Bioimpacts. 12(6):479-486.
doi: 10.34172/bi.2022.23477
Original Research
Enhanced control of bioactivity of tissue plasminogen activator (tPA) through domain-directed enzymatic oxidation of terminal galactose
Wael A. Mahdi 1  , Mohammad S. Absar 2, Suna Choi 2, Victor C. Yang 3, 4, Young M. Kwon 5, *
, Mohammad S. Absar 2, Suna Choi 2, Victor C. Yang 3, 4, Young M. Kwon 5, *
Author information:
1Department of Pharmaceutics, College of Pharmacy, King Saud University, Riyadh, Kingdom of Saudi Arabia
2Texas Tech University Health Sciences Center (TTUHSC), School of Pharmacy, Amarillo, TX 79106, USA
3Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnosis, School of Pharmacy, Tianjin Medical University Tianjin 300070, China
4University of Michigan, College of Pharmacy, MI 48109-1065, USA
5Department of Pharmaceutical Sciences, College of Pharmacy, Nova Southeastern University, Fort Lauderdale, FL, USA
Abstract
Introduction:
In targeted enzyme prodrug constructs, it is critical to control the bioactivity of the drug in its prodrug form. The preparation of such constructs often involves conjugation reactions directed to functional groups on amino acid side chains of the protein, which result in random conjugation and incomplete control of bioactivity of a prodrug, which may result in significant nontarget effect. Thus, more specific method of modification is desired. If the drug is a glycoprotein, enzymatic oxidation may offer an alternative approach for therapeutic glycoproteins.
Methods:
Tissue plasminogen activator (tPA), a model glycoprotein enzyme, was treated with galactose oxidase (GO) and horseradish peroxidase, followed by thiolation reaction and conjugation with low molecular weight heparin (LMWH). The LMWH-tPA conjugate was isolated by ion-exchange chromatography followed by centrifugal filtration. The conjugate was characterized for its fibrinolytic activity and for its plasminogen activation through an indirect amidolytic assay with a plasmin-specific substrate S-2251 when LMWH-tPA conjugate is complexed with protamine-albumin conjugate, followed by triggered activation in the presence of heparin.
Results:
LMWH-tPA conjugate prepared via enzymatic oxidation retained ~95% of its fibrinolytic activity with respect to native tPA. Upon complexation with protamine-albumin conjugate, the activity of LMWH-tPA was effectively inhibited (~90%) whereas the LMWH-tPA prepared by random thiolation exhibited ~55% inhibition. Addition of heparin fully generated the activities of both conjugates.
Conclusion:
The tPA was successfully modified via enzymatic oxidation by GO, resulting in enhanced control of its activity in the prodrug construct. This approach can be applied to other therapeutic glycoproteins.
Keywords: Pretargeting, Thrombolytic drug, Triggered release, Glycoprotein oxidation, tPA
Copyright and License Information
© 2022 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
There has been growing interest in targeted therapeutic protein delivery in the past decades because of their high potencies and target specificities.
1-3
However, it is highly probable that such drugs with high potency are prone to exhibit undesirable non-target effects. In order to alleviate this potential issue, pretargeting approach may be considered.
4-7
Pretargeting strategy aims at target-specific drug release with lessened non-target effects. In order to accomplish this, a macromolecular drug is given a ‘prodrug’ character which must remain significantly inactive when administered. If the construct is decorated with a targeting moiety, then the construct is allowed to accumulate at the intended target as an inactive form. Once significantly accumulated at the target, its active component will be released via a triggering mechanism.
8-11
Tissue-type plasminogen activator (tPA), a widely used thrombolytic drug, is a good example in which such strategy may be useful since the drug action is rapid while the side effect can be fatal.
12
The success of this approach hinges on whether or not the construct can stay inactive until triggered.
13
Macromolecular constructs for triggered delivery of tPA has previously been studied in which the drug was chemically modified.
13-15
By controlling the nature of the physical interaction between the drug and the covering molecules, the activity of tPA was triggered in vitro upon addition of unfractionated heparin (UFH) at its reported therapeutic plasma concentration range (0.2– 0.7 U/mL). In a rodent model of thrombosis, there was a statistically significant difference in clot lysis before and after triggering without increasing the activated partial thromboplastin time. This approach may be considered for coronary artery thrombosis, for which heparin is often used as a conjunctive agent.
16
Such scheme is illustrated in Fig. 1.
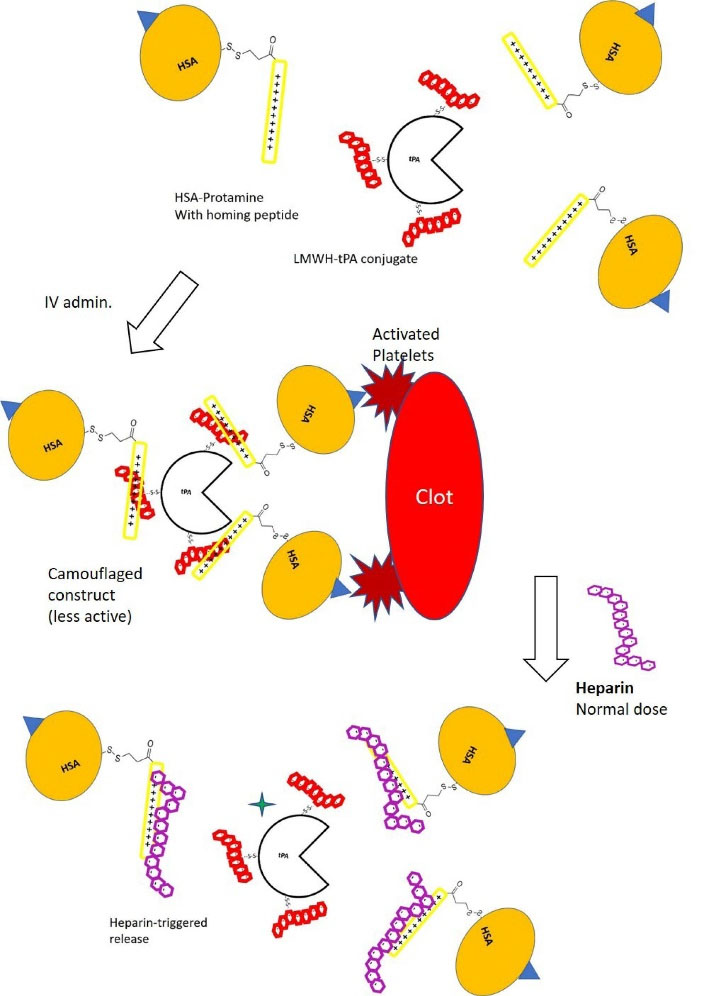
Fig. 1.
Schematic description of a pretargeting strategy for a thrombolytic drug (tPA) where low molecular weight heparin (LMWH) is conjugated to tPA. The carrier molecule consists of 1:1 conjugate of human serum albumin (HSA) and protamine peptide where albumin can also be conjugated with a targeting moiety such as the C-terminal peptide from fibrinogen gamma-chain as a homing device to target GPIIb/IIIa of activated platelets on a platelet-rich arterial thrombus. Protamine and LMWH from each conjugate form a reversible bond. The tPA molecule in the construct surrounded by 2-3 human serum albumins is sterically hindered so that systemic activation of plasminogen is attenuated during its transit to the target. The ‘inactive’ complex is then injected intravenously. Upon target accumulation, a triggering molecule (unfractionated heparin, UFH) is administered since UFH has stronger affinity to protamin than LMWH. The system can be tailored so that the therapeutic level of heparin (2-7 U/mL) is able to unleash the thrombolytic activity of the tPA in the vicinity of the plate-rich thrombus whereas a systemic lytic state and the subsequent increase of bleeding risk is avoided. However, randomly conjugated LMWH may create an imperfect ‘prodrug’ and the construct may still generate systemic effect.
.
Schematic description of a pretargeting strategy for a thrombolytic drug (tPA) where low molecular weight heparin (LMWH) is conjugated to tPA. The carrier molecule consists of 1:1 conjugate of human serum albumin (HSA) and protamine peptide where albumin can also be conjugated with a targeting moiety such as the C-terminal peptide from fibrinogen gamma-chain as a homing device to target GPIIb/IIIa of activated platelets on a platelet-rich arterial thrombus. Protamine and LMWH from each conjugate form a reversible bond. The tPA molecule in the construct surrounded by 2-3 human serum albumins is sterically hindered so that systemic activation of plasminogen is attenuated during its transit to the target. The ‘inactive’ complex is then injected intravenously. Upon target accumulation, a triggering molecule (unfractionated heparin, UFH) is administered since UFH has stronger affinity to protamin than LMWH. The system can be tailored so that the therapeutic level of heparin (2-7 U/mL) is able to unleash the thrombolytic activity of the tPA in the vicinity of the plate-rich thrombus whereas a systemic lytic state and the subsequent increase of bleeding risk is avoided. However, randomly conjugated LMWH may create an imperfect ‘prodrug’ and the construct may still generate systemic effect.
Construction of pretargeting system often involves covalent modification of the therapeutic cargo (e.g., proteins). However, popular chemical modification methods for proteins include the amine group-directed (via the ε-amine groups on lysine residues or the N-terminus) or carboxylic acid group-directed reactions (aspartic acid, glutamic acid residues, or the C-terminus).
17
These types of reactions are of random nature, which result in rather heterogeneous reaction outcomes. If these methods were used in a prodrug type construct for an enzyme drug, incomplete control of enzyme activities in the prodrug form could occur, which was evidenced by a significant residual activity from the constructs themselves prior to introducing triggering mechanisms.
12,14
This residual activity is undesirable and must be minimized through more site-specific chemical modification of the therapeutic agent.
It is noteworthy that modifying the side chains of amino acid residues (which constitutes the polypeptide chain) may be detrimental to the stability of the therapeutic protein, which may lead to loss of activity depending on the type of proteins modified. For instance, PEGylation of bacterial cocaine esterase enzyme via its accessible free sulfhydryl groups on its unpaired cysteine residues retained its thermal stability whereas PEGylation of the enzyme via its primary amine groups on the lysine residues resulted in the rapid loss of its enzymatic activity.
18
Therefore, for a glycoprotein, the function of a therapeutic protein may be better preserved upon chemical modification if conjugation reaction occurs at its specific saccharide moieties.
19
For instance, monoclonal antibodies are often modified via its sugar residues on the Fc region, thereby minimizing the likelihood of covalent modification in the vicinity of the epitope-binding Fab regions.
20
Mild oxidizing agents such as sodium periodate (1-100 mM) can open up terminal sugar moiety and introduce aldehyde groups, which subsequently participate in the conjugation step. Periodate oxidation requires different oxidizing strengths depending on the type of terminal saccharides (lower oxidant concentrations for sialic acid; higher oxidant concentrations for non-sialic acid sugars).
21
However, this type of reaction can be non-specific and may oxidatively damage certain amino acid residues on the polypeptide backbone,
22
which is highly undesirable.
On the contrary, oxidation of sugars using certain oxidase enzymes can create reactive functional groups without nonspecifically destroying the cyclic structure of sugar moieties or amino acid side chains. For example, galactose oxidase (GO) from Dactylium dendroides can preferentially act on the hydroxyl groups (C6 position) of terminal galactose (Gal) residues
23,24
to introduce an aldehyde group per terminal galactose.
Tissue plasminogen activator, tPA, is a multidomain glycoprotein consisting a finger domain, an epidermal growth factor (EGF)-like domain, Kringle domains (Kringle 1 and 2), and a domain analogous to a serine protease.
25
The tPA also has an unpaired Cys83 residue.
26
The N-linked glycosylation sites of tPA are mainly Asn117, Asn184, and Asn448. Asn117 is located in Kringle 1 domain, mannose-rich, and is known to be always glycosylated. Asn184 is found in Kringle 2 domain. However, glycosylation at this residue is only found at ~50% of the time. Asn448 is in the protease domain of tPA. This residue is always glycosylated and more importantly, terminal galactose residues are found.
27
Although tPA possesses complex glycosylation pattern,
28
terminal galactose residues are always found in the protease domain, which may be a valid modification site for controlling its catalytic activity without permanently damaging the active site. Thus, our hypothesis is that by directing the chemical modification to a terminal galactose moiety near the active site where a docking molecule can be introduced, the control of tPA is significantly enhanced to nearly an on/off manner.
Materials and Methods
Materials
Tissue Plasminogen Activator (manufactured by Genentech, Inc. (South San Francisco, CA, USA)) was purchased from Henry Schein (Melville, NY, USA). Low molecular weight heparin (LMWH, Fragmin®) was obtained from Eisai, Inc (Woodcliff lake, NJ, USA). The following chemicals were obtained from Sigma-Aldrich, Inc (St. Louis, MO, USA): thrombin, fibrinogen from human plasma, human plasminogen, protamine sulfate, horseradish peroxidase, galactose oxidase, sodium metaperiodate, calcium chloride, dithiothreitol (DTT), aldrithiol (2,2'-dithiopyridine), and N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide (EDC). The heterobifuctional crosslinkers N-succinimidyl 3-(2-pyridyldithio) propionate (SPDP) was purchased from CovaChem (Loves Park, IL) and 3-[2-Pyridyldithio] propionyl hydrazide (PDPH) was purchased from Thermo Fisher Scientific (Waltham, MA). The plasminogen specific chromogenic substrate S-2251 was purchased from Chromogenix (Lexington, MA). 2-(N-morpholino) ethanesulfonic acid (MES) was purchased from Acros Organics (Carlsbad, CA). All other chemicals were of reagent grade and used without further purification.
Methods
Oxidation of tPA by sodium periodate
Desalted tPA solution (1 mg/mL in phosphate buffer (pH 7)) was incubated in 1-, 5-, 10-, and 20-mM sodium meta-periodate for 30 minutes in an ice bath in darkness. The reaction mixtures were visually inspected for turbidity, as a sign of aggregation.
Thiolation of tPA via enzymatic oxidation of terminal galactose residue
Terminal galactose on tPA was oxidized using galactose oxidase/horseradish peroxidase followed by the addition of 3-[2-pyridyldithio] propionyl hydrazide (PDPH). An aliquot of 1 mg/mL of tPA was incubated with 7 U/mL of galactose oxidase from and 30 U/mL of horseradish peroxidase.
23
Shortly after mixing of enzymes, 1 mg of 3-[2-pyridyldithio] propionyl hydrazide (PDPH) dissolved in DMF was added to the reaction mixture. The pH was adjusted to ~6-6.5. Mild shaking at room temperature was maintained for 2 hours. The reaction mixture was centrifuged for 2 minutes at 10 000 rpm and the supernatant was injected to the FPLC equipped with CM-Sepharose-Sephadex G-25 combination cartridges to isolate the thiolated tPA.
The degree of thiolation was estimated by measuring the absorbance of the thiolated tPA at 280 nm followed by measuring the absorbance at 343 nm due to the release of pyridyl-2-thione upon the addition of 50 mM DTT.
29
Preparation of LMWH-PDP
LMWH (10 mg/mL in 50 mM MES, pH 6) reacted with 1 mg/mL of PDPH by linking the hydrazide groups of PDPH to the carboxylic groups on LMWH in the presence of EDC (1 mg/mL) at room temperature. DTT was added (to the final concentration of 50 mM) to the reaction mixture to reduce pyridyl disulfide bonds to yield thiolated LMWH (LMWH-SH). Since the average molecular weight of the LMWH used (Fragmin) was ~ 5kD, LMWH-SH was isolated by FPLC equipped with Sephadex G-25 desalting column (MW cutoff~5000) with freshly deoxygenated phosphate buffer (50 mM, pH=7.4).
LMWH-tPA conjugate preparation
The thiolated tPAs from both cases were further conjugated with thiolated LMWH (low-molecular weight heparin). The thiolated tPA was incubated with LMWH-SH (5-fold molar excess) for 2 hours at room temperature. The resulting LMWH-tPA conjugate was enriched with stepwise dilutions via centrifugal filtration device with 30,000 molecular weight cut-off (Millipore, Inc., Burlington, MA) to remove excess LMWH-SH. LMWH-tPA conjugate was isolated by ion-exchange chromatography using a Q-FF (Fast-Flow) column (GE Healthcare, Inc). The purified LMWH-tPA conjugate was stored at 4°C for further evaluations.
Preparation of HSA-protamine
The HSA-S-S-protamine conjugate was prepared according to the previously reported method.
30
In brief, the N-terminus of the protamine (proline residue) was modified by SPDP, followed by ion exchange chromatography with NaCl gradient elution. The resulting protamine-PDP was incubated with HSA in 50 mM phosphate buffered saline (HSA concentration = 1 mg/mL, HSA: protamine-PDP ratio = 5:1).
In vitro fibrin clot lysis assay (fibrin–agarose plate assay)
The fibrinolytic activities of tPA-conjugates were evaluated by a fibrin clot lysis assay using fibrin-containing agarose plates.
12,13
The fibrin–agarose gel plates were prepared as follows: in one container, 300 mg of agarose (low melting temperature) was dissolved in 10 mL Tris buffer (pH 7.3) at 45-55°C containing 25 μL of 200 units/mL of thrombin solution. In a separate container, 50 mg of fibrinogen was dissolved in 5 mL of Tris buffer and the solution was kept at 37°C. The fibrinogen solution was then mixed with melted agarose solution, which was stirred for 90 seconds. The mixture was poured into a square transparent plastic plate and was spread evenly. The mixture solidified within 2 hours at 37°C and a homogeneous fibrin gel was obtained. Sample reservoirs with 3-mm in diameter were created on the solidified agarose plate and an aliquot of 5 μL of plasminogen (1 mg/mL) was added to each well. Native tPA and LMWH-tPA conjugates (amounts in tPA equivalents) were added into wells and incubated at 37°C overnight for fibrinolysis. The fibrinolytic activity of each sample was determined by comparing the area of the lysed fibrin zone around 3-mm wells. The data was presented as % diameter with respect to that of native tPA.
Plasminogen activation assay
Plasminogen activation by tPA was determined spectrophotometrically by monitoring the rate of plasmin production using a plasmin-specific chromogenic substrate S-2251 (H-D-Val-Leu-Lys-pNA. 2HCl). Briefly, this assay consists of 0.24 μM plasminogen, 1.3 μM of S-2251, and 0.5 µg/mL tPA-conjugate (to native tPA), dissolved in 50 mM PBS containing 150 mM NaCl with 0.01% Tween 80 at pH 7.3. The initial rates of hydrolysis of S-2251 by plasmin, as a result of plasminogen activation by tPA-conjugate, camouflaged tPA (cam-tPA) by the addition of HSA-protamine (50-fold), and the camouflaged tPA with heparin (unfractionated), were evaluated by monitoring the absorbance at 405 nm using a microplate reader (Synergy H1 Hybrid reader, Biotek, Broadview, IL). The initial rates of S-2251 hydrolysis were determined from the slopes of absorbance at 405 against the square of time (∆A/min2)
31
via linear regression.
Statistical analysis
To compare the fibrinolytic activities and plasminogen activation activities, student t-test was carried out using GraphPad Prism version 7.0 (GraphPad Inc., La Jolla, USA). Differences were considered significant at P values less than 0.05.
Results
Synthesis and purification of LMWH- tPA conjugate
Oxidation of tPA by sodium periodate
The chemical oxidation method using sodium periodate caused irreversible aggregation of tPA at all periodate concentrations tried. Turbidity of the reaction mixture developed shortly after the addition of sodium periodate. Varying pH of the reaction mixture after the reaction did not change turbidity.
Thiolation of tPA
The LMWH-tPA conjugate preparation via enzymatic oxidation is depicted in Fig. 2. The reaction pH has an impact on thiolation of tPA and we have chosen pH 6.5, which was maintained during the reaction time. Pyridine-2-thione (P2T) released upon 50 mM DTT treatment assay was measured in order to calculate the degree of sulfhydryl modification of tPA per molecule. Our goal was to create ~1 thiolation per tPA via the enzymatic oxidation method. The assay estimated that the degree of modification was ~1.5 thiolation per tPA. On the other hand, the random thiolation method resulted in 2~3 thiolations per tPA at 20-fold molar excess of SPDP.
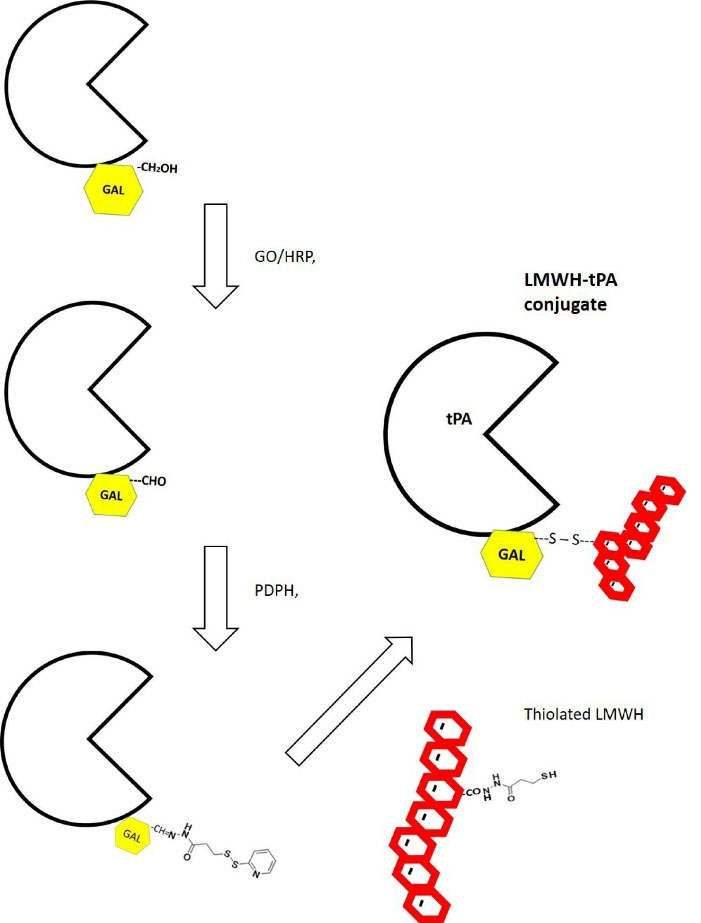
Fig. 2.
Preparation of LMWH-tPA conjugate via oxidation of C6 hydroxyl group of a terminal galactose by galactose oxidase (GO). The oxidation reaction is carried out in the presence of horseradish peroxidase (HRP) at room temperature. 3-[2-pyridyldithio] propionyl hydrazide (PDPH) is then added to the mixture and its reaction to the aldehyde group leads to the formation of a hydrazone linkage.
.
Preparation of LMWH-tPA conjugate via oxidation of C6 hydroxyl group of a terminal galactose by galactose oxidase (GO). The oxidation reaction is carried out in the presence of horseradish peroxidase (HRP) at room temperature. 3-[2-pyridyldithio] propionyl hydrazide (PDPH) is then added to the mixture and its reaction to the aldehyde group leads to the formation of a hydrazone linkage.
LMWH-SH was prepared separately by pyridyldithiol-activation of LMWH via PDPH and EDC, followed by treatment with DTT for 15 minutes in order to create free sulfhydryl groups. The level of sulfhydryl modification of LMWH-SH was also controlled to 1–2 sulfhydryl per average chain of LMWH, as determined by measuring P2T release upon 50 mM DTT treatment. After isolating LMWH-SH using a desalting column, LMWH-SH and tPA-PDP were incubated to form the final conjugate. The reaction kinetics of conjugating LMWH-SH to the tPA-PDP was monitored by the release of pyridine-2-thione at 343 nm and the conjugation was considered to be completed once pyridine-2-thione concentration reached its plateau. After removing the unreacted LMWH-SH by stepwise dilution and centrifugal enrichment (MW cutoff of 30 000 Da), LMWH-tPA conjugate was isolated from unreacted tPA, with sodium chloride gradient as shown in Fig. 3A.
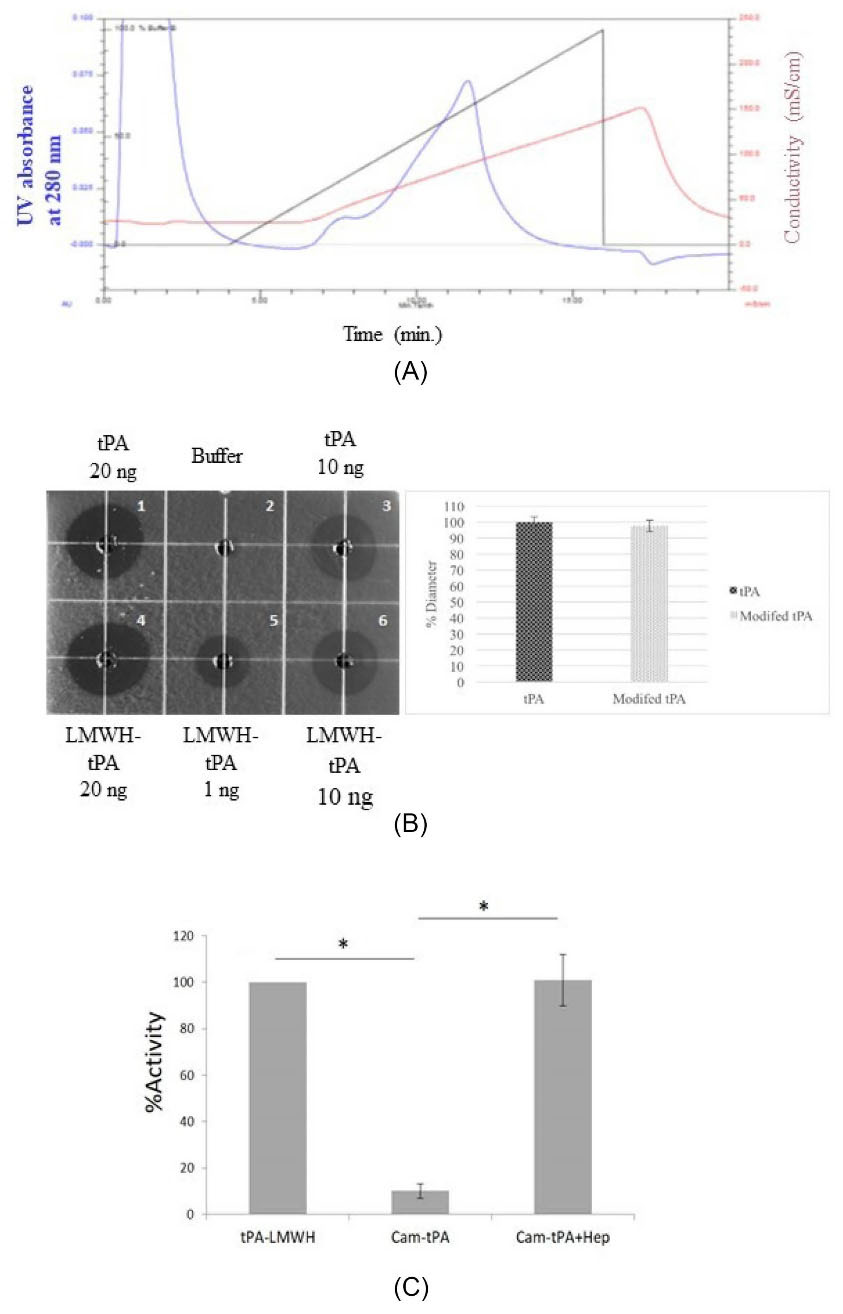
Fig. 3.
(A) Ion-exchange chromatography trace for purification of tPA-LMWH conjugate. The 1 mL reaction mixture was eluted on a Sepharose Q FF, with elution buffer A: 20 mM PBS, pH 7.4; B: 20 mM PBS+2 M NaCl, pH 7.4. The blue line: UV absorbance at 280 nm; Red line: Conductivity; Black line: Gradient showing %B concentration. Flow rate = 1 mL/min. (B) In-vitro Fibrin Clot Lysis Assay on a fibrin- agarose plate using LMWH-tPA conjugate (enzymatic oxidation) and native tPA (positive control). Left: Fibrinolysis observed after overnight incubation at 37°C; dot 1. tPA (20 ng), dot 2.Tris Buffer, dot 3. tPA (10 ng), dot 4. LMWH-tPA conjugates (20 ng), dot 5. LMWH-tPA conjugate (1 ng), dot 6. LMWH-tPA conjugate (10 ng); Right: Relative fibrinolytic activity of LMWH-tPA. Data represent mean ±SEM. n=3. P < 0.05 (n = 3). (C) Inhibition and regeneration of tPA activity in the camouflaged tPA (Cam-tPA) and after heparin addition (Cam-tPA+Hep) after LMWH conjugation of tPA via enzymatic oxidation of terminal galactose. Experimental condition: tPA 0.5 μg/mL, plasminogen 0.24 μM, S-2251 1.2 mM, HSA-protamine 0.2 μM and heparin (0.4 U/mL). Data represent mean±SD. P < 0.01 (n= 4).
.
(A) Ion-exchange chromatography trace for purification of tPA-LMWH conjugate. The 1 mL reaction mixture was eluted on a Sepharose Q FF, with elution buffer A: 20 mM PBS, pH 7.4; B: 20 mM PBS+2 M NaCl, pH 7.4. The blue line: UV absorbance at 280 nm; Red line: Conductivity; Black line: Gradient showing %B concentration. Flow rate = 1 mL/min. (B) In-vitro Fibrin Clot Lysis Assay on a fibrin- agarose plate using LMWH-tPA conjugate (enzymatic oxidation) and native tPA (positive control). Left: Fibrinolysis observed after overnight incubation at 37°C; dot 1. tPA (20 ng), dot 2.Tris Buffer, dot 3. tPA (10 ng), dot 4. LMWH-tPA conjugates (20 ng), dot 5. LMWH-tPA conjugate (1 ng), dot 6. LMWH-tPA conjugate (10 ng); Right: Relative fibrinolytic activity of LMWH-tPA. Data represent mean ±SEM. n=3. P < 0.05 (n = 3). (C) Inhibition and regeneration of tPA activity in the camouflaged tPA (Cam-tPA) and after heparin addition (Cam-tPA+Hep) after LMWH conjugation of tPA via enzymatic oxidation of terminal galactose. Experimental condition: tPA 0.5 μg/mL, plasminogen 0.24 μM, S-2251 1.2 mM, HSA-protamine 0.2 μM and heparin (0.4 U/mL). Data represent mean±SD. P < 0.01 (n= 4).
Determination of fibrinolytic activity of LMWH- tPA conjugates
The fibrinolytic activity of LMWH-tPA conjugate in vitro was determined by fibrin clot lysis assay. Active tPA has the ability to bind and cleave plasminogen into plasmin, which degrades fibrin resulting in a clear lysed zone (halo patterns) on the fibrin-agarose plate. All treatments were placed in the sample-well on the fibrin agarose-plate and incubated overnight at 37°C. The activity of tPA-LMWH conjugates was measured according to the diameter of the clear zone in the fibrin plate and then compared to native tPA. As shown in Fig. 3B, different amounts of LMWH-tPA conjugate (20 µg, 10 µg, and 1 µg) were applied in this experiment. LMWH-tPA conjugate exhibited >95% of diameter of halo patterns compared to that of native tPA, indicating that conjugating LMWH to tPA did not damage its activity. In addition, there was no statistically significant differences between tPA-frag conjugate and native tPA in terms of the diameter of the halo patterns, suggesting that the enzyme activity of the LMWH-tPA conjugate was similar to that of the native tPA (Fig. 3B).
Effect of the thiolation methods on controlling triggered prodrug delivery system
Heparin-triggered release construct for tPA was created with the aim of avoiding systemic bleeding complication associated with the treatment of tPA. LMWH was previously conjugated to tPA via the random thiolation method whereas in the current study a specific enzymatic oxidation reaction was carried out to conjugate LMWH to tPA. Specific site modification of tPA, which utilized direct enzymatic oxidation, was developed as a potential approach to better modulate the enzymatic activity of tPA.
The activity for plasminogen activation by both LMWH-tPA conjugates were evaluated using the chromogenic substrate S-2251. Camouflaged tPA was prepared by allowing binding interactions between LMWH-tPA conjugate and the HSA-protamine conjugate. In the LMWH-tPA conjugate prepared by enzymatic oxidation, protease domain-specific conjugation of LMWH on tPA demonstrated a substantially improved inhibition (~90%, < 10% residual activity) upon complexation with the carrier (Fig. 3C), whereas ~ 55% inhibition of tPA activity was observed in random thiolation method (~45% residual activity from the prodrug construct) from the previously published results.
16
The enzyme activities were fully recovered in both methods upon removal of the camouflage by introducing 0.4U/mL of heparin, which is equivalent to clinical steady state concentration of heparin in plasma upon heparin administration. This suggests that the modification via enzymatic oxidation method provided enhanced control of enzymatic activity of tPA compared to the random thiolation method.
Discussion
We have previously developed a tPA prodrug construct for targeted thrombolysis
14,16
: negatively charged docking molecule ((Glu)7Cys peptide or LMWH) on tPA molecule in a random conjugation method. Complexation of this conjugate with protamine-albumin conjugates (viz., camouflage) offered some control of tPA activity and a targetability of the construct through the attachment of platelet-targeting peptide on the albumin moiety. However, this approach suffered significant residual plasminogen activation activity in its complexation state (45%-50%) which is not ideal for a ‘prodrug’ construct since a true prodrug-type construct should be inactive before activation. Due to this limitation, therefore, an alternative approach for chemical modification of tPA was sought. Since tPA is a glycoprotein, with one glycosylation site in its protease domain (Asn447), oxidation of terminal saccharide moieties may offer more specific control of its activity.
Although oxidation of saccharide moiety on glycoproteins by sodium (meta)periodate is well-established for many years,
32
this method appears to be detrimental to the stability of tPA based on the observed aggregation during the reaction. Turbidity quickly developed as the oxidation reaction began, even at 1 mM of sodium periodate at which terminal sialic acid is known to be oxidized and tPA does have terminal sialic acid residues on the oligosaccharide on Asn 447 (protease domain).
28
To rule out the possibility of precipitation, pH of the reaction mixture was varied from 3 to 9 after the reaction. However, the turbidity did not disappear. Thus, this protein aggregate, for all practical purposes, was deemed irreversible and we did not pursue chemical oxidation of tPA any further.
The application of galactose oxidase from D. dendroides in bioconjugate synthesis technique has been highlighted in the recent decade
33
although the enzyme was discovered in 1959.
34
The glycosylation of tPA, as mentioned in the Introduction section, is known to occur at specific sites. Most notably, the glycosylation at Asn447 is in the protease domain and, despite its heterogenous glycosylation pattern, terminal Gal is present at higher probability compared to other sites on the molecule.
27
Prior to thiolation reaction, tPA was treated with aldrithiol (a disulfide dimer of PDP groups) to block any free thiol group because tPA is known to have an unpaired cysteine residue on its Kringle 1 domain (Cys83). Although the status of the unpaired Cys83 of tPA is known to be heterogenous,
26
the presence of unpaired -SH group will react with PDP groups of the thiolating agent, which is undesirable.
Random thiolation was carried out in 20-fold molar access of SPDP over tPA. The PDP-attached tPA has increased UV absorbance at 280 nm due to the molar extinction coefficient is known to be 5100 M -1cm -1.
35
In conjunction with the pyridyl-2-thione release assay by measuring the absorbance at 343 nm (molar extinction coefficient = 8080 M -1cm -1)
35
the number of PDP modification per tPA was calculated. Although tPA has lysine groups in abundance, even 20-fold SPDP (with an amine-reactive group), the net PDP/tPA ratio was less than 3. A higher feed ratio (40-fold SPDP) caused instability of tPA (observation). Lysine groups on the protein may contribute to the stabilization of a protein through salt bridge formation with glutamate,
36
and this may have contributed to the observed instability under excessive lysine modification. However, this is case-by-case as we have extensively PEGylated enzymes in the market, such as PEGylated asparaginases.
37
Specific oxidation of tPA using GO in the presence of horseradish peroxidase in the reported amount, followed by PDPH reaction, yielded ~1.5 PDP per tPA. After conjugation with thiolated LMWH and purification, the resulting conjugate retained its fibrinolytic activity, as shown in Fig. 3B. This suggests that conjugation of LMWH through terminal galactose moiety in the protease domain of tPA did not sterically interfere with its primary macromolecular substrate, plasminogen. Furthermore, complexation with protamine-albumin conjugate nearly abolished its activity in its prodrug state, despite the less degree of modification compared to the random thiolation method. This complexation was completely reversible in the presence of 0.4 U/mL of UFH. This reversal was rapid, which is attributed to the fact that the binding affinity UFH toward protamine is substantially higher compared to that of LMWH.
38
Research Highlights
What is the current knowledge?
√ Current thrombolytic enzymes (e.g., tPA) are associated with their potential for bleeding complications due to nonspecific plasmin generation upon systemic administration.
√ Targetable prodrug constructs for thrombolytic enzymes have been investigated. However, significant residual activities from the prodrug forms were observed.
What is new here?
√ Enzymatic oxidation of terminal galactose moieties on the protease domain of tPA allowed a more site-specific method of tPA modification that dramatically reduced the residual activity of its prodrug construct.
Conclusion
In summary, the LMWH-tPA conjugate prepared by a random thiolation method (resulting in 2-3 thiolations per tPA molecule) exhibited significant residual activity (55% inhibition, ~45% residual activity). On the other hand, the LMWH-tPA prepared by the enzymatic oxidation method using galactose oxidase has shown more specific modification (~1.5 thiolation per tPA), resulting in ~90% inhibition of its activity in its prodrug form. Those inhibitions were completely reversed by the presence of therapeutic concentration of heparin. This approach may be further utilized to create an ‘on/off’ switch on certain therapeutic enzymes with glycosylation, providing therapeutic proteins with enhanced control mechanisms toward improved target specific effects.
Acknowledgments
The authors acknowledge NSU College of Pharmacy for equipment and laboratory support.
Funding sources
This research was funded in part by Nova Southeastern University (NSU) Health Professions Division Grant, President’s Faculty Research and Development Grant (PFRDG, NSU), and in part by King Saud University through Saudi Arabian Cultural Mission (SACM) designated to WAM.
Ethical statement
None to be declared.
Conflict of Interests
The authors declare no conflict of interests.
Authors’ contribution
YMK, MSA, SC, VCY contributed to the conception and design of the work. WAM, YMK contributed to the acquisition, analysis, and interpretation of data. WAM prepared the manuscript draft. YMK finalized the manuscript. All authors have read and agreed to the published version of the manuscript.
References
- Kreitman RJ. Recombinant toxins for the treatment of cancer. CurrOpin Mol Ther 2003; 5:44-51. [ Google Scholar]
- Akbari B, Farajnia S, Ahdi Khosroshahi S, Safari F, Yousefi M, Dariushnejad H. Immunotoxins in cancer therapy: Review and update. Int Rev Immunol 2017; 36(#):207-19. doi: 10.1080/08830185.2017.1284211 [Crossref] [ Google Scholar]
- Dean SN, Turner KB, Medintz IL, Walper SA. Targeting and delivery of therapeutic enzymes. TherDeliv 2017; 8:577-95. doi: 10.4155/tde-2017-0020 [Crossref] [ Google Scholar]
- Xiong MP, Kwon GS. PEGylation of yeast cytosine deaminase for pretargeting. J Pharm Sci 2005; 94:1249-58. doi: 10.1002/jps.20354 [Crossref] [ Google Scholar]
- Ke S, Wright JC, Kwon GS. Avidin-biotin-PEG-CPA complexes as potential EPR-directed therapeutic protein carriers: preparation and characterization. Bioconjug Chem 2007; 18:1644-50. doi: 10.1021/bc700182t [Crossref] [ Google Scholar]
- Goodwin DA, Meares CF. Advances in pretargeting biotechnology. Biotechnol Adv 2001; 19:435-50. doi: 10.1016/s0734-9750(01)00065-9 [Crossref] [ Google Scholar]
- Hapuarachchige S, Artemov D. Theranostic Pretargeting Drug Delivery and Imaging Platforms in Cancer Precision Medicine. Front Oncol 2020; 10:1131. doi: 10.3389/fonc.2020.01131 [Crossref] [ Google Scholar]
- Kwon YM, Li YT, Liang JF, Park YJ, Chang LC, Yang VC. PTD-modified ATTEMPTS system for enhanced asparaginase therapy: a proof-of-concept investigation. J Control Release 2008; 130:252-8. doi: 10.1016/j.jconrel.2008.06.017 [Crossref] [ Google Scholar]
- Li YT, Kwon YM, Spangrude GJ, Liang JF, Chung HS, Park YJ. Preliminary in vivo evaluation of the protein transduction domain-modified ATTEMPTS approach in enhancing asparaginase therapy. J Biomed Mater Res A 2009; 91:209-20. doi: 10.1002/jbm.a.32204 [Crossref] [ Google Scholar]
- Shin MC, Zhang J, David AE, Trommer WE, Kwon YM, Min KA. Chemically and biologically synthesized CPP-modified gelonin for enhanced anti-tumor activity. J Control Release 2013; 172:169-78. doi: 10.1016/j.jconrel.2013.08.016 [Crossref] [ Google Scholar]
- Shin MC, Zhang J, Min KA, He H, David AE, Huang Y. PTD-Modified ATTEMPTS for Enhanced Toxin-based Cancer Therapy: An In Vivo Proof-of-Concept Study. Pharm Res 2015; 32:2690-703. doi: 10.1007/s11095-015-1653-y [Crossref] [ Google Scholar]
- Yang VC, Naik SS, Song H, Dombkowski AA, Crippen G, Liang JF. Construction and characterization of a t-PA mutant for use in ATTEMPTS: a drug delivery system for achieving targeted thrombolysis. J Control Release 2005; 110:164-76. doi: 10.1016/j.jconrel.2005.09.027 [Crossref] [ Google Scholar]
- Naik SS, Liang JF, Park YJ, Lee WK, Yang VC. Application of "ATTEMPTS" for drug delivery. J Control Release 2005; 101:35-45. doi: 10.1016/j.jconrel.2004.07.020 [Crossref] [ Google Scholar]
- Absar S, Choi S, Ahsan F, Cobos E, Yang VC, Kwon YM. Preparation and characterization of anionic oligopeptide-modified tissue plasminogen activator for triggered delivery: an approach for localized thrombolysis. Thromb Res 2013; 131:e91-9. doi: 10.1016/j.thromres.2012.11.030 [Crossref] [ Google Scholar]
- Absar S, Kwon YM, Ahsan F. Bio-responsive delivery of tissue plasminogen activator for localized thrombolysis. J Control Release 2014; 177:42-50. doi: 10.1016/j.jconrel.2013.12.036 [Crossref] [ Google Scholar]
- Absar S, Choi S, Yang VC, Kwon YM. Heparin-triggered release of camouflaged tissue plasminogen activator for targeted thrombolysis. J Control Release 2012; 157:46-54. doi: 10.1016/j.jconrel.2011.09.060 [Crossref] [ Google Scholar]
- Elzahhar P, Belal ASF, Elamrawy F, Helal NA, Nounou MI. Bioconjugation in Drug Delivery: Practical Perspectives and Future Perceptions. Methods Mol Biol 2019; 2000:125-82. doi: 10.1007/978-1-4939-9516-5_11 [Crossref] [ Google Scholar]
- Park JB, Kwon YM, Lee TY, Brim R, Ko MC, Sunahara RK. PEGylation of bacterial cocaine esterase for protection against protease digestion and immunogenicity. J Control Release 2010; 142:174-9. doi: 10.1016/j.jconrel.2009.10.015 [Crossref] [ Google Scholar]
- Wagh A, Law B. Methods for conjugating antibodies to nanocarriers. Methods Mol Biol 2013; 1045:249-66. doi: 10.1007/978-1-62703-541-5_15 [Crossref] [ Google Scholar]
- Hnasko RM. Bioconjugation of Antibodies to Horseradish Peroxidase (HRP). Methods Mol Biol 2015; 1318:43-50. doi: 10.1007/978-1-4939-2742-5_4 [Crossref] [ Google Scholar]
- HD A. Conjugation Methods. In: Wild D, editor. The immunoassay Handbook. 4th ed. Amsterdam: Elsevier; 2013. p. 310-3.
- Clamp JR, Hough L. The periodate oxidation of amino acids with reference to studies on glycoproteins. Biochem J 1965; 94:17-24. doi: 10.1042/bj0940017 [Crossref] [ Google Scholar]
- Avigad G, Amaral D, Asensio C, Horecker BL. The D-galactose oxidase of Polyporus circinatus. J Biol Chem 1962; 237:2736-43. [ Google Scholar]
- Toftgaard Pedersen A, Birmingham WR, Rehn G, Charnock SJ, Turner NJ, Woodley JM. Process Requirements of Galactose Oxidase Catalyzed Oxidation of Alcohols. Organic Process Research & Development 2015; 19:1580-9. doi: 10.1021/acs.oprd.5b00278 [Crossref] [ Google Scholar]
- Hébert M, Lesept F, Vivien D, Macrez R. The story of an exceptional serine protease, tissue-type plasminogen activator (tPA). Rev Neurol (Paris) 2016; 172:186-97. doi: 10.1016/j.neurol.2015.10.002 [Crossref] [ Google Scholar]
- Wu SL, Jiang H, Hancock WS, Karger BL. Identification of the unpaired cysteine status and complete mapping of the 17 disulfides of recombinant tissue plasminogen activator using LC-MS with electron transfer dissociation/collision induced dissociation. Anal Chem 2010; 82:5296-303. doi: 10.1021/ac100766r [Crossref] [ Google Scholar]
- Borisov OV, Field M, Ling VT, Harris RJ. Characterization of oligosaccharides in recombinant tissue plasminogen activator produced in Chinese hamster ovary cells: two decades of analytical technology development. Anal Chem 2009; 81:9744-54. doi: 10.1021/ac901498k [Crossref] [ Google Scholar]
- Spellman MW, Basa LJ, Leonard CK, Chakel JA, O'Connor JV, Wilson S. Carbohydrate structures of human tissue plasminogen activator expressed in Chinese hamster ovary cells. J Biol Chem 1989; 264:14100-11. [ Google Scholar]
- Schilke KF, Wilson KL, Cantrell T, Corti G, McIlroy DN, Kelly C. A novel enzymatic microreactor with Aspergillus oryzae β-galactosidase immobilized on silicon dioxide nanosprings. Biotechnol Prog 2010; 26:1597-605. doi: 10.1002/btpr.476 [Crossref] [ Google Scholar]
- Absar S, Nahar K, Choi S, Ahsan F, Yang VC, Kwon YM. Serum albumin-protamine conjugate for biocompatible platform for targeted delivery of therapeutic macromolecules. J Biomed Mater Res A 2014; 102:2481-90. doi: 10.1002/jbm.a.34916 [Crossref] [ Google Scholar]
- Christodoulides M, Boucher DW. The potency of tissue-type plasminogen activator (TPA) determined with chromogen and clot-lysis assays. Biologicals 1990; 18:103-11. doi: 10.1016/1045-1056(90)90019-v [Crossref] [ Google Scholar]
- Blakey DC, Watson GJ, Knowles PP, Thorpe PE. Effect of chemical deglycosylation of ricin A chain on the in vivo fate and cytotoxic activity of an immunotoxin composed of ricin A chain and anti-Thy 1.1 antibody. Cancer Res 1987; 47:947-52. [ Google Scholar]
- Henderson GE, Isett KD, Gerngross TU. Site-specific modification of recombinant proteins: a novel platform for modifying glycoproteins expressed in E. coli. Bioconjug Chem 2011; 22(#):903-12. doi: 10.1021/bc100510g [Crossref] [ Google Scholar]
- Cooper JA, Smith W, Bacila M, Medina H. Galactose oxidase from Polyporus circinatus, Fr. J Biol Chem 1959; 234:445-8. [ Google Scholar]
- York AW, Huang F, McCormick CL. Rational design of targeted cancer therapeutics through the multiconjugation of folate and cleavable siRNA to RAFT-synthesized (HPMA-s-APMA) copolymers. Biomacromolecules 2010; 11:505-14. doi: 10.1021/bm901249n [Crossref] [ Google Scholar]
- Donald JE, Kulp DW, DeGrado WF. Salt bridges: geometrically specific, designable interactions. Proteins 2011; 79:898-915. doi: 10.1002/prot.22927 [Crossref] [ Google Scholar]
- Radadiya A, Zhu W, Coricello A, Alcaro S, Richards NGJ. Improving the Treatment of Acute Lymphoblastic Leukemia. Biochemistry 2020; 59:3193-200. doi: 10.1021/acs.biochem.0c00354 [Crossref] [ Google Scholar]
- Dai S, Esson JM, Lutze O, Ramamurthy N, Yang VC, Meyerhoff ME. Bioanalytical applications of polyion-sensitive electrodes. J Pharm Biomed Anal 1999; 19:1-14. doi: 10.1016/s0731-7085(98)00134-4 [Crossref] [ Google Scholar]