Bioimpacts. 10(4):269-278.
doi: 10.34172/bi.2020.34
Original Research
Development of binary dispersions and nanocomposites of irbesartan with enhanced antihypertensive activity
Vivek Puri 1, Manju Nagpal 1, *  , Ameya Sharma 1, Gurjeet Singh Thakur 1, Manjinder Singh 1, Geeta Aggarwal 2
, Ameya Sharma 1, Gurjeet Singh Thakur 1, Manjinder Singh 1, Geeta Aggarwal 2
Author information:
1Chitkara College of Pharmacy, Chitkara University, Punjab, India
2Delhi Pharmaceutical Sciences and Research University, New Delhi-110017, India
Abstract
Introduction:
Irbesartan (IBS), an angiotensin II receptor (AT1 subtype) antagonist which blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selective binding to AT1 angiotensin II receptor. It belongs to BCS class II drug (low aqueous solubility and high permeability). Improvement of dissolution characteristics of the drug by formulating is being investigated in the current study.
Methods:
Solid dispersions (SD) formulations were prepared by the melting fusion technique and nanocomposites (NC) were prepared by a single emulsion technique. Eight batches of SD and three batches of NC were formulated in three ratios of drug to polymer (1:1, 1:2, and 1:3). The batches were evaluated for equilibrium solubility studies, Fourier-transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC), X-ray diffraction (XRD), scanning electron microscopy (SEM), field emission SEM (FESEM), transmission electron microscopy (TEM), and in vitro dissolution studies.
Results:
Solubility studies revealed maximum solubility at a 1:2 ratio of solid dispersions and a 1:1 ratio of nanocomposites. No drug-polymer interaction was observed in FTIR results. DSC, SEM, and XRD analysis revealed changes in drug crystallinity i.e. conversion to the amorphous state of drugs. Nanosize of particles in the NC1 batch was confirmed in TEM studies. Solid dispersions and nanocomposites showed significant enhancement of dissolution in comparison to that of the pure drug (100% drug release in approximately 1 hour).
Conclusion:
Nanocomposites proved superior carriers to solid dispersions in terms of the dissolution enhancement. Further, in vivo studies indicated that the induction of systolic and diastolic blood pressure in the optimized formulation (NC1) was significantly decreased in comparison to the disease control group (P <0.01) at all time intervals along with pure drug (P <0.05).
Keywords: Soluplus, Nanocomposites, Dissolution, Equilibrium solubility, Crystalline, Amorphous
Copyright and License Information
© 2020 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
The majority of new drug molecules in the pharmaceutical industry have deficient/poor solubility in water and reveal slow dissolution characteristics and thereby low bioavailability when given orally. This has posed a challenge for scientists to look out new ventures to conquer the repercussion of low solubility. Improving aqueous solubility of drug molecules by developing solid dispersions using hydrophilic polymers is the most promising one.
1-4
Solid dispersions tend to retain the amorphous state of drug molecules during their processing and also during storage. The amorphous state of the drug exhibits higher solubility i.e. about 10–1000 times to that of its crystalline form.
5
This amorphization of a drug is dependent on the drug concentration in solid dispersions, i.e., the drug may re-crystallize beyond a specific drug to polymer ratio.
6
Irbesartan which is chemically 2-butyl-3-[[2-(1H -tetrazole-5-yl)(1,1-biphenyl)-4-yl]-1,3-diazaspiro[4,4] non-1-en-4-one, antagonizes angiotensin I receptor leading to lowering of blood pressure. Although it exhibits good oral bioavailability (i.e., 60-80%), theoretically its bioavailability is limited by its solubility.
7
Several advanced techniques and carrier systems such as complexation with β-cyclodextrin,
8
crystal engineering technique,
9
self nano-emulsifying carriers,
10
liquisolid compacts,
11
solid dispersions,
12
wet granulation,
13
fusion and co-solvent technique,
14
solvent evaporation method
15
have been reported and of great interest to the pharmaceutical industry. Even as these existing strategies are gradually altered to tackle this issue, the future can still be bright with new technologies that deal with ultrafine particles in the nanoscale regime. Several innumerable nanotechnology-based formulations of irbesartan such as nano-crystals,
16
solid lipid nanoparticles,
17
polymeric nanoparticles,
18
polymeric self-assemblies and liposomes,
19
nano-crystalline suspensions
20
have also been worked out by researchers. Soluplus (amphiphilic graft co-polymer of polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol) exhibits amphiphilic properties and it acts as a carrier in matrices and also a good solubilizer.
21
Soluplus has been reported to increase bioavailability and solubility of poorly soluble drugs via hot-melt extrusion
22
with easy processing and excellent extrudability. It is perfect for the hot-melt extrusion technique and used to develop fourth-generation solid dispersions. Recently, Soluplus based composites
23
and films
24
has been studied for delivery of vitamin E, Soluplus-based micellar systems with enhanced permeability have been reported.
25,26
To continue with an escalating trend to look for nanotechnology-based solutions, the current study explored the use of a Soluplus matrix for the feasibility of developing irbesartan nanoparticles using a single emulsion technique with improved wetting characteristics with a decreased agglomeration of irbesartan nanoparticles. The study also explored solid dispersions of irbesartan using Soluplus-PEG (polyethylene glycol) hybrid polymer via the melting fusion method and evaluated dissolution characteristics.
Materials and Methods
Irbesartan was kindly gifted by Jubilant Life Sciences, Noida, U.P. Soluplus graft copolymer was kindly gifted by BASF, Mumbai. PEG 6000 was purchased from Central Drug House, New Delhi. All the other chemicals and solvents were of AR grade.
Methods
Preparation of solid dispersions via melting/fusion method
Accurately weighed amounts of polymer PEG 6000 and Soluplus were placed into a china dish and kept on a sand bath. The mixture was allowed to melt and melting occurs at a temperature range of 120-150˚C. The specified amount of drug was added in polymeric melt followed by vigorous stirring until complete mixing of the drug within the polymeric melt. The blend was solidified after cooling and further triturated using a mortar pestle and sieved through # 80. The obtained blend in powdered form was kept in a desiccator for further investigations.
Effect of pre-trituration
The drug, Soluplus, and PEG 6000 was triturated using a mortar pestle and melted in china dish using a hot plate. The SD batches were denoted as SD1b, SD2b, and SD3b.
Effect of amount of PEG 6000
The amount of PEG 6000 used was varied from 100-300 mg. The batches SD2c and SD3c contained 200 and 300 mg PEG respectively whereas SD1a and SD1b contain 100 mg PEG 6000.
Preparation of nanocomposites via single emulsion technique
Nanoemulsion of irbesartan using Soluplus was formulated via a single emulsion technique. The organic phase consisted of polymer-drug mixture dissolved in 20 mL dichloromethane (DCM) in various ratios (1:1, 2:1, and 3:1) and the aqueous phase consisted of 20 mL of distilled water with Tween 80 (0.05%) as the surfactant. The organic phase was slowly added dropwise into the aqueous one using a syringe pump with a flow rate of 0.3 mL/min, along with concurrent ultrasonication in an ice bath for effectual homogenization of both the phases. After that, the mixture was stirred magnetically for 24 hours for the complete elimination of solvent DCM. The mixture was further lyophilized at −40˚C for about 12 hours (Mini Lyodel, India) and softly ground to obtain free-flowing nanocomposite powder. The powders were then sieved through # 80 sieve and kept in desiccators. The three formulations were denoted as NC1, NC2, and NC3 for three drugs to polymer ratios (1:1, 1:2, and 1:3), respectively. The composition of various formulation batches is depicted in Table 1.
Table 1.
Composition of various solid dispersion batches and nanocomposites
|
Formulation code
|
Drug (mg)
|
Soluplus (mg)
|
PEG (mg)
|
| SD 1a (without pre-trituration) |
200 |
200 |
100 |
| SD1b (with pre-trituration) |
200 |
200 |
100 |
| SD 2a (without pre-trituration) |
200 |
400 |
100 |
| SD 2b (with pre-trituration) |
200 |
400 |
100 |
| SD 2c (with pre-trituration) |
200 |
400 |
200 |
| SD 3a (without pre-trituration) |
200 |
600 |
100 |
| SD 3b (with pre-trituration) |
200 |
600 |
100 |
| SD 3c (with pre-trituration) |
200 |
600 |
300 |
| NC1 |
200 |
200 |
- |
| NC2 |
200 |
400 |
- |
| NC3 |
200 |
600 |
- |
Evaluation of formulations
Equilibrium solubility studies
The equilibrium solubility was determined by dissolving prepared formulations (100 mg equivalent to the drug) in distilled water (25 mL) in a volumetric flask for 24 hours with discontinuous stirring in an orbital shaker. The samples were filtered and drug absorbance of various sample solutions was measured via UV spectrophotometer at 244 nm.
Drug encapsulation efficiency (DEE)
The DEE of IBS in NC1, NC2, and NC3 was determined by ultrafiltration using a centrifuge device. The filter was filled with 2 ml of NC formulation and centrifuged at 5000 rpm for 10 minutes. The unloaded drug concentration was determined spectrophotometrically. The EE was calculated using the following equation:
EE (%) = (CT-CF)/CT *100
Where, CT = total drug concentration in NC, CF = free drug concentration in the aqueous phase.
Fourier-transform infrared spectroscopy (FTIR) studies
KBr pellet method was applied for FTIR analysis (Perkin Elmer Spectrum 400). Pellets incorporating 10 mg samples are formed by compressing the mixture by using a hydraulic press. Transparent pellets were scanned over the range of 400–4000 cm-1.
Scanning electron microscopy (SEM) studies
Pure drug, polymer, and formulations SD2 and NC1 samples were mounted onto the stubs using double-sided adhesive tape and coated with gold-palladium alloy (150-200 Ao) using fine coat ion sputter (Joel, fine coat ion sputter, JFC-1100). The samples were subsequently analyzed for external morphology.
Transmission electron microscopy (TEM)
Samples were stained with phosphotungstic acid (PA) solution 1% (w/w). Excess of stain was removed using filter paper and the grid was dried to imaging. The images of samples were taken using a transmission electron microscope, Morgagni 268D, (Morgagni 268D, Fei Electron Optics, USA).
X-ray diffraction (XRD)
XRD patterns were traced employing the X-ray diffractometer using Ni filtered Cu (K-α) radiations, a voltage of 45 kV, a current of 40 mA. The samples were scanned over 2θ range of 0-50º with a scan step size of 0.0170º (2θ) and scan step time of 25 seconds.
Field emission scanning electron microscopy (FESEM) studies
Carbon tapes double sided were fixed on an aluminum stud and the powdered samples were sprinkled over the tape and gold-sputtered for 10 minutes. Then the aluminum stub was kept in a vacuum chamber of a scanning electron microscope (Carl Zeiss SMT-Super Ultra Model Gemini Ultra 55) operated at 4 kV. The surface characteristics of the particles were observed.
Differential scanning calorimetry (DSC) studies
Samples of pure drug irbesartan, polymer and formulations (SD2 and NC1)(approx. 6.5–10.9 mg) are heated under a nitrogen atmosphere using an aluminum pan at a rate of 10°C/min over 30 to 300°C temperature range. The DSC thermograms of samples were obtained using differential scanning calorimeter.
In vitro dissolution studies
In vitro drug release fromvarious batches of solid dispersions (SD1B, SD2B, and SD3B) and nanocomposites (NC1, NC2, and NC3) was carried out using type II USP dissolution test apparatus, at 37 ± 0.5°C and 100 rpm in 900 mL of 0.1N HCl. Aliquots (5 mL) were taken at determined time intervals (05, 15, 30, 45, 60, 90, and 120 minutes), filtered, suitably diluted and analyzed at 240 nm using UV-Visible Spectrophotometer (AU2701, Systronics, India).
In vitro release kinetics
To analyze the kinetics of drug release, various release kinetic models (zero-order, first-order, Higuchi, and Korsmeyer-Peppas model) were applied to in vitro release data.
In vivo studies
The standard laboratory diet of inbred male Wistar rats (230±20 g) was maintained (Ashirwad Feed Industry, Chandigarh) and kept only at tap water in the current study. The rats were set aside in the departmental animal house and were given 12 hours of light and dark cycle conditions. The experiments were conducted in a semi-sound proof laboratory.
In vivo anti-hypertensive study using adrenaline-induced hypertensive rats (non-invasive method)
Rats were anesthetized and 0.1 mL of adrenaline intraperitoneally (i.p.) was injected into rats for 10 consecutive days to induce hypertension.
27
To confirm the induction of hypertension, different hemodynamic parameters were calculated by using a non-invasive tail-cuff method with Biopac Polyrite 4 channel system.
Method to measure hemodynamic blood pressure parameters using BIOPAC Power lab with tail-cuff apparatus
The blood pressure was measured after the administration of dose to animal by non-invasive tail-cuff method.
28
Rats were kept in a strainer and the tail-cuff was applied on the tail of rat for determination of blood pressure. Normal blood pressure of all the rats was recorded as baseline blood pressure. After that, the animals were treated with individual drug treatments and again blood pressure was recorded in intervals of 30, 60, 90, and 120 minutes. The blood pressure as systolic blood pressure (SBP), diastolic blood pressure (DBP), were displayed on the monitor were recorded. To evaluate the anti-hypertensive effect of pure drug and formulation (NC1), adrenaline was injected after 5 minutes. Again, blood pressure was recorded. The difference between baseline blood pressure and blood pressure after various drug treatments was calculated and compared.
Statistical analysis
Data obtained from the study were statistically analyzed using one-way ANOVA followed by Tukey’s multiple range tests as post hoc analysis. A value of P < 0.05 was considered to be statistically significant.
Results and Discussion
Equilibrium solubility studies
SD batches with a different drug to polymer ratios (1:1, 1:2, and 1:3) were prepared. PEG was used as a plasticizer along with Soluplus in SD formulations. The effect of the amount of PEG and the effect of pre trituration of drug and Soluplus-PEG blend was also studied. The aqueous solubility of pure drug irbesartan was observed to be 91.35 µg/mL. Solid dispersions of drugs with Soluplus–PEG blend leads to significantly improved solubility of drug i.e. 133.34 µg/mL with SD1a, 211.9 µg/mL with SD2a and 177.45 µg/mL with SD3a in comparison to the pure drug (P value <0.05). However, a decline in solubility was observed with a higher ratio of Soluplus (as observed in SD3a) (Fig. 1).
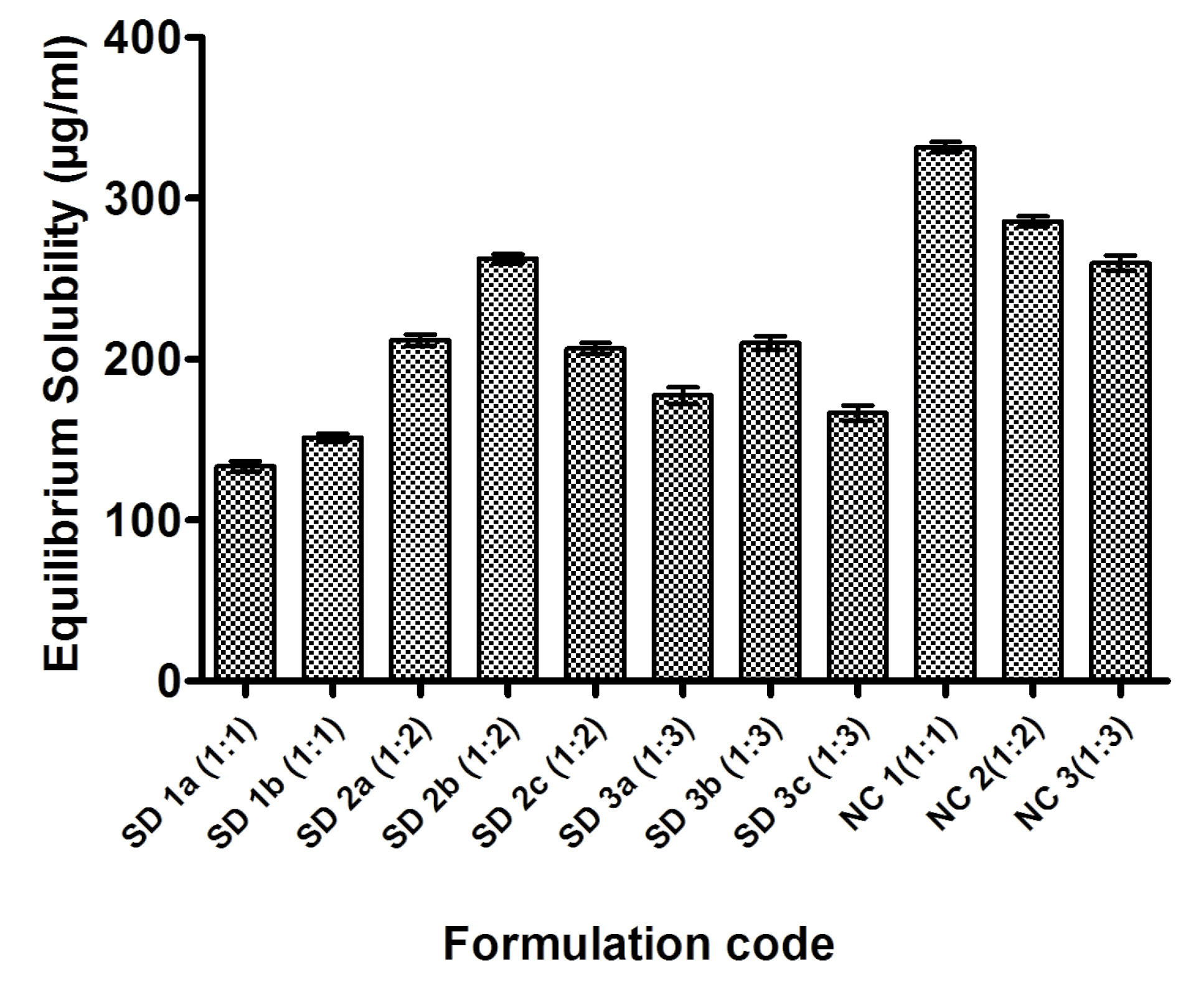
Fig. 1.
Equilibrium solubility of pure drug irbesartan, solid dispersions and nanocomposites.
.
Equilibrium solubility of pure drug irbesartan, solid dispersions and nanocomposites.
Effect of pre-trituration
Pre-trituration of Soluplus-PEG blend with drug leads to uniform drug dispersion in the blend and thereby further enhanced solubility of the drug in dispersions (155.35 µg/mL in SD1b, 262.41 µg/mL in SD2b and 210.18 µg/mL in SD3b in comparison to 133.34 µg/mL in SD1a, 211.9 µg/mL in SD2a and 177.45 µg/mL in SD3a respectively) (Fig. 1). Soluplus is most widely used in the hot-melt extrusion method. The formulation of solid dispersions of drug using Soluplus via the melting fusion method is not easy due to the thermal and mechanical properties Soluplus. The addition of PEG and pre-trituration lead to a uniform drug-polymer blend which can be easily triturated with easy sieving characteristics. Therefore prior trituration of drug with Soluplus and PEG before melting fusion resulted in uniform SD mixtures with significantly enhanced solubility characteristics.
Effect of addition of PEG and its concentration
Further, the amount of PEG was also assessed in the SD formulations, as it acts as a plasticizer and also affects the processing and solubility of SD formulations. As the amount of PEG increased (i.e., 200 mg in SD2c and 300 mg in SD3c) keeping drug to Soluplus ratio constant; the decrease in solubility (i.e. 206.76 µg/mL in SD2c and 166.52 µg/mL in SD3c) in comparison to SD2b and SD3b was observed (Fig. 1). Therefore, an optimum amount of PEG (100 mg) was sufficient enough to decrease the Tg of Soluplus in SD mixtures and to avoid drug degradation that can occur at higher processing temperatures. This would be a pre-requisite for uniform processing of solid dispersions with significantly enhanced solubility. Thus, plasticizers will be needed (in specified amount) to change thermal and mechanical properties in Soluplus® formulations to take full advantage of its potential as a solubilizer and solid dispersion carrier and this is also evidenced in literature.
29-32
Nanocomposites of drug with Soluplus in different drug to polymer ratios (1:1, 1:2, and 1:3) showed dramatically high drug solubility in comparison to solid dispersions (331.76 µg/mL with NC1, 285.53 µg/mL with NC2 and 259.84 µg/mL with NC3) (Fig. 1). The enhancement in solubility may be due to the amphiphilic nature of the polymer as well as the surface adsorption of the drug on the polymer. The low equilibrium solubility at high concentration of Soluplus (NC2 and NC3) reflects the spontaneous nature of drug solubilization beyond that ratio of drug to polymer.
Drug encapsulation efficiency (DEE)
The DEE of various nanocomposites (NC1, NC2, and NC3) was observed to be 86.5%, 78.72%, and 71.935%, respectively. It is suggested that increased Soluplus in the nanosystems require more organic solvent (DCM) to get solubilized and does not support the formulation of nanosystems. Therefore, it is revealed that the entrapment of drug decreases at a higher concentration of Soluplus.
Fourier transform infrared spectroscopy (FTIR) studies
The FTIR spectra of pure drug irbesartan, Soluplus, SD, and NC batches are depicted in Figs. 2-4. The characteristic absorption peaks of pure drug irbesartan were found at 3445 cm-1 (N-H stretch), 3033 cm-1 (C-H stretch), 1564 cm-1 (C=C aromatic). The FTIR of Soluplus also showed peaks at 3654.44 cm-1, 2965.98 cm-1, 1692.23 cm-1, 1477.21 cm-1(Fig. 2). The presence of characteristic peaks of drug (3448.49 cm-1, 1942.61 cm-1,1734.9 cm-1, 1629.7 cm-1) in solid dispersions (SD1, SD2, SD3) (Fig. 3) and (3445.5 cm-1, 1943.6 cm-1, 1732.1 cm-1, 1633.1 cm-1) in nanocomposites (NC1, NC2 and NC3) (Fig. 4) indicated no interaction between drug and Soluplus. However, the presence of additional peaks (2474.58 cm-1, 2931.14 cm-1) in the formulations indicated the overlapping of drugs with an excess of Soluplus.
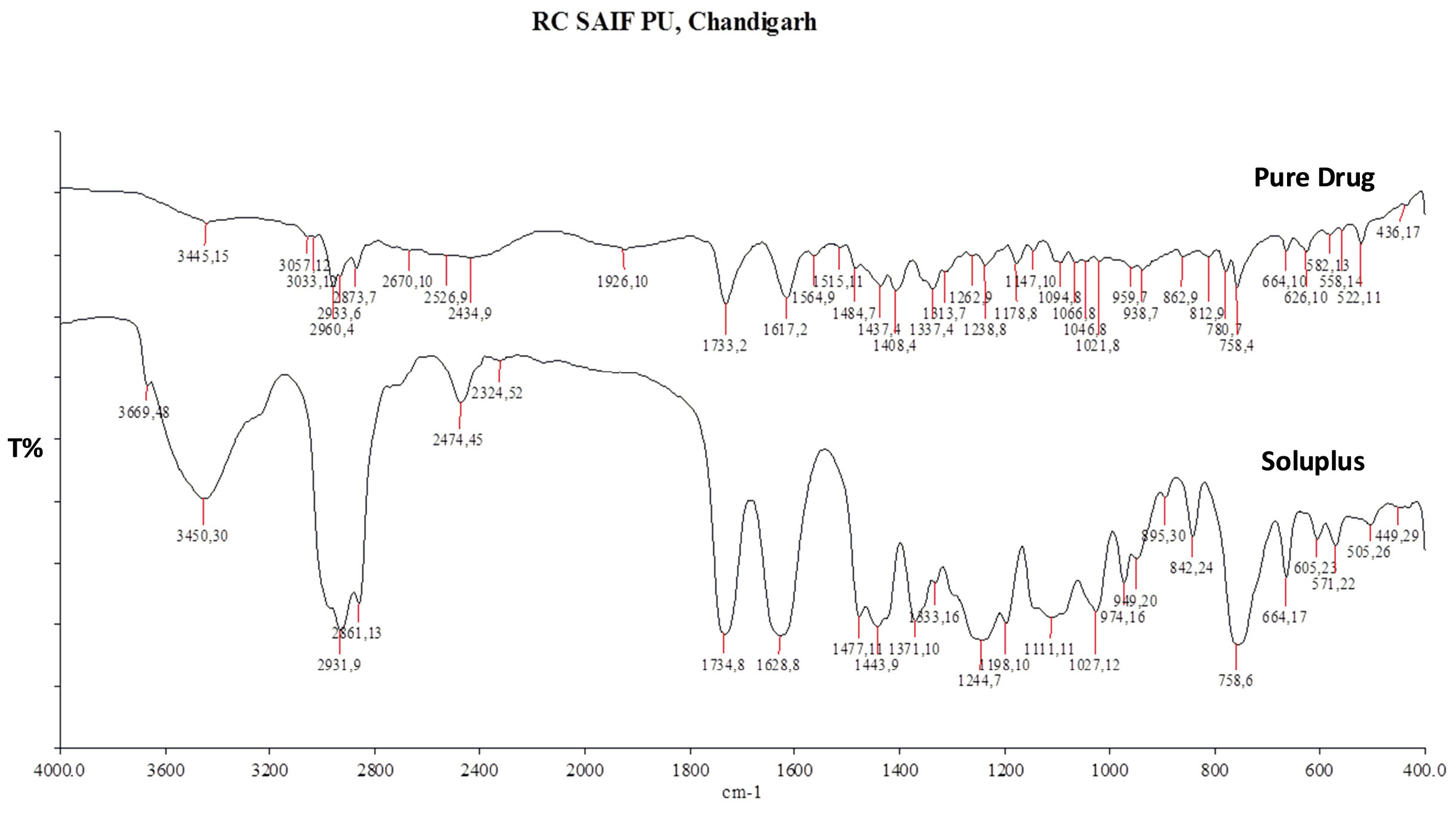
Fig. 2.
Overlay diagram of FTIR of pure drug irbesartan and Soluplus.
.
Overlay diagram of FTIR of pure drug irbesartan and Soluplus.
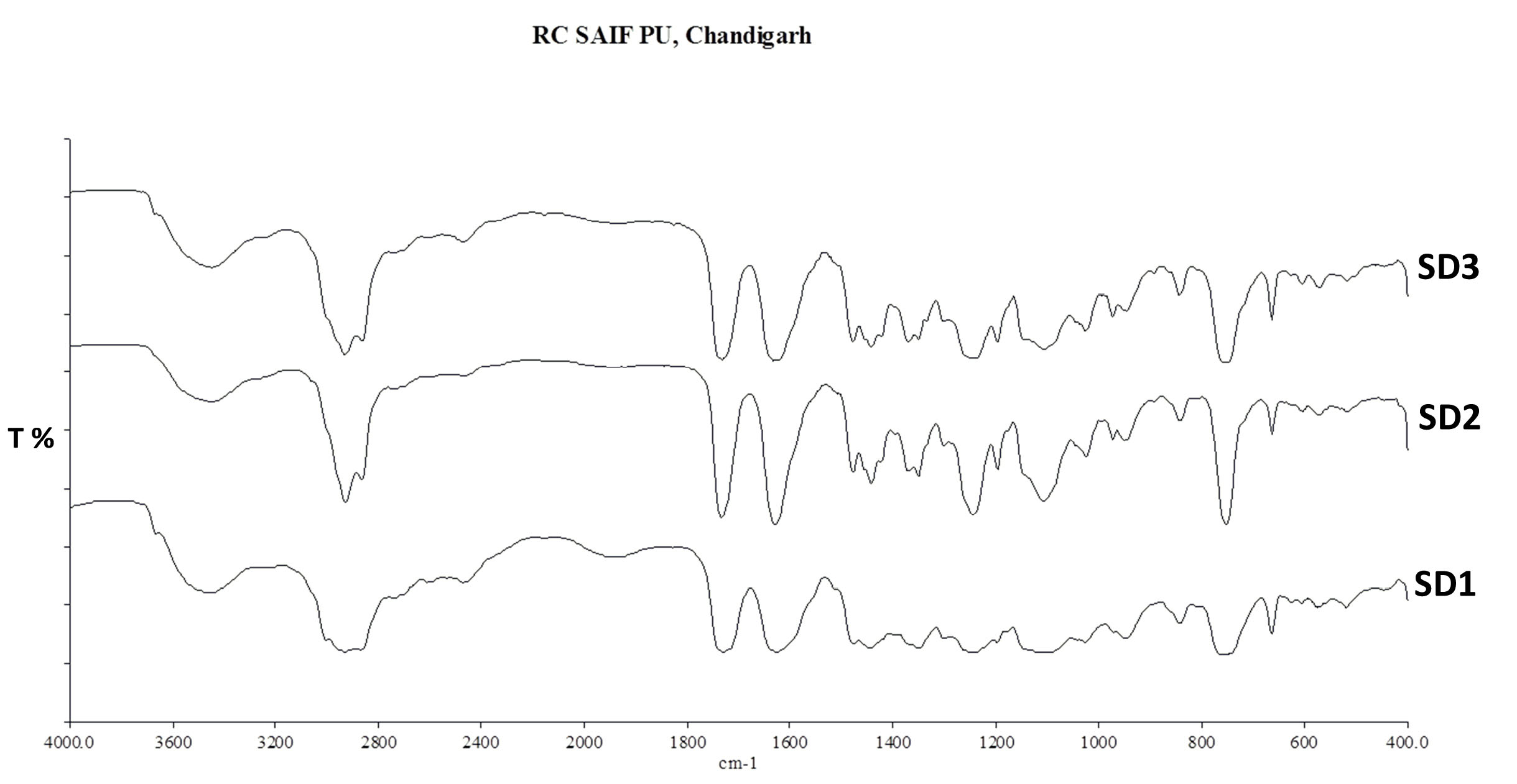
Fig. 3.
The overlayed diagram of the FTIR of solid dispersions (SD1, SD2, and SD3).
.
The overlayed diagram of the FTIR of solid dispersions (SD1, SD2, and SD3).
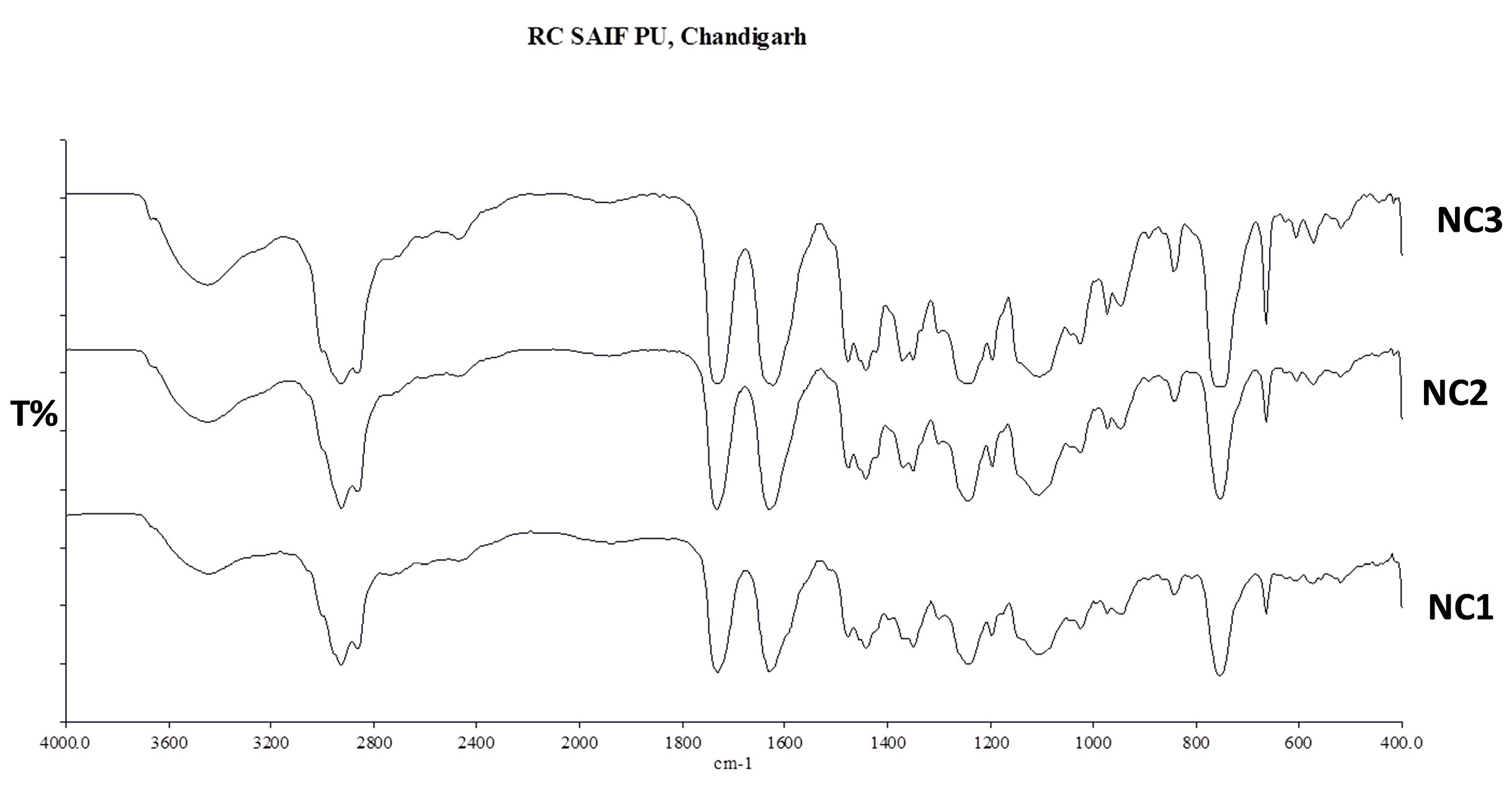
Fig. 4.
Overlay diagram of FTIR of nanocomposites (NC1, NC2, and NC3).
.
Overlay diagram of FTIR of nanocomposites (NC1, NC2, and NC3).
Scanning electron microscopy (SEM) studies
The SEM image of pure drug irbesartan and SD are shown in (Fig. 5). IBS particles showed specific shape in their morphology; SEM of Soluplus indicated the amorphous nature of the polymer. Changes in surface characteristics of SD2 and NC 1 particles were observed, which indicated amorphization of the drug in the polymer matrix.

Fig. 5.
Scanning electron micrographs of (A) pure drug irbesartan, (B) Soluplus, (C) solid dispersion (SD2), and (D) nanocomposites (NC1).
.
Scanning electron micrographs of (A) pure drug irbesartan, (B) Soluplus, (C) solid dispersion (SD2), and (D) nanocomposites (NC1).
Transmission electron microscopy (TEM) studies
The nanosized of particles in the NC1 batch was confirmed by TEM studies (Fig. 6). The nanosized droplets were seen as discreet particles of nano range (600-1000 nm).

Fig. 6.
TEM image of nanocomposites batch (NC1).
.
TEM image of nanocomposites batch (NC1).
The X-ray diffraction studies (XRD) studies
The XRD patterns of pure drug irbesartan, Soluplus, and SD are depicted in (Fig. 7). The characteristic peaks appeared in the XRD of the drug at diffraction angles (2θ) 4.73°, 4.98°, 12.65°, 19.65°, 21.36°, 23.35°, showing a typical crystalline pattern. However, all major characteristic crystalline peaks appeared in the diffractogram of SD2 and NC1 are of decreased intensity and the reduced area which suggested decreased crystallinity or amorphization of the drug in Soluplus matrix.
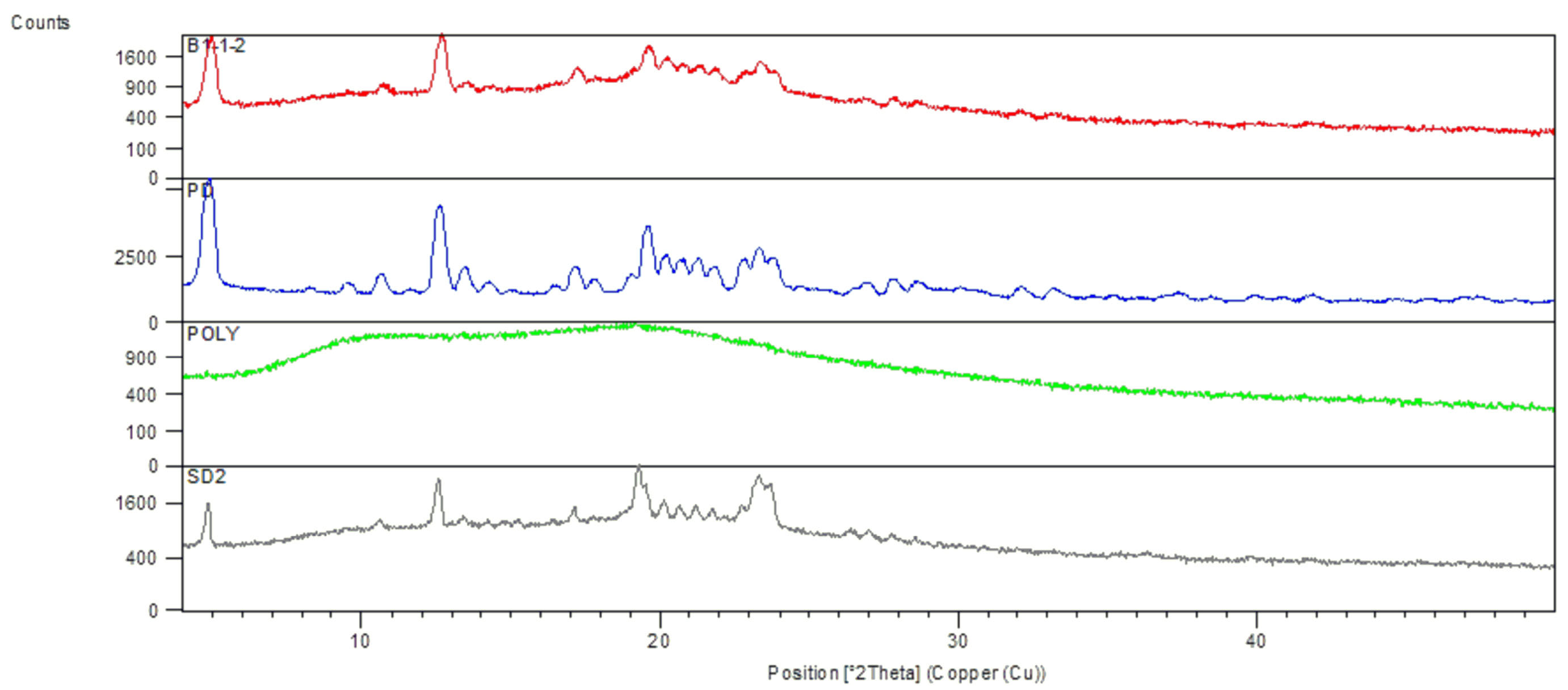
Fig. 7.
The overlayed diagram of The XRD of solid dispersion (SD2), Soluplus (Polymer), pure drug irbesartan (PD), and nanocomposite (NC1).
.
The overlayed diagram of The XRD of solid dispersion (SD2), Soluplus (Polymer), pure drug irbesartan (PD), and nanocomposite (NC1).
Field emission scanning electron microscopy (FESEM) studies
The FESEM image of the nanocomposite NC1 formulation is shown in (Fig. 8). The absence of distinct drug crystals in the FESEM image indicated that no crystallization occurred during the freeze-drying process and that the drug is present as an amorphous solid or as low crystalline moiety in the formulation NC1.

Fig. 8.
FESEM of nanocomposites (NC1).
.
FESEM of nanocomposites (NC1).
Differential scanning calorimetry (DSC) analysis
The endothermic peak of pure drug irbesartan was observed at 181°C corresponding to its melting point. The Soluplus also exhibited endotherm at 70°C, indicating its melting point. The complete disappearance of the endothermic peak in NC1 batch with a broad endotherm around 68°C suggested full incorporation of drugs in the Soluplus matrix revealing the amorphization of drugs. The results are seen in (Fig. 9).
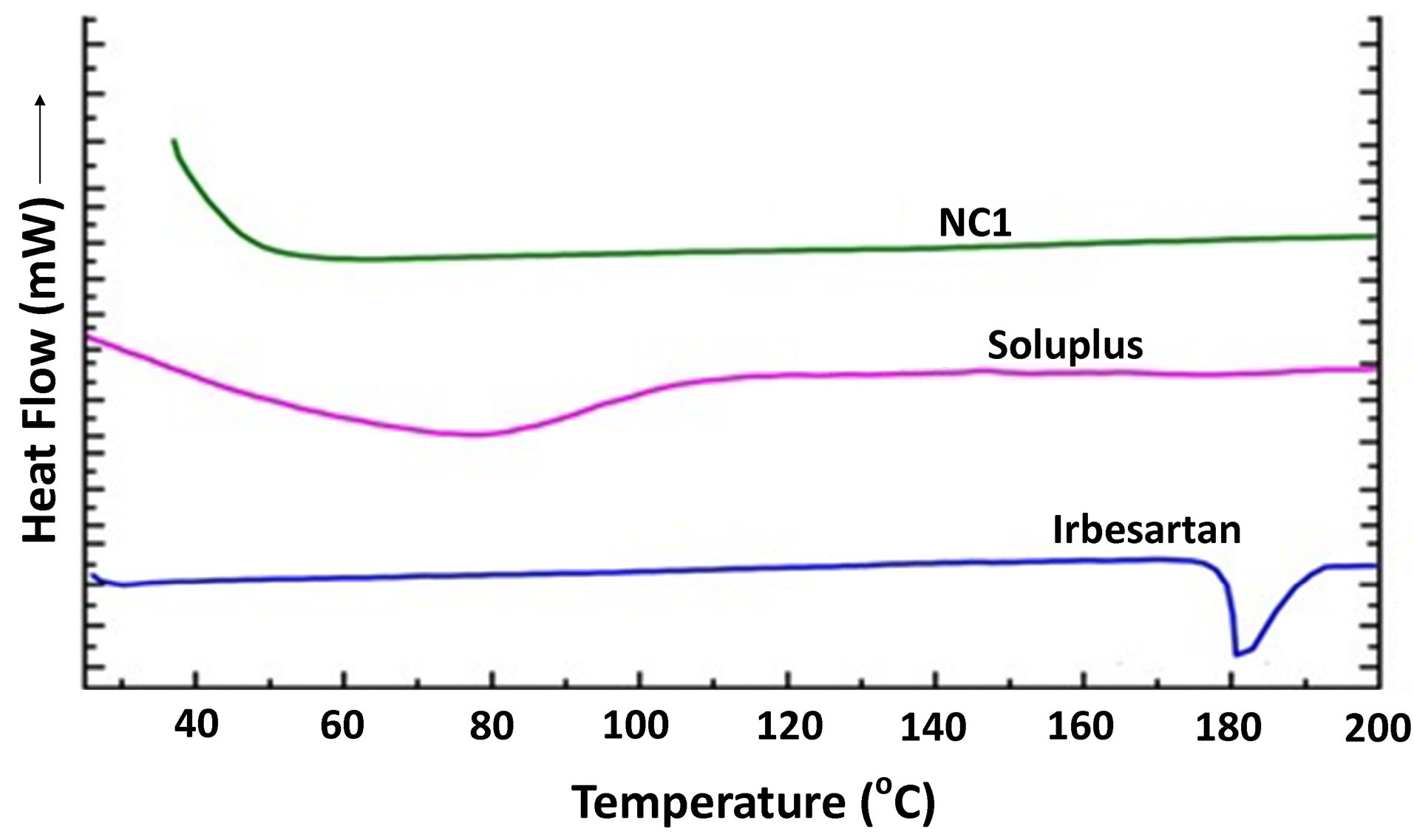
Fig. 9.
The overlayed DSC thermogram of pure drug irbesartan, Soluplus, and nanocomposite (NC1).
.
The overlayed DSC thermogram of pure drug irbesartan, Soluplus, and nanocomposite (NC1).
In vitro dissolution studies
In vitro drug release was studied using 0.1N HCl using USP II dissolution apparatus. Only optimized SD batches (SD1b, SD2b, and SD3b) based on solubility results were used for drug release studies. A significantly higher dissolution rate was observed in all SD batches (56.4% in SD1b, 78.4% in SD2b, and 63.4% in SD3b in 15 minutes) in comparison to the pure drug (25% release in 2 hours). More than 90% of drug release was achieved at the end of 2 hours in all SD batches. 1:2 ratio of drug to Soluplus revealed desired dissolution characteristics. The different batches of the formulations showed a higher release rate in the trend SD2b > SD3b > SD1b. This is in connection with the composition of the hybrid polymer matrices used for preparing various formulations. The release rate of the drug was higher for batch SD 2b where a higher concentration of Soluplus was incorporated in combination with PEG 4000 polymer. Suggested probable mechanisms for increased dissolution in SD can be the reduction of particle size; solubilization by the hybrid polymer; reduced or no aggregation of drug particles; improved dispersibility and wettability of the drug within the dispersion; dissolution of the drug in the polymeric matrix and conversion of crystalline drug to an amorphous state. As soon as the polymer comes in contact with the dissolution medium and gets dissolved, the insoluble fraction of the drug also comes in contact with the medium in the form of fine particles for more rapid dissolution.
33-34
In vitro results of NC batches showed remarkable enhancement in drug release characteristics Batch NC1 exhibited a higher rate and extent of dissolution achieved (84% in 15 minutes and 97% in 30 minutes) in comparison to NC2 and NC3. The nanoparticles were completely dispersed in the polymer matrix due to the proper miscibility of the drug with the hydrophobic part (caprolactam) of the polymer. When these composites of drug and polymer come in contact with water, the ethylene glycol (hydrophilic) part of the polymer gets hydrated quickly into the solution and simultaneously solubilize the dispersed nanoparticles as well. The reduced particle size of nanocomposites resulted in increased surface/volume ratio and the surface interactions, leading to drug-polymer miscibility and improved dissolution characteristics of the drug and finally the drug oral bioavailability.
35,36
Fig. 10 showed relative dissolution profile of pure drug irbesartan, solid dispersions and nanocomposites.
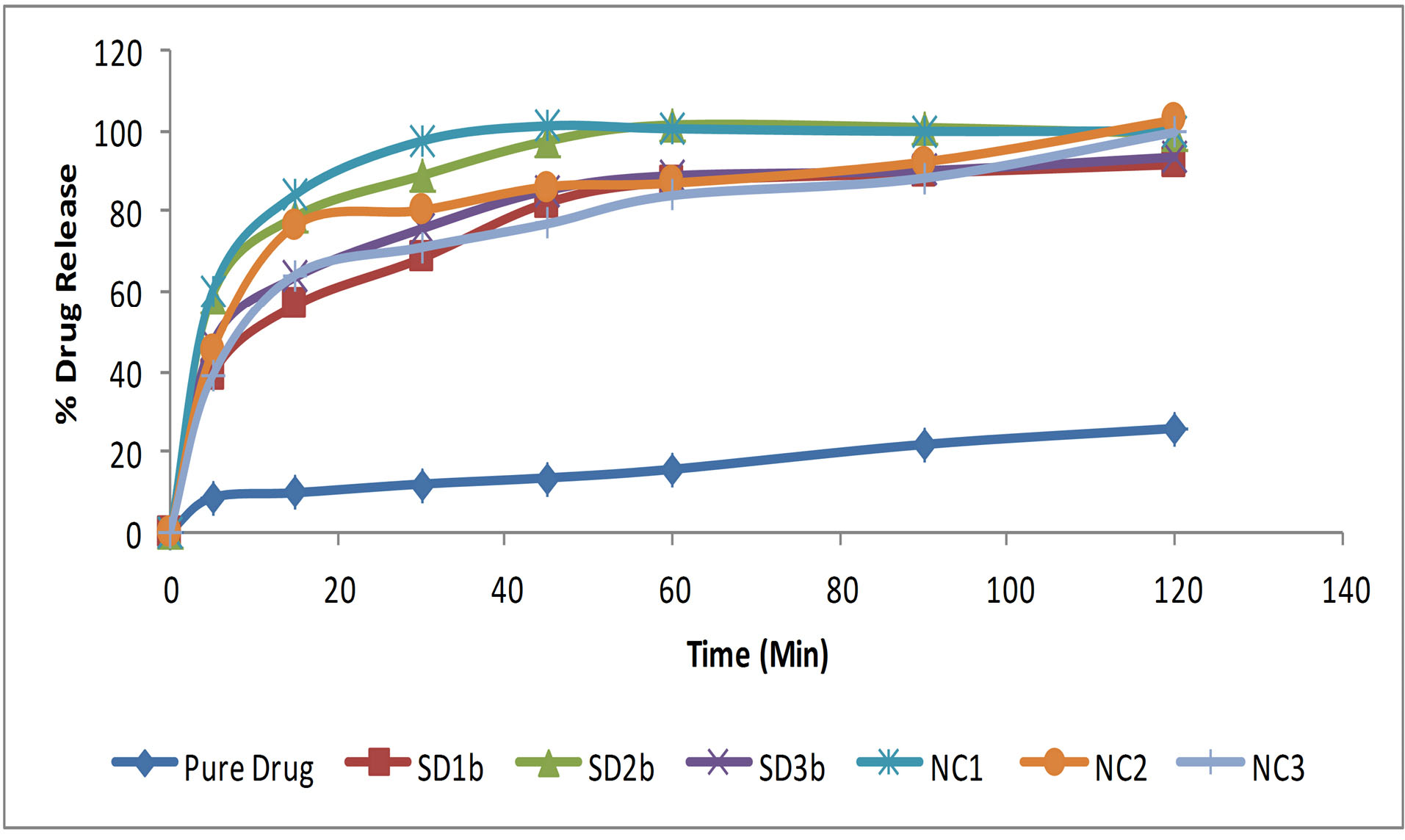
Fig. 10
.
Comparative in vitro dissolution profile of pure drug irbesartan and solid dispersions and nanocomposite batches..
.
Comparative in vitro dissolution profile of pure drug irbesartan and solid dispersions and nanocomposite batches..
In vitro release kinetics
In vitro release data of solid dispersions (SD2b) and nanocomposites (NC1) was fitted to various kinetic models. Higher R2 value suggests that drug release from various mixtures follows Koresmeyer Peppas (KP) model. The n value (less than 0.45) of the KP model is beyond the limits of the KP model. This is called the power-law model. The power-law model can only give limited insight into the exact release mechanism of the drug (Table 2).
Table 2.
R2 values of various kinetic models for SD2b and NC1
|
Formulation code
|
Zero Order
R
2
|
First Order
R
2
|
Higuchi Model
R
2
|
Korsmeyer-Peppas Model
|
|
R
2
|
n
|
| SD2b |
0.5869 |
0.5869 |
0.7689 |
0.9073 |
0.1732 |
| NC1 |
0.7982 |
0.7982 |
0.9077 |
0.9626 |
0.2308 |
In vivo studies
SBP and DBP were calculated in normal rats before and after drug treatment using tail-cuff apparatus with BIOPAC respectively. Induction of SBP and DBP, as well as percent inhibition in SBP and DBP, were measured which indicated antihypertensive action of Irbesartan (NC1) which was compared with a disease control group and with the pure drug (Table 3 and Table 4). From observation, induction of SBP and DBP in normal control, disease control, an optimized formulation of irbesartan and pure drug of irbesartan were measured, in which the induction of SBP and DBP in the optimized formulation was significantly decreased as compared to the disease control group (P < 0.01) at all time intervals along with pure drug (P < 0.05).
Table 3.
Systolic blood pressure in adrenaline-induced hypertension in rats
|
Groups
|
Before treatment SBP (mm Hg)
|
After adrenaline systolic blood pressure (mm Hg)
|
|
30 min
|
60 min
|
90 min
|
120 min
|
| Control |
132.5±3.1 |
133.4±3. |
133.8±2.9 |
133.9±2.5 |
133.4±3.2 |
| Disease control |
131.2±3.2 |
176.2±4.9 |
179.2±5.1 |
180.6±5.8 |
182.4±5.4 |
| Pure drug |
134.5±3.0 |
174.6±3.2 |
172.8±3.8 |
171.6±3.7 |
170.7±3.9 |
| Optimized formulation of irbesartan NC1 |
134.8±2.2 |
146.1±2.9#* |
143.±2.5#* |
144.4±3.1#* |
142.3±3.1#* |
|
|
|
Induction of systolic blood pressure (mm Hg)
|
|
30 min
|
60 min
|
90 min
|
120 min
|
| Control |
132.5±3.1 |
0.7±0.01 |
1.3±0.06 |
1.4±0.05 |
0.9±0.01 |
| Disease control |
131.2±3.2 |
45.9±4.4 |
48.4±3.9 |
49.4±3.8 |
51.1±3.1 |
| Pure drug |
134.5±3.0 |
40.1±2.9 |
38.3±3.8 |
37.1±2.75 |
36.2±2.62 |
| Optimized formulation of irbesartan NC1 |
134.8±2.2 |
11.3±0.8* |
9.0±0.51*# |
9.6±0.36*# |
7.5±0.60* |
* indicate a significant difference in the data disease compared to the control group and the level of significance was P < 0.001 ≈ highly significant. #indicate significant difference in the data compared to treatment groups and the level of significance was P < 0.05 ≈ significant.
Table 4.
Diastolic blood pressure in adrenaline-induced hypertension in rats
|
Groups
|
Before treatment DBP (mm Hg)
|
After adrenaline diastolic blood pressure (mm Hg)
|
|
30 min
|
60 min
|
90 min
|
120 min
|
| Control |
96.5±2.1 |
97.2±1.9 |
96.8±2.9 |
97.1±2.9 |
96.9±2.8 |
| Disease control |
101.1±3.0 |
135.7±3.4 |
136.2±3.8 |
136.8±4.4 |
137.2±4.6 |
| Pure drug |
103.2±3.5 |
130.4±2.9 |
129.5±3.6 |
128.2±3.8 |
127.40±3.7 |
| Optimized formulation of irbesartan NC1 |
99.4±3.1 |
104.2±2.4* |
103.8±2.3#* |
102.2±2.2#* |
100.4±2.8#* |
|
|
|
Induction of diastolic blood pressure (mm Hg)
|
|
30 min
|
60 min
|
90 min
|
120 min
|
| Control |
96.5±2.1 |
0.7±0.02 |
0.3±0.01 |
0.6±0.02 |
0.4±0.01 |
| Disease control |
101.1±3.0 |
34.6±3.3 |
35.1±3.4 |
35.7±3.8 |
36.3±3.2 |
| Pure drug |
103.2±2.5 |
27.2±2.80 |
26.3±2.90 |
28.0±2.98 |
24.2±3.1 |
| Optimized formulation of irbesartan NC1 |
99.4±2.1 |
4.8±.08*# |
4.4±0.08*# |
2.8±0.08#* |
1.0±0.09*# |
* indicate a significant difference in the data disease compared to the control group and the level of significance was P < 0.001 ≈ highly significant. #indicate significant difference in the data compared to treatment groups and the level of significance was P < 0.05 ≈ significant.
Conclusion
Soluplus has been a widely used synthetic carrier in solubility improvement of poorly aqueous soluble drugs. The hot-melt extrusion has been the most widely used method for Soluplus. The study revealed the use of Soluplus for dissolution enhancement of irbesartan by preparing solid dispersions via the fusion melting method. Soluplus-PEG hybrid proved to be potential carriers for improving the solubility of poorly soluble irbesartan. Further, Soluplus nanocomposites of the drug prepared via a single emulsion method proved to be more energetic amorphous forms as stable nanoparticles. The lyophilization process adopted during the development of these nanocomposites leads to the physical stability of the drug and the drug dissolution from these nanocarriers was far superior to that of pure drug. The in vivo pharmacodynamic evaluation of the antihypertensive activity of NC1 further proved the efficacy of these nanocomposites.
Acknowledgments
The authors are thankful to Dr. Madhu Chitkara, Vice-Chancellor, Chitkara University; Dr. Ashok Chitkara, Chancellor, Chitkara University; Dr. Sandeep Arora, Director, Chitkara College of Pharmacy for providing necessary facilities and support.
Funding sources
No funding by any agency.
Ethical statement
The institutional animal ethics committee approved the experimental protocol and care of the animals was carried out as per the guidelines of the Committee for Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Environment and Forest, Government of India (Chitkara College of Pharmacy Animal Facility Registration Number: 1181/PO/ReBi/S/08/CPCSEA).
Competing interests
The authors declared no conflicts of interest.
Authors’ contribution
MN designed the study. GA acted as a consultant. GS helped the in vivo studies. MS helped in the preparation and graphical presentation, VP and AS conducted all the lab works.
Research Highlights
What is the current knowledge?
simple
-
√ Soluplus-PEG polymeric mixture can be used to prepare solid dispersions via lab scale fusion melting method.
-
√ Soluplus alone may yield nanocomposite formulations via single emulsion techniques, which are better than solid dispersion formulations.
What is new here?
simple
-
√ In stead of using hot melt extrusion with soluplus (widely reported), current study used lab scale method for formulation of solid dispersions and nanocomposites for poorly aqueous soluble drugs.
-
√ Soluplus amount should be optimized for better formulation
development.
References
- Fouad EA, EL-Badry M, Mahrous GM, Alanazi FK, Neau SH, Alsarra IA. The use of spray-drying to enhance celecoxib solubility. Drug Dev Ind Pharm 2011; 37:1463-72. doi: 10.3109/03639045.2011.587428 [Crossref] [ Google Scholar]
- Al-Obaidi H, Buckton G. Evaluation of griseofulvin binary and ternary solid dispersions with HPMCAS. AAPS PharmSciTech 2009; 10:1172. doi: 10.1208/s12249-009-9319-x [Crossref] [ Google Scholar]
- Emara LH, Badr RM, Abd Elbary A. Improving the dissolution and bioavailability of nifedipine using solid dispersions and solubilizers. Drug Dev Ind Pharm 2002; 28:795-807. doi: 10.1081/DDC-120005625 [Crossref] [ Google Scholar]
- Ning X, Sun J, Han X, Wu Y, Yan Z, Han J. Strategies to improve dissolution and oral absorption of glimepiride tablets: solid dispersion versus micronization techniques. Drug Dev Ind Pharm 2011; 37:727-736. doi: 10.3109/03639045.2010.538061 [Crossref] [ Google Scholar]
- Lakshman JP, Cao Y, Kowalski J, Serajuddin AT. Application of melt extrusion in the development of a physically and chemically stable high-energy amorphous solid dispersion of a poorly water-soluble drug. Mol Pharm 2008; 5:994-1002. doi: 10.1021/mp8001073 [Crossref] [ Google Scholar]
- Weuts I, Van Dycke F, Voorspoels J, De Cort S, Stokbroekx S, Leemans R. Physicochemical properties of the amorphous drug, cast films, and spray dried powders to predict formulation probability of success for solid dispersions: Etravirine. J Pharm Sci 2011; 100:260-274. doi: 10.1002/jps.22242 [Crossref] [ Google Scholar]
- Boghra RJ, Kothawade PC, Belgamwar VS, Nerkar PP, Tekade AR, Surana SJ. Solubility, dissolution rate and bioavailability enhancement of irbesartan by solid dispersion technique. Chem Pharm Bull 2011; 59:438-41. doi: 10.1248/cpb.59.438 [Crossref] [ Google Scholar]
- Ganapuram BR, Alle M, Dadigala R, Kotu GM, Guttena V. Development, evaluation and characterization of surface solid dispersion for solubility and dispersion enhancement of irbesartan. J Pharm Res 2013; 7:472-477. doi: 10.1016/j.jopr.2013.06.012 [Crossref] [ Google Scholar]
- Shete AS, Yadav A, Murthy MS. Enhancement of dissolution rate of irbesartan by chitosan based crystal engineering technique. Indian J Pharm Educ Res 2012; 46:323-329. [ Google Scholar]
- Patel J, Patel A, Raval M, Sheth N. Formulation and development of a self-nanoemulsifying drug delivery system of irbesartan. J Adv Pharm Technol Res 2011; 2:9-16. doi: 10.4103/2231-4040.79799 [Crossref] [ Google Scholar]
- Boghra R, Patel A, Desai H, Jadhav A. Formulation and evaluation of irbesartan liquisolid tablets. Int J Pharm Sci Rev Res 2011; 9:32-37. [ Google Scholar]
- Kumar GA, Choudhary RK, Chaitanya CH. Enhancement of solubility and dissolution rate of irbesartan by solid dispersion technique. Asian J Pharm Clin Res 2011; 4:36-40. [ Google Scholar]
- Deepak G, Rahul R, Senthil A, Shantesh UM. Formulation and evaluation of irbesartan immediate Release Tablets. Int Res J Pharm 2012; 3:410-415. [ Google Scholar]
- Aruna MS, Babu AK, Thadanki M, Gupta ME. Solid dispersions–an approach to enhance the dissolution rate of Irbesartan. Int J Res Pharm Chem 2011; 1(4):780-8. [ Google Scholar]
- Ganapuram BR, Alle M, Dadigala R, Kotu GM, Guttena V. Development, evaluation and characterization of surface solid dispersion for solubility and dispersion enhancement of irbesartan. J Pharm Res 2013; 7:472-477. doi: 10.1016/j.jopr.2013.06.012 [Crossref] [ Google Scholar]
- Pokharkar VB, Malhi T, Mandpe L. Bicalutamide nanocrystals with improved oral bioavailability: in vitro and in vivo evaluation. Pharm Dev Techno 2013; 18:660-666. doi: 10.3109/10837450.2012.663391 [Crossref] [ Google Scholar]
- Potta SG, Minemi S, Nukala RK, Peinado C, Lamprou DA, Urquhart A. Development of solid lipid nanoparticles for enhanced solubility of poorly soluble drugs. J Biomed Nanotechnol 2010; 6:634-640. doi: 10.1166/jbn.2010.1169 [Crossref] [ Google Scholar]
- Shah S, Sailor G, Shah N, Chauhan S, Aundhia C, Seth AK. Formulation and Evaluation of irbesartan loaded solid lipid nanoparticles by solvent injection method. Int J Pharm Sci 2015; 6:159-170. [ Google Scholar]
- Chen H, Khemtong C, Yang X, Chang X, Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discov Today 2011; 16:354-360. doi: 10.1016/j.drudis.2010.02.009 [Crossref] [ Google Scholar]
- Meruva S, Thool P, Shah S, Karki S, Bowen W, Ghosh I. Formulation and performance of Irbesartan nanocrystalline suspension and granulated or bead-layered dried powders–Part. I Int J Pharm 2019; 568:118189. doi: 10.1016/j.ijpharm.2019.03.007 [Crossref] [ Google Scholar]
- Patnaik S, Aditha SK, Rattan T, Kamisetti V. Aceclofenac-Soluplus® nanocomposites for increased bioavailability. Scientific Research 2015; 5:54519. doi: 10.4236/snl.2015.52003 [Crossref] [ Google Scholar]
- Maniruzzaman M, Boateng JS, Snowden MJ, Douroumis D. A review of hot-melt extrusion: process technology to pharmaceutical products. ISRN Pharm 2012; 2012:436763. doi: 10.5402/2012/436763 [Crossref] [ Google Scholar]
- Salawi A, Nazzal S. The rheological and textural characterization of Soluplus®/Vitamin E composites. Int J Pharm 2018; 546:255-62. doi: 10.1016/j.ijpharm.2018.05.049 [Crossref] [ Google Scholar]
- Salawi A, Nazzal S. The physiochemical, mechanical, and adhesive properties of solvent-cast vitamin E/Soluplus® films. Int J Pharm 2018; 552:378-387. doi: 10.1016/j.ijpharm.2018.10.018 [Crossref] [ Google Scholar]
- Varela-Garcia A, Concheiro A, Alvarez-Lorenzo C. Soluplus micelles for acyclovir ocular delivery: Formulation and cornea and sclera permeability. Int J Pharm 2018; 552:39-47. doi: 10.1016/j.ijpharm.2018.09.053 [Crossref] [ Google Scholar]
- Zhu C, Gong S, Ding J, Yu M, Ahmad E, Feng Y. Supersaturated polymeric micelles for oral silybin delivery: the role of the Soluplus–PVPVA complex. Acta Pharm Sin B 2019; 9:107-17. doi: 10.1016/j.apsb.2018.09.004 [Crossref] [ Google Scholar]
- Adamczak M, Gross ML, Krtil J, Koch A, Tyralla K, Amann K. Reversal of glomerulosclerosis after high-dose enalapril treatment in subtotally nephrectomized rats. J Am Soc Nephrol 2003; 14:2833-42. doi: 10.1097/01.ASN.0000095248.91994.D3 [Crossref] [ Google Scholar]
- Vogel HG, Vogel WH. Drug discovery and evaluation. Biomed Pharmacother 1998; 52:47-47. [ Google Scholar]
- Hardung H, Djuric DE. Soluplus® A novel excipient for hot melt extrusion. Chimica Oggi-Chemistry Today 2010; 28:14-6. [ Google Scholar]
- Hughey JR, Keen JM, Brough C, Saeger S, McGinity JW. Thermal processing of a poorly water-soluble drug substance exhibiting a high melting point: the utility of KinetiSol® dispersing. Int J Pharm 2011; 419:222-230. doi: 10.1016/j.ijpharm.2011.08.007 [Crossref] [ Google Scholar]
- Repka MA, Gerding TG, Repka SL, McGinity JW. Influence of plasticizers and drugs on the physical-mechanical properties of hydroxypropylcellulose films prepared by hot melt extrusion. Drug Dev Ind Pharm 1999; 25:625-33. doi: 10.1081/DDC-100102218 [Crossref] [ Google Scholar]
- Repka MA, Battu SK, Upadhye SB, Thumma S, Crowley MM, Zhang F. Pharmaceutical applications of hot-melt extrusion: Part II. Drug Dev Ind Pharm 2007; 33:1043-57. doi: 10.1080/03639040701525627 [Crossref] [ Google Scholar]
- Reginald-Opara JN, Attama A, Ofokansi K, Umeyor C, Kenechukwu F. Molecular interaction between glimepiride and Soluplus®-PEG 4000 hybrid based solid dispersions: Characterisation and anti-diabetic studies. Int J Pharm 2015; 496:741-50. doi: 10.1016/j.ijpharm.2015.11.007 [Crossref] [ Google Scholar]
-
Aulton ME. The Science of Dosage Form Design. Churchill Livingstone; 2002.
- Merisko-Liversidge EM, Liversidge GG. Drug nanoparticles: formulating poorly water-soluble compounds. Toxicol Pathol 2008; 36:43-48. doi: 10.1177/0192623307310946 [Crossref] [ Google Scholar]
- Kim MS. Soluplus-coated colloidal silica nanomatrix system for enhanced supersaturation and oral absorption of poorly water-soluble drugs. Artif Cell Nanomed B 2013; 41:363-367. doi: 10.3109/21691401.2012.762369 [Crossref] [ Google Scholar]