Bioimpacts. 2025;15:26443.
doi: 10.34172/bi.2023.26443
Original Article
Mosaic vaccine design targeting mutational spike protein of SAR-SCoV-2: An immunoinformatics approach
Ysrafil Ysrafil Conceptualization, Funding acquisition, Methodology, Software, Writing – original draft, Writing – review & editing, 1, * 
Arlan K. Imran Data curation, Visualization, Writing – original draft, Writing – review & editing, 2
Prisca Syafriani Wicita Formal analysis, Writing – original draft, Writing – review & editing, 2
Vyani Kamba Project administration, Writing – original draft, Writing – review & editing, 2
Fihrina Mohamad Project administration, Writing – original draft, Writing – review & editing, 2
Ismail Ismail Resources, Writing – original draft, Writing – review & editing, 3
Ayyub Harly Nurung Validation, Writing – original draft, Writing – review & editing, 4
Noviyanty Indjar Gama Resources, Writing – original draft, Writing – review & editing, 5
Sari Eka Pratiwi Formal analysis, Writing – original draft, Writing – review & editing, 6
Indwiani Astuti Data curation, Writing – original draft, Writing – review & editing, 7
Firzan Nainu Conceptualization, Funding acquisition, Validation, Writing – original draft, Writing – review & editing, 8
Talha Bin Emran Investigation, Methodology, Software, Supervision, Writing – original draft, Writing – review & editing, 9
Author information:
1Department of Pharmacotherapy, Faculty of Medicine, Universitas Palangka Raya, Jekan Raya, Palangka Raya, Central Kalimantan 73111, Indonesia
2Department of Pharmacy, Health Polytechnic of Gorontalo, Kota Timur, Gorontalo, Gorontalo 96135, Indonesia
3Study Program of Pharmacy, Universitas Almarisah Madani, Makassar, Sulawesi Selatan 90245, Indonesia.
4Faculty of Pharmacy, Universitas Muslim Indonesia, Panakkukang, Makassar, South Sulawesi 90231, Indonesia
5Department of Clinical Pharmacy, Faculty of Pharmacy, University of Mulawarman, Samarinda Ulu, Samarinda, East Kalimantan 75242, Indonesia
6Department of Biology and Pathobiology Faculty of Medicine, Universitas Tanjungpura, Pontianak Tenggara, Pontianak, West Kalimantan 78124, Indonesia
7Department of Pharmacology and Therapy, Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada, Daerah Istimewa Yogyakarta 55281, Indonesia
8Department of Pharmacy, Faculty of Pharmacy, Hasanuddin University, Makassar 90245, Indonesia
9Department of Pharmacy, BGC Trust University Bangladesh, Chittagong 4381, Bangladesh
Abstract
Introduction:
Presently, the development of effective vaccines against SARS-CoV-2 is absolutely necessary, especially regarding the emergence of new variants that cause increasing morbidity and fatalities.
Methods:
In the present study we designed a mosaic vaccine targeting the mutational spike protein of COVID-19 using a bioinformatics approach. Various immunoinformatics tools were utilized to provide the highest potential for a mosaic vaccine that could activate immune responses against COVID-19.
Results:
The evaluation of the constructed vaccine revealed that it is antigenic and immunogenic as well as nonallergenic. The physicochemical properties also show promising characteristics, including being highly stable and hydrophilic. As expected, the vaccine shows strong interactions with several important receptors including angiotensin-converting enzyme 2 (ACE2), Toll-like receptor 3 (TLR3) and TLR8 by the lowest energy level, docking score and binding free energy. The vaccine binds to receptors via certain amino acids using various types of binding including salt bridges, hydrogen bonds, and other means. As shown in computationally derived models, the interactions promote activation of the immune response by eliciting the release of various cytokines, antibodies, memory B and T cells, as well as increasing of natural killer cell and dendrite cell counts.
Conclusion:
Therefore, the novel designed mosaic vaccine could be considered as a potential vaccine candidate for immediate production to stem the continuing and tragic effects of the COVID-19 pandemic. However, several advanced experimental studies should be conducted to ensure and verify the effectivity and safety against SARS‑CoV‑2 in vivo.
Keywords: SARS-CoV-2, Immunoinformatics, Mutation, Spike protein, Virus variant
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
Not applicable.
Introduction
The emergence of the COVID-19, which started in Wuhan, China at the end of 2019 has become a widespread global health emergency. The pandemic disease is caused by the SARS-CoV-2.1,2 As of October 13, 2023, there have been about 700 million people diagnosed with the disease and nearly 7 million deaths worldwide.3 The infected patients generally present with symptoms including a cold, cough, fever, shortness of breath, and loss of function of smell. In some patients who are treated intensively in the hospital, generally they also exhibit other comorbid conditions such as respiratory, cardiovascular, endocrine, anorexia, and gastrointestinal diseases.4-6 Several strategies have been applied to treat and break the chain of transmission of the novel Coronavirus. Development of effective vaccines using the important viral peptides that aim to induce immunological memory and production of neutralizing antibodies has become the highlighted and highly hoped for preventive measure.7
Phenotypically, SARS-CoV-2 is a positive sense RNA virus (non-segmented virus) that contains spike proteins which form homotrimers in its surface.2,8 This protein has 1,273 amino acids and specific receptor binding domains (RBD), that are the main means for viral entry into human cells with angiotensin-converting enzyme 2 (ACE2), making it the most targeted protein in vaccine development.9 The emerging of several recent variants of the spike protein in SARS-CoV-2 has raised the latest concerns for the development of more effective vaccines. Importantly, these current variants of concern include the alpha, beta, delta, and later omicron variants which were reported around the world. The mutations are located at specific regions of the spike protein including RBD, furin, and other cleavage sites that can affect sensitization of antibodies against the new variant.10,11 Some studies demonstrated that the emerging of spike mutations reduced the sensitivity of neutralizing antibodies against the full set of spike mutations.13,14 In this context, the development of an effective vaccine against these variants is direly needed.15
Advancements in bioinformatics have provided various facilities in the development of effective vaccines using computerized simulations. The approach known as immunoinformatics allows researchers to design a mosaic vaccine by combining multiple epitope-rich viral protein fragments of a virus using bioinformatics tools. The epitopes selected for construction are those that have good antigenicity, and could mediate cellular and humoral immune responses (both B-cell and T-cell epitopes), as well as lacking any side effects (non-allergenic and non-toxic).16-21
A vaccination with both B-cell and T-cell epitopes can effectively mediate humoral and cellular immune responses against a particular pathogen. The antigen will be presented by the major histocompatibility complex (MHC) class I and II of the antigen-presenting cells (APCs) to the B-cell and T-cell receptors. Accordingly, it is also essential to understand how epitopes interact with the MHC.18,22 Moreover, a vaccine's affinity for immunological receptors (TLR3 and 8) is required for successful vaccination protein transport into the APCs. TLR3 has been shown to induce a robust type I interferon-gamma (IFN-γ) dependent antiviral response in mice infected with the Coronavirus, while TLR8 exhibits antiviral inhibitory functions.23 Accordingly, in this study, we designed vaccines from mutational spike protein of SARS-CoV-2 using the immunoinformatics approach. The vaccines were further able to bind to ACE2, TLR 3 and 8 of host cells as they function in entering pathways used by the pathogen to induce the immunologic responses.
Methods
Retrieval of spike protein, antigenicity assessment and physicochemical characterization of protein selection
Spike proteins were retrieved from the National Center for Biotechnology Information (NCBI) in FASTA format. The spikes contain the current spreading mutations (variants of concern) including T19R, L18F, T20N, P26S, D138Y, G142D, 156del, 157del, R158G, R190S, K417N/T, L452R, T478K, E484K, N501Y, D614G, H655Y, P681R, D950N, T1027I, and V1176F. In the initial assessment, each mutational spike protein was evaluated for its antigenicity using VaxiJen v2.0 web server with a threshold value of 0.40.
Cytotoxic T lymphocytes epitopes prediction
Prediction of cytotoxic T lymphocytes (CTL) epitopes was conducted by using the Immune Epitopes Database (IEDB).20 The prediction method was modified from the previously published immunoinformatics guide by Ullah et al.17 CTL epitopes were predicted by the NetMHCpan EL 4.0 prediction method and set-up with 9 amino acids in length. The human leukocyte antigen (HLA) references were set in 14 library alleles including:
HLA-A*02:01, HLA-A*02:06, HLA-A*03:01,
HLA-A*11:01, HLA-A*23:01, HLA-A*24:02,
HLA-A*26:01, HLA-A*30:01, HLA-A*30:02,
HLA-A*31:01, HLA-A*32:01, HLA-A*33:03,
HLA-A*68:01, and HLA-A*68:02.17,24
Selected epitopes were continued for immunogenicity testing using the IEDB online analysis resource: http://tools.iedb.org/immunogenicity/. Epitopes showing positive score are considered to be immunogenic.
Helper T lymphocytes (HTL) epitopes prediction
Helper T lymphocyte (HTL) epitopes were predicted with the IEDB methods for prediction of HTL epitopes using the recommended 5 alleles including: HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*11:01 and HLA-DRB1*15:01 alleles with 15-mer in length. The epitopes for further analysis were selected based on a percentile rank that was lower than 2.
Allergenicity, antigenicity and toxicity prediction of epitopes
All epitope predictions were tested for their allergenicity, antigenicity and toxicity prediction. The allergenicity predictions were measured by online allergenicity prediction tools, AllergenFP v.1.0 and AllerTOP v.2.0 which are two predictors of allergenicity with good accuracy.25 Antigenicity predictions of all epitopes were done by VaxiJen v2.0 as the most reliable online predictor of antigenic tests.26 The measuring of antigenicity of epitopes was done on the tumor model with threshold 0.40.27 Meanwhile, the toxicity of all epitopes were analyzed by the ToxinPred (http://crdd.osdd.net/raghava/toxinpred/), with the default setting of the support-vector machine (SVM) (Swiss-Prot).28
Cytokine IFN-γ, IL-4 and IL-10 epitopes prediction
All predicted MHC-II epitopes were assessed for their possibility to induce IFN-γ, IL-4, and IL-10 releasing. To conduct the assessment, we used online IFN-γ, IL-4 and IL-10 prediction tools that were operated based on the SVM-based model with default value setting at the SVM threshold.17,18,29,30
Population coverage analysis of T-cell epitopes
Population coverage was used to measure the percentage of how, for a particular individual in a geographical area, the selected epitopes would stimulate the immune response, thereby indicating they are suitable for the multiple HLAs used in the prediction of the T-cell epitopes. The percentage of coverage in each country will certainly be different, because of the MHC polymorphism in each ethnicity and certain regions. This will also be an illustration of the response of the designed vaccine when used in the assessed areas. To assess this coverage, we used the online population coverage analysis tools from the IEDB (http://tools.iedb.org/population/).31
Vaccine construct
Constructions of vaccines from all selected best CTL, HTL, and B-cell lymphocyte (BCL) epitopes from mutation spike proteins were performed using modifications of previous immunoinformatics studies in vaccine design (template of construction is illustrated in Fig. 1).32 The epitopes were built with the following sequences: adjuvant (β-defensin), Pan HLA-DR reactive epitope (PADRE) sequence, CTL and HTL epitopes and some linkers including EAAAK to link the adjuvant and PADRE, AAY linkers for CTL epitopes, GPGPG linkers for HTL epitopes, and 6xHis-tag were added at the C terminal end of the vaccine construct.
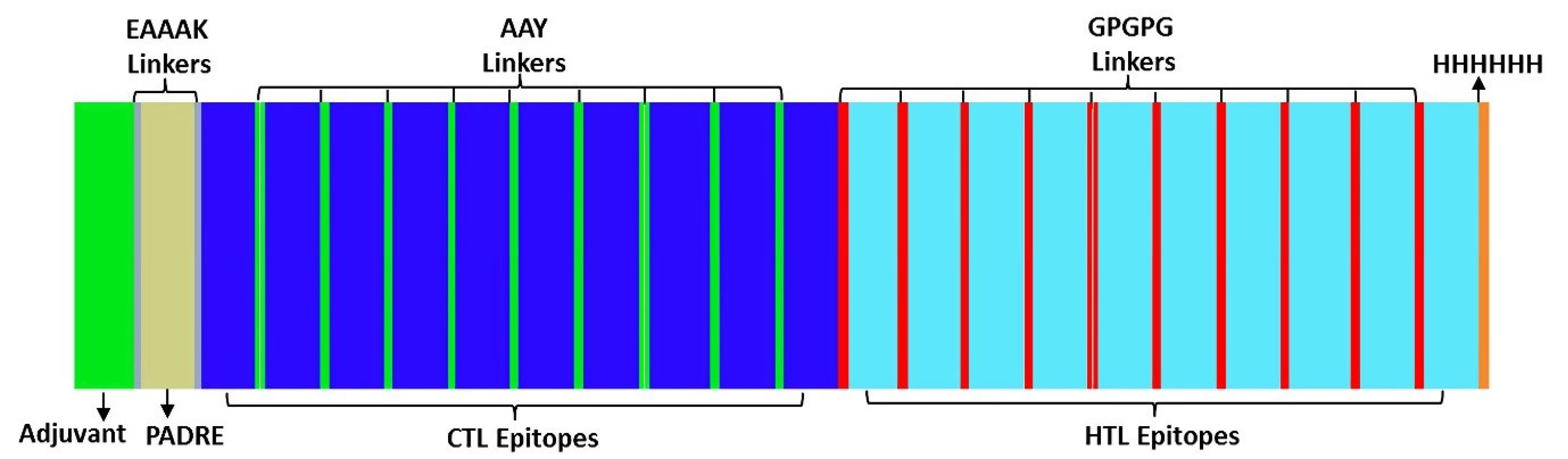
Fig. 1.
Schematic diagram of construction of mosaic vaccines.
.
Schematic diagram of construction of mosaic vaccines.
Evaluation of allergenicity, antigenicity and physicochemical characteristics of vaccine
Determination of allergenicity of the constructed vaccine was performed using VaxiJen v2.0 with threshold value 0.4 and tumor models. Meanwhile, to determine vaccine allergenicity and physicochemical properties of vaccines constructed using AllerTop v2.0, AllergenFP and ProtParam were selected, respectively.33 Furthermore, the solubility of the final construct was evaluated by the SOLpro server.34 Moreover, secondary structural predictions for the final vaccine protein sequences were conducted using the PSIPRED35 and SOPMA servers.36
Building of tertiary structure and refinement of vaccine construct
Vaccine constructs were further developed into their tertiary structures using an online 3D builder, trRosetta (https://yanglab.nankai.edu.cn/trRosetta/) that is known for fast and accurate protein structure prediction.37 Tertiary structures of the vaccines were then refined using the online refinement tools (e.g., GalaxyRefine).38 Furthermore, all tertiary vaccine constructs were visualized by PyMOL and USCF Chimera.
V alidation of vaccine construct
Final refined tertiary models of the vaccines were further verified by ERRAT, followed by structural validation by ProSA, and PROCHECK.39 The Ramachandran plot should result in over 90% residues in the most favored region to define it as a good quality model. Additionally, we also evaluated the structural flexibility of each vaccine using the CABS-Flex 2.0 server.40
Docking vaccine construct with Toll-like receptors (TLRs)
The constructed vaccines were docked with ACE2 (as the viral entry receptor) and TLRs that indicate the sensor pattern of viruses including Coronavirus to initiate immune responses. The PDB files of the protein included ACE2 (PDB ID: 1r42), TLR3 (PDB ID:1ziw), and TLR8 (PDB ID:3w3g) and were retrieved from the protein data bank, RCSB PDB. The molecular interaction predictions of the vaccine with the proteins were assessed by several online docking tools including ClusPro server, HDOCK, and HawkDock. All vaccine/TLR docking results were visualized by UCSF Chimera and PyMOL.41-43
Molecular dynamic and estimation of MM/GB-PBSA binding energy
The molecular docking results of the constructed vaccines were further analyzed for molecular dynamics and estimation of MM/GB-PBSA binding energy. The iMOD server (iMODS) (http://imods.chaconlab.org) was used for molecular dynamic analysis, while estimation of MM/GB-PBSA binding energy was performed by HawkDock (http://cadd.zju.edu.cn/hawkdock/).42,44,45
Codon optimization and in silico cloning
Codon optimization was conducted by Java Codon Adaptation Tool (JCat) for maximum expression of the vaccine. In this study we used the prokaryotic expression system Escherichia coli K-12 strain with selected time rho-independent transcription terminators, and prokaryotic ribosome binding sites. EcoRI and BamHI cleavage sites of restriction enzymes were avoided. The vaccine sequence was constructed in reverse to the DNA sequence. EcoRI and BamHI enzymes were selected for the N-terminal and C-terminal sites, respectively. Finally, the DNA sequences of the mosaic vaccine results were inserted into the pET-28a( + ) vector and further cloned by SnapGene tool.
Post translational modification (PTM) analysis
The post translational modifications (PTM) of the vaccine constructs were analyzed using MusiteDeep (https://www.musite.net/) as a deep-learning framework for protein post-translational modification site prediction. We used all of the prediction models in the tools to predict the possibility of PTM that could occur in the vaccine contructs.46
Immune simulation
Immune response simulations were conducted by C-IMMSIM server (https://kraken.iac.rm.cnr.it/C-IMMSIM/). In these simulations, we administrated three injections containing the constructed vaccine at 4-week intervals at 0, 28, and 56 days.
Results
Retrieval of spike protein
In the present study we designed vaccines from mutational spike proteins of SARS-CoV-2 which are included in the variants of concern that have recently emerged worldwide. This study was conducted in response to the imminent concerns over the impact of mutations in spike proteins. These mutations have caused increased transmissibility and antigenicity of the virus as well as contributing to the immune escape and evasion of the virus from antibodies formed against it.13,47 The spike proteins were retrieved from the NCBI database and followed antigenic evaluation to the selected spike by the VaxiJen server by following a procedure that was demonstrated in the literature.19 The results found that the protein is highly antigenic with the antigenic value > 0.4.
Cytotoxic T lymphocytes (CTL) epitopes prediction
CD8 + cytotoxic T lymphocyte (CTL) epitopes were predicted by screening the epitope which can bind to the major histocompatibility complex class I (MHC-I), which is a macromolecule that has the function of presenting foreign antigens to the CTL.48 The epitopes were tested with the 14 most common HLA Class I alleles including HLA-A*02:01, HLA-A*02:06, HLA-A*03:01, HLA-A*11:01, HLA-A*23:01, HLA-A*24:02, HLA-A*26:01, HLA-A*30:01, HLA-A*30:02, HLA-A*31:01, HLA-A*32:01, HLA-A*33:03, HLA-A*68:01, and HLA-A*68:02, which were used to generate potential epitopes with good binding affinities and characteristics.24 The obtained epitopes were further assessed for their antigenicity, immunogenicity, non-toxicity and non-allergenicity to generate the best epitopes that will be used in the construction of mosaic vaccines.19,32,49
From the 10 best epitopes identified, Table S1 shows one of the epitopes is located in RBD and specifically in RBM, namely 448NYNYLYRLF456, and another is in the furin protein namely 777NTQEVFAQV785. All of epitopes are putatively restricted to different HLA alleles. 1206QYIKWPWYI1214 attaches to 3 alleles: HLA-A*24:02, HLA-A*23:01, HLA-A*32:01, 775NTQEVFAQV783 can bind with 4 alleles: HLA-A*68:02, HLA-A*02:06, HLA-A*02:01, HLA-A*26:01, Meanwhile, 975SVLNDILSR983 attaches to 7 alleles: HLA-A*11:01, HLA-A*68:01, HLA-A*31:01, HLA-A*33:03, HLA-A*03:01, HLA-A*30:01, and HLA-A*26:01. The ability of the epitope to bind to several different HLA alleles will affect the higher population coverage of the epitope and vaccine.32
Helper T lymphocytes (HTL) epitopes prediction
The Helper T Lymphocytes (HTL) epitopes with 15-mer in length were predicted from the ability of foreign antigens to bind with MHC-II molecules that were tested with 5 different HLA Class II alleles including HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*11:01 and HLA-DRB1*15:01.24,50 Table S2 shows that the ten selected epitopes have good antigenicity, immunogenicity, non-toxicity and allergenicity profiles.
These HTL are generally recognized to be present in HTL epitopes in buds of MHC-II that are embedded on the surface of antigen-presenting cells (APC).18,51,52 Selected HTL epitopes were also tested for their ability to induce IFN-γ, IL-4, and IL-10 responses. As show in Table S2, most of HTL epitopes could induce the three cytokines tested. Induction of cytokines IFN-γ, IL-4 and IL-10 is important in the cascade of immune activation, such as the activation of CTL and other immune components in the body.53
Of the ten epitopes, three epitopes could induce all of the three cytokine responses, namely NLLLQYGSFCTQLNR, NTLVKQLSSNFGAIS, and LLLQYGSFCTQLNRA. The other epitopes could induce two of the cytokines, e.g., GNYNYLYRLFRKSNL (IFN-γ and IL-4), LQYGSFCTQLNRALT (IFN-γ and IL-10), and NTLVKQLSSNFGAIS (IL-4 and IL-10), as well as one type of cytokine, e.g. SFELLHAPATVCGPK (IL-4). This cytokine-inducing ability is important in the cellular and humoral immune system activation cascades. Several studies have claimed that IL-4 is involved in enhancing the immune response by inducing the development of naive CD + T cells to Th2.51,53 Meanwhile, IFN-γ has been reported to drive differentiation of CD + T cells to Th1 and Th2. IFN-γ is a type II IFN secreted by Natural Killer cells and T lymphocytes that play a role in the host defense mechanism against viruses through the activation of cellular and humoral immunity cells. In addition, the cytokines can also mediate CTL activity to increase virus killing activity.53,54
Population coverage of T-cell epitopes
Differences in the HLA alleles which are distributed in different ethnicities and regions affect the variety of vaccine effectiveness in different regions of the world. Therefore, the development of vaccines with broad-spectrum immunophenotypes or predictions of population coverage of constructed vaccines should be conducted to ensure suitable populations for coverage of certain vaccines.24,52,55 In the present study, we simulated the population coverage of our selected epitopes in several different geographical areas worldwide to show sufficient population that coverage of the selected epitopes. As shown in Fig. S1, the world average coverage of whole epitopes was 96.74%. Meanwhile, it appears that various ethnicities and regions in the world show different coverage percentages with North America (98.41%) becoming the highest coverage, followed by East Asia (98.07%), Europe (97.67%), South Asia (95.02%), West Indies (94.99%), and Southeast Asia (93.11%), that indicate the good qualities of the selected epitopes.
Lower population coverage occurs in Central Africa with only 15.11% coverage. This can be associated with the low level of this type of data analysis in that area which may affect the population coverage results. The difference in coverage of epitopes in various regions and ethnicities is similar to previous finding demonstrated by Kar et al in 2020,52 Qamar et al in 202056 and Chukwudozie et al in 2021.49
Construction of vaccine from selected epitopes
All selected peptides including CTL and HTL were further constructed to become mosaic vaccines. In the construction, we used several additional ingredients such as adjuvant and the pan and HLA-DR epitope peptide (PADRE sequence). These components are devoted to enhancing the response of the vaccines, antibody production, and to reduce the quantity of antigen input of the constructed vaccines while minimizing vaccine toxicity.57 We utilized β-defensin adjuvant as the most utilized in vaccine construction,48,56 that subsequently linked with PADRE and N-terminal of the mosaics; each of which is linked by EAAAK.58 In addition, several other linkers were used to link one epitope with another epitope including AAY and GPGPG for CTL and HTL linkers, respectively.58 The ends of the mosaic vaccine were tagged with 6 histidine (6xHis-tag) following the previously described procedure conducted by Yang et al in 2021,32 Bhattacharya et al. in 202,50 and Chukwudozie et al in 2021.49 The results of vaccine construction are listed in Table S3.
Evaluation of allergenicity, antigenicity and physicochemical characteristic of vaccine
The final constructed vaccines were checked for antigenicity by the Vaxijen 2.0 server59 that provided the score of 0.5396. With a threshold of 0.4, the score is higher and suggests that the vaccine has high antigenicity. The allergenicity profile was assessed using the AllergenFP 1.0 server and AllerTOP 2.0 webserver and showed that the vaccine is non-allergenic in nature. Furthermore, the physicochemical characteristics were evaluated by ExPASy ProtParam tools60 that provide information about molecular weight, isoelectrical point (pI), extension coefficient, thermostability (aliphatic index), instability index, and hydrophobicity value (GRAVY) of the constructed vaccine. The solubility of the constructed vaccine was also assessed using Solpro. This characterization is important in assessing the quality of the vaccine obtained.
As shown in the Protparam tool, the vaccine consists of 391 amino acids, molecular weight about 42.237 kDa and theoretical pI 9.54. The theoretical pI is the point at which the total liquid charge of an amino acid or protein molecule becomes zero based on a certain equilibrium point. Therefore, a value of 9.54 indicates that the protein is stable at pH 9.54.
The total negative (Asp + Glu) and positive (Arg + Lys) charges of the residue amino acids were 14 and 35, respectively. The total number of atoms was computed at 5,925 with chemical formula C1914H2939N531O529S12. Additional measuring at 280 nm obtained the extinction-coefficient of the vaccine of 66,990 M−1 cm−1 assuming all Cys residues were reduced. The half-life of the vaccine was estimated at 30 hours (in mammalian reticulocytes, in vitro), > 20 hours (in yeast, in vivo), and > 10 hours (in E. coli, in vivo). The stability index of protein indicated that the vaccine was stable with a score of 23.27. Meanwhile, two other calculated parameters were the aliphatic index and GRAVY, which were computed to be 85.19 and -0.131, respectively. The higher aliphatic index reflects the relative volume occupied by aliphatic amino acids in protein side chains, for example: alanine, valine, etc. This value also indicates that the vaccine protein is highly thermostable.61,62 Moreover, the GRAVY profiles depict that our vaccine is hydrophilic.17,61 The solubility result by SOLpro also indicates that the constructed vaccine has high solubility with score of 0.50.
Analysis of secondary structure of vaccine
α-Helices are the most abundant rod-like structure in proteins and are formed through the utilization of hydrogen bonds between the CO and NH groups of the main chain present in vaccine proteins. They play an important role in determining the overall structure and function of the proteins.63 Moreover, β-strands are part of the β-sheet of the protein. The β-sheets are formed by hydrogen bonds between the linear regions of the polypeptide molecular chain.64
In this study, secondary structures of the vaccine were elaborated by PSIPRED and SOPMA servers as shown in Figs. S2 and S3. In the PSIPRED results, of the 391 amino acids, 185 of them (47.1%) form alpha helixes, 31 (7.93%) amino acids were beta strands, 175 (44.76%) amino acids were coils. Meanwhile, as shown in the results, the SOPMA server results were 35.81% for alpha helixes, with extended stands 21.23%, β-turns 6.08%, and random coils 36.57%. These results indicate the good secondary structures of the vaccine.50 Another highlighted structure in this result is the β-turn, which is a structure formed by self-folding of two regions connected by hydrogen bonds.64
Three-dimensional structure building, refinement and validation
The tertiary structures of the three constructed vaccines were conducted by trRosetta which provides fast and accurate protein structure prediction. The 3D vaccine structures were further refined using the Galaxy refine server (Table S4). Of 5 refined structures, the fourth model was identified to have good quality characteristics and selected for further analysis (Fig. S4). According to the analysis, the Global Distance Test—High Accuracy (GDT-HA) score was computed higher at 0.9853 and indicated as the highest similarity. The root mean square deviation (RMSD) depicted the distance between atoms in the structure was 0.295 that confirms the selected model had a better stability. MolProbity of our chosen model was 1.664 which was lower than other models. These parameters depict crystallographic resolution of models that had lower critical errors.32 The Clash score and Poor rotamers showed results of 13.9 and 0.7. Furthermore, Rama favored regions were increased after refinement from the initial models from 96.9 to 97.9 and had an acceptable percentage of protein of 85%.
Another analysis to assess flexibility of vaccine was conducted by CABS-flex 2.0 server with 50 cycles and temperature range of 1.4˚C. The results shown in Fig. S5 depict the region near the C and N-terminals which are almost the same. The resultant contact-map reflects the less clear pattern of interactions between residues.56 Meanwhile, the Root Mean Square Fluctuation (RMSF) plot provides a fluctuation pattern of amino acids in the vaccine ranging 0.0 Å to 5 Å.
These results were also supported by validation using ERRAT and Ramachandran servers. As shown in Fig. 2, the refined model provides the high-quality factor of 97.6 as analyzed by the ERRAT server. The Ramachandran plot analysis reflected 94.3% of residues located in favored regions, 5.4% in allowed regions and 0.3 % in outer region. These results indicate the refined model has good quality.61
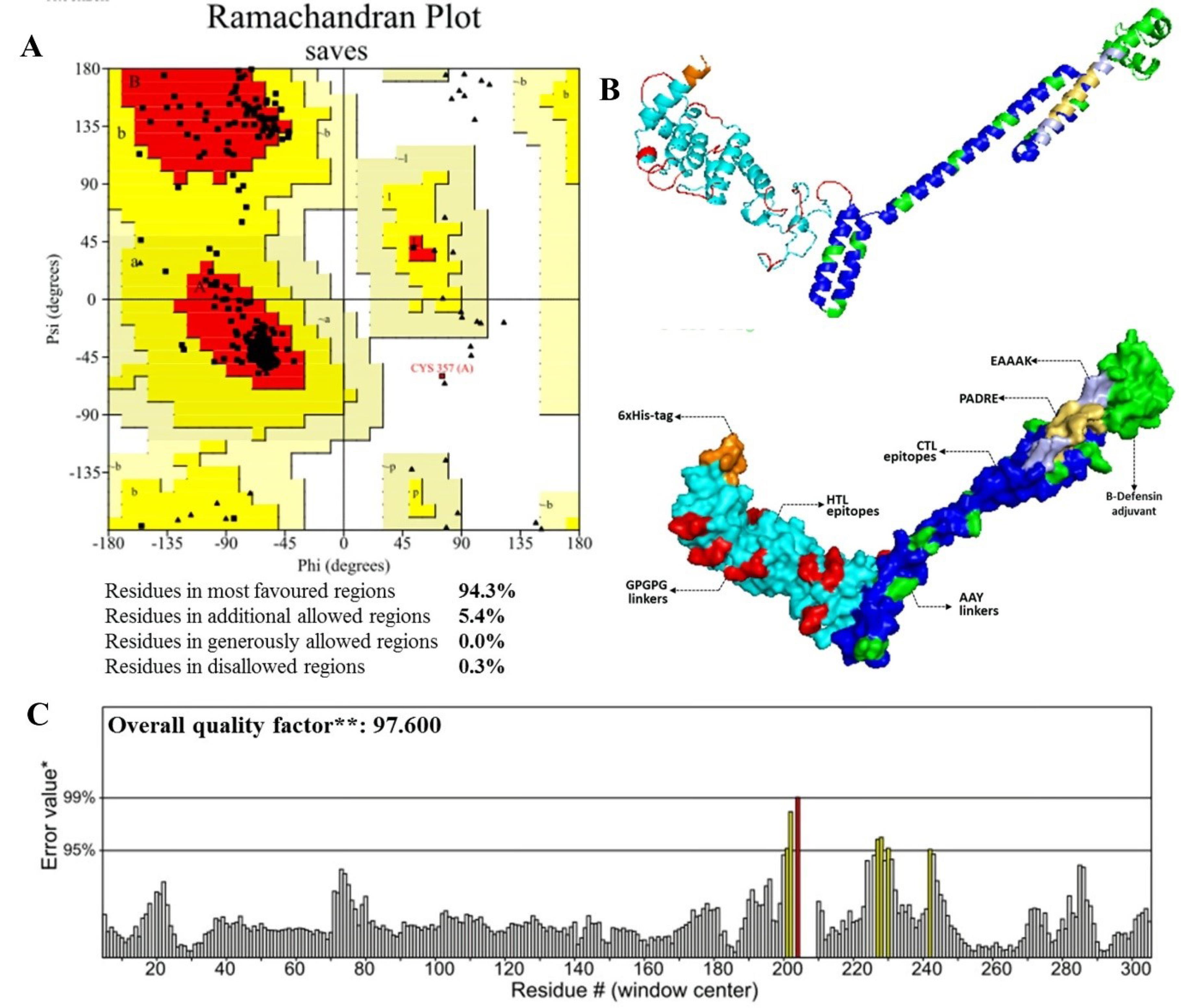
Fig. 2.
Results of vaccine validation. (A) Ramachandran plot of the vaccine (B) 3D structure of refined vaccine, and (C) Validation of the vaccine structure by ERRAT.
.
Results of vaccine validation. (A) Ramachandran plot of the vaccine (B) 3D structure of refined vaccine, and (C) Validation of the vaccine structure by ERRAT.
Additionally, we also conducted validation to confirm the overall quality of the selected models using the ProSA-web server. As show in Fig. 3, the vaccine reached Z-score of -6.33 that is located in the range close to native proteins with the comparable size and indicates the overall good quality model.32,61 A negative score indicates it is a good 3D protein model. We generated another local quality model using ProSA. This is represented by a significant plot, which indicated that the overall quality of the model was good. The results are closely similar with a previous study conducted by Yang et al in 2021.
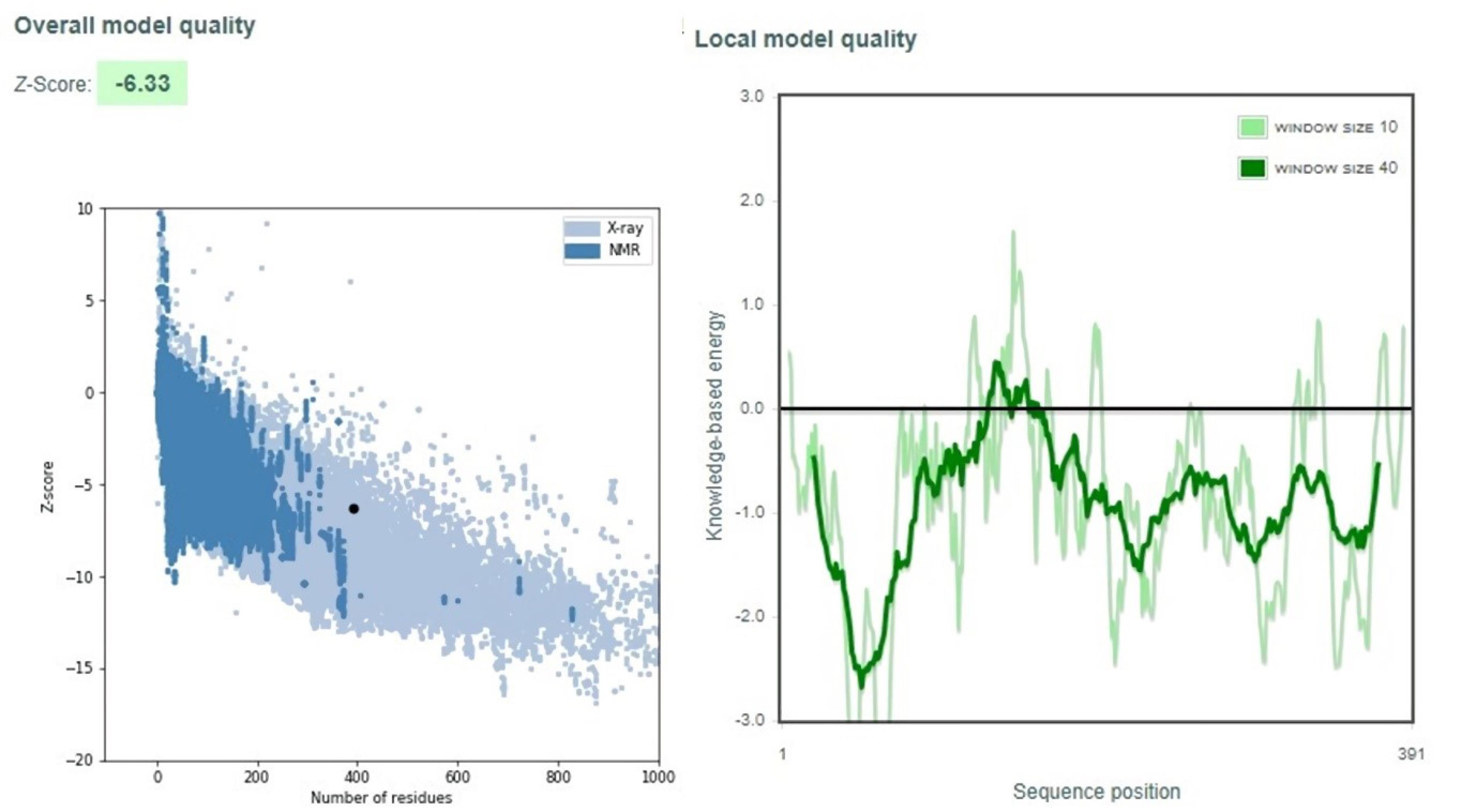
Fig. 3.
Validation of vaccine using ProSA.
.
Validation of vaccine using ProSA.
Docking of vaccine construct with ACE2 and TLRs
Exploring the interaction of the vaccine with ACE2
The 3D structures of the vaccine were exported in PDB format and further docked to certain important proteins related to the vaccine. ACE2 is identified as an important protein in SARS-CoV-2 infection and is an important target for vaccine development. We tried to assess the interaction and binding of the vaccine with ACE2 using several online docking tools including ClusPro, PatchDock, HDock and HawkDock. Overall, the results observed that the constructed vaccine can interact well with ACE2. This interaction reached Center and Lowest Energy of -1231.9 and -1231.9 in the ClusPro analysis (show in Table 1). Meanwhile, PatchDock analysis results for Global Energy, Attractive VdW, and Repulsive VdW were -39.19, -28.38, and 6.87, respectively, with the HDock results score and Ligand RMSD of -320.72 and 140.88 (Table 1), as well as the HawkDock results at -6096.45 in docking score (Table 2).
Table 1.
Results of molecular docking of vaccine with some receptors of ACE2, TLR3, and TLR8
|
Protein
|
ClusPro
|
PatchDock
|
HDock
|
|
Center
|
Lowest Energy
|
Global Energy
|
Attractive VdW
|
Repulsive VdW
|
ACE
|
HB
|
Docking Score
|
Ligand RMSD (Å)
|
| ACE2 |
-1231.9 |
-1231.9 |
-39.19 |
-28.38 |
6.87 |
1.64 |
-3.6 |
-320.72 |
140.88 |
| TLR3 |
-1069.6 |
-1069.6 |
-21.61 |
-24.70 |
10.10 |
10.69 |
-1.13 |
-365.43 |
101.02 |
| TLR8 |
-1205.6 |
-1411 |
-21.57 |
-40.83 |
49.07 |
-7.96 |
-0.97 |
-379.6 |
120.69 |
Note: ACE, angiotensin-converting enzyme; HB, hydrogen bond; RMSD, root mean square deviation; TLR, Toll-like receptor.
Table 2.
Molecular docking of vaccine with some protein and MM/GBSA binding free energy of complex Receptor-Vaccine
|
|
ACE2
|
TLR3
|
TLR8
|
| Docking Score |
-6096.45 |
-5607.81 |
-5031.26 |
| GBSA Score |
-91.25 kcal/mol |
-38.38 kcal/mol |
-62.91 kcal/mol |
|
Interaction
|
Rec
|
Lig
|
Rec
|
Lig
|
Rec
|
Lig
|
|
Residue
|
Score
|
Residue
|
Score
|
Residue
|
Score
|
Residue
|
Score
|
Residue
|
Score
|
Residue
|
Score
|
|
|
Asp-277 |
-8.01 |
Arg-14 |
13.03 |
Asp-488 |
3.31 |
Arg-265 |
8.73 |
Asp-478 |
7.4 |
Arg-42 |
-6.97 |
|
|
Ser-262 |
-6.17 |
Arg-38 |
-3.9 |
Asp-489 |
-2.77 |
Asn-304 |
-4.43 |
Pro-431 |
-4.84 |
Lys-45 |
-5 |
|
|
Asp-350 |
-4.76 |
Ile-30 |
-5.51 |
Phe-314 |
-2.75 |
Leu-119 |
-4.34 |
Thr-507 |
-3.81 |
Lys-39 |
-3.39 |
|
|
Asp-274 |
-4.01 |
Arg-42 |
-3 |
Pro-464 |
-2.14 |
Leu-123 |
-4.22 |
Asp-476 |
-3.07 |
Arg-38 |
-3.8 |
|
|
Gly-268 |
-3.71 |
Arg-36 |
-1.64 |
Phe-285 |
-2.08 |
Ile-122 |
-4.01 |
Asp-534 |
-2.51 |
Trp-57 |
-3.54 |
We further verified the possible amino acids responsible for the vaccine-receptor interactions and the type of binding that occurs using PDBsum. The results of this analysis are summarized in Fig. 4, and reveal that many of the amino acids of the two proteins involved in this interaction are using various types of bonds including: salt bridges, hydrogen bonds, as well as several other amino acids detected to non-bounded contacts.
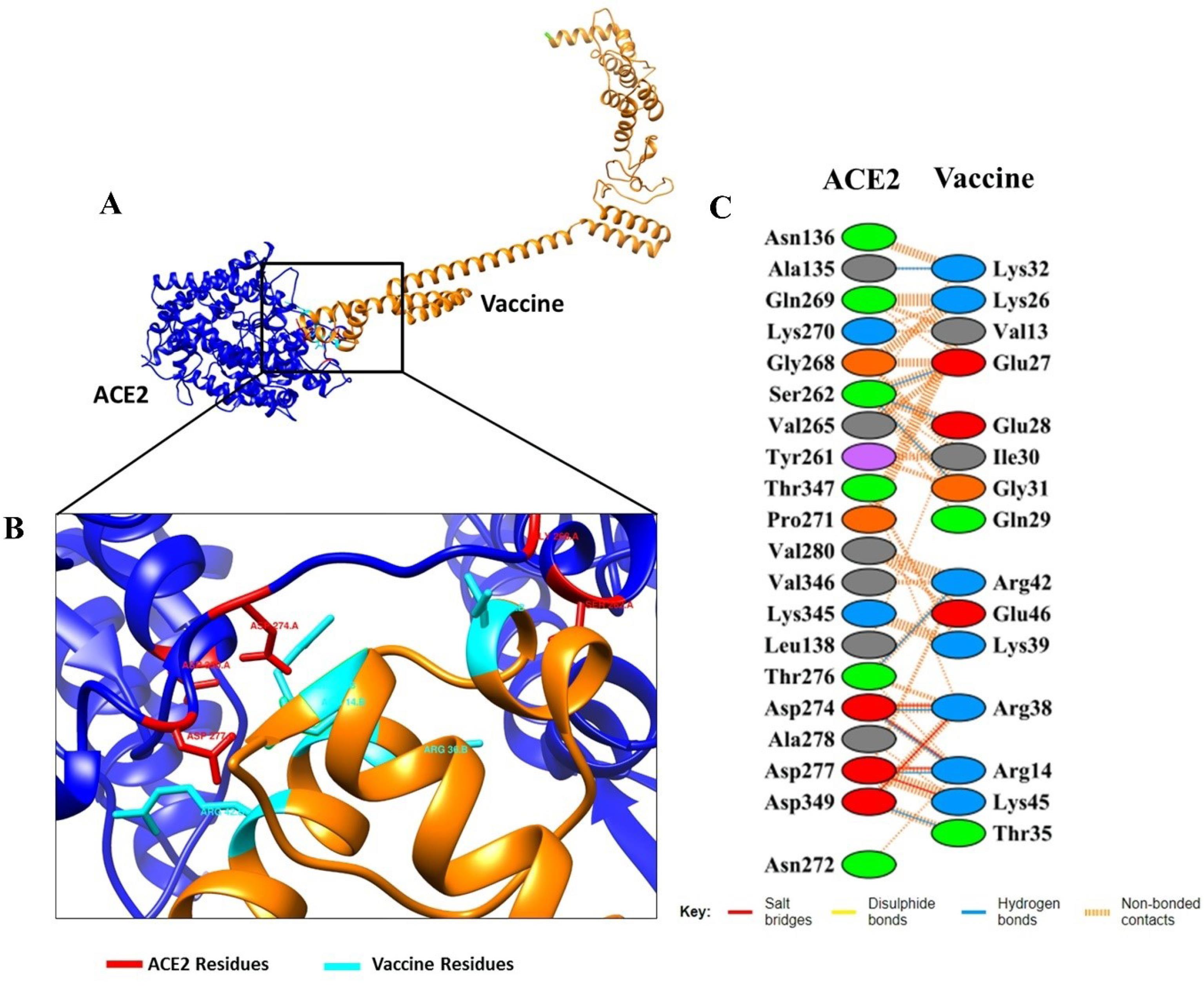
Fig. 4.
The docked complexes between vaccine with ACE2. (A) Animated representative of complex vaccine (orange) and ACE2 (blue), (B) Interacting residues of vaccine and ACE2, Residues in cyan color are vaccine residues and the red are ACE2’s, and (C) All interacting residues of vaccine-ACE2
.
The docked complexes between vaccine with ACE2. (A) Animated representative of complex vaccine (orange) and ACE2 (blue), (B) Interacting residues of vaccine and ACE2, Residues in cyan color are vaccine residues and the red are ACE2’s, and (C) All interacting residues of vaccine-ACE2
Exploring the interaction of the vaccine with TLRs
In the present study, we also analyzed the possibility for interaction of the vaccine with TLRs especially TLR-3 and TLR-8 as important pattern recognition receptors in immune activation. The interactions were computed by similar tools as the vaccine-ACE2 interaction analysis. We observed that the vaccine interacted well with both TLR-3 and TLR-8. The best-docked results for the two protein receptors showed the lowest energy scores of -1,069.6 and -1,205.6, respectively. The docked results used coefficient wattage (E = 0.40Erep + -0.40Eatt + 600Eelec + 1.00EDARS) to calculate the binding energy of the interactions.41 Meanwhile, the docking results using PatchDock and HDock are shown in Table 1. Molecular docking using HawkDock generated the docking scores of -5607.81 and -5031.26 for TLR-3 and TLR-8, respectively (Table 2).
For further analysis of detailed interactions of amino acids in Vaccine-TLRs, we found that the interactions of the vaccine with two receptors occur at several amino acids with different types of bonds. In the vaccine-TLR3 complex, this interaction involves several amino acids, such as Arg265-Asp489 (salt bridges), gly222-arg 438 (hydrogen bonds), Thr130-Asn338 (by non-bound interaction) and others (Fig. 5). Additionally, the vaccine-TLR8 interactions were in Arg14-Gly505 and Arg38-Ala504 (hydrogen bond), Lys45-Asp478 (salt bridges), as well as by non-bound interactions such as Cys33-Gly505, Arg38-Thr507 and Lys45-Val453 (Fig. 6). The width of the striped line in non-bound interactions is proportional to the number of atomic contacts.
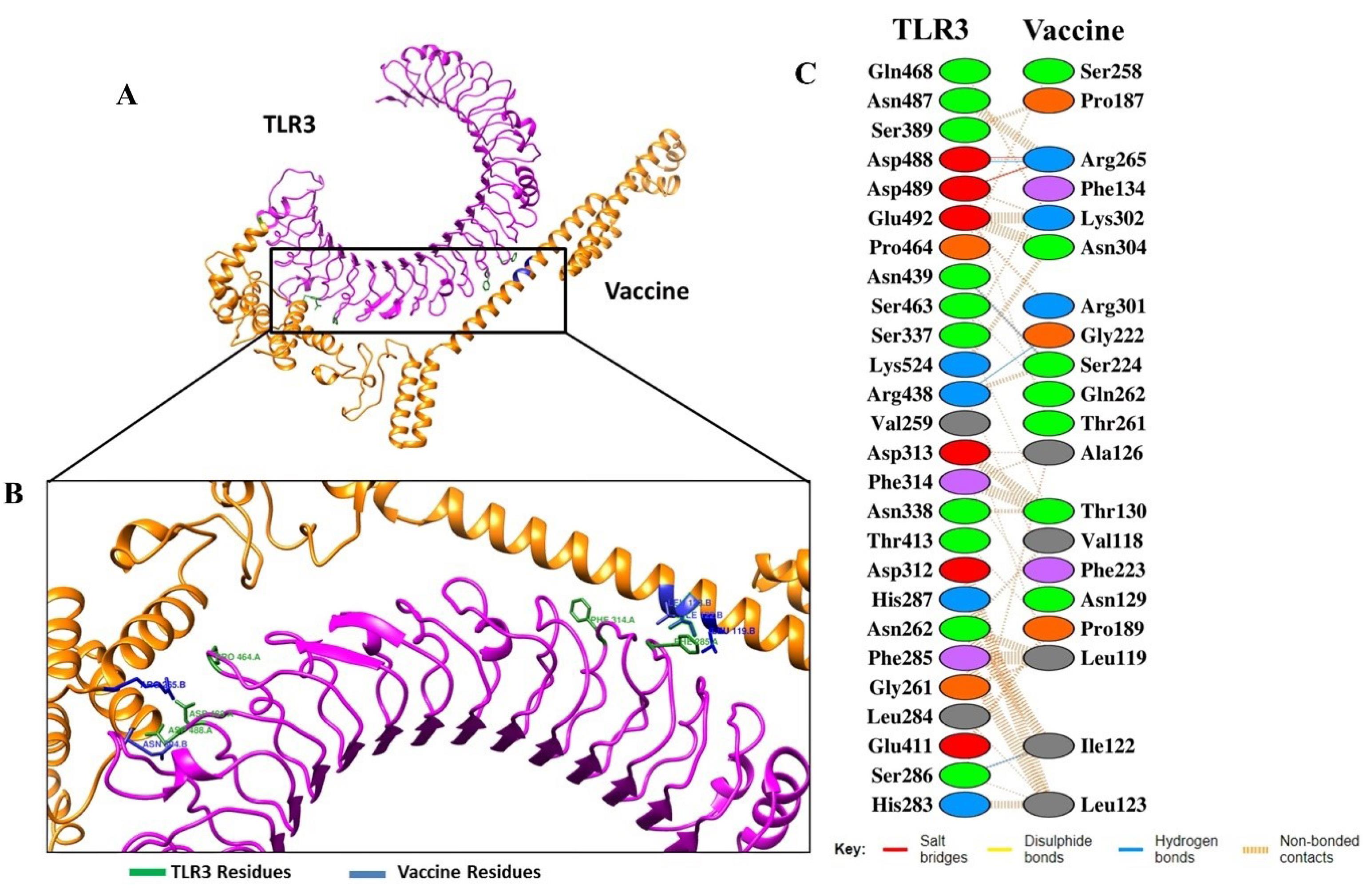
Fig. 5.
The docked complexes between vaccine with TLR3. (A) Animated representative of complex vaccine (orange) and TLR3 (magenta). (B) Interacting residues of vaccine (blue) and TLR3 (green), and (C) All interacting residues of vaccine-TLR3.
.
The docked complexes between vaccine with TLR3. (A) Animated representative of complex vaccine (orange) and TLR3 (magenta). (B) Interacting residues of vaccine (blue) and TLR3 (green), and (C) All interacting residues of vaccine-TLR3.
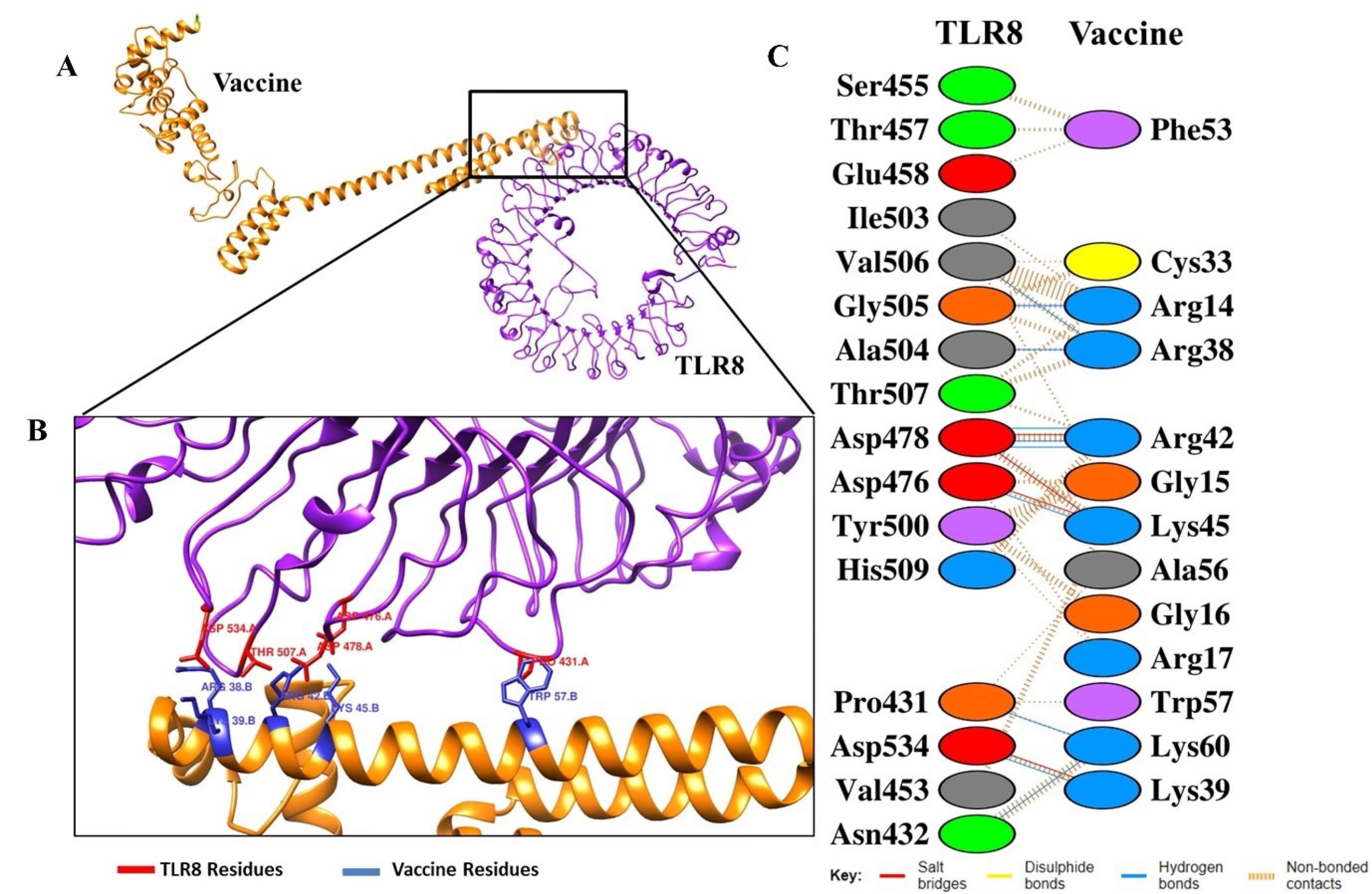
Fig. 6.
The docked complexes between vaccine with TLR8. (A) Animated representative of complex vaccine (orange) and TLR8 (purple), (B) Interacting residues of vaccine (blue) and TLR3 (red), and (C) all interacting residues of vaccine-TLR8.
.
The docked complexes between vaccine with TLR8. (A) Animated representative of complex vaccine (orange) and TLR8 (purple), (B) Interacting residues of vaccine (blue) and TLR3 (red), and (C) all interacting residues of vaccine-TLR8.
Molecular dynamic simulation and MM/GBSA binding free energy calculations of the complex receptor-vaccine
The rigidity of vaccine and receptor interaction was evaluated by the value of the radius of gyration (Rg). As follows in results (Figs. S6A, S7A and S8A), Rg value are 37.9325 Å, 43.3996 Å and 41.5319 Å for interaction vaccine with ACE2, TLR3 and TLR8, respectively throughout the 20 ps time span. We also analyzed the micro-interactions in terms of the physical movements of atoms and molecules of receptor-vaccine complex. The molecular dynamics were conducted using the iMods webserver as an online, fast and accurate tool for determining and measuring the protein flexibility.17,19 It utilizes normal mode analysis (NMA) to calculate internal coordinates by computing collective functional motions and generating a feasible transition path between two homologous structures on a biological macromolecule. The iMODS server is widely used to assess the interactivity of vaccines with receptors because it has the advantages of high speed and efficacy as well as other molecular dynamics tools such as GROMACS and NAMD.44,45,65
The results of molecular interactions of the vaccine with the TLR3 and TLR8 complex show almost the same deformability pattern, while ACE2 shows a different peak from the complex vaccine/TLRs.17 The locations with hinges in the deformability graph reflect higher deformability regions in the protein complex.17,32 β-factor graphs of the complex are shown in Fig. S6D, and Supplementals 7D and 8D. The parameters were calculated by comparison of normal mode analysis to root mean square, which reflects the calculated uncertainty of each atom. The eigenvalue showed remarkably good values of 1.183 x 10-6, 1.553 x 10-6, and 8.744 x 10-7 for the vaccine complex with ACE2, TLR3, and TLR8, respectively. Lower eigenvalues indicate easier deformation of the complex. Figs. S6D, S7D, and S8D depict covariant matrices of pair residues in the complex that reflect their correlation pattern. Red matrices indicated a correlated pair while white and blue are marked as uncorrelated and anti-correlated, respectively. Furthermore, the elastic network (Figs. S6F, S7F, and S8F) suggest connections between atoms and springs or stiffer regions in the complex.19,32
In the present study, we further analyzed the data regarding binding affinity of the complex by molecular mechanics/generalized born surface area (MM-GBSA) calculations. From the results generated by the HawkDock server (Table 2), total binding free energy of complex of ACE2, TLR3 and TLR8 were -91.25 kcal/mol, -38.38 kcal/mol, and -62.91 kcal/mol, respectively. Of these interactions, we obtained five residues with the best binding free energy scores both in the receptors and vaccine that are responsible in these interactions (Table 2)
Codon adaptation and in silico cloning of the vaccine
Codon adaptations were conducted to reverse the vaccine sequence into nucleotide sequences and performed using the JCat server.27 The codon sequences (cDNA) contain 1,173 nucleotides with GC content computed 55.92 that is considered a good sequence (40-60 in range). These reverse sequences generate the codon adaptation index (CAI) value of 0.898 (within the range 0.8–1.0).52,56 The value reflects that the DNA contains a high proportion of codons and is effectively expressed in the machinery organisms, E. coli strain K12.19
After the adaptation, the sequences were inserted into a selected vector for further cloning using the SnapGene tool. In the present study, we utilized the pET28a( + ) vector to insert cDNA of the constructed vaccine for further expression in the E. coli host. We also used buffer compatible restriction enzymes, with EcoRI in the start and BamHI in the end to cut the vector.32,66 As shown in Fig. 7, constructed vaccines were successfully expressed in the E. coli strain using the pET-28a vector. The codon sequences are red and the vector is seen in black with the total final length of the clone of 6542 bp.
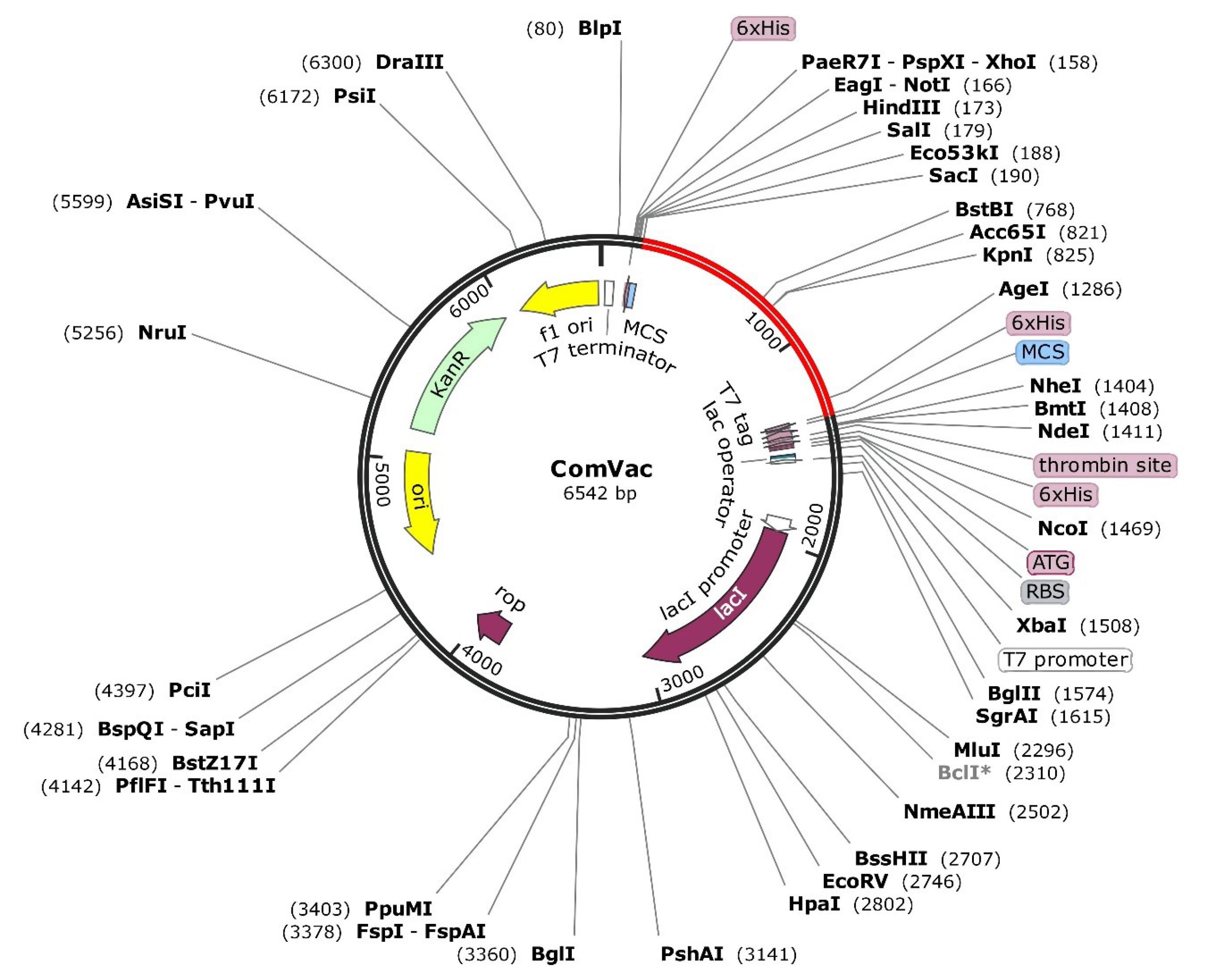
Fig. 7.
In silico cloning vaccine using pET28a( + ) expression vector. The red line is the inserted vaccine.
.
In silico cloning vaccine using pET28a( + ) expression vector. The red line is the inserted vaccine.
The results are in accordance with a previous study conducted by Safavi et al in 2019 that reported in order to optimize the codon using pET-28a expression vector, it is necessary to transfect the vaccine into E. coli before it can be produced and purified in the laboratory. In their published paper, they explained that the procedure could generate a good candidate vaccine that is effective in inducing immune responses in vitro and in vivo.67
Post-translational modification analysis
The PTM were analyze to assess possibility modification in vaccine construct in post translation phase. In this study, we found that there are some modifications at vaccine construct as follow at Fig. S9. Methylation (me) become most frequently namely 3 times each at amino acids R14, R265, and R345. Followed by acetylation (ac) twice in K8 and K52; glycosylation (gl) in N98 and S285; palmytoilation (pa) in C 40 and C41; ubiquitylation (ub) in K52 and K60 and several other PTMs in several amino acids such as pyrrolidone carboxylic acid (pc), phosphorylation (p) and hydroxylation (Hy).
Immune simulation of vaccine in human body administration
Immune simulation was conducted by the C-ImmSim server to compute the possible immune responses to the constructed vaccine after administration. As show in Figs. 8A and 8B, Natural Killer and dendrite cells acting as the first barrier in immune response were significantly increased after the vaccine administration. The proliferation was also identified in all of the antibody types, including IgM + IgG, IgG1 + IgG2, IgG1, IgG2 and IgM after the secondary and tertiary injections. Overall, secreted antibody peaks were higher at booster than main dose (Fig. 8C). IgM + IgG became the highest identified antibody, followed by IgG1 + IgG2. Meanwhile, the IgM appeared still higher than the IgG response. In a similar pattern response, the increase in IFN occurred in the first, second and third doses (Fig. 8D). However, the highest level of IFN occurred after the first dose administration and began to decrease around the 75th day. Several other cytokines such as IL-10, TNF-β, and IL-12 also appeared to increase after the first, second and third dose administration (Fig. 8D). A similar finding demonstrated by previous research indicated that various antibodies and cytokines such as IFN, TNF-β and IL-10 were increased after in silico injection of mosaic vaccines.49,56 The results were also consistent with the findings in the study conducted by Safavi et al. in 2019, which found an increase in antibodies (e.g. IgG1, IgG2a, IgG2a) and cytokines (e.g. IFN-γ) in the murine melanoma model after administration of mosaic vaccines.67
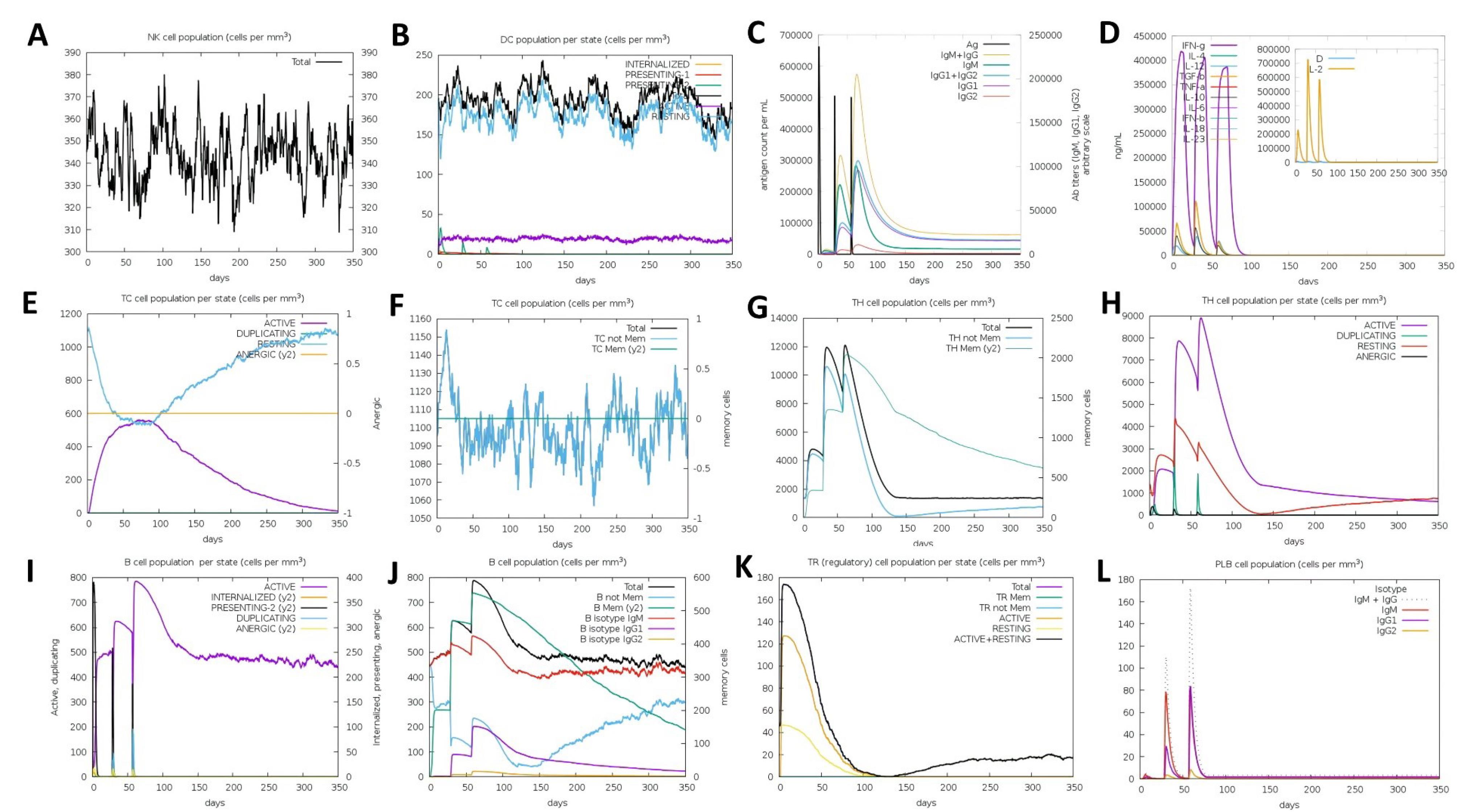
Fig. 8.
Immune simulation after three dose injections of vaccine at 0, 28 and 56 days. (A) Fluctuatitivity of total NK cell after injection of antigen vaccine (B) Show number of Dendritic cell population per state. DC cells are APCs that function to recognize and process antigens. (C) Antigen concentration and relative antibody responses. The injection of vaccine could induce increasing protective IgGs and IgM (D) Show the rise of releasing cytokine and interleukins count with Simpson index (E-F) Cytotoxic T-cell population counts and activation states (G-H) Helper T-cell population counts and activation states (I-J) B-cell population counts and activation states (K) T-Regulatory cell population per state (L) PLB cell population counts.
.
Immune simulation after three dose injections of vaccine at 0, 28 and 56 days. (A) Fluctuatitivity of total NK cell after injection of antigen vaccine (B) Show number of Dendritic cell population per state. DC cells are APCs that function to recognize and process antigens. (C) Antigen concentration and relative antibody responses. The injection of vaccine could induce increasing protective IgGs and IgM (D) Show the rise of releasing cytokine and interleukins count with Simpson index (E-F) Cytotoxic T-cell population counts and activation states (G-H) Helper T-cell population counts and activation states (I-J) B-cell population counts and activation states (K) T-Regulatory cell population per state (L) PLB cell population counts.
Concerning the T regulatory (Treg) related cytokines, the expressions of IL-10 and TNF-β were less dominant compared to IFN. CD4-T helper and CD8 T-cytotoxic lymphocytes (CTL) counts were also increased with injection of the vaccine (Fig. 8E-H). Contrary to the CD4-T helper (HTL) which increased to its peak and then decreased after the first and second doses, the active CTL count was consistently increasing and started to decrease on day 100. The increase in CTL and HTL counts can be attributed to an increase in the number of IFN cytokines as determinants of their activation. These results are in line with previous in vivo studies that revealed the administration of multi-epitope vaccines into the BALB/c mice model could generate improvement in immune and adaptive responses including functional antibodies and CD8 + and CD4 + T cell responses.68,69
Furthermore, the B-cell lymphocytes (BCLs) counts were increased since the initial dose administration of the vaccine. The increase occurred in almost all types including BCL memory, BCL isotype IgM, IgG1 and IgG2 (Fig. 8I-J). In this simulation, we also noted that Treg cell counts were immediately increased after the initial dose, and gradually decreased both before and after the second and third doses (Fig. 8K). Increased proliferation of B-cells, BCL memory, and BCL isotypes is important in generating long-term immunity of the immunized individuals and the general population.51,70
Discussion
The emerging variants of SARS-CoV-2 have raised new concerns about the virus that has caused almost 7 million deaths worldwide. Several recent reports have advocated that the mutations can lead to a reduction of the ability of neutralizing antibodies in post-vaccination serum.47 In this context, the development and updating of vaccine sequences to overcome the variants are earnestly continued by researchers and vaccine manufacturers. Moreover, several vaccine research teams in various countries are working to design and formulate more effective vaccines to stem this pandemic. Currently, growing evidence in the advancements of bioinformatics have prompted new methods in vaccine development. The immunoinformatics approach which provides a promising tool for designing and exploring potential vaccines has been adopted to construct vaccines from multiple epitope-rich SARS-CoV-2 protein fragments.71 The approach enables accurate prediction models to determine immunodominant epitopes and provides designed vaccines with promising characteristics. Additionally, the immunoinformatics approach can reduce excessive trial and error in the use of experimental animals, while providing a simple working approach.61,72 This approach can also be used to develop vaccines based on mutations in the immune-related genes, as well as linking single nucleotide polymorphisms of the HLA class I and II loci typing to vaccine coverage and person-specific resistance/susceptibility to diseases. The ultimate goal is to obtain more effective vaccines to create good immunity for patients against an infection-related disease including SARS-CoV-2.15
In the present study, we designed several vaccines using the mutational spike protein of SARS-CoV-2 as an important protein in the viral entry to host cells. Several computational tools in immunoinformatics were used to select immunodominant epitopes and further construct multi-epitope vaccines which have the ability to trigger activation of cellular and humoral immunity and prevent the excess antigenic load as well as allergenic reaction in the host.52,73 In this construction, we eventually resulted in a vaccine with 391 amino acid in length, that contain MHC class I epitopes and class II (as HTL epitopes), β-defensin (as adjuvant), PADRE sequence, linker, and 6xHis-tag.
The selection of MHC class I epitope prediction is due to their ability to elicit the immune response of CD8 + T-cytotoxic lymphocytes. Meanwhile, MHC class II epitopes were essential due to their role in mediating the immune response by TCD4 + lymphocytes activation and to further generate activation of T and B lymphocytes.61 Furthermore, we utilized β-defensin as an adjuvant due to its potential to activate primary innate immune responses and mediate other immunomodulatory activity against a number of viruses, including the Coronavirus.48 Recent evidence had demonstrated that adjuvants could lead to a longer lasting immune response and enhance the immunogenicity of the constructed vaccine. Moreover, linkers are added to maintain the function of each epitope and allow them to function independently, well and optimally in the human body.52,74,75 Addition of 6xHis-tag at the C-terminal of the vaccine is intended for purification and to identify the protein vaccine.32 The evaluations of multi-epitope vaccine show expected results that the vaccine is a potent and nonallergenic antigen. We also assessed that the vaccine was highly thermostable, and the hydrophobicity value (GRAVY) reflects that it is hydrophilic. A lower GRAVY score indicates better solubility of the protein. The protein vaccine was stable in pH 9.54 as its theoretical pI was calculated to be 9.54.17,19
The vaccines were docked with some important proteins both in the process of virus entry (i.e., ACE2) and in their recognition by the immune response namely TLRs. In the Vaccine/ACE2 docking, our results (Tables S1 and 2; Fig. 4) support our hypothesis that the vaccine or coronavirus antigen peptide can interact with ACE2.71 Several researchers have claimed that ACE2 is the most important protein in SARS-CoV-2 entry to the human cells.76 Furthermore, we also assessed that vaccine strongly interact with both of TLR3 and TLR8 (Tables 1 and 2; Figs. 5 and 6). TLRs are pattern recognition receptors that are widely reported to occupy the surface or endogenous regions of APCs. These interactions of the vaccine and TLRs will trigger activation of immune responses in both of innate and adaptive immune systems.61 Furthermore, some researchers have claimed that TLR3 is involved in the control of innate immunity during lung SARS-CoV-2 infection.77 Meanwhile, TLR8 is an endosomal TLR that plays a role in the recognition of antigens in the cytoplasm of APC. The recognition of antigens by these TLRs can trigger innate immune activation to release IFN type I which eventually leads to Th1 and Th2 responses.78
Expression of multi-epitope vaccine in E. coli provided the GC content 55.92 that indicated it was efficiently expressed. The cDNAs were further inserted via a vector expression of pET-28a. This array was used in accordance with a previous study conducted by Safavi et al in 2019 that reported in order to optimize the codon using pET-28a vector expression, it is necessary to transfect the vaccine into E. coli before it can be recombinated and purified in the laboratory. In their published paper, they explained that the procedure could generate a good candidate vaccine that is effective in inducing immune responses in vitro and in vivo.67
In this study, we also considered various post-translational modifications during their expression process. We obtained several PTMs in our vaccine protein synthesis process, including methylation, acetylation, glycosylation, palmytoilation, pyrrolidone carboxylic acid, phosphorylation and hydroxylation. Methylation or addition of methyl groups in protein synthesis will generally increase immunostimulatory activity and thus increase the potency of the vaccine constructed in this study. Furthermore, methylation generally occurs in palindromic CpG sequences in both bacterial and plasmid DNA.79,80 Additionally, acetylation usually occurs at the N-terminus, leading to cyclization of the epitope, ultimately leading to an increase in immunogenicity of the peptide vaccine,81 as well as several other NCDs that will play a role in the immune response generated by the vaccine peptide.80
This study’s immune simulation revealed that the mosaic vaccine could trigger an innate, cellular and humoral immune response at the time of the first, second, or third injection, which occurred in various ways (Fig. 8). We also noted the increasing of proliferation of B-cells, BCL memory, and BCL isotypes that are considered important in generating long-term immunity of the immunized individuals and the general population against the continued presence of the mutated SARS-CoV-2.51,70
Conclusion
In summary, we concluded that the construction of vaccines from mutational spike proteins using the immunoinformatics approach provides novel promising vaccine candidates. The physicochemical characteristics show parameters that are in accordance with the literature concerning the characteristics of a good vaccine. The vaccine was verified to effectively bind with receptors including ACE2, TLR3, and TLR8 with strong interactions for further activation of immune responses. As expected from the antigenicity and immunogenicity profiles, the administration could elicit strong immune responses by activation of innate immune responses such as Natural Killer and dendrite cells. As shown in the computational simulations, the vaccine could also mediate adaptive immune responses in both cellular and humoral immune systems, including the production of memory cells and isotyping B cells for long-term protection. These results suggest that the vaccine can effectively induce an immune response in humans. Nonetheless, this study is still limited to computerized analyses using the immunoinformatics approach, and it is necessary for advanced experimental research in the laboratory to further study the recombinants and prove the vaccine’s efficacy through in vitro, veterinary, and human studies.
Research Highlights
What is the current knowledge?
√ The SARS-CoV-2 has infected almost 700 million people and is responsible for nearly 7 million deaths worldwide.
√ Emerging mutations of SARS-CoV-2 especially in spike protein have led to the development of more effective vaccines to break the chain of transmission in the pandemic.
√ Development of vaccines can be done by immunoinformatics from an original spike protein.
What is new here?
√ Designing new vaccines can adopt the immunoinformatics approach using the mutational spike protein of SARS-CoV-2.
√ Mosaics of the constructed vaccines indicate they are antigenic, nonallegenic, and show good, promising vaccine characteristics.
√ The vaccines can generate strong interactions with some human receptors including TLR3, 8, and ACE2.
√ The constructed vaccines could induce immunological responses in both humoral and cellular systems.
Acknowledgments
Thanks for the support from the Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada, Yogyakarta, Indonesia in conducting this research.
Competing Interests
None declared.
Ethical Statement
Not applicable.
Supplementary files
Supplementary file 1 contains Tables S1-S4 and Fig. S1-S9.
(pdf)
References
- Hu B, Guo H, Zhou P, Shi ZL. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol 2021; 19:141-54. doi: 10.1038/s41579-020-00459-7 [Crossref] [ Google Scholar]
- Daniloski Z, Jordan TX, Wessels HH, Hoagland DA, Kasela S, Legut M, et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 2021; 184: 92-105.e16. 10.1016/j.cell.2020.10.030.
- Coronavirus (COVID-19) [database on the Internet]. WHO. 2021 [cited April 19, 2021]. Available from: https://who.sprinklr.com/.
- Chan JF, Yuan S, Kok KH, To KK, Chu H, Yang J. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 2020; 395:514-23. doi: 10.1016/s0140-6736(20)30154-9 [Crossref] [ Google Scholar]
- Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J. Clinical characteristics of 138 hospitalized patients with 2019 novel Coronavirus-infected pneumonia in Wuhan, China. Jama 2020; 323:1061-9. doi: 10.1001/jama.2020.1585 [Crossref] [ Google Scholar]
- Ysrafil Y, Astuti I, Mus R, Gama NI, Rahmaisyah D, Nur’amalia R. A summary of Coronavirus Disease 2019: what we should know?. Pharm Sci 2020; 26:S24-S35. [ Google Scholar]
- Kyriakidis NC, López-Cortés A, González EV, Grimaldos AB, Prado EO. SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines 2021; 6:28. doi: 10.1038/s41541-021-00292-w [Crossref] [ Google Scholar]
- Astuti I, Ysrafil Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes MetabSyndr 2020; 14:407-12. doi: 10.1016/j.dsx.2020.04.020 [Crossref] [ Google Scholar]
- Yu F, Jia R, Tang Y, Liu J, Wei B. SARS-CoV-2 infection and stem cells: Interaction and intervention. Stem Cell Res 2020; 46:101859. doi: 10.1016/j.scr.2020.101859 [Crossref] [ Google Scholar]
- Fontanet A, Autran B, Lina B, Kieny MP, Karim SSA, Sridhar D. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 2021; 397:952-4. doi: 10.1016/s0140-6736(21)00370-6 [Crossref] [ Google Scholar]
- Ysrafil Y, Mus R, Gama NI, Rahmaisyah D, Nur'amalia R. Emerging mutation in SARS-CoV-2 spike: Widening distribution over time in different geographic areas. Biomed J 2021; 44:570-81. doi: 10.1016/j.bj.2021.07.003 [Crossref] [ Google Scholar]
- Collier DA, De Marco A, Ferreira I, Meng B, Datir R, Walls AC. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 2021; 593:136-41. doi: 10.1038/s41586-021-03412-7 [Crossref] [ Google Scholar]
- Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021; 596:276-80. doi: 10.1038/s41586-021-03777-9 [Crossref] [ Google Scholar]
- Mengist HM, Kombe Kombe AJ, Mekonnen D, Abebaw A, Getachew M, Jin T. Mutations of SARS-CoV-2 spike protein: Implications on immune evasion and vaccine-induced immunity. Semin Immunol 2021; 55:101533. doi: 10.1016/j.smim.2021.101533 [Crossref] [ Google Scholar]
- Salemi A, Pourseif MM, Omidi Y. Next-generation vaccines and the impacts of state-of-the-art in-silico technologies. Biologicals 2021; 69:83-5. doi: 10.1016/j.biologicals.2020.10.002 [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Naghili B, Saeedi N, Parvizpour S, Nematollahi A. A novel in silico minigene vaccine based on CD4( + ) T-helper and B-cell epitopes of EG95 isolates for vaccination against cystic echinococcosis. Comput Biol Chem 2018; 72:150-63. doi: 10.1016/j.compbiolchem.2017.11.008 [Crossref] [ Google Scholar]
- Ullah MA, Sarkar B, Islam SS. Exploiting the reverse vaccinology approach to design novel subunit vaccines against Ebola virus. Immunobiology 2020; 225:151949. doi: 10.1016/j.imbio.2020.151949 [Crossref] [ Google Scholar]
- Khairkhah N, Aghasadeghi MR, Namvar A, Bolhassani A. Design of novel multiepitope constructs-based peptide vaccine against the structural S, N and M proteins of human COVID-19 using immunoinformatics analysis. PLoS One 2020; 15:e0240577. doi: 10.1371/journal.pone.0240577 [Crossref] [ Google Scholar]
- Sarkar B, Ullah MA, Johora FT, Taniya MA, Araf Y. Immunoinformatics-guided designing of epitope-based subunit vaccines against the SARS Coronavirus-2 (SARS-CoV-2). Immunobiology 2020; 225:151955. doi: 10.1016/j.imbio.2020.151955 [Crossref] [ Google Scholar]
- Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 2019; 47:D339-d43. doi: 10.1093/nar/gky1006 [Crossref] [ Google Scholar]
- Santra S, Liao HX, Zhang R, Muldoon M, Watson S, Fischer W. Mosaic vaccines elicit CD8 + T lymphocyte responses that confer enhanced immune coverage of diverse HIV strains in monkeys. Nat Med 2010; 16:324-8. doi: 10.1038/nm.2108 [Crossref] [ Google Scholar]
- Feng Y, Jiang H, Qiu M, Liu L, Zou S, Li Y. Multi-Epitope Vaccine Design Using an Immunoinformatic Approach for SARS-CoV-2. Pathogens 2021; 10:737. doi: 10.3390/pathogens10060737 [Crossref] [ Google Scholar]
- Yazdani Z, Rafiei A, Yazdani M, Valadan R. Design an Efficient Multi-Epitope Peptide Vaccine Candidate Against SARS-CoV-2: An in silico Analysis. Infect Drug Resist 2020; 13:3007-22. doi: 10.2147/idr.S264573 [Crossref] [ Google Scholar]
- Pourseif MM, Parvizpour S, Jafari B, Dehghani J, Naghili B, Omidi Y. A domain-based vaccine construct against SARS-CoV-2, the causative agent of COVID-19 pandemic: development of self-amplifying mRNA and peptide vaccines. Bioimpacts 2021; 11:65-84. doi: 10.34172/bi.2021.11 [Crossref] [ Google Scholar]
- Saha S, Raghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res 2006; 34:W202-9. doi: 10.1093/nar/gkl343 [Crossref] [ Google Scholar]
- Solanki V, Tiwari V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Sci Rep 2018; 8:9044. doi: 10.1038/s41598-018-26689-7 [Crossref] [ Google Scholar]
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 2005; 33:W526-31. doi: 10.1093/nar/gki376 [Crossref] [ Google Scholar]
- Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP. In silico approach for predicting toxicity of peptides and proteins. PLoS One 2013; 8:e73957. doi: 10.1371/journal.pone.0073957 [Crossref] [ Google Scholar]
- Fatoba AJ, Maharaj L, Adeleke VT, Okpeku M, Adeniyi AA, Adeleke MA. Immunoinformatics prediction of overlapping CD8( + ) T-cell, IFN-γ and IL-4 inducer CD4( + ) T-cell and linear B-cell epitopes based vaccines against COVID-19 (SARS-CoV-2). Vaccine 2021; 39:1111-21. doi: 10.1016/j.vaccine.2021.01.003 [Crossref] [ Google Scholar]
- Nagpal G, Usmani SS, Dhanda SK, Kaur H, Singh S, Sharma M. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci Rep 2017; 7:42851. doi: 10.1038/srep42851 [Crossref] [ Google Scholar]
- Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics 2006; 7:153. doi: 10.1186/1471-2105-7-153 [Crossref] [ Google Scholar]
- Yang Z, Bogdan P, Nazarian S. An in silico deep learning approach to multi-epitope vaccine design: a SARS-CoV-2 case study. Sci Rep 2021; 11:3238. doi: 10.1038/s41598-021-81749-9 [Crossref] [ Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook. Springer; 2005. p. 571-607.
- Magnan CN, Randall A, Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics 2009; 25:2200-7. doi: 10.1093/bioinformatics/btp386 [Crossref] [ Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics 2000; 16:404-5. doi: 10.1093/bioinformatics/16.4.404 [Crossref] [ Google Scholar]
- Geourjon C, Deléage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci 1995; 11:681-4. doi: 10.1093/bioinformatics/11.6.681 [Crossref] [ Google Scholar]
- Du Z, Su H, Wang W, Ye L, Wei H, Peng Z. The trRosetta server for fast and accurate protein structure prediction. Nat Protoc 2021; 16:5634-51. doi: 10.1038/s41596-021-00628-9 [Crossref] [ Google Scholar]
- Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res 2012; 40:W294-7. doi: 10.1093/nar/gks493 [Crossref] [ Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 1993; 26:283-91. doi: 10.1107/S0021889892009944 [Crossref] [ Google Scholar]
- Kuriata A, Gierut AM, Oleniecki T, Ciemny MP, Kolinski A, Kurcinski M. CABS-flex 20: a web server for fast simulations of flexibility of protein structures. Nucleic Acids Res 2018; 46:W338-w43. doi: 10.1093/nar/gky356 [Crossref] [ Google Scholar]
- Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C. The ClusPro web server for protein-protein docking. Nat Protoc 2017; 12:255-78. doi: 10.1038/nprot.2016.169 [Crossref] [ Google Scholar]
- Weng G, Wang E, Wang Z, Liu H, Zhu F, Li D. HawkDock: a web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res 2019; 47:W322-w30. doi: 10.1093/nar/gkz397 [Crossref] [ Google Scholar]
- Yan Y, Tao H, He J, Huang SY. The HDOCK server for integrated protein-protein docking. Nat Protoc 2020; 15:1829-52. doi: 10.1038/s41596-020-0312-x [Crossref] [ Google Scholar]
- Lopéz-Blanco JR, Garzón JI, Chacón P. iMod: multipurpose normal mode analysis in internal coordinates. Bioinformatics 2011; 27:2843-50. doi: 10.1093/bioinformatics/btr497 [Crossref] [ Google Scholar]
- López-Blanco JR, Aliaga JI, Quintana-Ortí ES, Chacón P. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res 2014; 42:W271-6. doi: 10.1093/nar/gku339 [Crossref] [ Google Scholar]
- Wang D, Liu D, Yuchi J, He F, Jiang Y, Cai S. MusiteDeep: a deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res 2020; 48:W140-w6. doi: 10.1093/nar/gkaa275 [Crossref] [ Google Scholar]
- Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol 2021; 19:409-24. doi: 10.1038/s41579-021-00573-0 [Crossref] [ Google Scholar]
- Behmard E, Soleymani B, Najafi A, Barzegari E. Immunoinformatic design of a COVID-19 subunit vaccine using entire structural immunogenic epitopes of SARS-CoV-2. Sci Rep 2020; 10:20864. doi: 10.1038/s41598-020-77547-4 [Crossref] [ Google Scholar]
- Chukwudozie OS, Gray CM, Fagbayi TA, Chukwuanukwu RC, Oyebanji VO, Bankole TT. Immuno-informatics design of a multimeric epitope peptide based vaccine targeting SARS-CoV-2 spike glycoprotein. PLoS One 2021; 16:e0248061. doi: 10.1371/journal.pone.0248061 [Crossref] [ Google Scholar]
- Bhattacharya M, Sharma AR, Ghosh P, Lee SS, Chakraborty C. A Next-Generation Vaccine Candidate Using Alternative Epitopes to Protect against Wuhan and All Significant Mutant Variants of SARS-CoV-2: An Immunoinformatics Approach. Aging Dis 2021; 12:2173-95. doi: 10.14336/ad.2021.0518 [Crossref] [ Google Scholar]
- Murphy K, Janeway C, Weaver C. Janeway's Immunobiology. 9th ed. New York: Garland Science; 2017.
- Kar T, Narsaria U, Basak S, Deb D, Castiglione F, Mueller DM. A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep 2020; 10:10895. doi: 10.1038/s41598-020-67749-1 [Crossref] [ Google Scholar]
- Renu K, Subramaniam MD, Chakraborty R, Myakala H, Iyer M, Bharathi G. The role of Interleukin-4 in COVID-19 associated male infertility - A hypothesis. J Reprod Immunol 2020; 142:103213. doi: 10.1016/j.jri.2020.103213 [Crossref] [ Google Scholar]
- Gadotti AC, de Castro Deus M, Telles JP, Wind R, Goes M, Garcia Charello Ossoski R. IFN-γ is an independent risk factor associated with mortality in patients with moderate and severe COVID-19 infection. Virus Res 2020; 289:198171. doi: 10.1016/j.virusres.2020.198171 [Crossref] [ Google Scholar]
- López D. Predicted HLA Class I and Class II Epitopes From Licensed Vaccines Are Largely Conserved in New SARS-CoV-2 Omicron Variant of Concern. Front Immunol 2022; 13:832889. doi: 10.3389/fimmu.2022.832889 [Crossref] [ Google Scholar]
- Tahir Ul Qamar M, Rehman A, Tusleem K, Ashfaq UA, Qasim M, Zhu X. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS One 2020; 15:e0244176. doi: 10.1371/journal.pone.0244176 [Crossref] [ Google Scholar]
- Wu CY, Monie A, Pang X, Hung CF, Wu TC. Improving therapeutic HPV peptide-based vaccine potency by enhancing CD4 + T help and dendritic cell activation. J Biomed Sci 2010; 17:88. doi: 10.1186/1423-0127-17-88 [Crossref] [ Google Scholar]
- Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng 2001; 14:529-32. doi: 10.1093/protein/14.8.529 [Crossref] [ Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 2007; 8:4. doi: 10.1186/1471-2105-8-4 [Crossref] [ Google Scholar]
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 2003; 31:3784-8. doi: 10.1093/nar/gkg563 [Crossref] [ Google Scholar]
- Droppa-Almeida D, Franceschi E, Padilha FF. Immune-Informatic Analysis and Design of Peptide Vaccine From Multi-epitopes Against Corynebacterium pseudotuberculosis. Bioinform Biol Insights 2018; 12:1177932218755337. doi: 10.1177/1177932218755337 [Crossref] [ Google Scholar]
- Rahman N, Ali F, Basharat Z, Shehroz M, Khan MK, Jeandet P. Vaccine Design from the Ensemble of Surface Glycoprotein Epitopes of SARS-CoV-2: An Immunoinformatics Approach. Vaccines (Basel) 2020; 8:1-17. doi: 10.3390/vaccines8030423 [Crossref] [ Google Scholar]
- Haimov B, Srebnik S. A closer look into the α-helix basin. Sci Rep 2016; 6:38341. doi: 10.1038/srep38341 [Crossref] [ Google Scholar]
- Murray K, Rodwell V, Bender D, Botham KM, Weil PA, Kennelly P. Harper's Illustrated Biochemistry. 28 ed. New York: McGraw-Hill Education; 2009.
- Rouzbahani AK, Kheirandish F, Hosseini SZ. Design of a multi-epitope-based peptide vaccine against the S and N proteins of SARS-COV-2 using immunoinformatics approach. Egypt J Med Hum Genet 2022; 23:1-18. doi: 10.1186/s43042-022-00224-w [Crossref] [ Google Scholar]
- Nain Z, Karim MM, Sen MK, Adhikari UK. Structural basis and designing of peptide vaccine using PE-PGRS family protein of Mycobacterium ulcerans-An integrated vaccinomics approach. Mol Immunol 2020; 120:146-63. doi: 10.1016/j.molimm.2020.02.009 [Crossref] [ Google Scholar]
- Safavi A, Kefayat A, Sotoodehnejadnematalahi F, Salehi M, Modarressi MH. Production, purification, and in vivo evaluation of a novel multiepitope peptide vaccine consisted of immunodominant epitopes of SYCP1 and ACRBP antigens as a prophylactic melanoma vaccine. Int Immunopharmacol 2019; 76:105872. doi: 10.1016/j.intimp.2019.105872 [Crossref] [ Google Scholar]
- Xu Z, Wise MC, Chokkalingam N, Walker S, Tello-Ruiz E, Elliott STC. In Vivo Assembly of Nanoparticles Achieved through Synergy of Structure-Based Protein Engineering and Synthetic DNA Generates Enhanced Adaptive Immunity. Adv Sci (Weinh) 2020; 7:1902802. doi: 10.1002/advs.201902802 [Crossref] [ Google Scholar]
- Ghaffari-Nazari H, Tavakkol-Afshari J, Jaafari MR, Tahaghoghi-Hajghorbani S, Masoumi E, Jalali SA. Improving Multi-Epitope Long Peptide Vaccine Potency by Using a Strategy that Enhances CD4 + T Help in BALB/c Mice. PLoS One 2015; 10:e0142563. doi: 10.1371/journal.pone.0142563 [Crossref] [ Google Scholar]
- Palm AE, Henry C. Remembrance of Things Past: Long-Term B Cell Memory After Infection and Vaccination. Front Immunol 2019; 10:1787. doi: 10.3389/fimmu.2019.01787 [Crossref] [ Google Scholar]
- Naz A, Shahid F, Butt TT, Awan FM, Ali A, Malik A. Designing Multi-Epitope Vaccines to Combat Emerging Coronavirus Disease 2019 (COVID-19) by Employing Immuno-Informatics Approach. Front Immunol 2020; 11:1663. doi: 10.3389/fimmu.2020.01663 [Crossref] [ Google Scholar]
- Ysrafil Y, Sapiun Z, Astuti I, Anasiru MA, Slamet NS, Hartati H. Designing multi-epitope based peptide vaccine candidates against SARS-CoV-2 using immunoinformatics approach. Bioimpacts 2022; 12:359-70. doi: 10.34172/bi.2022.23769 [Crossref] [ Google Scholar]
- Sanami S, Zandi M, Pourhossein B, Mobini GR, Safaei M, Abed A. Design of a multi-epitope vaccine against SARS-CoV-2 using immunoinformatics approach. Int J Biol Macromol 2020; 164:871-83. doi: 10.1016/j.ijbiomac.2020.07.117 [Crossref] [ Google Scholar]
- Pandey RK, Bhatt TK, Prajapati VK. Novel Immunoinformatics Approaches to Design Multi-epitope Subunit Vaccine for Malaria by Investigating Anopheles Salivary Protein. Sci Rep 2018; 8:1125. doi: 10.1038/s41598-018-19456-1 [Crossref] [ Google Scholar]
- Shamriz S, Ofoghi H, Moazami N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput Biol Med 2016; 76:24-9. doi: 10.1016/j.compbiomed.2016.06.015 [Crossref] [ Google Scholar]
- Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020; 181: 271-80.e8. 10.1016/j.cell.2020.02.052.
- Bortolotti D, Gentili V, Rizzo S, Schiuma G, Beltrami S, Strazzabosco G. TLR3 and TLR7 RNA Sensor Activation during SARS-CoV-2 Infection. Microorganisms 2021; 9:1820. doi: 10.3390/microorganisms9091820 [Crossref] [ Google Scholar]
- Webb LM, Lundie RJ, Borger JG, Brown SL, Connor LM, Cartwright AN. Type I interferon is required for T helper (Th) 2 induction by dendritic cells. Embo j 2017; 36:2404-18. doi: 10.15252/embj.201695345 [Crossref] [ Google Scholar]
- Abdulhaqq SA, Weiner DB. DNA vaccines: developing new strategies to enhance immune responses. Immunol Res 2008; 42:219-32. doi: 10.1007/s12026-008-8076-3 [Crossref] [ Google Scholar]
- Ojha R, Prajapati VK. Cognizance of posttranslational modifications in vaccines: A way to enhanced immunogenicity. J Cell Physiol 2021; 236:8020-34. doi: 10.1002/jcp.30483 [Crossref] [ Google Scholar]
- Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov 2007; 6:404-14. doi: 10.1038/nrd2224 [Crossref] [ Google Scholar]