Bioimpacts. 12(6):515-531.
doi: 10.34172/bi.2022.23433
Original Research
Evaluation of poly (lactic-co-glycolic acid) nanoparticles to improve the therapeutic efficacy of paclitaxel in breast cancer
Laura Cabeza 1, 2, 3  , Mazen M. El-Hammadi 4 , Raul Ortiz 1, 2, 3 , Maria D. Cayero-Otero 4, Julia Jiménez-López 1, 3 , Gloria Perazzoli 1, 3 , Lucia Martin-Banderas 4 , Jose M. Baeyens 5 , Consolación Melguizo 1, 2, 3, * , Jose Prados 1, 2, 3
, Mazen M. El-Hammadi 4 , Raul Ortiz 1, 2, 3 , Maria D. Cayero-Otero 4, Julia Jiménez-López 1, 3 , Gloria Perazzoli 1, 3 , Lucia Martin-Banderas 4 , Jose M. Baeyens 5 , Consolación Melguizo 1, 2, 3, * , Jose Prados 1, 2, 3
Author information:
1Institute of Biopathology and Regenerative Medicine (IBIMER), Center of Biomedical Research (CIBM), University of Granada, 18100 Granada, Spain
2Department of Anatomy and Embryology, Faculty of Medicine, University of Granada, 18071 Granada, Spain
3Biosanitary Institute of Granada (ibs.GRANADA), SAS-University of Granada, 18014 Granada, Spain
4Department of Pharmacy and Pharmaceutical Technology, Faculty of Pharmacy, University of Seville, 41012 Sevilla, Spain
5Department of Pharmacology, Institute of Neuroscience, Biomedical Research Center (CIBM), University of Granada, 18100, Granada, Spain
Abstract
Introduction:
Paclitaxel (PTX) is a cornerstone in the treatment of breast cancer, the most common type of cancer in women. However, this drug has serious limitations, including lack of tissue-specificity, poor water solubility, and the development of drug resistance. The transport of PTX in a polymeric nanoformulation could overcome these limitations.
Methods:
In this study, PLGA-PTX nanoparticles (NPs) were assayed in breast cancer cell lines, breast cancer stem cells (CSCs) and multicellular tumor spheroids (MTSs) analyzing cell cycle, cell uptake (Nile Red-NR-) and α-tubulin expression. In addition, PLGA-PTX NPs were tested in vivo using C57BL/6 mice, including a biodistribution assay.
Results:
PTX-PLGA NPs induced a significant decrease in the PTX IC50 of cancer cell lines (1.31 and 3.03-fold reduction in MDA-MB-231 and E0771 cells, respectively) and CSCs. In addition, MTSs treated with PTX-PLGA exhibited a more disorganized surface and significantly higher cell death rates compared to free PTX (27.9% and 16.3% less in MTSs from MCF-7 and E0771, respectively). PTX-PLGA nanoformulation preserved PTX’s mechanism of action and increased its cell internalization. Interestingly, PTX-PLGA NPs not only reduced the tumor volume of treated mice but also increased the antineoplastic drug accumulation in their lungs, liver, and spleen. In addition, mice treated with PTX-loaded NPs showed blood parameters similar to the control mice, in contrast with free PTX.
Conclusion:
These results suggest that our PTX-PLGA NPs could be a suitable strategy for breast cancer therapy, improving antitumor drug efficiency and reducing systemic toxicity without altering its mechanism of action.
Keywords: Paclitaxel, PLGA, Breast cancer, Cancer stem cells, Mice xenografts
Copyright and License Information
© 2022 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Paclitaxel (PTX) or Taxol® is a chemotherapy drug that is used to treat a variety of cancers, including breast cancer, the most common type of cancer in women.
1
This drug acts by binding to cell microtubules and stabilizing them as a result of depolymerization inhibition, with subsequent cell cycle arrest at the G2/M phase, which ultimately leads to apoptosis.
2-4
Treatment by PTX is associated with (i) a lack of specificity, which results in significant adverse effects (e.g. neuropathic pain and hypersensitivity reactions),
5
and (ii) side effects related to the commercial preparation of PTX (Taxol®) and its poor water solubility (<0.1 µg/mL). Consequently, alternative formulations for PTX have been developed,
6
as is the case with Cremophor® EL free formulations.
7
However, they can produce acute hypersensitivity reactions and systemic immunostimulation.
8
Therefore, new strategies are required to enhance the efficiency and specificity of PTX.
In recent years, new strategies for breast cancer treatment have been investigated using nanoparticles (NPs) as drug carriers. These molecules have been shown to improve drug solubility and cell internalization, avoid multidrug resistance mechanisms (e.g. P-glycoprotein) and enable surface functionalization for specific targeting to tumor cells.
6,9-13
In addition, the accumulation of NPs in tumor tissues is higher than in healthy tissues. This differential behavior can be explained by the small size and physicochemical properties of NPs, as well as by the enhanced permeability and retention (EPR) effect, or passive targeting. This increased EPR in tumors has been related to inefficient lymphatic drainage and the presence of fenestrated and more permeable blood vessels.
14-16
Currently, different nanodrugs for breast cancer treatment have been developed
17
and approved by the Food and Drug Administration (FDA) (Abraxane®, Doxil®, and Lipusu®), and some others are currently under research (LEP-ETU®, EndoTAGR®-I, and NK105).
17-21
In this context, some of the most commonly assayed degradable polymers for cancer treatment include polylactic acid (PLA), polyglycolic acid (PGA), and their copolymer poly (lactic-co-glycolic acid, PLGA). These polymers are biodegradable, biocompatible and non-toxic, and provide controlled and sustained release profiles after parenteral administration.22–24 In addition, they are internalized into cells via clathrin-mediated endocytosis and liquid phase pinocytosis, escaping rapidly to the cytoplasm and initiating a sustained drug release.
22,25
However, PLGA NPs also have some problems that need to be solved, such as low stability after compound preparation and poor drug loading (≈1%). The latter limitation significantly hinders their clinical use, making it necessary to administer a large number of NPs to achieve therapeutic doses. Finally, PLGA NPs show a fast-initial release followed by a sustained release of the drug (biphasic release), probably caused by drug adsorption on the surface of the NPs. Therefore, only a proportion of the drug can reach the tumor site
22,24,26
to produce its antitumor effect.
This work aimed to test a previously synthesized PTX-loaded PLGA system to improve breast cancer treatment. This Cremophor® EL-free nanodrug was tested in tumor and non-tumor cells from breast epithelium to compare the relative inhibition of cell proliferation in comparison with free PTX. In addition, the modulation of the mechanism of action of PLGA-PTX and its internalization were determined by quantitative and qualitative cellular uptake studies and cell cycle analyses. Finally, in vivo studies with breast tumor-bearing mice were conducted to determine tumor volume modulation, mouse survival, PTX biodistribution, and possible treatment toxicity.
Materials and methods
Cell culture and cytotoxicity study
The human breast cancer cell lines MCF-7, MDA-MB-231, SK-BR-3 and T47D, (with different expression patterns of estrogens receptors) and breast epithelial cell line (MCF-10A) were purchased and grown following American Type Culture Collection (ATCC, Manassas, VA) recommendations.
27
The E0771 mouse breast cancer cell line was provided by Robin Anderson (Peter MacCallum Cancer Center, East Melbourne, Australia) and was grown in a similar way. Previous developed PTX-loaded PLGA system by a modified nanoprecipitation method.
28
Briefly, PLGA was dissolved with Span® 60 in acetone until reach 1.5% (w/v) concentration and 5mL of this solution were added to 15 mL of an aqueous solution of Pluronic® F-68 (0.5% w/v). PTX was added into de PLGA organic solution (5, 10, and 15% w/w) and also Nile red by adding 100 μL of an acetonic solution (10mM). After the evaporation of the organic solvent, the NP suspension was centrifuged (10 000 rpm, 30 minutes, 4°C), washed, re-suspended in a trehalose solution (5% w/v) and lyophilized (Cryodos freeze-drier, Telstar Industrial S.L., Spain). The morphology of the nanoformulation (Scanning Electron Microscopy, Philips XL30, Philips, GER) and size, mean diameter, size distribution, and zeta potential (ZP) (Zetasizer Nano ZS, Malvern Instruments, UK) were investigated. The PTX loaded and released from the nanoformulations were measured by HPLC as described previously.
28
PTX-loaded PLGA was assayed in breast cancer cells to determine its antiproliferative effect. Cell lines were seeded in 24-well plates (1.5×103 for MCF-7 and E0771, 13×103 for MDA-MB-231, and T47D, and 20×103 for MCF-10A and SK-BR-3) in 400 µL of culture medium, incubated overnight, and then exposed to PTX-loaded PLGA NPs, blank PLGA NPs, and free PTX (from 0.1 to 100 nM during 4 days) with a renewal of the culture medium and the treatments at 48 hours. A colorimetric modified assay of sulforhodamine B (SRB) was performed
29
and the optical density (OD) of the dye was measured with a Titertek multiscan colorimeter (Flow, Irvine, California) at 492 nm. The percentages of proliferation (Pf %) and cytotoxicity (Ct %) were calculated with Eq.1. Pf%= (sample OD/negative control OD)×100 and Eq. 2. Ct%=100-Pf%.
Cell culture and cytotoxicity study in CSC
Cancer stem cells (CSCs) tumorspheres from MCF-7 cells were obtained using the protocol published previously by our group.
27
Briefly, cells were cultured in 6-well plates precoated with 1% agarose to create a low attachment surface in 2 mL of the induction medium: DMEM/Nutrient Mixture F-12 Ham (Sigma Aldrich) with B27 (1X) (Gibco, Spain), 1% of an antibiotics mixture (penicillin-streptomycin), Heparin (4 μg/mL), bFGF (20 ng/mL) and EGF (20 ng/mL) (Sigma Aldrich). Every 48 hours optical microscopy images of the cells were taken and 1/3 of the culture medium was replaced with a new induction medium. When the CSC tumorspheres were formed (two weeks) we use the qPCR to determine cell population markers. The cell markers used previously were CD133, CD44, OCT4, DNMT1 and SOX2 to identify CSCs phenotype whose increase in expression is related to cell aggressiveness and invasiveness. This a method frequently used for the induction and characterization of CSC.
30-34
CSCs tumorspheres were collected after two weeks, trypsinized, grown in 96-well plates (5000 cells/well) and treated with PTX, PLGA-PTX and PLGA blank NPs. After 72 hours, cell proliferation was determined with Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan) following the manufacturer protocol.
Cell cycle analysis
Cell lines were seeded (1 × 105cells/well) in 6-well plates with 2 mL of culture medium. After 24 hours, the medium was replaced overnight with a volume of 2 mL of serum-free culture medium to induce cell cycle arrest and allow the cells to start from the same phase of the cycle. Cells were treated with a half-maximal inhibitory concentration (IC50) dose of free PTX and PTX-loaded PLGA NPs for 48 hours of incubation. Cell cycle analysis was performed by flow cytometry with propidium iodide (PI) and RNase solution (PI/RNase) from Immunostep (Salamanca, Spain). Cells were collected and then fixed with 70% ethanol-PBS for 30 minutes at 4ºC. Then, cells were centrifuged (5000 rpm, 5 minutes) and the pellet was resuspended and incubated in 500 µL of PI/RNase solution for 15 minutes at room temperature. The suspension was analyzed with FACScan (Becton Dickinson, San Jose, USA).
Immunofluorescence microscopy analysis
MCF-7, MDA-MB-231, E0771, and MCF-10A cell lines were grown on 8-well chamber slides, and after arresting the cell cycle (see previous sections), were treated with PTX, PLGA-PTX, and blank PLGA NPs (IC50 and 100 nM). Elapsed 48 hours, cells were washed three times with PBS, fixed with 4% paraformaldehyde (20 minutes at room temperature) and washed three times with 10× pH 7.4 Tris-buffered saline solution (TBS; Fisher Scientific, Madrid, Spain) with 0.1% Tween-20 (TBST) and one time with TBS with 0.1% Triton X-100. Subsequently, blocking buffer (TBST, 0.1% Triton X-100 and 0.5% goat serum) was added (1 hour) and cells were incubated with monoclonal anti-α-tubulin antibody (clone DM1A, ascites fluid, Sigma-Aldrich) (1:500) 1 hour at room temperature and washed with TBST. Next, goat anti-mouse IgG, Alexa Fluor® 488 (Thermo Fisher Scientific, San Jose, CA, U.S.A.) (1:500) was added 1 hour at room temperature and washed with TBST, cell nucleus was stained with Hoechst 33258 (Sigma-Aldrich) (5 μg/mL for 5 minutes). Cells were observed in a fluorescence microscope (Nikon eclipse 50i).
Nile red-loaded PLGA NPs cell uptake
PLGA NPs cell uptake was analyzed in MCF-7 cells seeded in 6-well plates (1 × 105 cells/well) in 2 mL of culture medium. After 24 hours, cells were incubated with free Nile Red (NR) (0.5 μM) and NR-loaded PLGA NPs for 0.5, 1, 2, 4, and 6 hours. Then, cells were washed with PBS, trypsinized, and centrifuged (5000 rpm, 5 minutes) and the supernatant was discarded. After two washes with PBS, cells were resuspended in 200 µL of PBS. FACScan (Becton Dickinson, San Jose, USA) was used to measure NR fluorescence. Simultaneously, cells were observed at the same exposure times in the fluorescent microscope Leica DM IL LED (Leica Microsystems S.L.U., Barcelona, Spain). In addition, a confocal microscopy analysis (Leica TCS-SP5 II) of the uptake of PLGA NPs was carried out in MCF-7 cells seeded (1 × 105 on a sterile coverslip into 6-well plates) and treated with NR and NR-PLGA NPs at 0.05 µM (0.5, 1, 2, and 4 hours) and fixed with a 4% formaldehyde solution (20 minutes, room temperature, in darkness). The cell nucleus was stained with 5 μg/mL of Hoechst 33258 (Sigma-Aldrich).
PTX-loaded PLGA NPs cell uptake
To verify if there are differences in the intracellular concentration of the free drug and the drug-loaded in PLGA NPs, we followed the modified procedure of Leiva et al and Li et al.
27,35,36
Briefly, the cell lines MCF-7, MDA-MB-231, E0771, and MCF-10A were grown in 6-well plates in a volume of 2 mL of culture medium and treated with 500 nM of PTX, PLGA-PTX, and blank PLGA NPs for 0.5, 1, 2, 4, and 6 hours. After two washes with PBS, cells were lysed (800 μL of a lysis solution in each well) and by sonication. Afterward, 500 μL of Methyl tert-butyl ether (Sigma-Aldrich) and 15 μL of docetaxel (internal standard) were added to the cell lysate. The mixture was vortexed and incubated (5 minutes). Then, the samples were centrifuged, and the organic phase was collected and left to dry. The dry residue was resuspended in methanol and analyzed with a triple quadrupole mass spectrometer analyzer (triple-quad) (WATERS, XEVO TQ-S) and ultra-high-pressure liquid chromatography (UPLC) (WATERS, ACQUITY H CLASS) (UPLC-MS/MS) following the premises of Fernández-Peralbo et al.
35
Part of the cell lysate was used to analyze the protein concentration of the samples by the Bradford method.
Multicellular tumor spheroid assays
MCF-7 and E0771 multicellular tumor spheroids (MTSs) were generated using our previously described protocol
37
and treated with PTX, PTX-PLGA and PLGA NPs (6.3, 12.7, 25.4 and 50.8 nM for MCF-7; 74.6, 150, 300 and 600 nM for E0771). Equivalent concentrations of PLGA NPs were used in both cell lines. A group of MTSs without treatment was used as a negative control. Growth of the MTSs was monitored every 2 days using an inverted phase-contrast microscope to obtain a median relative volume (V, µm3) with the formula Eq. 3. V(mm3)= (a × b2× π)/6, where “a” is the longest diameter and “b” is the shortest diameter.
38
Spherocytes (25×103 of cells in the case of MCF-7) were also used to compare the proliferation rate of treated MTSs with respect to negative controls using Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan). The percentage of proliferation (Pf%) was calculated using the formula in the section "Cell culture and cytotoxicity study". Finally, a TUNEL kit (Roche Mannheim, Germany) was used to determine apoptosis in MTSs after treatments. MTSs were fixed with 4% paraformaldehyde at room temperature (3 hours) and the TUNEL kit was applied following the manufacturer’s protocol. Cell nuclei were stained with Hoechst 33258 (Sigma-Aldrich) (5 μg/mL for 10 minutes). MTSs were observed by confocal microscopy (Nikon A1, Nikon Corporation, Tokyo, Japan).
In vivo treatment
To induce breast subcutaneous tumors, five groups (13 mice in each group) of the immunocompetent C57BL/6 mice were inoculated with half-million E0771 mice breast tumor cells subcutaneously in the right hind flank in a total volume of 200 µL of PBS. After 14 days, each group was treated every three days, for a total of four times by intravenous administration through the tail vein, with a dose of 10 mg/kg of PTX, PTX-PLGA NPs and the equivalent concentrations of PLGA blank NPs, Cremophor® EL and saline solution for the negative control group. Besides, the mice's survival, weight, and tumor volume were measured. The following formula was used to calculate the tumor volume: Eq.3. V(mm3) = (a × b2 × π)/6, where “a” is the largest diameter of the tumor, and “b” is the largest diameter perpendicular to the previous. After the administration of four doses of the different treatments, blood samples from four randomly selected mice were obtained in 1.5 mL EDTA-coated Eppendorf tubes and blood populations were analyzed (Mythic 22CT C2 Hematology Analyzer, Orphée SA, Switzerland). The final point of the experiment was 34 days from the initial inoculation of tumor cells.
Biodistribution assay
A total of 33 C57BL/6 mice were inoculated with half-million E0771 cells to induce subcutaneous breast tumors as described in the previous section. These mice were randomly divided into three groups. Two groups were treated intravenously through the tail vein with a single dose of 10 mg/kg of PTX and PTX-PLGA NPs. The third group was treated with saline as a negative control group. After 0.5, 1, 6, 12, and 24 hours, mice were sacrificed to collect different tissues: tumor, liver, spleen, kidneys, heart, lungs, and brain. All the tissues and plasma were stored at -80°C. A modified protocol
35
was used to measure PTX in the samples by LC-MS/MS. Briefly, 0.9% NaCl aqueous solution was added to the tissues (twice the weight) before being homogenized (T10 Basic ULTRA-TURRAX, Germany). A volume of 200 µL of each tissue was collected and the internal standard (docetaxel) was added to reach a final concentration of 150 ng/mL. After that, a volume of 500 µL of methyl tert-butyl ether (MTBE) was added. Samples were vortexed vigorously for 1 minute, and after 5 minutes of incubation at room temperature, they were vortexed again for 5 minutes and centrifuged at 16000 g, 5 minutes at room temperature. The upper phase is collected in a new 1.5 mL tube and left to evaporate in a vacuum. Finally, the dried residue was resuspended in 50 µL and analyzed by a triple quadrupole mass spectrometer (triple-quad) (WATERS, XEVO TQ-S) and ultra-high-pressure liquid chromatography (UPLC) (WATERS, ACQUITY H CLASS) (UPLC-MS/MS).
Statistical analysis
All the results were expressed as the mean ± standard deviation (SD). Statistical analysis was performed by the Student’s t test and the one-way ANOVA with post-hoc Tukey test (SPSS v.15, SPSS, Chicago, USA). Mice survival was evaluated with the Kaplan–Meier method. Finally, the logrank test was used to compare the proportion of living mice between groups. Values of P < 0.05 were considered significant.
Results
Antitumor activity of PTX-PLGA in breast cancer cells and breast cancer stem cells
Although both free PTX and PTX-loaded PLGA NPs showed a dose-dependent response, PTX-PLGA led to a significant increase in PTX cytotoxicity in all the breast cancer cell lines tested (P < 0.05) (Fig. 1, Table 1). In fact, PTX-PLGA was associated with a 1.74 and 3.03-fold reduction in the IC50 of PTX in human MCF-7 and mouse E0771 cells, respectively (Fig. 1A and F). PTX-PLGA also reduced the IC50 of PTX in the MCF-10A human breast cell line (Fig. 1E). Analysis of the PLGA blank NPs showed no toxicity in the cell lines investigated (Fig. S1, Supplementary file 1). In addition,CSCs from MCF-7 were exposed to PTX and PTX-loaded NPs to determine the cytotoxic effect using the CCK-8 assay (Fig. 1G and H). The IC50 of PTX in MCF-7 CSCs was higher (16.5 nM) than in MCF-7 cells (9.13 nM), indicating a greater resistance of the latter cells to the cytotoxic action of PTX. However, as shown in Figure 1G, there were no major differences in the IC50 of PTX versus PTX-PLGA NPs in MCF-7 CSCs. Only at high doses (above 10 nM), PTX-loaded PLGA NPs show a significantly greater cytotoxic effect compared to PTX (P < 0.05). Finally, no cytotoxicity was observed with PLGA NPs in these CSCs (Fig. 1G).
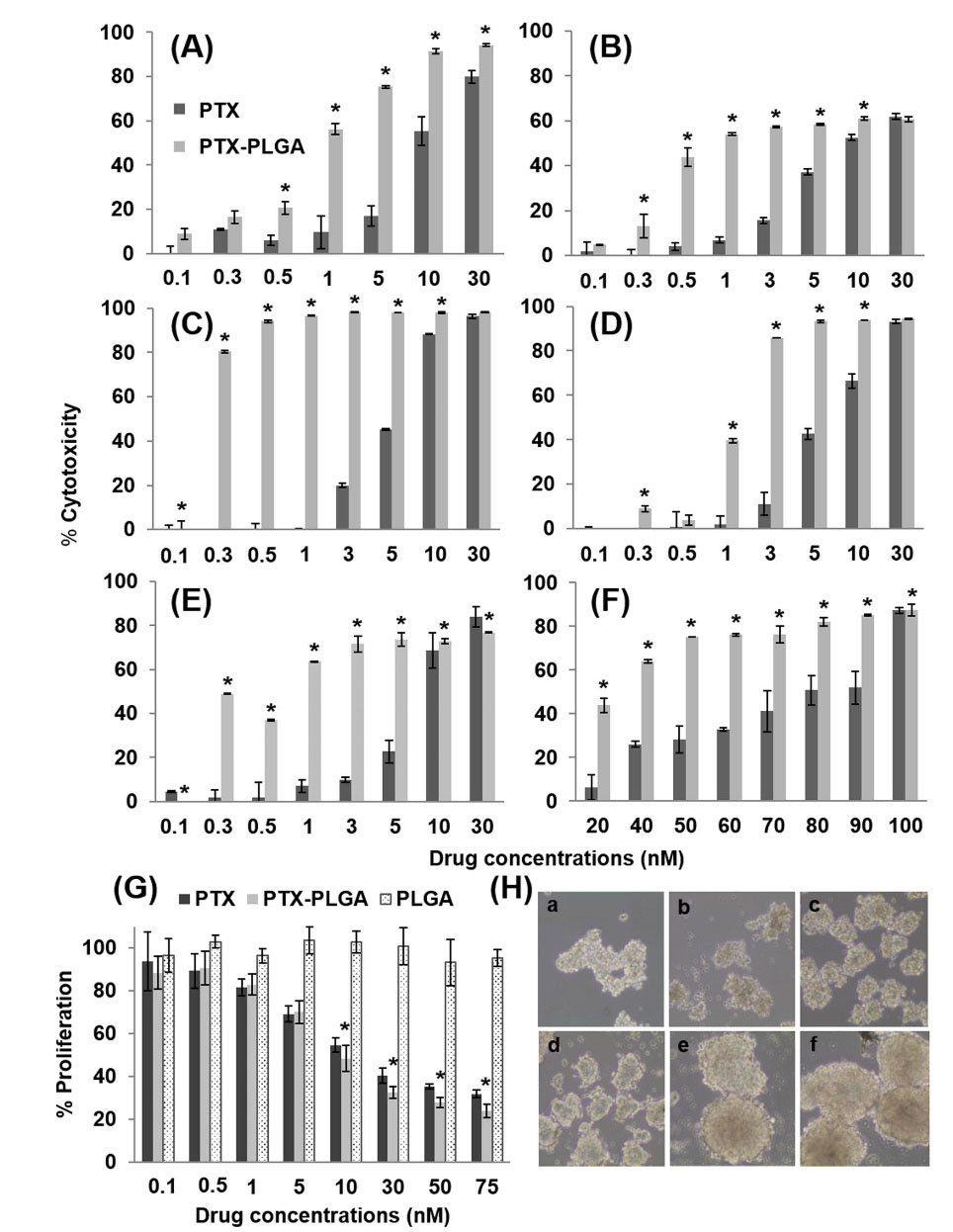
Fig. 1.
PTX-loaded PLGA NPs proliferation assay. Cell lines MCF-7 (A), MDA-MB-231(B), T47D (C), SK-BR-3 (D), MCF-10A (E) and E0771 (F) were treated with PTX-loaded PLGA NPs and free PTX. Cell cultures without treatment were used as a control. The percentage of cytotoxicity was measured after 4 days of incubation with the treatments (0.1 to 100 nM). Data were represented as the mean value ± SD of triplicate cultures. In addition, CSCs from MCF-7 were treated with the same treatments (G). Data were represented as the mean value ± SD of sextuplicate cultures. Optical microscope analysis showed the tumor spheres formation during the induction of the CSCs at 3 (a),5 (b),7 (c), 10 (d),12 (e) and 14 (f) days in culture (10× magnification) (H). “*” represent significant differences.
.
PTX-loaded PLGA NPs proliferation assay. Cell lines MCF-7 (A), MDA-MB-231(B), T47D (C), SK-BR-3 (D), MCF-10A (E) and E0771 (F) were treated with PTX-loaded PLGA NPs and free PTX. Cell cultures without treatment were used as a control. The percentage of cytotoxicity was measured after 4 days of incubation with the treatments (0.1 to 100 nM). Data were represented as the mean value ± SD of triplicate cultures. In addition, CSCs from MCF-7 were treated with the same treatments (G). Data were represented as the mean value ± SD of sextuplicate cultures. Optical microscope analysis showed the tumor spheres formation during the induction of the CSCs at 3 (a),5 (b),7 (c), 10 (d),12 (e) and 14 (f) days in culture (10× magnification) (H). “*” represent significant differences.
Table 1.
Half maximal inhibitory concentration of PTX and PLGA-PTX in breast cell lines from tumor and healthy tissues of humans and mice
|
Cell line
|
PTX IC
50
(nM)
|
PLGA-PTX IC
50
(nM)
|
Sensitivity index (SI)*
|
| MCF-7 |
9.13 |
5.24 |
1.74 |
| MDA-MB-231 |
20.08 |
15.36 |
1.31 |
| T47D |
5.6 |
3.96 |
1.41 |
| SK-BR-3 |
6.7 |
4.94 |
1.36 |
| MCF-10A |
8 |
3.6 |
2.23 |
| E0771 |
74.6 |
24.6 |
3.03 |
*Times that PTX-loaded nanoparticles reduces the IC50 value of free PTX
Modulation of the cell cycle by PTX-PLGA NPs
As shown in Figs. 2 and 3 and Table 2 PTX-PLGA NPs induced a cell cycle arrest in the G2/M phase similar to that found with free PTX. In fact, only SK-BR-3 cells showed a higher percentage (26.7% more) of cells in the G2/M phase after treatment with PTX-PLGA NPs, as compared to free PTX. In addition, MCF-7 cells showed a mild increase (7.05%) in the proportion of cells in the G2/M phase after exposure to PTX-PLGA NPs while in T47D and SK-BR-3 cell lines treated with PTX-PLGA NPs, a slight increase in the subG1 phase was observed in comparison with free PTX (9.74% and 14.3% increase, respectively). Cells treated with blank PLGA NPs showed no significant differences in the cell cycle profile compared to non-treated (i.e., control) cells.
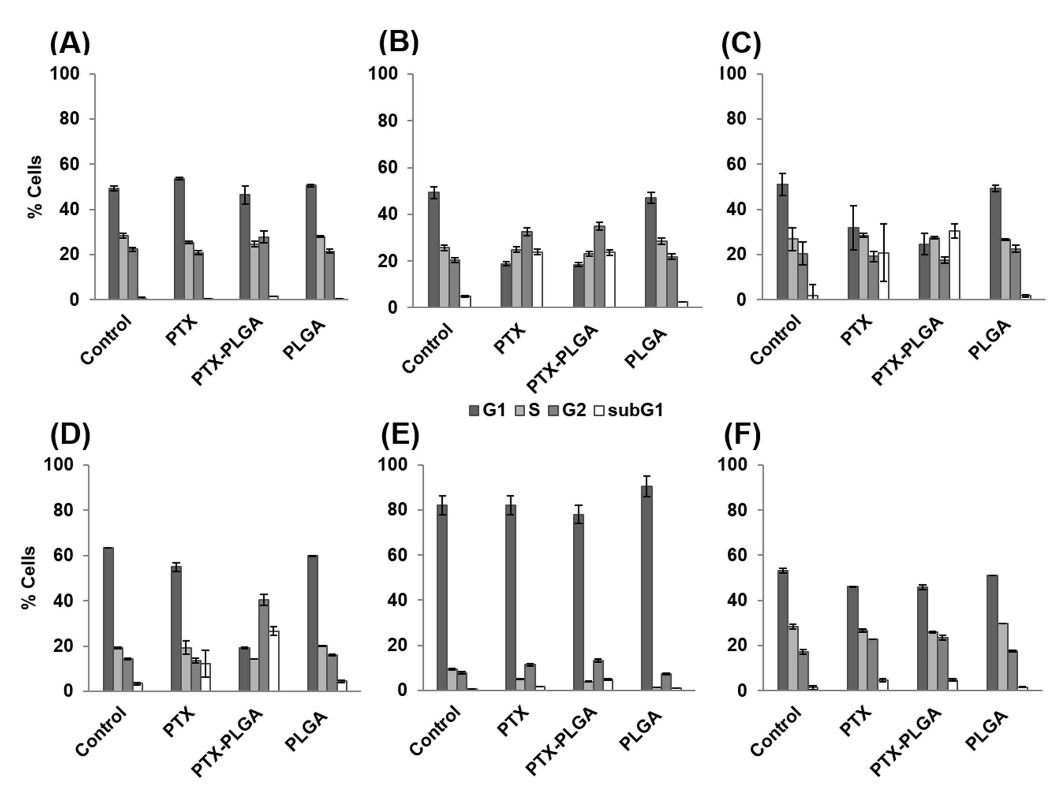
Fig. 2.
Modulation of cell cycle analysis by PTX-loaded PLGA NPs. MCF-7 (A), MDA-MB-231(B), T47D (C), SK-BR-3 (D), MCF-10A (E), E0771 (F) cells were treated with free PTX, PTX-loaded PLGA NPs and blank PLGA NPs at a dose of IC50 value during 48 hours. Cell cultures without treatment were used as a negative control. Results are expressed as the percentage of labelled cells in each cell cycle phase. Data represented as the mean value ± SD of triplicate cultures.
.
Modulation of cell cycle analysis by PTX-loaded PLGA NPs. MCF-7 (A), MDA-MB-231(B), T47D (C), SK-BR-3 (D), MCF-10A (E), E0771 (F) cells were treated with free PTX, PTX-loaded PLGA NPs and blank PLGA NPs at a dose of IC50 value during 48 hours. Cell cultures without treatment were used as a negative control. Results are expressed as the percentage of labelled cells in each cell cycle phase. Data represented as the mean value ± SD of triplicate cultures.
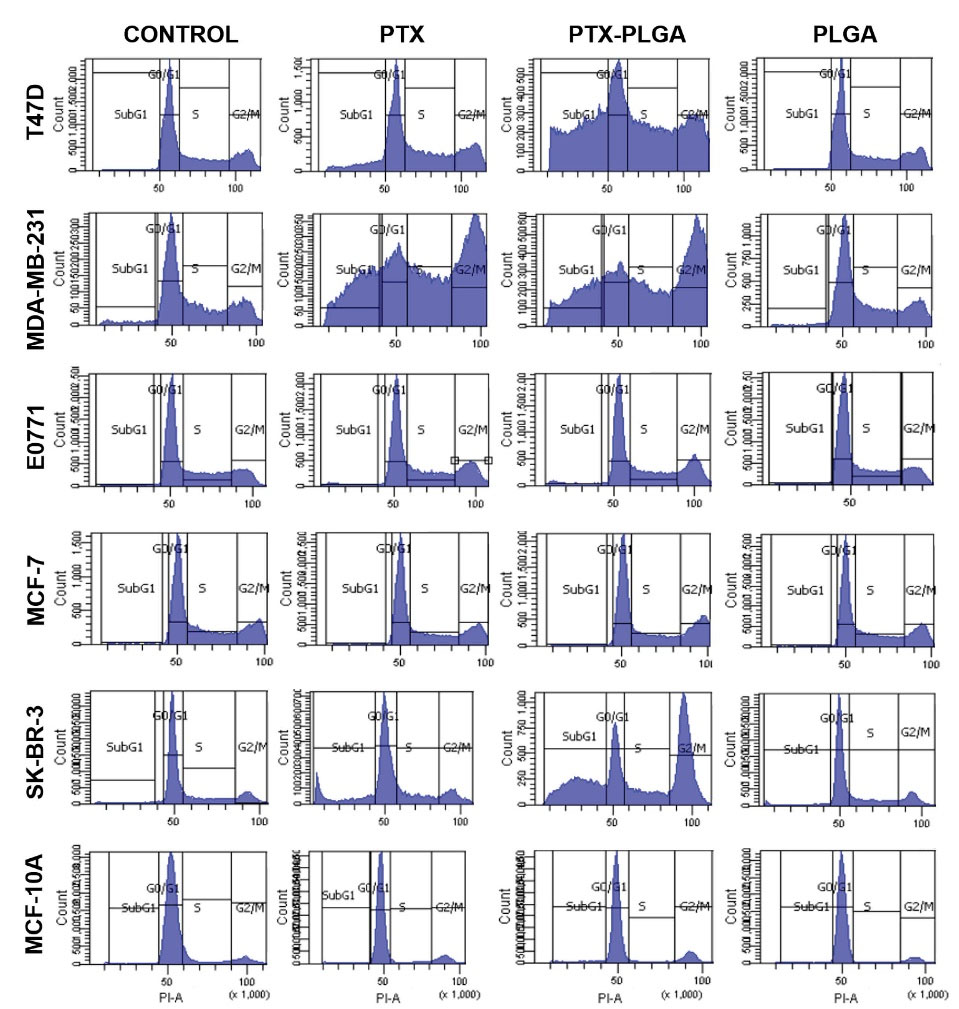
Fig. 3.
Flow cytometry charts of the modulation of cell cycle analysis by PTX-loaded PLGA NPs. Cells were treated with free PTX, PTX-loaded PLGA NPs and blank PLGA NPs at a dose of IC50 value during 48 h. Cell cultures without treatment were used as a negative control. Results are expressed as the percentage of labelled cells in each cell cycle phase. Data represented as the mean value ± SD of triplicate cultures.
.
Flow cytometry charts of the modulation of cell cycle analysis by PTX-loaded PLGA NPs. Cells were treated with free PTX, PTX-loaded PLGA NPs and blank PLGA NPs at a dose of IC50 value during 48 h. Cell cultures without treatment were used as a negative control. Results are expressed as the percentage of labelled cells in each cell cycle phase. Data represented as the mean value ± SD of triplicate cultures.
Table 2.
Quantitative values of cell cycle analysis modulated by PTX-loaded PLGA NPs
|
Cell line
|
Treatment
|
SubG1
|
G0/G1
|
S
|
G2/M
|
| T47D |
Control |
40203 |
55761 |
76249 |
105131 |
| PTX |
36977 |
56203 |
76203 |
105663 |
| PTX-PLGA |
32976 |
55710 |
76902 |
105590 |
| PLGA |
45521 |
55840 |
76309 |
105114 |
| MDA-MB-231 |
Control |
23427 |
48999 |
66170 |
91676 |
| PTX |
27180 |
48529 |
67846 |
94191 |
| PTX-PLGA |
26763 |
48492 |
67187 |
95013 |
| PLGA |
28495 |
50269 |
66301 |
92974 |
| E0771 |
Control |
32411 |
50222 |
71399 |
94034 |
| PTX |
21685 |
50940 |
70262 |
95847 |
| PTX-PLGA |
20284 |
52649 |
72455 |
98878 |
| PLGA |
32089 |
44948 |
62482 |
84891 |
| MCF-7 |
Control |
23192 |
49192 |
66622 |
93186 |
| PTX |
28924 |
49647 |
66828 |
93171 |
| PTX-PLGA |
22022 |
50287 |
6705 |
94251 |
| PLGA |
29362 |
49462 |
66971 |
92871 |
| SK-BR-3 |
Control |
25323 |
48566 |
68547 |
92418 |
| PTX |
19904 |
49825 |
67849 |
94051 |
| PTX-PLGA |
27989 |
50979 |
68596 |
95047 |
| PLGA |
25082 |
49404 |
67312 |
93665 |
| MCF-10A |
Control |
246648 |
51787 |
65132 |
98394 |
| PTX |
18515 |
47481 |
70504 |
89622 |
| PTX-PLGA |
29489 |
48628 |
67300 |
92321 |
| PLGA |
30883 |
49280 |
71769 |
93380 |
Results are expressed as fluorescence intensity (PI-A) in each cell cycle phase.
Immunofluorescence analysis of microtubules
As in the case of the cell cycle, possible modulation of the mechanism of action of PTX-loaded PLGA NPs was investigated. As shown in Fig. 4, both free PTX and PTX-PLGA NPs induced an important accumulation of microtubules. The density of microtubules was particularly intense around the cell nuclei, which could be better observed using a higher concentration (100 nM) (Fig. 5). However, no significant differences were observed between free PTX and PTX-PLGA. Interestingly, in E0771 cells, microtubule density was already increased at the IC50 dose. Finally, blank PLGA NPs did not modify the pattern of microtubule density.
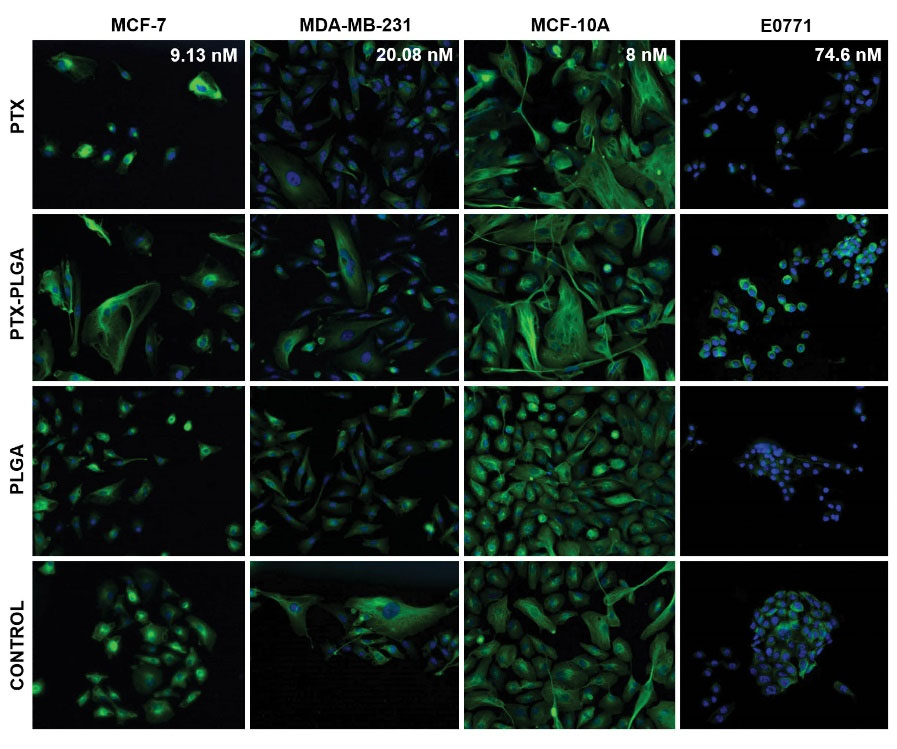
Fig. 4.
Effect of free PTX, PTX-PLGA NPs and blank PLGA on microtubules in MCF-7, MDA-MB-231, MCF-10A and E0771 cell lines. The cells were treated with their respective IC50 drug dose for 48 hours after a cell cycle synchronization and stained with an anti-α-tubulin antibody and Hoechst 33258 (see Material and Methods). Images at 20× of magnification.
.
Effect of free PTX, PTX-PLGA NPs and blank PLGA on microtubules in MCF-7, MDA-MB-231, MCF-10A and E0771 cell lines. The cells were treated with their respective IC50 drug dose for 48 hours after a cell cycle synchronization and stained with an anti-α-tubulin antibody and Hoechst 33258 (see Material and Methods). Images at 20× of magnification.
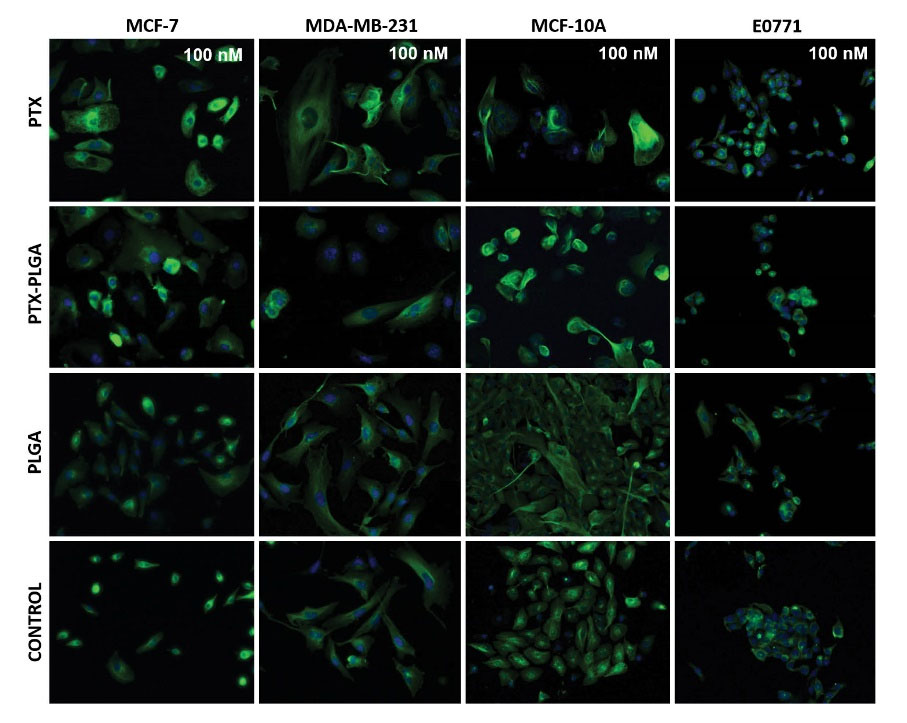
Fig. 5.
Effect of free PTX, PTX-PLGA NPs and blank PLGA on microtubules in MCF-7, MDA-MB-231, MCF-10A and E0771 cell lines. The cells were treated with 100 nM drug dose for 48 hours after a cell cycle synchronization and stained with an anti-α-tubulin antibody and Hoechst 33258 (see Material and Methods). Images at 20× of magnification.
.
Effect of free PTX, PTX-PLGA NPs and blank PLGA on microtubules in MCF-7, MDA-MB-231, MCF-10A and E0771 cell lines. The cells were treated with 100 nM drug dose for 48 hours after a cell cycle synchronization and stained with an anti-α-tubulin antibody and Hoechst 33258 (see Material and Methods). Images at 20× of magnification.
Cell uptake of PLGA and PTX-loaded PLGA NPs
PLGA NPs loaded with NR were used to verify their internalization in breast cancer (MCF-7) cells. Flow cytometry analysis showed that cell uptake in MCF-7 cells was very similar between NR and NR-PLGA NPs in short exposure times (0.5 hours), whereas the uptake of NR-PLGA NPs was higher after 1 hour of exposure (Fig. 6A and 7). Specifically, the uptake of NR-PLGA NPs increased by 24.5%, 62.5%, 70.1%, 63%, and 51.3% with respect to the uptake of free NR for exposition times of 0.5, 1, 2, 4 and 6 hours, respectively. These results were corroborated by fluorescence microscopy (Fig. 6B) and by confocal microscopy (Fig. 6C), which showed a higher fluorescence intensity of cells treated with NR-loaded PLGA NPs at all exposure times and a similar cytoplasmic location of NR (Fig. 6B).
Then, LC-MS/MS was used to detect differences in cellular internalization of free PTX versus PTX-loaded PLGA NPs. Cells were exposed to PTX and PTX-PLGA (500 nM) for 0.5, 1, 2, 4, and 6 hours. As shown in Fig. 8, much higher cell uptake of PTX was detected when PTX-PLGA NPs were used, even after the first half-hour exposure to the drug. The maximum intracellular concentration of PTX and PTX-PLGA in MCF-7 cells was 0.2 and 2.24 µg PTX/mg cell protein, respectively. Similar differences were also observed in MDA-MB-231 (0.13 and 0.37), MCF-10A (0.2 and 0.65) and E0771 (0.16 and 0.53) breast cancer cells.
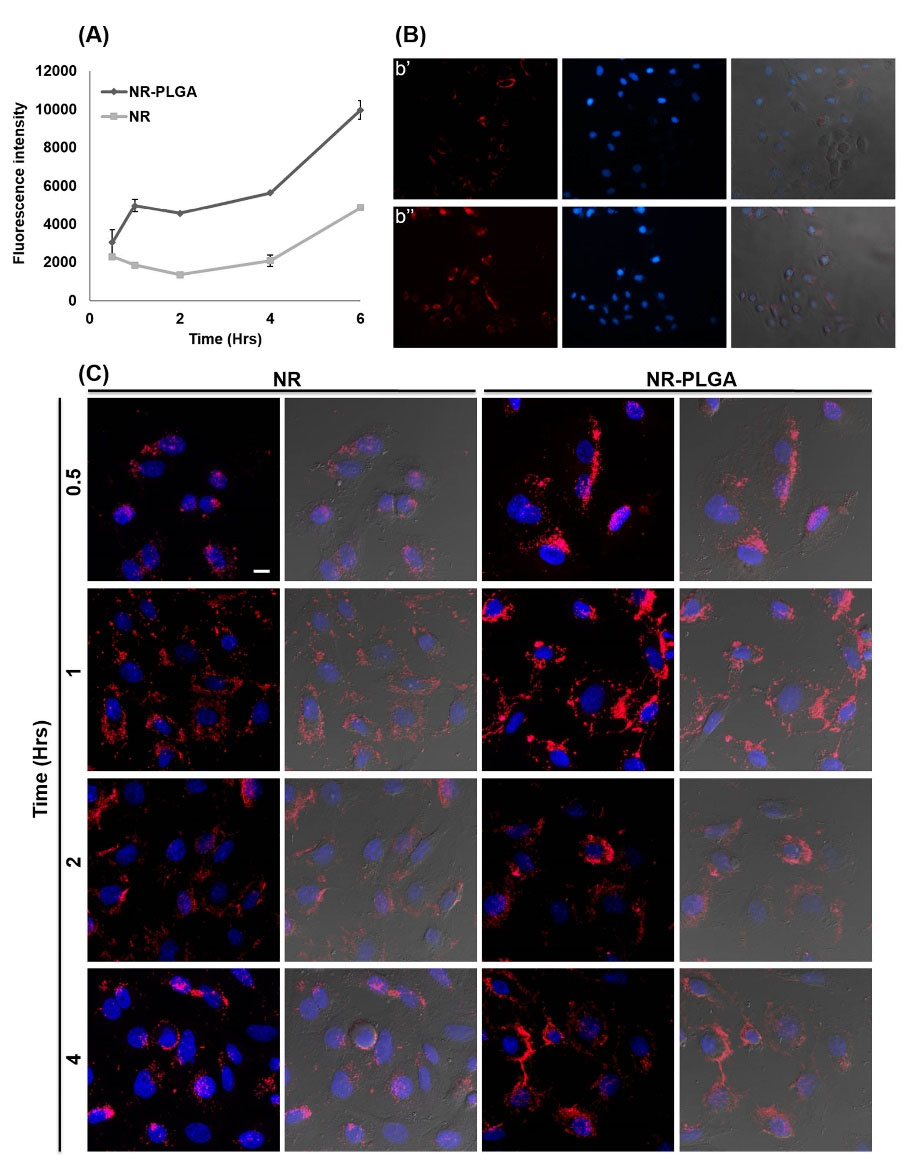
Fig. 6.
Cell uptake of NR-loaded PLGA NPs. Cell uptake was quantitatively measured by flow cytometry with MCF-7 cells treated with a solution of NR and NR-loaded PLGA NPs (0.5 μM) from 0.5 to 6 hours (A). Assay was performed in duplicate (mean value ± SD). Cell uptake was qualitatively measured by fluorescence microscopy in MCF-7 cell line (B) treated with free NR (b’) and NR-loaded PLGA NPs (b’’) (0.05 μM). The images showed were taken at 4 hours of exposition, at 20× of magnification. Confocal microscopy images of MCF-7 cells treated with NR-loaded PLGA NPs. MCF-7 cells were treated with NR-loaded PLGA NPs and a NR solution (0.05 μM) at different times (from 0.5 to 4 hours) and their cell nuclei were stained with 5 μg/mL of Hoechst 33258 (0.05 μM). The figure shows merged images of the treatments at different exposure times with NR or the NPs NR-PLGA and Hoechst 33258 (images with dark background) and also with the superposition of images taken in bright field (images with gray background) (bar scale: 10 μm) (C).
.
Cell uptake of NR-loaded PLGA NPs. Cell uptake was quantitatively measured by flow cytometry with MCF-7 cells treated with a solution of NR and NR-loaded PLGA NPs (0.5 μM) from 0.5 to 6 hours (A). Assay was performed in duplicate (mean value ± SD). Cell uptake was qualitatively measured by fluorescence microscopy in MCF-7 cell line (B) treated with free NR (b’) and NR-loaded PLGA NPs (b’’) (0.05 μM). The images showed were taken at 4 hours of exposition, at 20× of magnification. Confocal microscopy images of MCF-7 cells treated with NR-loaded PLGA NPs. MCF-7 cells were treated with NR-loaded PLGA NPs and a NR solution (0.05 μM) at different times (from 0.5 to 4 hours) and their cell nuclei were stained with 5 μg/mL of Hoechst 33258 (0.05 μM). The figure shows merged images of the treatments at different exposure times with NR or the NPs NR-PLGA and Hoechst 33258 (images with dark background) and also with the superposition of images taken in bright field (images with gray background) (bar scale: 10 μm) (C).
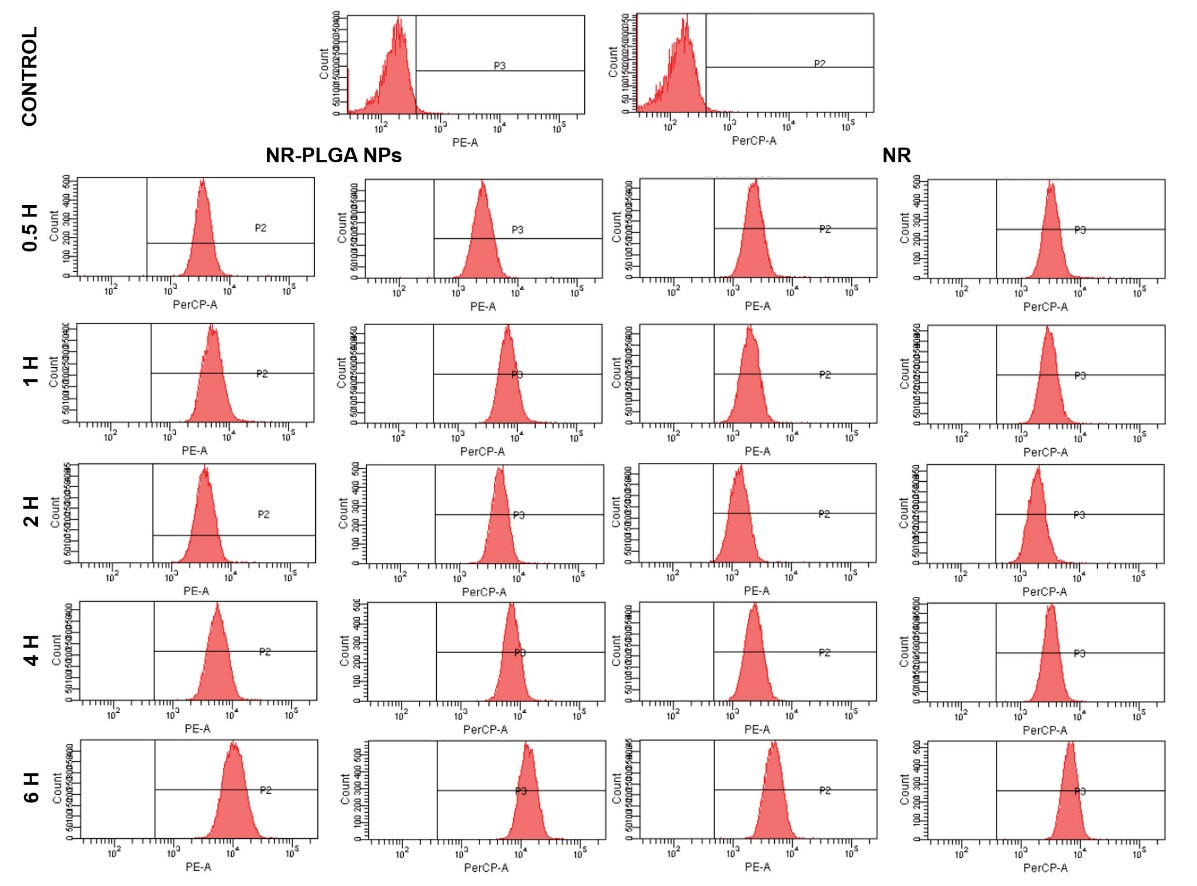
Fig. 7.
Flow cytometry charts of MCF-7 cells treated with a solution of NR and NR-loaded PLGA NPs (0.5 μM) from 0.5 to 6 hours. Assay was performed in duplicate (mean value ± SD).
.
Flow cytometry charts of MCF-7 cells treated with a solution of NR and NR-loaded PLGA NPs (0.5 μM) from 0.5 to 6 hours. Assay was performed in duplicate (mean value ± SD).
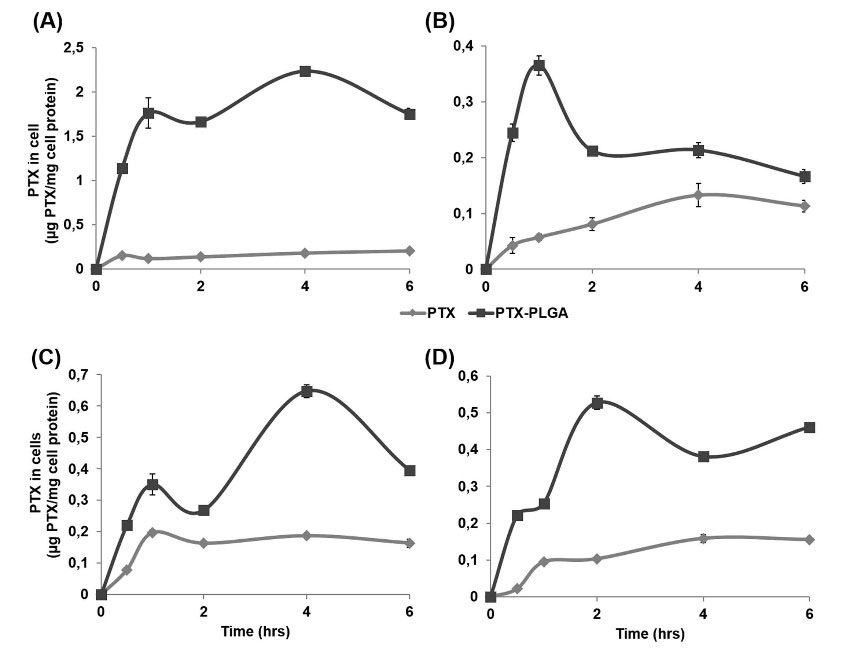
Fig. 8.
PTX-loaded PLGA NPs cell uptake measured by LC-MS/MS. MCF-7 (A) MDA-MB-231 (B) MCF-10A (C) E0771 (D) cells were treated with 500 nM of PTX and PTX-PLGA NPs for 0.5, 1, 2, 4 and 6 hrs. After the exposition time, cells were lysed and the PTX was extracted and measured by LC-MS/MS to obtain the intracellular concentration of PTX (μg) in relation with cell proteins (mg). Data were represented as the mean value ± SD of triplicate cultures.
.
PTX-loaded PLGA NPs cell uptake measured by LC-MS/MS. MCF-7 (A) MDA-MB-231 (B) MCF-10A (C) E0771 (D) cells were treated with 500 nM of PTX and PTX-PLGA NPs for 0.5, 1, 2, 4 and 6 hrs. After the exposition time, cells were lysed and the PTX was extracted and measured by LC-MS/MS to obtain the intracellular concentration of PTX (μg) in relation with cell proteins (mg). Data were represented as the mean value ± SD of triplicate cultures.
Growth modulation of MTSs by PTX-PLGA NPs
To determine the effectiveness of PTX-PLGA NPs in a system that limits drug access to all tumor cells, MTSs were generated from MCF-7 and E0771 tumor cells. In comparison with free PTX, PTX-PLGA NPs induced a significant reduction in cell proliferation in MCF-7 (27.9 %) and E0771 (16.3%) from the first dose administered (P < 0.05) (Figs. 9B and 10B). Exposure of MCF-7 and E0771 MTSs to blank PLGA NPs was not associated with significant changes in cell viability. On the other hand, no significant differences in the volume of MCF-7 or E0771 MTSs treated with free PTX and PTX-PLGA were detected (Figs. 9A, C and 10A, C). However, analysis of the evolution of MTSs by light microscopy showed clear differences in the degree of surface compaction. In fact, MTSs treated with PTX-PLGA showed earlier surface disorganization (including greater disintegration) than MTSs treated with free PTX, which were characterized by a compact surface (Figs. 9C and 10C). In MCF-7 MTSs in particular, early surface disintegration after PTX-PLGA treatment (faster than with PTX treatment) could be related to the non-significant volume reduction and the decrease in proliferation previously observed. MTSs treated with PLGA and control MTSs are similar because the PLGA NPs were not toxic. However, light microscopy images showed that they are quite different from those MTSs treated with PTX and PTX-PLGA. The latter is considerably smaller especially at 96 hours with the edge of the sphere much more irregular than in the case of PLGA and control MTSs whose edge is much flatter and there is much less cellular debris around, especially when were compared to PTX-PLGA-treated MTSs. This is just one more piece of evidence of what happens in the cell proliferation experiment with CCK8: PTX and PTX-PLGA were toxic for MTSs and reduced their volume compared to those treated with PLGA and untreated MTSs. Finally, the TUNEL assay showed an increased rate of apoptosis in E0771 MTSs treated with PLGA-PTX NPs versus free PTX, which was less evident in MCF-7 MTSs (Figs. 9D and 10D).

Fig. 9.
MTS from MCF-7 human breast cancer cell line treated with PTX-loaded PLGA NPs. Representative images of the volume evolution of MTS after treatment (drug dose of 36,5 nM) (Scale bar: 100 μm; 10×magnification) (A). Proliferation assay of the MTS after treatment. Non treated MTS were used as controls. Data represented as the mean ± SD octuplicate cultures (B). Progression of the tumor volume of the treated MTS (C). Confocal microscopy images of the MTS labeled (TUNEL kit) to detect apoptotic cells (red labeling) (Scale bar: 100 μm) (D). “*” represent significant differences.
.
MTS from MCF-7 human breast cancer cell line treated with PTX-loaded PLGA NPs. Representative images of the volume evolution of MTS after treatment (drug dose of 36,5 nM) (Scale bar: 100 μm; 10×magnification) (A). Proliferation assay of the MTS after treatment. Non treated MTS were used as controls. Data represented as the mean ± SD octuplicate cultures (B). Progression of the tumor volume of the treated MTS (C). Confocal microscopy images of the MTS labeled (TUNEL kit) to detect apoptotic cells (red labeling) (Scale bar: 100 μm) (D). “*” represent significant differences.
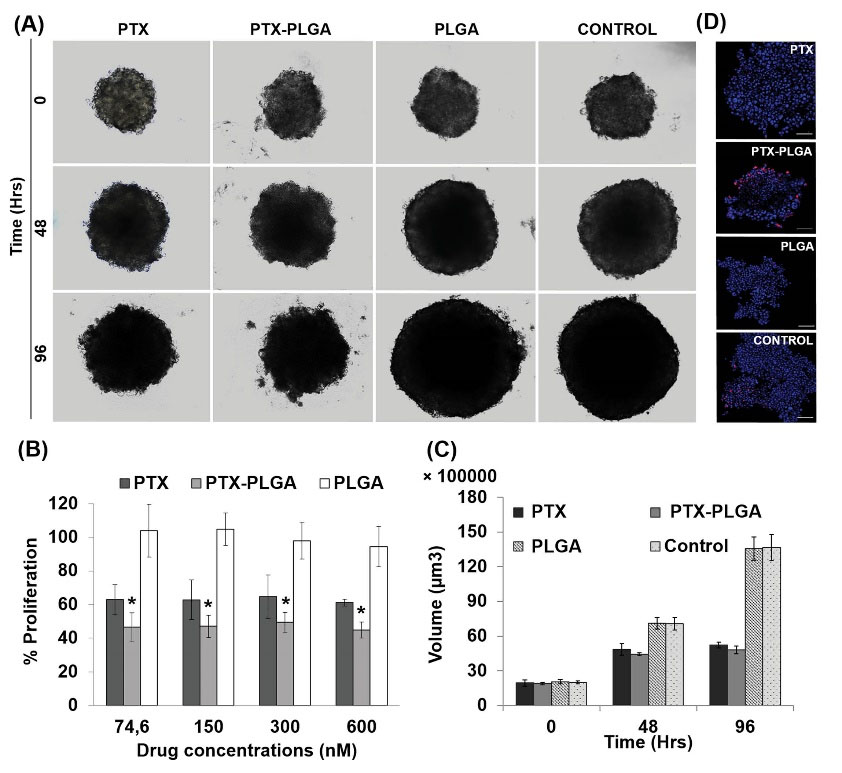
Fig. 10.
MTS from E0771 mice breast cancer cell line treated with PTX-loaded PLGA NPs. Representative images of the volume evolution of MTS after treatment (drug dose of 300 nM) (Scale bar: 100 μm; 10×magnification) (A). Proliferation assay of the MTS after treatment. Non treated MTS were used as controls. Data represented as the mean ± SD octuplicate cultures (B). Progression of the tumor volume of the treated MTS (C). Confocal microscopy images of the MTS labeled (TUNEL kit) to detect apoptotic cells (red labeling) (Scale bar: 100 μm) (D). “*” represent significant differences.
.
MTS from E0771 mice breast cancer cell line treated with PTX-loaded PLGA NPs. Representative images of the volume evolution of MTS after treatment (drug dose of 300 nM) (Scale bar: 100 μm; 10×magnification) (A). Proliferation assay of the MTS after treatment. Non treated MTS were used as controls. Data represented as the mean ± SD octuplicate cultures (B). Progression of the tumor volume of the treated MTS (C). Confocal microscopy images of the MTS labeled (TUNEL kit) to detect apoptotic cells (red labeling) (Scale bar: 100 μm) (D). “*” represent significant differences.
Antitumor activity and biodistribution of PTX-PLGA NPs in vivo
As shown in Fig. 11, a reduction in tumor volume in mice treated with PLGA-PTX compared to free PTX was detected (Fig. 11A). Nevertheless, no significant differences were detected between the two treatments. In addition, no significant differences in weight or survival of mice were found (Fig. 11B and C). In addition, biodistribution analysis demonstrated that PTX-PLGA induced higher time-dependent concentrations of PTX in the spleen, lungs, and liver, in comparison with free PTX (Fig. 11D). Interestingly, blood analyses showed no significant differences between PTX-PLGA NPs and the control group (Table 3). On the contrary, mice treated with free PTX showed significantly higher percentages of lymphocytes and lower percentages of monocytes and neutrophils. Therefore, the in vivo treatment with PTX-PLGA NPs did not significantly alter the anti-tumor properties of free PTX while leading to reduced blood toxicity, as reflected by non-significant alterations in the blood parameters (i.e. similar to the control group).
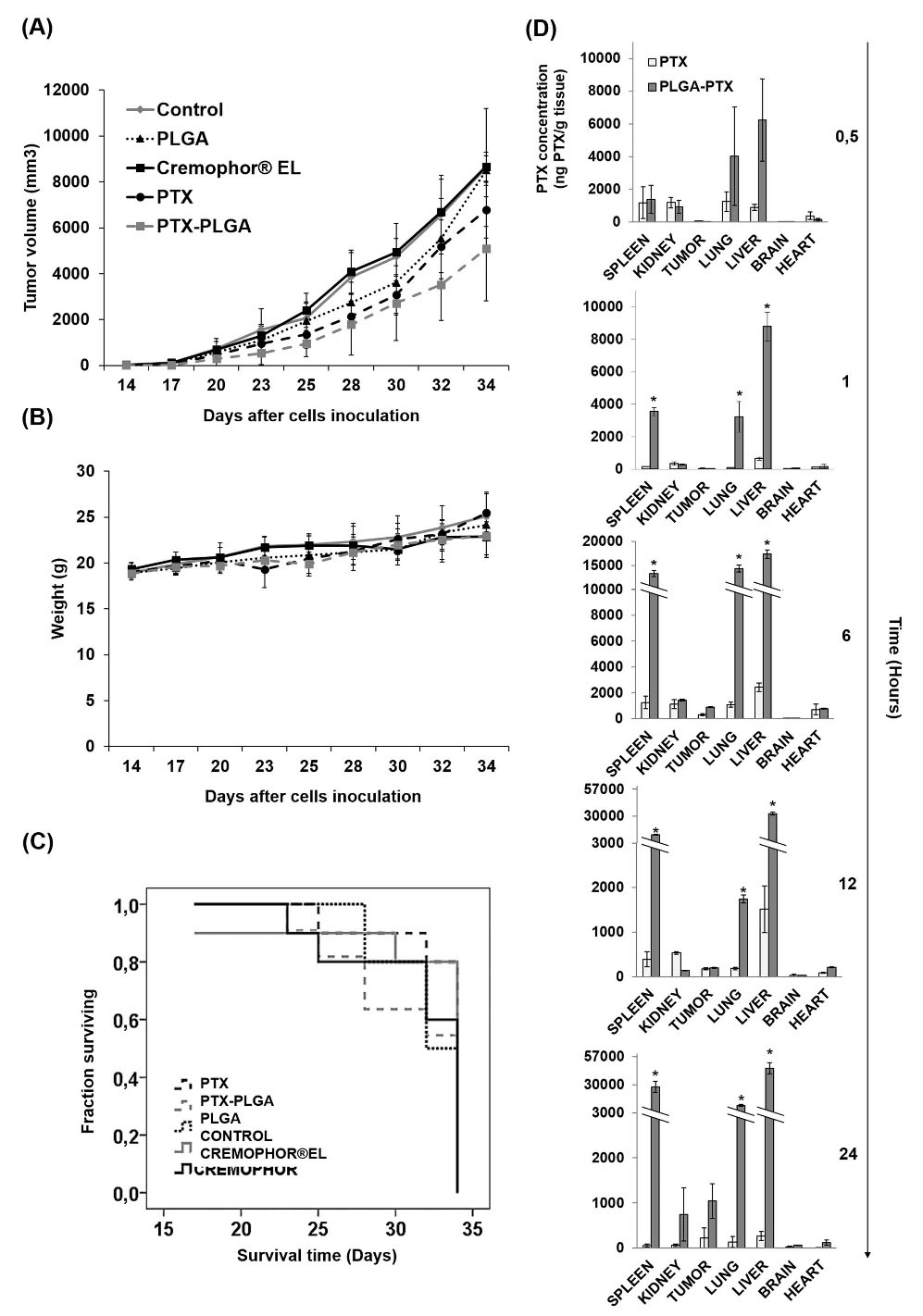
Fig. 11.
Breast cancer xenografts treated with PTX-PLGA NPs. Graphical representation of the tumor volume progression after treatments with free PTX, PTX-PLGA NPs, blank PLGA NPs and Cremophhor®EL (i.v. administration) into C57BL/6 mice bearing subcutaneous breast tumors (A). Body weight variation (B). Kaplan-Meier curve of mice survival rates (C). Data were represented as the mean ± S.D. (n = 13). PTX biodistribution in mice treated intravenously with PTX and PTX-PLGA NPs (D). Data were represented as the mean ± S.D. (n = 3).
.
Breast cancer xenografts treated with PTX-PLGA NPs. Graphical representation of the tumor volume progression after treatments with free PTX, PTX-PLGA NPs, blank PLGA NPs and Cremophhor®EL (i.v. administration) into C57BL/6 mice bearing subcutaneous breast tumors (A). Body weight variation (B). Kaplan-Meier curve of mice survival rates (C). Data were represented as the mean ± S.D. (n = 13). PTX biodistribution in mice treated intravenously with PTX and PTX-PLGA NPs (D). Data were represented as the mean ± S.D. (n = 3).
Table 3.
Analysis of blood parameters in breast tumors-bearing mice treated with PTX, PTX-PLGA, PLGA, and Cremofor®EL
|
Blood parameters
|
Control
|
Cremofor® EL
|
PTX
|
PTX-PLGA
|
|
WBC (103/uL)
|
2.75±0.07 |
1.95±0.21* |
1.8±0.53 |
2.13±0.06 |
|
RBC (106/uL)
|
2.47±0.09 |
7.65±0.39* |
3.26±0.21 |
2.71±0.4 |
| LYM (%) |
83.55±1.2 |
97.5±2.26* |
95.65±1.1* |
84±5.07 |
| MON (%) |
7.25±0.21 |
0.3±0.05* |
1.53±0.3* |
5.6±1.45 |
| NEU (%) |
6.1±1.13 |
1±0.99* |
1.53±0.6* |
3.87±1.01 |
| EOS (%) |
0.38±0.17 |
0.2±0.05 |
0.43±0.25 |
0.27±0.21 |
| BAS (%) |
2.3±0.42 |
0.3±0.28* |
1.4±0.00 |
4.8±1.98 |
WBC, white blood cell; RBC, red blood cell; LYM, lymphocyte; MON, monocyte; NEU, neutrophil; EOS, eosinophil; BAS, basophil. The data represent the mean value ± S.D. (n =4). * P ˂ 0.05 compared to control.
Discussion
We assayed PTX-PLGA NPs in breast cancer cell lines and breast cancer-bearing mice in order to (i) increase cell drug internalization in the tumor mass, (ii) avoid the need to use toxic solvents, and (iii) improve drug efficacy in this type of tumor. Our results showed that this nanoformulation may represent an important advance in the current therapeutic arsenal for the treatment of breast cancer.
We used the PLGA polymer based on its biocompatibility, biodegradability, and physicochemical properties, which are especially useful for drugs with low solubility. In fact, our blank PLGA NPs showed no significant toxicity in any of the breast cell lines investigated, probably due to their simple degradation to lactic and glycolic acids, which can be easily metabolized.
22,39-41
As previously stated,
28
the PTX-PLGA NPs were homogeneous in size, with a mean diameter of 250 nm and coefficients of variation and polydispersity indexes of less than 15% and 0.3%, respectively. The surface charge was negative in the water (mean ζ value ~ −30 mV). Regarding PTX encapsulation and release, the encapsulation efficiency was from 85.0 ± 10.7 to 91.6 ± 5.1, which is quite high compared to the PLGA nanoformulations described by Danhier et al
42
(70%), Duan et al
43
(31.3 ± 2.3% and 32.3 ± 2.4%) and Venugopal et al
44
(85.42 ± 2.6% and 85.58 ± 1.2%) which represented a notable advantage. When PLGA NPs were loaded with PTX, greater antitumor activity in breast cancer cell lines was observed, specifically 1.74 times higher than the free drug in MCF-7 cells. In a similar way, Parveen and Sahoo observed a decrease in the IC50 of PTX in MCF-7 (10.2 and 0.8 ng/L with PTX and PEG/chitosan-coated PTX-PLGA, respectively) after 5 days of treatment.
45
A 2.2-fold reduction of the IC50 of PTX was also observed in MCF-7 cells using a copolymer of PLGA and dextran, being effective in MCF-7/Adr (7.8-fold IC50 decrease as compared to free PTX) and not being associated with alterations in the expression of P-glycoprotein.
46,47
With regard to other breast cancer cell lines such as MDA-MB-231, Vicari et al reported a 25% increase in PTX cytotoxicity using PTX-PLGA NPs.
48
Similarly to other studies, we detected an increase in the antiproliferative activity of PTX-loaded NPs when they were used in the non-tumor MCF-10A cell line. In fact, Zubris et al reported similar toxicity in MCF-10A with free PTX and PTX-loaded NPs,
49
despite the fact that these nanoformulations had high anti-tumor activity in vivo due to the EPR effect.
14-16,50
Interestingly, our PTX-PLGA NPs also induced a significant decrease in CSC growth compared to PTX at high doses. CSCs have been associated with tumor recurrence due to their high drug resistance, self-renewal capacity, and aggressiveness in terms of metastasis formation.
51
Thus, our results indicate that PLGA NPs can cause a greater cytotoxic effect in MCF7 CSCs, which are normally more resistant to chemotherapeutic drugs (PTX IC50: 16.5 nM in MCF7 CSCs versus 9.13 in MCF7). The development of novel nanoformulations that allow the elimination of CSCs is a crucial step in achieving success in anticancer therapy. In this context, Das et al. demonstrated that PLGA NPs loaded with the antitumor drug wedelolactone improved CSCs’ uptake and enhanced drug cytotoxicity.
52
These results show that PLGA NPs could be a good strategy to transport and release anti-tumor therapeutic drugs into CSCs.
On the other hand, analysis of the internalization of our NPs using NR revealed increased fluorescence intensity in cells treated with NR-PLGA NPs. This increase was detected even after short exposure times, indicating a high internalization efficiency that may be related to the physicochemical properties of the NPs investigated, particularly the surface charge. In fact, their positive charge could favor the interaction between the cell membranes and the NPs, and thus their internalization.
22,25,40
In addition, LC-MS/MS confirmed a high degree of PTX internalization when it was loaded into PLGA NPs, even after the first half-hour of drug exposure. However, while free PTX showed a relatively linear pattern of internalization that increased over time, PTX carried in PLGA NPs showed a two-peak pattern: one during the first 2 hours of drug exposure, and another one after 4 hours. Other authors reported maximum intracellular PTX concentrations at 2 hours of treatment.
36,53
Li et al found that cells not only capture the PTX loaded into PLGA NPs but also the extracellular free PTX present in the medium, persisting even when the internalization of NPs decreases.
36
This may indicate that the uptake of NPs and drugs is carried out through two independent processes, although they must be considered at the same time. We hypothesize that at high doses (500 nM) of PTX-PLGA, some cells die after the first cell uptake (i.e., the first peak in the graph), showing a decrease in the intracellular concentration of PTX (i.e., after the first peak in the graph). Then, the PTX released into the medium by the dead cells would be internalized by the cells that remain alive (second peak). Complementary studies showed no significant differences between cells treated with PTX and PTX-PLGA NPs in their cell cycle profiles. Only SK-BR-3, T47D, and MCF-7 showed a slight increase in the percentage of cells in the G2/M or subG1 phases, a behavior that was also demonstrated in other studies.
54-57
In addition, immunofluorescence images revealed a similar tubulin pattern in cells treated with PTX and PTX-PLGA. Increased microtubule density was detected only in E0771 cells. Overall, these results indicate that the mechanism of action of the internalized drug was preserved and even improved, similarly to what other authors have verified using other PTX-loaded nanoformulations.
58,59
The in vitro studies were completed with analyses of the effect of PTX-PLGA on three-dimensional models of tumor cells (MTSs), which allowed a closer approximation to its behavior in in vivo models. The surface area of MTSs is more exposed to the drug, generating an inner gradient of oxygen, nutrients, and catabolites, analogous to primary tumors inserted into the tissue.
60,61
We observed greater disorganization of the spherocytes treated with PTX-PLGA in comparison with those treated with PTX, which were smaller and more compact in the early stages of treatment. This phenomenon could be due to the death of cells on the MTSs surface (first exposed), allowing drug entry into inner areas of the MTSs and thus facilitating its diffusion into the structure.
62,63
Moreover, cell death rates were higher in MTSs treated with PTX-PLGA, as demonstrated by the CCK-8 test. Lee et al found that co-administering DOX and PTX in multilayer PLLA/PLGA microparticles with poly(1,6-bis(p carboxyphenoxy) hexane) (PCPH) resulted in a lower cytotoxic effect in 3-D tumor spheroids.
64
In addition, PTX-loaded NPs of trimethylene carbonate showed poor results in glioblastoma MTSs from the U87-MG cell line, which could only be improved (i.e., leading to spherocyte disintegration) with the use of a specific peptide for integrin-rich tumors.
38
In vivo assays showed that both PTX-PLGA and free PTX achieved similar results in terms of mouse survival and variations in mouse weight. In addition, PTX-PLGA clearly induced a greater tumor volume reduction than free PTX (with no significant differences). Interestingly, mice treated with PTX-PLGA showed higher PTX concentrations in the spleen, lungs and liver, and similar PTX concentrations in tumor tissues in comparison with a previous study in mice with subcutaneous lung tumors.
28
Hence, this could represent a therapeutic advantage considering the frequent presence of metastases derived from breast cancer in some of these organs. On the other hand, although we observed a reduction in breast tumor volume, this was lower than in lung cancer. The dissimilar results in tumor volume progression could be due to different tissue architecture between breast and lung tumors. In fact, the accumulation of NPs in the tumor occurs passively due to the EPR effect. Thus, variations in the distribution of blood vessels could affect the retention of macromolecules within the tumor. Moreover, breast tumors were larger than lung tumors and more heterogeneous, with a central region having no EPR effect. It has been demonstrated that tumors larger than 2 cm have fewer vascularized areas and thus a greater number of necrotic areas.
65-67
In this line, specific strategies to improve the EPR effect in NP-based therapy have been proposed, such as vasodilation of tumor blood vessels using nitric oxide or blood pressure elevation.
66,68-70
Finally, the side effects of PTX included alterations in hematological parameters.
71-73
In particular, free PTX was associated with significantly higher percentages of lymphocytes and lower percentages of monocytes and neutrophils as compared to the control mice. By contrast, our PTX-PLGA NPs improved drug side effects, obtaining blood parameters similar to the control mice. This could be explained by rapid PTX tissue accumulation when the drug is transported in the NPs, reducing its neutropenic effect. Other PTX-loaded nanoformulations such as nab-PTX
74
were also associated with fewer side effects (neutropenia and hypersensitivity) compared to the free drug.
75
Therefore, the use of PLGA NPs to transport PTX could help reduce the side effects of this drug.
Research Highlights
What is the current knowledge?
√ Paclitaxel has many limitations in its administration in patients.
√ PTX-PLGA NPs have been designed as a Cremophor® EL free formulation
What is new here?
√ NPs were tested in breast cancer tumor models.
√ NPs increase the antitumor effect of PTX without modifying its mechanism of action.
√ PLGA-NPs improve drug internalization into tumor cells.
√ NPs treatment reduce tumor volume in breast cancer-bearing C57BL/6 mice.
Conclusion
In conclusion, our PTX-PLG NPs were able to increase the internalization and antitumor activity of PTX in in vitro models of breast cancer. In addition, PLGA NPs were shown to be a safer and biocompatible alternative for the administration of PTX, avoiding the use of toxic solvents (e.g., Cremophor® EL). Moreover, in vitro experiments revealed that PTX-PLGA did not alter the mechanism of action of PTX. In vivo experiments showed a similar antitumor effect of PTX-PLGA in comparison with the free drug, which could be related to the lack of vascularization and the EPR effect within the tumor. However, this nanoformulation was able to reduce the side effects of PTX in mice. Therefore, our NPs could be a good strategy for the treatment of breast cancer, based on the reduction of drug toxicity and subsequent decrease in side effects. Nevertheless, further studies would be desirable to analyze their behavior in large tumors that lack the EPR effect or to explore their functionalization for active drug targeting.
Acknowledgments
We thank the Microscopy Service of CITIUS (University of Seville) for their technical assistance.
Funding sources
Financial support from the V Plan Propio (University of Seville). This work was also supported by Consejería de Salud de la Junta de Andalucía (PI-0102-2017 and P18-HO-3882) and Instituto de Salud Carlos III (ISCIII) (Project PI19/01478) (FEDER).
Ethical statement
The animal studies were approved by the Ethics Committee on Animal Experimentation at the University of Granada.
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
Participated in research design: LC, ROz, LM-B, JMB, CM, JP. Conducted experiments: LC, RO, ME-H, MDC-O, JJ-L, GP. Performed data analysis: LC, RO, ME-H, MDC-O, JJ-L, GP. Wrote or contributed to the writing of the manuscript: LC, LM-B, RO, CM, JP.
Supplementary files
Supplementary file 1 contains Figure S1.
(pdf)
References
- Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Intern Med 2013; 274:113-26. doi: 10.1111/joim.12084 [Crossref] [ Google Scholar]
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004; 4:253-65. doi: 10.1038/nrc1317 [Crossref] [ Google Scholar]
- Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer 2010; 10:194-204. doi: 10.1038/nrc2803 [Crossref] [ Google Scholar]
- Prota AE, Bargsten K, Zurwerra D, Field JJ, Díaz JF, Altmann K-H. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013; 339:587-90. doi: 10.1126/science.1230582 [Crossref] [ Google Scholar]
- Gornstein E, Schwarz TL. The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology 2014; 76:175-83. doi: 10.1016/j.neuropharm.2013.08.016 [Crossref] [ Google Scholar]
- Nehate C, Jain S, Saneja A, Khare V, Alam N, Dubey RD. Paclitaxel formulations: challenges and novel delivery options. Curr Drug Deliv 2014; 11:666-86. doi: 10.2174/1567201811666140609154949 [Crossref] [ Google Scholar]
- Adams JD, Flora KP, Goldspiel BR, Wilson JW, Arbuck SG, Finley R. Taxol: a history of pharmaceutical development and current pharmaceutical concerns. J Natl Cancer Inst Monogr 1993; 15:141-7. [ Google Scholar]
- Weiszhár Z, Czúcz J, Révész C, Rosivall L, Szebeni J, Rozsnyay Z. Complement activation by polyethoxylated pharmaceutical surfactants: Cremophor-EL, Tween-80 and Tween-20. Eur J Pharm Sci 2012; 45:492-8. doi: 10.1016/j.ejps.2011.09.016 [Crossref] [ Google Scholar]
- Allahverdiyev AM, Parlar E, Dinparvar S, Bagirova M, Abamor EŞ. Current aspects in treatment of breast cancer based of nanodrug delivery systems and future prospects. Artif Cells Nanomedicine Biotechnol 2018; 46:S755-62. doi: 10.1080/21691401.2018.1511573 [Crossref] [ Google Scholar]
-
Klochkov SG, Neganova ME, Nikolenko VN, Chen K, Somasundaram SG, Kirkland CE, et al. Implications of nanotechnology for the treatment of cancer: Recent advances. Semin Cancer Biol 2019. 10.1016/j.semcancer.2019.08.028.
- Tanaka T, Decuzzi P, Cristofanilli M, Sakamoto JH, Tasciotti E, Robertson FM. Nanotechnology for breast cancer therapy. Biomed Microdevices 2009; 11:49-63. doi: 10.1007/s10544-008-9209-0 [Crossref] [ Google Scholar]
- Tharkar P, Madani AU, Lasham A, Shelling AN, Al-Kassas R. Nanoparticulate carriers: an emerging tool for breast cancer therapy. J Drug Target 2015; 23:97-108. doi: 10.3109/1061186X.2014.958844 [Crossref] [ Google Scholar]
- Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release 2015; 200:138-57. doi: 10.1016/j.jconrel.2014.12.030 [Crossref] [ Google Scholar]
- Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release 2001; 74:47-61. doi: 10.1016/s0168-3659(01)00309-1 [Crossref] [ Google Scholar]
- Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm 2009; 71:409-19. doi: 10.1016/j.ejpb.2008.11.010 [Crossref] [ Google Scholar]
- McGuire WP, Markman M. Primary ovarian cancer chemotherapy: current standards of care. Br J Cancer 2003; 89:S3-8. doi: 10.1038/sj.bjc.6601494 [Crossref] [ Google Scholar]
- Barkat MA, Beg S, Pottoo FH, Ahmad FJ. Nanopaclitaxel therapy: an evidence based review on the battle for next-generation formulation challenges. Nanomed 2019; 14:1323-41. doi: 10.2217/nnm-2018-0313 [Crossref] [ Google Scholar]
- Barenholz Y. Doxil®--the first FDA-approved nano-drug: lessons learned. J Control Release 2012; 160:117-34. doi: 10.1016/j.jconrel.2012.03.020 [Crossref] [ Google Scholar]
- Bernabeu E, Cagel M, Lagomarsino E, Moretton M, Chiappetta DA. Paclitaxel: What has been done and the challenges remain ahead. Int J Pharm 2017; 526:474-95. doi: 10.1016/j.ijpharm.2017.05.016 [Crossref] [ Google Scholar]
- Sofias AM, Dunne M, Storm G, Allen C. The battle of “nano” paclitaxel. Adv Drug Deliv Rev 2017; 122:20-30. doi: 10.1016/j.addr.2017.02.003 [Crossref] [ Google Scholar]
- Yamamoto Y, Kawano I, Iwase H. Nab-paclitaxel for the treatment of breast cancer: efficacy, safety, and approval. OncoTargets Ther 2011; 4:123-36. doi: 10.2147/OTT.S13836 [Crossref] [ Google Scholar]
- Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release 2012; 161:505-22. doi: 10.1016/j.jconrel.2012.01.043 [Crossref] [ Google Scholar]
- Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(D,L-lactide-co-glycolide) and its derivatives. J Control Release 2008; 125:193-209. doi: 10.1016/j.jconrel.2007.09.013 [Crossref] [ Google Scholar]
- Sadat Tabatabaei Mirakabad F, Nejati-Koshki K, Akbarzadeh A, Yamchi MR, Milani M, Zarghami N. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac J Cancer Prev 2014; 15:517-35. doi: 10.7314/apjcp.2014.15.2.517 [Crossref] [ Google Scholar]
- Vasir JK, Labhasetwar V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv Drug Deliv Rev 2007; 59:718-28. doi: 10.1016/j.addr.2007.06.003 [Crossref] [ Google Scholar]
- Khan J, Alexander A, Ajazuddin Ajazuddin, Saraf S, Saraf S. Exploring the role of polymeric conjugates toward anti-cancer drug delivery: Current trends and future projections. Int J Pharm 2018; 548:500-14. doi: 10.1016/j.ijpharm.2018.06.060 [Crossref] [ Google Scholar]
- Leiva MC, Ortiz R, Contreras-Cáceres R, Perazzoli G, Mayevych I, López-Romero JM. Tripalmitin nanoparticle formulations significantly enhance paclitaxel antitumor activity against breast and lung cancer cells in vitro. Sci Rep 2017; 7:13506. doi: 10.1038/s41598-017-13816-z [Crossref] [ Google Scholar]
- Jiménez-López J, El-Hammadi MM, Ortiz R, Cayero-Otero MD, Cabeza L, Perazzoli G. A novel nanoformulation of PLGA with high non-ionic surfactant content improves in vitro and in vivo PTX activity against lung cancer. Pharmacol Res 2019; 141:451-65. doi: 10.1016/j.phrs.2019.01.013 [Crossref] [ Google Scholar]
- Cabeza L, Ortiz R, Prados J, Delgado ÁV, Martín-Villena MJ, Clares B. Improved antitumor activity and reduced toxicity of doxorubicin encapsulated in poly(ε-caprolactone) nanoparticles in lung and breast cancer treatment: An in vitro and in vivo study. Eur J Pharm Sci 2017; 102:24-34. doi: 10.1016/j.ejps.2017.02.026 [Crossref] [ Google Scholar]
- Pourbagher R, Akhavan-Niaki H, Jorsaraei SGA, Fattahi S, Sabour D, Zabihi E. Targeting LA7 breast cancer stem cells of rat through repressing the genes of stemness-related transcription factors using three different biological fluids. Gene 2020; 734:144381. doi: 10.1016/j.gene.2020.144381 [Crossref] [ Google Scholar]
- Karakas D, Cevatemre B, Aztopal N, Ari F, Yilmaz VT, Ulukaya E. Addition of niclosamide to palladium(II) saccharinate complex of terpyridine results in enhanced cytotoxic activity inducing apoptosis on cancer stem cells of breast cancer. Bioorg Med Chem 2015; 23(17):5580-6. doi: 10.1016/j.bmc.2015.07.026 [Crossref] [ Google Scholar]
- Liu C, Dong L, Sun Z, Wang L, Wang Q, Li H. Esculentoside A suppresses breast cancer stem cell growth through stemness attenuation and apoptosis induction by blocking IL-6/STAT3 signaling pathway. Phytother Res 2018; 32:2299-311. doi: 10.1002/ptr.6172 [Crossref] [ Google Scholar]
- Chen S-F, Chang Y-C, Nieh S, Liu C-L, Yang C-Y, Lin Y-S. Nonadhesive culture system as a model of rapid sphere formation with cancer stem cell properties. PloS One 2012; 7:e31864. doi: 10.1371/journal.pone.0031864 [Crossref] [ Google Scholar]
- Al-Dhfyan A, Alhoshani A, Korashy HM. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and β-Catenin and Akt activation. Mol Cancer 2017; 16:1-14. doi: 10.1186/s12943-016-0570-y [Crossref] [ Google Scholar]
- Fernández-Peralbo MA, Priego-Capote F, Luque de Castro MD, Casado-Adam A, Arjona-Sánchez A, Muñoz-Casares FC. LC-MS/MS quantitative analysis of paclitaxel and its major metabolites in serum, plasma and tissue from women with ovarian cancer after intraperitoneal chemotherapy. J Pharm Biomed Anal 2014; 91:131-7. doi: 10.1016/j.jpba.2013.12.028 [Crossref] [ Google Scholar]
- Li M, Czyszczon EA, Reineke JJ. Delineating intracellular pharmacokinetics of paclitaxel delivered by PLGA nanoparticles. Drug Deliv Transl Res 2013; 3:551-61. doi: 10.1007/s13346-013-0162-y [Crossref] [ Google Scholar]
- Prados J, Melguizo C, Rama AR, Ortiz R, Segura A, Boulaiz H. Gef gene therapy enhances the therapeutic efficacy of doxorubicin to combat growth of MCF-7 breast cancer cells. Cancer Chemother Pharmacol 2010; 66:69-78. doi: 10.1007/s00280-009-1135-1 [Crossref] [ Google Scholar]
- Jiang X, Sha X, Xin H, Xu X, Gu J, Xia W. Integrin-facilitated transcytosis for enhanced penetration of advanced gliomas by poly(trimethylene carbonate)-based nanoparticles encapsulating paclitaxel. Biomaterials 2013; 34:2969-79. doi: 10.1016/j.biomaterials.2012.12.049 [Crossref] [ Google Scholar]
- Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev 2011; 63:170-83. doi: 10.1016/j.addr.2010.10.008 [Crossref] [ Google Scholar]
- Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces 2010; 75:1-18. doi: 10.1016/j.colsurfb.2009.09.001 [Crossref] [ Google Scholar]
- Mir M, Ahmed N, Rehman AU. Recent applications of PLGA based nanostructures in drug delivery. Colloids Surf B Biointerfaces 2017; 159:217-31. doi: 10.1016/j.colsurfb.2017.07.038 [Crossref] [ Google Scholar]
- Danhier F, Lecouturier N, Vroman B, Jérôme C, Marchand-Brynaert J, Feron O. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release 2009; 133:11-17. doi: 10.1016/j.jconrel.2008.09.086 [Crossref] [ Google Scholar]
- Duan T, Xu Z, Sun F, Wang Y, Zhang J, Luo C. HPA aptamer functionalized paclitaxel-loaded PLGA nanoparticles for enhanced anticancer therapy through targeted effects and microenvironment modulation. Biomed Pharmacother 2019; 117:109121. doi: 10.1016/j.biopha.2019.109121 [Crossref] [ Google Scholar]
- Venugopal V, Krishnan S, Palanimuthu VR, Sankarankutty S, Kalaimani JK, Karupiah S. Anti-EGFR anchored paclitaxel loaded PLGA nanoparticles for the treatment of triple negative breast cancer. In-vitro and in-vivo anticancer activities. PLoS One 2018; 13:e0206109. doi: 10.1371/journal.pone.0206109 [Crossref] [ Google Scholar]
- Parveen S, Sahoo SK. Long circulating chitosan/PEG blended PLGA nanoparticle for tumor drug delivery. Eur J Pharmacol 2011; 670:372-83. doi: 10.1016/j.ejphar.2011.09.023 [Crossref] [ Google Scholar]
- Liu P, Situ J-Q, Li W-S, Shan C-L, You J, Yuan H. High tolerated paclitaxel nano-formulation delivered by poly (lactic-co-glycolic acid)-g-dextran micelles to efficient cancer therapy. Nanomedicine Nanotechnol Biol Med 2015; 11:855-66. doi: 10.1016/j.nano.2015.02.002 [Crossref] [ Google Scholar]
- Sahay G, Batrakova EV, Kabanov AV. Different internalization pathways of polymeric micelles and unimers and their effects on vesicular transport. Bioconjug Chem 2008; 19:2023-9. doi: 10.1021/bc8002315 [Crossref] [ Google Scholar]
- Vicari L, Musumeci T, Giannone I, Adamo L, Conticello C, De Maria R. Paclitaxel loading in PLGA nanospheres affected the in vitro drug cell accumulation and antiproliferative activity. BMC Cancer 2008; 8:212. doi: 10.1186/1471-2407-8-212 [Crossref] [ Google Scholar]
- Zubris KAV, Liu R, Colby A, Schulz MD, Colson YL, Grinstaff MW. In vitro activity of Paclitaxel-loaded polymeric expansile nanoparticles in breast cancer cells. Biomacromolecules 2013; 14:2074-82. doi: 10.1021/bm400434h [Crossref] [ Google Scholar]
- Liu R, Gilmore DM, Zubris KAV, Xu X, Catalano PJ, Padera RF. Prevention of nodal metastases in breast cancer following the lymphatic migration of paclitaxel-loaded expansile nanoparticles. Biomaterials 2013; 34:1810-9. doi: 10.1016/j.biomaterials.2012.11.038 [Crossref] [ Google Scholar]
- Gasch C, Ffrench B, O’Leary JJ, Gallagher MF. Catching moving targets: cancer stem cell hierarchies, therapy-resistance & considerations for clinical intervention. Mol Cancer 2017; 16:43. doi: 10.1186/s12943-017-0601-3 [Crossref] [ Google Scholar]
-
Das S, Mukherjee P, Chatterjee R, Jamal Z, Chatterji U. Enhancing Chemosensitivity of Breast Cancer Stem Cells by Downregulating SOX2 and ABCG2 Using Wedelolactone-encapsulated Nanoparticles. Mol Cancer Ther 2019;18:680-92. 10.1158/1535-7163.MCT-18-0409
- Kuh HJ, Jang SH, Wientjes MG, Au JL. Computational model of intracellular pharmacokinetics of paclitaxel. J Pharmacol Exp Ther 2000; 293:761-70. [ Google Scholar]
- Cui Y, Zhang M, Zeng F, Jin H, Xu Q, Huang Y. Dual-Targeting Magnetic PLGA Nanoparticles for Codelivery of Paclitaxel and Curcumin for Brain Tumor Therapy. ACS Appl Mater Interfaces 2016; 8:32159-69. doi: 10.1021/acsami.6b10175 [Crossref] [ Google Scholar]
- Markeb AA, El-Maali NA, Sayed DM, Osama A, Abdel-Malek MAY, Zaki AH. Synthesis, Structural Characterization, and Preclinical Efficacy of a Novel Paclitaxel-Loaded Alginate Nanoparticle for Breast Cancer Treatment. Int J Breast Cancer 2016; 2016:7549372. doi: 10.1155/2016/7549372 [Crossref] [ Google Scholar]
- Ruttala HB, Ramasamy T, Shin BS, Choi H-G, Yong CS, Kim JO. Layer-by-layer assembly of hierarchical nanoarchitectures to enhance the systemic performance of nanoparticle albumin-bound paclitaxel. Int J Pharm 2017; 519:11-21. doi: 10.1016/j.ijpharm.2017.01.011 [Crossref] [ Google Scholar]
- Xing L, Shi Q, Zheng K, Shen M, Ma J, Li F. Ultrasound-Mediated Microbubble Destruction (UMMD) Facilitates the Delivery of CA19-9 Targeted and Paclitaxel Loaded mPEG-PLGA-PLL Nanoparticles in Pancreatic Cancer. Theranostics 2016; 6:1573-87. doi: 10.7150/thno.15164 [Crossref] [ Google Scholar]
- Hariri G, Edwards AD, Merrill TB, Greenbaum JM, van der Ende AE, Harth E. Sequential targeted delivery of paclitaxel and camptothecin using a cross-linked “nanosponge” network for lung cancer chemotherapy. Mol Pharm 2014; 11:265-75. doi: 10.1021/mp400432b [Crossref] [ Google Scholar]
- Lai C-H, Chang T-C, Chuang Y-J, Tzou D-L, Lin C-C. Stepwise Orthogonal Click Chemistry toward Fabrication of Paclitaxel/Galactose Functionalized Fluorescent Nanoparticles for HepG2 Cell Targeting and Delivery. Bioconjug Chem 2013; 24:1698-709. doi: 10.1021/bc400219t [Crossref] [ Google Scholar]
- Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller-Klieser W, Kunz-Schughart LA. Multicellular tumor spheroids: an underestimated tool is catching up again. J Biotechnol 2010; 148:3-15. doi: 10.1016/j.jbiotec.2010.01.012 [Crossref] [ Google Scholar]
- McMahon KM, Volpato M, Chi HY, Musiwaro P, Poterlowicz K, Peng Y. Characterization of changes in the proteome in different regions of 3D multicell tumor spheroids. J Proteome Res 2012; 11:2863-75. doi: 10.1021/pr2012472 [Crossref] [ Google Scholar]
- Lu Z, Tsai M, Lu D, Wang J, Wientjes MG, Au JL-S. Tumor-penetrating microparticles for intraperitoneal therapy of ovarian cancer. J Pharmacol Exp Ther 2008; 327:673-82. doi: 10.1124/jpet.108.140095 [Crossref] [ Google Scholar]
- Mehta G, Hsiao AY, Ingram M, Luker GD, Takayama S. Opportunities and Challenges for use of Tumor Spheroids as Models to Test Drug Delivery and Efficacy. J Control Release 2012; 164:192-204. doi: 10.1016/j.jconrel.2012.04.045 [Crossref] [ Google Scholar]
- Lee WL, Guo WM, Ho VHB, Saha A, Chong HC, Tan NS. Inhibition of 3-D tumor spheroids by timed-released hydrophilic and hydrophobic drugs from multilayered polymeric microparticles. Small 2014; 10:3986-96. doi: 10.1002/smll.201400536 [Crossref] [ Google Scholar]
- Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 2011; 63:136-51. doi: 10.1016/j.addr.2010.04.009 [Crossref] [ Google Scholar]
- Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev 2015; 91:3-6. doi: 10.1016/j.addr.2015.01.002 [Crossref] [ Google Scholar]
- Nagamitsu A, Greish K, Maeda H. Elevating Blood Pressure as a Strategy to Increase Tumor-targeted Delivery of Macromolecular Drug SMANCS: Cases of Advanced Solid Tumors. Jpn J Clin Oncol 2009; 39:756-66. doi: 10.1093/jjco/hyp074 [Crossref] [ Google Scholar]
-
Islam W, Fang J, Imamura T, Etrych T, Subr V, Ulbrich K, et al. Augmentation of the Enhanced Permeability and Retention Effect with Nitric Oxide-Generating Agents Improves the Therapeutic Effects of Nanomedicines. Mol Cancer Ther 2018;17:2643-53. 10.1158/1535-7163.MCT-18-0696
- Matsunaga T, Matsunaga N, Kusunose N, Ikeda E, Okazaki H, Kakimoto K. Angiotensin-II regulates dosing time-dependent intratumoral accumulation of macromolecular drug formulations via 24-h blood pressure rhythm in tumor-bearing mice. Biochem Biophys Res Commun 2018; 498:86-91. doi: 10.1016/j.bbrc.2017.11.162 [Crossref] [ Google Scholar]
- Tahara Y, Yoshikawa T, Sato H, Mori Y, Zahangir MH, Kishimura A. Encapsulation of a nitric oxide donor into a liposome to boost the enhanced permeation and retention (EPR) effect. MedChemComm 2017; 8:415-21. doi: 10.1039/c6md00614k [Crossref] [ Google Scholar]
- Abu Samaan TM, Samec M, Liskova A, Kubatka P, Büsselberg D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019; 9:789. doi: 10.3390/biom9120789 [Crossref] [ Google Scholar]
- Ibrahim NK, Samuels B, Page R, Doval D, Patel KM, Rao SC. Multicenter Phase II Trial of ABI-007, an Albumin-Bound Paclitaxel, in Women With Metastatic Breast Cancer. J Clin Oncol 2005; 23:6019-26. doi: 10.1200/JCO.2005.11.013 [Crossref] [ Google Scholar]
- Spencer CM, Faulds D. Spencer CM, Faulds DPaclitaxelA review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the treatment of cancer. Drugs 1994; 48:794-847. doi: 10.2165/00003495-199448050-00009 [Crossref] [ Google Scholar]
- Kundranda MN, Niu J. Albumin-bound paclitaxel in solid tumors: clinical development and future directions. Drug Des Devel Ther 2015; 9:3767-77. doi: 10.2147/DDDT.S88023 [Crossref] [ Google Scholar]
- Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol 2005; 23:7794-803. doi: 10.1200/JCO.2005.04.937 [Crossref] [ Google Scholar]