Bioimpacts. 13(6):439-455.
doi: 10.34172/bi.2023.27521
Review
The application, safety, and future of ex vivo immune cell therapies and prognosis in different malignancies
Katelyn R. Einloth Conceptualization, Visualization, Writing – original draft, Writing – review & editing, 1 
Scott Gayfield Writing – original draft, Writing – review & editing, 1
Thomas McMaster Writing – original draft, 1
Alexander Didier Writing – original draft, 1
Lance Dworkin Supervision, 1
Justin Fortune Creeden Conceptualization, Project administration, Resources, Writing – original draft, Writing – review & editing, 2, 3, *
Author information:
1Department of Medicine, University of Toledo College of Medicine and Life Sciences, Toledo, OH 43614, USA
2Department of Cancer Biology, University of Toledo College of Medicine and Life Sciences, Toledo, OH, USA
3Department of Neurosciences, University of Toledo College of Medicine and Life Sciences, Toledo, OH, USA
Abstract
Introduction:
Immunotherapy has revolutionized how cancer is treated. Many of these immunotherapies rely on ex vivo expansion of immune cells, classically T cells. Still, several immunological obstacles remain, including tumor impermeability by immune cells and the immunosuppressive nature of the tumor microenvironment (TME). Logistically, high costs of treatment and variable clinical responses have also plagued traditional T cell-based immunotherapies.
Methods:
To review the existing literature on cellular immunotherapy, the PubMed database was searched for publications using variations of the phrases "cancer immunotherapy", "ex vivo expansion", and "adoptive cell therapy". The Clinicaltrials.gov database was searched for clinical trials related to ex vivo cellular therapies using the same phrases. The National Comprehensive Cancer Network guidelines for cancer treatment were also referenced.
Results:
To circumvent the challenges of traditional T cell-based immunotherapies, researchers have developed newer therapies including tumor infiltrating lymphocyte (TIL), chimeric antigen receptor (CAR), T cell receptor (TCR) modified T cell, and antibody-armed T cell therapies. Additionally, newer immunotherapeutic strategies have used other immune cells, including natural killer (NK) and dendritic cells (DC), to modulate the T cell immune response to cancers. From a prognostic perspective, circulating tumor cells (CTC) have been used to predict cancer morbidity and mortality.
Conclusion:
This review highlights the mechanism and clinical utility of various types of ex vivo cellular therapies in the treatment of cancer. Comparing these therapies or using them in combination may lead to more individualized and less toxic chemotherapeutics.
Keywords: Ex vivo expansion, Cancer immunotherapy, Adoptive cell therapy, DC vaccination, T cell immunotherapy, Circulating tumor cells
Copyright and License Information
© 2023 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
The authors have no funding to disclose.
Introduction
Globally, one in five men and one in six women will be diagnosed with cancer in their lifetime.1 Depending on the type and stage of cancer, treatments may target specific portions of the body, known as local therapy, or may affect the entire body, known as systemic therapy. Examples of local therapy include surgery and radiation therapy, while drug treatments such as chemotherapy and immunotherapy are examples of systemic treatments.2 Of these, systemic immunotherapeutic interventions are most productively enhanced by ex vivo (i.e., outside the body) laboratory techniques that allow for the expansion of therapeutic or prognostic biological material.
Adoptive cell therapy (ACT) is perhaps the most widely recognized type of immunotherapy relying on ex vivo cellular preparation. ACT requires ex vivo modification and expansion of autologous cells before these cells are infused back into the patient.3 The earliest forms of ACT used T cells to treat cancers. Today, there are multiple types of T cell therapy, including tumor infiltrating lymphocyte (TIL) therapy, chimeric antigen receptor (CAR) therapy, T cell receptor (TCR) modified T cell therapy, and therapies combining T cells with antibodies against specific tumor markers. Recent studies involving CAR-T cell therapy have explored refocusing CAR-Ts towards cells expressing specific molecules, such as EGFR, which allows for more targeted cancer therapy.4,5 To increase specificity further, a combination of T cell and antibody therapies has been developed, known as bispecific antibody-armed T cells. Though still in the early stages of development, this therapy has been used to treat some B-cell leukemias.6
In addition to T cell therapies, researchers are investigating other ex vivo anti-cancer therapeutics using immune cells that modulate T cell responses. Dendritic cells (DCs), which present antigens to T cells to trigger cytotoxic T lymphocyte (CTL) responses, can be loaded with tumor-specific antigens ex vivo and administered as cellular vaccinations.7,8 Similarly, natural killer (NK) cells create robust anti-tumor responses with a better safety profile than CAR-T cells and have been explored in a wide variety of cancers.9-15
Ex vivo cellular expansion techniques also demonstrate diagnostic and prognostic utility. Circulating tumor cells (CTCs) can be expanded ex vivo to discover tumor-specific biomarkers, identify therapeutic drug sensitivities, and prognosticate survival outcomes in cancer patients.16-18 Current research involving CTCs aims to find the most efficient ways of isolating CTCs from tumor samples as well as testing various culture conditions in which CTCs are expanded ex vivo.19
The technique of using ex vivo expanded T cells to treat cancer has been prevalent for several decades, yet it remains a promising area of cancer research. The expansion of other immune cells, such as dendritic and NK cells, to kill cancer has opened even more avenues of discovery (Fig. 1). While ex vivo cell therapy is effective in hematological malignancies, cancer researchers now turn their attention towards treating solid cancers with ex vivo cell therapy.20,21 Additionally, ex vivo expansion of CTCs provides an exciting new avenue of cancer prognostication based on samples of CTCs obtained from patients and personalized treatment through mouse models.
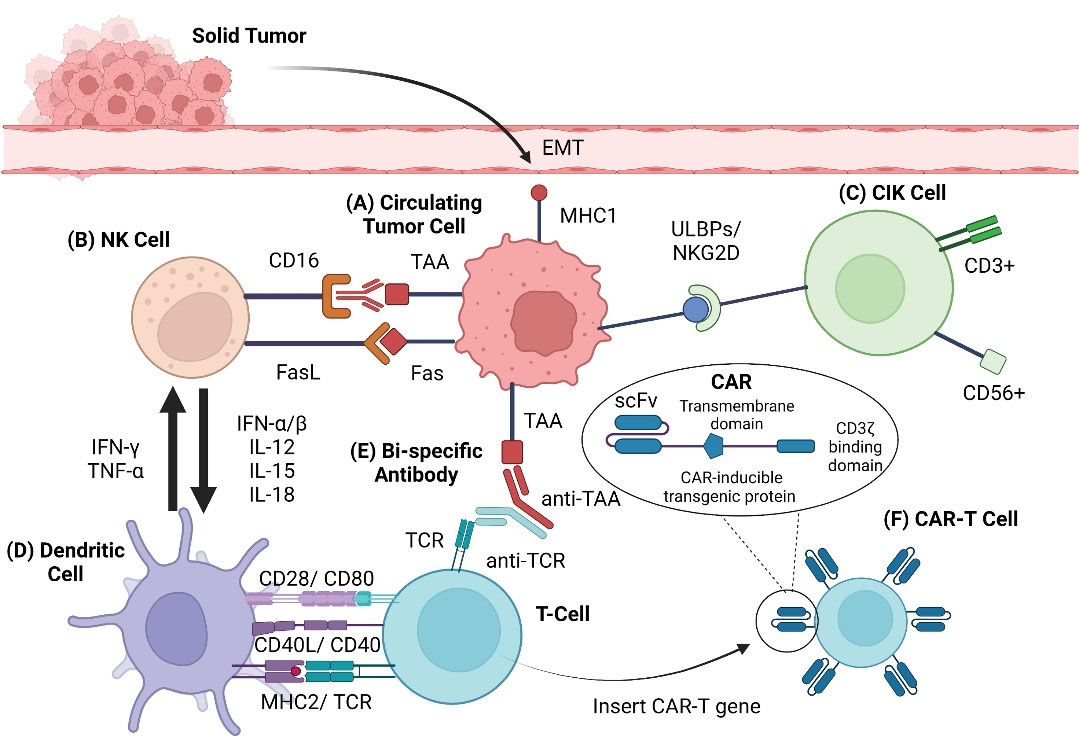
Fig. 1.
Graphical overview of therapies. Schematic of immune cellular interactions. (A) Tumor cells undergo epithelial-mesenchymal-transition, causing cells to lose cell-cell adhesions and apical-basal polarity. Tumor cells are then able to migrate into circulation, becoming circulating tumor cells (CTCs). (B) Natural killer (NK) cells lyse tumor cells through the Fas-FasL death receptor pathway and antibody-dependent cellular cytotoxicity (ADCC) (IgG- CD16 / IgG receptor III). NK cells activate and attract DCs via IL-12, IL-15, and IL-18, among other cytokines. (C) Interactions between cytokine-induced killer (CIK) cells and tumor cells via natural killer group 2 member D (NKG2D) and ligand UL16 binding proteins (ULBPs) lead to perforin-mediated tumor cell lysis. (D) DCs proliferate and activate NK cells through the production of IFN-γ and TNF-α. DCs assist in the initiation and regulation of immunity through multiple interactions with T cells (CD28-CD80, CD40L-CD40, MHC2-TCR). (E) Bispecific antibodies activate T cells and promote tumor cell death by simultaneously binding to tumor antigens and T cells. (F) T cells are altered by adding the gene for the specific CAR. First generation CARs consisted of an extracellular domain, scfv (single-chain fragment variable), a transmembrane domain, and an intracellular signaling domain, such as CD3ζ. Later generations of CAR-T cells are more complex.
.
Graphical overview of therapies. Schematic of immune cellular interactions. (A) Tumor cells undergo epithelial-mesenchymal-transition, causing cells to lose cell-cell adhesions and apical-basal polarity. Tumor cells are then able to migrate into circulation, becoming circulating tumor cells (CTCs). (B) Natural killer (NK) cells lyse tumor cells through the Fas-FasL death receptor pathway and antibody-dependent cellular cytotoxicity (ADCC) (IgG- CD16 / IgG receptor III). NK cells activate and attract DCs via IL-12, IL-15, and IL-18, among other cytokines. (C) Interactions between cytokine-induced killer (CIK) cells and tumor cells via natural killer group 2 member D (NKG2D) and ligand UL16 binding proteins (ULBPs) lead to perforin-mediated tumor cell lysis. (D) DCs proliferate and activate NK cells through the production of IFN-γ and TNF-α. DCs assist in the initiation and regulation of immunity through multiple interactions with T cells (CD28-CD80, CD40L-CD40, MHC2-TCR). (E) Bispecific antibodies activate T cells and promote tumor cell death by simultaneously binding to tumor antigens and T cells. (F) T cells are altered by adding the gene for the specific CAR. First generation CARs consisted of an extracellular domain, scfv (single-chain fragment variable), a transmembrane domain, and an intracellular signaling domain, such as CD3ζ. Later generations of CAR-T cells are more complex.
T cell therapy
T cell overview
T cells originate from lymphoid stem cells and play an essential role in the adaptive immune response. Once antigens are displayed to T cells by antigen-presenting cells (APCs), T cells produce cytokines which recruit and activate appropriate cell types to defend against the protected antigen.22 ACT is a branch of cancer immunotherapy that manipulates this immune response to impact the sustainability and progression of a tumor. Strategies for ACT involve either (i) ex vivo expansion of naturally-occurring TILs from surgically resected tumor samples or (ii) ex vivo genetic modification of T cells isolated from peripheral blood23 to form CAR, tumor-associated antigen, and bispecific antibody T cells (Fig. 2). Recent advances in ACT show significant promise for the future clinical application of immunotherapy in hematologic and solid tumor malignancies.
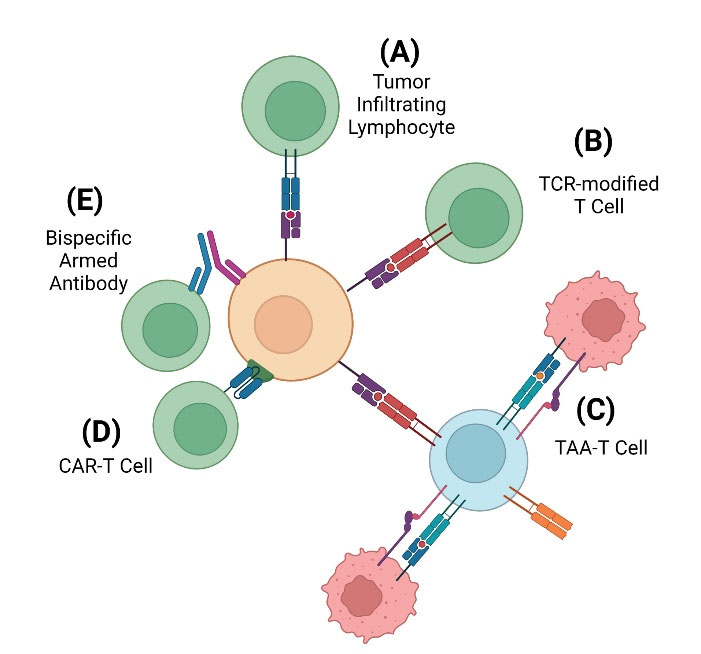
Fig. 2.
Types of adoptive T cell therapy. There are multiple methods developed for ACT approaches. (A) The original therapy utilized was TIL therapy, which utilized unmodified T cells found within a surgically excised tumor that were expanded ex vivo prior to re-infusion. The unmodified TCR (blue) interacts directly with the MHC (purple) on the tumor cell to begin signaling the immune response. (B) TCR-modified T cells are T cells that were obtained from the patient via leukapheresis prior to ex vivo genetic modification. TCR-modification (red) attempts to improve the therapeutic strength of T cells through altering the expression and pairing of TCR α and β chains. (C) TAA-T cells are T cells exposed to multiple, selected tumor antigens via DCs. When the DCs present these selected antigens to the native T cell via MHC and co-stimulation of B7 protein to the T cell via TCR and CD28 binding, the T cell will mount an immune response to each of those antigens. While using multiple antigen targets will increase the effectiveness of the therapy, there is an increased risk of on-target off-tumor toxicity. (D) CAR-modified T cells are like TCR-modified T cells, except CARs (blue) are synthetic receptors that utilize antigen-binding portions of immunoglobulin that bind a unique antigen. CARs will bind directly to a specific TAA (green) on the tumor cell. (E) Bispecific antibody (BsAb) armed T cells are genetically engineered antibodies that utilize two arms. One arm will bind a T cell while the other arm binds a specific antigen that is being targeted. This technique does not involve genetic modification but will rather guide T cells towards the antigen that is being targeted by the engineered antibody.
.
Types of adoptive T cell therapy. There are multiple methods developed for ACT approaches. (A) The original therapy utilized was TIL therapy, which utilized unmodified T cells found within a surgically excised tumor that were expanded ex vivo prior to re-infusion. The unmodified TCR (blue) interacts directly with the MHC (purple) on the tumor cell to begin signaling the immune response. (B) TCR-modified T cells are T cells that were obtained from the patient via leukapheresis prior to ex vivo genetic modification. TCR-modification (red) attempts to improve the therapeutic strength of T cells through altering the expression and pairing of TCR α and β chains. (C) TAA-T cells are T cells exposed to multiple, selected tumor antigens via DCs. When the DCs present these selected antigens to the native T cell via MHC and co-stimulation of B7 protein to the T cell via TCR and CD28 binding, the T cell will mount an immune response to each of those antigens. While using multiple antigen targets will increase the effectiveness of the therapy, there is an increased risk of on-target off-tumor toxicity. (D) CAR-modified T cells are like TCR-modified T cells, except CARs (blue) are synthetic receptors that utilize antigen-binding portions of immunoglobulin that bind a unique antigen. CARs will bind directly to a specific TAA (green) on the tumor cell. (E) Bispecific antibody (BsAb) armed T cells are genetically engineered antibodies that utilize two arms. One arm will bind a T cell while the other arm binds a specific antigen that is being targeted. This technique does not involve genetic modification but will rather guide T cells towards the antigen that is being targeted by the engineered antibody.
Tumor infiltrating T cells
TILs are naturally occurring lymphocytes, most commonly T cells, that infiltrate tumor tissue. TILs capable of recognizing tumor-associated antigens can be isolated from surgically resected tumors and expanded ex vivo to create clinically appreciable numbers of TILs.24 Tumor fragments are plated in media containing high doses of IL-2 for two to four weeks to allow for initial cell expansion. Cells demonstrating the strongest anti-tumor effects undergo further expansion for another two weeks. These cells then undergo preconditioning with cyclophosphamide and fludarabine to induce a temporary lymphodepletion, a step which has been shown to increase the persistence of TILs following infusion.25 Finally, the cells are infused back into the patient’s blood stream where they travel to the tumor and stimulate an anti-cancer immune response (Fig. 3). This method has been successful in treating refractory metastatic melanoma.
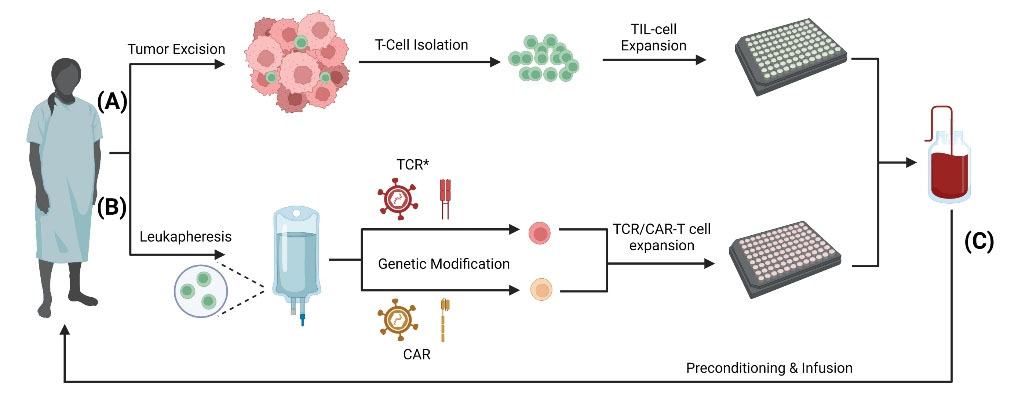
Fig. 3.
T cell isolation and ex vivo expansion. For ACT therapy, T cells require ex vivo manipulation in the form of expansion and/or genetic modification. (A) TILs are isolated from a surgically excised tumor after which they undergo ex vivo expansion. (B) Genetically modified T cells, including TCR-modified (TCR*), CAR-T, and TAA-T start initially with native T cells isolated from the patient’s blood via leukapheresis. Following isolation, the T cells will undergo ex vivo genetic modification through various routes including mRNA transfection, viral vectors, transposons, or homologous recombination.23 Following genetic modification, the T cells will be expanded ex vivo. (C) Following expansion, the cells will then be packaged into an appropriate concentration and dosage for immunotherapy. The patient will undergo preconditioning with cyclophosphamide and fludarabine prior to re-infusion of T cells.25
.
T cell isolation and ex vivo expansion. For ACT therapy, T cells require ex vivo manipulation in the form of expansion and/or genetic modification. (A) TILs are isolated from a surgically excised tumor after which they undergo ex vivo expansion. (B) Genetically modified T cells, including TCR-modified (TCR*), CAR-T, and TAA-T start initially with native T cells isolated from the patient’s blood via leukapheresis. Following isolation, the T cells will undergo ex vivo genetic modification through various routes including mRNA transfection, viral vectors, transposons, or homologous recombination.23 Following genetic modification, the T cells will be expanded ex vivo. (C) Following expansion, the cells will then be packaged into an appropriate concentration and dosage for immunotherapy. The patient will undergo preconditioning with cyclophosphamide and fludarabine prior to re-infusion of T cells.25
One popular modification to this approach uses “minimally cultured” TILs. Minimally cultured TILs forego selection of cells demonstrating pronounced anti-tumor effects and rapidly expand all harvested T cells, regardless of anti-tumor activity. This significantly reduces the cost and total culture time associated with traditional ex vivo expansion techniques.26 The “minimally cultured” approach also allows for a more generalized anti-tumor response. Because this approach targets a wide array of antigens, the TILs are more likely to remain effective even as the tumor inevitably mutates.
In many cancers, the presence of TILs in tumors is associated with a more favorable prognosis compared to those without.27 Because TIL therapy relies on cells that are naturally occurring, they are generally well tolerated with few adverse effects. A recent phase II clinical trial of TIL therapy with post-infusion low-dose IL-2 infusions in metastatic melanoma was well-tolerated by participants with most of the noted adverse effects being related to the IL-2 infusions. The observed adverse effects presented as grade 1-3 events that were able to be managed in a non-ICU setting.28
TILs have shown promise for prognostic and therapeutic effects in many cancers, including metastatic melanoma, ovarian, cervical, head and neck squamous cell cancers, and non-small cell lung cancer.27 One form of TIL-based ACT, Ln-145 was recently awarded breakthrough therapy designation for cervical cancer, something that no other cellular immunotherapy for solid tumors has done before. By achieving breakthrough therapy designation, this therapy will receive expedited development and review by the FDA. However, unlike other forms of ACT such as CAR-T cell therapy, there are no FDA-approved TIL-based therapies for use in cancer presently. Despite these successes, research and clinical trial efforts have slowed due to difficulties overcoming mutating tumor antigens and the immunosuppressive tumor microenvironment (TME). Instead, research efforts have focused on other types of ACT such as TCR and CAR-T therapies.
Chimeric antigen receptor and T cell receptor modified T cells
In the last decade, CAR and TCR modified T cell therapies have demonstrated increased therapeutic potential. This form of ACT involves the isolation of T cells from peripheral blood, followed by ex vivo genetic modification and expansion. Finally, these cells will be infused into the patient following preconditioning. By engineering ACTs specifically to the tumor of interest, this generally offers a more specific and potentially more potent therapeutic effect against the tumor. This strategy offers advantages in situations where TILs not utilizing genetic modification fail to provide adequate anti-tumor effects.29
TCR-modified ACT enhances T cell specificity by altering the expression and pairing of the TCR alpha and beta chains, leading to a more potent tumor antigen-specific TCR.29 CAR-modified ACT generates CARs by combining the antigen-binding portions of an immunoglobulin with the signal-producing component of immunoreceptors and appropriate co-stimulatory molecules. CARs are designed to combine the high specificity of antibodies with the therapeutic potential of T cells. Both TCR and CAR-modified ACTs involve similar production methods.23
Hematologic malignancies have successfully been treated by CAR-T cell therapy. In 2017, tisagenlecleucel, which targets CD19, was approved by the FDA for use in patients less than 25 years of age with relapsing or refractory B-cell precursor acute lymphoblastic leukemia (ALL).30 Since then, a total of six CAR-T cell therapies have been FDA approved for various hematological malignancies as outlined in Table 1. Several ongoing clinical trials for hematological malignancies utilizing various CD molecules that are well-summarized by Vandghanooni et al.31
Table 1.
FDA-approved CAR-T cell immunotherapies
|
Approved CAR-T therapy
|
Target antigen
|
Disease
|
Age indications
|
| Tisagenlecleucel 30 (Kymriah) |
CD19 |
B-cell acute lymphocytic leukemia |
Up to 25 years |
| B-cell non-Hodgkin’s lymphoma |
18 and older |
| Axicabtagene ciloleucel 32 (Yescarta) |
CD19 |
B-cell non-Hodgkin’s lymphoma |
18 and older |
| Follicular lymphoma |
| Brexucabtagene autoleucel 33 (Tecartus) |
CD19 |
Mantle cell lymphoma |
18 and older |
| B-cell acute lymphocytic leukemia |
| Lisocabtagene maraleucel 34 (Breyanzi) |
CD19 |
B-cell non-Hodgkin’s lymphoma |
18 and older |
| Idecabtagene vicleucel 35 (Abecma) |
BCMA |
Multiple myeloma |
18 and older |
| Citacabtagene autoleucel36 (Carvykti) |
BCMA |
Multiple myeloma |
18 and older |
While these techniques demonstrate success in the treatment of hematologic malignancies, solid tumors have been more difficult to target due primarily to a lack of unique tumor target antigens, limited trafficking ability of CAR-T cells to tumor sites, tumor heterogeneity, and antigen loss throughout disease progression.37 However, there have been recent clinical trials utilizing various specific tumor markers for solid tumors that have shown promising results. These trials, several of which are well-summarized in Table 1 from Bagley et al,29 have studied CAR-T cells in gastrointestinal (colorectal, pancreatobiliary), genitourinary (renal cell carcinoma, prostate, ovarian), breast, and lung cancers.
With the overall promise of these therapies, bioengineering is at the forefront of guiding the future of CAR-T cell therapy. Researchers are now studying the use of nanotechnology to develop antigen-recognizing CAR-T cells in vivo. These synthetic nanoparticles can be designed to express CAR genes which bind to molecules of interest, as well as proteins that selectively bind to T cells and help to insert the CAR genes into the cell’s DNA. Currently, this therapy is being explored in pre-clinical animal models and researchers have successfully generated therapeutic levels of T cells by using this in situ delivery system.38 This type of system could potentially reduce costs and ease storage difficulties seen in ex vivo therapies but will require further laboratory research prior to use.
Tumor associated antigen T cells
Tumor-associated antigen cytotoxic T cells (TAA-Ts) are another alteration of the original CAR-T cell therapy. While CAR-T cell therapy selectively targets one antigen, TAA-T cell therapy aims to express multiple tumor antigens. This is done by exposing ex vivo white blood cells to DCs with selected TAAs. By introducing these TAAs via MHC to the native TCR, the T cells can mount an immune response against multiple TAAs. These cells are then expanded prior to infusion. In a recent phase I trial, TAA-Ts were evaluated for treatment of relapsed/refractory solid tumors utilizing the following targets: Wilms tumor gene 1 (WT1), preferentially expressed antigen of melanoma (PRAME), and survivin (BIRC5).39 WT1 has been shown to be overexpressed in Wilms tumor,40,41 sarcomas,42 ovarian,43 and prostate cancers.44 PRAME, involved in cell proliferation and survival,45 is associated with advanced disease and poor prognosis.45-48 BIRC5, a gene that is highly expressed during fetal development and is absent in most mature tissues,49 is overexpressed in many malignancies50-52 and associated with disease characteristics including chemotherapy resistance, disease recurrence, and decreased survival.39 In this phase I trial, fifteen patients received infusions of the TAA-T cell therapy, of which 11/15 (73%) responded favorably. Overall, the treatment was tolerated well without dose-limiting toxicities or infusion-related adverse events.
While TAA-T cell therapy needs to be investigated further, the utilization of multi-antigen targeting may mitigate some of the previously discussed limitations of CAR-T cell therapy. By utilizing TAA-T cells that employ multiple unique targets rather than only one target as in CAR-T therapy, the probability that the therapy will be able to effectively recognize and target heterogenous tumor cells increases substantially. Another benefit is that this multi-target treatment has a greater chance of withstanding the rapid evolution of a tumor as individual targets mutate. Finally, by targeting multiple antigens at once, the treatment eliminates the logistical and financial complications associated with repeating T cell expansion several times.39
Despite the advantages of having multiple targets, the specific antigens for TAA-T cell therapy have to be carefully selected to lessen the on-target off-target toxicities. The lack of dose-limiting toxicities, infusion-related adverse events, and cytokine release syndrome (CRS) in the Hont et al phase I clinical trial shows promise that selecting appropriate targets and doses can mitigate adverse events while providing effective immunotherapy.39 While there are no FDA-approved TAA-T cell therapies, several clinical trials are ongoing, as summarized in Table 2.
Table 2.
Current TAA-T cell therapy clinical trials
|
NCT # |
Targeted condition or disease
|
Antigen of interest
|
Phase
|
| NCT02475707 |
Acute lymphoblastic leukemia |
WT1, PRAME, Survivin |
Phase 1 |
| NCT04679194 |
Acute myeloid leukemia, myelodysplastic syndrome |
WT1, PRAME, Survivin |
Phase 1 |
| NCT02494167 |
Acute myeloid leukemia, myelodysplastic syndrome |
WT1, PRAME, NY-ESO-1, Survivin |
Phase 1 |
| NCT03843294 |
Hodgkin’s lymphoma, diffuse large B cell lymphoma |
WT1, PRAME, Survivin |
Phase 1 |
NCT05134740
NCT01333046 |
Hodgkin’s lymphoma, non-Hodgkin’s lymphoma |
NY-ESO-1, MAGE A4, PRAME, Survivin, SSX |
Phase 1 |
| NCT02291848 |
Multiple Myeloma, high risk MGUS/ Smoldering myeloma |
NY-ESO-1, MAGE A4, PRAME, Survivin, SSX |
Phase 1 |
| NCT02203903 |
Relapsed/ refractory hematological malignancies, acute myeloid leukemia, myelodysplastic syndromes |
WT1, PRAME, Survivin |
Phase 1 |
| NCT02789228 |
Solid tumors (Ewing sarcoma, neuroblastoma, rhabdomyosarcoma, soft tissue sarcomas, osteosarcoma, adenocarcinoma, esophageal carcinoma, renal cell carcinoma) |
WT1, PRAME, Survivin |
Phase 1 |
| NCT05238792 |
Solids tumors (Ewing Sarcoma, Wilms tumor, neuroblastoma, rhabdomyosarcoma, soft tissue sarcoma, osteosarcoma) |
WT1, PRAME, Survivin |
Phase 1 |
| NCT03192462 |
Pancreatic cancer |
NY-ESO-1, MAGE A4, PRAME, Survivin, SSX2 |
Phase 1/2 |
| NCT04511130 |
Acute myeloid leukemia |
WT1, PRAME, NY-ESO-1, Survivin |
Phase 2 |
| NCT03093350 |
Breast cancer (metastatic or locally recurrent unresectable disease) |
NY-ESO-1, MAGE A4, PRAME, Survivin, SSX2 |
Phase 2 |
| NCT03535246 |
Cancers positive for at least one of the following antigens: GD2, mesothelin, P16, MMP, Melan A, MAGE A1, MAGE A3, MAGE A4 |
GD2, mesothelin, P16, MMP, Melan P, MAGE A1/A3/A4 |
Phase 2 |
Bispecific antibody armed T cells
A more recent development in ex vivo T cell therapy is the development of bispecific antibodies (BsAbs), which utilize a single molecule that has combined the binding sites of two monoclonal antibodies. This BsAb is engineered to bind both an activated cytotoxic T cell as well as a unique TAA, effectively “arming” the cytotoxic T cell to a specific TAA. Thus, these BsAb treatments increase perforin and granzyme activity towards tumors expressing unique TAAs.53 The “arming” of T cells enhances their therapeutic effect while minimizing adverse effects of T cell therapy previously discussed. BsAb treatment strategies represent what is known as “off the shelf” therapies, meaning that collecting patient cells is not required, unlike other forms of ACT. Rather, BsAb treatments serve as an immediately available therapeutic option, in stark contrast to CAR/TCR-modified T cell therapy which often requires weeks to develop and may lose viability by the time of their use due to tumor heterogeneity.29 Compared to the living and self-expanding CAR-T cells, BsAbs persist for a shorter duration and rely more heavily on frequent application.54 As of 2015, the first BsAb therapy, blinatumomab, was fully approved for use in acute B-cell lymphoblastic leukemia.6 A list of FDA-approved bispecific antibody therapies is seen below in Table 3.
Table 3.
FDA-approved bispecific antibody immunotherapies
|
Bispecific antibody immunotherapy
|
Targeted condition or disease
|
Target antigens
|
Age indications
|
| Blinatumomab (Blincyto)55 |
B-cell acute lymphoblastic leukemia |
CD3/CD19 |
All ages |
| Amivantamab (Rybrevant)56 |
Non-small cell lung cancer |
EGFR/MET |
18 and older |
| Tebentafusp (Kimmtrak) 57 |
Metastatic uveal melanoma |
CD3/gp100 |
18 and older |
Due to the short longevity of BsAbs in the body, there has been a surge in the use of BsAbs that are “armed” to activated T cells, effectively combining the strengths of BsAb and CAR-T cell therapy. While still in early development, one group investigated the use of BsAb ex vivo armed T cells (EATs) in various cancer cell lines and patient-derived xenograft mouse models. The generated EATs used anti-CD3 single-chain variable fragment (scFv) attached to the light chain of a tumor-binding IgG (IgG-[L]-scFv). They found that compared to isolated BsAb injections, EATs infiltrated tumors more quickly and were associated with more prolonged survival. Additionally, compared to BsAb therapy alone, EATs released significantly fewer cytokines, particularly TNF-α, in vitro without affecting trafficking ability or tumoricidal activity of the EATs in vitro or in vivo.54 Therefore, these generated EATs show promise in promoting tumoricidal effects while preventing CRS, a well-known adverse effect of ex vivo T cell54 and BsAb therapy.6
Neurotoxicity is another obstacle for both CAR-T cell and BsAb therapy alike. The neurotoxic effects of EAT and CAR-T cells have not been compared directly. However, previous studies have shown that while GD2-CAR-T cells cause neurological damage in mouse models, GD2-BsAbs and GD2-EATs did not.58 This suggests BsAb and EAT therapies may display reduced neurotoxic side effects compared to traditional CAR-T cell therapy. By combining the strengths of BsAbs and CAR-T cell therapy, EATs further increase the cancer therapeutic arsenal. While more research is required for full clinical application, early results suggest that EATs generate increased tumoricidal effects while minimizing common adverse effects such CRS and neurotoxicity.
NK cell therapy
NK cell overview
NK cells, which are lymphocytes derived from a common progenitor of T and B cells, have also shown promise as cancer therapeutics. These cells initiate and regulate both the innate and adaptive immune systems. As part of the innate immune system, NK cells are important for tumor surveillance because they have the unique ability to kill tumor cells without T and B cells.59,60 NK cells also play an important role in the adaptive immune system because of their ability to engage in reciprocal interactions with DCs, macrophages, and T cells.
The immune surveillance that NK cells provide is invaluable, and NK cell deficiencies are associated with increased occurrence of malignancies.61 Since NK cell anti-tumor properties were first discovered in the 1970s,59 the potential for NK cell-based immunotherapies against cancer continues to be actively investigated.
NK cell therapies have several advantages over traditional T cell therapies, including fewer side effects, faster clinical response, and a greater ability to communicate with other immune cells.62 While NK cell immunotherapy has achieved success in treating hematological cancers,63 this branch of cellular immunotherapy must overcome many of the issues other cellular therapies face.64,65 NK cells struggle to penetrate solid tumors, which may be due to their inability to travel to the tumor site, as well as the immunosuppressive effect of the TME,66,67 as depicted in Fig. 4. Additionally, the use of NK cells has been limited by difficulty in obtaining sufficient cell numbers due to their rarity in the peripheral blood and poor ex vivo expansion methods. With the knowledge of how tumors evade immunosurveillance, researchers have turned toward developing therapies that restore the cytolytic abilities of NK cells. These therapies, which include cytokine-induced memory-like natural killer cells (CIMLs), CAR modified NK cells, and cytokine-induced killer (CIK) cells were developed to improve NK cell tumor infiltration and activation.68,69
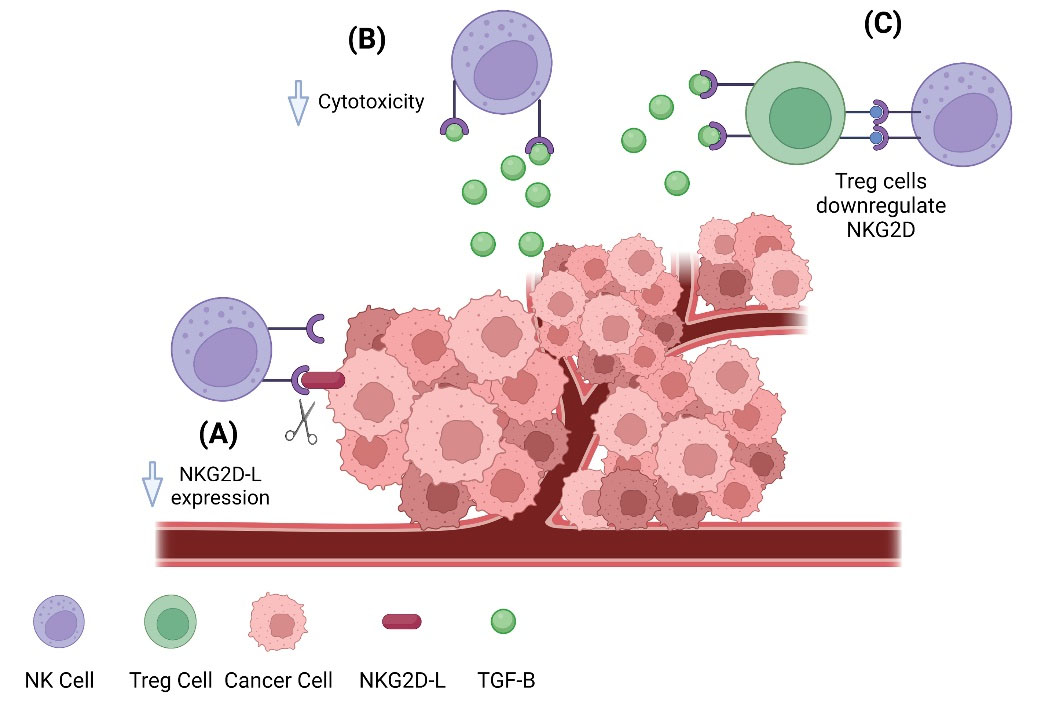
Fig. 4.
Tumor suppressing NK cell activity. Tumor cells utilize a variety of tools to suppress the apoptotic actions of NK cells, including destroying NKG2D-L, releasing TGF-β, and recruiting regulatory T cells to downregulate NKG2D receptors. (A) The natural killer group 2, member D ligand (NKG2D-L) expressed on the tumor cell surface binds to the natural killer group 2, member D receptor (NKG2D) and activates natural killer cells, which allows them to destroy cancerous cells. Tumor cells can release metalloproteinases that function to cleave the NKG2D-L from their surface, preventing NK cells cytotoxic activation. (B) The cytokine transforming growth factor-β (TGF-β) is released by tumor cells during periods of growth and progression to malignancy. TGF-β functions as an immunosuppressant and can interrupt the NKG2D/NKG2DL system on NK cells by downregulating NKG2D 148. The cytokine transforming growth factor-β (TGF-β) is released by tumor cells during periods of growth and progression to malignancy. TGF-β functions as an immunosuppressant and can interrupt the NKG2D/NKG2DL system on NK cells by downregulating NKG2D.148 (C)Regulatory T cells (Tregs) play a role in suppressing the cytotoxic function of NK cells. This mechanism also relies on the immunosuppressant cytokine TGF-β, which activates Tregs who in turn inactivate NK cells, thus decreasing their ability to destroy cancerous cells.
.
Tumor suppressing NK cell activity. Tumor cells utilize a variety of tools to suppress the apoptotic actions of NK cells, including destroying NKG2D-L, releasing TGF-β, and recruiting regulatory T cells to downregulate NKG2D receptors. (A) The natural killer group 2, member D ligand (NKG2D-L) expressed on the tumor cell surface binds to the natural killer group 2, member D receptor (NKG2D) and activates natural killer cells, which allows them to destroy cancerous cells. Tumor cells can release metalloproteinases that function to cleave the NKG2D-L from their surface, preventing NK cells cytotoxic activation. (B) The cytokine transforming growth factor-β (TGF-β) is released by tumor cells during periods of growth and progression to malignancy. TGF-β functions as an immunosuppressant and can interrupt the NKG2D/NKG2DL system on NK cells by downregulating NKG2D 148. The cytokine transforming growth factor-β (TGF-β) is released by tumor cells during periods of growth and progression to malignancy. TGF-β functions as an immunosuppressant and can interrupt the NKG2D/NKG2DL system on NK cells by downregulating NKG2D.148 (C)Regulatory T cells (Tregs) play a role in suppressing the cytotoxic function of NK cells. This mechanism also relies on the immunosuppressant cytokine TGF-β, which activates Tregs who in turn inactivate NK cells, thus decreasing their ability to destroy cancerous cells.
NK cell therapy
NK cells may be harvested using donor cells, autologous cells, umbilical cord blood70 or pluripotent stem cells.71 Presently, most clinical trials utilize peripherally derived NK cells. NK cells are first separated from other peripheral cells using CD3- CD56+ markers to avoid life-threatening illnesses following cell transplants, such as graft-versus-host-disease (GVHD) and post-transplant lymphoproliferative disorder.72 Cells are then plated in medium containing IL-2, a step which has bene shown to improve cytotoxicity and viability.72,73 NK cells can also be co-cultured with other immune cells, including monocytes, macrophages, and DCs, to produce an even more cytotoxic NK response.74
Ultimately, the efficacy of NK cell immunotherapy on solid tumors depends on the infused cell’s ability to reach the tumor site and penetrate the hypovascular, dense network of extracellular matrix within the TME.75,76 Chemokines such as CCR2, CCR5, and CXCR3 play an essential role in the migration of NK cells and the manipulation of the chemokine receptor profile on NK cells has been shown to improve NK cell infiltration into the TME.77 Genetically altering NK cell chemokine receptors has yielded promising results in pre-clinical models of several different cancers, including ovarian, renal cell, and lung cancer.78-81
Cytokine-induced memory-like NK cells
NK cells were initially thought to be short-lived and incapable of remembering previous exposures to antigens. However, Cooper et al observed that murine NK cells, after activation with IL-12, IL-15, and IL-18, exhibited memory-like properties by producing high levels of IFN-γ when stimulated weeks later. These cells became known as cytokine-induced memory-like (CIML) NK cells.82 CIML NK cells became an attractive option for adoptive cellular therapy because of their anti-tumor response and ability to persist in vivo in pre-clinical models of melanoma and hematological malignancies.83,84
Based on these promising pre-clinical results, a phase 1 clinical trial of CIML NK cells in patients with relapsed/ refractory AML was completed. Patients were lymphodepleted using fludarabine and cyclophosphamide before receiving the CIML NK cells, which were generated using haploidentical donors. Results of this study demonstrated robust proliferation and expansion of the CIML NK cells without major toxicities such as CRS or GVHD.85 Currently, researchers are continuing to explore the safety and efficacy of CIML NK cell immunotherapy, and have begun to study their use in solid tumors. A list of ongoing clinical trials involving CIML NK cells can be found below in Table 4.
Table 4.
Ongoing CIML NK clinical trials
|
NCT#
|
Targeted cancer or disease
|
Phase
|
| NCT04024761 |
Acute myeloid leukemia, myelodysplastic syndromes, juvenile myelomonocytic leukemia |
Phase 1 |
| NCT04290546 |
Head and neck squamous cell carcinoma |
Phase 1 |
| NCT05580601 |
Acute myeloid leukemia |
Phase 1/2 |
| NCT04634435 |
Multiple myeloma |
Phase 1/2 |
| NCT04354025 |
Acute myeloid leukemia |
Phase 2 |
Chimeric antigen receptor NK cells in immunotherapy
Similar to CAR-T cells, CAR-NK cells have been genetically engineered to express CARs uniquely selected to target specific antigens. CAR-NK cells are thought to have several advantages over CAR-T cells, including the ability to trigger tumor cell death independent of the tumor antigen via NK cell receptors stimulation.86 CAR-NK cells have also demonstrated a decreased risk of GVHD compared to CAR-T cells because the NK cells are recognized as “self” cells, protecting them from the alloreactive effects of T cells. This in turn improves their safety profile.87,88 Additionally, CAR-NK cells have been shown to destroy tumor cells using both CAR-independent and CAR-dependent mechanisms.89 Thus, it may be less likely that tumor cells can evade CAR-NK cells when compared with CAR-T cells, despite the tumors’ ability to downregulate CAR target antigen. Further, NK cells lack the ability to produce autocrine growth factors, limiting their lifespan and thus decreasing the risk of negative off-target effects.90
Many preclinical studies have examined the efficacy of CAR-NK cells in hematologic malignancies and demonstrated strong favorable results.91 To date, there has only been one clinical trial published examining the efficacy in CAR-NK cells used in relapsed or refractory CD-19 positive cancers.92 These cancers include chronic lymphocytic leukemia (CLL) and NHL. In this study (n=11), 8 patients (73%) treated with CAR-NK cells responded to the therapy, and of those, all but one had a complete remission. Importantly, none of the patients experienced any of the well-known side effects of CAR-T cell therapy, including GVHD, CRS, or neurotoxicity. These results are promising and should serve to highlight the excellent safety profile of CAR-NK cell therapy. Interestingly, results of another small pilot study examining the use of CAR-NK cells in acute myeloid leukemia (AML) yielded different results. In this study (n=3), none of the three patients with relapsed or refractory AML had a clinical response to CAR-NK cell therapy targeting CD-33.93 However, small sample sizes in both pilot studies limit the power of these studies and further investigation is needed to fully understand the efficacy of CAR-NK therapy.
While many studies of CAR-NK cells focus on their use in hematologic malignancies, this therapy shows encouraging, although statistically underpowered, results in solid tumors. In a small-scale pilot study (n=3), Xiao et al found CAR-NK cells to be effective in the treatment of metastatic colon cancer. This study featured CAR-NKs targeting NK group 2 member D ligand (NKG2DL), a molecule often expressed by tumor cells which activates NK cells. Two of these patients with malignant ascites were treated with allogeneic haploidentical NK cells via direct local injection into the peritoneal cavity. Both patients experienced significant reductions in the number of cancer cells in ascites fluid as well as reduction in volumes of ascites fluid. The third patient received ultrasound-guided percutaneous allogeneic haploidentical CAR-NK cell injection. Following therapy, the size of the tumor decreased by 40% as measured by Doppler ultrasound.91 While the small sample size of this trial limits the impact of the results, this evidence supports continued research and development of safe CAR-NK cell therapies. At the present time, this is the only study published using CAR-NK cells in solid tumor disease, although there are multiple ongoing clinical trials focusing on solid malignancies as outlined in Table 5.
Table 5.
Ongoing clinical trials studying CAR-NK therapy in solid tumors
|
NCT#
|
Targeted cancer or disease
|
Antigen of interest
|
Phase
|
| NCT05194709 |
Advanced solid tumors |
5T4 |
Early Phase 1 |
| NCT03692663 |
Prostate cancer |
PSMA |
Early Phase 1 |
NCT05213195
NCT05248048 |
Colorectal cancer |
NKG2D |
Phase 1 |
| NCT05528341 |
Relapsed/ refractory solid tumors |
NGK2D |
Phase 1 |
| NCT05507593 |
Small cell lung cancer |
DLL3 |
Phase 1 |
| NCT05410717 |
Ovarian cancer, testicular cancer, endometrial cancer |
CLDN6 |
Phase 1/2 |
| NCT04847466 |
Gastroesophageal junction cancers, head and neck squamous cell carcinoma |
PD-L1 |
Phase 2 |
These and other clinical trials have demonstrated the exciting potential for NK cells to be used as cancer therapy (Fig. 4). Currently, most studies are testing these therapies using autologous NK cells, meaning cells from the patient’s body. In the future, researchers may use allogeneic NK cells, or cells from a matched donor, to produce NK cell therapeutics on a larger scale at a potentially lower cost. Ideally, a standardized method of cell harvesting and infusion will simplify the logistics of this process and reduce variability regarding clinical outcomes, allowing researchers to better compare therapies.87
Cytokine-induced killer cells in immunotherapy
CIK cells are a heterogenous group of immune cells with both NK and T cell properties. CD3+ CD56+ CIK cells are able to kill both hematological and solid tumor cells in an MHC-unrestricted manner based on interactions between NKG2D molecules on CIK cells and UBLPs on tumor cells (Fig. 4). Using CIK cells to treat cancer is a relatively inexpensive method and does not rely on feeder or accessory cells, unlike NK cells. Another benefit is that the heterogeneity of the CIK cell population allows cells to attack a wide variety of tumor antigens.94
Many clinical trials have demonstrated that CIKs have anti-tumor effects in a variety of cancers. In a meta-analysis of clinical trials assessing the efficacy of CIK therapy in patients with hepatocellular carcinoma, CIK therapy significantly prolonged progression-free survival and improved quality of life when used alongside the standard of care therapy.95 Other clinical trials have demonstrated that the use of CIK therapy prolongs median overall survival and has a favorable effect on progression-free survival in colon, lung, pancreatic, and ovarian cancers.96-99
These trials demonstrate the utility of CIK cells as a form of immunotherapy while also raising questions regarding the optimal length of maintenance treatments and frequency of CIK infusions. Further studies will be needed to compare efficacy between autologous and allogeneic CIK cells. Currently, researchers are continuing to explore the utility of CIK therapy individually or in combination with DC therapies.100
Dendritic cells
Dendritic cells overview
Dendritic cells are another tool used to enhance the immune response to tumor cells. DCs are cells of the innate immune system, scanning peripheral blood and tissues for pathogens such as viruses. Once in contact with a presentable antigen, it is loaded onto an MHC Class I or II molecule, which is then recognized by a TCR to activate helper T cells and cytotoxic T cells.101 As the DC matures, several changes occur that allow the cell to induce a T cell response more effectively, including an increase in the expression of chemokine receptors and surface MHC I/II molecules.101 Different subsets of DCs can be found in virtually any organ, including the lungs, stomach, and intestines. Clinical studies have focused on blood and skin derived DCs due to accessibility via peripheral blood samples or skin samples, such as from dermatome-cut skin preparation.102
Dendritic cell immunotherapy
Most DC-based immunotherapies have been studied in the form of DC-based anti-tumor vaccines. These vaccines have been shown to be safe although their clinical efficacy has been inconsistent.103 To develop vaccines, DCs are extracted from a patient and matured ex vivo with a cocktail of toll-like-receptor ligands and prostaglandins. Then, the DCs are “loaded” with tumor-associated antigens. Some examples of tumor-associated antigens include synthetic long peptides and melanoma-associated antigens.104,105 Finally, the DC vaccine is administrated to the patient. From there, the DCs can travel to lymph nodes and stimulate an anti-tumor T cell response.106
DC vaccines have been used in the treatment of cancer for decades, but they are especially efficacious in the treatment of melanoma. In one study, patients with stage III and IV melanoma were administered monocyte derived DCs loaded with keyhole limpet hemocyanin and mRNA encoding gp100 and tyrosinase tumor antigens. Patients received either intranodal or combined intradermal/intravenous injections. Using delayed-type hypersensitivity (DTH) skin tests, tumor antigen-specific responses were detected following either type of injection. Stronger immune responses as determined by the DTH tests were correlated with longer OS in patients with stage IV melanoma. Despite these promising results, side effects of the vaccinations included hepatotoxicity and pneumonitis, potentially due to the use of Bacillus Calmette-Guérin (BCG). A retrospective cohort of stage IV melanoma patients found that patients with TAA-specific CD8 T cells within skin-infiltrating lymphocytes had significantly improved OS.107
The concept of pre-conditioning the vaccination site with a potent recall antigen, such as tetanus/diphtheria (Td) toxoid, is also being explored to improve the clinical efficacy of DC vaccinations. One trial studied the efficacy of pre-conditioning the vaccination site with mature DCs versus Td toxoid. Following pre-conditioning, patients were administered DCs pulsed with pp65, an antigen expressed in over 90% of glioblastoma multiforme tumors. Patients receiving pre-conditioning with the toxoid demonstrated improved DC migration and OS.108 Given the small sample size used in this study (n=12), this method of DC-based vaccination will need to be investigated in future trials.
Future directions for DC therapy
Undoubtedly, DC vaccine immunotherapy represents an exciting avenue of personalized cancer therapy. Moving forward, researchers have a multitude of questions that need to be explored to ensure the efficacy and safety of this treatment. The maturation period and tumor-antigen cocktail will need to be optimized, as well as the most ideal site of delivery of DC vaccines for each malignancy. While intranodal injection may work for cancers such as melanoma, this approach may not be most advantageous for patients with glioblastoma. Combining DC therapy with other treatments, such as radiotherapy or chemotherapy, is a promising route that is currently being explored.
Interestingly, in addition to direct cytotoxicity, radiotherapy can have both immunosuppressive and immunostimulatory effects.109 As the tumor cells are irradiated and destroyed, they release a storm of tumor antigens that can be taken up by circulating DCs. These antigens can be used as a blueprint for a tumor-specific attack by endogenous immune cells.110 This suggests a potential synergistic effect between radiation therapy and DC-based immunotherapy. This concept has been previously studied in vivo111 and in clinical trials for patients with HER2/neu overexpressing ductal carcinoma in situ of the breast, which yielded hopeful results.112 In their study, Czerniecki et al used breast microcalcifications, as measured by MRI, to estimate the extent of disease. The majority (6/11) of patients who received DC vaccines prior to resection had tumors over 50% smaller than the area predicted by microcalcifications. This suggests DC vaccines may improve prognosis and lead to tumor regression in these patients. Currently this concept is being investigated with larger cohorts of patients and there are over 250 ongoing clinical trials featuring the use of anti-tumor DC-based vaccines, several of which are described in Table 6.
Table 6.
Ongoing clinical trials studying dendritic cell therapy in solid tumors
|
NCT#
|
Targeted cancer or disease
|
Phase
|
| NCT05504707 |
Breast cancer |
Phase 1 |
| NCT04078269 |
Non-small cell lung cancer |
Phase 1 |
| NCT02919644 |
Colorectal cancer |
Phase 2 |
| NCT00458536 |
Renal cell carcinoma |
Phase 1 |
| NCT01197625 |
Prostate cancer |
Phase 2 |
| NCT00799110 |
Ovarian cancer |
Phase 2 |
| NCT02496273 |
Gastric cancer |
Phase 1 |
At present, DC-based monotherapy and combination therapies have demonstrated longitudinal anti-tumor responses.113,114 However, multiple challenges need to be addressed to improve outcomes in cancer. These challenges include proper selection of synergistic treatment modalities and effective inclusion of sensitizing agents within the vaccines. Additionally, optimal treatment lengths and delivery strategies for of these vaccines remain unresolved. Finally, clinical trials utilizing DCs have been inconsistent and costly.115 Learning how to best utilize DC-based vaccines represents an inspiring frontier in immunotherapy research.
Circulating tumor cells
CTC overview
CTCs are cancer cells released from solid tumors into the bloodstream.116 First identified in 1869 by Thomas Ashworth, CTCs are thought to separate from tumors, either as single CTCs or in clusters, through a process known as epithelial-mesenchymal transition. From there, they travel to distant sites within the body where they become “seeds” for distant metastasis.117,118 Therefore, researchers believe that CTCs may play an invaluable role in cancer diagnosis and prognosis. However, several challenges have limited their utility. CTCs are sparse within the blood, making it difficult to collect enough cells in a blood sample. CTCs are also heterogenous in that they differ in which biomarkers are expressed on their surface. Thus, a variety of methods which exploit chemical and molecular properties of these cells must be used to harness their power.
CTC isolation and enumeration
CTCs must first be isolated and enumerated from a whole blood sample, using either label dependent (affinity-based) or label-independent selection.119 The label-dependent method relies on positive selection using cell surface markers targeted towards CTCs, such as epithelial cell adhesion molecule (EpCAM) or cytokeratin (CK) markers.120 The label-independent method relies on negative selection by size or other biophysical properties.119
Presently, one of the most powerful tools used for identification and enumeration of CTCs is the CellSearch CTC system, the only FDA-approved platform for CTC isolation. CellSearch isolates CTCs using a label-dependent process. Anti-EpCAM antibodies are used to positively select for CTCs before cells are stained for epithelial markers (CK8, CK18, CK19), a leukocyte marker (CD45), and a nuclear marker (6-diamidino-2-phenylindole or DAPI). Cells are considered CTCs if they are CK+, CD45-, and have a nucleus.121 While CellSearch has great potential for a variety of analyses, it is only officially approved by the FDA for in vitro diagnostic use and monitoring patients with breast, prostate, and colorectal cancers.122
Recently, several platforms have been used to successfully isolate CTC clusters, in addition to individual CTCs. One such platform consists of a 3D scaffold chip coated with thermosenstivie gelatin. This chip can capture individual CTCs and clusters and, when heated to 37 ˚C, the gelatin dissolves without destroying the integrity of the clusters and maintaining viability.123 In 2015, the Cluster Chip, a microfluidic platform capable of capturing clusters made of only two CTCs, was created.123 Several microfluidic methods have further improved our ability to capture and isolate CTC clusters. Even newer techniques are being developed to allow separation of single CTCs from CTC clusters, which may provide valuable insight into in vivo cancer development.124 Nonetheless, further pre-clinical and clinical studies are required to ascertain the utility of these platforms.
Preclinical applications of CTCs
CTCs have been used to model small cell lung cancer (SCLC) patients using circulating tumor cell-derived explants (CDXs). These CDXs are created by taking CTCs from patients with SCLC and injecting the cells into immunocompromised mouse models.116 When treated with etoposide and platinum therapies, the CDXs therapeutic response mirrored that of the patients from which the CTCs were taken (Fig. 5). This was found to be true for patients with chemosensitive SCLC as well as those with chemorefractory disease. This method is beneficial because it serves as a less invasive method of studying rapidly-progressive cancers such as SCLC.125 Thus, CDXs may provide a platform to screen new treatments, identify predictive and pharmacodynamic biomarkers, and investigate mechanisms of resistance to better understand the progression of cancer.126
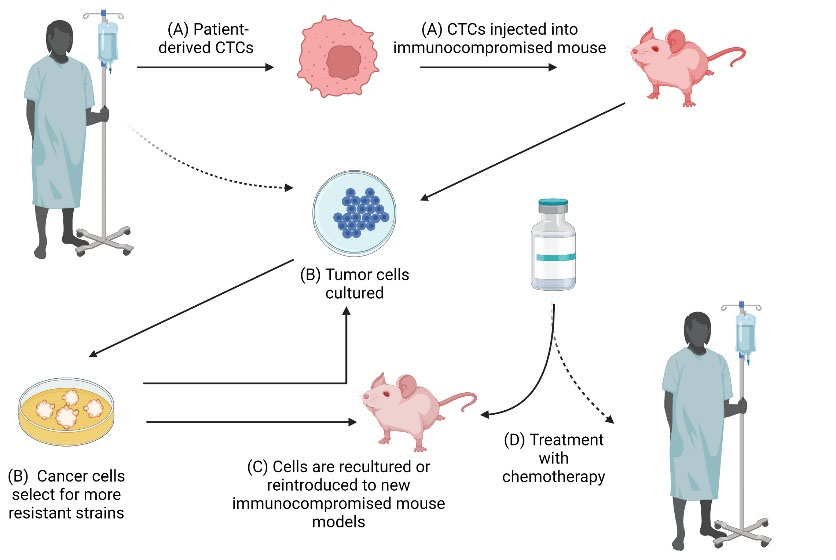
Fig. 5.
Mouse models and CTCs. Immunocompromised mouse models can be used to generate large numbers of CTC samples. Studying CTCs in mouse models can help to predict how patients with metastatic cancers will respond to certain treatments. (A) Patient-derived CTCs are injected into immunocompromised mice to form patient-derived xenografts (“xenopatients”). (B) Tumor cells are then cultured from the mice. Over time, the cultured cancer cells begin to select for more resistant strains. Cells are cultured for roughly four weeks to simulate a human patient’s internal environment (symbolized by dashed arrow) without drastic clonal changes, as have been seen in more long-term cultures. (C) Cultured tumor cells can then either be reintroduced into a new mouse model or can be re-cultured. (D) Chemotherapy can then be administered to these mouse models. Studies have shown that mice display a similar sensitivity to chemotherapy compared to their human counterparts (as shown by the dashed arrow).
.
Mouse models and CTCs. Immunocompromised mouse models can be used to generate large numbers of CTC samples. Studying CTCs in mouse models can help to predict how patients with metastatic cancers will respond to certain treatments. (A) Patient-derived CTCs are injected into immunocompromised mice to form patient-derived xenografts (“xenopatients”). (B) Tumor cells are then cultured from the mice. Over time, the cultured cancer cells begin to select for more resistant strains. Cells are cultured for roughly four weeks to simulate a human patient’s internal environment (symbolized by dashed arrow) without drastic clonal changes, as have been seen in more long-term cultures. (C) Cultured tumor cells can then either be reintroduced into a new mouse model or can be re-cultured. (D) Chemotherapy can then be administered to these mouse models. Studies have shown that mice display a similar sensitivity to chemotherapy compared to their human counterparts (as shown by the dashed arrow).
The use of CTCs in pre-clinical models also has potential to develop therapeutic targets in vivo. If CTCs are eliminated in vivo, the risk of metastasis may be reduced, and survival outcomes may improve. This was studied in animal models by injecting green fluorescent protein (GFP)-expressing CTCs into mice. Photodynamic therapy was used to clear these GFP-expressing CTCs, and then evaluated the therapeutic effect of CTC clearance (Fig. 5). In cultured cells, apoptotic agents such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), have been used to target CTCs to reduce the probability of metastasis.127 These studies demonstrate the possible utility of CTC-targeting cancer therapies.
CTCs and CDX give researchers insight into the severity of cancer as well as a window into how treatment will impact these diseased cells. However, the samples can only be used for limited amounts of time in an ex vivo setting, as the most resilient clones to survive in culture will begin to predominate which will reflect the patient’s true tumor biology less accurately. Studies done on SCLC derived CDX in culture for 4 weeks maintained their neuroendocrine phenotype and were reversible when reimplanted in vivo.126 In addition to a limited timeframe, researchers must also be conscious of CTC biopsy quality. A quality biopsy is necessary to create an assay that captures a representative tumor sample while maintaining tumor specificity.128
Clinical applications of CTCs
Clinical research analyzing the utility of CTCs can be categorized broadly as related to either CTC enumeration or molecular phenotype. For over a decade, it has been shown that CTC counts may be able to predict cancer diagnosis and prognosis. In patients without cancer screened for CTCs, the presence of CTCs has been associated with higher risk of malignancy. In patients with cancer, higher levels of CTCs and CTC clusters are associated with worse clinical outcomes. Conversely, stable or declining levels of CTCs are associated with improved clinical outcomes (Fig. 6).129,130 For patients previously treated for cancer, the detection of CTCs several years after diagnosis is associated with higher rates of relapse and an unfavorable prognosis.131,132 Interestingly, CTCs are even being used to stage breast cancer: the stage cM0(i+) refers to cases in which CTCs are detected but there is no clinical or radiological evidence of metastasis.133
Researchers hope that studying the molecular phenotype of CTCs may serve as a less invasive method of assessing the tumor’s molecular characteristics, which may guide diagnosis and treatment. Unfortunately, studies related to CTCs at the molecular level are fewer and have yielded less consistent results. Fehm et al found a limited correlation between CTC HER2 status assessed by PCR and HER2 status of the tumor determined by histopathological methods.134 Pestrin et al attempted to treat HER2- breast cancer patients with HER2+ CTCs with the HER2 inhibitor, lapatinib. Only 7% of patients with detectable CTCs had HER2+ CTCs and, despite being treated with lapatinib, all but one of those patients experienced progressive disease.135 Despite these results, there are several ongoing clinical trials, including the DETECT trials, which are attempting to further study the clinical applications of CTCs based on their molecular phenotype.136
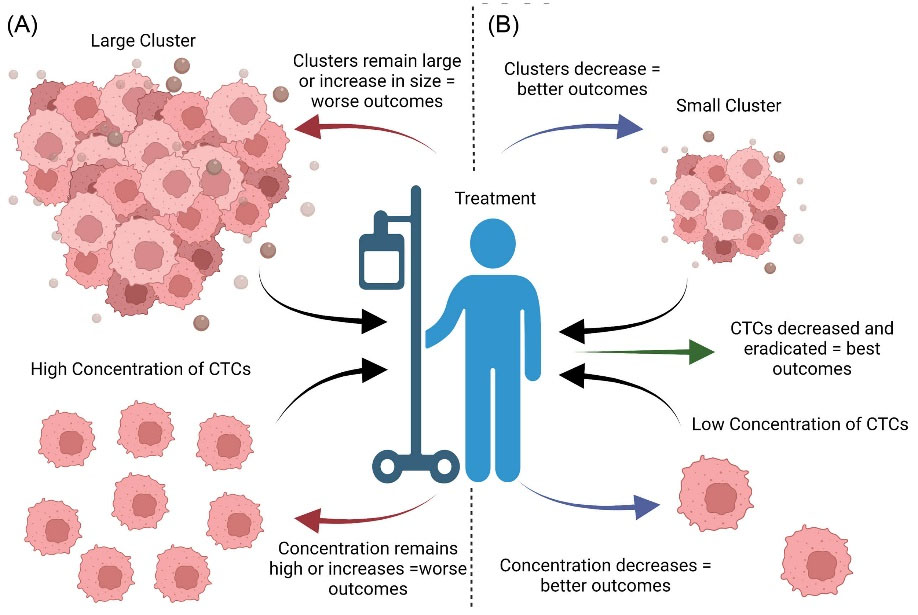
Fig. 6.
Impacts of CTC size and concentration on prognosis. The impact of treatment on CTC concentration and size. (A) Patients with biopsies containing larger CTCs or higher concentrations of CTCs have worse survival outcomes. Those who continue to have larger CTCs or higher concentrations with treatment or develop these characteristics despite treatment have worse outcomes. (B) Individuals with smaller CTCs or smaller concentrations of CTCs have better survival outcomes. Patients with CTCs that shrink or decrease in concentration following medical therapy have better outcomes. Those who start out with lower CTC burdens and respond to therapy are more likely to be cured of their cancer.
.
Impacts of CTC size and concentration on prognosis. The impact of treatment on CTC concentration and size. (A) Patients with biopsies containing larger CTCs or higher concentrations of CTCs have worse survival outcomes. Those who continue to have larger CTCs or higher concentrations with treatment or develop these characteristics despite treatment have worse outcomes. (B) Individuals with smaller CTCs or smaller concentrations of CTCs have better survival outcomes. Patients with CTCs that shrink or decrease in concentration following medical therapy have better outcomes. Those who start out with lower CTC burdens and respond to therapy are more likely to be cured of their cancer.
Conclusion
The success of T cell therapy in treating hematological malignancies has been well documented for several decades.137 Attempts at treating solid tumors have been less successful in part due to cancer’s immunosuppressive effects and antigen variability.138,139 To circumnavigate these difficulties, researchers continue to develop new methods to redirect and expand T cells. Several of these methods, including TILs, TCR-modified and CAR-T cells, tumor-associated antigen T cells (TAATs), and bispecific armed T cells, are created through ex vivo expansion, activation, and proliferation of T cells.39,140
Furthermore, harnessing the power of other immune cells, such as NK, cytokine-induced killer, and DCs, in the place of or in addition to T cell therapy may elicit robust, long-lasting tumor cell death.141 NK cells have proven to be a valuable therapeutic with limited toxicities, their ability to persist within the TME, and their efficacious use in various cancers including hematological malignancies and sarcomas.63 Variations of NK cell therapy, including CIML, CAR-NK, and CIK cells are also currently being studied in clinical trials to treat both hematological and solid malignancies.142-146 DC vaccines, which have demonstrated clinical safety, are currently being explored as adjunct chemotherapeutics. However, choosing DC type (autologous versus allogeneic), determining vaccination dose and route, and selecting the ideal antigen are all ongoing challenges of DC vaccination.147 Finally, though CTCs are presently used for screening, diagnostic, and prognostic purposes, they have exciting potential therapeutic value.147 This value largely comes in the form of explanted CTCs into mouse models with the final goal of being able to treat CTCs, and thus metastatic cancer, with the most efficacy and least side effects.116,126,127 By comparing and combining these ex vivo expanded therapeutics, researchers may be able to develop more personalized, less toxic chemotherapeutics for the treatment of solid and hematological malignancies.
Research Highlights
What is the current knowledge?
√ Many oncological cellular therapies rely on ex vivo expansion of immune cells to fight cancer.
√ Traditional T cell therapies have yielded inconsistent clinical results and have potentially life-threatening toxicities.
What is new here?
√ Exciting new methods of expanding the T cell response to solid and hematological cancers are highlighted.
√ Applications and limitations of NK, cytokine-induced killer, and DC therapies are reviewed.
√ The use of CTCs as screening, diagnostic, and prognostic tools is discussed.
Acknowledgements
The authors would like to acknowledge Dr. John Nemunaitis for his valuable and constructive suggestions during the planning and development of this research. They would also like to thank Laura Nejedlik for facilitating collaboration among the authors. Figures created with Created with BioRender.com.
Competing Interests
The authors have no conflicts of interest to disclose.
Ethical Statement
Not applicable.
References
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68:394-424. doi: 10.3322/caac.21492 [Crossref] [ Google Scholar]
- Palumbo MO, Kavan P, Miller WH Jr, Panasci L, Assouline S, Johnson N. Systemic cancer therapy: achievements and challenges that lie ahead. Frontiers in pharmacology 2013; 4:57. doi: 10.3389/fphar.2013.00057 [Crossref] [ Google Scholar]
- Rohaan MW, Wilgenhof S, Haanen J. Adoptive cellular therapies: the current landscape. Virchows Arch 2019; 474:449-61. doi: 10.1007/s00428-018-2484-0 [Crossref] [ Google Scholar]
- Zhang Y, Zhang Z, Ding Y, Fang Y, Wang P, Chu W. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J Cancer Res Clin Oncol 2021; 147:3725-34. doi: 10.1007/s00432-021-03613-7 [Crossref] [ Google Scholar]
- Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci 2016; 59:468-79. doi: 10.1007/s11427-016-5023-8 [Crossref] [ Google Scholar]
- Viardot A, Bargou R. Bispecific antibodies in haematological malignancies. Cancer Treat Rev 2018; 65:87-95. doi: 10.1016/j.ctrv.2018.04.002 [Crossref] [ Google Scholar]
- Leone DA, Rees AJ, Kain R. Dendritic cells and routing cargo into exosomes. Immunology & Cell Biology 2018; 96:683-93. doi: 10.1111/imcb.12170 [Crossref] [ Google Scholar]
- van Willigen WW, Bloemendal M, Gerritsen WR, Schreibelt G, de Vries IJM, Bol KF. Dendritic Cell Cancer Therapy: Vaccinating the Right Patient at the Right Time. Front Immunol 2018; 9:2265. doi: 10.3389/fimmu.2018.02265 [Crossref] [ Google Scholar]
- Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020; 59:102975. doi: 10.1016/j.ebiom.2020.102975 [Crossref] [ Google Scholar]
- Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood 2014; 123:3855-63. doi: 10.1182/blood-2013-10-532531 [Crossref] [ Google Scholar]
- Passweg JR, Tichelli A, Meyer-Monard S, Heim D, Stern M, Kühne T. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia 2004; 18:1835-8. doi: 10.1038/sj.leu.2403524 [Crossref] [ Google Scholar]
- Modak S, Le Luduec JB, Cheung IY, Goldman DA, Ostrovnaya I, Doubrovina E. Adoptive immunotherapy with haploidentical natural killer cells and Anti-GD2 monoclonal antibody m3F8 for resistant neuroblastoma: Results of a phase I study. Oncoimmunology 2018; 7:e1461305. doi: 10.1080/2162402x.2018.1461305 [Crossref] [ Google Scholar]
- Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P. A Pilot Trial of Humanized Anti-GD2 Monoclonal Antibody (hu1418K322A) with Chemotherapy and Natural Killer Cells in Children with Recurrent/Refractory Neuroblastoma. Clin Cancer Res 2017; 23:6441-9. doi: 10.1158/1078-0432.Ccr-17-0379 [Crossref] [ Google Scholar]
- Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother 2015; 38:24-36. doi: 10.1097/cji.0000000000000059 [Crossref] [ Google Scholar]
- Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011; 13:98-107. doi: 10.3109/14653249.2010.515582 [Crossref] [ Google Scholar]
- Kapeleris J, Kulasinghe A, Warkiani ME, Oleary C, Vela I, Leo P. Ex vivo culture of circulating tumour cells derived from non-small cell lung cancer. Transl Lung Cancer Res 2020; 9:1795-809. doi: 10.21037/tlcr-20-521 [Crossref] [ Google Scholar]
- Shishido SN, Carlsson A, Nieva J, Bethel K, Hicks JB, Bazhenova L. Circulating tumor cells as a response monitor in stage IV non-small cell lung cancer. Journal of Translational Medicine 2019; 17:294. doi: 10.1186/s12967-019-2035-8 [Crossref] [ Google Scholar]
- Tan Y, Wu H. The significant prognostic value of circulating tumor cells in colorectal cancer: A systematic review and meta-analysis. CurrProbl Cancer 2018; 42:95-106. doi: 10.1016/j.currproblcancer.2017.11.002 [Crossref] [ Google Scholar]
- Maheswaran S, Haber DA. Ex Vivo Culture of CTCs: An Emerging Resource to Guide Cancer Therapy. Cancer Res 2015; 75:2411. doi: 10.1158/0008-5472.CAN-15-0145 [Crossref] [ Google Scholar]
- Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther 2021; 12:81. doi: 10.1186/s13287-020-02128-1 [Crossref] [ Google Scholar]
- Bear AS, Fraietta JA, Narayan VK, O’Hara M, Haas NB. Adoptive Cellular Therapy for Solid Tumors. In: American Society of Clinical Oncology Educational Book. 2021. p. 57-65.
- Sprent J. Direct stimulation of naive T cells by antigen-presenting cell vesicles. Blood Cells Mol Dis 2005; 35:17-20. doi: 10.1016/j.bcmd.2005.04.004 [Crossref] [ Google Scholar]
- Met O, Jensen KM, Chamberlain CA, Donia M, Svane IM. Principles of adoptive T cell therapy in cancer. Semin Immunopathol 2019; 41:49-58. doi: 10.1007/s00281-018-0703-z [Crossref] [ Google Scholar]
- Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 2003; 26:332-42. doi: 10.1097/00002371-200307000-00005 [Crossref] [ Google Scholar]
- Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 2005; 23:2346-57. doi: 10.1200/JCO.2005.00.240 [Crossref] [ Google Scholar]
- Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother 2008; 31:742-51. doi: 10.1097/CJI.0b013e31818403d5 [Crossref] [ Google Scholar]
- Lin B, Du L, Li H, Zhu X, Cui L, Li X. Tumor-infiltrating lymphocytes: Warriors fight against tumors powerfully. Biomed Pharmacother 2020; 132:110873. doi: 10.1016/j.biopha.2020.110873 [Crossref] [ Google Scholar]
- Nguyen LT, Saibil SD, Sotov V, Le MX, Khoja L, Ghazarian D. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol Immunother 2019; 68:773-85. doi: 10.1007/s00262-019-02307-x [Crossref] [ Google Scholar]
- Bagley SJ, O'Rourke DM. Clinical investigation of CAR T cells for solid tumors: Lessons learned and future directions. PharmacolTher 2020; 205:107419. doi: 10.1016/j.pharmthera.2019.107419 [Crossref] [ Google Scholar]
- Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018; 378:439-48. doi: 10.1056/NEJMoa1709866 [Crossref] [ Google Scholar]
- Vandghanooni S, Eskandani M, Sanaat Z, Omidi Y. Recent advances in the production, reprogramming, and application of CAR-T cells for treating hematological malignancies. Life Sci 2022; 309:121016. doi: 10.1016/j.lfs.2022.121016 [Crossref] [ Google Scholar]
- Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017; 377:2531-44. doi: 10.1056/NEJMoa1707447 [Crossref] [ Google Scholar]
- Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med 2020; 382:1331-42. doi: 10.1056/NEJMoa1914347 [Crossref] [ Google Scholar]
- Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 2020; 396:839-52. doi: 10.1016/S0140-6736(20)31366-0 [Crossref] [ Google Scholar]
- Munshi NC, Anderson LD, Jr Jr. Munshi NC, Anderson LD, Jr, Shah N, Madduri D, Berdeja J, Lonial S, et alIdecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med 2021; 384:705-16. doi: 10.1056/NEJMoa2024850 [Crossref] [ Google Scholar]
- Chekol Abebe E, Yibeltal Shiferaw M, Tadele Admasu F, Asmamaw Dejenie T. Ciltacabtagene autoleucel: The second anti-BCMA CAR T-cell therapeutic armamentarium of relapsed or refractory multiple myeloma. Front Immunol 2022; 13:991092. doi: 10.3389/fimmu.2022.991092 [Crossref] [ Google Scholar]
- Wang Z, Wu Z, Liu Y, Han W. New development in CAR-T cell therapy. J Hematol Oncol 2017; 10:53. doi: 10.1186/s13045-017-0423-1 [Crossref] [ Google Scholar]
- Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol 2017; 12:813-20. doi: 10.1038/nnano.2017.57 [Crossref] [ Google Scholar]
- Hont AB, Cruz CR, Ulrey R, O'Brien B, Stanojevic M, Datar A. Immunotherapy of Relapsed and Refractory Solid Tumors With Ex Vivo Expanded Multi-Tumor Associated Antigen Specific Cytotoxic T Lymphocytes: A Phase I Study. J Clini Oncol 2019; 37:2349-59. doi: 10.1200/JCO.19.00177 [Crossref] [ Google Scholar]
- Hylander B, Repasky E, Shrikant P, Intengan M, Beck A, Driscoll D. Expression of Wilms tumor gene (WT1) in epithelial ovarian cancer. Gynecol Oncol 2006; 101:12-7. doi: 10.1016/j.ygyno.2005.09.052 [Crossref] [ Google Scholar]
- Lee SB, Haber DA. Wilms tumor and the WT1 gene. Exp Cell Res 2001; 264:74-99. doi: 10.1006/excr.2000.5131 [Crossref] [ Google Scholar]
- Kim A, Park EY, Kim K, Lee JH, Shin DH, Kim JY. Prognostic significance of WT1 expression in soft tissue sarcoma. World J Surg Oncol 2014; 12:214. doi: 10.1186/1477-7819-12-214 [Crossref] [ Google Scholar]
- Barbolina MV, Adley BP, Shea LD, Stack MS. Wilms tumor gene protein 1 is associated with ovarian cancer metastasis and modulates cell invasion. Cancer 2008; 112:1632-41. doi: 10.1002/cncr.23341 [Crossref] [ Google Scholar]
- Brett A, Pandey S, Fraizer G. The Wilms' tumor gene (WT1) regulates E-cadherin expression and migration of prostate cancer cells. Mol Cancer 2013; 12:3. doi: 10.1186/1476-4598-12-3 [Crossref] [ Google Scholar]
- Yin B. PRAME: from diagnostic marker and tumor antigen to promising target of RNAi therapy in leukemic cells. Leuk Res 2011; 35:1159-60. doi: 10.1016/j.leukres.2011.04.018 [Crossref] [ Google Scholar]
- Tan P, Zou C, Yong B, Han J, Zhang L, Su Q. Expression and prognostic relevance of PRAME in primary osteosarcoma. BiochemBiophys Res Commun 2012; 419:801-8. doi: 10.1016/j.bbrc.2012.02.110 [Crossref] [ Google Scholar]
- Toledo SR, Zago MA, Oliveira ID, Proto-Siqueira R, Okamoto OK, Severino P. Insights on PRAME and osteosarcoma by means of gene expression profiling. J Orthop Sci 2011; 16:458-66. doi: 10.1007/s00776-011-0106-7 [Crossref] [ Google Scholar]
- Liu SH, Hong Y, Markowiak S, Sanchez R, Creeden J, Nemunaitis J. BIRC5 is a target for molecular imaging and detection of human pancreatic cancer. Cancer Lett 2019; 457:10-9. doi: 10.1016/j.canlet.2019.04.036 [Crossref] [ Google Scholar]
- Shinozawa I, Inokuchi K, Wakabayashi I, Dan K. Disturbed expression of the anti-apoptosis gene, survivin, and EPR-1 in hematological malignancies. Leuk Res 2000; 24:965-70. doi: 10.1016/s0145-2126(00)00065-5 [Crossref] [ Google Scholar]
- Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Mol Cancer Ther 2006; 5:1087-98. doi: 10.1158/1535-7163.MCT-05-0375 [Crossref] [ Google Scholar]
- Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res 1998; 58:5315-20. [ Google Scholar]
- Coughlin CM, Fleming MD, Carroll RG, Pawel BR, Hogarty MD, Shan X. Immunosurveillance and survivin-specific T-cell immunity in children with high-risk neuroblastoma. J Clin Oncol 2006; 24:5725-34. doi: 10.1200/JCO.2005.05.3314 [Crossref] [ Google Scholar]
- Bhutani D, Lum LG. Activated T cells armed with bispecific antibodies kill tumor targets. CurrOpinHematol 2015; 22:476-83. doi: 10.1097/MOH.0000000000000176 [Crossref] [ Google Scholar]
- Park JA, Santich BH, Xu H, Lum LG, Cheung NV. Potent ex vivo armed T cells using recombinant bispecific antibodies for adoptive immunotherapy with reduced cytokine release. J Immunother Cancer 2021; 9. 10.1136/jitc-2020-002222.
- Gokbuget N, Dombret H, Bonifacio M, Reichle A, Graux C, Faul C. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018; 131:1522-31. doi: 10.1182/blood-2017-08-798322 [Crossref] [ Google Scholar]
- Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J Clin Oncol 2021; 39:3391-402. doi: 10.1200/JCO.21.00662 [Crossref] [ Google Scholar]
- Nathan P, Hassel JC, Rutkowski P, Baurain JF, Butler MO, Schlaak M. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N Engl J Med 2021; 385:1196-206. doi: 10.1056/NEJMoa2103485 [Crossref] [ Google Scholar]
- Santich BH, editor. PA8.4 41BB or CD28 driven disialoganglioside (GD2)-specific CAR-T, but not T-cell engagingbispecific antibody, induces fatal neurotoxicity in mice Advances in Neuroblastoma Research Symposium; 2021; Amsterdam.
- Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumorsIDistribution of reactivity and specificity. Int J Cancer 1975; 16:216-29. doi: 10.1002/ijc.2910160204 [Crossref] [ Google Scholar]
- Kiessling R, Klein E, Pross H, Wigzell H. "Natural" killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cellsCharacteristics of the killer cell. Eur J Immunol 1975; 5:117-21. doi: 10.1002/eji.1830050209 [Crossref] [ Google Scholar]
- Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol 2013; 132:515-25. doi: 10.1016/j.jaci.2013.07.020 [Crossref] [ Google Scholar]
- Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front Immunol 2015; 6:368. doi: 10.3389/fimmu.2015.00368 [Crossref] [ Google Scholar]
- Lee DA. Cellular therapy: Adoptive immunotherapy with expanded natural killer cells. Immunol Rev 2019; 290:85-99. doi: 10.1111/imr.12793 [Crossref] [ Google Scholar]
- Lee HR, Baek KH. Role of natural killer cells for immunotherapy in chronic myeloid leukemia (Review). Oncol Rep 2019; 41:2625-35. doi: 10.3892/or.2019.7059 [Crossref] [ Google Scholar]
- Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. Semin Immunol 2017; 31:37-54. doi: 10.1016/j.smim.2017.07.009 [Crossref] [ Google Scholar]
- Habif G, Crinier A, Andre P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol 2019; 16:415-22. doi: 10.1038/s41423-019-0224-2 [Crossref] [ Google Scholar]
- Zou W. Mechanistic insights into cancer immunity and immunotherapy. Cell Mol Immunol 2018; 15:419-20. doi: 10.1038/s41423-018-0011-5 [Crossref] [ Google Scholar]
- Lian G, Mak TS, Yu X, Lan HY. Challenges and Recent Advances in NK Cell-Targeted Immunotherapies in Solid Tumors. Int J Mol Sci 2021; 23. 10.3390/ijms23010164.
- Zhang C, Hu Y, Shi C. Targeting Natural Killer Cells for Tumor Immunotherapy. Front Immunol 2020; 11:60. doi: 10.3389/fimmu.2020.00060 [Crossref] [ Google Scholar]
- Cany J, Dolstra H, Shah N. Umbilical cord blood-derived cellular products for cancer immunotherapy. Cytotherapy 2015; 17:739-48. doi: 10.1016/j.jcyt.2015.03.005 [Crossref] [ Google Scholar]
- Knorr DA, Kaufman DS. Pluripotent stem cell-derived natural killer cells for cancer therapy. Transl Res 2010; 156:147-54. doi: 10.1016/j.trsl.2010.07.008 [Crossref] [ Google Scholar]
- Granzin M, Wagner J, Köhl U, Cerwenka A, Huppert V, Ullrich E. Shaping of Natural Killer Cell Antitumor Activity by Ex Vivo Cultivation. Front Immunol 2017; 8:458. doi: 10.3389/fimmu.2017.00458 [Crossref] [ Google Scholar]
- Koehl U, Brehm C, Huenecke S, Zimmermann SY, Kloess S, Bremm M. Clinical grade purification and expansion of NK cell products for an optimized manufacturing protocol. Front Oncol 2013; 3:118. doi: 10.3389/fonc.2013.00118 [Crossref] [ Google Scholar]
- Newman KC, Riley EM. Whatever turns you on: accessory-cell-dependent activation of NK cells by pathogens. Nat Rev Immunol 2007; 7:279-91. doi: 10.1038/nri2057 [Crossref] [ Google Scholar]
- Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov 2014; 4:522-6. doi: 10.1158/2159-8290.Cd-13-0985 [Crossref] [ Google Scholar]
- Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol 2014; 44:1582-92. doi: 10.1002/eji.201344272 [Crossref] [ Google Scholar]
- Yao X, Matosevic S. Chemokine networks modulating natural killer cell trafficking to solid tumors. Cytokine Growth Factor Rev 2021; 59:36-45. doi: 10.1016/j.cytogfr.2020.12.003 [Crossref] [ Google Scholar]
- Ng YY, Tay JCK, Wang S. CXCR1 Expression to Improve Anti-Cancer Efficacy of Intravenously Injected CAR-NK Cells in Mice with Peritoneal Xenografts. Mol TherOncolytics 2020; 16:75-85. doi: 10.1016/j.omto.2019.12.006 [Crossref] [ Google Scholar]
- Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother Cancer 2017; 5:73. doi: 10.1186/s40425-017-0275-9 [Crossref] [ Google Scholar]
- Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol Immunother 2015; 64:225-35. doi: 10.1007/s00262-014-1629-5 [Crossref] [ Google Scholar]
- Yang L, Huang C, Wang C, Zhang S, Li Z, Zhu Y. Overexpressed CXCR4 and CCR7 on the surface of NK92 cell have improved migration and anti-tumor activity in human colon tumor model. Anticancer Drugs 2020; 31:333-44. doi: 10.1097/cad.0000000000000868 [Crossref] [ Google Scholar]
- Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A 2009; 106:1915-9. doi: 10.1073/pnas.0813192106 [Crossref] [ Google Scholar]
- Leong JW, Chase JM, Romee R, Schneider SE, Sullivan RP, Cooper MA. Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transplant 2014; 20:463-73. doi: 10.1016/j.bbmt.2014.01.006 [Crossref] [ Google Scholar]
- Ni J, Hölsken O, Miller M, Hammer Q, Luetke-Eversloh M, Romagnani C. Adoptively transferred natural killer cells maintain long-term antitumor activity by epigenetic imprinting and CD4(+) T cell help. Oncoimmunology 2016; 5:e1219009. doi: 10.1080/2162402x.2016.1219009 [Crossref] [ Google Scholar]
- Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med 2016; 8:357ra123. doi: 10.1126/scitranslmed.aaf2341 [Crossref] [ Google Scholar]
- Habib S, Tariq SM, Tariq M. Chimeric Antigen Receptor-Natural Killer Cells: The Future of Cancer Immunotherapy. Ochsner J 2019; 19:186-7. doi: 10.31486/toj.19.0033 [Crossref] [ Google Scholar]
- Lupo KB, Matosevic S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers (Basel) 2019; 11:769. doi: 10.3390/cancers11060769 [Crossref] [ Google Scholar]
- Craig DJ, Creeden JF, Einloth KR, Gillman CE, Stanbery L, Hamouda DM. Resident Memory T Cells and Their Effect on Cancer. Vaccines 2020; 8:562. [ Google Scholar]
- Hu Y, Tian ZG, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin 2018; 39:167-76. doi: 10.1038/aps.2017.125 [Crossref] [ Google Scholar]
- Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 2015; 5:1282-95. doi: 10.1158/2159-8290.CD-15-1020 [Crossref] [ Google Scholar]
- Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol Ther 2019; 27:1114-25. doi: 10.1016/j.ymthe.2019.03.011 [Crossref] [ Google Scholar]
- Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med 2020; 382:545-53. doi: 10.1056/NEJMoa1910607 [Crossref] [ Google Scholar]
- Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front Immunol 2019; 10:1205. doi: 10.3389/fimmu.2019.01205 [Crossref] [ Google Scholar]
- Sharma A, Schmidt-Wolf IGH. 30 years of CIK cell therapy: recapitulating the key breakthroughs and future perspective. J Exp Clin Cancer Re 2021; 40:388. doi: 10.1186/s13046-021-02184-2 [Crossref] [ Google Scholar]
- Ma Y, Xu YC, Tang L, Zhang Z, Wang J, Wang HX. Cytokine-induced killer (CIK) cell therapy for patients with hepatocellular carcinoma: efficacy and safety. Exp Hematol Oncol 2012; 1:11. doi: 10.1186/2162-3619-1-11 [Crossref] [ Google Scholar]
- Zhao H, Wang Y, Yu J, Wei F, Cao S, Zhang X. Autologous Cytokine-Induced Killer Cells Improves Overall Survival of Metastatic Colorectal Cancer Patients: Results From a Phase II Clinical Trial. Clin Colorectal Cancer 2016; 15:228-35. doi: 10.1016/j.clcc.2016.02.005 [Crossref] [ Google Scholar]
- Chen D, Sha H, Hu T, Dong S, Zhang J, Liu S. Cytokine-induced killer cells as a feasible adoptive immunotherapy for the treatment of lung cancer. Cell Death Dis 2018; 9:366. doi: 10.1038/s41419-018-0404-5 [Crossref] [ Google Scholar]
- Wang H, Cui Y, Wang S, Zhao R, Sun M. Curative Effects of Dendritic Cells Combined with Cytokine-Induced Killer Cells in Patients with Malignant Pericardial Effusion. Med Sci Monit 2016; 22:4159-63. doi: 10.12659/msm.897657 [Crossref] [ Google Scholar]
- Zhou Y, Chen C-l, Jiang S-w, Feng Y, Yuan L, Chen P. Retrospective analysis of the efficacy of adjuvant CIK cell therapy in epithelial ovarian cancer patients who received postoperative chemotherapy. OncoImmunology 2019; 8:e1528411. doi: 10.1080/2162402X.2018.1528411 [Crossref] [ Google Scholar]
- Garofano F, Gonzalez-Carmona MA, Skowasch D, Schmidt-Wolf R, Abramian A, Hauser S. Clinical Trials with Combination of Cytokine-Induced Killer Cells and Dendritic Cells for Cancer Therapy. Int J Mol Sci 2019; 20:4307. doi: 10.3390/ijms20174307 [Crossref] [ Google Scholar]
- Nussenzweig MC, Steinman RM, Gutchinov B, Cohn ZA. Dendritic cells are accessory cells for the development of anti-trinitrophenyl cytotoxic T lymphocytes. J Exp Med 1980; 152:1070-84. doi: 10.1084/jem.152.4.1070 [Crossref] [ Google Scholar]
- Gunawan M, Jardine L, Haniffa M. Isolation of Human Skin Dendritic Cell Subsets. Methods Mol Biol 2016; 1423:119-28. doi: 10.1007/978-1-4939-3606-9_8 [Crossref] [ Google Scholar]
- Butterfield LH. Dendritic cells in cancer immunotherapy clinical trials: are we making progress?. Front Immunol 2013; 4:454. doi: 10.3389/fimmu.2013.00454 [Crossref] [ Google Scholar]
- Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, van der Burg SH, Offringa R. Superior induction of anti‐tumor CTL immunity by extended peptide vaccines involves prolonged, DC‐focused antigen presentation. Eur J Immunol 2008; 38:1033-42. [ Google Scholar]
- Sundarasetty BS, Chan L, Darling D, Giunti G, Farzaneh F, Schenck F. Lentivirus-induced 'Smart' dendritic cells: Pharmacodynamics and GMP-compliant production for immunotherapy against TRP2-positive melanoma. Gene Ther 2015; 22:707-20. doi: 10.1038/gt.2015.43 [Crossref] [ Google Scholar]
- Bol KF, Aarntzen EH, Pots JM, Olde Nordkamp MA, van de Rakt MW, Scharenborg NM. Prophylactic vaccines are potent activators of monocyte-derived dendritic cells and drive effective anti-tumor responses in melanoma patients at the cost of toxicity. Cancer Immunol Immunother 2016; 65:327-39. doi: 10.1007/s00262-016-1796-7 [Crossref] [ Google Scholar]
- Aarntzen EH, Bol K, Schreibelt G, Jacobs JF, Lesterhuis WJ, Van Rossum MM. Skin-test infiltrating lymphocytes early predict clinical outcome of dendritic cell-based vaccination in metastatic melanoma. Cancer Res 2012; 72:6102-10. doi: 10.1158/0008-5472.CAN-12-2479 [Crossref] [ Google Scholar]
- Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015; 519:366-9. doi: 10.1038/nature14320 [Crossref] [ Google Scholar]
- Bhattacharyya T, Purushothaman K, Puthiyottil SS, Bhattacharjee A, Muttah G. Immunological interactions in radiotherapy-opening a new window of opportunity. Ann Transl Med 2016; 4:51. doi: 10.3978/j.issn.2305-5839.2015.10.44 [Crossref] [ Google Scholar]
- Burnette B, Weichselbaum RR. Radiation as an immune modulator. Semin Radiat Oncol 2013; 23:273-80. doi: 10.1016/j.semradonc.2013.05.009 [Crossref] [ Google Scholar]
- Nikitina EY, Gabrilovich DI. Combination of gamma-irradiation and dendritic cell administration induces a potent antitumor response in tumor-bearing mice: approach to treatment of advanced stage cancer. Int J Cancer 2001; 94: 825-33. 10.1002/1097-0215(20011215)94:6<825::aidijc1545>3.0.co;2-5.
- Czerniecki BJ, Koski GK, Koldovsky U, Xu S, Cohen PA, Mick R. Targeting HER-2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst secretion. Cancer Res 2007; 67:1842-52. doi: 10.1158/0008-5472.CAN-06-4038 [Crossref] [ Google Scholar]
- Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med 1998; 4:328-32. doi: 10.1038/nm0398-328 [Crossref] [ Google Scholar]
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010; 363:411-22. doi: 10.1056/NEJMoa1001294 [Crossref] [ Google Scholar]
- Engell-Noerregaard L, Hansen TH, Andersen MH, Thor Straten P, Svane IM. Review of clinical studies on dendritic cell-based vaccination of patients with malignant melanoma: assessment of correlation between clinical response and vaccine parameters. Cancer Immunol Immunother 2009; 58:1-14. doi: 10.1007/s00262-008-0568-4 [Crossref] [ Google Scholar]
- Harouaka R, Kang Z, Zheng SY, Cao L. Circulating tumor cells: advances in isolation and analysis, and challenges for clinical applications. PharmacolTher 2014; 141:209-21. doi: 10.1016/j.pharmthera.2013.10.004 [Crossref] [ Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2:442-54. doi: 10.1038/nrc822 [Crossref] [ Google Scholar]
- Aceto N, Toner M, Maheswaran S, Haber DA. En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer 2015; 1:44-52. [ Google Scholar]
- Habli Z, AlChamaa W, Saab R, Kadara H, Khraiche ML. Circulating Tumor Cell Detection Technologies and Clinical Utility: Challenges and Opportunities. Cancers (Basel) 2020; 12:1930. doi: 10.3390/cancers12071930 [Crossref] [ Google Scholar]
- Eslami SZ, Cortés-Hernández LE, Alix-Panabières C. Epithelial Cell Adhesion Molecule: An Anchor to Isolate Clinically Relevant Circulating Tumor Cells. Cells 2020; 9:1836. doi: 10.3390/cells9081836 [Crossref] [ Google Scholar]
- Andree KC, van Dalum G, Terstappen LW. Challenges in circulating tumor cell detection by the CellSearch system. Mol Oncol 2016; 10:395-407. doi: 10.1016/j.molonc.2015.12.002 [Crossref] [ Google Scholar]
- Swennenhuis JF, van Dalum G, Zeune LL, Terstappen LW. Improving the CellSearch(R) system. Expert Rev Mol Diagn 2016; 16:1291-305. doi: 10.1080/14737159.2016.1255144 [Crossref] [ Google Scholar]
- Cheng S-B, Xie M, Chen Y, Xiong J, Liu Y, Chen Z. Three-Dimensional Scaffold Chip with Thermosensitive Coating for Capture and Reversible Release of Individual and Cluster of Circulating Tumor Cells. Anal Chem 2017; 89:7924-32. doi: 10.1021/acs.analchem.7b00905 [Crossref] [ Google Scholar]
- Edd JF, Mishra A, Dubash TD, Herrera S, Mohammad R, Williams EK. Microfluidic concentration and separation of circulating tumor cell clusters from large blood volumes. Lab Chip 2020; 20:558-67. doi: 10.1039/c9lc01122f [Crossref] [ Google Scholar]
- Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med 2014; 20:897-903. doi: 10.1038/nm.3600 [Crossref] [ Google Scholar]
- Lallo A, Gulati S, Schenk MW, Khandelwal G, Berglund UW, Pateras IS. Ex vivo culture of cells derived from circulating tumour cell xenograft to support small cell lung cancer research and experimental therapeutics. Br J Pharmacol 2019; 176:436-50. doi: 10.1111/bph.14542 [Crossref] [ Google Scholar]
- Kim YR, Yoo JK, Jeong CW, Choi JW. Selective killing of circulating tumor cells prevents metastasis and extends survival. J Hematol Oncol 2018; 11:114. doi: 10.1186/s13045-018-0658-5 [Crossref] [ Google Scholar]
- Alix-Panabières C, Pantel K. Circulating Tumor Cells: Liquid Biopsy of Cancer. Clinical Chemistry 2013; 59:110-8. doi: 10.1373/clinchem.2012.194258 [Crossref] [ Google Scholar]
- Krebs MG, Hou JM, Ward TH, Blackhall FH, Dive C. Circulating tumour cells: their utility in cancer management and predicting outcomes. Ther Adv Med Oncol 2010; 2:351-65. doi: 10.1177/1758834010378414 [Crossref] [ Google Scholar]
- Costa C, Muinelo-Romay L, Cebey-Lopez V, Pereira-Veiga T, Martinez-Pena I, Abreu M. Analysis of a Real-World Cohort of Metastatic Breast Cancer Patients Shows Circulating Tumor Cell Clusters (CTC-clusters) as Predictors of Patient Outcomes. Cancers (Basel) 2020; 12:1111. doi: 10.3390/cancers12051111 [Crossref] [ Google Scholar]
- Sparano J, O'Neill A, Alpaugh K, Wolff AC, Northfelt DW, Dang CT. Association of Circulating Tumor Cells With Late Recurrence of Estrogen Receptor-Positive Breast Cancer: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol 2018; 4:1700-6. doi: 10.1001/jamaoncol.2018.2574 [Crossref] [ Google Scholar]
- Kantara C, O'Connell MR, Luthra G, Gajjar A, Sarkar S, Ullrich RL. Methods for detecting circulating cancer stem cells (CCSCs) as a novel approach for diagnosis of colon cancer relapse/metastasis. Lab Invest 2015; 95:100-12. doi: 10.1038/labinvest.2014.133 [Crossref] [ Google Scholar]
- Chauhan A, Kaur R, Ghoshal S, Pal A. Exploration of Circulating Tumour Cell (CTC) Biology: A Paradigm Shift in Liquid Biopsy. Indian J Clin Biochem 2021; 36:131-42. doi: 10.1007/s12291-020-00923-4 [Crossref] [ Google Scholar]
- Fehm T, Müller V, Aktas B, Janni W, Schneeweiss A, Stickeler E. HER2 status of circulating tumor cells in patients with metastatic breast cancer: a prospective, multicenter trial. Breast Cancer Research and Treatment 2010; 124:403-12. doi: 10.1007/s10549-010-1163-x [Crossref] [ Google Scholar]
- Pestrin M, Bessi S, Puglisi F, Minisini AM, Masci G, Battelli N. Final results of a multicenter phase II clinical trial evaluating the activity of single-agent lapatinib in patients with HER2-negative metastatic breast cancer and HER2-positive circulating tumor cellsA proof-of-concept study. Breast Cancer Research and Treatment 2012; 134:283-9. doi: 10.1007/s10549-012-2045-1 [Crossref] [ Google Scholar]
- Vasseur A, Kiavue N, Bidard FC, Pierga JY, Cabel L. Clinical utility of circulating tumor cells: an update. Mol Oncol 2021; 15:1647-66. doi: 10.1002/1878-0261.12869 [Crossref] [ Google Scholar]
- Halliday GM, Patel A, Hunt MJ, Tefany FJ, Barnetson RS. Spontaneous regression of human melanoma/nonmelanoma skin cancer: association with infiltrating CD4+ T cells. World J Surg 1995; 19:352-8. doi: 10.1007/bf00299157 [Crossref] [ Google Scholar]
- Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012; 21:309-22. doi: 10.1016/j.ccr.2012.02.022 [Crossref] [ Google Scholar]
- Zhang E, Gu J, Xu H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol Cancer 2018; 17:7. doi: 10.1186/s12943-018-0759-3 [Crossref] [ Google Scholar]
- Park JA, Santich BH, Xu H, Lum LG, Cheung N-KV. Potent ex vivo armed T cells using recombinant bispecific antibodies for adoptive immunotherapy with reduced cytokine release. J Immunother Cancer 2021; 9:e002222. doi: 10.1136/jitc-2020-002222 [Crossref] [ Google Scholar]
- Patel S, Burga RA, Powell AB, Chorvinsky EA, Hoq N, McCormack SE. Beyond CAR T Cells: Other Cell-Based Immunotherapeutic Strategies Against Cancer. Frontiers in oncology 2019; 9:196. doi: 10.3389/fonc.2019.00196 [Crossref] [ Google Scholar]
- Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy 2012; 14:830-40. doi: 10.3109/14653249.2012.671519 [Crossref] [ Google Scholar]
- Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol 2014; 8:297-310. doi: 10.1016/j.molonc.2013.12.001 [Crossref] [ Google Scholar]
- Chu Y, Yahr A, Huang B, Ayello J, Barth M, M SC. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. Oncoimmunology 2017; 6:e1341031. doi: 10.1080/2162402X.2017.1341031 [Crossref] [ Google Scholar]
- Chung MJ, Park JY, Bang S, Park SW, Song SY. Phase II clinical trial of ex vivo-expanded cytokine-induced killer cells therapy in advanced pancreatic cancer. Cancer Immunology, Immunotherapy 2014; 63:939-46. doi: 10.1007/s00262-014-1566-3 [Crossref] [ Google Scholar]
- Li R, Wang C, Liu L, Du C, Cao S, Yu J. Autologous cytokine-induced killer cell immunotherapy in lung cancer: a phase II clinical study. Cancer Immunol Immunother 2012; 61:2125-33. doi: 10.1007/s00262-012-1260-2 [Crossref] [ Google Scholar]
- Sadeghzadeh M, Bornehdeli S, Mohahammadrezakhani H, Abolghasemi M, Poursaei E, Asadi M. Dendritic cell therapy in cancer treatment; the state-of-the-art. Life Sci 2020; 254:117580. doi: 10.1016/j.lfs.2020.117580 [Crossref] [ Google Scholar]
- Nemunaitis JJ, Gillman CE, Creeden JF, Dworkin LD, Stanbery L, Atkinson RL. The Role of TGFβ in Clinical Cancer Response. Computers & Operations Research 2020; 2020:1-8. [ Google Scholar]