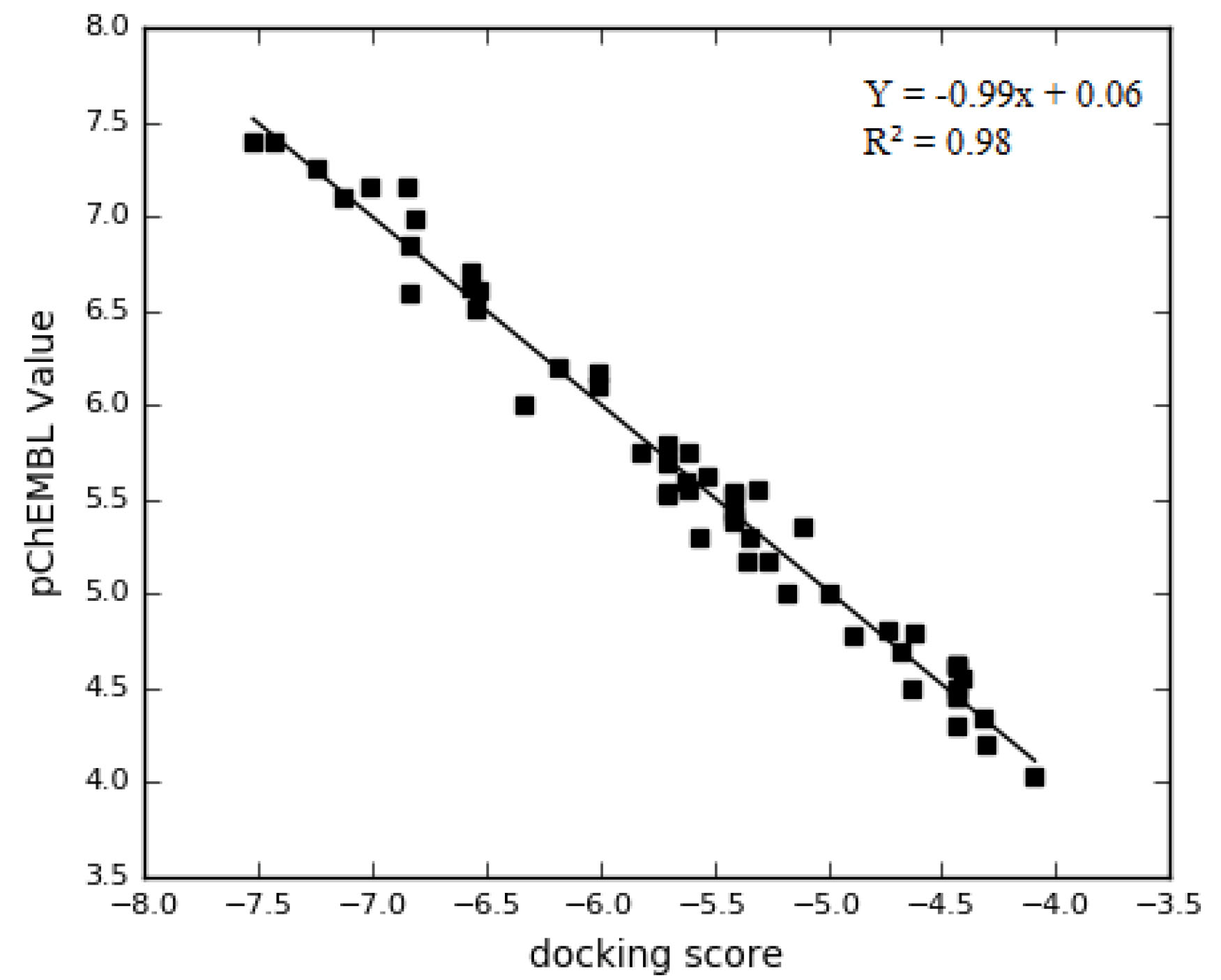
Fig. 1.
The correlation coefficient graph between the experimentally determined pIC50 of 1FIQ and their docked score with R2 of 0.98 indicating that docking experiment can replicate the experimentally determined values of the inhibitors.
.
The correlation coefficient graph between the experimentally determined pIC50 of 1FIQ and their docked score with R2 of 0.98 indicating that docking experiment can replicate the experimentally determined values of the inhibitors.
Pharmacology parameters
Schrödinger QikProp was employed to calculate the Absorption, Distribution, Metabolism and Excretion (ADME) properties of selected phytoconstituents along with the standard compound, for their pharmacological properties such as drug-likeness by checking for any violation of Lipinski’s rule of five (ROF) and several pharmacological parameters including Human Oral Absorption, Predicted octanol/water partition coefficient and prediction of binding to human serum albumin.
Results and Discussion
The present study features in silico experimental procedures described in Fig. 2 to evaluate the molecular interactions, binding mode, pharmacological profile, and inhibitory potential of novel XO inhibitor.
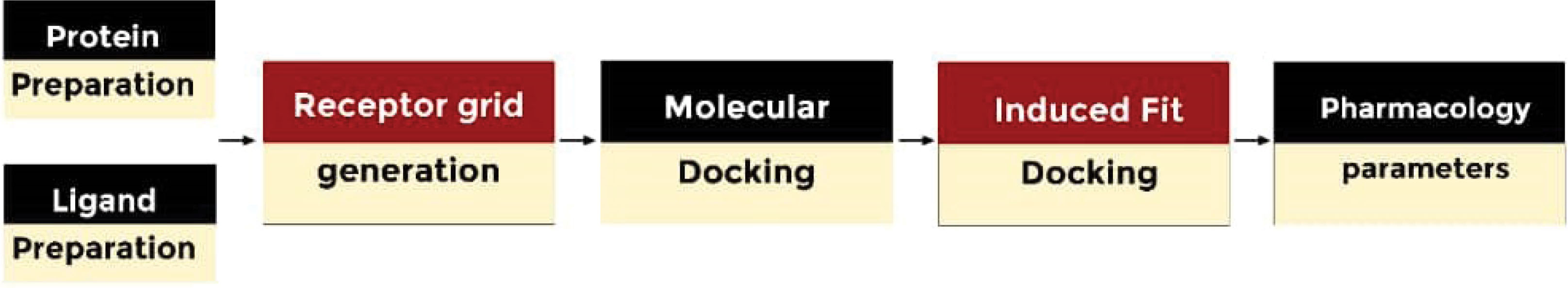
Fig. 2.
Flow chat for identifying potential inhibitors against xanthine oxidase.
.
Flow chat for identifying potential inhibitors against xanthine oxidase.
Molecular docking
Protein docking is a significant tool in the study and provision of a proper understanding of protein-ligand interactions. Protein docking is regularly used in various stages to facilitate the design of potentially active leads.
27
The library of compounds generated was subjected to Glide and IFD to determine compounds with high binding mode and docking scores when compared with the small molecule inhibitor of XO from the protein data bank, Allopurinol. Glide docking scores make use of the basic assumption of a rigid receptor; its result may not be accurate if the ligands induce conformational changes in the target receptor. For this reason, IFD was used to generate flexible receptors (only for hit compounds).
28
The top five hit compounds of glide docking yield flavones scaffold (as shown in Table 1 and Fig. 3), which can also be called ‘skeleton key’ as it plays an important role in numerous compounds that act at different targets to produce pharmacological properties with various substitution pattern.
29
According to the study, flavones scaffold report an induced fit score of -1526.86 kcal/mol, -1527.99 kcal/mol, -1525.25 kcal/mol, -1525.65 kcal/mol and -1525.80 kcal/mol for compounds 1, 2, 3, 4 and 5, respectively. The Glide Gscore and IFD scores of flavones scaffold are lower when compared to that of the standard, Allopurinol which recorded an IFD score of -1519.00 kcal/mol. Based on these results, it is obvious that the flavones scaffold has a relatively better inhibitory than allopurinol. These results are consistent with earlier reports gotten from both in silico and in vitro experiments.
30,31
The docking scores calculated by glide XP-scoring function and IFD are shown in Table 2.
Table 1.
Flavones scaffold generated from scaffold analysis of Chromolaena odorata
|
|
Compound Name
|
| Compound 1 |
2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxychromen-4-one |
| Compound 2 |
2-(3,4-dihydroxyphenyl)-5,7-dihydroxychromen-4-one |
| Compound 3 |
2-(3,4-dihydroxyphenyl)-3,5,6,7-tetrahydroxychromen-4-one |
| Compound 4 |
3,5,7-trihydroxy-2-(3-hydroxy-4-methoxyphenyl)chromen-4-one |
| Compound 5 |
5,7-dihydroxy-2-(3,4,5-trimethoxyphenyl)chromen-4-one |
Table 2.
Docking score calculated by glide XP-scoring function and induced fit docking (IFD)
|
Phyto-constituents
|
Glide XP-Docking Score (kcal/mol)
|
Induced Fit Docking
|
|
Glide Gscore (kcal/mol)
|
IFD score (kcal/mol)
|
| Compound 1 |
-11.339 |
-14.614 |
-1526.86 |
| Compound 2 |
-10.872 |
-14.130 |
-1527.99 |
| Compound 3 |
-11.804 |
-16.303 |
-1525.25 |
| Compound 4 |
-11.185 |
-13.343 |
-1525.65 |
| Compound 5 |
-10.081 |
-12.948 |
-1525.80 |
| 5281702 |
-9.999 |
|
|
| 5280443 |
-9.717 |
|
|
| 1794427 |
-9.298 |
|
|
| 5281697 |
-8.834 |
|
|
| Allopurinol |
-4.109 |
-9.003 |
-1519.00 |
IFD was calculated for hit compounds and standard compound (allopurinol).
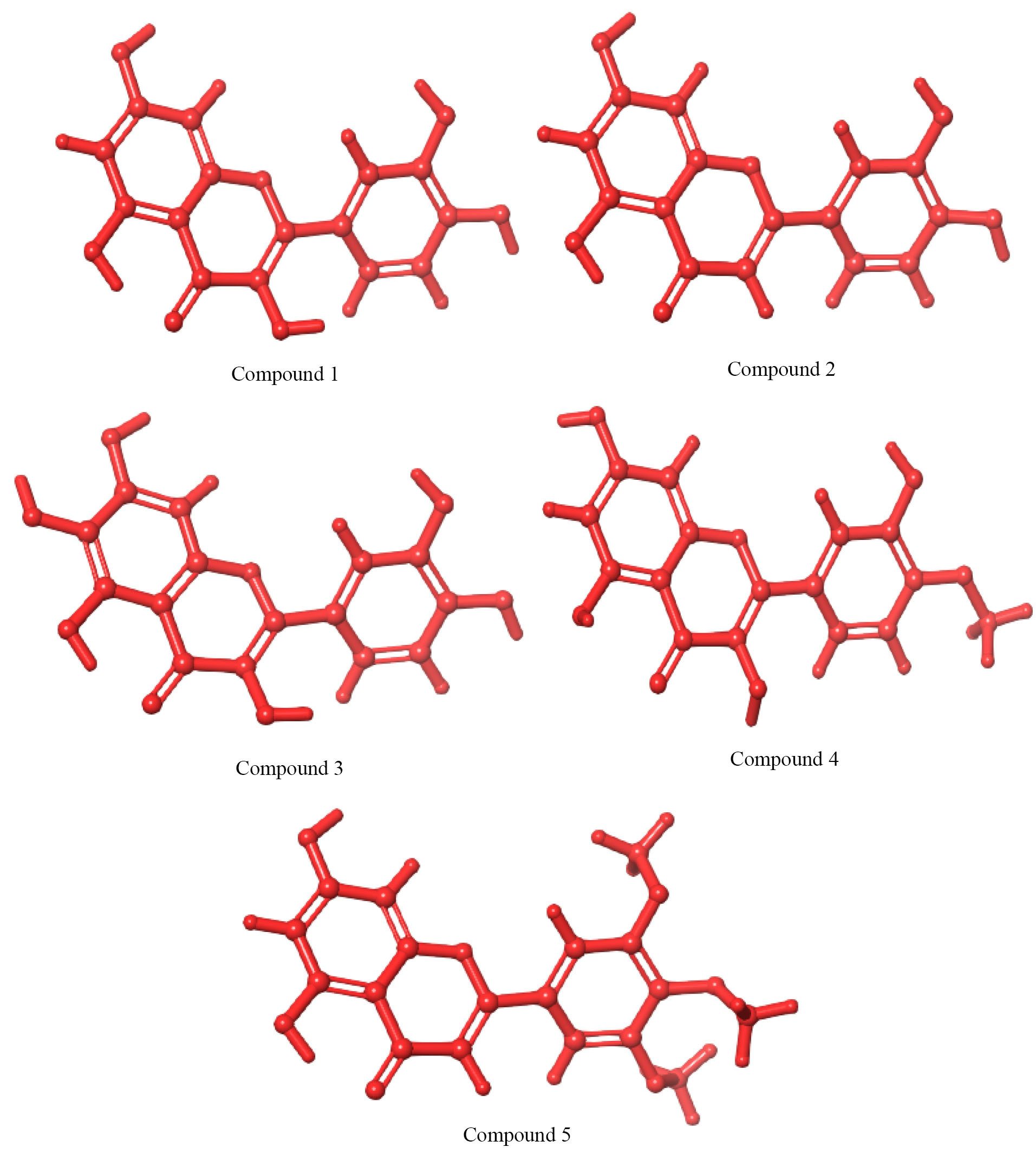
Fig. 3.
3D chemical structure of compounds 1-5
.
3D chemical structure of compounds 1-5
Interaction profiling and scaffold analysis
The active site of XO is believing to contain several amino acid residues. GLU1216, ARG880 and GLU802 residues in the molybdenum center of the gorge play crucial roles in the Xanthine oxidation reaction. At the entrance of the cavity, LEU648, PHE649, PHE914, PHE1009, VAL1011, PHE1013 and LEU1014 regulate the entry of small molecules.
32
Interestingly, Figs. 4-9 shows that flavones scaffold interacted through the formation of pi-pi stacking, hydrogen bond and hydrophobic interaction with the molybdenum atomic domain and some other amino acid residues of XO, which are crucial for the enzymatic activity and hence blocked catalytic activities that is similar to the report of Yan et al.
30
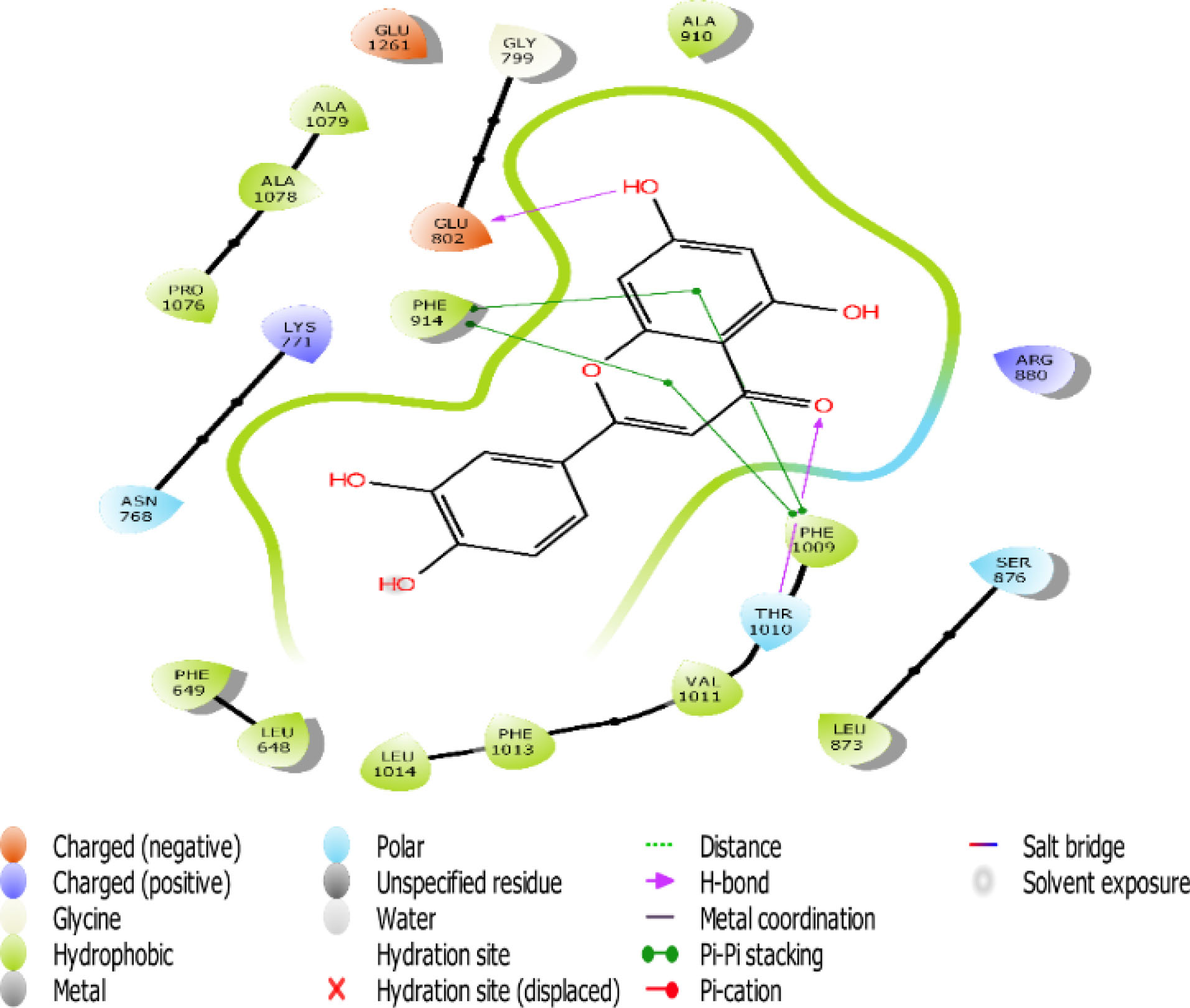
Fig. 4.
The 2D interaction view of compound 1 with amino acid residues at the active site of XO.
.
The 2D interaction view of compound 1 with amino acid residues at the active site of XO.
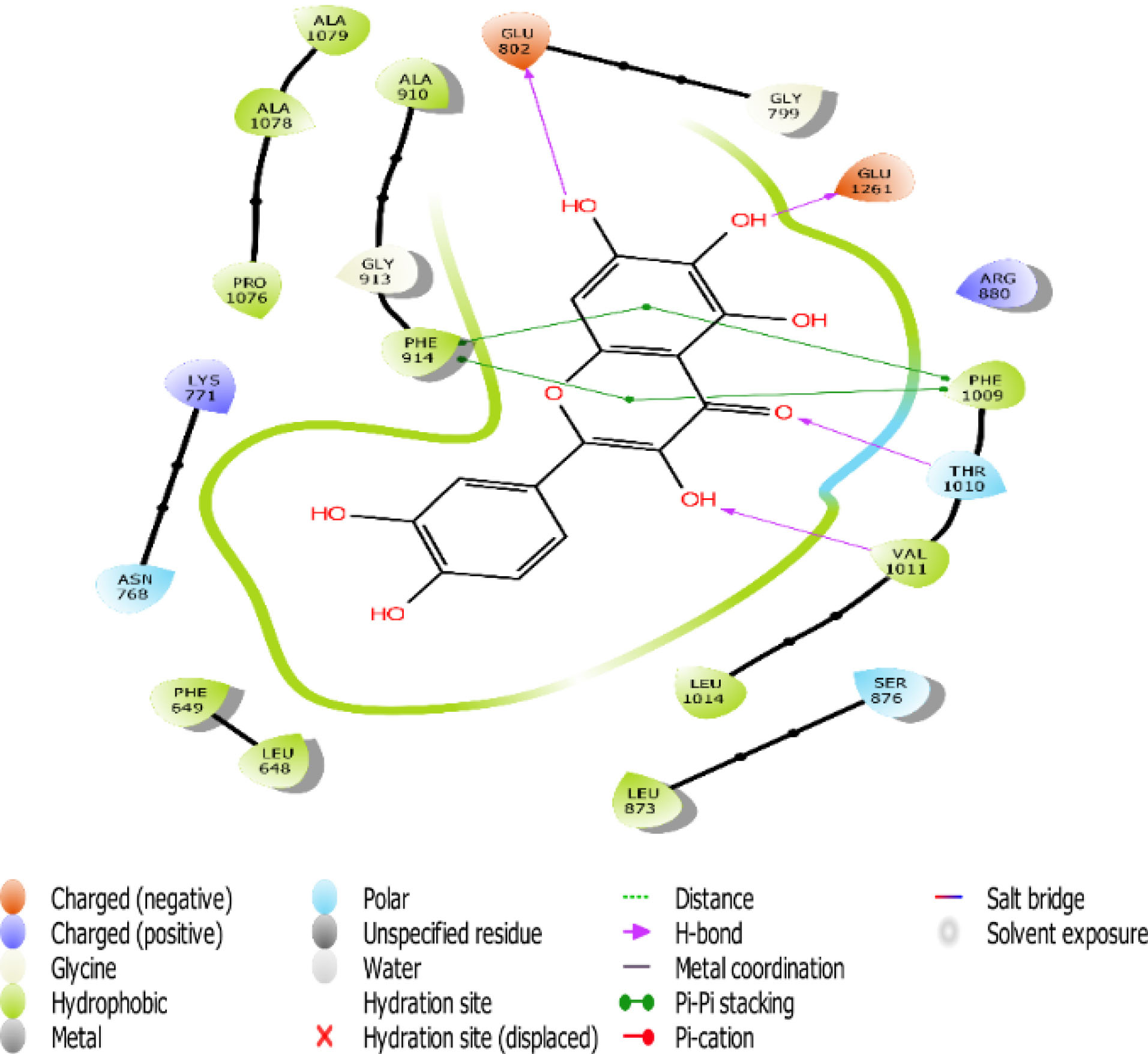
Fig. 5.
The 2D interaction view of compound 2 with amino acid residues at the active site of XO.
.
The 2D interaction view of compound 2 with amino acid residues at the active site of XO.
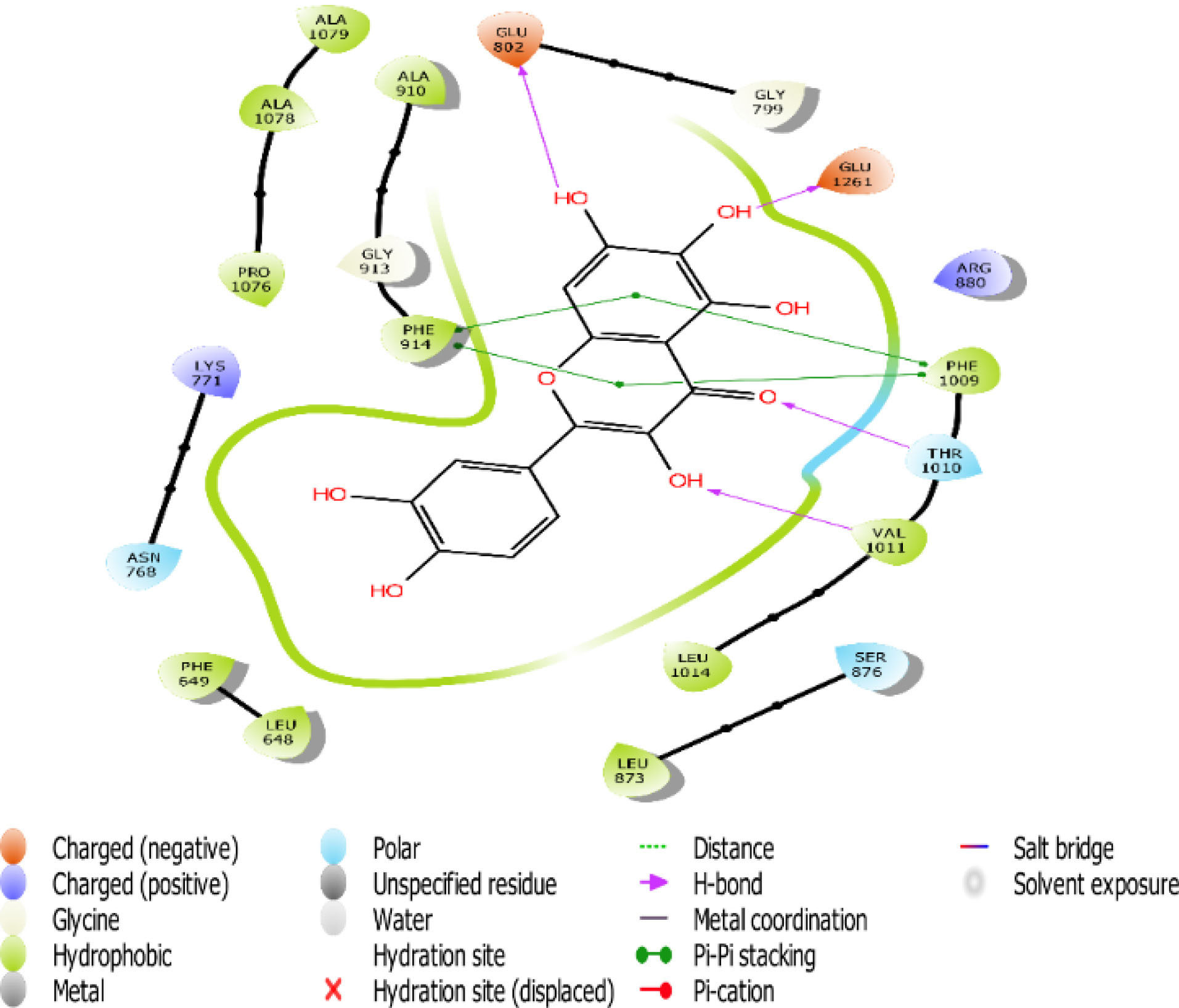
Fig. 6.
The 2D interaction view of compound 3 with amino acid residues at the active site of XO.
.
The 2D interaction view of compound 3 with amino acid residues at the active site of XO.
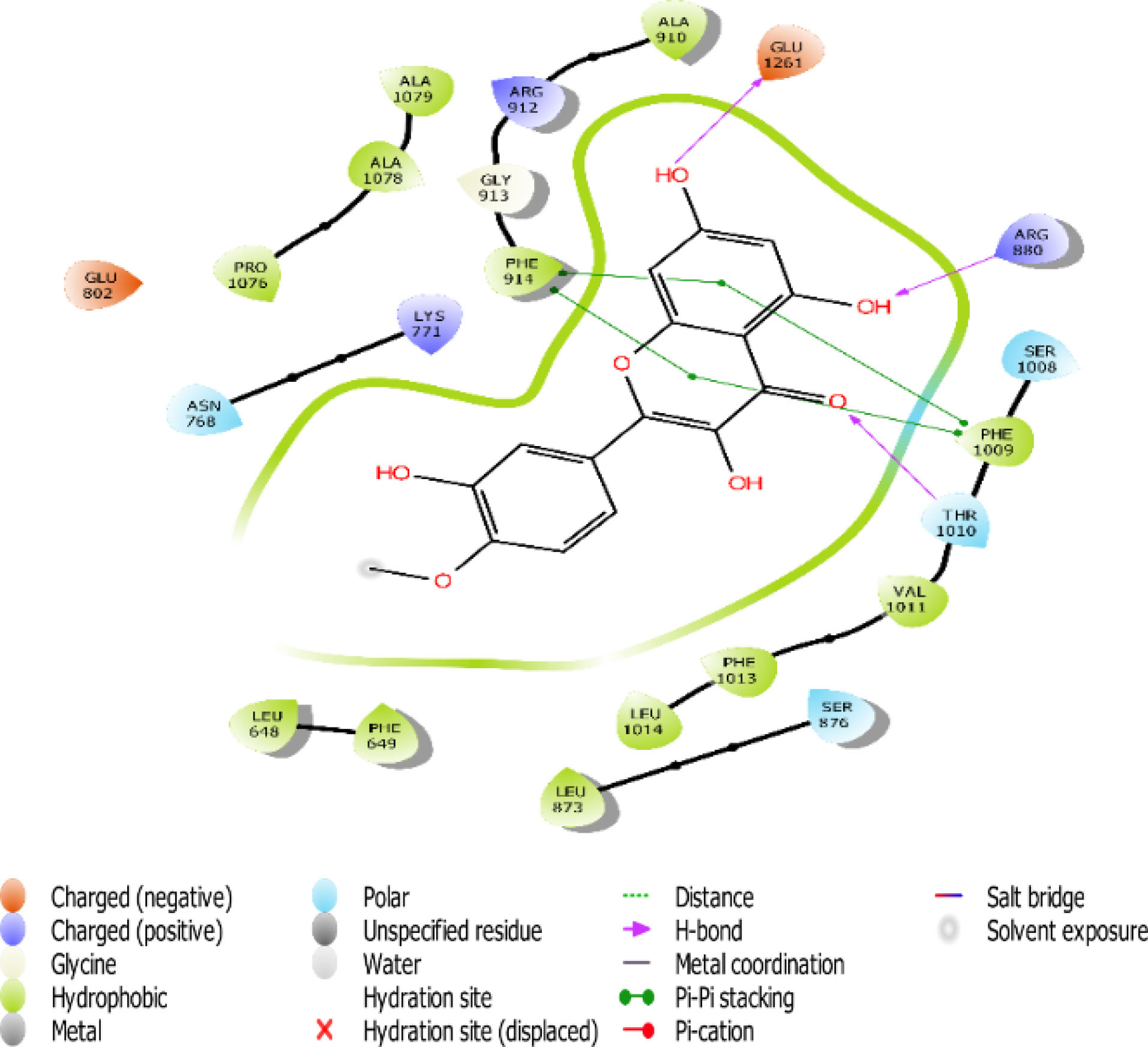
Fig. 7.
The 2D interaction view of compound 4 with amino acid residues at the active site of XO.
.
The 2D interaction view of compound 4 with amino acid residues at the active site of XO.
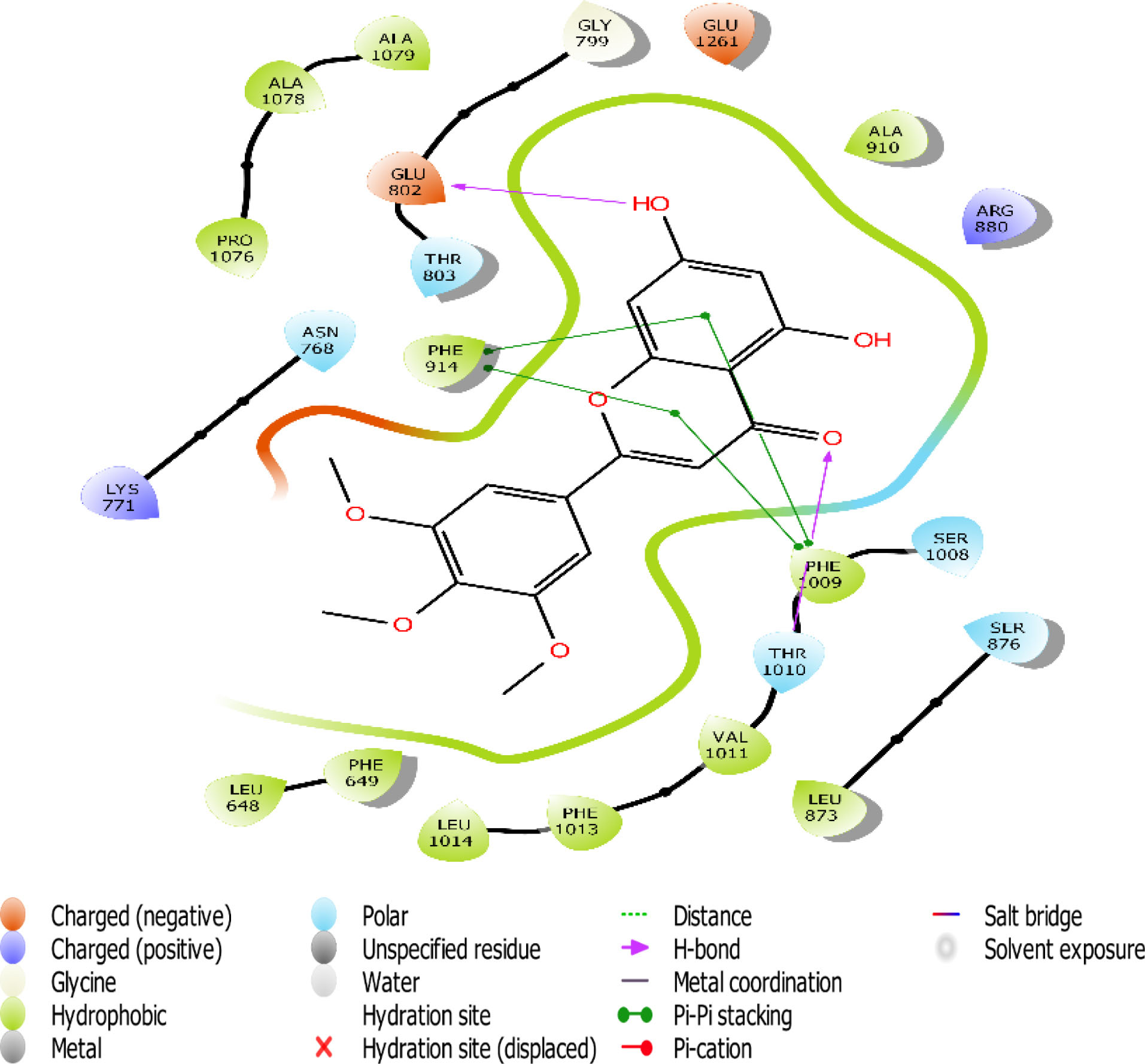
Fig. 8.
The 2D interaction view of compound 5 with amino acid residues at the active site of XO.
.
The 2D interaction view of compound 5 with amino acid residues at the active site of XO.
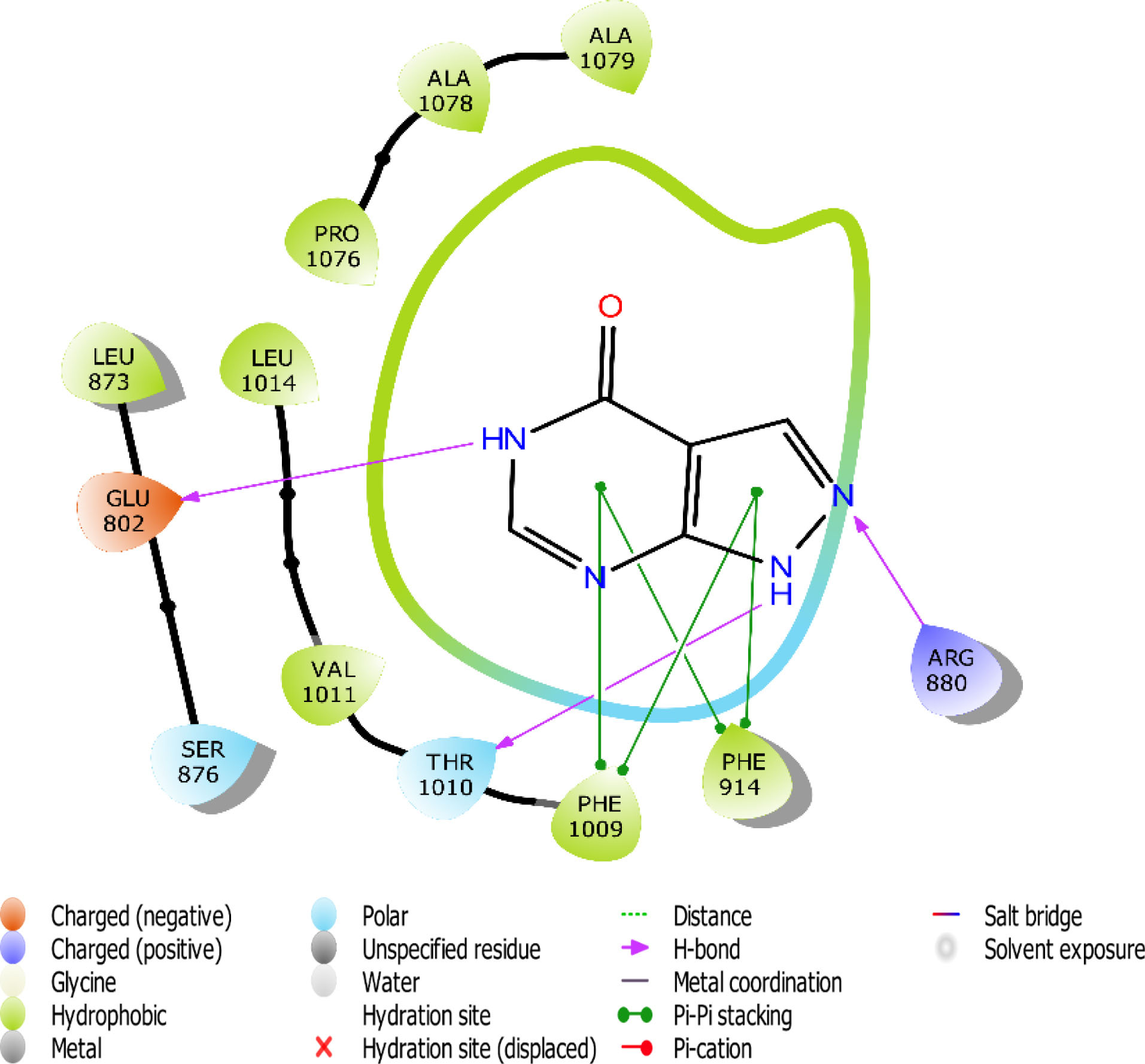
Fig. 9.
The 2D interaction view of Allopurinol with amino acid residues at the active site of XO.
.
The 2D interaction view of Allopurinol with amino acid residues at the active site of XO.
Flavones scaffold consist of phenyl rings A and B joined by a heterocyclic Ring C. The phenyl Ring A and heterocyclic Ring C of flavones scaffold, were buried deep in the hydrophobic side chains, therefore contributing to the stability of the compounds at the hydrophobic pocket by forming pi-pi staking with PHE 914 and PHE1009. The direct staking interactions between PHE 914 and PHE 1009 seem to be a general feature in the binding of substrates in the active site of XO and constitute a good starting point for consideration of competitive inhibition of the enzyme.
33
Hydrogen bond also has a key role in enzyme catalysis and structural stability of many biological molecules. Hydrogen bond interactions exist between flavones scaffold and XO as shown in Figs. 4-8. Compounds 1, 2, 3, and 5 form hydrogen bond interaction with the hydroxyl group at their phenyl ring A with GLU802 and, VAL1011 and GLU1261 in the case of compounds 1 and 3, respectively. Compound 5 also forms hydrogen bond interaction with the hydroxyl group at its phenyl ring A with ARG 880 and GLU1261. Compounds 1, 3, and 4 form hydrogen bond using the carbonyl group at their ring C with polar THR1010. Compounds 2 and 5 also form hydrogen bonds using the carbonyl group at ring C with hydrophobic PHE1009.
Scaffold and substituent analysis were carried out for the top five compounds, and the essentiality of the flavonoid 3-ring structure was revealed both spatially and chemically. Spatially, the 3-ring structure was essential for optimal occupancy of the active site, thus enabling the observed binding pose within the active site. The number and positioning of hydroxyl groups on Ring A have a role to play in displacing the ALA910, a crucial residue for maintaining the hydrophobic shell surrounding it. Only compound 3 with three OH- substituents on Ring A was able to form two hydrogen bonds with the polar GLU802 and GLU 1261, thus displacing ALA910 effectively. Another important observation is the relationship between solvent (water) exposure and the nature of substituents on Ring B. Although they did not seem to engage in any interaction, they play an essential role in the reversible binding of the ligand by disrupting the strong hydrophobicity that surrounds them. Compounds 1, 2, and 3 share similar Ring B nature. Improved active site opening was observed with compound 5 where polar 3 carbonyl groups readily interact with water molecules. The reverse is seen in the case of allopurinol which enjoys a tight hydrophobic space, thus discouraging reversibility, a consequence of desolvation.
34
The result is a zero-order binding kinetic which may have been linked to allopurinol accumulation and toxicity.
35
ADME/Tox
ADME properties of molecules obtained by the Glide docking were checked, using Schrödinger QikProp. Qikprop helps in analyzing the pharmacokinetics properties of molecules by checking the drug-like properties. The predicted ADME properties include the human oral absorption, the number of rotatable bonds, the molecular weight of the molecule, the number of hydrogen bond acceptors, the prediction of binding to human serum albumin, the number of hydrogen bond donors, the predicted hexadecane/gas partition coefficient, and the number of violations of Lipinski’s Ro5.
36
Lipinski’s Ro5 helps to evaluate the drug-likeness, and determine the prospect of small molecules in becoming an orally active drug for human. The rule permits a molecular weight <500 Da, octanol-water partition coefficient < 5, hydrogen bond donor ≤ 5, and hydrogen bond acceptor ≤ 10. Prospective drug candidates that obey the Ro5 tend to have lower attrition rates at the stage of clinical trials and for this reason, it has an increased chance of becoming and staying marketable.
36
Compounds of C. odorata have shown excellent result and are in accordance with this rule. Therefore, they can be developed as a promising lead in the design of XO inhibitors. The results of the ADME properties are shown in Table 3.
Table 3.
QikProp results of selected compounds based on docking score with Standard compound
|
Phyto-constituents
|
#rotor
A
|
Mol_MW
B
|
donorHB
C
|
acceptHB
D
|
QPlogPo /w
E
|
QPlogKhsa
F
|
HOA
G
|
ROF
H
|
| Compound 1 |
5 |
302.240 |
4.000 |
5.250 |
0.315 |
-0.364 |
2 |
0 |
| Compound 2 |
4 |
286.240 |
3.000 |
4.500 |
0.926 |
-0.200 |
3 |
0 |
| Compound 3 |
6 |
318.239 |
5.000 |
6.000 |
-0.306 |
-0.501 |
2 |
1 |
| Compound 4 |
5 |
316.267 |
3.000 |
5.250 |
1.152 |
-0.175 |
3 |
0 |
| Compound 5 |
5 |
344.320 |
1.000 |
5.250 |
2.688 |
0.172 |
3 |
0 |
| Allopurinol |
0 |
136.113 |
2.000 |
4.500 |
-0.551 |
-0.741 |
3 |
0 |
A. Number of rotatable bonds (Range: 0 to 15) B. Molecular weight of the molecule (Range: 130.0 to 725.0) C. Number of hydrogen bond donors (Range: 0.0 to 6.0) D. Number of hydrogen bond acceptors (Range: 2.0 to 20.0) E. Predicted octanol/water partition coefficient. (Range: -2.0 to 6.5) F. Prediction of binding to human serum albumin (Range: -1.5 to 1.5) G. Human Oral Absorption (Range: 1, 2, or 3 for low, medium, or high) H. Number of violations of Lipinski’s rule of five (Range: maximum is 4).
Conclusion
The docking study and pharmacology parameters revealed that flavones scaffold could be a useful drug candidate for preventing and treating gout, flavones scaffold has a higher binding affinity with XO, interacting through the formation of pi-pi stacking, hydrogen bond, and hydrophobic interaction. Therefore, in vivo and or in vitro assays could be examined to further demonstrate its potential to inhibit XO.
Acknowledgments
Authors like to acknowledge the Centre for Biocomputing and Drug Development (CBDD), AAUA.
Ethical statement
No ethical issue to be declared.
Competing interests
Authors declare no conflicts of interest.
Authors’ contribution
KB brought the presented idea. KB, OAJ, IOK and ANS designed the computational flow chat. MDS and IOK provided the study equipment and materials. ANS, DTI and OOO retrieved ligands and protein from their respective database for molecular docking studies. KB and ITT wrote the manuscript with input from all the authors. OOI reviewed the article before submission.
Research Highlights
What is the current knowledge?
simple
-
√ Flavones scaffold interacts with the molybdenum atomic
domain and some other amino acid residues of XO that are
essential for enzymatic action and hence blocked catalytic
activities.
What is new here?
simple
-
√ Flavones scaffold has relatively better inhibitory than
allopurinol and may poses lesser side effects in the treatment
of gout.
References
- Gill T. Epidemiology and health impact of obesity: an Asia Pacific perspective. Asia Pac J Clin Nutr 2006; 15:3-14. [ Google Scholar]
- Dalbeth N, Lauterio TJ, Wolfe HR. Mechanism of action of colchicine in the treatment of gout. Clin Ther 2014; 36:1465-79. doi: 10.1016/j.clinthera.2014.07.017 [Crossref] [ Google Scholar]
- Nile SH, Kumar B, Park SW. In vitro evaluation of selected benzimidazole derivatives as an antioxidant and xanthine oxidase inhibitors. Chem Biol Drug Des 2013; 82:290-5. doi: 10.1111/cbdd.12141 [Crossref] [ Google Scholar]
- Symons MC, Taiwo AF, Petersen LR. Electron addition to xanthine oxidase An electron spins resonance study of the effects of ionizing radiation. J Chem Soci 1989; 85:4063-74. doi: 10.1039/F19898504063 [Crossref] [ Google Scholar]
- Zarepour M, Kaspari K, Stagge S, Rethmeier R, Mendel R, Bittner F. Xanthine dehydrogenase AtXDH1 from Arabidopsis thaliana is a potent producer of superoxide anions via its NADH oxidase activity. Plant Mol Biol 2010; 72:301-10. doi: 10.1007/s11103-009-9570-2 [Crossref] [ Google Scholar]
- Egwim EC, Vunchi MA, Egwim PO. Comparison of xanthine oxidase activities in cow and goat milks. Biokemistri 2005; 17:1-6. doi: 10.4314/biokem.v17i1.32581 [Crossref] [ Google Scholar]
- Borges F, Feranandes E, Roleira F. Progress towards the discovery of xanthine oxidase inhibitors. Curr Med Chem 2002; 9:195-217. doi: 10.2174/0929867023371229 [Crossref] [ Google Scholar]
- Teh CL, Chew KF, Ling GR. Acute gout in hospitalized patients in Sarawak general hospital. Med J Malaysia 2014; 69:126-8. [ Google Scholar]
- Nguyen MT, Awale S, Tezuka Y, Le Tran Q, Watanabe H, Kadota S. Xanthine oxidase inhibitory activity of Vietnamese medicinal plants. Biol Pharm Bull 2004; 27:1414-21. doi: 10.1248/bpb.27.1414 [Crossref] [ Google Scholar]
- Strazzullo P, Puig JG. Uric acid and oxidative stress: relative impact on cardiovascular risk. Nutr Metab Cardiovasc Dis 2007; 17:409-14. doi: 10.1016/j.numecd.2007.02.011 [Crossref] [ Google Scholar]
- Pacher P, Nivorozhkin A, Szabó Szabó, C C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev 2006; 58:87-114. doi: 10.1124/pr.58.1.6 [Crossref] [ Google Scholar]
- Recalls P, Reporting AR, Update P, Advisory MA. Anticonvulsant hypersensitivity syndrome. Prescriber Update 1998; 16:28-31. [ Google Scholar]
- Unno T, Sugimoto A, Kakuda T. Xanthine oxidase inhibitors from the leaves of Lagerstroemia speciosa (L) Pers. J Ethnopharmacol 2004; 93:391-5. doi: 10.1016/j.jep.2004.04.012 [Crossref] [ Google Scholar]
- Tapsell LC, Hemphill I, Cobiac L, Sullivan DR, Fenech M, Patch CS. Supplement-health benefits of herbs and spices: the past, the present, the future. Med J Aust 2006; 185:4-24. [ Google Scholar]
- Triggiani V, Resta F, Guastamacchia E, Sabba C, Licchelli B, Ghiyasaldin S. Role of antioxidants, essential fatty acids, carnitine, vitamins, phytochemicals and trace elements in the treatment of diabetes mellitus and its chronic complications. Endocr Metab Immune Disord Drug Targets 2006; 6:77-93. doi: 10.2174/187153006776056611 [Crossref] [ Google Scholar]
- Srivastava S, Lal VK, Pant KK. Polyherbal formulations based on Indian medicinal plants as antidiabetic phytotherapeutics. Phytopharmacology 2012; 2:1-5. [ Google Scholar]
- Ojieh AE, Adegor EC, Lawrence EO. Preliminary phytochemical screening, analgesic and anti-inflammatory properties of Celosia isertii. European J Med Plants 2013; 3:369-80. doi: 10.9734/EJMP/2013/3257 [Crossref] [ Google Scholar]
- Iwu Iwu, M M. M Handbook of African Medicinal Plants CRC Press. Beca Raton 1993:181-2.
- Asomugha RN, Okafor PN, Ijeh II, Orisakwe OE, Asomugha AL, Ndefo JC. Toxicological evaluation of aqueous leaf extract of Chromolaena odorata in male wistar albino rats. J Appl Pharm Sci 2013; 3:89. [ Google Scholar]
- Bahadori MB, Asghari B, Dinparast L, Zengin G, Sarikurkcu C, Abbas-Mohammadi M. Salvia nemorosa L: A novel source of bioactive agents with functional connections. LWT 2017; 75:42-50. doi: 10.1016/j.lwt.2016.08.048 [Crossref] [ Google Scholar]
- Hanhineva K, Törrönen R, Bondia-Pons I, Pekkinen J, Kolehmainen M, Mykkänen H. Impact of dietary polyphenols on carbohydrate metabolism. Int J Mol Sci 2010; 11:1365-402. doi: 10.3390/ijms11041365 [Crossref] [ Google Scholar]
- Surabhi S, Singh BK. COMPUTER AIDED DRUG DESIGN: AN OVERVIEW. J Drug Deliv Ther 2018; 8:504-9. doi: 10.22270/jddt.v8i5.1894 [Crossref] [ Google Scholar]
- Dinparast L, Valizadeh H, Bahadori MB, Soltani S, Asghari B, Rashidi MR. Design, synthesis, α-glucosidase inhibitory activity, molecular docking and QSAR studies of benzimidazole derivatives. J Mol Struct 2016; 1114:84-94. [ Google Scholar]
- Caliceti C, Capriotti AL, Calabria D, Bonvicini F, Zenezini Chiozzi R, Montone CM. Peptides from Cauliflower By-Products, obtained by an Efficient, Ecosustainable, and Semi-Industrial Method, Exert Protective Effects on Endothelial Function. Oxid Med Cell Longev 2019; 2019:1046504. doi: 10.1155/2019/1046504 [Crossref] [ Google Scholar]
- Greenwood JR, Calkins D, Sullivan AP, Shelley JC. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J Comput Aided Mol Des 2010; 24:591-604. doi: 10.1007/s10822-010-9349-1 [Crossref] [ Google Scholar]
- Zhong H, Tran LM, Stang JL. Induced-fit docking studies of the active and inactive states of protein tyrosine kinases. J Mol Graph model 2009; 28:336-46. doi: 10.1016/j.jmgm.2009.08.012 [Crossref] [ Google Scholar]
- Huang SY, Zou X. Advances and Challenges in Protein-Ligand docking. Int J Mol Sci 2010; 11:3016-34. doi: 10.3390/ijms11083016 [Crossref] [ Google Scholar]
- Banerjee K, Gupta U, Gupta S, Wadhwa G, Gabrani R, Sharma SK. Molecular docking of glucosamine-6-phosphate synthase in Rhizopus oryzae. Bioinformation 2011; 7:285. doi: 10.6026/007/97320630007285 [Crossref] [ Google Scholar]
- Singh M, Kaur M, Silakari O. Flavones: An important scaffold for medicinal chemistry. Eur J Med Chem 2014; 84:206-39. doi: 10.1016/j.ejmech.2014.07.013 [Crossref] [ Google Scholar]
- Yan J, Zhang G, Hu Y, Ma Y. Effect of luteolin on xanthine oxidase: Inhibition kinetics and interaction mechanism merging with docking simulation. Food chem 2013; 141:3766-73. doi: 10.1016/j.foodchem.2013.06.092 [Crossref] [ Google Scholar]
- Umamaheswari M, Prabhu P, Asokkumar K, Sivashanmugam T, Subhadradevi V, Jagannath P. In silico docking studies and in vitro xanthine oxidase inhibitory activity of commercially available terpenoids. Int J Phytopharm 2012; 4:3460-2. doi: 10.7439/ijpp.v2i5.617 [Crossref] [ Google Scholar]
- Okamoto K, Matsumoto K, Hille R, Eger BT, Pai EF, Nishino T. The crystal structure of xanthine oxidoreductase during catalysis: implications for reaction mechanism and enzyme inhibition. Proc Natl Acad Sci 2004; 101:7931−6. doi: 10.1073/pnas.0400973101 [Crossref] [ Google Scholar]
-
Pauff JM. Structure-Function Studies of Xanthine Oxidoreductase. [PhD dissertation]. The Ohio State University, 2008.
- Enroth C, Eger BT, Okamoto K, Nishino T, Nishino T, Pai EF. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: structure-based mechanism of conversion. Proc Natl Acad Sci 2000; 97:10723-8. doi: 10.1073/pnas.97.20.10723 [Crossref] [ Google Scholar]
- Vauquelin G, Charlton SJ. Long‐lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br J Pharmcol 2010; 61:488-508. doi: 10.1111/j.1476-5381.2010.00936.x [Crossref] [ Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews 1997; 46:3-26. doi: 10.1016/S0169-409X(96)00423-1 [Crossref] [ Google Scholar]