Bioimpacts. 14(2):27780.
doi: 10.34172/bi.2023.27780
Original Article
Essential role of CD38 in platelet aggregation through the PKC-mediated internalization and activation
Mazhar Mushtaq Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing, 1, * 
Maira Mahmood Formal analysis, Methodology, Resources, Software, Writing – review & editing, 2
Uzma Jabbar Formal analysis, Investigation, Resources, Software, Writing – review & editing, 3
Uh-Hyun Kim Conceptualization, Project administration, Supervision, Validation, Visualization, Writing – original draft, 4
Author information:
1Basic Medical Sciences, Sulaiman Al Rajhi University, Al-Qaseem, Kingdom of Saudi Arabia
2Department of Biochemistry, FMH College of Medicine and Dentistry, Lahore, Pakistan
3Department of Biochemistry, Gujranwala Medical College, Gujranwala, Pakistan
4Department of Biochemistry, Chonbuk National University, Chonbuk, South Korea
Abstract
Introduction:
CD38 is a multifunctional enzyme with a potent Ca2+ mobilizing effect, cyclic ADP-ribose (cADPR), and nicotinic acid adenine dinucleotide phosphate (NAADP). Here, we aimed to demonstrate the role of CD38 in platelets via protein kinase C (PKC)-mediated internalization and activation.
Methods:
Mouse platelets were used in this study. Thrombin, an agonist of platelet function, provoked a prompt and long-lasting increase in intracellular Ca2+ concentration ([Ca2+]i), resulting from an interplay of multifold Ca2+ mobilizing messengers.The signaling pathway was delineated using different inhibitors and techniques such as platelet aggregation assay, intracellular calcium measurements, immunoprecipitation, immunoblotting, and flow cytometry.
Results:
We observed a sequential formation of cADPR and NAADP through CD38 activation by PKC of non-muscle myosin heavy chain IIA (MHCIIA), resulting in phospholipase C (PLC) activation in the thrombin-stimulated platelets. These findings reveal that PKC is fundamental in activating CD38 and elicits a physiological response in the murine platelets.
Conclusion:
PKC is involved in many signaling pathways. Specifically, PKC is involved in the internalization of CD38 via MHCIIA in CD38+/+ wild-type (WT) and CD38-/- knockout mice (KO). CD38 generates calcium-mobilizing agents that act on specific receptors of the calcium stores. Calcium triggered platelet aggregation while serving as a secondary messenger.
Keywords: CD38, Platelets, cADPR, Calcium, NAADP, PKC
Copyright and License Information
© 2024 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
The primary function of platelets is to aggregate at injury sites regardless of the stimuli. Many agonists trigger platelet aggregation; however, ultrasound-like modalities cause platelets to change their intercellular calcium levels leading to aggregation.1 Thus, a surge in cytosolic free calcium ion concentrations ([Ca2+]i) is a prerequisite for aggregation and is achieved by the combined action of inositol 1,4,5-trisphosphate (IP3), cyclic ADP-ribose (cADPR), and nicotinic acid adenine dinucleotide phosphate (NAADP) on specific intracellular stores.2
The [Ca2+]i signal represents a balance between activation of the secretory and the avoidance of elevated [Ca2+]i levels that are toxic to the cell. Such toxic levels can raise/reduce the physiological processes of any cell type. The optimum level of resting [Ca2+]i is thus preserved by the action of the Ca2+ plasma membrane-bound pump.3
Many cell lines use cADPR and NAADP as Ca2+-mobilizing nucleotides. These second messengers are produced by the type II transmembrane glycoprotein CD38 with the action of NAD+. They act on distinct intracellular calcium stores. Besides, cADPR mobilizes calcium from the ER and NAADP from acidic organelles, the receptor of which are in the vicinity of the plasma membrane to control Ca2+ influx3 to the cell and release from the ER.4-7
The NAADP-sensitive, acidic, lysosome-related granules possess NAADP receptors.8,9 NAADP has been shown to mobilize Ca2+ in other species, from plants to animals, including various mammalian cells.10-13 NAADP is involved in initiating and coordinating Ca2+ signaling,6,7 enabling many extracellular signals to use Ca2+ as a second messengerwhile preserving the specificity of the received signal.
The presence of CD38 on the platelet and production of cADPR was first reported in 1996.14 Recently, we elucidated the importance of NAADP and cADPR in platelets and their synthesis upon the activation of CD38.15 CD38 association with the cytoskeleton of platelets upon thrombin stimulation was equally demonstrated by other groups.16 The cytoskeletal reorganization of platelets upon their stimulation involves the phosphorylation of many proteins.17 PKC is vital in the phosphorylation of cytoskeletal proteins. Four PKC isoforms (α, β, δ, θ) are expressed in platelets,18-21 and each isoform is involved in platelet function.22,23 Hence, demonstrating the interaction/activation of CD38 with PKC is vital in understanding the process.16
We, therefore, aimed to elucidate the role of CD38 as a positive modulator of platelet aggregation through the activation of PKC. We used rodent platelets to perform this study. However, little or no differences in the functioning of human and mouse platelets have been documented.
Materials and Methods
Reagents
Thrombin, RO318220, Xestospongin C, U73122, Wortaminnin, and collagen were all purchased from Sigma. Fura-2 AM was obtained from Invitrogen.
Antibodies
CD38 monoclonal antibody (mAb) was from BD Biosciences, and anti-MHCIIA polyclonal antibody (pAb) was purchased from Covance.
Platelet preparation
Mice were bred and kept at the animal house of the university. All animal studies were performed according to a protocol approved by the Institutional Animal Care and Use Committee of Chonbuk National University Medical School. WT mice were mated with KO to get the appropriate breed of WT and KO. Six hundred microliters to 800 μL of blood was obtained from each mouse through cardiac puncture into Acid Citrate Dextrose (ACD) (20 mM citric acid, 110 mM sodium citrate, and 5 mM glucose) buffer at a 1:10 v/v ratio. Platelets were prepared as described earlier.15 ACD blood was centrifuged at 2000 RPM for 6 minutes at room temperature. The supernatant was separated in Tyrode Buffer (TB). All the supernatant collected in one tube was spun again at 5000 RPM for 30 seconds to remove RBCs and WBCs. The supernatant containing the platelets was re-suspended and washed in TB. As previously described, the optical method adjusted the platelet count to 0.5 × 109 per mL.24 Platelets collected in TB were allowed to rest for 60 minutes at 37 °C before use. For aggregation, 0.1% BSA was supplemented during resting. For the calcium study, 0.02 U of apyrase was added to minimize the effect of ADP and ATP and to maximize the effect of thrombin. Calcium was added if stated, or a calcium-free buffer was used.
Platelet aggregation assay
Aggregation assays, as previously described25,26, were performed using a Thermomax microplate reader (Molecular Devices Spectra max plus) connected to a computer with SOFTmax plus 4.0. The equipment was turned on at least 30 minutes before and pre-warmed to 37 °C. Briefly, in a transparent flat-bottomed 96-well plate, we added agonist, antagonist, or the same volume of vehicle solution in appropriate volumes of TB-suspended platelets using a multichannel pipette. The plate was then stirred in the instrument, and the turbidity change at 650 nm was measured as % of light transmission (% LT). The data produced by the software were plotted, time against %LT.
Measurements of intracellular calcium
TB-washed platelet was incubated by adding 1% BSA and 5 µM Fura 2-AM for 40 minutes. In some experiments, an antagonist or inhibitor was used to incubate platelets. After the specific incubation time, 1 mL of Fura 2-loaded platelets were administered to the magnetically stirred cuvette of PTI Spectrofluorometer for calcium measurement systems at 37 °C. Excitatory wavelengths of 340 and 380 nm were used, and the system captured emitted wavelength of 500 nm. Changes in [Ca2+]i were monitored using a 340 nm/380 nm fluorescence ratio of Fura-2.
Immunoprecipitation and western blotting
Thrombin-stimulated platelets were incubated for 5 minutes. The reaction was stopped by adding ice-cold cell lysis buffer. The rest of the protocol remains as described in our previous publication.15 Briefly, a selected amount of proteins was subjected to immunoprecipitation with anti-CD38 mAb and precipitated protein and were subjected to SDS-PAGE on 10% gel and then transferred to a nitrocellulose membrane. One percent BSA in PBS is used as a blocking agent. After a series of washes, it was incubated with the primary antibody, MHCIIA. After rinsing and washing, it was incubated with horseradish peroxidase-conjugated Ab using a chemiluminescence kit (Amersham Biosciences AB) and exposed to LAS Image Reader. A series of photos were saved, and the cellulose membrane was further washed, rinsed, blocked, and a CD38 antibody was used to check the precipitation of CD38 as a control.
In vitro thrombus formation
We assessed thrombus formation on the surface of collagen in vitro. In 4-well cell culture dishes, 100 μL each of 50 μg/mL collagen was added and left for 3 hours in the incubator. The excess amount of collagen was removed and 100 μL of TB-suspended platelets were added to each well and stimulated with an agonist or the same volume of the vehicle in control. Dishes were left on a slow-moving shaker for 10 minutes. Then, excess platelets were aspirated. Reactions were stopped by adding 1 mL of 3% ice-cold formalin. Adhered platelet/thrombus on the bottom of the dish was studied using the phase contrast technique. Images from six random microscopic fields were acquired using Nikon Eclipse TE2000-S at magnification 200X. The images were acquired using Focus Lite Version 2.88 software. Thrombi seen in the images were visually counted.
Flow cytometry
Incubated platelets were stimulated with thrombin for 5 minutes at 37 °C. After the specific time, the reactions were stopped by adding 1 mL of 3% ice-cold formalin. After 15 minutes of fixation on ice, the tubes were spun at 13000 RPM for 5 minutes at 4 °C and washed once with ice-cold PBS. Then, 500 µL of 3% BSA in PBS was used as a blocking buffer, and the tubes were left inside the rotor in the cold room for 1 hour. Tubes were centrifuged again at 13000 RPM for 5 minutes at 4 °C and washed once with ice-cold PBS, and then 500 µL of 1% BSA was added along CD38 Ab conjugated with FITC for 2 hours in a cold and dark room. Further, the platelets were subjected to FACS along with a control that only contained a FITC antibody. A total of 10 000 events per sample were collected.
Statistical analyses
Data were analyzed using SigmaPlot 9 via an unpaired Student’s t test. The collected parameters were expressed as mean, and P< 0.05 was considered significant, validating positive findings. Each experiment was repeated, and n indicated the number of experiments.
Results
Ablation of CD38 markedly attenuates thrombus formation in vivo
To begin with, we determined the bleeding time of two genotypes, CD38+/+ and CD38-/- mice. Although CD38-/- mice did not display any evidence of spontaneous bleeding, we observed a significant difference in the bleeding time across the two groups (Fig. 1A). Fig. 1B shows that RBC sedimentation is significantly higher in CD38-/- mice.
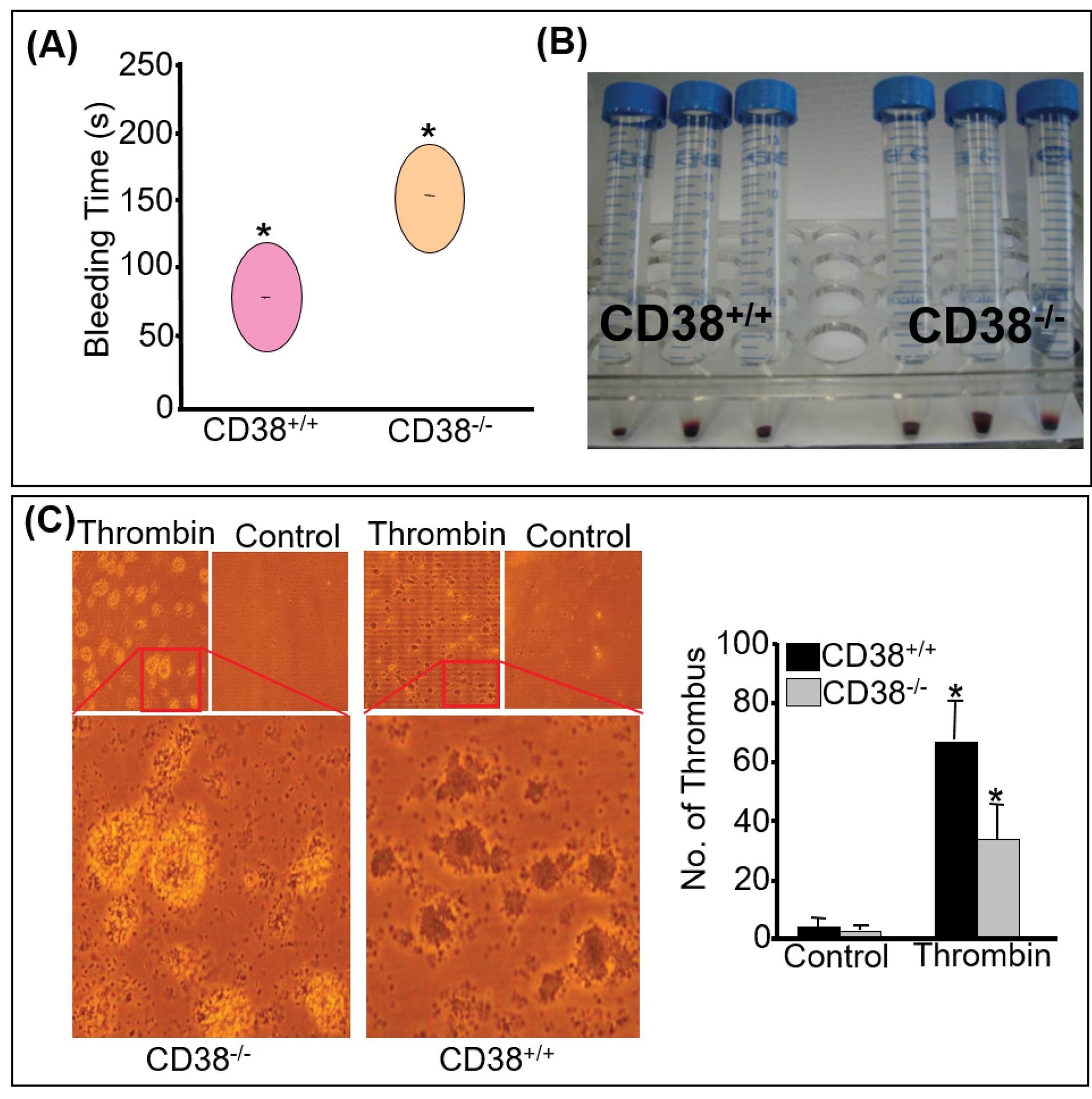
Fig. 1.
Ablation of CD38 markedly attenuates thrombus formation in vivo and in vitro. (A) In vivo thrombus formation was assessed between the two genotypes by the tail-cutting method. Significant difference is observed between CD38+/+ and CD38-/-. The area of the circle represents the average time *P<0.05. (B) Hand-held tubes were spun, and amounts of blood precipitated were compared. The data presented are from two independent experiments with three to four mice in each group. (C) Sedimented platelets of CD38+/+ and CD38-/- were suspended in TB and added to the pre-incubated 4-well plates with collagen. Agonist, 0.5U of thrombin or the same volume of vehicle was added in each well. Thrombus formed was visually counted and plotted. A significantly reduced number of tightly packed thrombi was seen in CD38-/- with thrombin *P<0.05.
.
Ablation of CD38 markedly attenuates thrombus formation in vivo and in vitro. (A) In vivo thrombus formation was assessed between the two genotypes by the tail-cutting method. Significant difference is observed between CD38+/+ and CD38-/-. The area of the circle represents the average time *P<0.05. (B) Hand-held tubes were spun, and amounts of blood precipitated were compared. The data presented are from two independent experiments with three to four mice in each group. (C) Sedimented platelets of CD38+/+ and CD38-/- were suspended in TB and added to the pre-incubated 4-well plates with collagen. Agonist, 0.5U of thrombin or the same volume of vehicle was added in each well. Thrombus formed was visually counted and plotted. A significantly reduced number of tightly packed thrombi was seen in CD38-/- with thrombin *P<0.05.
In vitro thrombus formation was assessed on a collagen-coated surface. We observed a noticeably reduced thrombus formation in CD38-/- platelets treated with thrombin compared to CD38+/+ platelets. Fig. 1C displayed a densely packed platelet thrombus upon thrombin treatment, revealing that platelets lacking CD38 loosely interacted on the collagen surface. These findings revealed that CD38-/- platelets cannot form tightly packed thrombi despite their large surface area. They were arrested at primary aggregation and could not proceed to secondary aggregation.
Platelet dependency on CD38 for aggregation and calcium signal
Platelets of WT and KO were suspended in an appropriate volume of Ca2+-free TB for aggregation and were stimulated with 0.5 units of thrombin. An orthodox pattern of aggregation with the agonist is displayed in Fig. 2A. However, platelets from CD38-/- mice showed a significantly diminished aggregation in response to thrombin; the aggregation of CD38-/- platelets did not reach the baseline, and the oscillation pattern was evident, revealing the interplay of other pathways. Similarly, [Ca2+]i mobilization showed a significant difference between the two genotypes upon thrombin stimulation due to a defect in two calcium-mobilizing messengers (Fig. 2B).
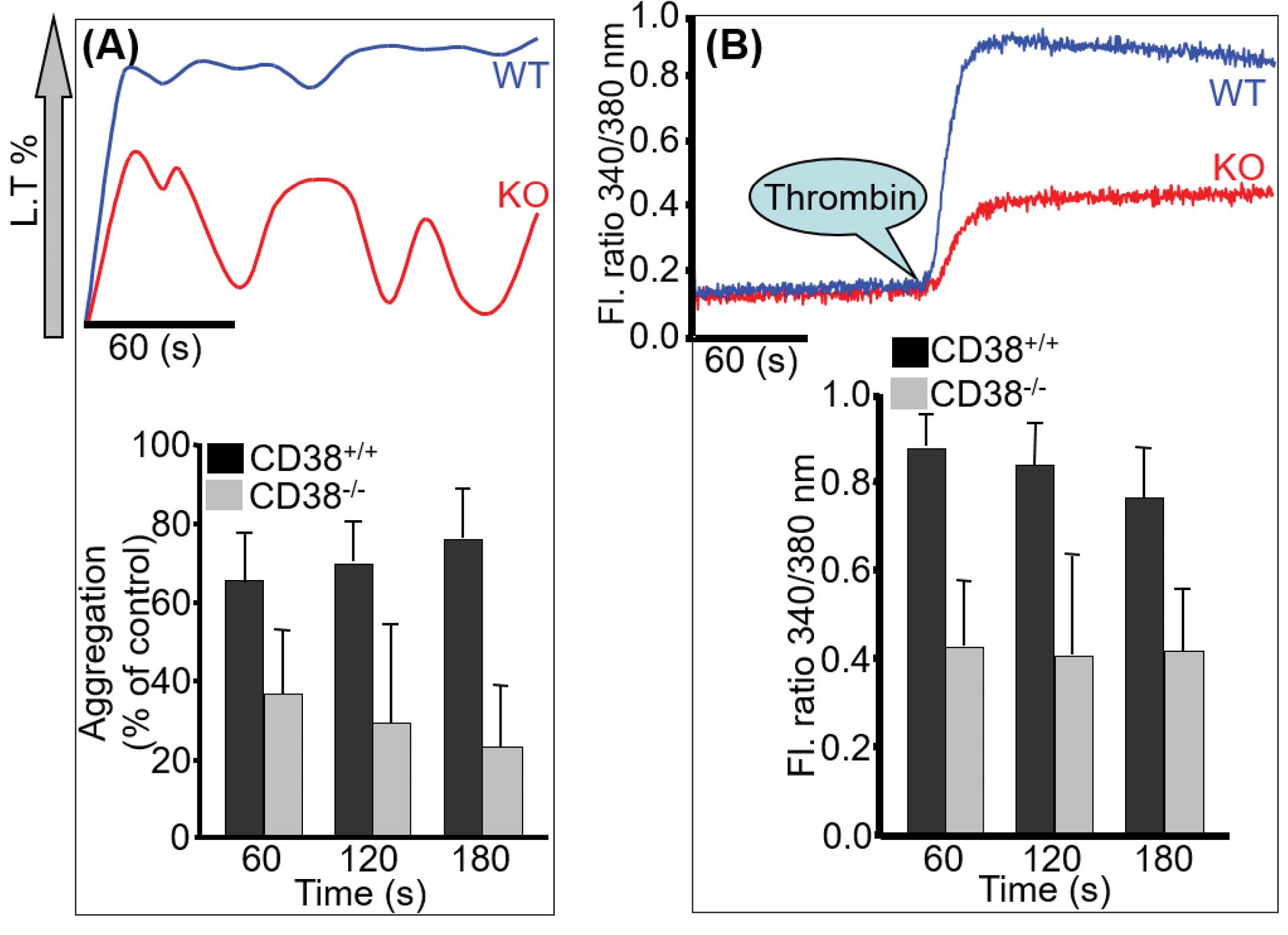
Fig. 2.
Platelet dependency on CD38 for aggregation and calcium signal. Platelets of WT and KO were simultaneously processed and were stimulated with 0.5 U of thrombin for aggregation (A) and [Ca2+]i measurement (B). A significant difference is observed between CD38+/+ and CD38-/-. The bar graph describes quantitative data (mean ± SD) from seven experiments.
.
Platelet dependency on CD38 for aggregation and calcium signal. Platelets of WT and KO were simultaneously processed and were stimulated with 0.5 U of thrombin for aggregation (A) and [Ca2+]i measurement (B). A significant difference is observed between CD38+/+ and CD38-/-. The bar graph describes quantitative data (mean ± SD) from seven experiments.
PKC is required for intact calcium signaling via CD38
Platelets were pre-incubated with RO318220 (R136), a potent PKC inhibitor, and then stimulated with thrombin to assess aggregation and calcium measurement. Aggregation and calcium signal were abolished (Fig. 3A, B respectively). After pre-incubating the platelets with R136 or blebbistatin, they were stimulated with thrombin, and cell extracts were subjected to immunoprecipitation using anti-CD38 mAb. Treatment of platelets with thrombin significantly increased co-immunoprecipitation of MHCIIA with CD38 compared to control, while this association decreased in the presence of R136 or blebbistatin (Fig. 3C). R136 alleviates the thrombin-induced CD38 internalization (Fig. 3D). These findings demonstrate that thrombin activates platelets to internalize CD38 using MHCIIA via PKC signaling.
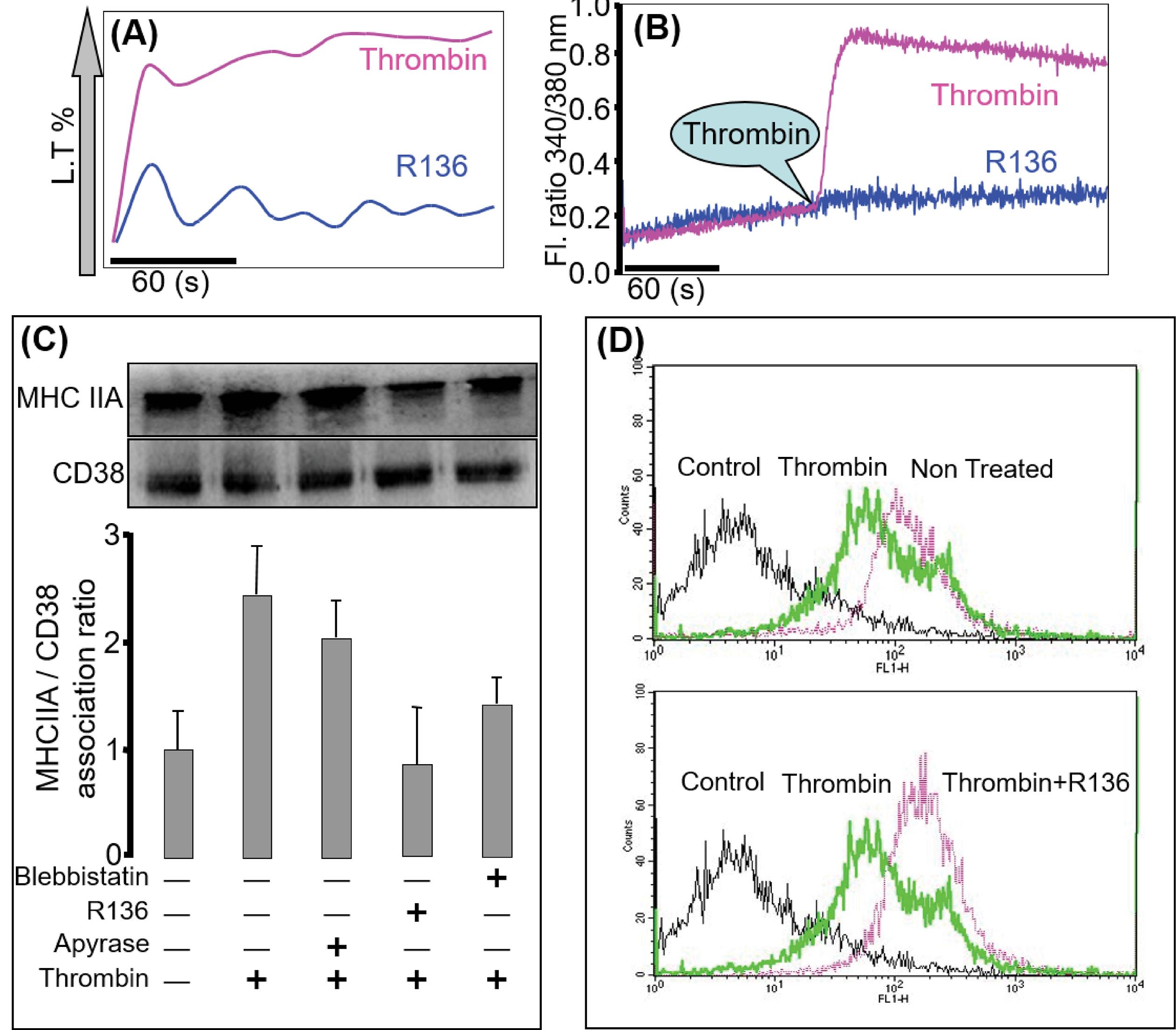
Fig. 3.
PKC is required for intact calcium signaling via CD38. Effects of PKC inhibitor on thrombin-induced aggregation (A; n=4) and [Ca2+]i measurement (B; n=5) in platelets, a concentration of 5 µg/ml for 25 minutes of PKC inhibitor abolishes the aggregation and the calcium signal (C; n=3). Washed platelets were incubated with 5 µg/mL of PKC inhibitor for 25 minutes, apyrase 0.02U (used as control as it was used in the calcium study), and MHCIIA inhibitor; 50 µM blebbistatin for 30 minutes and then activated with 0.5U of thrombin for 5 minutes. After a specific time, the activated platelets were extracted with lysis buffer for immunoprecipitation using anti-CD38 mAb. The immunoprecipitated proteins were analyzed by immunoblotting with anti-CD38 mAb or anti-MHCIIA pAb. The bar graph represents the association ratio of CD38 with MHCIIA (D; n=3). Pre-incubation of washed platelets with PKC inhibitor was followed by thrombin stimulation for 5 minutes; CD38 fluorescence intensity was measured by flow cytometry. PKC inhibitor substantially reduced the internalization of CD38, as observed with a right shift of peak.
.
PKC is required for intact calcium signaling via CD38. Effects of PKC inhibitor on thrombin-induced aggregation (A; n=4) and [Ca2+]i measurement (B; n=5) in platelets, a concentration of 5 µg/ml for 25 minutes of PKC inhibitor abolishes the aggregation and the calcium signal (C; n=3). Washed platelets were incubated with 5 µg/mL of PKC inhibitor for 25 minutes, apyrase 0.02U (used as control as it was used in the calcium study), and MHCIIA inhibitor; 50 µM blebbistatin for 30 minutes and then activated with 0.5U of thrombin for 5 minutes. After a specific time, the activated platelets were extracted with lysis buffer for immunoprecipitation using anti-CD38 mAb. The immunoprecipitated proteins were analyzed by immunoblotting with anti-CD38 mAb or anti-MHCIIA pAb. The bar graph represents the association ratio of CD38 with MHCIIA (D; n=3). Pre-incubation of washed platelets with PKC inhibitor was followed by thrombin stimulation for 5 minutes; CD38 fluorescence intensity was measured by flow cytometry. PKC inhibitor substantially reduced the internalization of CD38, as observed with a right shift of peak.
Internalization of CD38 via PKC requires the PLC pathway, not PI3K
Washed platelets were stimulated with thrombin after incubation with wortmannin or U73122 or co-incubation. Incubation with wortmannin or U73122 alone did not reduce the thrombin-induced aggregation, while incubation with wortmannin and U73122 completely abolished aggregation (Fig. 4A). However, [Ca2+]i mobilization was impaired in the presence of U73122 and not wortmannin (Fig. 4B).
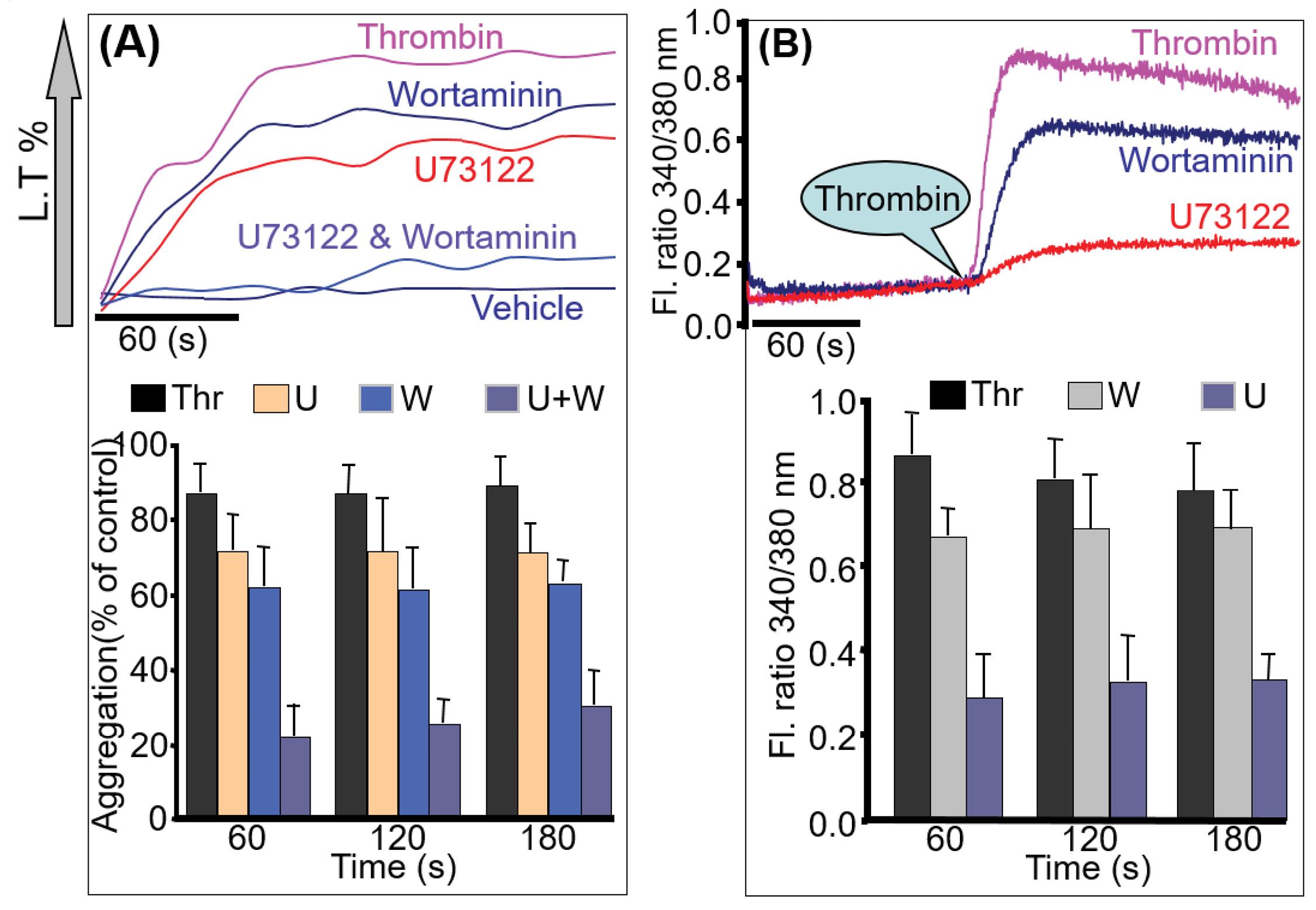
Fig. 4.
CD38 activation pathway. Washed platelets were incubated with 2 µM of U73122, 100 nM of Wortaminnin, and the same concentration was co-incubated for 30 minutes and then activated with thrombin for aggregation (A) and calcium measurement (B). Co-incubation significantly abolishes aggregation. U73122 knocked down the calcium signal while wortmannin did not alter the calcium signal. Quantitative data (mean ± SD) from three experiments are described in the bar graph (C).
.
CD38 activation pathway. Washed platelets were incubated with 2 µM of U73122, 100 nM of Wortaminnin, and the same concentration was co-incubated for 30 minutes and then activated with thrombin for aggregation (A) and calcium measurement (B). Co-incubation significantly abolishes aggregation. U73122 knocked down the calcium signal while wortmannin did not alter the calcium signal. Quantitative data (mean ± SD) from three experiments are described in the bar graph (C).
Discussion
The biphasic aggregation of platelet complexity is widely discussed in the literature as primary and secondary aggregations. These are calcium-dependent processes. However, the primary release of calcium from ER is under the guidance of an agonist, resulting in “platelet shape change.” Two nucleotides that control calcium release, cADPR and NAADP, are produced from CD38. In our previous study, we demonstrated the production of both nucleotides in a time-dependent manner15 and showed that calcium released under their influence came from different sources.5-7
CD38 is involved in the mobilization of intracellular calcium. However, CD38 requires its internalization to produce a physiological response, despite it being an ecto-enzyme. The involvement of CD38 with PKC induces a physiological response in LAK cells from the spleen,13 isolated cardiomyocytes,27 islets of Langerhans,28 and various macrophages. Similarly, CD38 in platelets under the influence of PKC is required to complete the second aggregation phase. CD38 is internalized by its association with MHCIIA and is involved in the synthesis of cADPR.29 PKC phosphorylates MHCIIA30-34 at the Ser-1917 of the C-terminal end.35 Hence, we examined the relationship between these molecules using R136 (a PKC inhibitor) and blebbistatin (an MHCIIA inhibitor).36-38 Molecular mechanisms for activating CD38 under the PKC and subsequent physiological response in platelets observed in our study align with those proposed by others.
When G-protein-coupled receptors are stimulated, simultaneous activation of PLC and phosphatidylinositol 3-kinase (PI3K) follows. Amongst them, Gα interacts with PLC and Gβγ regulates the activation of PI3K. These pathways reveal substantial redundancy in platelet signaling pathways as far as aggregation is concerned. However, the difference lies in their ability to mobilize intracellular calcium. The use of inhibitors of these pathways showed that the activation of PKC is under the influence of PLC. Many other research groups have shown similar results.20,21 Moreover, PI3K is vital in platelets' cytoskeletal dynamics, and calcium mobilization is only through PLC.
MHCIIA phosphorylation with PKC leads to the translocation of CD38 from the plasma membrane to the cytosol, and successively, the development of cADPR and NAADP in platelets takes place after its internalization.
Conclusion
CD38 is a positive modulator of platelet aggregation under the activation of PKC. Our findings revealed that CD38 is the primary molecule on the platelet surface that mobilizes intracellular calcium stored in different compartments, as platelets from CD38-/- mice do not show peaked and sustained calcium signals. Furthermore, using PKC inhibitors confirmed our hypothesis in CD38-/- mice.
Research Highlights
What is the current knowledge?
√ Various research groups have shown the inevitable involvement of CD38 in platelets and other cell lines. Principally physiological involvement of CD38 in any cell line is the movement/release of intercellular calcium ions.
What is new here?
√ CD38’s ectoenzyme nature requires internalization to perform its physiological function. In platelets, this mechanism has not been completely highlighted.
√ In this study, we demonstrated the role of PKC Albeit, we were the first group to show the significance of CD38 on the platelets in our previous published result.
Competing interests
The authors declare no conflict of interest.
Ethical Statement
Not applicable.
Funding
Nil.
References
- Samal AB. Platelet aggregation and change in intracellular Ca2+ induced by low frequency ultrasound in vitro. Eur J Ultrasound 2000; 11:53-59. doi: 10.1016/S0929-8266(99)00077-4 [Crossref] [ Google Scholar]
- Lee HC, Aarhus R. Functional visualization of the separate but interacting calcium stores sensitive to NAADP and cyclic ADP-ribose. J Cell Sci 2000; 113:4413-4420. doi: 10.1242/jcs.113.24.4413 [Crossref] [ Google Scholar]
- Petersen OH. Calcium signalling and secretory epithelia Cell. Cell Calcium 2014; 55:282-289. doi: 10.1016/j.ceca.2014.01.003 [Crossref] [ Google Scholar]
- Moccia F, Lim D, Nusco GA, Ercolano E, Santella L. NAADP activates a Ca2+ current that is dependent on F-actin cytoskeleton. FASEB J 2003; 17(13):1907-1909. doi: 10.1096/fj.03-0178fje [Crossref] [ Google Scholar]
- Mareike Steen, Tanja Kirchberger, Andreas H. Guse2NAADP Mobilizes Calcium from the Endoplasmic Reticular Ca2+ Store in T-lymphocytes. J Biol Chem 2007; 282:8864-1887. doi: 10.1074/jbc.M610925200 [Crossref] [ Google Scholar]
- Raffaello A, Mammucari C, Gherardi G, Rizzuto R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem Sci 2016; 41:1035-1049. doi: 10.1016/j.tibs.2016.09.001 [Crossref] [ Google Scholar]
- Ronco V, Potenza DM, Denti F, Vullo S, Gagliano G, Tognolina M. A novel Ca2+-mediated cross-talk between endoplasmic reticulum and acidic organelles: Implications for NAADP-dependent Ca2+ signaling. Cell Calcium 2015; 57:89-100. doi: 10.1016/j.ceca.2015.01.001 [Crossref] [ Google Scholar]
- Galione A. NAADP Receptors. Cold Spring Harb Perspect Biol 2019; 11:a035071. doi: 10.1101/cshperspect.a035071 [Crossref] [ Google Scholar]
- Zhang J, Guan X, Shah, K, Yan J. Lsm12 is an NAADP receptor and a two-pore channel regulatory protein required for calcium mobilization from acidic organelles. Nat Commun 2021. 12: 4739. 10.1038/s41467-021-24735-z.
- Cancela JM, Churchill Churchill, GC GC, Galione A. Coordination of agonist induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells. Nature 1999; 398:74-76. doi: 10.1038/18032 [Crossref] [ Google Scholar]
- Patel S, Churchill GC, Galione A. Unique kinetics of nicotinic acid-adenine dinucleotide phosphate (NAADP) binding enhances the sensitivity of NAADP receptors for their ligand. Biochem J 2000; 352:725-729. [ Google Scholar]
- Churchill GC, Galione A. Spatial control of Ca2+ signaling by nicotinic acid adenine dinucleotide phosphate diffusion and gradients. J Biol Chem 2000; 275:38687-38692. doi: 10.1074/jbc.M005827200 [Crossref] [ Google Scholar]
- Rah SY, Park KH, Han MK, Im MJ, Kim UH. Activation of CD38 by interleukin-8 Signaling regulates intracellular Ca2+ level and motility of lymphokines activated Killer cells. J Biol Chem 2005; 280:2888-2895. doi: 10.1074/jbc.M409592200 [Crossref] [ Google Scholar]
- Ramaschi G, Torti M, Festetics ET, Sinigaglia F, Malavasi F, Balduini C. Expression of cADPR synthetizing CD38 molecule on Human platelet membrane. Blood 1996; 87:2308-2313. doi: 10.1182/blood.V87.6.2308.bloodjournal8762308 [Crossref] [ Google Scholar]
- Mazhar M, Tae-Sik N, Uh-Hyun K. Critical Role for CD38-mediated Calcium Signaling in Thrombin-induced Procoagulant Activity of Mouse Platelets and Hemostasis. J Biol Chem 2011; 286:12952-12958. doi: 10.1074/jbc.M110.207100 [Crossref] [ Google Scholar]
- Mauro T, Enrico TF, Alessandra B, Fabiola S, Cesare B. Thrombin induces the association of cyclic ADP-ribose-synthesizing CD38 with the platelet cytoskeleton. FEBS Lett 1998; 428:200-204. doi: 10.1016/S0014-5793(98)00516-X [Crossref] [ Google Scholar]
- Phillips DR, Prasad KS, Manganello J, Bao M, Nannizzi-Alaimo Nannizzi-Alaimo, L L. Integrin tyrosine phosphorylation in platelet signaling. Curr Opin Cell Biol 2001; 13:546-554. doi: 10.1016/s0955-0674(00)00250-7 [Crossref] [ Google Scholar]
- Crosby Crosby, D D. Crosby, DAlastair WPInteraction of Bruton’s tyrosine kinase and protein kinase C theta in plateletsCross-talk between tyrosine and serine/ threonine kinases. J Biol Chem 2002; 277:9958-9965. doi: 10.1074/jbc.M108965200 [Crossref] [ Google Scholar]
- Pula G, Schuh K, Nakayama K, Nakayama K, Walter U, Alastair WP. PKC δ regulates collagen induced platelet aggregation through inhibition of VASP mediated filopodia formation. Blood 2006; 108:4035-4044. doi: 10.1182/blood-2006-05-023739 [Crossref] [ Google Scholar]
- Khan WA, Blobe G, Halpern A, Taylor W, Wetsel WC, Burns D. Selective regulation of protein kinase C isoenzymes by oleic acid in human platelets. J Biol Chem 1993; 268:5063-5068. doi: 10.1016/S0021-9258(18)53502-X [Crossref] [ Google Scholar]
- Brian E, Xiaoping D. New Concepts and Mechanisms of Platelet Activation Signaling. Physiology 2017; 32:162-177. doi: 10.1152/physiol.00020.2016 [Crossref] [ Google Scholar]
- Liverani E, Kilpatrick E, Mondrinos MJ, Sun S, Kunapuli SP, Laurie EK. PLoS One 2018; 13: e0195379. 10.1371/journal.pone.0195379.
- Harper MT, Poole AW. Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J Thromb Hemost 2010; 8:454-462. doi: 10.1111/j.1538-7836.2009.03722.x [Crossref] [ Google Scholar]
- Kitamura Y, Suzuki M, Tsukioka T, Isobe K, Tsujino T, Watanabe T. Spectrophotometric determination of platelet counts in platelet-rich plasma. Int J Implant Dent 2018; 4:29. doi: 10.1186/s40729-018-0140-8 [Crossref] [ Google Scholar]
- Chan MV, Armstrong PC, Warner TD. 96-well plate-based aggregometry. Platelets 2018; 29:650-655. doi: 10.1080/09537104.2018.1445838 [Crossref] [ Google Scholar]
- Ribeiro JM, Francischetti IM. Platelet-activating-factor-hydrolyzing phospholipase C in the salivary glands and saliva of the mosquito Culex quinquefasciatus. J Exp Biol 2001; 204:3887-3894. doi: 10.1242/jeb.204.22.3887 [Crossref] [ Google Scholar]
- Gul R, Kim SY, Park KH, Kim BJ, Kim SJ, Im MJ. A novel signaling pathway of ADP-ribosyl cyclase activation by angiotensin II in adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol 2008; 295:77-88. doi: 10.1152/ajpheart.01355.2007 [Crossref] [ Google Scholar]
- Shawl AI, Park KH, Kim BJ, Higashida C, Higashida H, Kim UH. Involvement of actin filament in the generation of Ca2+ mobilizing messengers in glucose-induced Ca2+ signaling in pancreatic β-cells. Islets 2012; 4:145-151. doi: 10.4161/isl.19490 [Crossref] [ Google Scholar]
- Rah SY, Park KH, Nam TS, Kim SJ, Kim H, Im MJ. Association of CD38 with non-muscle myosin heavy chain IIA and Lck is essential for the internalization and activation of CD38. J Biol Chem 2007; 23:5653-5660. doi: 10.1074/jbc.M609478200 [Crossref] [ Google Scholar]
- Helfman D, Kim E, Lukanidin E. The metastasis associated protein S100A4: role in tumour progression and metastasis. Br J Cancer 2005; 92:1955-1958. doi: 10.1038/sj.bjc.6602613 [Crossref] [ Google Scholar]
- Kiss B1, Duelli A, Radnai L, Kékesi KA, Katona G, Nyitray L. Crystal structure of the S100A4-nonmuscle myosin IIA tail fragment complex reveals an asymmetric target binding mechanism. Proc Natl Acad Sci USA 2012; 109:6048-6053. doi: 10.1073/pnas.1114732109 [Crossref] [ Google Scholar]
- Mark G, Turner J, Bowling KE, Vandenberg SR, Gear ARL. The role of protein kinase C in the initial events of platelet activation by thrombin assessed with a selective inhibitor. Thromb Res 1993; 69:113-124. doi: 10.1016/0049-3848(93)90008-C [Crossref] [ Google Scholar]
- Satoshi K, Ikebe M. The Phosphorylation of Myosin II at the Ser1 and Ser2 Is Critical for Normal Platelet-derived Growth Factor–induced Reorganization of Myosin Filaments. Mol Biol Cell 2007; 18:5081-5090. doi: 10.1091/mbc.e06-12-1076 [Crossref] [ Google Scholar]
- Straussman S, Even L, Ravid S. Myosin II heavy chain isoforms are phosphorylated in an EGF-dependent manner: involvement of protein kinase C. J Cell Sci 2001; 114:3047-3057. doi: 10.1242/jcs.114.16.3047 [Crossref] [ Google Scholar]
- Mohammad AR, Marko U, Dilson ER, Alf M. Blebbistatin Effects Expose Hidden Secrets in the Force-Generating Cycle of Actin and Myosin. Biophys J 2018; 115:386-397. doi: 10.1016/j.bpj.2018.05.037 [Crossref] [ Google Scholar]
- Mihály K, Judit T, Csaba H, András MC, James RS. Mechanism of Blebbistatin Inhibition of Myosin II. J Biol Chem 2004; 279:35557-35563. doi: 10.1074/jbc.M405319200 [Crossref] [ Google Scholar]
- Ramamurthy B, Yengo CM, Straight AF, Mitchison TJ, Sweeney HL. Kinetic Mechanism of Blebbistatin Inhibition of Nonmuscle Myosin IIB. Biochemistry 2004; 43:14832-14839. doi: 10.1021/bi0490284 [Crossref] [ Google Scholar]
- Horváth ÁI, Gyimesi M, Várkuti BH. Effect of allosteric inhibition of non-muscle myosin 2 on its intracellular diffusion. Sci Rep 2020; 10:13341. doi: 10.1038/s41598-020-69853-8 [Crossref] [ Google Scholar]