Bioimpacts. 13(5):405-413.
doi: 10.34172/bi.2022.24208
Original Article
Vandetanib alters the tumoricidal capacity of human breast cancer stem cells via inhibiting vasculogenic capacity
Sanya Haiaty Investigation, Methodology, Writing – original draft, 1, 2, 3, 4 
Mohammad-Reza Rashidi Conceptualization, 1
Maryam Akbarzadeh Investigation, Methodology, 1, 5
Ahad Bazmany Investigation, Methodology, 6, 3
Mostafa Mostafazadeh Investigation, Methodology, 2
Saba Nikanfar Investigation, Methodology, 2
Roya Shabkhizan Investigation, Methodology, 7
Rostam Rezaeian Investigation, Methodology, 3
Reza Rahbarghazi Conceptualization, Supervision, Writing – review & editing, 8, 9, *
Mohammad Nouri Funding acquisition, Supervision, 1, 2, 8, *
Author information:
1Stem Cell and Regenerative Medicine Institute, Tabriz University of Medical Sciences, Tabriz, Iran
2Department of Biochemistry and Clinical Laboratories, Tabriz University of Medical Sciences, Tabriz, Iran
3Research Center of Infectious Diseases and Tropical Medicine, Tabriz University of Medical Science, Tabriz, Iran
4Student Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran
5Department of Biochemistry, Erasmus University Medical Center, Rotterdam, the Netherlands
6Department of Pathobiology, Faculty of Veterinary Medicine, Ferdowsi University Of Mashhad, Mashhad, Iran
7Department of Medical Genetics, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran
8Stem Cell Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
9Departmnt of Applied Cell Sciences, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences, Tabriz, Iran
Abstract
Introduction:
The inhibition of vascularization into tumor stroma as well as dynamic cell growth is the center of attention. Here, we aimed to examine the role of vandetanib on angiogenesis capacity of breast cancer stem cell (CSCs).
Methods:
MDA-MB-231 cells were exposed to different doses of vandetanib and survival rate was monitored. Stimulatory effects of vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and epidermal growth factor (EGF) were evaluated in vandetanib-treated MDA-MB-231 cells. In vitro tubulogenesis capacity was studied on the Matrigel surface. The synergistic effects of vandetanib on cell survival were also assessed after PI3K and/or Wnt3a inhibition. Vascular endothelial (VE)-cadherin, matrix metalloproteinase-2 (MMP-2), -9, Wnt3a, and p-Akt/Akt ratio were measured using western blotting.
Results:
Vandetanib reduced survival rate in a dose-dependent manner (P<0.05). Proliferative effects associated with VEGF, FGF, and EGF were blunted in these cells pre-exposed to vandetanib (P<0.05). The microcirculation pattern’s triple-negative breast cancer (TNBC) was suppressed by 1, 5 µM of vandetanib (P<0.05). Hence 1, 5 µM of vandetanib potentially decreased the population of CD24– cells. 1 and 5 µM of vandetanib inhibited cell proliferation by blocking PI3K and Wnt3a pathways and decreased the p-Akt/Akt ratio, Wnta3 protein levels (P<0.05). 1 and 5 µM vandetanib combined with PI3K inhibitor diminished metastatic markers including, MMP-2, and MMP-9. The concurrent treatment (PI3K, inhibitor+ 1, 5 µM vandetanib) also considerably reduced epithelial-mesenchymal transition (EMT) markers such as VE-cadherin (P<0.05).
Conclusion:
Vandetanib suppressed vasculogenic mimicry (VM) networking through blunting stemness properties, coincided with suppression of VE-cadherin in CSCs.
Keywords: Vandetanib, Vasculogenic mimicry, MDA-MB-231 cells, Wnt3a-PI3K signaling pathways
Copyright and License Information
© 2023 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Breast cancer is the most prevalent anaplastic changes in females with high-rate mortality.1,2 Patients with triple-negative breast cancer (TNBC) exhibit poor outcomes compared to other anaplastic changes.3 TNBC that has the most aggressive behavior, metastasizes to remote sites.4 Unfortunately, TNBC cells are resistant to commercially available chemotherapeutic drugs.4,5 The release of pro-angiogenesis cytokines like vascular endothelial growth factor (VEGF) helps tumor mass growth via the promotion of de novo vascular formation and hence facilitates tumor cell metastasis.6 Besides these properties, multiple studies have been conducted on the inhibition of the angiogenesis signaling pathway such as VEGFR via neutralizing antibodies.7-9 However, several recent studies have shown that the abortion of VEGF signaling could not completely affect the regression of all cancer types. For instance, upon the administration of sunitinib with a tyrosine kinase inhibitor activity, tumor regrowth, and metastasis limit its application in cancer patients.4 Although the suppression of angiogenesis could lead to a reduction of vascular density in tumor stroma from one point of view, the continuous lack of oxygen and nutrients causes special angiogenesis event so-called vasculogenic mimicry (VM).10,11 In 1999, Maniotis et al discovered VM phenomenon in melanoma cells.12 It is thought that VM development is promoted by the participation of tumor cells and their differentiation toward endothelial cells (ECs).4 of note, hepatocellular, prostate, inflammatory breast and ovarian carcinoma, glioblastoma, gastrointestinal stromal tumors exhibit VM channel formation.12-16 VM can determines the metastasis behavior of cancer cells. This structure consists of several types of cells such as mesenchymal stem cells, tumor-related macrophages, and other stem cell types.12 Cancer stem cell (CSCs) participate in the formation of VM units.4 CSCs exist within tumor parenchyma with distinct multipotentiality and self-renewal properties.17 CSCs could promote the formation of VM through an engaging mechanism entitled epithelial-mesenchymal transition (EMT) process.18-20 EMT is a dedifferentiation process in which epithelial marker mainly E-cadherin down-regulated coincides with expression of stemness marker vimentin, snail, Zeb along with vascular endothelial (VE)- cadherin.21
Thus, several mechanisms such as CSCs and EMT as well as various signaling pathways, including VE-cadherin, PI3K, Wnt3a, matrix metalloproteinases (MMPs), VEGFR1, focal adhesion kinase, cyclic adenosine monophosphate and hypoxia-inducible factor (HIF)-1α are involved in VM network formation.22,23 Therefore, VM has been touted as a potential alternative for tumor growth inhibition. Some researchers have examined the effect of several classical anti-angiogenic agents on VM networks inhibition, and finally inconsistent results were obtained. There are contradictory results on these drugs. For example, TNP-470 and Endostatin do not suppress VM network formation while sunitinib increases these structures, but Thalidomide, Tivantinib, Rapamycin, and Trastuzumab inhibits the VM network formation.10,19,21 Besides, the efficacy of other anti-angiogenic drugs like vandetanib on VM networking is unknown yet, so future studies are needed. Vandetanib possesses an inhibitory effect on receptors with serine-threonine and tyrosine kinases activity like VEGFR-2, and epidermal growth factor receptor (EGFR).24,25 Indeed, vandetanib has been proved to use in clinical trials against various types of cancers.24,25 Despite progress in the field of tumor biology and treatment, the potency of vandetanib on VM properties of human breast CSCs and its differentiation potential is not found so far. Whether vandetanib can alter VM of MDA-MB-231 CSCs remains to be elucidated. This study was conducted to examine anti-VM properties of vandetanib on MDA-MB-231 cells by focusing on the PI3K/Akt and Wnt/β-catenin axes.
Material and Methods
Chemicals and antibodies
Vandetanib (Cat no: 14706) was purchased from Cayman (USA). Vandetanib dissolved in dimethylsulfoxide (DMSO), and the final concentration of DMSO was below 0.1%. Cell culture medium (RPMI-1640), fetal bovine serum (FBS), and enzymatic solution (0.25% Trypsin-EDTA) were obtained from Gibco (USA). 3- [4, 5-dimeylthiazol-2-yl]-2, 5 diphenyl tetrazolium bromide (MTT, Cas No: 298-93-1), PI3K-specific inhibitor (Ly294002, CAS no: 934389-88-5), FGF-2 (Cat no: F0291) and epidermal growth factor (EGF) (Cat no: SRP3027) were purchased from Sigma-Aldrich (USA). Wnt3a -specific inhibitor (LGK974, Cat no: 14072) was obtained from Cayman (USA). VEGF (Cat no: C64423) was obtained from Promocell (USA). Growth factor free-Matrigel® (Cat no: 356230) were purchased from Corning. FITC-conjugated CD24 antibody (Order no: 130-095-952) was from Miltenyi Biotec (Germany). Primary antibodies against β-actin (Cat no: sc-47778), VE-cadherin (Cat no: sc-52751), phospho-Akt (Dilution: 1:100; Cat no: sc-271966), MMP-2 (Cat no: sc-10736), MMP-9 (Cat no: sc-393859), Wnt-3a (Cat no: sc-74537), and HRP conjugated anti-rabbit secondary antibody (Cat no: sc-2357) were provided from Santa Cruz Biotechnology Inc. (USA). Primary antibody against Akt (Cat no: E-AB-30471) was obtained from Elabscience Biotechnology Inc. (USA).
Cell culture protocol
MDA-MB-231 cells were obtained from Pasture Institute (Iranian Cell Bank, Tehran) and expanded using RPMI-1640 culture medium with FBS (10% v/v) and Pen-Strep (1% v/v) under standard condition [37°C, 95% relative humidity, 5% CO2]. Passages from 3 to 6 were used for several analyses.
MTT assay
The spectrophotometric measurement of mitochondrial dehydrogenase activity was done in MDA-MB-231 cells using MTT assay. Briefly, 104 CSCs were plated in each well of a 96-well plates and exposed to various concentrations of vandetanib (0.195, 0.390, 0.781, 1.562, 3.125. 6.25, 12.5, 25, 50, and 100 µM) for 48 hours. After that, 30 μL MTT solution [5 mg/mL] was poured onto each well, and plates kept at 37˚C for 4 hours. Then, supernatant was discarded and 100 μL DMSO added onto each well. Using a microplate reader (Hiperion MPR4+, Germany), samples were read at 570 nm with a reference wavelength of 630 nm. The experiments were conducted in triplicate and cell viability was expressed as % relative to control group.
Furthermore, we investigated the combined effect of two inhibitors [PI3K inhibitor (Ly294002; 10 µM), and Wnt inhibitor (LGK974; 10 µM)] with vandetanib on CSC survival rate.26,27 The similar procedure was done as above-mentioned. After 24-hour incubation time, supernatant was discarded and cells were exposed to 5 µM vandetanib for the next 48 hours.
To assess whether the addition of vandetanib to the culture medium could blunt the effects of VEGF, EGF, and EGF, we measured cell viability after being exposed to vandetanib plus growth factors. For this purpose, cells were pre-treated with vandetanib (1 and/or 5 µM) for 48 hours followed by incubation in a culture medium containing 10 ng/mL of each growth factor for 24 hours. In this study, the final concentration of solvent was below 1%.
Tubulogenesis assay
The inhibitory effect of vandetanib on CSCs VM potential was examined using in vitro tube formation assay.28 Each well of a 96-well plates was filled 50 µL pre-cooled Matrigel and maintained at 37˚C for 2 hours until gelation. Thereafter, 2.5 × 104 cells pretreated with vandetanib (1 and 5 µM) were incubated in 200 μL culture medium and placed on the Matrigel surface and kept under a conventional culture system. During 8-24 hours, the formation of tubular units was monitored and photographs were taken using an inverted microscope (Optika IM3, Italy). The total number, length, and intersection of tubules were measured using ImageJ software (Version 1.52a).
Stemness feature
To assess the impact of vandetanib on CSC stemness, flow cytometric analysis was done for the evaluation of cell membrane CD24. Cells pre-treated with 1 and 5 µM vandetanib were washed with PBS. Then, 100 μL cell suspension containing 106 cells was prepared and FITC-conjugated CD24 antibody (1 µg/mL) was added. The samples were kept at 4˚C for 30 minutes and washed with PBS. CD24+ cells were detected using the BD FACSCalibur system and FlowJo software (ver.7.6.1).
Western blotting
Pellets of vandetanib-treated MDA-MB-231cells were lysed in a protein extraction buffer containing protease cocktail inhibitor. Lysates were centrifuged at 12000×g for 20 minutes at 4˚C. Protein concentration was calculated using Bradford assay. About 10 µg/mL proteins were electrophoresed with 10% SDS-PAGE gels and transferred to the PVDF membrane. The procedure was continued with the blocking of membranes in 2% non-fat milk for 75 minutes at RT. Then, membranes were incubated with primary antibodies as follows: β-actin (dilution: 1:300), VE-cadherin (dilution: 1:200), Akt (dilution: 1:1000), p-Akt (dilution: 1:100), MMP2 (dilution: 1:200), MMP9 (dilution: 1:100), Wnt-3a (dilution: 1:100). The membranes were maintained at 4°C overnight. After three-time PBS washes (each for 10 minutes), an appropriate HRP-conjugated secondary antibody (1:1000) was used. The immunoblots were detected with an ECL reagent and the intensity of immunoblots calculated using ImageJ software (Ver 1.4, NIH).
Statistical analysis
GraphPad Prism [ver. 8] was applied for comparison between groups using One-Way ANOVA with Tukey post hoc analysis. Values below P< 0.05 was considered statistically significant.
Results
Vandetanib diminished cells viability
MDA-MB-231 CSCs were incubated with different doses of vandetanib ranging from 0.195 to 100 µM for 48 hours (Fig. 1A). Data revealed the inhibitory effect and dose-dependent activity of vandetanib on human breast CSCs. The inhibitory impact was seen in the group that received 3.125 µM vandetanib but these changes did not yield significant differences (P> 0.05). Vandetanib at higher doses, more than 6.250 µM, decreased prominently the survival rate of MDA-MB-231 cells (P< 0.001; Fig. 1A). These data exhibited a dose- and time-dependent activity of vandetanib on MDA-MB-231 CSCs (Fig. 1A). Consistent with the previous data and our results, we selected 1 and 5 µM vandetanib for subsequent analyses.29,30 Along with these data, we monitored morphological adaptation of MDA-MB-231 CSCs after being exposed to vandetanib. Bright-field imaging indicated that MDA-MB-231 CSCs acquired a rounded morphology after treatment with vandetanib, showing the reduction of adherence to the plastic surface (Fig. 1B).
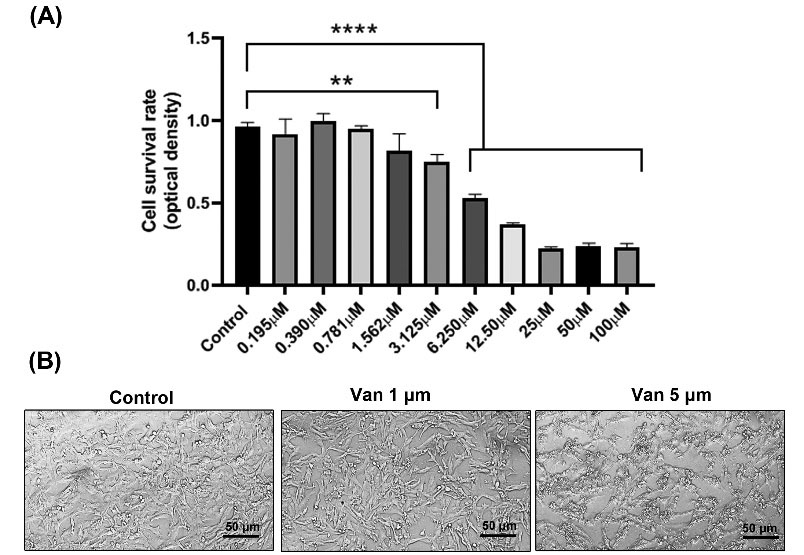
Fig. 1.
Dose-dependent growth inhibitory effects of Vandetanib on triple-negative breast cancer cell lines. MDA-MB-231 cells were treated with Consecutive concentrations of vandetanib and incubated for 48 h, and MTT assays were performed (A). Phase-contrast microscopy indicated morphological changes after treatment with vandetanib (B). **P < 0.01, ****P < 0.0001 represent level of significance with respect to control.
.
Dose-dependent growth inhibitory effects of Vandetanib on triple-negative breast cancer cell lines. MDA-MB-231 cells were treated with Consecutive concentrations of vandetanib and incubated for 48 h, and MTT assays were performed (A). Phase-contrast microscopy indicated morphological changes after treatment with vandetanib (B). **P < 0.01, ****P < 0.0001 represent level of significance with respect to control.
Vandetanib inhibited VM capacity
MDA-MB-231 CSCs pretreated with 1 and 5 µg/mL vandetanib were placed on Matrigel substrate to assess VM capacity (Fig. 2A-B). In this study, data showed that the control MDA-MB-231 CSCs were aligned and juxtaposed to form vessel-like structures with a relatively thick wall. Of note, incubation of MDA-MB-231 CSCs with 1 and 5 μM vandetanib aborted VM properties. According to our obtained data, all parameters like tube area, number, and perimeter, were markedly blunted (P< 0.05; Fig. 2A-B). In the vandetanib-treated groups, the MDA-MB-231 CSCs lost cell-to-cell connection which is important for vasculogenic activity and finally exhibited focal cell microaggregates. Considering our data, we noted the dose-dependent activity of vandetanib to inhibit VM capacity in breast CSCs after 48 hours, showing the anti-vasculogenic activity of vandetanib MDA-MB-231 CSCs.
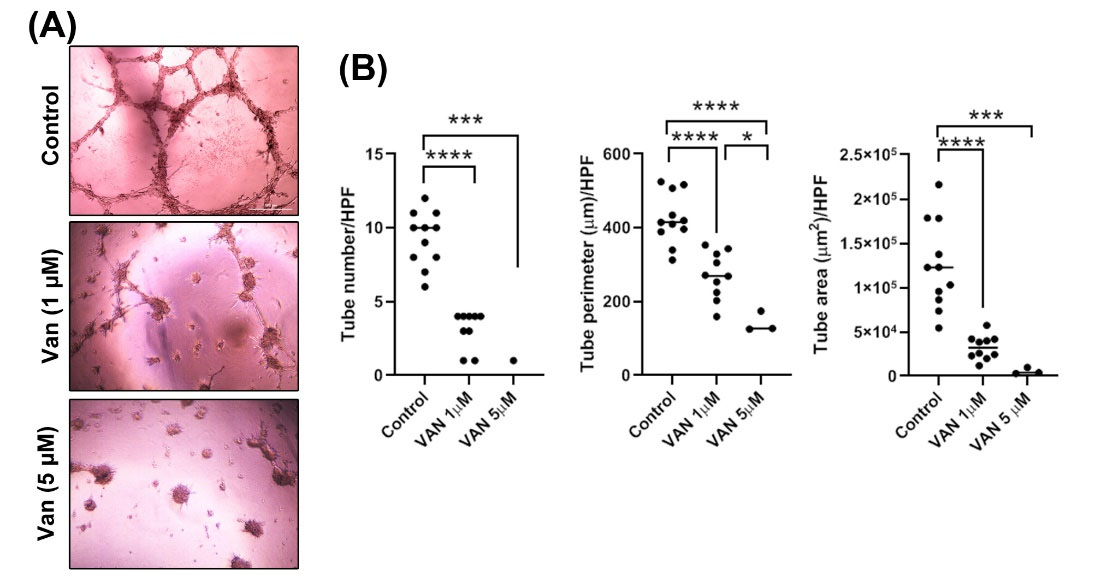
Fig. 2.
The effect of 1, 5 µM vandetanib on VM network formation by breast cancer cells on Matrigel. Phase-contrast microscopy showed that MDA-MB-231 cells did not form VM channels when was treated with1, 5 µM vandetanib (A). Tube number, tube perimeter, and tube area evaluated using ImageJ software (B). *P<0.05, ***P<0.001 and ****P<0.0001 represent the level of significance concerning control.
.
The effect of 1, 5 µM vandetanib on VM network formation by breast cancer cells on Matrigel. Phase-contrast microscopy showed that MDA-MB-231 cells did not form VM channels when was treated with1, 5 µM vandetanib (A). Tube number, tube perimeter, and tube area evaluated using ImageJ software (B). *P<0.05, ***P<0.001 and ****P<0.0001 represent the level of significance concerning control.
Vandetanib diminished stemness feature properties
The multipotentiality was analyzed by the density of the surface marker CD24 (Fig. 3). Data showed the significant increase of CD24 in the MDA-MB-231 CSCs from 28.8 ± 1.95% (control levels) to 98.1 ± 1.05% and 99.1 ± 0.62% in the 1 and 5 µM vandetanib groups, respectively (P< 0.05; Fig.3). Non-significant differences were achieved regarding the number of CD24+ cells in groups received 1 and 5 µM vandetanib (Fig.3). Vandetanib can reduce viability and stemness feature of MDA-MB-231 CSCs.
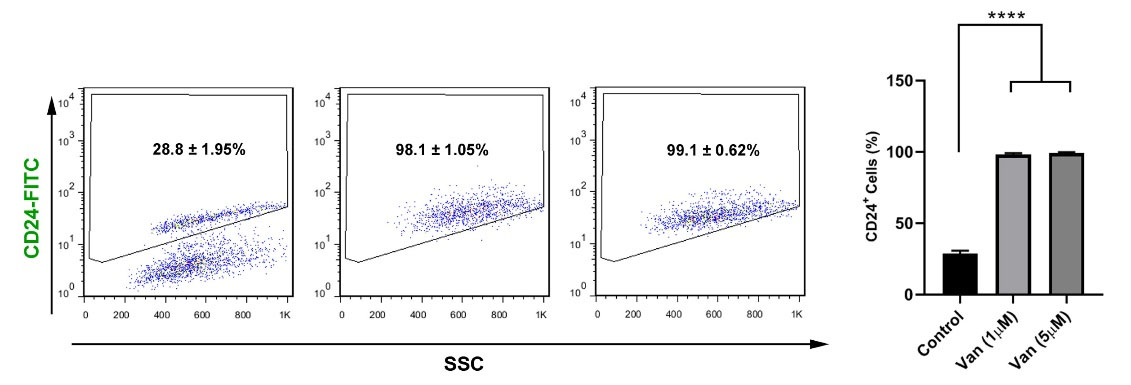
Fig. 3.
Representative flow cytometric analysis of the CD24-populations of MDA-MB-231 cells after treatment with 1, 5 µM vandetanib. The increase in the CD24- population showed after treatment with 1, 5 µM vandetanib. ***P < 0.001; ****P < 0.0001 represent level of significance with respect to control.
.
Representative flow cytometric analysis of the CD24-populations of MDA-MB-231 cells after treatment with 1, 5 µM vandetanib. The increase in the CD24- population showed after treatment with 1, 5 µM vandetanib. ***P < 0.001; ****P < 0.0001 represent level of significance with respect to control.
Vandetanib exhibited synergy with PI3K, Wnt 3a inhibitors to reduce MDA-MB-231 CSCs viability
MTT assay was done to compare the inhibitory effect of vandetanib on MDA-MB-231 CSCs survival rate in comparison with the suppression of PI3K, and Wnt 3a (Fig. 4). We noted that the inhibition of PI3K did not reduce the viability of CSCs compared to the control group (P> 0.05). The incubation of CSCs with 1 and 5 µM vandetanib reduced significantly survival rate as compared to the control CSCs. These effects were more evident in group received 5 µM vandetanib. According to our data, the combination of vandetanib at a higher concentration (5 µM) with PI3K inhibitor decreased the survival rate related to cells exposed to only 10 µM PI3K inhibitor (P< 0.0001; Fig. 4). Non-significant difference was achieved between the groups given PI3K inhibitor + 1 µM vandetanib and PI3K inhibitor alone (Fig. 4). Again, inhibition of Wnt3a caused non-significant changes in the viability of CSCs after 48 hours (P> 0.05; Fig. 4). Compared to the Wnt3a inhibitor group, the combination of Wnt3a inhibitor and 5 µM vandetanib reduced significantly the survival rate (P< 0.0001). We found non-significant differences in the percent of viable cells between groups 5 µM vandetanib and Wnt3a inhibitor. These data showed the superiority of vandetanib at higher concentrations to decrease the viability of breast CSCs compared to the Wnt3a and PI3K inhibitors.
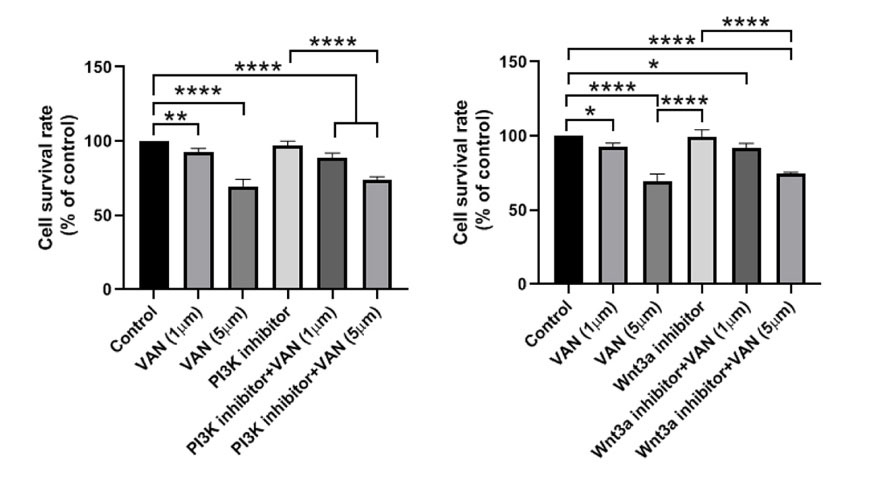
Fig. 4.
Representative figure of MTT assay of MDA-MB-231 cells treated with 1, 5 µM vandetanib and/or PI3Kand Wnt3a inhibitors along with control. Higher dosage of vandetanib (5 µM) combined with inhibitors significantly decreased cell proliferation as compared to the control group. The significant differences were shown as the mean ± SD as follows *P < 0.05, **P < 0.01, and ****P < 0.0001.
.
Representative figure of MTT assay of MDA-MB-231 cells treated with 1, 5 µM vandetanib and/or PI3Kand Wnt3a inhibitors along with control. Higher dosage of vandetanib (5 µM) combined with inhibitors significantly decreased cell proliferation as compared to the control group. The significant differences were shown as the mean ± SD as follows *P < 0.05, **P < 0.01, and ****P < 0.0001.
Vandetanib blunted stimulatory effect of growth factors
We monitored the stimulatory impact of VEGF, EGF, and FGF on the CSCs pre-treated with 1, and 5 µM vandetanib using MTT assay. Data demonstrated an enhanced survival rate in groups that received 10 ng/mL growth factors (P< 0.05; Fig. 5). Treatment of CSCs with 1, and 5 µM vandetanib blunted VEGF, EGF, and FGF effects in comparison with matched control groups. Data showed the superiority of 5 µM vandetanib to inhibit the stimulatory effect of VEGF, FGF, and EGF compared to the 1 µM vandetanib group (Fig. 5). These data highlight the modulatory effect of vandetanib to reduce CSCs sensitivity to VEGF, FGF, and EGF.
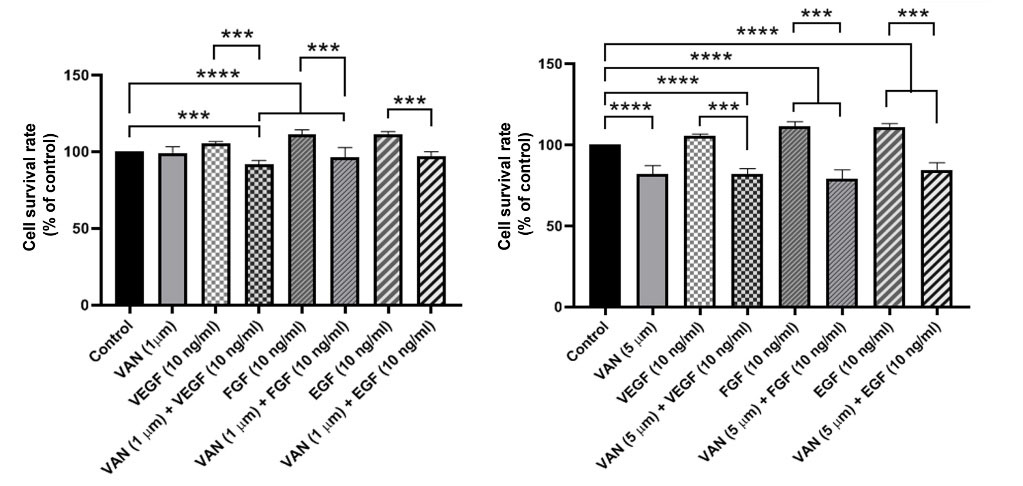
Fig. 5.
The inhibitory effect of 1, 5 µM vandetanib in exposure to growth factors such as VEGF, EGF, and FGF was examined through MTT assay Data revealed that non of the growth factors could not increase the cell proliferation in MDA-MB-231 cells treated with 1, 5 µM vandetanib. Results are expressed as mean ± SD. ***P < 0.001 and ****P < 0.0001.
.
The inhibitory effect of 1, 5 µM vandetanib in exposure to growth factors such as VEGF, EGF, and FGF was examined through MTT assay Data revealed that non of the growth factors could not increase the cell proliferation in MDA-MB-231 cells treated with 1, 5 µM vandetanib. Results are expressed as mean ± SD. ***P < 0.001 and ****P < 0.0001.
Vandetanib decreased endothelial differentiation and migration capacity
Anti-angiogenic and anti-migratory properties of vandetanib on MDA-MB-231 CSCs were assed using western blotting (Fig. 6). Our data showed that vandetanib alone (1 and 5 µM) could not significantly decrease Wnt3a. Co-treatment of cells with Wnt3a inhibitor and vandetanib, either 1 or 5 µM blunted the expression of Wnt3a compared to the Wnt3a inhibitor alone (Fig. 6). Incubation of MDA-MB-231 CSCs with PI3K inhibitor, but not Wnt3a inhibitor, suppressed VE-cadherin synthesis (Fig. 6). The combination of vandetanib and Wnt3a and PI3K inhibitors suppressed VE-cadherin related to control group. Non-significant differences were found between the protein levels of VE-cadherin in vandetanib + Wnt3a and vandetanib + PI3K inhibitors, and the groups received inhibitors only. The combination of vandetanib with inhibitors significantly suppressed the p-Akt/Akt ratio compared to the control cells (P< 0.05). Again, non-significant differences were achieved between the groups that received vandetanib + inhibitors and those receiving inhibitors only. Moreover, the results show that CSCs treated with 1 and 5 µM vandetanib had reduced MMP-2, but not MMP-9 (P< 0.01; Fig. 6). PI3K inhibitor plus vandetanib suppressed MMP-2 and -9 in CSCs. Of note, we found a significant reduction of MMP-9 in the CSCs treated with the combination of Wnt3a inhibitor and vandetanib. Vandetanib could regulate the angiogenic and metastatic behavior of breast CSCs and these effects were intensified along with Wnt3a and PI3K inhibitors.
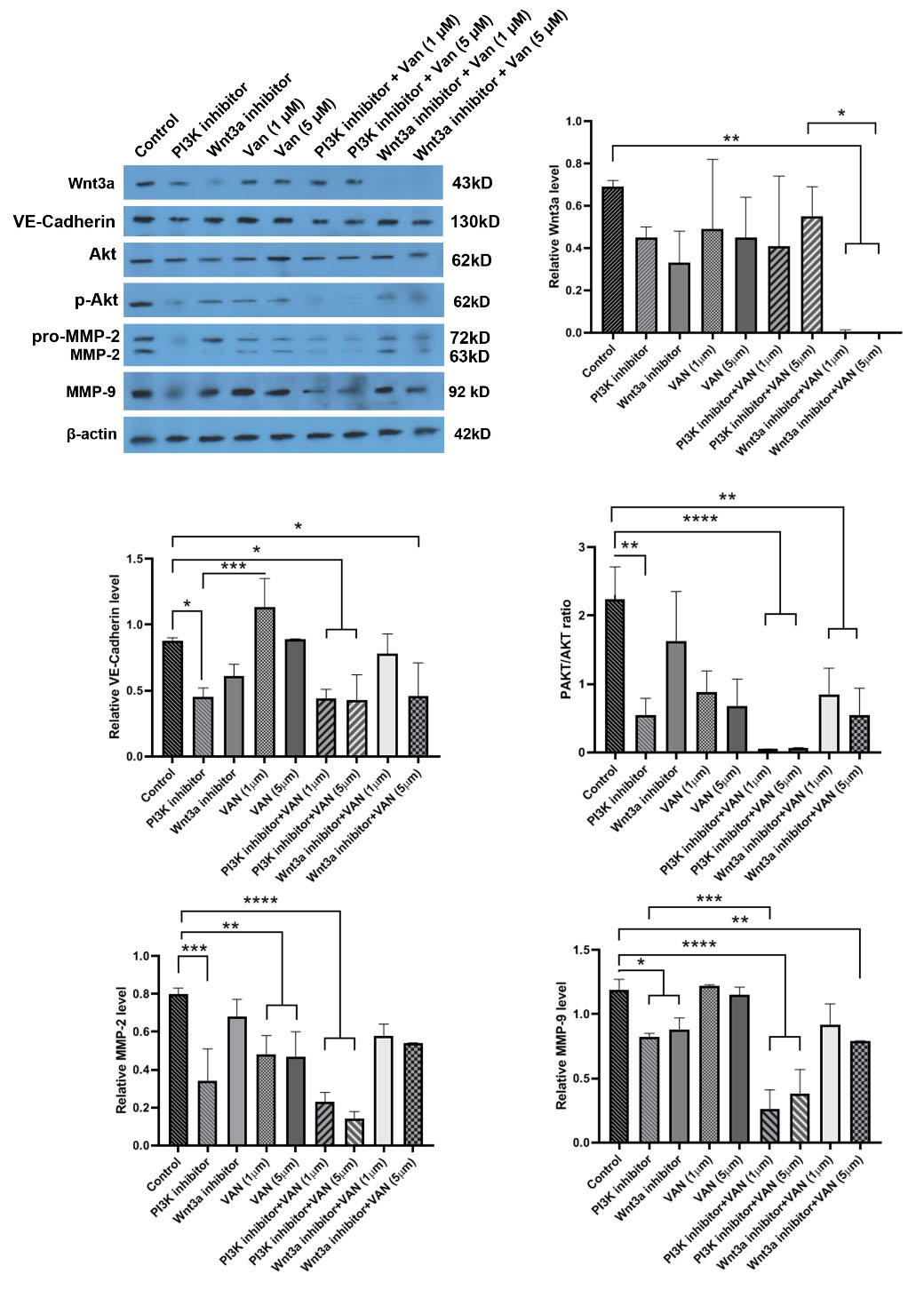
Fig. 6.
Western blot analyses of VM associated markers by human MDA-MB-231 cells. Western blot results and the densitometry analysis revealed that 1, 5 µM vandetanib combined inhibitors could decrease the protein levels of VE-cadherin, MMP-2, MMP-9, Wnt3a, and p-Akt/Akt ratio with a dose-dependent effect. The significant differences were shown as the mean ± SD as follows *P < 0.05, **P < 0.01; ***P < 0.001 and ****P < 0.0001.
.
Western blot analyses of VM associated markers by human MDA-MB-231 cells. Western blot results and the densitometry analysis revealed that 1, 5 µM vandetanib combined inhibitors could decrease the protein levels of VE-cadherin, MMP-2, MMP-9, Wnt3a, and p-Akt/Akt ratio with a dose-dependent effect. The significant differences were shown as the mean ± SD as follows *P < 0.05, **P < 0.01; ***P < 0.001 and ****P < 0.0001.
Discussion
The term VM stands for the formation of a vascular-like network by the progenitor cells residing in the most aggressive and metastatic tumor types.10 High mortality rates and poor outcomes mostly occur in patients with VM behavior.31 Previously Zhang and colleagues indicated a close association between VM and TNBC cells advertised as CSCs.4 It has been thought that CSCs possess VM capacity via being trans-differentiated into endothelial-like cells in hepatocellular carcinomas, glioblastomas, and breast cancers, which consequently leads to strong tumorigenicity and increased resistance to medication.32,33
Vandetanib, as a commercially available tyrosine kinase inhibitor, is administrated orally.24,25 Various signaling pathways related to tumor growth, progression, and angiogenesis have been studied after the administration of vandetanib for different cancer types. However, the effect of vandetanib in association with the angiogenic behavior of CSCs has been neglected, termed here as VM capacity.24 Herein, the effect of vandetanib on the VM capacity of breast TNBC cells was investigated. As a result, we found that vandetanib exerts dose-dependent cytotoxic effects on human breast CSCs after 48 hours.
In the present study, we found that the decreased CSC viability concurred with multipotentiality removal (an increase of surface CD24) after being exposed to vandetanib. The treatment of breast CSCs with vandetanib decreased the vasculogenic capacity of MDA-MB-231 CSCs. To achieve the vasculogenic activity, the expression of specific endothelial adhesion molecules like VE-Cadherin is critical for VM network development via aligning ECs.15,22,34-36 Additionally, PI3K/Akt and Wnt3a/β-catenin axes are considered as the main pathways in CSC, EMT, and VM networks formation processes.18,19,21 Along with these comments, in the current study, we also monitored VE-Cadherin, MMP-2, -9, Wnt3a, and p-Akt/Akt ratio. As a result, we found an efficient suppression of the Wnt3a factor in the group co-treated with vandetanib with Wnt3a inhibitor, showing the synergistic effect of vandetanib with a chemical inhibitor on blocking the Wnt3a signaling pathway. Vandetanib alone reduced the level of MMP-2, but not that of MMP-9, and the combination of vandetanib with PI3K and Wnt3a blockers caused a higher inhibitory effect on MMPs synthesis. The data showed that the migration of human breast CSCs decreased by the treatment with vandetanib and chemical blockers. Vandetanib alone could stop the synthesis of distinct MMP types. To achieve better therapeutic effects, a simultaneous combination of vandetanib with other blockers is mandatory. According to our obtained data, vandetanib did not decrease VE-Cadherin alone while the combined regime of vandetanib and PI3K and Wnt3a blockers diminished the synthesis of VE-cadherin. Calling attention, in vitro tubulogenesis assay showed that vandetanib prevented the human breast CSCs VM tube formation capacity on the Matrigel surface.
This study revealed that co-treatment of CSCs with vandetanib and PI3K and Wnt3a inhibitors can diminish VE-cadherin and differentiation into ECs. Both PI3K and Wnt3a signaling pathways are the main mechanisms involved in the anti-vasculogenic property of vandetanib. Several cascades such as PI3K/Akt/MMPs and Wnt3a/β-catenin are important in tumor growth and progression via the regulation of EMT and vascular network formation.37 Previous data have confirmed that the existence of EMT cancer cells can lead to VM.38 It is touted that EMT is a dedifferentiation process in which epithelial marker mainly E-cadherin is down-regulated coincides with expression of stemness marker vimentin, snail, Zeb.21 To assess whether vandetanib in combination with PI3K and Wnt3a inhibitors can decrease angiogenic potential, VE-cadherin, an endothelial lineage factor, were monitored. Vandetanib plus PI3K and/or Wnt3a inhibitors diminished CSCs differentiation toward cells expressing VE-cadherin. It has been shown that VE-cadherin can stabilize the intercellular junctions between ECs and promote VM capacity.37 Furthermore, the critical association of VE-cadherin with PI3K/Akt signaling pathway has been known in erythropoietin-producing hepatocellular carcinoma-A2.39 The suppression PI3K/Akt axis can result in pleiotropic effects such as inhibition of MMPs as reported here.40
According to our obtained data, we found that pre-treatment of cells with vandetanib blunted normal activity of cytokines like VEGF, EGF, and FGF on CSCs. These data show that vandetanib inhibited the vasculogenic capacity of CSCs possibly via the inhibition of VEGF, EGF, and FGF signaling pathways and regulation of VE-Cadherin activity. Again, p-Akt/Akt ratio was not altered by vandetanib while the combination of vandetanib and inhibitors decreased the phosphorylation of Akt.
VEGF, EGF, and FGF are proangiogenic factors that stimulated cancer cell proliferation, neoangiogenesis, and tumor progression.41 It has been elucidated that some anti-angiogenic agents such as thalidomide and rapamycin inhibited VM networking by targeting VEGF.42,43 Besides, according to studies, VEGF could modulate the invasion and metastasis in different malignant tumors by targeting the EMT process.44 Mandal et al showed that vandetanib inhibited EGF-induced cell proliferation in breast cancer cell lines.25
Conclusion
Vandetanib suppressed the CSCs' VM networks formation and metastasis via the inhibition of VEGF, FGF, and EGF signaling pathways. This drug has the potential to decrease the stemness feature of breast CSCs. Therefore, it seems that a combination of PI3K and Wnt3a inhibitors could increase the tumoricidal effect of vandetanib.
Research Highlights
What is the current knowledge?
√ Anti-angiogenic effect of vandetanib was studied on breast CSCs.
What is new here?
√ Vandetanib can suppress the angiogenic capacity of breast CSCs.
√ Both PI3K and Wnt signaling pathways are affected in cells exposed to vandetanib.
√ Vandetanib decreased angiogenic differentiation of CSCs into endothelial lineage.
Acknowledgments
We special thanks to the Research Center of Infectious Diseases and Tropical Medicine for their assistance in the progression of my thesis.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Ethical statement
All procedures of this study were approved by the Local Ethics Committee of Tabriz University of Medical Sciences (59575).
Funding
This study financially was supported by the Stem Cell and Regenerative Medicine Institute (SCARM) and Research Vice-chancellor of Tabriz University of Medical Sciences, Tabriz, Iran (Grant Ph.D. thesis no. 59575).
Competing interests
The authors declare that they have no competing interests.
References
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65:87-108. doi: 10.3322/caac.21262 [Crossref] [ Google Scholar]
- Zamani ARN, Mashayekhi MR, Jadid MFS, Faridvand Y, Tajalli H, Rahbarghazi R. Photo-modulation of zinc phthalocyanine-treated breast cancer cell line ZR-75-1 inhibited the normal tumor activity in vitro. Lasers Med Sci 2018; 33:1969-78. doi: 10.1007/s10103-018-2563-0 [Crossref] [ Google Scholar]
- Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res 2007; 13:2329-34. doi: 10.1158/1078-0432.CCR-06-1109 [Crossref] [ Google Scholar]
- Zhang D, Sun B, Zhao X, Ma Y, Ji R, Gu Q. Twist1 expression induced by sunitinib accelerates tumor cell vasculogenic mimicry by increasing the population of CD133+ cells in triple-negative breast cancer. Mol Cancer 2014; 13:207. doi: 10.1186/1476-4598-13-207 [Crossref] [ Google Scholar]
- Dianat-Moghadam H, Heidarifard M, Jahanban-Esfahlan R, Panahi Y, Hamishehkar H, Pouremamali F. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J Control Release 2018; 288:62-83. doi: 10.1016/j.jconrel.2018.08.043 [Crossref] [ Google Scholar]
- Dong F, Ruan S, Wang J, Xia Y, Le K, Xiao X. M2 macrophage-induced lncRNA PCAT6 facilitates tumorigenesis and angiogenesis of triple-negative breast cancer through modulation of VEGFR2. Cell Death Dis 2020; 11:1-14. [ Google Scholar]
- Cao Y, Langer R. Optimizing the delivery of cancer drugs that block angiogenesis. Sci Transl Med 2010; 2:15ps3. doi: 10.1126/scitranslmed.3000399 [Crossref] [ Google Scholar]
- Cao Y, Zhong W, Sun Y. Improvement of antiangiogenic cancer therapy by understanding the mechanisms of angiogenic factor interplay and drug resistance. Semin Cancer Biol 2009; 19:338-43. doi: 10.1016/j.semcancer.2009.05.001 [Crossref] [ Google Scholar]
- Gampenrieder SP, Westphal T, Greil R. Antiangiogenic therapy in breast cancer. Memo 2017; 10:194-201. doi: 10.1007/s12254-017-0362-0 [Crossref] [ Google Scholar]
- Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res 2012; 18:2726-32. doi: 10.1158/1078-0432.CCR-11-3237 [Crossref] [ Google Scholar]
- Paulis YW, Soetekouw PM, Verheul HM, Tjan-Heijnen VC, Griffioen AW. Signalling pathways in vasculogenic mimicry. BiochimBiophys Acta 2010; 1806:18-28. doi: 10.1016/j.bbcan.2010.01.001 [Crossref] [ Google Scholar]
- Haiaty S, Rashidi MR, Akbarzadeh M, Maroufi NF, Yousefi B, Nouri M. Targeting vasculogenic mimicry by phytochemicals: A potential opportunity for cancer therapy. IUBMB Life 2020; 72:825-41. doi: 10.1002/iub.2233 [Crossref] [ Google Scholar]
- Liu R, Yang K, Meng C, Zhang Z, Xu Y. Vasculogenic mimicry is a marker of poor prognosis in prostate cancer. Cancer Biol Ther 2012; 13:527-33. doi: 10.4161/cbt.19602 [Crossref] [ Google Scholar]
- Shirakawa K, Kobayashi H, Sobajima J, Hashimoto D, Shimizu A, Wakasugi H. Inflammatory breast cancer: vasculogenic mimicry and its hemodynamics of an inflammatory breast cancer xenograft model. Breast Cancer Res 2003; 5:136-9. doi: 10.1186/bcr585 [Crossref] [ Google Scholar]
- Sun T, Zhao N, Zhao XL, Gu Q, Zhang SW, Che N. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry. Hepatology 2010; 51:545-56. doi: 10.1002/hep.23311 [Crossref] [ Google Scholar]
- Wang JY, Sun T, Zhao XL, Zhang SW, Zhang DF, Gu Q. Functional significance of VEGF-a in human ovarian carcinoma: role in vasculogenic mimicry. Cancer Biol Ther 2008; 7:758-66. [ Google Scholar]
- Fathi F, Rezabakhsh A, Rahbarghazi R, Rashidi MR. Early-stage detection of VE-cadherin during endothelial differentiation of human mesenchymal stem cells using SPR biosensor. BiosensBioelectron 2017; 96:358-66. doi: 10.1016/j.bios.2017.05.018 [Crossref] [ Google Scholar]
- Liu Q, Qiao L, Liang N, Xie J, Zhang J, Deng G. The relationship between vasculogenic mimicry and epithelial-mesenchymal transitions. J Cell Mol Med 2016; 20:1761-9. doi: 10.1111/jcmm.12851 [Crossref] [ Google Scholar]
- Qiao L, Liang N, Zhang J, Xie J, Liu F, Xu D. Advanced research on vasculogenic mimicry in cancer. J Cell Mol Med 2015; 19:315-26. doi: 10.1111/jcmm.12496 [Crossref] [ Google Scholar]
- Pouyafar A, Zadi Heydarabad M, Aghdam SB, Khaksar M, Azimi A, Rahbarghazi R. Resveratrol potentially increased the tumoricidal effect of doxorubicin on SKOV3 cancer stem cells in vitro. J Cell Biochem 2019; 120:8430-7. [ Google Scholar]
- Luo Q, Wang J, Zhao W, Peng Z, Liu X, Li B. Vasculogenic mimicry in carcinogenesis and clinical applications. J Hematol Oncol 2020; 13:19. doi: 10.1186/s13045-020-00858-6 [Crossref] [ Google Scholar]
- Sun B, Zhang D, Zhao N, Zhao X. Epithelial-to-endothelial transition and cancer stem cells: two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget 2017; 8:30502-10. doi: 10.18632/oncotarget.8461 [Crossref] [ Google Scholar]
- Wang M, Zhao X, Zhu D, Liu T, Liang X, Liu F. HIF-1α promoted vasculogenic mimicry formation in hepatocellular carcinoma through LOXL2 up-regulation in hypoxic tumor microenvironment. J Exp Clin Cancer Res 2017; 36:60. doi: 10.1186/s13046-017-0533-1 [Crossref] [ Google Scholar]
- Morabito A, Piccirillo MC, Falasconi F, De Feo G, Del Giudice A, Bryce J. Vandetanib (ZD6474), a dual inhibitor of vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases: current status and future directions. Oncologist 2009; 14:378-90. doi: 10.1634/theoncologist.2008-0261 [Crossref] [ Google Scholar]
- Sarkar S, Mazumdar A, Dash R, Sarkar D, Fisher PB, Mandal M. ZD6474, a dual tyrosine kinase inhibitor of EGFR and VEGFR-2, inhibits MAPK/ERK and AKT/PI3-K and induces apoptosis in breast cancer cells. Cancer Biol Ther 2010; 9:592-603. doi: 10.4161/cbt.9.8.11103 [Crossref] [ Google Scholar]
- Fraveto A, Cardinale V, Bragazzi MC, Giuliante F, De Rose AM, Grazi GL. Sensitivity of Human Intrahepatic Cholangiocarcinoma Subtypes to Chemotherapeutics and Molecular Targeted Agents: A Study on Primary Cell Cultures. PLoS One 2015; 10:e0142124. doi: 10.1371/journal.pone.0142124 [Crossref] [ Google Scholar]
- Iskender B, Izgi K, Canatan H. Novel anti-cancer agent myrtucommulone-A and thymoquinone abrogate epithelial-mesenchymal transition in cancer cells mainly through the inhibition of PI3K/AKT signalling axis. Mol Cell Biochem 2016; 416:71-84. doi: 10.1007/s11010-016-2697-y [Crossref] [ Google Scholar]
- Rezabakhsh A, Montazersaheb S, Nabat E, Hassanpour M, Montaseri A, Malekinejad H. Effect of hydroxychloroquine on oxidative/nitrosative status and angiogenesis in endothelial cells under high glucose condition. Bioimpacts 2017; 7:219-26. doi: 10.15171/bi.2017.26 [Crossref] [ Google Scholar]
- Addison CL, Pond GR, Cochrane B, Zhao H, Chia SK, Levine MN. Correlation of baseline biomarkers with clinical outcomes and response to fulvestrant with vandetanib or placebo in patients with bone predominant metastatic breast cancer: An OCOG ZAMBONEY sub-study. J Bone Oncol 2015; 4:47-53. doi: 10.1016/j.jbo.2015.04.001 [Crossref] [ Google Scholar]
- Sarkar S, Mazumdar A, Dash R, Sarkar D, Fisher PB, Mandal M. ZD6474 enhances paclitaxel antiproliferative and apoptotic effects in breast carcinoma cells. J Cell Physiol 2011; 226:375-84. doi: 10.1002/jcp.22343 [Crossref] [ Google Scholar]
- Sun BC, Zhang SW, Zhao XL, Hao XS. [Study on vasculogenic mimicry in malignant melanoma]. Zhonghua Bing Li Xue Za Zhi 2003; 32:539-43. [ Google Scholar]
- Liu TJ, Sun BC, Zhao XL, Zhao XM, Sun T, Gu Q. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013; 32:544-53. doi: 10.1038/onc.2012.85 [Crossref] [ Google Scholar]
- Yao X, Ping Y, Liu Y, Chen K, Yoshimura T, Liu M. Vascular endothelial growth factor receptor 2 (VEGFR-2) plays a key role in vasculogenic mimicry formation, neovascularization and tumor initiation by Glioma stem-like cells. PLoS One 2013; 8:e57188. doi: 10.1371/journal.pone.0057188 [Crossref] [ Google Scholar]
- Maniotis AJ, Chen X, Garcia C, DeChristopher PJ, Wu D, Pe'er J. Control of melanoma morphogenesis, endothelial survival, and perfusion by extracellular matrix. Lab Invest 2002; 82:1031-43. doi: 10.1097/01.lab.0000024362.12721.67 [Crossref] [ Google Scholar]
- Ma JL, Han SX, Zhu Q, Zhao J, Zhang D, Wang L. Role of Twist in vasculogenic mimicry formation in hypoxic hepatocellular carcinoma cells in vitro. BiochemBiophys Res Commun 2011; 408:686-91. doi: 10.1016/j.bbrc.2011.04.089 [Crossref] [ Google Scholar]
- Zhu QQ, Ma C, Wang Q, Song Y, Lv T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumour Biol 2016; 37:185-97. doi: 10.1007/s13277-015-4450-7 [Crossref] [ Google Scholar]
- Rezaei M, Martins Cavaco AC, Stehling M, Nottebaum A, Brockhaus K, Caliandro MF, et al. Extracellular Vesicle Transfer from Endothelial Cells Drives VE-Cadherin Expression in Breast Cancer Cells, Thereby Causing Heterotypic Cell Contacts. Cancers (Basel) 2020; 12. 10.3390/cancers12082138.
- Shuai Q, Cao L, Qin Z, Zhang Y, Gu Z, Yang J. VE-cadherin fusion protein substrate enhanced the vasculogenic mimicry capability of hepatocellular carcinoma cells. J Mater Chem B 2020; 8:1699-712. doi: 10.1039/c9tb02790d [Crossref] [ Google Scholar]
- Haiaty S, Rashidi MR, Akbarzadeh M, Bazmani A, Mostafazadeh M, Nikanfar S. Thymoquinone inhibited vasculogenic capacity and promoted mesenchymal-epithelial transition of human breast cancer stem cells. BMC Complement Med Ther 2021; 21:83. doi: 10.1186/s12906-021-03246-w [Crossref] [ Google Scholar]
- Lu XS, Sun W, Ge CY, Zhang WZ, Fan YZ. Contribution of the PI3K/MMPs/Ln-5gamma2 and EphA2/FAK/Paxillin signaling pathways to tumor growth and vasculogenic mimicry of gallbladder carcinomas. Int J Oncol 2013; 42:2103-15. doi: 10.3892/ijo.2013.1897 [Crossref] [ Google Scholar]
- Mostofa AGM, Hossain MK, Basak D, Bin Sayeed MS. Thymoquinone as a Potential Adjuvant Therapy for Cancer Treatment: Evidence from Preclinical Studies. Front Pharmacol 2017; 8:295. doi: 10.3389/fphar.2017.00295 [Crossref] [ Google Scholar]
- Su M, Feng YJ, Yao LQ, Cheng MJ, Xu CJ, Huang Y. Plasticity of ovarian cancer cell SKOV3ip and vasculogenic mimicry in vivo. Int J Gynecol Cancer 2008; 18:476-86. doi: 10.1111/j.1525-1438.2007.01034.x [Crossref] [ Google Scholar]
- Zhang S, Li M, Gu Y, Liu Z, Xu S, Cui Y. Thalidomide influences growth and vasculogenic mimicry channel formation in melanoma. J Exp Clin Cancer Res 2008; 27:60. doi: 10.1186/1756-9966-27-60 [Crossref] [ Google Scholar]
- Yan JD, Liu Y, Zhang ZY, Liu GY, Xu JH, Liu LY. Expression and prognostic significance of VEGFR-2 in breast cancer. Pathol Res Pract 2015; 211:539-43. doi: 10.1016/j.prp.2015.04.003 [Crossref] [ Google Scholar]