Bioimpacts. 14(4):27680.
doi: 10.34172/bi.2023.27680
Original Article
New approach to generating of human monoclonal antibodies specific to the proteolytic domain of botulinum neurotoxin A
Marina Vladimirovna Silkina Conceptualization, Data curation, Writing – original draft, Writing – review & editing, 
Alena Sergeevna Kartseva Conceptualization, Data curation, Formal analysis, Writing – original draft, Writing – review & editing,
Alena Konstantinovna Riabko Methodology,
Mariia Aleksandrovna Makarova Formal analysis, Investigation, Methodology,
Metkhun Madibronovich Rogozin Investigation,
Yana Olegovna Romanenko Investigation,
Igor Georgievich Shemyakin Funding acquisition, Resources, Writing – review & editing,
Ivan Alekseevich Dyatlov Funding acquisition, Resources, Supervision,
Victoria Valerievna Firstova Project administration, Writing – review & editing, , *
Author information:
State Research Center for Applied Microbiology and Biotechnology (SRCAMB), Obolensk 142279, Russia
Abstract
Introduction:
Botulinum neurotoxins (BoNTs) cause botulism and are the most potent natural toxins known. Immunotherapy with neutralizing monoclonal antibodies (MAbs) is considered to be the most effective immediate response to BoNT exposure. Hybridoma technology remains the preferred method for producing MAbs with naturally paired immunoglobulin genes and with preserved innate functions of immune cells. The affinity-matured human antibody repertoire may be ideal as a source for antibody therapeutics against BoNTs. In an effort to develop novel BoNT type A (BoNT/A) immunotherapeutics, sorted by flow cytometry plasmablasts and activated memory B cells from a donor repeatedly injected with BoNT/A for aesthetic botulinum therapy could be used due to obtain hybridomas producing native antibodies.
Methods:
Plasmablasts and activated memory B-cells were isolated from whole blood collected 7 days after BoNT/A injection and sorted by flow cytometry. The sorted cells were then electrofused with the K6H6/B5 cell line, resulting in a producer of native human monoclonal antibodies (huMAbs). The 3 antibodies obtained were then purified by affinity chromatography, analyzed for binding by Western blot assay and neutralization by FRET assay.
Results:
We have succeeded in creating 3 hybridomas that secrete huMAbs specific to native BoNT/A and the proteolytic domain (LC) of BoNT/A. The 1B9 antibody also directly inhibited BoNT/A catalytic activity in vitro.
Conclusion:
The use activated plasmablasts and memory B-cells isolated at the peak of the immune response (at day 7 of immunogenesis) that have not yet completed the terminal stage of differentiation but have undergone somatic hypermutation for hybridization allows us to obtain specific huMAbs even when the immune response of the donor is weak (with low levels of specific antibodies and specific B-cells in blood). A BoNT/A LC-specific antibody is capable of effectively inhibiting BoNT/A by mechanisms not previously associated with antibodies that neutralize BoNT. Antibodies specific to BoNT LC can be valuable components of a mixture of antibodies against BoNT exposure.
Keywords: Botulism, Botulinum neurotoxin, Clostridium botulinum, SNAP25, human monoclonal antibody, K6H6/B5
Copyright and License Information
© 2024 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Botulism is BoNT-mediated life-threatening disease.1 BoNTs comprise seven serotypes, from A to G, of protein neurotoxins generated by anaerobic Gram-positive spore-forming bacterium Clostridium botulinum.2 BoNTs from A, B, E, and (more rarely) F serotypes (BoNT/A, B, E, and F) cause human disease. BoNT/A produces the most severe syndrome, with the highest proportion of patients requiring mechanical ventilation.3-5
Depending on the route by which BoNT enters the human body, there are four different forms of botulism: foodborne botulism, which results from eating food that already contains the toxin; wound botulism, which results from bacterial colonization of the wound or intestine (infant botulism and adult intestinal colonization botulism); and iatrogenic botulism, which results from highly concentrated cosmetic or therapeutic injections of toxin. BoNT enters the vascular circulation and is transported to peripheral nerve terminals, neuromuscular junctions and peripheral ganglia.6 As a result of blocking the transmission of acetylcholine through the neuromuscular junction by inhibiting the release of acetylcholine from the presynaptic end of the motor neuron, paralysis occurs.7 Through similar pharmacological mechanisms at the neuromuscular junction share and a similar structure all BoNT types produce a similar clinical syndrome of cranial nerve palsies that can be followed by bilateral flaccid paralysis, affecting limb musculature, that might progress to respiratory failure and death.3,7,8
The BoNT molecule is a heterodimer synthesised as a single polypeptide chain with a molecular weight of 150 kDa, consisting of a 100 kDa heavy chain (HC) and a 50 kDa light chain (LC) linked by a disulfide bond.9 HC consists of two functionally distinct regions: the receptor-binding C-terminal domain (HС50) ~50 kDa and the translocation N-terminal domain (HN). The C-terminal half of the HC polypeptide is a receptor-binding domain (HC50) specific for a ganglioside and a protein receptor on the surface of neuronal cells and initiating endocytosis, the first stage of cellular intoxication caused by BoNT. The N-terminal half of the HC polypeptide is a translocation domain responsible for the formation of a transmembrane channel in endosomes for the movement of the LC polypeptide into the cytosol, where LC exhibits its enzymatic activity that determines the clinical picture of botulism.10,11
Treatment of botulism includes supportive care, intubation and, if necessary, mechanical ventilation. Antibiotic therapy is ineffective in treating botulism.12 There have been theoretical concerns about increased release of BoNT from lysed Clostridia organisms after antimicrobial treatment.13 To date, the most effective specific treatment for botulism is botulinum antitoxin.12 Mortality and duration of treatment are reduced by timely administration of botulinum antitoxin.14,15 The antitoxin is a preparation of antibodies derived from horses that bind and neutralize BoNT in the bloodstream. Bering botulism antitoxin (Novartis Vaccines and Diagnostics GmbH and Co. KG) is used to treat types A, B and E, while heptavalent botulism antitoxin (BAT, Cangene Corporation) is used to treat types A, B, C, D, E, F and G. However, as concentrated preparations of foreign proteins, they can trigger immune reactions, including anaphylaxis.15,16 A formulation of a human immune serum called BabyBIG (California Department of Public Health) has recently become available, but in limited supply and for treating only childhood botulism.17,18 In addition, immune serum production involves complex and time-consuming production processes and quality management. Therefore, the development of safe, effective, and standardized antibodies is required.
Several studies have reported the development of monoclonal antibodies against various BoNT types.19-26 Existing MAbs against various BoNTs serotypes that are generated by phage display,27,28 humanization,29 chimerisation30 as well via hybridoma technology,9,31 are preferable. HuMAbs are safe, standardized, and more effective in treating human diseases than antibodies obtained from other animal species.12,32
Hybridoma technology remains the preferred method for producing MAbs with naturally paired immunoglobulin genes and with preserved innate functions of immune cells. Unlike other technologies, when used to generate huMAbs, hybridoma technology yields antibodies with naturally paired regions of heavy and light antibody chains as well as a constant region with a naturally switched immunoglobulin class.33,34 For years, the use of hybridoma technology to generate huMAbs producers has been limited due to the lack of suitable fusion partners. The use of murine myeloma cells to generate huMAbs-producing hybridomas resulted in active segregation of the human chromosome following B cell fusion with the murine myeloma cell.35 As a result, special cell lines were developed to generate huMAbs-producing hybridomas. Previously, we have shown that the use of the K6H6/B5 line for hybridization with human B cells makes it possible to obtain a stable producer of huMAbs.36
The use of hybridoma technology to generate huMAbs also proves to be challenging due to the main pool of specifically differentiated B cells (i.e., memory B cells and plasma cells) being found in the lymphoid organs. For example, the anti-tetanus IgG antibody-producing B cells comprise about are only 1×10-4 of circulating B-cellsafter 2-4 weeks of booster injection. This low amount of B cells in blood, combined with the fact that typically B cells account for 2-10% of all lymphocytes as well as the low fusion efficiency obtained in the hybrid technique (i.e., 10-5 to 10-6), suggest that the likelihood of generating antigen-specific human hybridoma is only about 10–9 to 10–10.37
In this article, we propose a novel approach to generate huMAbs specific for the BoNT/A LC via cytometry-based sorting of human plasmablast and activated memory B cells isolated from the peripheral blood of donors who received aesthetic botulinum therapy and human hybridoma technique.
Materials and Methods
Native BoNT /A preparation
The Clostridium botulinum ATCC 19397 strain was used to produce native BoNT/A, and the non-pathogenic Clostridium difficile ATCC 43255 strain was used as a negative control. The strains were grown in Clostridial agar medium (Merck, USA) and cultivated under anaerobic conditions at 35 ºС for 48 hours. Isolated typical colonies were suspended in PBS (until the optical density was adjusted to the McFarland standard of 4) followed by heating at 80 °C for 10 minutes to inactivate the vegetative cells. The heated culture (1 mL) was inoculated into the toxin production broth medium containing 30 g/L Trypton T (Type III, HiMedia, India), 5 g/L glucose, 20 g/L yeast extract and 0.5 g/L L-cysteine HCl, pH 7.4. The strain was cultivated under anaerobic conditions for five days. The resulting culture was autoclaved at 1 atm for 20 minutes. Bacterial cells were separated by centrifugation at 5000× g for 30 minutes at 10 °C. The proteins from the culture filtrate were concentrated by acid precipitation at pH 3.8 for 45 min. The precipitate was separated by centrifugation at 12000× g for 30 minutes and dissolved in 0.2 M sodium phosphate buffer with pH 5.6. Next, ammonium sulfate was added to 60% saturation and the precipitate was collected by centrifugation. Finally, the precipitate was redissolved in 0.05 M citrate-phosphate buffer with pH 5.5.38 The resulting native BoNT/A was used for Western blot and FRET assay.
Cloning, expression and purification of recombinant proteins ( BoNT /A HC50 and LC)
The pET-22b(+) plasmid (Novagen, Germany) was used to express the recombinant BoNT/A HC50 and BoNT/A LC proteins. Modifications in the N-terminal translation region were introduced into the pET-22b(+) plasmid: an amino acid MASMT translation enhancer sequence, six amino acid histidine residues for protein purification by metal-chelated chromatography, a double tandem c-myc epitope for recognition by specific antibodies.39
The BoNT/A HC50 protein was represented by a region of the R861 – L1296 amino acid sequence of the precursor protein BoNT/A (NCBI Reference Sequence accession number: WP_011948511.1). The BoNT/A LC protein was represented by a region of the P2-N418 amino acid sequence of the precursor protein BoNT/A (UniProtKB database, P0DPI1). The stability of BoNT/A LC was ensured by the deletion of 31 amino acid residues in the C-terminal region that are not involved in the proteolytic function.40 The BoNT/A HC50 and BoNT/A LC coding sequences were cloned into modified pET-22b(+) plasmids by restriction BamHI and XhoI sites using FastDigest (Thermo Fisher, USA) and alkaline phosphatase (Fermetas, USA) restriction enzymes. Plasmids were ligated using the Rapid DNA Ligation Kit (Roche, Germany) according to the manual of the manufacturer.
Competent E. coli ElectroMAX DH12S cells (Thermo Fisher, USA) transformed with pET-HC50 and pET-LC plasmids by electroporation using an Electroporator 2510 (Eppendorf, Germany). The transformants were grown at 37°C overnight on 2×YT agar medium containing 100 μg/mL ampicillin and 1% glucose for plasmid selection and maintenance. pET-HC50-positive and pET-LC-positive clones were confirmed by PCR followed by sequencing of the plasmid expression cassette with standard T7 primers (Evrogen, Russian Federation).
E. coli BL21(DE3) strain (NEB, UK) was transformed with pET-HC50 and pET-LC plasmids by electroporation and used to express recombinant proteins. Trasformed cells were grown at 37 °C overnight in 2×YT agar medium supplemented with 100 μg/mL ampicillin and 1% glucose. All cultures were transferred in 2×YT broth medium supplemented with 100 μg/mL ampicillin and 0.1% glucose and cultured in an air shaker (200 rpm) at 37 °C until they reached an absorbency of 0.5–0.6 (OD600 nm). Protein expression was induced by addition of isopropyl-β-D thiogalactoside at a final concentration of 0.4 мМ. Cells were allowed to grow further at 25 °C and 4 hours later cells were harvested by centrifugation. The cell pellets were resuspended in buffer containing 20 mM Tris-HCl, 100 mM NaCl, pH 7.4 and lysed step-by-step with a cocktail of cOmplete EDTA-free protease inhibitors (Roche, Germany), lysozyme (AppliChem, Germany), Triton X-100 detergent and DNase I (Sigma-Aldrich, USA). The suspension was then sonicated and centrifuged at 12 000 × g for 20 minutes. The supernatant was loaded onto a cOmpleteTM His-Tag Purification Resin (Roche, Germany) column by using an AKTA Pure chromatography system (Cytiva, USA). Proteins of interest were eluted with elution buffer containing 20 mM Tris-HCl, 100 mM NaCl, 250 mM imidazole, pH 7.4. Proteins were concentrated using Amicon Ultra-15 centrifuge tubes (Merk Millipore, USA) and were loaded onto a SuperdexÔ 75 10/300 GL (GE Healthcare, UK) column, which was previously equilibrated with buffer containing 137 мМ NaCl, 2.7 мМ KCl, 10 мМ Na2HPO4, 1.76 мМ KH2PO4, рН 7.4 by using chromatography system.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) was carried out to estimate the purity of the isolated proteins and formed according to Laemmli.41 The electrophoresis was carried out on a PowerPacTM Basic System (BioRad, USA) using a 10% polyacrylamide gel. Under different conditions non-reducing SDS 2 × sample buffer (0.125 M Tris, 4% SDS, 20% glycerol, 0.02% bromophenol blue, pH 6.8) аnd reducing SDS 2 × sample buffer (0.125 M Tris, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, pH 6.8) heating at 95 °C for 5 minutes. After electrophoresis, protein bands were detected by Coomassie blue R250 staining.
Verification of proteolytic activity of the recombinant BoNT/A LC protein
Proteolytic activity of the recombinant BoNT/A LC was measured using the commercially available fluorescence resonance energy transfer (FRET) substrate SNAPtide® Botulinum Toxin A Substrate, Fluorogenic (Calbiochem, USA). The reaction was run in a buffer solution (20 mM HEPES pH 8.0, 5 mM DTT, 0.3 mM ZnSO4, and 0.1% Tween-20) using OptiPlate-96, white opaque 96-well microplate (Perkin Elmer, USA). The total volume of solution in each well was 250 μL. The working concentration of the FRET substrate in a well was 10 μM. To run the experiment, the following samples were used: 10 μL culture fluid collected after Clostridium botulinum ATCC 19397 (producer BoNT/A) cultivation; 10 μL culture fluid collected after cultivation of a non-pathogenic Clostridium difficile ATCC 43255 strain; and 10 μg solution of the resulting recombinant BoNT/A LC protein. A well filled with reaction buffer without FRET substrate and a well filled with 10 μL of FRET substrate and 240 μL of reaction buffer were used as negative controls. The fluorescent signal was recorded using the EnSpire multimode plate reader (Perkin Elmer, USA) once a 1 hour for a day. Ex/Em = 320 nm/420 nm (Fig. 1).
Fig. 1.
Verification of the specific proteolytic activity of the recombinant BoNT/A LC using commercially available FRET substrate (SNAPtide).
.
Verification of the specific proteolytic activity of the recombinant BoNT/A LC using commercially available FRET substrate (SNAPtide).
Donors
Written informed consent to donate blood was obtained from each volunteer. The date of birth, the number of injections administered previously, and the brand of BoNT/A used were all recorded. The study was received and approved by the Local Bioethics Committee of the Federal State Budgetary Institution of Science "Kirov Research Institute of Hematology and Blood Transfusion of the Federal Medical and Biological Agency" with the Ethics Committee Approval No. 3 dated 20 January 2021.
The peripheral blood of women who were injected with BoNT/A for aesthetic botulinum therapy was used to generate the BoNT/A-specific huMAbs producing cells. The three women (age 45±4.4 years) who were injected Relatox® (NPO Microgen, Russian Federation) at a dose of 30 units at least twice a year for 10 years were included in the study. Women who received low-dose BoNT/A and less than 2 times a year were excluded from the study. Peripheral blood was sampled 7 days after the last injection with the baseline donor serum anti-BoNT/A HC50 and LC antibody titers being measured. The blood of 5 healthy women aged 42.8±10.1 years who did not receive BoNT/A injections was used as a negative control for the detection of non-specific B-lymphocyte binding to recombinant BoNT/A HC50 and LC proteins by flow cytometry and detection of specific antibodies by ELISA.
Detection of specific antibodies in serum by ELISA
96-well ELISA Microplates (Corning, USA) were coated with 100 μL of recombinant BoNT/A HC50 and LC proteins (1 μg/well) in phosphate-buffered saline (PBS) and incubated in a shaker (300 rpm) for 2 hours at 37 °C. The wells were washed three times with PBS containing 0.05% Tween-20 (PBS-Tw) and blocked with 0.5% skim milk powder (SERVA Electrophoresis GmbH, Germany) in PBS-Tw at 37 °C for 1 hour. The plates were then washed three times with PBS-Tw. Donor sera were diluted at 1:5 to 1:640 with PBS using a twofold dilution process and incubated in a shaker for 1 hour at 37 °C. The wells were washed three times with PBS-Tw and incubated with 100 μL (1:20 000) of Rabbit anti-human IgG (Sigma-Aldrich, USA) conjugated to HRP diluted in PBS-Tw for 1 hour at 37˚C. The plates were washed six times and were incubated with 100 μL of the substrate-indicator mixture ready-made 3,3',5,5'-tetramethylbenzidine solution (TMB, Sigma-Aldrich, USA) for 5 minutes at RT. Finally, the reaction was stopped by adding 50 μL per well of 4 N H2SO4, and the plates were read at 450 nm using an xMark microplate absorbance spectrophotometer (Bio-Rad, USA). Serum from 5 healthy donors who had never been injected with BoNT/A was used as a negative control.
B cell enrichment
To increase the efficiency of plasmablasts and activated memory B cells isolation from donor peripheral blood, B cell enrichment was performed using a commercially available RosetteSepTM Human B Cell Enrichment Cocktail kit (Stemcell Technologies, Canada) according to the manufacturer’s manual. The resulting B cell fraction was resuspended in medium RPMI 1640 (Sigma-Aldrich, USA) containing 2 mM glutamine (PanEco, Russia), 10 mM HEPES (Sigma-Aldrich, USA), 25 μM 2-mercaptoethanol (Sigma-Aldrich, USA) and 10% fetal bovine serum (FBS; Gibco, USA). Cell viability was assessed using a TC-20 automated cell counter (Bio-Rad, USA) upon trypan blue (Invitrogen, USA) staining.
PE-labeled recombinant proteins (BoNT/A HC50 and LC) generation
To assess the specific binding of donor B cells to recombinant BoNT/A HC50 and LC proteins, the proteins were biotinylated and labelled with R-phycoerythrin (R-PE). Biotinylating reagent solution was prepared by dissolving 2.5 mg sodium salt of biotin 3-sulfo-N-hydroxysuccinimide ester (Biotin-Sulfo-NHS, Sigma-Aldrich) in 20 µl DMSO and diluted to 250 µl with sterile PBS. Resulting solution (50 μL) was added to each of the recombinant protein solutions (BoNT/A HC50 and LC) with a concentration of 1 mg/mL, and incubated for 1 h at RT in a shaker. Biotinylated proteins were loaded onto a Superdex 75 column (GE Healthcare, UK) and purified from unbound biotin by chromatography. The biotinylation efficiency was evaluated by Western blot assay. Streptavidin binding to biotin was visualized using an enzymatic color test.
R-PE (1 mg/mL) conjugated with Neutravidin (Invitrogen, A2660, USA) was used to label biotinylated recombinant proteins. The R-PE conjugate of neutravidin was used in a significant deficit relative to biotinylated recombinant proteins. R-PE conjugate with neutravidin (100 µL) was added to each of the protein solutions and incubated in a dark place for 30 minutes at RT in a shaker. Amicon Ultra centrifugal filter units (Merck Millipore) were used to remove unlabeled proteins. The PE-free protein was removed by successive cycles of centrifugation in PBS. The concentrated solution of the PE-labeled protein was resuspended in 100 μL of PBS, transferred to Eppendorf tubes, and diluted to 250 μL.
B cell phenotyping
Plasmablast and activated memory B cell subpopulations were identified using the panel of fluorochrome-conjugated monoclonal antibodies against CD antigens by BD Biosciences (USA) according to the manual of the manufacturer: anti-CD19 APC (HIB19 clone), anti-CD38 PE-cy7 (HIT2 clone), and anti-CD27 BB515 (M-T271 clone). CD19+ CD38hi CD27hi cells were identified as a plasmablast subpopulation, CD19+ CD27+ CD38+ cells were identified as an activated memory B cell subpopulation. Immunophenotype and sorting of target B cell populations were performed on the day of blood sampling using with the FACS Aria III flow cytometer, and the analysis was performed with the program BD FACSDiva v 8.0 (BD Biosciences, USA). The results were expressed as absolute cell counts of the target B cell population. The gating strategy of memory B cells and plasmablasts is shown in Fig. 2.
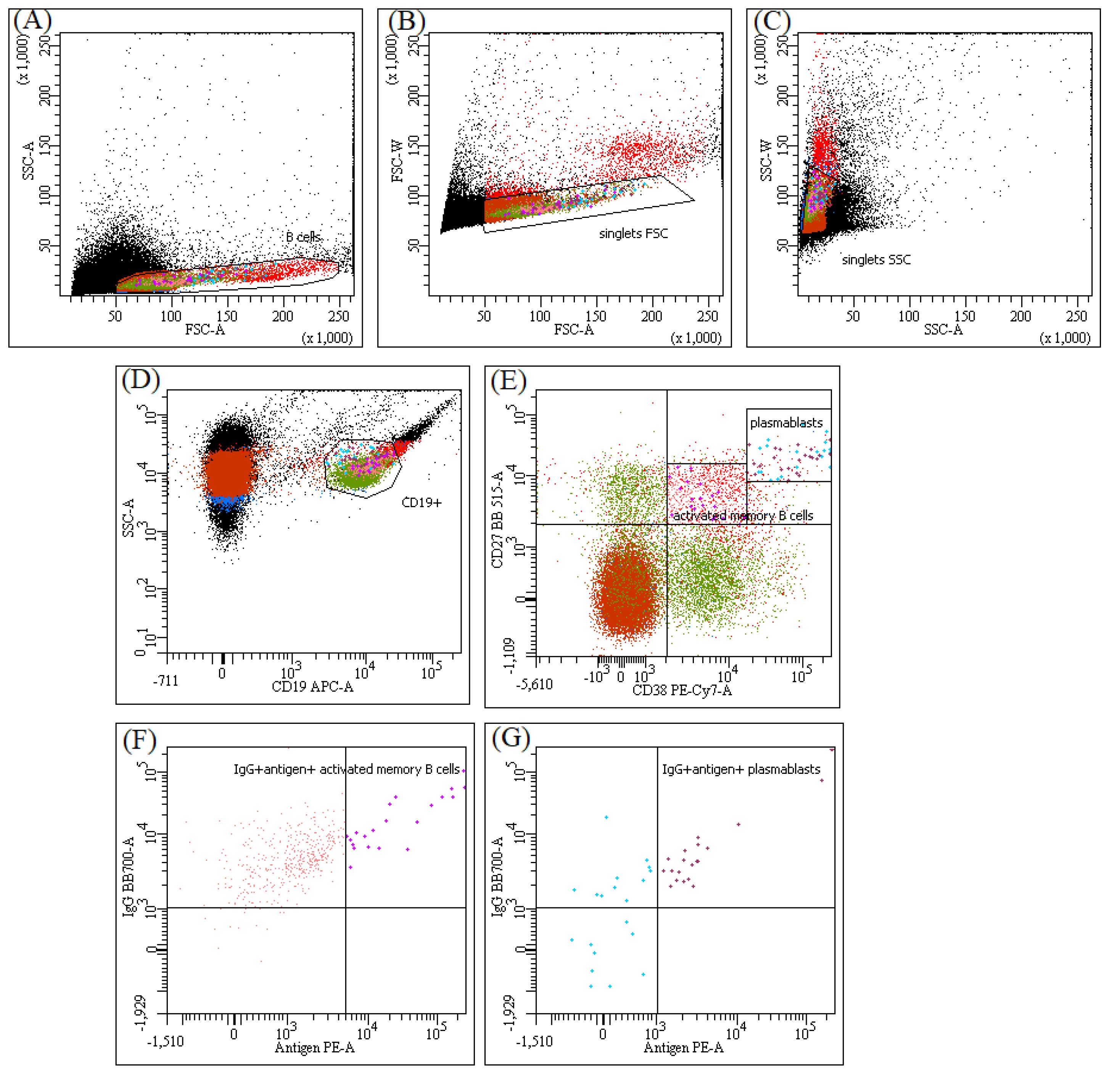
Fig. 2.
The gating strategy for identification of plasmablasts and activated memory B cells. The cells were analysed in a BD FACS ARIA III. B cells (A) gated through the FSC-A vs SSC-A plot were further interrogated by the ratios of width to area in forward scatter (B) and side scatter (C) to eliminate doublets. Then CD19+ B cells were selected for analysis as shown in D. Subpopulations of plasmablasts and activated memory B cells were based on the expression of CD 27 vs CD 38 and gated on (E). CD19+ CD27hi CD38hi and CD19+ CD27+ CD38+ cells from histogram E were sorted for fusion with K6H6/B5 cell line. Membrane-bound IgG and antigens (recombinants BoNT LC or HC50) staining allowed a gate antigen-specific memory B cells (F) and plasmablasts (G) for comparative analyses of test and control groups donors.
.
The gating strategy for identification of plasmablasts and activated memory B cells. The cells were analysed in a BD FACS ARIA III. B cells (A) gated through the FSC-A vs SSC-A plot were further interrogated by the ratios of width to area in forward scatter (B) and side scatter (C) to eliminate doublets. Then CD19+ B cells were selected for analysis as shown in D. Subpopulations of plasmablasts and activated memory B cells were based on the expression of CD 27 vs CD 38 and gated on (E). CD19+ CD27hi CD38hi and CD19+ CD27+ CD38+ cells from histogram E were sorted for fusion with K6H6/B5 cell line. Membrane-bound IgG and antigens (recombinants BoNT LC or HC50) staining allowed a gate antigen-specific memory B cells (F) and plasmablasts (G) for comparative analyses of test and control groups donors.
Anti-human IgG-BB700-conjugated (clone18-145, BD Biosciences) monoclonal antibody and PE-labeled recombinant BoNT/A HC50 and LC were added to the above panel of CD antigens to screen for BoNT/A-specific plasmablasts and activated memory B cells in the peripheral donor blood.
Heterohybridomas generation
To generate human hybridomas, human whole blood СD19+ CD27hi CD38hi plasmablasts and activated memory B cells (СD19+ CD27+ CD38+) were fused with the myeloma cell line K6H6/B5 (ATCC® CRL1823TM).
Cell line cultivation
The cell line K6H6/B5 (ATCC CRL-1823) were used as electrofusion partners. The cell line K6H6/B5 were grown in RPMI 1640 (Sigma-Aldrich, USA) medium supplemented with 10% FBS. Cell lines were cultivated in culture flasks with a growth surface of 150 cm2 at 37 ºC in a humid 5% CO2 atmosphere. For electrofusion, these cell lines were used in the exponential growth phase.
Electrofusion
Hybridization was performed using a cell electrofusion system consisting of a BTX ECM 2001 electrofusion and electroporation generator and a Model 453 microslide (BTX, Harvard Bioscience, USA). We mixed both cells in the ratio of 1:2 at the concentration 3 × 105 cells/mL for B cells (plasmablasts and activated memory B cells) and 6 × 105 cells/mL for K6H6/B5 cells. The resulting cell cocktail was washed twice by centrifugation at 400×g for 5 minutes in an excess of BTXpress Cytofusion Medium C electrofusion buffer (BTX, Harvard Bioscience, USA). Then the cell cocktail was resuspended in 500 μL of BTXpress Cytofusion Medium C buffer, transferred into an electrofusion cuvette and electroporated under the following electric field conditions: field strength – 1560 V/cm, cell dielectrophoresis duration – 60 s; and electric pulse conditions: a 30 μs-long double pulse with an amplitude of 500 V.
Cultivation of heterohybridomas
Upon fusion, the cells were inoculated into 96-well microplates with Hybri-Care medium (ATCC 46-XTM) supplemented with 20% FBS (Gibco, USA) and hypoxanthine-aminopterin-thymidine (HAT) supplement (Gibco, USA) and incubated at 37 °C in 5% CO2 atmosphere. After 3-4 days, the culture medium in each well was replaced. Hybrid colonies were selected using selective HAT and HT supplements (Thermo Fisher, USA). On Day 14, the HAT-supplemented medium was replaced with HT-supplemented medium and on Day 20 with the regular culture medium.
Screening of heterohybridhuMAbs-producing cells
The positive heterohybrid clones producing huMAbs to BoNT/A HC50 and LC were selected by ELISA. 96-well ELISA Microplates (Corning, USA) were coated with 100 μL of recombinant BoNT/A HC50 and LC proteins (0.5 μg/well) in PBS and incubated in a shaker (300 rpm) for 2 hours at 37 °C. The wells were washed three times with PBS-Tw and blocked with 0.5% skim milk powder (SERVA Electrophoresis GmbH, Germany) in PBS-Tw at 37 °C for 1 hour. The plates were then washed three times with PBS-Tw. Then, hybridoma supernatants (100 μL) were added into each well and incubated for a 1 hour at 37 °C. The wells were washed three times with PBS-Tw and incubated with 100 μL (1:20 000) of Rabbit anti-human IgG (Sigma-Aldrich, USA) conjugated to HRP diluted in PBS-Tw for 1 hour at 37 ˚C. The plates were washed six times and were incubated with 100 μL of TMB (Sigma-Aldrich, USA) for 10 minutes at RT. Finally, the reaction was stopped by adding 50 μL per well of 4 N H2SO4, and the plates were read at 450 nm using an xMark microplate absorbance spectrophotometer (Bio-Rad, USA). PBS and culture medium were used as a negative control.
Scaling up hybridoma supernatants and huMAbs generation
The heterohybrid specific huMAbs-producing cells were cloned by the limiting dilution. The heterohybrid cell line clones were cultured in 96-well microplates and then scaled-up in 24-well microplates and Corning® T-25, T-75 and T-150 culture flasks containing medium Hybri-Care (ATCC® 46-XTM) supplemented with 20% FBS at 37 °C in 5% CO2 atmosphere. The subcultivation cell counts in the culture flasks were maintained in the range of 1×105 to 1×106 per 1 mL of culture medium. The culture medium was replaced every 2-3 days.
When replacing the medium, 100 μL hybridoma supernatants were taken for ELISA assay of the specific activity to BoNT/A HC50 and LC. Scaling-up was carried out only in case of stable cell growth and stable production of antibodies with specific activity (the stability of huMAbs production were assessed based on at least three hybridoma supernatants samples taken at different time points). Stable heterohybridomas were adapted to a serum-free Hybridoma-SFM medium (Gibco, UK) and cultured for 5 days in a Corning® T-150 culture flask at an initial density of 5×105 cells/mL for the subsequent isolation and purification of huMAbs.
HuMAbs purification and characterization
HuMAbs were isolated from hybridoma supernatants by affinity chromatography in a protein Protein G-Sepharose column (HiTrapTM Protein G, Sweden). After applying the samples, the column was washed to remove unbound components, and immunoglobulins were eluted with 0.05 N glycine buffer, 0.15 N NaCl, pH 2.3. The isolated immunoglobulins were transferred into PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) by gel filtration on a Superdex 200 10/300 GL (GE Healthcare, UK). The purity of the resulting immunoglobulin fractions was assessed by denaturing SDS-PAGE. Immunoglobulin concentrations were measured spectrophotometrically at 280 nm (Smart Spec Plus spectrophotometer, Bio Rad, USA).
Measurement of specific antibody activity by western blot
The specificity of monoclonal antibody clones to the native BoNT/A, BoNT/A HC50 and LC was performed by Western blot assay. BoNT/A HC50 and LC proteins and native BoNT/A underwent denaturing electrophoresis in 10% polyacrylamide gel, and then the proteins were horizontally transferred from the gel onto a Hybond-C Extra nitrocellulose membrane (GE Healthcare, UK) in a blotter (Trans-Blot® TurboTM Transfer System, Bio-Rad, USA). After transfer, membranes were blocked with 0.5% skim milk powder (SERVA Electrophoresis GmbH, Germany) for 1 h at RT. Then, the membranes were washed three times with PBS-Tw. The blocked and washed membranes were cut into strips so that each strip contained a molecular weight marker and all the test proteins (recombinant BoNT/A HC50 and LC, native BoNT/A). Each strip was incubated separately with 1 μg/mL huMAbs solution in PBS for 1 hour at 37 °C. The strips were washed three times with PBS-Tw and incubated with Rabbit anti-human IgG (Sigma-Aldrich, USA) conjugated to HRP diluted in PBS-Tw (1:40000) for 1 hour at 37 ˚C in a shaker. The strips were washed six times with PBS-Tw. The reaction was visualized using a diaminobenzidine substrate solution based on 0.05% diaminobenzidine (Sigma-Aldrich, USA) and 0.015% H2O2 in PBS, pH 7.4. The reaction was stopped by washing with distilled water.
Measurement of the neutralizing activity of huMAbs by FRET assay
FRET assay was used to measure the neutralizing activity of huMAbs. The underlying principle of the assay is to inhibit the proteolytic activity of the recombinant BoNT/A LC by huMAbs using the commercially available FRET substrate SNAPtide® Botulinum Toxin A Substrate, Fluorogenic (Sigma-Aldrich, USA).
For this, a 5 mM stock substrate solution in dimethyl sulfoxide was prepared. Then, a 250 μM solution was prepared using a reaction buffer containing 20 mM HEPES pH 8.0, 5 mM DTT, 0.3 mM ZnSO4, and 0.1% Tween-20. The reaction was run in an OptiPlate-96 microplate (Perkin Elmer). The total volume of solution per well was 250 μL. The total volume of solution in each well was 250 μL. The working concentration of the FRET substrate in a well was 10 μM. 10 μg of recombinant protein BoNT/A LC and 60 μg of huMAbs were added to each well, diluted to 240 μL with reaction buffer and incubated in a thermostated shaker at 37 C for 60 min. Immediately before transferring the plate into the reader Multimode Plate Reader EnSpire (Perkin Elmer, USA), 10 μL of 250 μM FRET substrate solution was added into the wells. Three samples were used as a negative control: the sample containing buffer and SNAPtide protein (negative control 1); the sample containing buffer (negative control 2); the sample containing buffer and studied huMAbs (negative control 3). As a positive control was used the sample containing BoNT/A LC and SNAPtide protein. The fluorescent signal was recorded every 10 min at Ex/Em = 320 nm/420 nm.
The inhibitory activity of the studied huMAbs was calculated as follows:
where RFU (studied huMAbs) is relative fluorescent units of the sample with the investigated huMAbs;
RFU (negative control 1) is relative fluorescent units of the sample containing buffer and SNAPtide protein;
RFU (positive control) is relative fluorescent units of the sample containing LC BoNT/A and SNAPtide protein.
Statistical analysis
Statistical analysis and visualization of data was performed using GraphPad Prism 6 software. Cytometric results were expressed as Me (Q25; Q75). The data of FRET assay and ELISA were presented as the means ± SD.
Results
Generation of recombinant BoNT /A HC50 and LC proteins
Recombinant BoNT/A HC50 and LC proteins were generated in a bacterial expression system, purified by chromatography and verified electrophoresis in 10% SDS-PAGE with Coomassie staining (Fig. 3).
Fig. 3.
Electrophoretic separation of the purified recombinant BoNT/A HC50 and LC proteins. Proteins were separated by SDS-PAGE under reducing conditions (2×sample buffer containing 0.125 M Tris, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, 0.02% bromphenol blue, pH 6.8 and heating at 95°C for 5 min) on a 5% stacking, 10% running gel. Protein bands were detected by Coomassie blue staining. Note: 1 – molecular weight marker PageRulerTM Prestained Protein Ladder SM0671 (Fermentas, USA); 2 – BoNT/A HC50 protein, MW 56.3 kDa); 3 – BoNT/A LC protein, MW 52.8 kDa.
.
Electrophoretic separation of the purified recombinant BoNT/A HC50 and LC proteins. Proteins were separated by SDS-PAGE under reducing conditions (2×sample buffer containing 0.125 M Tris, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, 0.02% bromphenol blue, pH 6.8 and heating at 95°C for 5 min) on a 5% stacking, 10% running gel. Protein bands were detected by Coomassie blue staining. Note: 1 – molecular weight marker PageRulerTM Prestained Protein Ladder SM0671 (Fermentas, USA); 2 – BoNT/A HC50 protein, MW 56.3 kDa); 3 – BoNT/A LC protein, MW 52.8 kDa.
Detection of specific antibodies against BoNT /A HC50 and LC proteins in donor serum
We analyzed specific antibodies to BoNT/A HC50 and LC for effective isolation of antigen-specific plasmablasts and activated memory B cells from donor peripheral blood after BoNT/A injections (Fig. 4).
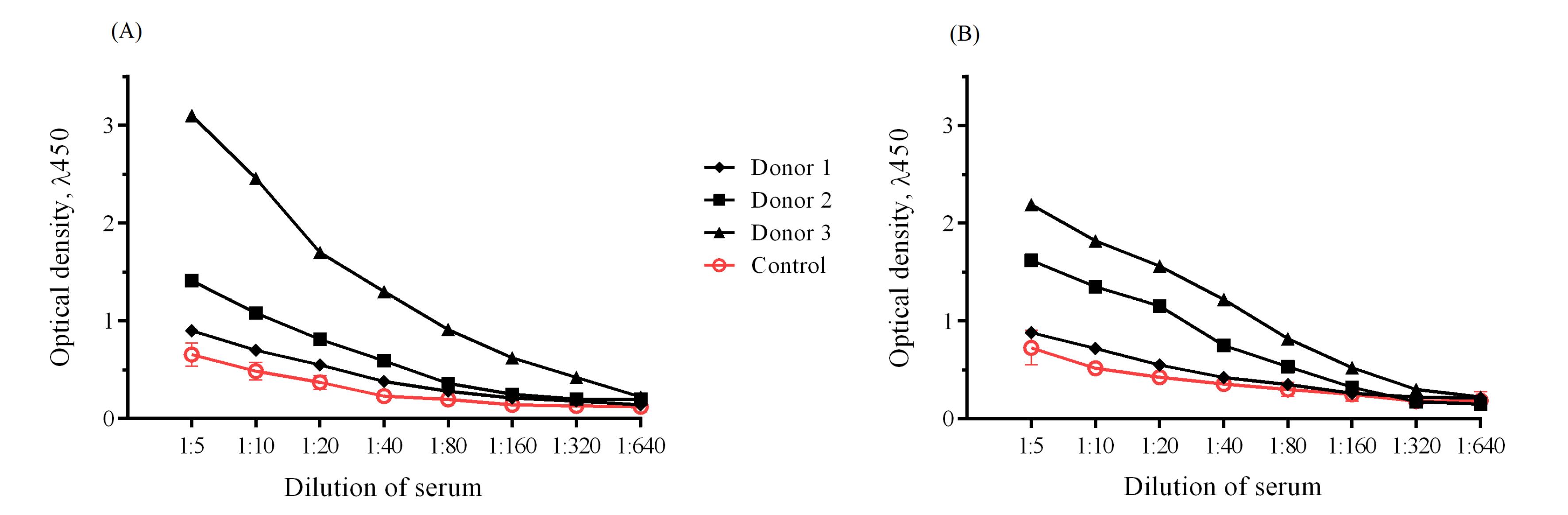
Fig. 4.
Detection of specific antibodies against BoNT/A HC50 (A) and BoNT/A LC (B) in donor serum.
.
Detection of specific antibodies against BoNT/A HC50 (A) and BoNT/A LC (B) in donor serum.
Specific antibodies were detected in two of the three donors who were injected with BoNT/A. In the control serum samples, specific antibodies specific against BoNT/A did not exceed the threshold values.
Assessment of specific binding of donor B cells to fluorochrome-labeled recombinant BoNT /A HC50 and LC proteins
Further, we carried out a comparative assessment of the content of specific IgG+ B cells in the peripheral blood of donors. To measure the cell counts of cells specific to BoNT/A HC50 and BoNT/A LC in the total plasmablasts and activated memory B cells pool, a mixture of PE-labeled recombinant BoNT/A HC50 and BoNT/A LC proteins was added into the phenotyping panel. The previous studies showed that a short-term release of specific plasmablast and activated memory B cells into the bloodstream on day 7 post-immunization or infection with the pathogen.34,42
Therefore, on day 7 post the scheduled BoNT/A injection, blood was sampled from the donors. After B cell phenotyping by multiple gating, a subpopulation of IgG+ plasmablasts and activated memory B cells was isolated and the cell counts of cells expressing specific membrane-bound anti-BoNT/A HC50 and LC IgG antibodies were measured in these subpopulations (Fig. 5).
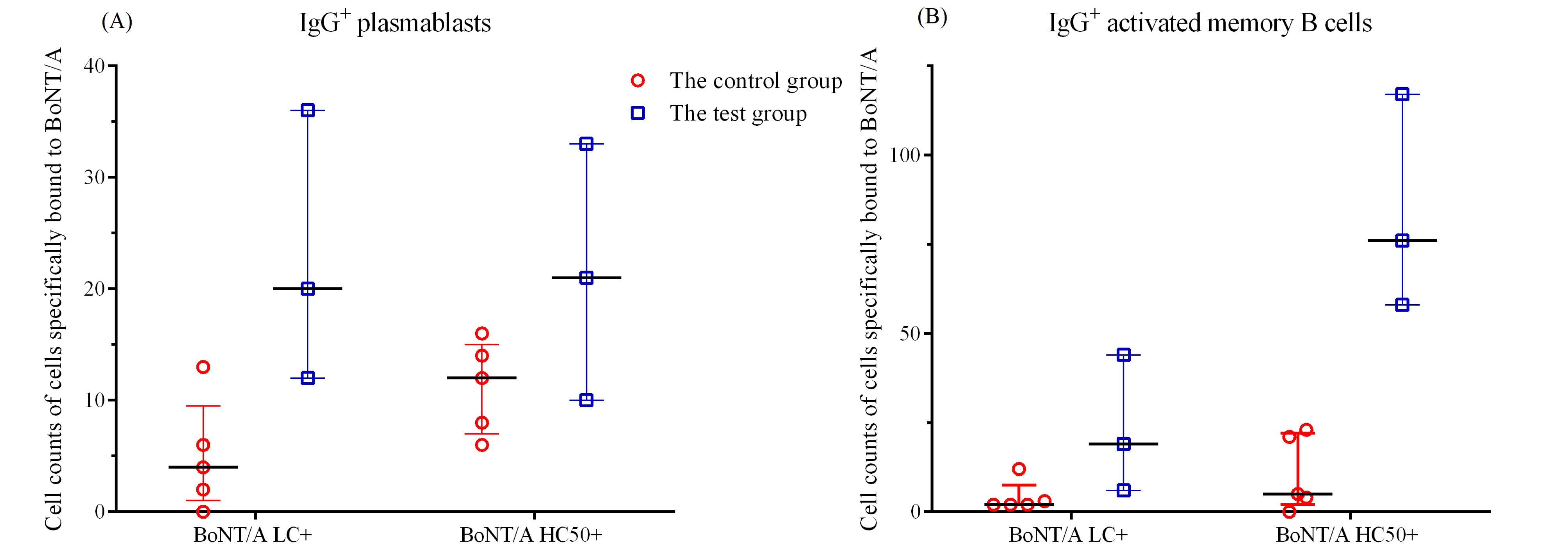
Fig. 5.
IgG+ plasmablasts (A) and IgG+ activated memory B cells (B) expressing specific membrane-bound anti-BoNT/A LC and HC50 antibodies in the test and control groups.
.
IgG+ plasmablasts (A) and IgG+ activated memory B cells (B) expressing specific membrane-bound anti-BoNT/A LC and HC50 antibodies in the test and control groups.
Cytometric analysis showed the absence of specific binding of peripheral blood B cells of control donors to BoNT/A HC50 or BoNT/A LC. Donor 3 had the highest titers of specific Ig antibodies and the content of specific B cells. Therefore, donor 3 blood was selected for sorting plasmablasts and activated memory B-cells to obtain hybridomas.
BoNT /A-specific heterohybridomas generation
Heterohybridoma clones producing huMAbs to recombinant BoNT/A HC50 or LC were generated by electrofusing plasmablasts (CD19+ CD27hi CD38hi) and activated memory B cells (CD19+ CD27+ CD38+) derived from an immune donor with maximum antibody titers and the amount of specific IgG+ B cells to BoNT/A with the cell line K6H6/B5. Primary heterohybridoma screening for their ability to produce huMAbs to the BoNT/A HC50 and LC was performed 10 days post fusion by ELISA assay. Based on the primary screening, 10 clones with a high ELISA signal value were selected that indicated specific immunoglobulin content in the hybridoma supernatants.
To generate stable single hybridoma clones, all selected clones were subcloned twice by limiting dilutions. After each subloning step, a new testing run was performed to identify the specific interaction of heterohybridoma clone-produced huMAbs with the recombinant BoNT/A HC50 or LC by ELISA assay (Table 1).
Table 1.
Selection data of heterohybridoma clones producing specific huMAbs to BoNT/A HC50 or LC
|
Heterohybridoma clones |
Primary screening (optical density (OD450) by ELISA)
|
Post cloning ELISA data
|
Post subcloning ELISA data
|
ELISA data in scaling-up
(OD450)
|
Assay I
(OD450)
|
Assay II
(OD450)
|
Assay I
(OD450)
|
Assay II
(OD450)
|
| 1В9 |
0.687 |
0.632 |
0.598 |
0.591 |
0.607 |
0.613 |
| 1D3 |
0.907 |
0.807 |
0.635 |
0.523 |
0.394 |
0.15 |
| 1В5 |
0.581 |
0.532 |
0.6 |
0.574 |
0.597 |
0.589 |
| 2С11 |
0.81 |
0.189 |
– |
– |
– |
– |
| 2С10 |
0.742 |
0.808 |
0.798 |
0.765 |
0.754 |
0.732 |
| 3D7 |
1.4 |
0.603 |
0.171 |
– |
– |
– |
| 4F5 |
0.72 |
0.75 |
0.324 |
0.19 |
– |
– |
| 5С8 |
1.04 |
0.837 |
0.817 |
0.425 |
0.145 |
– |
| 5H3 |
0.475 |
0.437 |
0.517 |
0.325 |
0.113 |
0.124 |
| 6D9 |
1 |
0.753 |
0.432 |
0.240 |
– |
– |
Based on these experiments, some heterohybridoma clones were removed from the study because they had lost their ability to produce antibodies during cultivation. 3 of 10 heterohybridoma clones produced stably specific immunoglobulins (i.e., 1B9, 1B5, and 2C10) and were scaled-up and cryoconserved to establish a master cell bank.
huMAbs purification
Anti-human IgG-HRP-conjugated polyclonal antibodies were used to detect the binding of huMAbs to BoNT/A HC50 or LC proteins by ELISA. The presence of the reaction confirmed the human nature of MAbs and protein G-binding ability of immunoglobulins. Therefore, the huMAbs were isolated from the hybridoma supernatants and purified by affinity chromatography in a HiTrapTM Protein G (GE Healthcare Bio-Sciences, Sweden) followed by gel filtration on a column Superdex 75 10/300 GL (GE Healthcare Bio-Sciences, Sweden). The resulting huMAbs purity was evaluated by SDS-PAGE. The light and heavy antibody chains were detected in a soluble protein preparation as bands migrating in polyacrylamide gel with molecular weights of 23-26 kDa and 53-55 kDa, respectively (Fig. 6).
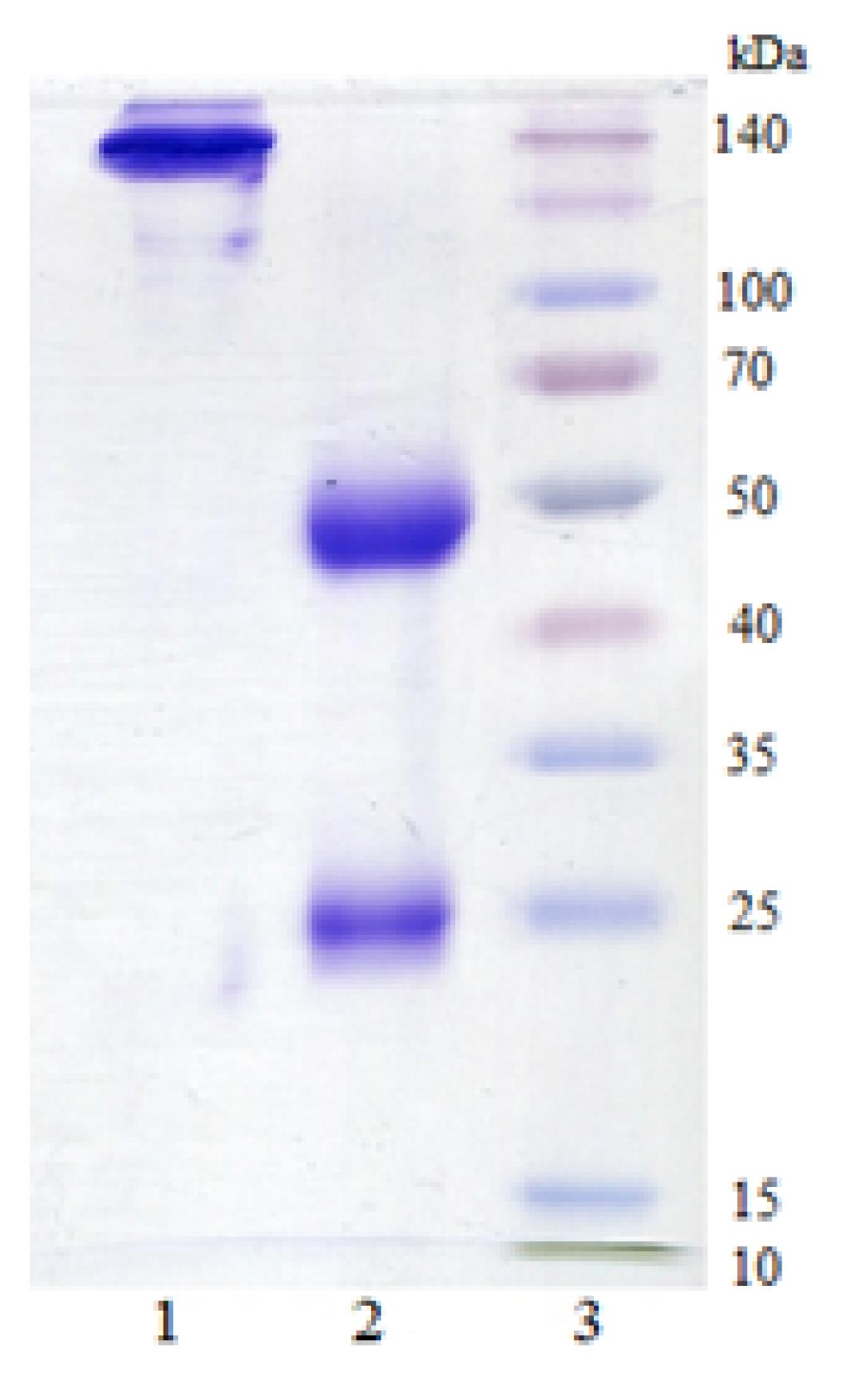
Fig. 6.
Electrophoretic separation of the huMAb (1B9). Antibodies were separated by SDS-PAGE under non-reducing conditions (2×sample buffer containing 0.125 M Tris, 4% SDS, 20% glycerol, 0.02% bromphenol blue, pH 6.8) and reducing conditions (2×sample buffer containing 0.125 M Tris, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, 0.02% bromphenol blue, pH 6.8 and heating at 95°C for 5 min) on a 5% stacking, 10% running gel. Protein bands were detected by Coomassie blue staining. Note: 1 – huMAb sample under non-reducing conditions; 2 – huMAb sample under reducing conditions; 3 – molecular weight marker SpectraTM Multicolor Broad Range Protein Ladder (Thermo Scientific, USA).
.
Electrophoretic separation of the huMAb (1B9). Antibodies were separated by SDS-PAGE under non-reducing conditions (2×sample buffer containing 0.125 M Tris, 4% SDS, 20% glycerol, 0.02% bromphenol blue, pH 6.8) and reducing conditions (2×sample buffer containing 0.125 M Tris, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, 0.02% bromphenol blue, pH 6.8 and heating at 95°C for 5 min) on a 5% stacking, 10% running gel. Protein bands were detected by Coomassie blue staining. Note: 1 – huMAb sample under non-reducing conditions; 2 – huMAb sample under reducing conditions; 3 – molecular weight marker SpectraTM Multicolor Broad Range Protein Ladder (Thermo Scientific, USA).
huMAbs specificity determination
The BoNT/A specific huMAbs activity was measured by Western blot assay. All three huMAbs had specific activity to native BoNT/A and BoNT/A LC (Fig. 7).
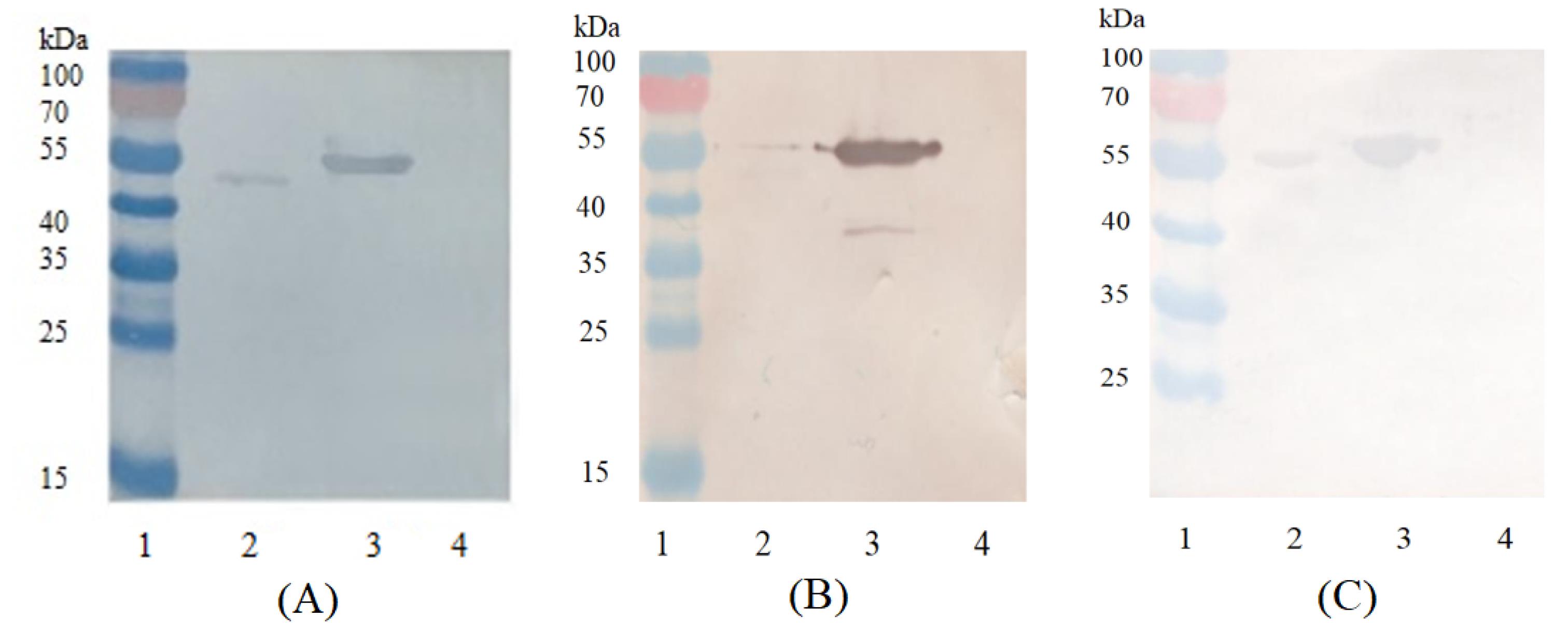
Fig. 7.
Western blot assay of 1B9 (A), 1B5 (B) and 2C10 (C) huMAbs binding to native BoNT/A, recombinant BoNT/A HC50 and LC proteins. Proteins (native BoNT/A, recombinant HC50 and LC BoNT/A) were separated by SDS-PAGE under reducing conditions (2×sample buffer containing 0.125 M Tris, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, 0.02% bromphenol blue, pH 6.8 and heating at 95°C for 5 min) on a 5% stacking, 10% running gel. Following transfer onto a nitrocellulose membrane the immunoblot were probed with one of huMAbs (1B9, 1B5 or 2C10) and detected by an anti-human IgG-HRP-conjugated rabbit antibody. Bands were visualized using a diaminobenzidine substrate solution based on 0.05% diaminobenzidine, 0.015% H2O2 in PBS, pH 7.4. Notes: 1 – molecular weight marker PageRuler TM Prestained Protein Ladder SM0671 (Fermentas, USA); 2 – native BoNT/A; 3 –BoNT/A LC; 4 –BoNT/A HC50.
.
Western blot assay of 1B9 (A), 1B5 (B) and 2C10 (C) huMAbs binding to native BoNT/A, recombinant BoNT/A HC50 and LC proteins. Proteins (native BoNT/A, recombinant HC50 and LC BoNT/A) were separated by SDS-PAGE under reducing conditions (2×sample buffer containing 0.125 M Tris, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, 0.02% bromphenol blue, pH 6.8 and heating at 95°C for 5 min) on a 5% stacking, 10% running gel. Following transfer onto a nitrocellulose membrane the immunoblot were probed with one of huMAbs (1B9, 1B5 or 2C10) and detected by an anti-human IgG-HRP-conjugated rabbit antibody. Bands were visualized using a diaminobenzidine substrate solution based on 0.05% diaminobenzidine, 0.015% H2O2 in PBS, pH 7.4. Notes: 1 – molecular weight marker PageRuler TM Prestained Protein Ladder SM0671 (Fermentas, USA); 2 – native BoNT/A; 3 –BoNT/A LC; 4 –BoNT/A HC50.
Neutralizing activity of huMAbs in vitro
We analyzed the neutralization activity of the 1B9, 1B5 and 2C10 antibodies using FRET assay, which measures cleavage of a synthetic SNAP-25 peptide (SNAPtide), the proteolytic substrate of the BoNT/A LC (Fig. 8).43 HuMAbs were incubated with SNAPtide and then the fluorescence intensity was measured.
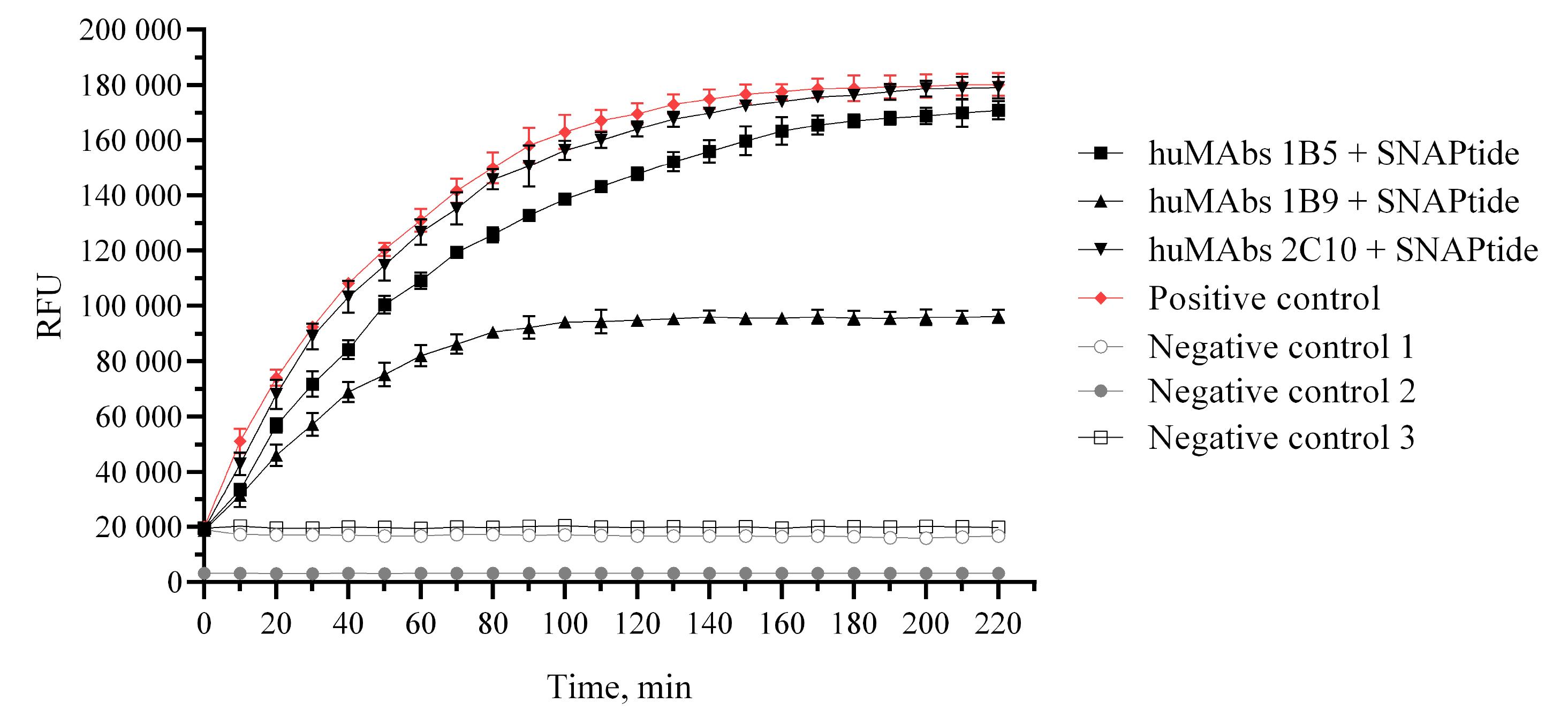
Fig. 8.
Determination of huMAbs ability to inhibit the proteolytic activity of BoNT/A LC. Notes: Negative control 1 – sample containing reaction buffer and SNAPtide protein; negative control 2 – sample containing reaction buffer; negative control 3 – sample containing reaction buffer and mix studied huMAbs; positive control – sample containing BoNT/A LC and SNAPtide protein.
.
Determination of huMAbs ability to inhibit the proteolytic activity of BoNT/A LC. Notes: Negative control 1 – sample containing reaction buffer and SNAPtide protein; negative control 2 – sample containing reaction buffer; negative control 3 – sample containing reaction buffer and mix studied huMAbs; positive control – sample containing BoNT/A LC and SNAPtide protein.
Antibodies 1B5 and 2C10 did not interfere with cleavage of SNAPtide, yielding 170834 ± 3312 fluorescence units (RFU) and 179059 ± 3904 RFU, respectively, compared to 180150 ± 4121 RFU for BoNT/A LC, while antibody 1B9 inhibited cleavage of SNAPtide (96206 ± 2368 RFU). Therefore, 1B9 huMAb is able to inhibit the activity of the BoNT/A metalloproteinase with an inhibitory activity of 48.6%.
Discussion
Hybridoma technology remains the preferred method for producing huMAbs with naturally paired immunoglobulin genes.33,34 The development of recombinant technologies for producing antibodies, such as chimerization and humanization, has increased the potential of hybridoma technology and made it possible to overcome barriers at the species level and has shown success in the generation of hybridomas between phylogenetically different species.32
Hybridoma technology is based on the fusion of antigen-specific B cells and myeloma cells. The main problem is obtaining antigen-specific B cells from donated blood. B cell subsets of terminally-differentiated plasma cell are found in the bone marrow (a relatively stable population of long-lived plasma cells) or in the spleen and other secondary lymphoid organs (short-lived plasma cells). In the peripheral blood, plasma cells circulate at a very low frequency. In a secondary immune response, the precursors of plasma cells are activated memory B cells and/or plasmablasts, which are already capable of producing specific membrane-bound antibodies.44-46 It has been shown that 7-8 days after immunization or disease, there is a transient release of plasmablasts and activated memory B cells into the bloodstream.34,42,47
In our work, the precursors of plasma cells, plasmablasts and memory B cells, were isolated by flow cytometry from the blood of donors on the 7th day after the next immunization with Relatox. The enrichment population of B cells was labeled with monoclonal antibodies, and the populations of activated memory B cells and plasmablasts were sorted according to the CD19+ CD27+ CD38+ and CD19+ CD27hi CD38hi phenotypes, respectively.
CD 27 is a universal marker of human memory B cells, whose detection on the cell membrane indicates the transition of B cells into a more mature state of memory cells.48,49 СD38 molecule allows for identification of CD38+ and CD27+ activated memory cells as well as of CD38hi and CD27hi plasmablasts.50 As a result of sorting 3×105 cells with both phenotypes were collected and used for further somatic cell hybridization.
The cell line K6H6/B5 was used as fusion partner cells. K6H6/B5 derived by a fusion of NS-1-Ag4 myeloma cells with cells from a human nodular lymphoma. Originally the cells secreted human IgM with a lambda light chain; however, with continuous passage they spontaneously lost the ability to secrete immunoglobulin. They are used as a fusion partner for human B cells.
To name a few, K6H6/B5 were used to generate hybridomas producing human anti-hepatitis C virus antibodies.35
Another limiting factor in the production of human hybridoma is the efficiency of fusion, which can be increased by choosing the optimal conditions and parameters.36
Known huMAbs to BoNT were obtained by fusion of myeloma cells with PMBCs31 or B cells9 isolated from the peripheral blood of donors having high titers of antibodies to BoNT. We assume that the predominance of B cells and the subsequent sorting of subpopulations of specifically activated memory B cells and plasmablasts makes it possible to obtain a pool of cells that have not yet completed the terminal stage of differentiation, but have undergone somatic hypermutation of Ig-variable regions in the germinal centers after encountering with the antigen.51-53 The use for somatic cell hybridization of plasmablasts and memory B cells isolated at the peak of the immune response (on the 7th day of immunogenesis) contributes to an increase in the efficiency of obtaining specific hybridomas even with a weak immune response of the donor (with low specific antibody and specific B cells in the blood). The huMAbs produced by in vitro methods54,55 do not provide a dependable evaluation of the specificity of human-generated epitopes in vivo. As a result, the use of these methods is limited in fields such as epitope discovery, vaccine development, and evaluation.
The donor of specific lymphocytes for subsequent fusion should be actively infected patients or those who have been exposed to antigen.32 Due to ethical considerations, immunizing people is usually impossible. Vaccines developed against botulism are currently not being used56 even for people at risk of occupational exposure to BoNT due to the low efficacy.57 However, low doses of BoNT/A are increasingly used therapeutically with clinical or cosmetic benefits.58-62
In this study, donors had a minimum of 15 injections with BoNT/A (Relatox®) for aesthetic botulinum therapy for at least 10 years.
The possibility of antibody formation in such patients has been previously shown in a number of publications.63,64 The human immunological response to BoNT/A is usually low and not observed in all people, which is probably due to the product doses injected being low.65-68
Variations in the immune systems of the donors used can lead to differences in the quantity of antibodies generated. Certain donors may respond poorly to the vaccination, potentially yielding only 10-30% of antigen-specific antibody-secreting cells, resulting in a low production of antigen-specific antibodies. Therefore, in the endeavour to produce huMAbs, we initially analysed the serum of donors and identified donors possessing the greatest antibody titres to BoNT/A as a potential source of specific plasmablasts and activated memory B cells.
A total 3 of stable hybridoma producing huMAbs against BoNT/A LC, but not BoNT/A HC were obtained after electrofusion of the K6H6/B5 cell line with plasmablasts and activated memory B cells. It seems we lost hybridoma clones that produced antibodies specific to BoNT/A HC, because antibodies and activated B-cells/plasmablasts specific to BoNT/A-HC were found in the blood of the donor.
The purpose of our study was to show the possibility of obtaining huMAbs to BoNT/A using the above technique.
The BoNT/A LC is the catalytic domain of BoNT/A acquires proteolytic activity as it is transported across the vesicle membrane into the neuron cytosol. Through cleavage of tethering proteins, the BoNT/A LC prevents the neuron from releasing acetylcholine in response to neural stimulation. As the obtained huMAbs directly binds to the BoNT/A LC, further in our study we investigated whether it can inhibit the catalytic function of the LC. Further analysis was performed using the synaptosomal-associated protein, 25-kD (SNAP-25) substrate. SNAP-25 is a membrane protein and a component of soluble NSF attachment receptor (SNARE) protein complex, which docks synaptic vesicles to the neuron’s presynaptic membrane and fuses them with subsequent neurotransmitter release. BoNT/A LC, which is a zinc (Zn2+) metalloprotease, penetrates into the cytosol and cleaves the components of the SNARE complex. In response to BoNT/A, SNAP-25 is specifically cleaved and inactivated via removal of 9-amino acids from the C-terminal peptide.69 Acetylcholine penetration into the neuromuscular synapse, therefore, becomes impossible, which leads to paralysis. In our research using SNAP-25 substrate, we have shown that each of obtained huMAbs is capable neutralize the BoNT/A.
The majority of studies related to the generation of antibodies that neutralize BoNT/A are aimed at producing BoNT/A HC-binding antibodies. This is due to the fact that it is through BoNT/A HC involvement that BoNT binds to presynaptic receptors and BoNT/A LC translocates across the endosome membrane. Therefore, by inhibiting the BoNT/A penetration via blocking the BoNT/A HC binding sites by MAbs, the manifestation of the BoNT toxicity can be prevented. At the same time, the kinetic studies have revealed that combinations of antibodies work together to enhance the potency of neutralization while maintaining the stability of the antibody-toxin complex.70 A theoretical advantage of incorporating a BoNT/A LC-specific huMAb into a mixture of neutralizing huMAbs is the added safeguard against BoNT/A through intracellular neutralization.2 Additionally, pharmacokinetics research has indicated that in the blood circulation, immune complexes formed between BoNT and polyclonal antisera quickly trap the toxin in the liver and spleen.71
MAbs specific for BoNT/A LC are able to inhibit the BoNT/A molecule at the final stages of the intoxication process, after the penetration of BoNT/A into the cell, but before the cleavage of the intracellular substrate SNAP-25. A number of works have described the mechanisms that determine the inhibitory activity of LC-specific MAbs: retention of LC in endosomes; return of BoNT to the cell surface by recycling of synaptic vesicles72; prevention of BoNT/A LC translocation across the endosomal plasma membrane.73 The BoNT/A LC is an important target in the creation of BoNT/A-neutralizing huMAbs.74 The mechanism of action that determines the ability of huMAb 1B9 to neutralize the enzymatic activity of BoNT/A LC is unknown and requires further research. An antibody which is specific to BoNT/A LC is capable of potently inhibiting BoNT/A in vivo and in vitro, utilizing mechanisms that have not previously been associated with BoNT-neutralizing antibodies. Antibodies which are specific to BoNT LC have the potential to be valuable components of an antibody antidote for BoNT exposure.9
Conclusion
In summary, in our study we proposed a new approach to the creation of huMAb-producing hybridomas. The new approach is that we combined the cytometric sorting method, which made it possible to concentrate plasmablasts and memory B cells, and hybridoma technology. As a result of carrying out hybridization by electrofusion method and using as partner cells heteromyeloma cell line K6H6/B5, 10 clones were obtained. 3 of 10 clones produced BoNT/A LC-specific huMAbs, and one clone produced huMAbs capable of neutralizing BoNT/A enzymatic activity. This approach makes it possible to obtain huMAbs from donors with a weak immune response.
Research Highlights
What is the current knowledge?
√ Hybridoma technology is the most popular technique to obtain mouse monoclonal antibodies. Recent technical advances in expanding the starting number of human antigen-specific B cells, improving fusion efficiency, and isolating new myeloma partners and new cell cloning methods have enabled the development of protocols that make the isolation of human monoclonal antibodies from blood samples feasible.
What is new here?
√ To generating a producer of huMAbs specific to the proteolytic domain of BoNT/A via hybridoma technology, subpopulations of specifically activated memory B cells and plasmablasts can be used isolated from the peripheral blood of donors who received aesthetic botulinum therapy and their subsequent electrofusion with K6H6/В5 fusion partner cells.
Competing Interests
The authors declare no conflict of interest.
Ethical Statement
This study was approved by the Local Bioethics Committee of the Federal State Budget Institution of Science "Kirov Research Institute of Hematology and Blood Transfusion of the Federal Medical and Biological Agency", ethics committee approval number № 3 from 20 January 2021 year.
Funding
This study was financially supported by a grant of the Ministry of Science and Higher Education of the Russian Federation (Agreement No. 075-15-2019-1671 of October 31, 2019).
References
- Rao AK, Sobel J, Chatham-Stephens K, Luquez C. Clinical guidelines for diagnosis and treatment of botulism. MMWR Recomm Rep 2021; 70:1-30. doi: 10.15585/mmwr.rr7002a1 [Crossref] [ Google Scholar]
- Pirazzini M, Rossetto O, Eleopra R, Montecucco C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol Rev 2017; 69:200-235. doi: 10.1124/pr.116.012658 [Crossref] [ Google Scholar]
- Chatham-Stephens K, Fleck-Derderian S, Johnson SD, Sobel J, Rao AK, Meaney-Delman D. Clinical features of foodborne and wound botulism: a systematic review of the literature, 1932–2015. Clin Infect Dis 2017; 6(suppl_1):S11-S16. doi: 10.1093/cid/cix811 [Crossref] [ Google Scholar]
- Hughes JM, Blumenthal JR, Merson MH, Lombard GL, Dowell Jr VR, Gangarosa EJ. Clinical features of types A and B food-borne botulism. Ann Intern Med 1981; 95(4):442-445. doi: 10.7326/0003-4819-95-4-442 [Crossref] [ Google Scholar]
- Woodruff BA, Griffin PM, McCroskey LM, Smart JF, Wainwright RB, Bryant RG, Hutwagner LC, Hatheway CL. Clinical and laboratory comparison of botulism from toxin types A, B, and E in the United States, 1975–1988. J Infect Dis 1992; 166(6):1281-1286. doi: 10.1093/infdis/166.6.1281 [Crossref] [ Google Scholar]
- Smith LD. Botulism. The organism, its toxins, the disease. Illinois, USA: Charles C. Thomas; 1977.
- Humeau Y, Doussau F, Grant NJ, Poulain B. How botulinum and tetanus neurotoxins block neurotransmitter release. Biochimie 2000; 82(5):427-446. doi: 10.1016/s0300-9084(00)00216-9 [Crossref] [ Google Scholar]
- Rao AK, Lin NH, Jackson KA, Mody RK, Griffin PM. Clinical characteristics and ancillary test results among patients with botulism—United States, 2002–2015. Clin Infect Dis 2017; 66(suppl_1):S4-10. doi: 10.1093/cid/cix935 [Crossref] [ Google Scholar]
- Adekar SP, Takahashi T, Jones RM, Al-Saleem FH, Ancharski DM, Root MJ. Neutralization of botulinum neurotoxin by a human monoclonal antibody specific for the catalytic light chain. PloS One 2008; 3:e3023. doi: 10.1371/journal.pone.0003023 [Crossref] [ Google Scholar]
- Zhang Y, Gardberg AS, Edwards TE, Sankaran B, Robinson H, Varnum SM. Structural insights into the functional role of the Hcn sub-domain of the receptor-binding domain of the botulinum neurotoxin mosaic serotype C/D. Biochimie 2013; 95:1379-1385. doi: 10.1016/j.biochi.2013.03.006 [Crossref] [ Google Scholar]
- Blasi J, Chapman ER, Link E, Binz T, Yamasaki S, De Camilli P, Jahn R. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 1993; 365:160-163. doi: 10.1038/365160a0 [Crossref] [ Google Scholar]
- O'Horo JC, Harper EP, El Rafei A, Ali R, DeSimone DC, Sakusic A. Efficacy of antitoxin therapy in treating patients with foodborne botulism: a systematic review and meta-analysis of cases, 1923–2016. Clin Infect Dis 2017; 66:43-56. doi: 10.1093/cid/cix815 [Crossref] [ Google Scholar]
- Robinson RF, Nahata MC. Management of botulism. Ann Pharmacother 2003; 37(1):127-131. doi: 10.1345/aph.1C034 [Crossref] [ Google Scholar]
- Griese SE, Kisselburgh HM, Bartenfeld MT, Thomas E, Rao AK, Sobel J. Pediatric botulism and use of equine botulinum antitoxin in children: a systematic review. Clin Infect Dis 2017; 66(suppl_1):S17-29. doi: 10.1093/cid/cix812 [Crossref] [ Google Scholar]
- Yu PA, Lin NH, Mahon BE, Sobel J, Yu Y, Mody RK, Rao AK. Safety and improved clinical outcomes in patients treated with new equine-derived heptavalent botulinum antitoxin. Clin Infect Dis 2017; 66:57-64. doi: 10.1093/cid/cix816 [Crossref] [ Google Scholar]
- Schussler E, Sobel J, Hsu J, Yu P, Meaney-Delman D, Grammer LC 3rd, et al. Workgroup Report by the Joint Task Force Involving American Academy of Allergy, Asthma & Immunology (AAAAI); Food Allergy, Anaphylaxis, Dermatology and Drug Allergy (FADDA) (Adverse Reactions to Foods Committee and Adverse Reactions to Drugs, Biologicals, and Latex Committee); and the Centers for Disease Control and Prevention Botulism Clinical Treatment Guidelines Workgroup-Allergic Reactions to Botulinum Antitoxin: A Systematic Review. Clin Infect Dis. 2017;66(suppl_1):S65-S72. 10.1093/cid/c
- Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS. Botulinum toxin as a biological weapon: medical and public health management. JAMA 2001; 285(8):1059-1070. doi: 10.1001/jama.285.8.1059 [Crossref] [ Google Scholar]
- Arnon Arnon, SS; Schechter, R; Maslanka, SE; Jewell, NP NP. Hatheway, CLHuman Botulism Immune Globulin for the Treatment of Infant Botulism. N.Engl.J.Med 2006; 354(5):462-471. doi: 10.1056/NEJMoa051926 [Crossref] [ Google Scholar]
- Chen C, Wang S, Wang H, Mao X, Zhang T, Ji G. Potent neutralization of botulinum neurotoxin/B by synergistic action of antibodies recognizing protein and ganglioside receptor binding domain. PLoS One 2012; 7(8):e43845. doi: 10.1371/journal.pone.0043845 [Crossref] [ Google Scholar]
- Nayak SU, Griffiss JM, McKenzie R, Fuchs EJ, Jurao RA, An AT. Safety and pharmacokinetics of XOMA 3AB, a novel mixture of three monoclonal antibodies against botulinum toxin A. Antimicrob Agents Chemother 2014; 58(9):5047-5053. doi: 10.1128/AAC.02830-14 [Crossref] [ Google Scholar]
- Fan Y, Dong J, Lou J, Wen W, Conrad F, Geren IN. Monoclonal antibodies that inhibit the proteolytic activity of botulinum neurotoxin serotype/B. Toxins (Basel) 2015; 7(9):3405-3423. doi: 10.3390/toxins7093405 [Crossref] [ Google Scholar]
- Fan Y, Garcia-Rodriguez C, Lou J, Wen W, Conrad F, Zhai W. A three monoclonal antibody combination potently neutralizes multiple botulinum neurotoxin serotype F subtypes. PloS one 2017; 12(3):e0174187. doi: 10.3390/toxins15050316 [Crossref] [ Google Scholar]
- Garcia-Rodriguez C, Razai A, Geren IN, Lou J, Conrad F, Wen WH. A three monoclonal antibody combination potently neutralizes multiple botulinum neurotoxin serotype E subtypes. Toxins (Basel) 2018; 10(3):105. doi: 10.3390/toxins10030105 [Crossref] [ Google Scholar]
- Snow DM, Riling K, Kimbler A, Espinoza Y, Wong D, Pham K. Safety and pharmacokinetics of a four monoclonal antibody combination against botulinum C and D neurotoxins. Antimicrob Agents Chemother 2019; 63(12):10-128. doi: 10.1128/AAC.01270-19 [Crossref] [ Google Scholar]
- Tomic MT, Espinoza Y, Martinez Z, Pham K, Cobb RR, Snow DM. Monoclonal antibody combinations prevent serotype A and serotype B inhalational botulism in a guinea pig model. Toxins (Basel) 2019; 11(4):208. doi: 10.3390/toxins11040208 [Crossref] [ Google Scholar]
- Espinoza Y, Wong D, Ahene A, Der K, Martinez Z, Pham J. Pharmacokinetics of human recombinant anti-botulinum toxin antibodies in rats. Toxins (Basel) 2019; 11(6):345. doi: 10.3390/toxins11060345 [Crossref] [ Google Scholar]
- Amersdorfer P, Wong C, Smith T, Chen S, Deshpande S, Sheridan R. Genetic and immunological comparison of anti-botulinum type A antibodies from immune and non-immune human phage libraries. Vaccine 2002; 20:1640-1648. doi: 10.1016/s0264-410x(01)00482-0 [Crossref] [ Google Scholar]
- Kalb SR, Santana WI, Geren IN, Garcia-Rodriguez C, Lou J, Smith TJ. Extraction and inhibition of enzymatic activity of botulinum neurotoxins/B1,/B2,/B3,/B4, and/B5 by a panel of monoclonal anti-BoNT/B antibodies. BMC Biochem 2011; 12:1-11. doi: 10.1186/1471-2091-12-58 [Crossref] [ Google Scholar]
- Derman Y, Selby K, Miethe S, Frenzel A, Liu Y, Rasetti-Escargueil C. Neutralization of botulinum neurotoxin type E by a humanized antibody. Toxins (Basel) 2016; 8(9):257. doi: 10.3390/toxins8090257 [Crossref] [ Google Scholar]
- Rhéaume C, Cai BB, Wang J, Fernández-Salas E, Aoki KR, Francis J. A highly specific monoclonal antibody for botulinum neurotoxin type A-cleaved SNAP25. Toxins (Basel) 2015; 7:2354-2370. doi: 10.3390/toxins7072354 [Crossref] [ Google Scholar]
- Matsumura T, Amatsu S, Misaki R, Yutani M, Du A, Kohda T. Fully human monoclonal antibodies effectively neutralizing botulinum neurotoxin serotype B. Toxins (Basel) 2020; 12:302. doi: 10.3390/toxins12050302 [Crossref] [ Google Scholar]
- Parray H A, Shukla S, Samal S, Shrivastava T, Ahmed S, Sharma C. Hybridoma technology a versatile method for isolation of monoclonal antibodies, its applicability across species, limitations, advancement and future perspectives. Int Immunopharmacol 2020; 85:106639. doi: 10.1016/j.intimp.2020.106639 [Crossref] [ Google Scholar]
- Zaroff S, Tan G. Zaroff S, Tan GHybridoma technology: the preferred method for monoclonal antibody generation for in vivo applications. Biotechniques 2019; 67:90-92. doi: 10.2144/btn-2019-0054 [Crossref] [ Google Scholar]
- Lushova AA, Biazrova MG, Prilipov AG, Sadykova GK, Kopylov TA, Filatov AV. Next-generation techniques for discovering human monoclonal antibodies. Mol Biol 2017; 51:782-787. doi: 10.1134/S0026893317060103 [Crossref] [ Google Scholar]
- da Silva Cardoso M, Siemoneit K, Sturm D, Krone C, Moradpour D, Kubanek B. Isolation and characterization of human monoclonal antibodies against hepatitis C virus envelope glycoproteins. J Med Virol 1998; 55:28-34. [ Google Scholar]
- Silkina MV, Kartseva AS, Ryabko AK, Marin MA, Romanenko YO, Kalmantaeva OV. Optimization of electrofusion parameters for producing hybridomas synthesizing human monoclonal antibodies. Applied Biochemistry and Microbiology 2022; 58(9):984-991. doi: 10.1134/S0003683822090095 [Crossref] [ Google Scholar]
- Yu X, McGraw PA, House FS, Crowe JE. JrAn optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J Immunol Methods 2008; 156:5-10. doi: 10.1016/j.jim.2008.04.008 [Crossref] [ Google Scholar]
- Malizio CJ, Goodnough MC, Johnson EA. Purification of Clostridium botulinum type A neurotoxin. Methods Mol Biol 2000; 145:27-39. doi: 10.1385/1-59259-052-7:27 [Crossref] [ Google Scholar]
- Kolesnikov AV, Kozyr AV, Ryabko AK, Shemyakin IG. Ultrasensitive detection of protease activity of anthrax and botulinum toxins by a new PCR-based assay. Pathog Dis 2016; 74(1):ftv112. doi: 10.1093/femspd/ftv112 [Crossref] [ Google Scholar]
- Ryabko AK, Kozyr’ AV, Kolesnikov AV, Khlyntseva AE, Zharnikova IV, Shemyakin IG. Strategies for upgrading analyte detection in immuno-PCR studied on identification of type A botulinum neurotoxin. Appl Biochem Microbiol 2016; 52:110-120. doi: 10.1134/S0003683816010117 [Crossref] [ Google Scholar]
- Laemmli UK. Laemmli UKCleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227(5259):680-685. doi: 10.1038/227680a0 [Crossref] [ Google Scholar]
- Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008; 453:667-671. doi: 10.1038/nature06890 [Crossref] [ Google Scholar]
- Boldt GE, Kennedy JP, Hixon MS, McAllister LA, Barbieri JT, Tzipori S, et al. Synthesis, characterization and development of a high-throughput methodology for the discovery of botulinum neurotoxin a inhibitors. J Comb Chem. 2006;8. 10.1021/cc060010h.
- Sanz I, Wei C, Lee FE, Anolik J. Sanz I, Wei C, Lee FE, Anolik JPhenotypic and functional heterogeneity of human memory B cells. Semin Immunol 2008; 20(1):67-82. doi: 10.1016/j.smim.2007.12.006 [Crossref] [ Google Scholar]
- Palm AE, Henry C. Palm AE, Henry CRemembrance of Things Past: Long-term B cell memory after infection and vaccination. Front Immunol 2019; 10:1787. doi: 10.3389/fimmu.2019.01787 [Crossref] [ Google Scholar]
- Ellebedy AH, Jackson KJ, Kissick HT, Nakaya HI, Davis CW, Roskin KM. Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat Immunol 2016; 17(10):1226-1234. doi: 10.1038/ni.3533 [Crossref] [ Google Scholar]
- Silkina MV, Kartseva AS, Zeninskaya NA, Maryin MA, Ryabko AK, Muntyan YaO, et al. Analysis of the content of plasmablasts in the blood of people at different times after immunization with the live dry anthrax vaccine. Immunology. 2019;40. 10.24411/0206-4952-2019-12004.
- Luning Prak ET, Ross J, Sutter J, Sullivan KE. Age-related trends in pediatric B-cell subsets. Pediatr Dev Pathol 2011; 14(1):45-52. doi: 10.2350/10-01-0785-OA.1 [Crossref] [ Google Scholar]
- Agematsu K, Hokibara S, Nagumo H, Komiyama A. CD27: a memory B-cell marker. Immunol Today 2000; 21(5):204-206. doi: 10.1016/s0167-5699(00)01605-4 [Crossref] [ Google Scholar]
- Funaro A, Morra M, Calosso L, Zini MG, Ausiello CM, Malavasi F. Role of the human CD38 molecule in B cell activation and proliferation. Tissue Antigens 1997; 49(1):7-15. doi: 10.1111/j.1399-0039.1997.tb02703.x [Crossref] [ Google Scholar]
- Mc Adam AJ, Schweitzer AN, Sharpe AH. The role of B7 co-stimulation in activation and differentiation of CD4+ and CD8+ T cells. Immunol Rev 1998; 165:231-247. doi: 10.1111/j.1600-065x.1998.tb01242.x [Crossref] [ Google Scholar]
- Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol 2015; 15(3):149-159. doi: 10.1038/nri3802 [Crossref] [ Google Scholar]
- Moir S, Fauci AS. Moir S, Fauci ASB cells in HIV infection and disease. Nat Rev 2009; 9(4):235-245. doi: 10.1038/nri2524 [Crossref] [ Google Scholar]
- Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 2009; 326(5950):285-289. doi: 10.1126/science.1178746 [Crossref] [ Google Scholar]
- Huang J, Doria-Rose NA, Longo NS, Laub L, Lin CL, Turk E. Isolation of human monoclonal antibodies from peripheral blood B cells. Nat Protoc 2013; 8(10):1907-1915. doi: 10.1038/nprot.2013.117 [Crossref] [ Google Scholar]
- Rusnak JM, Smith LA. Rusnak JM, Smith LABotulinum neurotoxin vaccines: past history and recent developments. Hum Vaccin 2009; 5(12):794-805. doi: 10.4161/hv.9420 [Crossref] [ Google Scholar]
- Pirazzini M, Rossetto O. Pirazzini M, Rossetto OChallenges in searching for therapeutics against . Botulinum NeurotoxinsExpert Opin Drug Discov 2017; 12(5):497-510. doi: 10.1080/17460441.2017.1303476 [Crossref] [ Google Scholar]
- Kaji R. Kaji RNew and emerging indications of botulinum toxin therapy. Parkinsonism RelatDisord 2011; 17(1):25-7. doi: 10.1016/j.parkreldis.2011.06.017 [Crossref] [ Google Scholar]
- Fonfria E, Maignel J, Lezmi S, Martin V, Splevins A, Shubber S. The Expanding Therapeutic Utility of Botulinum Neurotoxins. Toxins (Basel) 2018; 10(5):208. doi: 10.3390/toxins10050208 [Crossref] [ Google Scholar]
- Jankovic J. Jankovic JAn update on new and unique uses of botulinum toxin in movement disorders. Toxicon 2018; 147:84-88. doi: 10.1016/j.toxicon.2017.09.003 [Crossref] [ Google Scholar]
- Dressler D. Dressler DClinical applications of botulinum toxin. CurrOpin Microbiol 2012; 15(3):325-236. doi: 10.1016/j.mib.2012.05.012 [Crossref] [ Google Scholar]
- Dressler D, Roggenkaemper P. Dressler D, Roggenkaemper PA brief history of neurological botulinum toxin therapy in Germany. J Neural Transm (Vienna) 2017; 124(10):1217-1221. doi: 10.1007/s00702-017-1762-3 [Crossref] [ Google Scholar]
- Dolimbek BZ, Aoki KR, Steward LE, Jankovic J, Atassi MZ. Mapping of the regions on the heavy chain of botulinum neurotoxin A (BoNT/A) recognized by antibodies of cervical dystonia patients with immunoresistance to BoNT/A. Mol Immunol 2007; 44:1029-1041. doi: 10.1016/j.molimm.2006.03.011 [Crossref] [ Google Scholar]
- Atassi MZ, Jankovic J, Steward LE, Aoki KR, Dolimbek BZ. Molecular immune recognition of botulinum neurotoxin BThe light chain regions that bind human blocking antibodies from toxin-treated cervical dystonia patientsAntigenic structure of the entire BoNT/B molecule. Immunobiology 2012; 217:17-27. doi: 10.1016/j.imbio.2011.08.009 [Crossref] [ Google Scholar]
- Dressler D, Rothwell JC. Dressler D, Rothwell JCElectromyographic quantification of the paralysing effect of botulinum toxin in the sternocleidomastoid muscle. Eur Neurol 2000; 43:13-16. doi: 10.1159/000008122 [Crossref] [ Google Scholar]
- Dressler D, Bigalke H. Dressler D, Bigalke HBotulinum toxin type B de novo therapy of cervical dystonia. J Neurol 2005; 252:904-907. doi: 10.1007/s00415-005-0774-3 [Crossref] [ Google Scholar]
- Naumann M, Boo LM, Ackerman AH, Gallagher CJ. Immunogenicity of botulinum toxins. J Neural Transm (Vienna) 2013; 120:275-290. doi: 10.1007/s00702-012-0893-9 [Crossref] [ Google Scholar]
- Mehanna R, Jankovic J. Botulinum Neurotoxins as a Therapeutic. Handbook of Neurotoxicity 2021;1-48. 10.1007/978-3-030-71519-9_169-1.
- Blasi J, Chapman ER, Link E, Binz T, Yamasaki S, De Camilli P. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 1993; 365:160-163. doi: 10.1038/365160a0 [Crossref] [ Google Scholar]
- Marks JD. Marks JDDeciphering antibody properties that lead to potent botulinum neurotoxin neutralization. Mov Disord 2004; 19(S8):S101-8. doi: 10.1002/mds.20023 [Crossref] [ Google Scholar]
- Ravichandran E, Gong Y, Al Saleem FH, Ancharski DM, Joshi SG, Simpson LL. An initial assessment of the systemic pharmacokinetics of botulinum toxin. J Pharmacol Exp Ther 2006; 318:1343-1135. doi: 10.1124/jpet.106.104661 [Crossref] [ Google Scholar]
- Verderio C, Rossetto O, Grumelli C, Frassoni C, Montecucco C, Matteoli M. Entering neurons: botulinum toxins and synaptic vesicle recycling. EMBO Rep 2006; 7(10):995-9. doi: 10.1038/sj.embor.7400796 [Crossref] [ Google Scholar]
- Fischer A, Montal M. Fischer A, Montal MSingle molecule detection of intermediates during botulinum neurotoxin translocation across membranes. Proc Natl Acad Sci U S A 2007; 104(25):10447-52. doi: 10.1073/pnas.0700046104 [Crossref] [ Google Scholar]
- Rasetti-Escargueil C, Popoff MR. Rasetti-Escargueil C, Popoff MRAntibodies and Vaccines against Botulinum Toxins: Available Measures and Novel Approaches. Toxins (Basel) 2019; 11(9):528. doi: 10.3390/toxins11090528 [Crossref] [ Google Scholar]