Bioimpacts. 12(6):487-499.
doi: 10.34172/bi.2022.24113
Original Research
Graph theoretical network analysis, in silico exploration, and validation of bioactive compounds from Cynodon dactylon as potential neuroprotective agents against α-synuclein
Raja Rajeswari Rajeshkumar 1  , Banoth Karan Kumar 2 , Pavadai Parasuraman 3 , Theivendren Panneerselvam 4 , Krishnan Sundar 1 , Damodar Nayak Ammunje 5 , Sureshbabu Ram Kumar Pandian 1 , Sankaranarayanan Murugesan 2 , Shanmugampillai Jeyarajaguru Kabilan 1 , Selvaraj Kunjiappan 1, *
, Banoth Karan Kumar 2 , Pavadai Parasuraman 3 , Theivendren Panneerselvam 4 , Krishnan Sundar 1 , Damodar Nayak Ammunje 5 , Sureshbabu Ram Kumar Pandian 1 , Sankaranarayanan Murugesan 2 , Shanmugampillai Jeyarajaguru Kabilan 1 , Selvaraj Kunjiappan 1, *
Author information:
1Department of Biotechnology, Kalasalingam Academy of Research and Education, Krishnankoil-626126, Tamil Nadu, India
2Medicinal Chemistry Research Laboratory, Department of Pharmacy, Birla Institute of Technology & Science Pilani, Pilani Campus, Vidya Vihar, Pilani-333031, Rajasthan, India
3Department of Pharmaceutical Chemistry, Faculty of Pharmacy, M.S. Ramaiah University of Applied Sciences, M S R Nagar, Bengaluru-560054, Karnataka, India
4Department of Pharmaceutical Chemistry, Swamy Vivekanandha College of Pharmacy, Elayampalayam, Tiruchengodu-637205, Tamil Nadu, India
5Department of Pharmacology, Faculty of Pharmacy, M.S. Ramaiah University of Applied Sciences, M S R Nagar, Bengaluru-560054, Karnataka, India
Abstract
Introduction:
Parkinson's disease (PD) is a chronic, devastating neurodegenerative disorder marked by the death of dopaminergic neurons in the midbrain's substantia nigra pars compacta (Snpc). In alpha-synuclein (α-Syn) self-aggregation, the existence of intracytoplasmic inclusion bodies called Lewy bodies (LBs) and Lewy neurites (LNs) causes PD, which is a cause of neuronal death.
Methods:
The present study is aimed at finding potential bioactive compounds from Cynodon dectylon that can degrade α-Syn aggregation in the brain, through in silico molecular docking investigations. Graph theoretical network analysis was used to identify the bioactive compounds that target α-Syn and decipher their network as a graph. From the data repository, twenty-nine bioactive chemicals from C. dactylon were chosen and their structures were retrieved from Pubchem. On the basis of their docking scores and binding energies, significant compounds were chosen for future investigation. The in silico prediction of chosen compounds, and their pharmacokinetic and physicochemical parameters were utilized to confirm their drug-likeness profile.
Results:
During molecular docking investigation the bioactive compounds vitexin (-7.3 kcal.mol-1) and homoorientin (-7.1 kcal.mol-1) showed significant binding energy against the α-Syn target protein. A computer investigation of molecular dynamics simulation study verifies the stability of the α-Syn-ligand complex. The intermolecular interactions assessed by the dynamic conditions indicate that the bioactive compound vitexin has the potency to prevent α-Syn aggregation.
Conclusion:
Interestingly, the observed results indicate that vitexin is a potential lead compound against α-Syn aggregation, and in vitro and in vivo studies are warranted to confirm the promising therapeutic capability.
Keywords: α-Synuclein, Cynodon dactylon, Neuroprotective agents, Molecular docking, Nolecular dynamics, In silico ADMET
Copyright and License Information
© 2022 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Parkinson's disease (PD) is a complex devastating neurodegenerative illness that is the second most prevalent movement disorder, next to Alzheimer's disease (AD) in persons over the age of 65.
1
It affects 1-2 percent of people in the 65-69 age range and its incidence has risen by 1%-3% in those over the age of 80.
2
According to a World Health Organization (WHO) study, PD affects approximately 6.1 million individual’s worldwide.
3
There are two kinds of impaired behaviors that develop in people with PD: motor and non-motor impairments. Tremors, muscle stiffness, and bradykinesia are the most prevalent motor deficits. Cognitive decline, autonomic impairment, rapid eye movement, behavioral sleep problems, painful cramps, constipation, and other behavioral abnormalities are examples of non-motor impairments.
4
PD is connected to the death of dopaminergic neurons in the substantia nigra pars compacta (Snpc), which is situated in the midbrain's basal ganglia and is responsible for transmitting signals to the spinal cord thereby controlling muscular contraction, addiction and coordinating movement.
5
The loss of dopaminergic neurons in Snpc leads to the formation of intracytoplasmic inclusions called as Lewy bodies (LBs) and Lewy neurites (LNs) that contain alpha-synuclein (α-Syn) which causes neuronal cell death.
6
α-Synuclein (14.5 kDa, 140 amino acids), is an intraneuronal and intraglial protein that controls neurotransmitter release.
7
The structure of α-Syn reveals the existence of three domains: the N-terminal domain, a non-amyloid-beta plaque component and the C-terminal domain.
8
α-Syn is believed to be present as an intrinsically disordered monomer or helically folded tetramer in its natural state.
9
On the other hand, at the neuropathological level, the α-Syn protein misfolds and accumulates in the brain through protein aggregation, causing neuronal cell damage.
10,11
A53T point mutation on chromosome 4q21-23 was originally discovered as the connection between PD and α-Syn in the year 1996. The family type of PD is affected by a mutation in the α-Syn gene.
12,13
In autosomal dominant PD patients, the α-Syn gene is linked to the PARK1 locus on chromosome 4q21.
14
In addition, four mutations in PD have been discovered so far: A30P, E46K, H50Q and G51D.
15
All these mutations cause an increase in the rate of aggregation, a change in the oligomeric state or a reduction in the normal tetramer: monomer ratios, allowing these alterations to occur.
16
There is no secondary structure in the natively unfolded state of α-Syn. Changes in the environment such as agitation, ionic strength and pH, make amyloid-like fibrils and α-Syn aggregates.
17
α-Syn, on the other hand, is more cytotoxic than amyloid proteins, which produce amyloid-like fibrils.
18
By targeting α-Syn, it is possible to inhibit protein aggregation or breakdown in the treatment of PD. Levodopa is a dopamine replacement medication used to treat PD. Levodopa induces the production of dopamine, which sends messages between regions of the brain and nerves to efficiently regulate bradykinetic movement.
19
While this medication has been used for a long time, it has caused various adverse effects such as fluctuations, dyskinesias, toxicity and effectiveness loss. Furthermore, present treatment methods mostly concentrate on symptom management and postponing the disease's development rather than preventing it.
20
Interestingly, metabolites of plants have been shown to exhibit neuroprotective benefits. Tobacco, coffee, Mucuna pruriens, green tea, Ginkgo biloba, Panax ginseng, Curcuma longa, Bacopa monnieri and Salvia officinalis are just a few examples.
21
The discovery of plants having antioxidant properties has received a lot of interest recently. In this view, bioactive chemicals from Cynodon dectylon (Family: Poaceae) were selected for evaluating their neuroprotective effects and other movement problems in this study.
22
Carbohydrates, proteins, calcium, iron, potassium, phosphorus, beta-carotene, cynodine, triticin, furfural, levoglucosenone and other nutrients are abundant in C. dactylon.
23
The metabolites are used in the treatment of cough, cancer, snakebites, kidney stones, skin illnesses, bronchitis, asthma and neurological disorders in traditional medicine.
24
Furthermore, many studies have shown that C. dectylon extract has potential antioxidant capabilities for neuroprotective benefits.
22,25
Therefore, this study was designed to analyze protein aggregation in the brain by targeting the α-Syn. Using graph theoretical network analysis, we build the signaling network of genes implicated in the α-Syn protein. The network that was created has been utilized to identify the optimal medication target for successful neuroprotection. The binding effect of the identified bioactive chemicals with the specified target was predicted using molecular docking. The pharmacokinetic (absorption, distribution, metabolism, and excretion) toxicity properties of the chosen bioactive chemicals were also studied. The molecular stability and binding mechanisms of the chosen bioactive molecules with α-Synuclein protein were investigated using molecular dynamics (MD) simulation as well.
Materials and Methods
Network analysis
The protein signaling network of (HSA: 05012) Homo sapiens was screened using graph theoretical network analysis (Cytoscape software 3.7.1) to find the influential genes involved in the target protein (derived from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database).
26,27
In the present research, the α-Syn signaling network was recreated as a graph, containing several entities of genes, proteins (nodes) and their connections (edges). The network includes one node and 134 edges, as shown in Fig. 1, based on centrality characteristics such as degree, proximity, eccentricity, eigenvector, and radiality. The beginning value of all measurements, as well as important nodes in the network organisation has been shown using the measured values of degree (11), proximity (0.0418), eccentricity (0.6216), eigenvector (0.06054), radiality (61.29496) and stress (4914) (Supplementary file 1, Table S1).
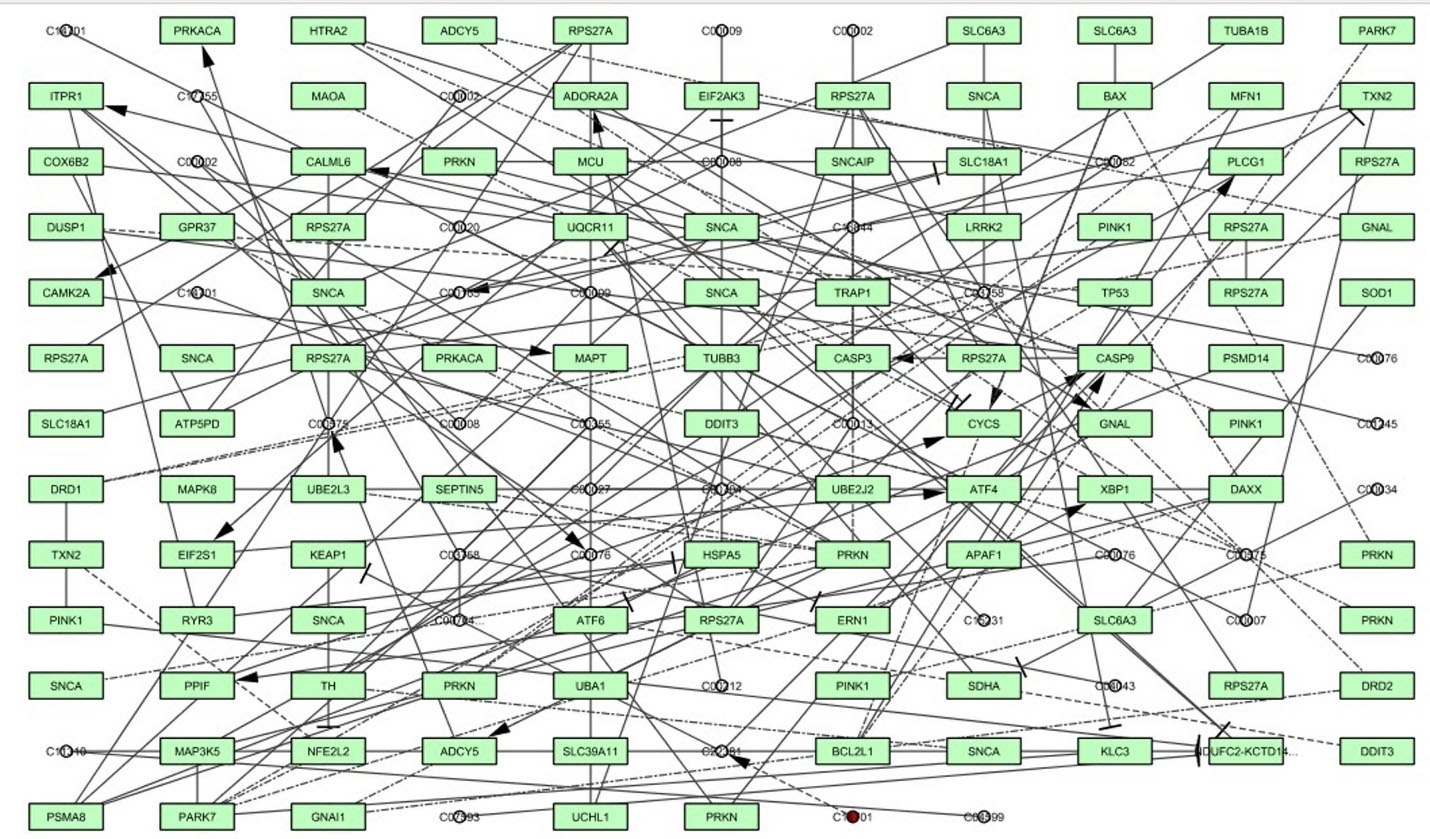
Fig. 1.
The α-synuclein signaling network was developed through graph theoretical network analysis using Cytoscape software 3.7.1.
.
The α-synuclein signaling network was developed through graph theoretical network analysis using Cytoscape software 3.7.1.
Protein receptor preparation
The crystal structure of α-Syn, PDB entry ID: 1XQ8, was downloaded and utilized as a target protein from the RCSB Protein Data Bank (PDB: http://www.rcsb.org/pdb). Before the molecular screening, optimize the protein structure by adding missing residues through Swiss-PDB Viewer v4.1.0's protein process tool.
28
The file was given the name target.pdb and was stored for future investigation. The protein of interest was then prepared using the Discovery studio visualizer 2020 (BIOVIA). All the water molecules occupying the protein were examined and eliminated. The ligands and ions that were bound were also removed. PDB proteins are devoid of hydrogen atoms in general. As a consequence, hydrogen atoms were added to the protein to give it a more conventional feel. Optimization and minimization techniques were employed to make the target protein ready for docking.
Preparation of ligands
The Indian Medicinal Plants, Phytochemistry and Therapeutics (IMPPAT) database contains twenty-nine bioactive chemicals from C. dactylon.
29
For virtual screening, the 3D SDF structures of identified bioactive components were acquired from the chemical database PubChem (http://pubchem.ncbi.nlm.nih.gov).
Molecular docking
The AutoDock Vina program included in the PyRx software was used to perform molecular docking calculations of the selected bioactive molecules from C. dactylon with the target protein, α-Syn.
30
The protein-ligand interaction profiler (https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index) was used to analyze the active site in the target and the resulting receptor protein was imported into the workspace. Using PyRx software, polar hydrogen atoms and Kollman partial charges were added to the three-dimensional structure of the protein. AutoDock Vina program estimated the energy affinity values of up to 10 distinct docking sites for each ligand. The ligand conformation at the active binding site was used to determine the complex affinity energies, with the RMSD between the original and subsequent docked structures taken into account. The Discovery Studio Visualizer, which provides information on compounds and interaction graphics (2D and 3D), was used to estimate information on hydrogen bonds and non-covalent interactions for each ligand-protein complex.
Physicochemical and ADME properties prediction
Pharmacokinetic (absorption, distribution, metabolism, and excretion) and physicochemical features are critical in the drug identification of new treatments since many suggested compounds fail in the development phase. The SwissADME online tool was used to examine molecular weight, molar refractivity, solubility, bioavailability, bioavailability radar map, egg-boiled plot, brain permeation and human gastrointestinal absorption properties for the selected bioactive substances (https://www.swissadme.ch).
31
Toxicity prediction
Small molecule toxicology prediction is crucial for anticipating the safety or hazardous profile of proposed bioactive substances before they are physiologically appropriated. As a consequence, to predict toxicity, and carcinogenicity profiles of the selected bioactive compounds, the canonical SMILES (simplified molecular-input line-entry system) of the selected bioactive compounds were uploaded to the web-based server called pkCSM-pharmacokinetics (http://biosig.unimelb.edu.au/pkcsm/prediction).
32
Molecular dynamics simulation
The MD simulation is a useful tool in current molecular modeling techniques for understanding macromolecular structure-to-function correlations. MD modeling was used to examine the binding stability, conformation, and interaction mechanisms between the chosen bioactive compounds (ligands) and protein α-Syn. Using GROMACS 2019.2 software, the best binding-free energy of ligand-α-Syn complex files was chosen for MD simulation investigations.
33-35
The topology of the selected bioactive compounds (ligand) was retrieved from the PRODRG website.
36
All of the complex systems were prepared as previously described. In MD simulation, the initial vacuum was lowered using the steepest descent approach for 5000 steps. Using a simple point charge (SPC) water model, the complex structure was solvated in a cubic periodic box of 0.5 nm. After then, enough Na+ and Cl - counter ions were injected to keep the salt concentration in the complex system at 0.15M. From the NPT (Isothermal-Isobaric, constant number of particles, pressure, and temperature) equilibration, each complex was assigned a simulation time of 50 ns for the final run. Using the internet server "WebGRO” for macromolecular simulations (https://simlab.uams.edu/), the root mean square deviation (RMSD) and root mean square fluctuation (RMSF) trajectories structure was executed in the GROMACS program.
Molecular mechanics Poisson–Boltzmann surface area (MMPBSA) calculation
The protein-ligand binding free energy of each complex was calculated using the MMPBSA technique. The g_mmpbsa tool created for GROMACS was used to calculate the binding free energy.
37,38
Density functional theory (DFT)
The Gaussian 03W program and the Gauss View molecular visualization tools were used to perform computational calculations of the selected bioactive compounds.
39
Using a 6-311G (d,p) basis set, the DFT/Becke-3–Lee–Yang–Parr (B3LYP) approach was utilized to optimize the molecular structures of the chosen bioactive compound. Using the optimized structures, the frontier molecular orbital energies of both compounds (lowest unoccupied molecular orbital [ELUMO], highest occupied molecular orbital [EHOMO], and their energy gap [Eg]) were computed. Gauss View, a molecular visualization tool, was used to display the created molecular orbital energy diagrams of the selected bioactive chemicals.
Results
Bioactive compounds retrieval and preparation
The IMPPAT database was searched for the available bioactive chemicals of the selected plant from C. dactylon. Table 1 shows a list of twenty-nine key bioactive chemicals and their structures found in C. dactylonwere retrieved from the database.
Table 1.
Bioactive compounds from Cynodon dectylon and their binding affinity against α-Syn
|
Ligand
|
Compound Id
|
Docking score (kcal.mol
-1
)
|
| Vitexin |
CID:5280441 |
-7.3 |
| Homoorientin |
CID:114776 |
-7.1 |
| Friedlein |
CID:244297 |
-6.8 |
| Beta-carotene |
CID:5280489 |
-6.6 |
| Orientin |
CID:5281675 |
-6.4 |
| Triterpenoids |
CID:71597391 |
-6.3 |
| Ergonovine |
CID:443884 |
-6.3 |
| Luteolin |
CID:5280445 |
-6.3 |
| Ergometrinine |
CID:5486180 |
-6.2 |
| Phytosterols |
CID:12303662 |
-6.2 |
| 2''-O-glycosylisovitexin |
CID:101698596 |
-6.2 |
| Arundoin |
CID:12308619 |
-6.2 |
| Apigenin |
CID:5280443 |
-6.1 |
| Tricin |
CID:5281702 |
-5.6 |
| Beta-Ionene |
CID:53249763 |
-4.9 |
| 2-Coumarinate |
CID:5280841 |
-4.9 |
| Triglochinin |
CID:5281124 |
-4.8 |
| Ferulic acid |
CID:445858 |
-4.5 |
| Phenyl acetaldehyde |
CID:998 |
-4.1 |
| Syringic acid |
CID:10742 |
-4.1 |
| Vanillic acid |
CID:8468 |
-4.0 |
| L-ascorbic acid |
CID:54670067 |
-3.9 |
| Phytol |
CID:145386 |
-3.9 |
| Palmitic acid |
CID:985 |
-3.6 |
| Docosanoic acid |
CID:8215 |
-3.6 |
| Hexadecanal |
CID:984 |
-3.5 |
| Tritriacontane |
CID:12411 |
-3.4 |
| Furfuryl alcohol |
CID:7361 |
-3.2 |
| Furfural |
CID:7362 |
-3.1 |
Molecular docking
The structure-based molecular docking method was used to evaluate the efficacy of identified putative bioactive chemicals against the α-Syn protein which could offer neuroprotective effects. Twenty-nine bioactive chemicals were docked against the α-Syn protein using AutoDock Vina tools in PyRx software. Thirteen bioactive metabolites out of 29 have a greater binding affinity (-6 kcal.mol -1) with the target protein and the results are presented in Table 1. Two of the bioactive compounds, vitexin (-7.3 kcal.mol -1) and homoorientin (-7.1 kcal.mol -1) which exhibited higher binding energy values were selected for further studies. The intermolecular interactions between the α-Syn protein with vitexin (Fig. 2), homoorientin (Fig. 3), and other compounds are presented in Table S2. The Maestro 11.7 version of the Schrodinger suite was used to depict the interactions between the ligands (vitexin and homoorientin) and the α-Syn protein. Vitexin (VIT) created three hydrophobic contacts with LYS43A (3.68 Å), VAL48A (3.68 Å) and VAL48A (3.73 Å), while LYS32A (2.68 Å), VAL40A (3.06 Å) and LYS45A (3.08 Å) formed three hydrogen bonds. Homoorientin (HOM) formed four hydrogen bonds with LYS32A (3.15 Å), GLU35A (2.19 Å), GLY36A (2.42 Å), LYS43A (2.91 Å) and one π-Stacking with TYR39A (5.43 Å).
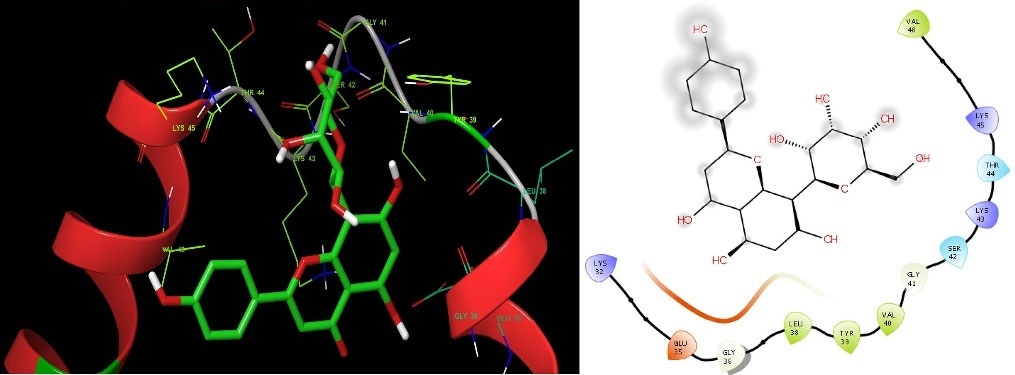
Fig. 2.
Representation of intermolecular interaction between the bioactive compound vitexin with α-Syn protein. The left side shows three-dimensional and the right side shows the two-dimensional structure of protein-ligand interaction.
.
Representation of intermolecular interaction between the bioactive compound vitexin with α-Syn protein. The left side shows three-dimensional and the right side shows the two-dimensional structure of protein-ligand interaction.
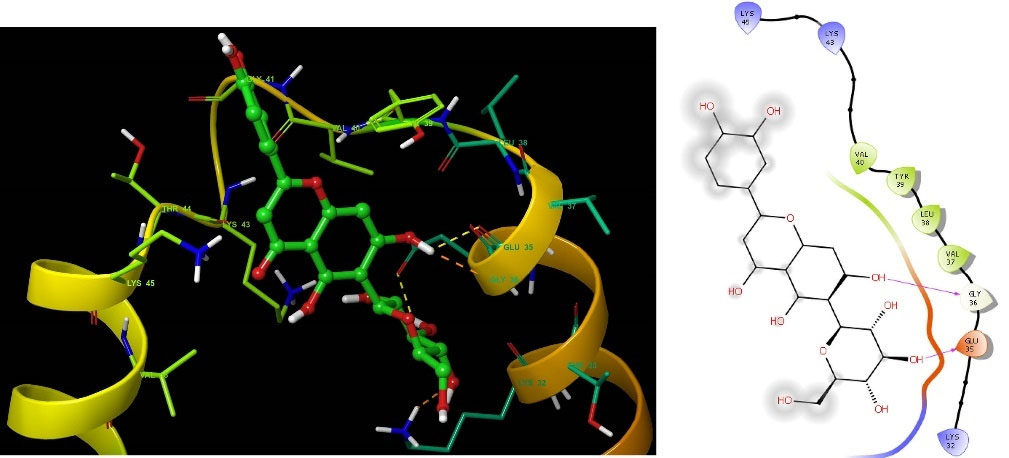
Fig. 3.
Representation of intermolecular interaction between the bioactive compound homoorientin with α-Syn protein. The left side shows three-dimensional and the right side shows the two-dimensional structure of protein-ligand interaction.
.
Representation of intermolecular interaction between the bioactive compound homoorientin with α-Syn protein. The left side shows three-dimensional and the right side shows the two-dimensional structure of protein-ligand interaction.
In silico prediction of physicochemical and ADME properties
SwissADME (http://www.swissadme.ch/) online tool was used to evaluate the pharmacokinetics (ADME) and physicochemical characteristics of the bioactive molecules from C. dactylon and the results are shown in Table 2 and Table S3. As the data in Table 2 indicates, the molecular weights of VIT and HOM are 432.38 g.mol -1 & 448.38 g.mol -1 respectively and this was found to contradict Lipinski's rule of five as the molecular weights are higher (MW >350). The two bioactive molecules, VIT and HOM have a polar surface area of 181.05 Å2 and 201.28 Å2 respectively. The anticipated findings also revealed that the bioactive chemicals VIT and HOM had a lower rate of gastrointestinal (GI) absorption in humans. The greater the amount of H-bonds, the more likely they are engaged in protein-ligand interaction. As a result, VIT, the discovered bioactive molecule, has a higher probability of becoming a drug-relevant candidate with neuroprotective potential. Both the chosen bioactive molecules and the conventional drug. The synthetic accessibility scores were determined to be 5.12 (VIT), and 5.04 (HOM), which showed that the bioactive molecules, VIT and HOM are difficult to manufacture.
Table 2.
In silico predicted physicochemical and ADME parameters of vitexin, and homoorientin
|
Parameter
|
Vitexin
|
Homoorientin
|
| Formula |
C21H20O10
|
C21H20O11
|
|
MW (g.mol -1)
|
432.38 |
448.38 |
| Number of heavy atoms |
31 |
32 |
| Number of aromatic heavy atoms |
16 |
16 |
| Fraction Csp3 |
0.29 |
0.29 |
| Number of rotatable bonds |
3 |
3 |
| Number of H-bond acceptors |
10 |
11 |
| Number of H-bond donors |
7 |
8 |
| Molar refractivity |
106.61 |
108.63 |
|
TPSA (Å2)
|
181.05 |
201.28 |
| Solubility class |
Soluble |
Soluble |
| Gastro Intestinal absorption |
Low |
Low |
| Blood Brain Barrier crossing |
No |
No |
| Violation of Lipinski’s rule of five |
1 |
2 |
| Bioavailability score |
0.55 |
0.17 |
| Synthetic accessibility |
5.12 |
5.04 |
Fig. 4 depicts a bioavailable radar map of drug-like properties of the selected chemicals VIT and HOM. The pink zone within the hexagon signifies the optimal range for the compounds. The drug-like compound's authorized range was unsaturation (INSATU): rotatable bonds (FLEXI): no more than 9 rotatable bonds, molecular weight (SIZE): between 150 and 500 g.mol -1, polar surface area (POLAR): between 20 and 130 g.mol -1 and polar surface area (POLAR): between 20 and 130 Å2.
40
The red slanted hexagon off-shoot of the vertex displays drug-like properties of the bioactive chemicals VIT and HOM (Fig. 4). In addition, using an egg-boiled model, the pharmacokinetic characteristics of both the bioactive chemical VIT and HOM were examined. The egg-boiled model proved useful in predicting two important pharmacokinetic characteristics at the same time, namely, passive gastrointestinal absorption (HIA) and blood-brain barrier (BBB) penetration.
29
The chemical contained in the yolk (i.e., yellow area) indicates very likely BBB permeation, whereas albumin (i.e., white region) represents highly possible HIA absorption in the egg-shaped organization plot. The bioactive components, VIT and HOM were detected outside the boiled egg in Fig. 5, indicating poor gastrointestinal absorption. The bioactive molecules VIT and HOM have significant potential to be drug-like agents for neuroprotective treatment, as indicated by the aforementioned expected findings.
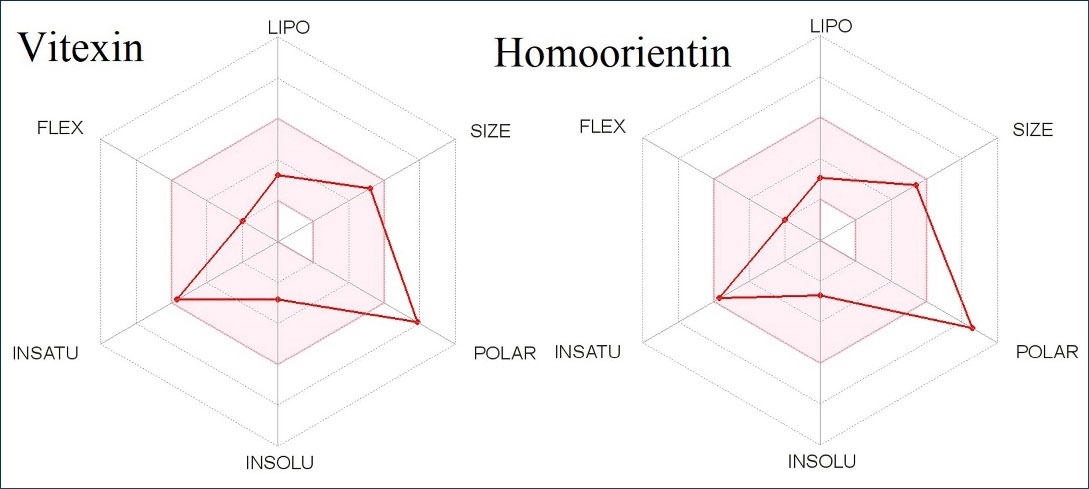
Fig. 4.
The oral bioavailability radar map of the bioactive molecule vitexin, and homoorientin.
.
The oral bioavailability radar map of the bioactive molecule vitexin, and homoorientin.
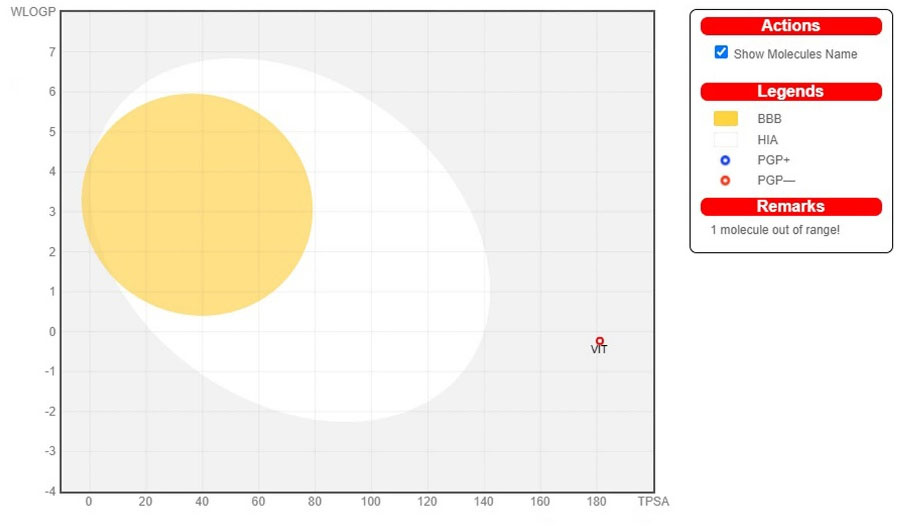
Fig. 5.
The EGG-BOILED model for vitexin and homoorientin.
.
The EGG-BOILED model for vitexin and homoorientin.
Toxicity prediction
Using the pkCSM-pharmacokinetics web-based platform, in silico toxicity prediction of the selected bioactive molecules, VIT, and HOM was conducted. Table 3 and Table S4 depicted the results of the predictions for AMES toxicity, drug-induced hERG toxicity, LD50 (median fatal dosage), hepatotoxicity, skin sensitization, Tetrahymena pyriformis (TP) toxicity, and minnow toxicity of selected (higher binding energy) and other bioactive compounds of C. dactylon, respectively. The results found that selected bioactive compounds VIT and HOM were no unfavorable effects such as hepatotoxicity, carcinogenicity, and skin sensitization. The LD50 specifies the instant or acute toxicity of molecules that were determined to be the most effective in the exploration.
Table 3.
Toxicity profile of top scored bioactive molecules
|
Compound
|
AMES toxicity
|
Maximum tolerated dose (human)
|
hERG inhibition
|
LD50
|
Hepatotoxicity
|
Carcinogenicity
|
Skin Sensitisation
|
T. pyriformis
toxicity
|
Minnow toxicity
|
| Vitexin |
Yes |
1.089 |
No |
2.996 |
No |
No |
No |
0.285 |
2.865 |
| Homoorientin |
Yes |
0.7 |
No |
2.963 |
No |
No |
No |
0.285 |
3.579 |
Molecular dynamics
While protein-ligand docking is extensively used and effective, it only offers a snapshot depiction of the ligand's binding posture at the receptor's active area. MD must be used to mimic the dynamics of atoms in the system as a function of time while also integrating Newton's equations of motion.
30
The results of 100-ns MD simulations were analyzed for two complexes produced from docking studies against α-Syn: VIT, and HOM. To investigate the stability and fluctuations of these complexes, MD trajectory analysis was performed to calculate the RMSD (Root Mean Square Deviation), RMSF (Root Mean Square Fluctuation), RGY (Radius of gyration), and SASA (Solvent Accessible Surface Area).
The RMSD is an important metric for analyzing the equilibration of MD trajectories and ensuring the stability of complex systems during simulation. To evaluate structural conformation changes, the RMSD of the protein backbone atoms was plotted vs time. With no substantial differences in the results, a stable conformation was attained between 31 and 100 ns (Fig. 6A). The test VIT complex's backbone RMSD ranged from 3.6 to 30 ns. With no substantial differences in the data, the stable conformation was attained in 31-100 ns (Fig. 6A). The test HOM complex had backbone RMSD variations ranging from 3.6 to 40 ns. With no substantial fluctuations in the measurements, a stable conformation was attained in 41-100 ns (Fig. 6A). This suggests that throughout the early stages of the simulations, the protein undertook small structural changes in all of the complexes.
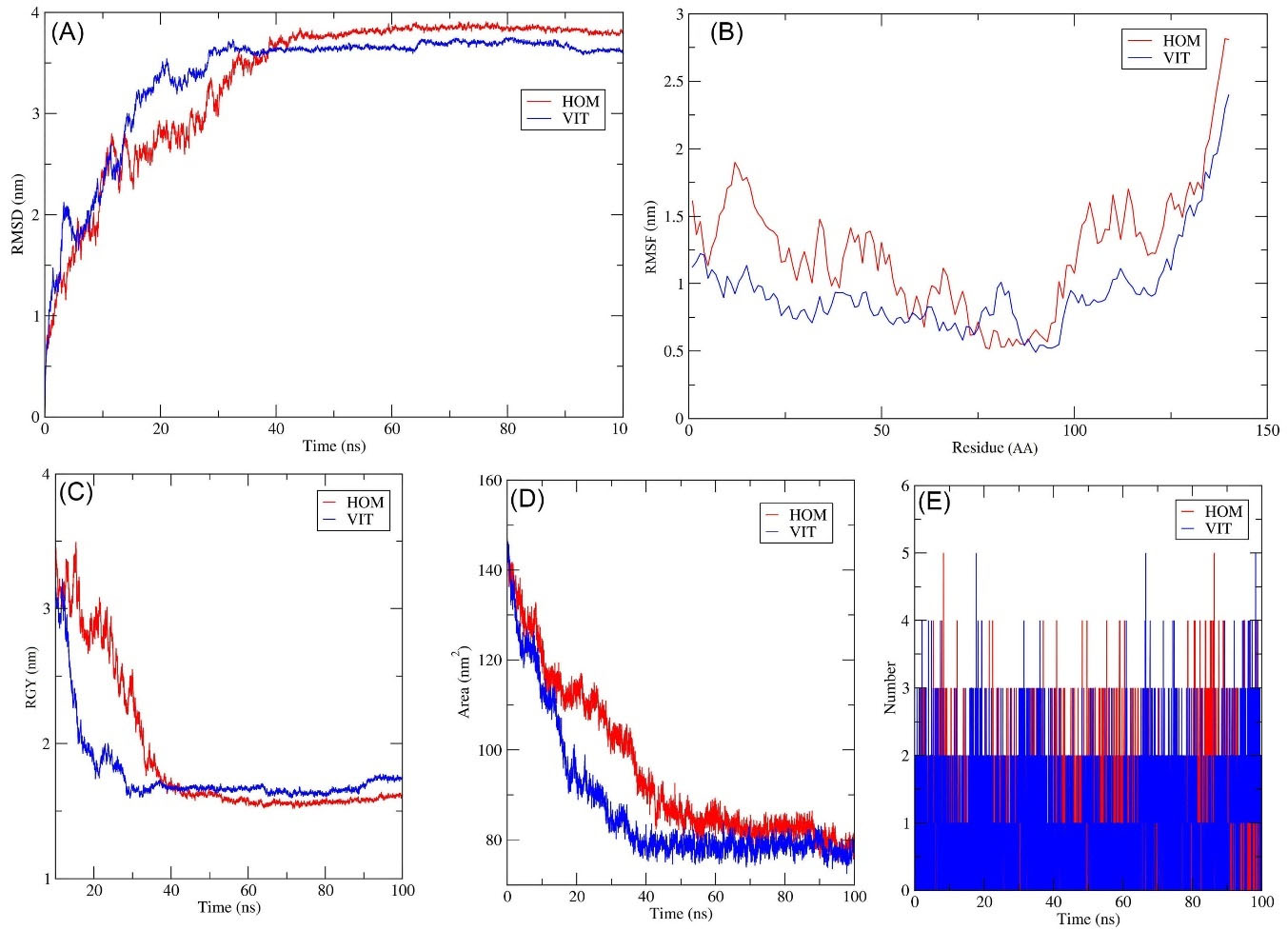
Fig. 6.
Time evaluation of RMSD plots for MD simulation of VIT (Blue) and HOM (Red) (A); Time evaluation of RMSF plots for MD Simulation. Chain of α-Syn-VIT (Blue) and α-Syn- HOM (Red) (B); Time evaluation of Radius of gyration plot for MD Simulation of VIT (Blue) and HOM (Red) (C); Time evaluation of SASA plots for MD Simulation of VIT (Blue) and HOM (Red) (D); Stability of intermolecular hydrogen bonding plot for MD Simulation of VIT (Blue) and HOM (Red).
.
Time evaluation of RMSD plots for MD simulation of VIT (Blue) and HOM (Red) (A); Time evaluation of RMSF plots for MD Simulation. Chain of α-Syn-VIT (Blue) and α-Syn- HOM (Red) (B); Time evaluation of Radius of gyration plot for MD Simulation of VIT (Blue) and HOM (Red) (C); Time evaluation of SASA plots for MD Simulation of VIT (Blue) and HOM (Red) (D); Stability of intermolecular hydrogen bonding plot for MD Simulation of VIT (Blue) and HOM (Red).
When modeling the stability and flexibility of complex systems, the RMSF is a crucial metric to consider
41
. When a target protein interacts with a ligand, the RMSF was utilized to see how the behavior of its amino acid residues changed. The C atoms' RMSF values were considered and plotted versus the residues (Fig. 6B).
The VIT complex fluctuated a little bit during the simulation time shown in the image, but the HOM complex fluctuated a lot. These results reveal that the binding of the VIT ligand does not affect the protein's flexibility.
The gyration radius (RGY) of complicated systems was also examined. The RMS distance between the atoms of the protein and the rotation axis is denoted by RGY. It is one of the most important parameters for determining the complete change in the compactness and dimensions of the protein structure over time.
42
Higher RGY values imply a less compact and flexible protein, while low RGY values indicate a compact and rigid protein. RGY values of protein backbone atoms were plotted against time to evaluate differences in structural compactness.
Between 31 and 100 ns, there were no notable differences and the value of 2 nm remained fairly constant. After VIT binding, the backbone RGY values dropped 35 ns. Between 36 and 100 ns, there were no significant changes in duration and the value of 1.7 nm remained nearly constant. Following HOM binding, the backbone RGY values dropped by 40 ns. Between 41 and 100 ns, there were no significant changes and the value of 1.6 nm remained relatively constant (Fig. 6C). The compounds cause no significant structural changes in the protein, according to the results of the entire investigation.
A Solvent Accessible Surface Area (SASA) analysis was also performed on all of the complexes.
43
SASA is an important parameter for determining the degree of receptor exposure to the surrounding solvent molecules during simulation. In general, ligand binding can impact the structural integrity of the receptor, affecting the region in contact with the solvent. The surface area changes in protein SASA measurements were plotted against time.
Except for a few periods throughout the simulation, minute alterations were seen throughout. The average value of SASA was found to be 85 nm2. The VIT complex showed a decrease in values until 35 ns in its trajectory. Around 80 nm2 was found to be the average SASA value. Until 45 ns, the HOM complex's trajectory showed a decrease in values. Except for a few periods throughout the simulation, minute alterations were seen throughout. The average SASA value was 83 nm2, as shown in Fig. 6D. According to the study, the surface area of protein in all complexes shrank during the simulation.
The MD trajectories were analyzed to determine the number of hydrogen bond formations throughout the simulation and the results are shown in Fig. 6E to investigate the ligands' binding energy to the target protein. Almost, three hydrogen bonds were established regularly throughout the simulation, showing the stability of the complex. The steadiness in making three hydrogen bonds with a maximum of five bonds for the VIT complex was maintained at specific time intervals. The HOM complex was found to be consistent in forming two hydrogen bonds with a maximum of four bonds at various time intervals. As can be seen, the top phytochemicals bind to the target protein with greater affinity.
Binding free energy analysis / MM/PBSA
The binding free energy (G bind) for VIT and HOM complexes was determined using the MM/PBSA technique for the final 20 ns (80-100 ns) of simulated trajectories with dt 1000 frames. VIT and HOM complexes each had a ΔG bind of -126.3011.215 kJ.mol -1 and -114.2717.66 kJ.mol -1, respectively. These low negative free binding energies indicate that the test ligands have a strong affinity for binding to α-Syn protein. The RMSD, RMSF, RGY, and SASA were used to verify the complexes and the lead complex Vitexin was determined to be stable throughout the simulations.
Density functional theory analysis
In that HOMO is a crude estimate of a compound's electron-donating capacity, the frontier-orbital energies (highest occupied molecular orbital (HOMO) & lowest unoccupied molecular orbital (LUMO)) of bioactive compounds play a major influence on bioactivities.
26
The energies and HOMO–LUMO energy gap of selected bioactive chemical vitexin and homooreintein were measured by B3LYP level using 6-311G (d, p) basis set and the HOMO–LUMO diagram of vitexin, and homooreintein was given in Table 4. The HOMO-LUMO energy gap values of the bioactive molecule vitexin were 0.12863 and for homooreintein were 0.12543. The findings of the energy gap values revealed that vitexin, homooreintein are very stable in nature.
Table 4.
EHOMO and ELUMO and ΔE values of Vitexin and Homoorientin
|
Drug
|
Homo
|
EHomo (eV)
|
LUMO
|
ELUMO (eV)
|
Energy gap
|
| Vitexin |
|
-0.31164 |
|
-0.18301 |
0.12863 |
| Homooreintin |
|
-0.31063 |
|
-0.18520 |
0.12543 |
Discussion
The most common reason for the pathological condition of dopaminergic neurons of PD is an abnormal aggregation of α-Syn protein in the form of Lewy bodies and Lewy neuritis in neurons.
44
Oxidative stress plays a critical role in the degradation of dopaminergic neurons in PD.
45
Several studies were shown that bioactive compounds from plants have neuroprotective effects against α-Syn aggregation and oxidative stress on PD.
46,47
Hence, the objective of this present study was to find out the effective neuroprotective agents from a plant source to prevent α-Syn protein aggregation that occurs in the midbrain’s Snpc. Among the medicinal plants, C. dactylonpossesses enormous medicinal valued bioactive compounds that are commonly used as antioxidants and neuroprotective purposes.
48
In silico molecular docking tools have been used to narrow down these medicinally valued bioactive compounds, particularly for the prevention of α-Syn protein aggregation in a neuron.
In silico molecular docking helps in accelerating the drug discovery process because the screening of the bioactive compounds that bind to the target protein can be predicted in a short duration. This along with in silico evaluation of pharmacokinetic, physiochemical, and pharmacodynamics properties assists in the identification of a few lead compounds that can be developed into a drug that can lead to disease prevention. In silico-based virtual screening, molecular docking, and molecular dynamics simulation experiments were employed in many studies and the results obtained were later confirmed by in vitro and in vivo activity evaluation studies.
49
This upholds the value of in silico studies in the drug discovery processes. Researchers may be able to find therapeutic solutions for specific diseases by better understanding how the compounds bind to, interact with, and down or up-regulate the associated proteins.
Pathway analysis, functional analysis and essentiality prediction can help in the identification of key genes and proteins in the biomolecular networks.
50
In this study, a graph theoretical network was constructed with the help of centrality measures such as closeness, betweenness, eigenvector, eccentricity, stress, radiality, and a degree in order to explore the roles and responsibilities of genes, proteins and enzymes that are essential for the development of biological pathway (α-Syn protein pathway). The created network comprising of some points (nodes) linked in a pattern by a set of edges, follows the structures of actual biological systems. The re-constructed biomolecular network, α-Syn protein was recognized as an ideal drug target for neurological disease “PD”. Historically, plant extracts and natural phytoconstituents were majorly utilized for drug discovery and development.
51
As a result, the goal of this study was to find out bioactive chemicals from C. dactylon by inhibiting α-Syn aggregation in the Snpc of the midbrain. Using IMPPAT database, a total of twenty-nine phytocompounds present in C. dactylon were selected. The docking scores of the chosen phytocompounds against α-Syn protein range from -3.1 kcal.mol -1 to -7.3 kcal.mol -1. Two of the compounds, Vitexin (-7.3 kcal.mol -1) and homoorientin (-7.1 kcal.mol -1) were chosen for further study because they exhibited significant binding energy. Similarly, Thangavel et al also used in silico analysis to find out effective derivatives of 7,8-dihydroxyflavone (DHF) for inhibiting α-Syn aggregation previously.
11
A new drug discovery process also involves the assessment of pharmacokinetics (absorption, distribution, metabolism and excretion) pharmacodynamics, and physicochemical properties, because they are directly linked with the effectiveness of phytocompounds.
52
Several highly active compounds failed during the quality control process due to unsatisfactory pharmacokinetics and toxicity properties.
53
Further, biologically active phytocompounds must reach their desired target location in sufficient quantity and remain in a stable form for long enough to trigger biological reactions.
54
Therefore, pharmacokinetic and physicochemical properties of phytocompounds must be evaluated during the drug development stages in order to pass routine clinical investigations and be regarded as promising drug candidates. Further, the Lipinski rule of 5 methodologies appears to be a helpful tool to predict the drug-likeness properties of phytocompounds.
55
Molecular weight and topological polar surface area (TPSA) have been observed under pharmacokinetic properties that govern the permeability of bioactive compounds across the biological barriers (i.e. blood-brain barrier).
56
Permeability of a phytocompounds may be reduced due to higher molecular weight and lower TPSA.
57
Solubility impacts the absorption of phytocompounds in the body.
58
The selected bioactive compounds (HOM and VIT) from this investigation show lower gastrointestinal absorption which could be attributed to the higher molecular weight of these two compounds. A few phytochemicals are known to be absorbed into the circulatory system through the small intestine, whereas others are absorbed by the colon and are believed to be transformed by the gut microbiota, and released back into the blood along with some microbial metabolites. These are known to exhibit substantial pharmacological action.
59
The number of hydrogen bond acceptors/donors present in a phytochemical molecule is also known to influence their permeability. Phytocompounds that have 12 or a smaller number of hydrogen bond donor provides good permeability across the biological barriers.
60
The bioactive molecules evaluated in this study, VIT and HOM have 10 and 11 hydrogen bond acceptors and 7 and 8 hydrogen bond donors respectively, these results indicate the moderate permeability of biological barriers. Moreover, good oral bioavailability indicates 10 or fewer rotatable bonds of phytocompounds.
61
The boiled-egg analysis was carried out to establish the human intestinal absorption and blood-brain barrier permeability of identified phytocompounds.
30
Since both the bioactive compounds, VIT and HOM were detected outside the boiled egg (Fig. 5), these were considered to have poor gastrointestinal absorption.
In this study, all the selected phytocompounds were analyzed for pharmacokinetics and physicochemical properties, and further assessment studies were conducted based on the findings. The toxicity of phytocompounds must be determined in order to explore their unwanted toxic effects, which can harm human beings. It is also one of the important phases in drug design and development.
62
The most common causes of drug failure in late-stage drug development include toxicity and serious adverse reactions.
63
Studies on animal models necessary to investigate the toxic profile of phytocompounds were constrained due to the longer time taken for the studies, ethical consideration, cost, and complexity of the studies.
64
As a result, in silico toxicity method that uses computational tools for predicting the toxicity of identified phytocompounds is regarded as useful. The toxicity levels of chosen bioactive molecules were evaluated using in silico methods in this study. In silico analyses were used to predict the hERG (ion channel), AMES, hepatotoxicity, carcinogenicity, and skin-irritating properties of chosen drugs. VIT and HOM were found to score negatively for hERG toxicity, carcinogenicity, and skin irritation and exhibited favorable logBB values. The LD50 represents the speedy or acute toxicity of the drugs that were shown to be the most effective in the study. Molecular dynamics simulation, a method for examining the physical movements of atoms and molecules,
65
confirms the stability and fluctuation of a drug candidate at the active site of the target protein. The α-Syn–ligand (VIT and HOM) complexes were validated by interpreting the RMSD, RMSF, RGY, hydrogen bond interactions, and SASA during the simulations.
Research Highlights
What is the current knowledge?
√ Parkinson's disease is a complex progressive neurodegenerative disorder, next to Alzheimer's disease.
√ Plants and their extracts possessing antioxidant properties has received a lot of interest recently.
√
Cynodon dectylon and their extracts were traditionally used for neuroprotective effects and other motor neuron problems
What is new here?
√ Ideal target via graph theoretical network analysis (alpha-synuclein (has: 05012)) and translate their signalling route, a network was built.
√ Molecular dynamics simulation verified the stability of bioactive compound vitexin at the alpha-synuclein binding pockets.
√ The density functionality theory revealed that bioactive compound vitexin was very stable in nature.
√
In silico pharmacokinetic prediction analyse showed the druggability and safety profile of the lead compound vitexin.
Conclusion
Plant extracts and plant-derived bioactive substances have traditionally been used to help suppress chronic disease, protect health, lower treatment expenses, and enhance the quality of life. α-Syn protein is a key component during the earlier stages of development of neurodegeneration in PD. The present study looked for a possible neuroprotective agent in C. dactylon that can prevent α-Syn protein aggregation. Vitexin, an unreported bioactive compound showed significant binding energy (-7.3 kcal mol -1) towards the α-Syn protein among the twenty-nine compounds that were evaluated. The pharmacokinetics and physicochemical properties of vitexin were found non-toxic and drug-like. The stability of the ligand-receptor complex was examined using molecular dynamics simulation and it was discovered that vitexin with α-Syn protein complexes was stable throughout the simulation. The density functionality theory findings also revealed that vitexin is very stable in nature. Overall, the bioactive chemical vitexin from C. dactylon could be a potential lead molecule for future testing as a possible neuroprotective agent acting on α-Syn aggregation using both in vitro and in vivo assays.
Acknowledgments
The authors are thankful to Biorender (https://biorender.com/) for the development of the graphical presentation of data.
Funding sources
This work was carried out under the grant of the Department of Biotechnology, Indo-Spain, New Delhi (Ref. No: BT/IN/Spain/39/SM/2017- 2018), and the Ministry of Tribal Affairs, Government of India for providing financial assistance (Award no-201920-NFST-TEL- 01497). KS would like to acknowledge the financial support from the Science and Engineering Research Board of India (EMR/2016/003035) and SK and KS thank the Department of Biotechnology, New Delhi for financial assistance (BT/PR36633/TRM/120/277/2020).
Ethical Statement
There is none to be declared.
Competing interests
The authors declare no conflict of interest in publishing this paper. This study not supported by any grant money from a pharmaceutical company or for-profit organization.
Authors’ contribution
Conceptualization: RRR and SK; Methodology, RRR, BKK, SM, PP and SK; Validation: RRR, SRK, DNA, KSJ and SK; Formal analysis: SK, PP, KS, SM and TP; Investigation: RRR and SK; Resource: RRR and SK; Data curation: KS, RRR and SK; Writing-original draft preparation: RRR, PP and SK; Writing-review and editing: RRR, KS and SK; Visualization: RRR, KS and SK; Supervision: SK. All authors have read and agreed to the published version of the manuscript.
Supplementary files
Supplementary files 1 contains Tables S1-S4.
(pdf)
References
-
Sam R, Han T-U, Sidransky E, Chen Y. Progress in generating iPSC-derived dopaminergic neurons as accurate models of neurodegenerative disease. In: Current Progress in iPSC-derived Cell Types. Elsevier; 2021. p. 181-203. 10.1016/B978-0-12-823884-4.00011-0.
- Nussbaum RL, Ellis CE. Alzheimer's disease and Parkinson's disease. N Engl J Med 2003; 348:1356-64. doi: 10.1056/NEJM2003ra020003 [Crossref] [ Google Scholar]
- Menken M, Janca A, World Health Organization. Parkinson's disease and public health: educational and management implications. WHO; 1997.
- Pedroso JL, Franca Jr MC, Braga‐Neto P, D'Abreu A, Saraiva‐Pereira ML, Saute JA. Nonmotor and extracerebellar features in Machado‐Joseph disease: a review. Mov Disord 2013; 28:1200-8. doi: 10.1002/mds.25513 [Crossref] [ Google Scholar]
- Kinoshita K-i, Tada Y, Muroi Y, Unno T, Ishii T. Selective loss of dopaminergic neurons in the substantia nigra pars compacta after systemic administration of MPTP facilitates extinction learning. Life Sci 2015; 137:28-36. doi: 10.1016/j.lfs.2015.07.017 [Crossref] [ Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012; 338:949-53. doi: 10.1126/science.1227157 [Crossref] [ Google Scholar]
- Jayaraj RL, Elangovan N. In silico identification of potent inhibitors of alpha-synuclein aggregation and its in vivo evaluation using MPTP induced Parkinson mice model. Biomed Aging Pathol 2014; 4:147-52. doi: 10.1016/j.biomag.2014.01.002 [Crossref] [ Google Scholar]
- Blumenstock S. Misfolded proteins affect synaptic plasticity and network activity [Dissertation]. Munich: Ludwig Maximilian University; 2017.
- Moretti P, Mariani P, Ortore MG, Plotegher N, Bubacco L, Beltramini M. Comprehensive Structural and Thermodynamic Analysis of Prefibrillar WT α-Synuclein and Its G51D, E46K, and A53T Mutants by a Combination of Small-Angle X-ray Scattering and Variational Bayesian Weighting. J Chem Inf Model 2020; 60:5265-81. doi: 10.1021/acs.jcim.0c00807 [Crossref] [ Google Scholar]
- Bourdenx M, Koulakiotis NS, Sanoudou D, Bezard E, Dehay B, Tsarbopoulos A. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: Examples of amyloidopathies, tauopathies and synucleinopathies. Prog Neurobiol 2017; 155:171-93. doi: 10.1016/j.pneurobio.2015.07.003 [Crossref] [ Google Scholar]
- Mohankumar T, Chandramohan V, Lalithamba HS, Jayaraj RL, Kumaradhas P, Sivanandam M. Design and molecular dynamic investigations of 7, 8-dihydroxyflavone derivatives as potential neuroprotective agents against alpha-synuclein. Sci Rep 2020; 10:1-10. doi: 10.1038/s41598-020-57417-9 [Crossref] [ Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2001; 2:492-501. doi: 10.1038/35081564 [Crossref] [ Google Scholar]
- Spillantini M. Parkinson's disease, dementia with Lewy bodies and multiple system atrophy are α-synucleinopathies. Parkinsonism Relate Disord 1999; 5:157-62. doi: 10.1016/S1353-8020(99)00031-0 [Crossref] [ Google Scholar]
-
Krüger R, Menezes Vieira‐Saecker AM, Kuhn W, Berg D, Müller T, Kühnl N, et al. Increased susceptibility to sporadic Parkinson's disease by a certain combined α‐synuclein/apolipoprotein E genotype. Ann Neurol 1999; 45: 611-7.10.1002/1531-8249(199905)45:5<611::AID-ANA9>3.0.CO;2-X
- Ruf VC, Nübling GS, Willikens S, Shi S, Schmidt F, Levin J. Different effects of α-synuclein mutants on lipid binding and aggregation detected by single molecule fluorescence spectroscopy and ThT fluorescence-based measurements. ACS Chem Neurosci 2019; 10:1649-59. doi: 10.1021/acschemneuro.8b00579 [Crossref] [ Google Scholar]
- Meade RM, Fairlie DP, Mason JM. Alpha-synuclein structure and Parkinson’s disease–lessons and emerging principles. Mol Neurodegener 2019; 14:1-14. doi: 10.1186/s13024-019-0329-1 [Crossref] [ Google Scholar]
- Ghosh D, Singh PK, Sahay S, Jha NN, Jacob RS, Sen S. Structure based aggregation studies reveal the presence of helix-rich intermediate during α-Synuclein aggregation. Sci Rep 2015; 5:1-15. doi: 10.1038/srep09228 [Crossref] [ Google Scholar]
- El-Agnaf OM, Jakes R, Curran MD, Middleton D, Ingenito R, Bianchi E. Aggregates from mutant and wild-type α-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of β-sheet and amyloid-like filaments. FEBS Lett 1998; 440:71-5. doi: 10.1016/S0014-5793(98)01418-5 [Crossref] [ Google Scholar]
- Obeso JA, Merello M, Rodríguez‐Oroz MC, Marin C, Guridi J, Alvarez L. Levodopa‐induced dyskinesias in Parkinson's disease. Handbook Clin Neurol 2007; 84:185-218. doi: 10.1016/S0072-9752(07)84040-1 [Crossref] [ Google Scholar]
- Jankovic J, Aguilar LG. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr Dis Treat 2008; 4:743. doi: 10.2147/ndt.s2006 [Crossref] [ Google Scholar]
- Dey A, De JN. Possible anti-Parkinson’s disease therapeutics from nature: A review. Stud Natural Prod Chem 2015; 44:447-520. doi: 10.1016/B978-0-444-63460-3.00009-2 [Crossref] [ Google Scholar]
- Sharma N, Bafna P. Effect of Cynodon dactylon on rotenone induced Parkinson’s disease. Orient Pharm Exp Med 2012; 12:167-75. doi: 10.1007/s13596-012-0075-1 [Crossref] [ Google Scholar]
- Rad AK, Rajaei Z, Mohammadian N, Valiollahi S, Sonei M. The beneficial effect of Cynodon dactylon fractions on ethylene glycol-induced kidney calculi in rats. Urol J 2011; 8:179. [ Google Scholar]
- Khatun P, Das SK, Khulna B. Medicinal and Versatile Uses of an Amazing, Obtainable and Valuable Grass: Cynodon dactylon. Int J Pharm Med Res 2020; 8:1-11. [ Google Scholar]
- Poojary R, Kumar NA, Kumarchandra R, Vinodini N, Bhagyalakshmi K, Sanjeev G. Cynodon dactylon extract ameliorates cognitive functions and cerebellar oxidative stress in whole body irradiated mice. Asian Pac J Trop Biomed 2019; 9:278. doi: 10.4103/2221-1691.261763 [Crossref] [ Google Scholar]
- Saravanan G, Panneerselvam T, Kunjiappan S, Parasuraman P, Alagarsamy V, Udayakumar P. Graph theoretical analysis, in silico modeling, prediction of toxicity, metabolism and synthesis of novel 2‐(methyl/phenyl)‐3‐(4‐(5‐substituted‐1, 3, 4‐oxadiazol‐2‐yl) phenyl) quinazolin‐4 (3H)‐ones as NMDA receptor inhibitor. Drug Dev Res 2019; 80:368-85. doi: 10.1002/ddr.21511 [Crossref] [ Google Scholar]
-
Aoki-Kinoshita KF, Kanehisa M. Gene annotation and pathway mapping in KEGG. In: Comparative genomics. Springer; 2007. p. 71-91. 10.1007/978-1-59745-515-2_6.
- Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K. The protein data bank. Acta Crystallogr. D Acta Crystallogr D 2002; 58:899-907. doi: 10.1107/S0907444902003451 [Crossref] [ Google Scholar]
- Mohanraj K, Karthikeyan BS, Vivek-Ananth R, Chand RB, Aparna S, Mangalapandi P. IMPPAT: A curated database of I ndian M edicinal P lants, P hytochemistry A nd T herapeutics. Sci Rep 2018; 8:1-17. doi: 10.1038/s41598-018-22631-z [Crossref] [ Google Scholar]
- Kalimuthu AK, Panneerselvam T, Pavadai P, Pandian SRK, Sundar K, Murugesan S. Pharmacoinformatics-based investigation of bioactive compounds of Rasam (South Indian recipe) against human cancer. Scic Rep 2021; 11:1-19. doi: 10.1038/s41598-021-01008-9 [Crossref] [ Google Scholar]
- Islam MA, Pillay TS. Identification of promising anti-DNA gyrase antibacterial compounds using de novo design, molecular docking and molecular dynamics studies. J Biomol Struct Dyn 2020; 38:1798-809. doi: 10.1080/07391102.2019.1617785 [Crossref] [ Google Scholar]
- Jia C-Y, Li J-Y, Hao G-F, Yang G-F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov Today 2020; 25:248-58. doi: 10.1016/j.drudis.2019.10.014 [Crossref] [ Google Scholar]
-
Markov S, Petkov P, Pavlov V, editors. Large-Scale Molecular Dynamics Simulations on Modular Supercomputer Architecture with Gromacs. In: Dimov I, Fidanova S, eds. Advances in High Performance Computing. HPC 2019. Studies in Computational Intelligence, vol 902. Cham: Springer; 2021. 10.1007/978-3-030-55347-0_30.
- Zai Y, Xi X, Ye Z, Ma C, Zhou M, Chen X. Aggregation and Its Influence on the Bioactivities of a Novel Antimicrobial Peptide, Temporin-PF, and Its Analogues. Int J Mol Sci 2021; 22:4509. doi: 10.3390/ijms22094509 [Crossref] [ Google Scholar]
- Rajendran BK, Suresh MX, Bhaskaran SP, Harshitha Y, Gaur U, Kwok HF. Pharmacoinformatic approach to explore the antidote potential of phytochemicals on bungarotoxin from Indian Krait, Bungarus caeruleus. Comput Struct Biotech J 2018; 16:450-61. doi: 10.1016/j.csbj.2018.10.005 [Crossref] [ Google Scholar]
- Van Aalten DM, Bywater R, Findlay JB, Hendlich M, Hooft RW, Vriend G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J Comput Aided Mol Des 1996; 10:255-62. doi: 10.1007/BF00355047 [Crossref] [ Google Scholar]
- Rajagopal G, Nivetha A, Sundar M, Panneerselvam T, Murugesan S, Parasuraman P. Mixed phytochemicals mediated synthesis of copper nanoparticles for anticancer and larvicidal applications. Heliyon 2021; 7:e07360. doi: 10.1016/j.heliyon.2021.e07360 [Crossref] [ Google Scholar]
- Sharma A, Datta D, Balasubramaniam R. Molecular dynamics simulation to investigate the orientation effects on nanoscale cutting of single crystal copper. Computl Mat Sci 2018; 153:241-50. doi: 10.1016/j.commatsci.2018.07.002 [Crossref] [ Google Scholar]
- Frisch ME, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 16 Release S. 3: MacroModel. 2014.
- Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A 2001; 105:9954-60. doi: 10.1021/jp003020w [Crossref] [ Google Scholar]
- Kumar B, Parasuraman P, Murthy TPK, Murahari M, Chandramohan V. In silico screening of therapeutic potentials from Strychnos nux-vomica against the dimeric main protease (Mpro) structure of SARS-CoV-2. J Biomol Struct Dyn 2021; 40:7796-7814. doi: 10.1080/07391102.2021.1902394 [Crossref] [ Google Scholar]
- Rahman MM, Saha T, Islam KJ, Suman RH, Biswas S, Rahat EU. Virtual screening, molecular dynamics and structure–activity relationship studies to identify potent approved drugs for Covid-19 treatment. J Biomol Struct Dyn 2021; 39:6231-41. doi: 10.1080/07391102.2020.1794974 [Crossref] [ Google Scholar]
- Ghosh SK, Saha B, Banerjee R. Insight into the sequence-structure relationship of TLR cytoplasm’s Toll/Interleukin-1 receptor domain towards understanding the conserved functionality of TLR 2 heterodimer in mammals. J Biomol Struct Dyn 2021; 39:5348-57. doi: 10.1080/07391102.2020.1786457 [Crossref] [ Google Scholar]
- Mandal PK, Pettegrew JW, Masliah E, Hamilton RL, Mandal R. Interaction between Aβ peptide and α synuclein: molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochem Res 2006; 31:1153-62. doi: 10.1007/s11064-006-9140-9 [Crossref] [ Google Scholar]
- Janda E, Isidoro C, Carresi C, Mollace V. Defective autophagy in Parkinson’s disease: role of oxidative stress. Mol Neurobiol 2012; 46:639-61. doi: 10.1007/s12035-012-8318-1 [Crossref] [ Google Scholar]
- Limanaqi F, Biagioni F, Mastroiacovo F, Polzella M, Lazzeri G, Fornai F. Merging the multi-target effects of phytochemicals in neurodegeneration: From oxidative stress to protein aggregation and inflammation. Antioxidants 2020; 9:1022. doi: 10.3390/antiox9101022 [Crossref] [ Google Scholar]
- Fazel Nabavi S, Braidy N, Habtemariam S, Sureda A, Manayi A, Mohammad Nabavi S. Neuroprotective effects of fisetin in Alzheimer’s and Parkinson’s Diseases: From chemistry to medicine. Curr Top Med Chem 2016; 16:1910-5. doi: 10.2174/1568026616666160204121725 [Crossref] [ Google Scholar]
- Al-Snafi AE. Chemical constituents and pharmacological effects of Cynodon dactylon-A review. IOSR J Pharm 2016; 6:17-31. [ Google Scholar]
- Vora J, Patel S, Sinha S, Sharma S, Srivastava A, Chhabria M. Structure based virtual screening, 3D-QSAR, molecular dynamics and ADMET studies for selection of natural inhibitors against structural and non-structural targets of Chikungunya. J Biomol Struct Dyn 2019; 37:3150-61. doi: 10.1080/07391102.2018.1509732 [Crossref] [ Google Scholar]
- Mistry D, Wise RP, Dickerson JA. DiffSLC: A graph centrality method to detect essential proteins of a protein-protein interaction network. PloS One 2017; 12:e0187091. [ Google Scholar]
- Mushtaq S, Abbasi BH, Uzair B, Abbasi R. Natural products as reservoirs of novel therapeutic agents. EXCLI Journal 2018; 17:420. doi: 10.1371/journal.pone.0187091 [Crossref] [ Google Scholar]
- Sharma A, Sharma S, Gupta M, Fatima S, Saini R, Agarwal SM. Pharmacokinetic profiling of anticancer phytocompounds using computational approach. Phytochem Anal 2018; 29:559-68. doi: 10.1002/pca.2767 [Crossref] [ Google Scholar]
- Wang J, Urban L. The impact of early ADME profiling on drug discovery and development strategy. Drug Discov World 2004; 5:73-86. [ Google Scholar]
- Sabaragamuwa R, Perera CO, Fedrizzi B. Centella asiatica (Gotu kola) as a neuroprotectant and its potential role in healthy ageing. Trends Food Sci Technol 2018; 79:88-97. doi: 10.1016/j.tifs.2018.07.024 [Crossref] [ Google Scholar]
- Prabhavathi H, Dasegowda K, Renukananda K, Lingaraju K, Naika HR. Exploration and evaluation of bioactive phytocompounds against BRCA proteins by in silico approach. J Biomol Struct Dyn 2021; 39:5471-85. doi: 10.1080/07391102.2020.1790424 [Crossref] [ Google Scholar]
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017; 7:42717. doi: 10.1038/srep42717 [Crossref] [ Google Scholar]
- Ullah A, Prottoy NI, Araf Y, Hossain S, Sarkar B, Saha A. Molecular docking and pharmacological property analysis of phytochemicals from Clitoria ternatea as potent inhibitors of cell cycle checkpoint proteins in the cyclin/CDK pathway in cancer cells. Comput Mol Biosci 2019; 9:81. doi: 10.4236/cmb.2019.93007 [Crossref] [ Google Scholar]
- Rajeev R, Marathe SD, Niranjan V, Sharma B, Sarojini S. In silico Analysis of Stigmasterol from Saraca asoca as a Potential Therapeutic Drug Against Alzheimer’s Disease. J Biol Act Prod Nat 2021; 11:516-29. doi: 10.1080/22311866.2021.1970021 [Crossref] [ Google Scholar]
- Dey P. Gut microbiota in phytopharmacology: A comprehensive overview of concepts, reciprocal interactions, biotransformations and mode of actions. Pharmacol Res 2019; 147:104367. doi: 10.1016/j.phrs.2019.104367 [Crossref] [ Google Scholar]
- Hossain S, Sarkar B, Prottoy MNI, Araf Y, Taniya MA, Ullah MA. Thrombolytic activity, drug likeness property and ADME/T analysis of isolated phytochemicals from ginger (Zingiber officinale) using in silico approaches. Modern Res Inflammation 2019; 8:29-43. doi: 10.4236/mri.2019.83003 [Crossref] [ Google Scholar]
-
Mandal M, editor. Phytochemicals as potential inhibitors for novel coronavirus 2019-nCoV/SARS-CoV-2: a graph-based computational analysis. The 12th International Conference on Advances in Information Technology; 2021. 10.1145/3468784.3468886.
- Zhang C, Cheng F, Li W, Liu G, Lee PW, Tang Y. In silico prediction of drug induced liver toxicity using substructure pattern recognition method. Mol Inform 2016; 35:136-44. doi: 10.1002/minf.201500055 [Crossref] [ Google Scholar]
- Dykens JA, Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov Today 2007; 12:777-85. doi: 10.1016/j.drudis.2007.07.013 [Crossref] [ Google Scholar]
-
Santos DI, Saraiva JMA, Vicente AA, Moldão-Martins M. Methods for determining bioavailability and bioaccessibility of bioactive compounds and nutrients. Innovative thermal and non-thermal processing, bioaccessibility and bioavailability of nutrients and bioactive compounds. Elsevier; 2019. p. 23-54. 10.1016/B978-0-12-814174-8.00002-0.
- Van Gunsteren WF, Berendsen HJ. Computer simulation of molecular dynamics: methodology, applications, and perspectives in chemistry. Angew Chem Int Ed 1990; 29:992-1023. doi: 10.1002/anie.199009921 [Crossref] [ Google Scholar]