Bioimpacts. 14(5):27783.
doi: 10.34172/bi.2024.27783
Original Article
Computational studies and structural insights for discovery of potential natural aromatase modulators for hormone-dependent breast cancer
Snehal Aditya Arvindekar Conceptualization, Data curation, Investigation, Methodology, Resources, Software, Validation, 1, * 
Sanket Rathod Software, 1
Prafulla Balkrishna Choudhari Formal analysis, Supervision, 1
Pradnya Kiran Mane Formal analysis, 1
Aditya Umesh Arvindekar Supervision, Visualization, Writing – original draft, Writing – review & editing, 2, *
Suraj Narayan Mali Data curation, Resources, Software, Writing – review & editing, 3
Bapu Thorat Formal analysis, Supervision, Visualization, 4
Author information:
1Department of Pharmaceutical Chemistry, Bharati Vidyapeeth College of Pharmacy, Kolhapur, (M.S.), India
2Y. D. Mane Institute of Pharmacy, Kagal, (M.S.), India
3Department of Pharmaceutical Sciences & Technology, Birla Institute of Technology, Mesra, Ranchi, India
4Department of Chemistry, Government College of Arts and Science, Aurangabad (M.S.), India
Abstract
Introduction:
The aromatase enzyme plays an important role in the progress of hormone-dependent breast cancer, especially in estrogen receptor-positive (ER+) breast cancers. In case of postmenopausal women, the aromatization of androstenedione to estrone in adipose tissue is the most important source of estrogen. Generally 60%-75% of pre- and post-menopausal women suffer from estrogen-dependent breast cancer, and thus suppressing estrogen has been recognized to be a successful therapy. Hence, to limit the stimulation of estrogen, aromatase inhibitors (AIs) are used in the second-line treatment of breast cancer.
Methods:
The present computational study employed an in silico approach in the identification of natural actives targeting the aromatase enzyme from a structurally diverse set of natural products. Molecular docking, QSAR studies and pharmacophore modeling were carried out using the VLife Molecular Design Suite (version 4.6). The stability of the compounds was confirmed by molecular dynamics.
Results:
From molecular docking and analysis of interactions with the amino acid residues of the binding cavity, it was found that the amino acid residues interacting with the non-steroidal inhibitors exhibited π-stacking interactions with PHE134, PHE 221, and TRP 224, while the steroidal drug exemestane lacked π-stacking interactions. QSAR studies were performed using the flavonoid compounds, in order to identify the structural functionalities needed to improve the anti-breast cancer activity. Molecular dynamics of the screened hits confirmed the stability of compounds with the target in the binding cavity. Moreover, pharmacophore modelling presented the pharmacophoric features of the selected scaffolds for aromatase inhibitory activity.
Conclusion:
The results presented 23 hit compounds that can be developed as anti-breast cancer modulating agents in the near future. Additionally, anthraquinone compounds with minor structural modification can also serve to be potential aromatase inhibitors. The in silico protocol utilised can be useful in the drug discovery process for development of new leads from structurally diverse set of natural products that are comparable to the drugs used clinically in breast cancer therapy.
Keywords: In silico studies, Natural products, Aromatase inhibitors, QSAR, Aromatase inhibitors, Breast cancer
Copyright and License Information
© 2024 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
No founding sources.
Introduction
Despite tremendous studies on the prevention and treatment, breast cancer remains a leading cause of cancer deaths in women globally. At present radiotherapy, surgery, chemotherapy, and hormonal therapy outline a general combination for the treatment of breast cancer, which has to be modulated as per the need of every individual cancer treatment.1 In recent times, repurposing drugs such as alkylating agents, anthracyclines, antifolates, CDK4/6 inhibitors, aromatase inhibitors, mTOR inhibitors and mitotic inhibitors have been repositioned and successfully used in the breast cancer treatment. However, the repurposed drugs usually do not work in mono-therapy, but in combinations. Furthermore, the toxicity of repurposed drugs in patients with combination therapy is unknown.2
Aromatase is a catalyzing enzyme present in adipose tissue that plays a vital role in the biosynthesis of estrogen - a steroidal hormone,1 and is a potential target for new drug development in the treatment of ER dependent breast cancer.3 In case of postmenopausal women, aromatization of androstenedione to estrone in adipose tissue is the most important source of estrogen.4 Approximately, 60% of women in pre-menopausal and 75% women of post-menopausal stage generally have estrogen-dependent breast cancer, and thus inhibiting or suppressing estrogen has been recognized to be a successful tumour therapy.2,3 To limit the stimulation of the steroidal hormone-estrogen in breast cancer, aromatase inhibitors (AIs) have been effectively used as a second-line treatment to females with advanced-stage of estrogen-dependent breast carcinoma, since AIs have the capacity to change the hormonal environment by averting the conversion of androgens to estrogen.4,7 In clinical practice, the non-selective AIs include aminoglutethimide and testolactone, while the selective AIs include anastrozole, letrozole, exemestane, vorozole, formestane, and fadrozole that reduce the production of estrogen in the peripheral tissues and cancer cells by inhibiting the aromatase enzyme activity with some toxicity. AIs are associated with unwanted side effects like atypical lipid metabolism, loss of bone density, etc., which may be attributed to the nonspecific reduction of the aromatase activity in other tissues. Even though numerous synthetic compounds and ligands of the nuclear receptor are identified to reduce the action of the tumor-specific aromatase inhibition, the development of effective drugs without toxic effects is still necessary.1
Phytoestrogens from natural sources may contribute to the aversion of estrogen-dependent breast cancers by several similar mechanisms of antiestrogens of pharmaceutical origin. There are several reports on natural products belonging to the class of indoles, alkaloids, lignans, flavonoids, phenols, and glycosides found in fruits, vegetables, and other natural sources to possess anti-cancerproperties.5,8-10 Since estrogens are well-known cancer promoters mostly in breast cancer, natural actives with aromatase inhibitory activity would be of great importance in breast cancer prevention. It is therefore of great interest to identify new phytoconstituents with mechanism-based aromatase inhibitory activity. The accessibility of the structure of estrogen receptor-α (ER-α) implies the application of molecular docking protocol to screen the possible natural actives from natural sources as aromatase inhibitors. In this pursuit, various in silico techniques molecular docking, pharmacophore modeling, and 3D-QSAR have been performed in the identification of ligands against aromatase.
Two-dimensional and three-dimensional structure-activity relationship techniques are helpful in discovering the molecular properties and their connection with the pharmacological activity. The QSAR protocols are often supported by the pharmacophore models, which give an idea about the structural features essential for the development of new compounds.
In the present study, natural anti-breast cancer actives were screened from previously reported literature, followed by virtual screening using VLife Molecular Design Suite (version 4.6). The interactions of obtained natural compounds by virtual screening were analysed to get an idea of the important structural features that are necessary for protein-ligand interactions of the aromatase inhibition. Validation of natural hits was achieved by molecular dynamics studies. Since, flavonoids were found to exhibit potential aromatase interactions, QSAR studies (2D and 3D) were conducted on 33 flavonoids to develop a possible quantitative relationship between the flavonoid nucleus and aromatase inhibitory activity. Additionally, pharmacophore modelling was performed using letrozole as a reference molecule to obtain the essential structural features responsible for aromatase inhibition against estrogen-dependent breast cancers.
Materials and Methods
Collection of data
In the present study, 246 anti-breast cancer natural actives, including alkaloids, flavonoids, glycosides, phenols, quinones, and miscellaneous compounds, were selected as ligands by reviewing reported literature (Table S1).10-20 The X-ray diffracted crystal structure of the human placental aromatase complexed with the steroidal breast cancer drug exemestane with PDB ID: 3S7S, having 3.21 A0 resolutions was downloaded in.pdb format from the RCSB protein databank (http://www.rcsb.org).
Molecular docking, QSAR studies and pharmacophore modeling were performed using the Molecular Design Suite – version 4.6, developed by VLife Sciences (VLifeMDS, Pune). The structures of the ligands used in this study were derived from the National Library of Medicine (PubChem) and the structures which were not available in the PubChem database were drawn using ChemDraw software (version 8). The VLife Engine module was employed to convert all 2D (.mol) structures to 3D (.mol2) format and to minimize energy to attain the lowest free energy. The MMFF method with 0.01 kcal/mol A0 of RMS gradient and an iteration limit of 100 000 was employed in the ligand optimization.
Preparation of target structure (PDB ID: 3S7S) for docking study and Ramchandran plot
The crystal structure of the human placental aromatase (3S7S) was prepared by the removal of water molecules and adding hydrogen atoms to the atomic coordinates of the aromatase structure using the BioPredicta module. Using BioPredicta module, the binding cavity for the ligands present on aromatase enzyme structure was identified, and the structure of steroidal aromatase inhibitor-exemestane was extracted from the enzyme target and used as a reference ligand.
The interacting cavities present in the aromatase enzyme structure were identified using the BioPredicta module and the active site was identified and used for virtual screening. Using the same module, Ramchandran plot was generated to get insights of the distribution of amino acid residues in both allowed and disallowed spaces. A 2D scatter plot was generated to give an idea about the conformational angles, viz., phi and psi, along with the name of the amino acid residue.
Molecular docking studies
We attempted to find the potential aromatase inhibitors by a virtual screening of the natural products against the active conformation of the aromatase enzyme. The GRIP batch docking application was used for the molecular docking study of 246 natural actives (ligands) with 3S7S. Docking was performed by keeping the receptor in rigid mode and the ligand in flexible mode. The convergence factor was set to 0.0001 for 10000 generations and the fitness function criteria as the docking score. All other parameters were set to default values. On completion of the docking process, the interaction energy of the ligand-receptor complex was obtained in kcal/mol.20The natural actives with good docking scores were compared with the reference ligands and the hits were chosen and relative study with the clinically used non-steroidal aromatase inhibitors, viz., letrozole, anastrozole, vorozole, and fadrozole.
Post-docking analysis and validation
Interactions of the hit compounds based on the docking scores were analyzed using the Analyze tool in the BioPredicta module. Specifically, the study of hydrophobic, charge, van der Waal’s, hydrogen, and π-stacking interactions was done. The distance involving the functional groups of the hit compounds and the amino acid residues of the aromatase enzyme was studied. The amino acid residues present around exemestane (a steroidal aromatase inhibitor) and binding cavity were identified. In order to validate the results of the obtained hits, a comparative assessment of the interactions of the steroidal and non-steroidal aromatase inhibitors was performed.
2D and 3D QSAR studies
2D and 3D QSAR dataset
Generally, most of the hits obtained from the virtual screening procedures contain six-member rings with carbonyl group in common and belong to the flavonoid or quinone class of compounds. As a result, the compounds with a benzopyran nucleus that showed cytotoxicity against the ER+ MCF-7breast cancer cell lines were employed for QSAR study.
In the 3D QSAR study, all compounds in the dataset were aligned and subjected to generate a common rectangular grid. The electrostatic and steric interaction energies were calculated at the lattice points of the grid by using Gasteiger and Marsili charge type. These descriptors were utilized for the generation of the model and to decide the similitude between the molecules.10
Model generation
Benzopyran structure was used as the template in VLifeMDS version-4.6. The physicochemical constitutional, electrostatic, and alignment-independent 2D descriptors were computed for the 2D QSAR study. The MMFF94 force field was used to optimize the compound geometries for energy minimization and Gasteiger-Marsili as an atom charge. The 3D QSAR module was employed to achieve a proper alignment of selected compounds, followed by a common rectangular grid around the compounds. The steric, electrostatic and hydrophobic descriptors were generated and used as independent variables. For QSAR, the random method of training and test set selection was employed. The 2D and 3D QSAR models were obtained using the multiple linear regression analysis. The superiority of the regression model obtained was evaluated using statistical parameters, including q2, r2(F test), etc. The models with q2 below 0.7 were not considered. Furthermore, external predictivity of compounds was checked and the values of predicted Vs observed anticancer activity was reported.
Molecular dynamics simulation
The MD simulations study of the identified hits were conducted using the Desmond 2020.1 software of Schrödinger, LLC. The simulations were done using the OPLS-2005 force field.21 TIP3P water box explicitly represented the solvent environment, and the entire system was enclosed within a periodic boundary box measuring 10 Å x 10 Å x 10 Å. To neutralize charges, Na+ ions were added at a concentration of 0.15 M.22 Additionally, NaCl solutions were introduced to mimic physiological conditions. The equilibration process was initiated with a 10 ns simulation under the NVT ensemble, allowing the protein-ligand complexes to stabilize.23 This was followed by a brief equilibration and energy minimization phase using an NPT ensemble for 12 ns. The NPT ensemble utilized the Nose-Hoover chain coupling approach, maintaining a pressure of 1 bar with a relaxation time of 1.0 ps for temperature adjustments.24 The time step for all simulations was set at 2 fs. Pressure control was maintained using the Martyna-Tuckerman-Klein chain coupling scheme barostat method with a relaxation time of 2 ps. Long-range electrostatic interactions were computed using the particle mesh Ewald method, while the Coulomb interaction radius was consistently set at 9 Å. For bonded forces, a RESPA integrator was employed with a time step of 2 fs per trajectory.25 This integrator was also used for calculating bonded forces with the same time step. A final production run lasting 100 ns was carried out to capture the dynamic behaviour of the system.26 The MD trajectory analysis was done with statistical metrics including root mean square deviation (RMSD), radius of gyration (Rg), root mean square fluctuation (RMSF), and intermolecular interactions.27
Binding free energy (MMGBSA) calculations
The computation of binding free energies was carried out using the MMGBSA approach. The Prime MMGBSA method for binding free energy calculation was determined through the utilization of the thermal_mmgbsa.py Python script.3,28 Calculation was performed on the last 50 frames of the each simulation trajectory, with a 1-step sampling size. The binding free energy (expressed in kcal/mol) was evaluated using an additive principle, wherein various energy components (ΔGbind, ΔGbindCoulomb, ΔGbindCovalent, ΔGbindHbond, ΔGbindLipo, ΔGbindSolvGB, and ΔGbindvdW) were combined.29,30 The calculation of ΔGbind was executed using the following equation:
ΔGbind = ΔGMM + ΔGSolv - ΔGSA
Where ΔGbind signifies the overall binding free energy, ΔGMM denotes the difference between the free energies and the combined energies of the protein and ligand in their isolated forms, ΔGSolv represents the difference in the solvation energies obtained from the generalized born surface area (GBSA) method compared to the cumulative solvation energies of the unbound protein and ligand, ΔGSA refers to the difference in the surface area energies contributed by the protein and the ligand.
Pharmacophore modeling
For pharmacophore modeling, the MolSign module of VLifeMDS was employed. A series of obtained hit compounds were aligned on the reference compound- letrozole. Pharmacophore model was analysed for three-dimensional features necessary for a clinically active molecule. A minimum three-feature pharmacophore was selected with a tolerance limit of 10 A˚. Also, the maximum allowed distance between the two features was set to10 A˚.20
Results
Binding cavity and Ramchandran plot
Using the BioPredicta module of VLife MDS, it was observed that the selected aromatase enzyme (PDB id: 3S7S) consisted of 6 cavities (Fig. S1). Cavity 1 of aromatase (represented in pink colour in Fig. S1) was identified as a binding cavity.
Structure-based pharmacophore analysis was performed using the drug exemestane, which showed the presence of a total of following 31 amino acid residues close to the reference ligand suggesting its active site, viz., GLY121, LEU122, ILE133, SER153, ASP186, VAL187, LEU188, THR189, GLN218, LEU227, LEU228, ASP309, MET318, MET364, ARG365, PRO368, VAL369, VAL370, ASP371, LEU372, VAL373, MET374, GLY439, LYS440, TYR441, ILE442, GLN472, SER478, LEU479, ASP482, and GLU489.
The amino acid residues distribution was allowed and the disallowed region of 3S7S was analysed by the Ramachandran plot analysis indicating 1.11% of disallowed amino acid residues count, thereby suggesting the stability of conformers. Fig. S2 provides the following insights: Core Count: 375/450 (83.33%), Allowed Count: 58/450 (12.89%), Generously Allowed Count: 12/450 (2.67%), Disallowed Count: 5/450 (1.11%) and all counts including the GLY & PRO residues.
Molecular docking study
Table 1 represents the docking scores of the reference ligand exemestane (a steroidal aromatase inhibitor) and clinically used non-steroidal aromatase inhibitors. Exemestane exhibited a docking score of -62.29. The clinically reported non-steroidal AIs having triazole as the base nucleus like letrozole, anastrozole, vorozole, and fadrozole showed docking scores of -60.06, -71.44, -79.44, and -48.76, respectively.
Table 1.
Docking scores and interactions of clinically used drugs
|
Ligand
|
Docking score
|
Pi
|
Hydrogen Bond
|
|
Amino acids
|
Distance (A0)
|
Amino acids
|
Distance (A0)
|
| Letrozole |
-60.06 |
PHE221
TRP224 |
4.889
5.247 |
ARG115
MET374 |
1.701
2.272 |
| Anastrozole |
-71.44 |
PHE134
TRP224 |
5.450
5.179 |
ARG115
MET374 |
2.394
2.453 |
| Vorozole |
-79.44 |
PHE134
PHE221 |
4.623, 4.630
5.099 |
ARG115
MET374 |
2.278
2.508
1.582 |
| Fadrozole |
-48.76 |
TRP224 |
5.328 |
ARG115
MET374 |
2.197
1.504 |
| Exemestane |
- 73.10 |
- |
- |
ARG115 |
1.719 |
| Aminoglutethimide |
-69.74 |
TRP224 |
5.429 |
ARG115 |
2.162 |
Among screened 246 molecules, a docking score cut-off for the at –40 and the similitude of interactions were set as the selection criteria for screened compounds with reference to drugs in clinical practice. The computational values of the obtained hit compounds are shown in Table 2 wherein, delphinidin 3-glucoside exhibited a lowest docking score (−80.77), while emodin showed a maximum score (−52.50). The hit compounds having the docking scores close to that of the clinically used compounds were selected subsequently for the interaction analysis.
Table 2.
Docking scores and interactions of the obtained hits
|
Ligand
|
Docking score
|
π-Stacking
|
Hydrogen Bond
|
|
Amino acids
|
Distance (A0)
|
Amino acids
|
Distance (A0)
|
| 1-Acetoxy, 9, 10-anthraquinone |
-54.07 |
PHE134 |
4.515 |
ARG115 |
2.475 |
| 7,8-Dihydroxy flavone |
-59.12 |
PHE134 |
5.470 |
ARG115
MET374 |
2.526, 2.595
2.086 |
| 7-Hydroxy flavone |
-56.45 |
PHE134 |
5.256 |
MET374 |
2.337 |
| Isoquercetin |
-71.55 |
PHE134
TRP224 |
5.220
5.254 |
ARG115
MET374 |
1.710, 2.182
1.861, 2.299, 1.739 |
| Naringenin |
-56.69 |
- |
- |
ARG115
PRO429 |
2.312
2.366 |
| Centaureidin |
-64.80 |
- |
- |
ARG115 |
1.604, 2.127 |
| Taxifolin |
-57.43 |
PHE134 |
4.802 |
ARG115
ALA306 |
2.227
2.443 |
| Nobiletin |
-71.34 |
- |
- |
ARG115
MET374 |
2.243
2.176 |
| Epicatechin |
-55.19 |
PHE134 |
5.212 |
ARG115
MET374 |
2.173, 2.368, 1.784 |
| 2-Carbomethoxy, 1,4 naphthoquinone |
-63.57 |
PHE134 |
5.113 |
ARG115 |
2.177 |
| 3,5,7,3',4'-Glucopyranoside |
-71.50 |
PHE134
TRP224 |
5.454
5.120 |
ARG115
ARG435 |
2.177
2.382 |
| 6-Gingerol |
-64.76 |
PHE134 |
5.473 |
ARG115
MET374 |
2.585
1.908 |
| 8-Prenylnaringenin |
-61.95 |
TRP224 |
4.672 |
ARG115
ASP309
LEU372 |
2.358, 1.942, 1.847, 1.475, 2.245 |
| Cimicifugic acid G |
-72.77 |
TRP224 |
5.189 |
ARG115 |
2.220 |
| Delphinidin 3-glucoside |
-80.77 |
PHE134 |
5.214 |
ARG115
MET374
ARG375 |
2.249, 2.216
2.501, 2.272
2.367, 2.129, 2.435 |
| Denbinobin |
-61.49 |
PHE134 |
5.452 |
ARG115
MET374 |
2.406
1.850 |
| Emodin |
-52.50 |
PHE134 |
5.402 |
ARG115
MET374 |
2.403, 2.567
1.656 |
| Epigallocatechin 3-gallate |
-76.63 |
TRP224 |
5.050 |
ARG115 |
1.637 |
| Flemingin B |
-63.13 |
PHE134 |
4.486 |
ARG115
MET374 |
2.336, 1.730, 1.484
2.488 |
| Honokiol |
-74.13 |
PHE134
TRP224 |
5.382
4.999 |
ARG115
MET374 |
2.206, 2.412
2.147 |
| Phyllanthin |
-72.58 |
PHE134
PHE148 |
4.997
4.783 |
ARG115
MET374 |
1.956
2.545 |
| Physcion |
-54.04 |
TRP224 |
4.383 |
ARG115 |
1.915, 2.525 |
| Rhein |
-62.62 |
TRP224 |
5.430 |
ARG115
MET374 |
2.121
1.493 |
Interaction analysis of clinically used drugs with aromatase
To validate the probable action of the clinically used AIs mentioned in Table 1; we analysed the van der Waals, hydrophobic, charge, π-stacking, and hydrogen bond interactions between the molecules and receptor. The amino acid residues involved in the van der Waals and hydrophobic interactions are not mentioned as they were common amino acid residues of the active site and the screened hit compounds. The residues exhibiting the hydrogen bonding and π-stacking interactions were only considered for the study.
Aromatase inhibitors interaction analysis
The cavity 1 in the aromatase enzyme consisted of 31 amino acid residues, wherein the drugs letrozole, vorozole, and anastrozole showed π-stacking interactions with PHE221, PHE134, and TRP224, and hydrogen bond interaction with ARG115 and MET374 to bind with the aromatase target (Table 1, Fig. 1).
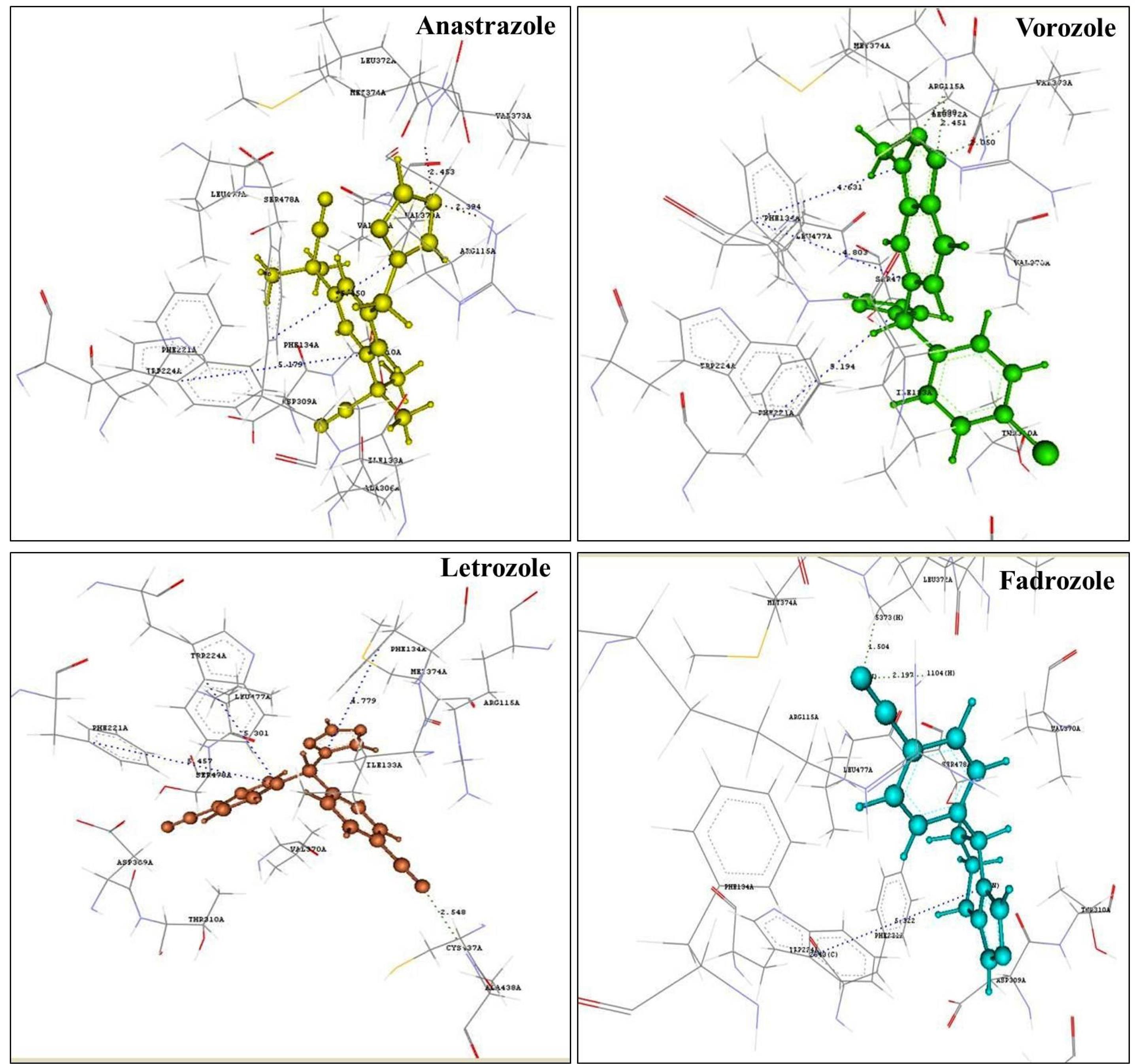
Fig. 1.
Binding pose of aromatase inhibitors.
.
Binding pose of aromatase inhibitors.
As against to this binding commonality, fadrozole showed a π-stacking interaction only with TRP224 and not with PHE221 or PHE134 like other aromatase inhibitors.
Interaction analysis of screened hit compounds with aromatase enzyme and in silico validation
Table 2 represents the binding energy scores and interactions of 23 screened hits with aromatase enzyme (PDB ID: 3S7S).
Subsequently, the interactions of the amino acid residues of the screened hits were compared to that of the drugs used in clinical regimen. This could assist the understanding of the putative mechanisms of action. Based on the resemblance of the interactions of hit compounds and the clinically used AIs with the target, the natural product hit compounds were categorized into the different mechanisms of action:
Natural product as hit compounds of AIs group
A comparison of the interactions of the screened hit compounds and the Ais showed that for a hit compound to be an anastrozole-like aromatase inhibitor, the prerequisites were π-stacking interaction with the PHE134 and TRP 224 with a distance below 5.5 Å and hydrogen bond interactions with ARG115 and MET374 with distance less than 2.5 Å. Among the obtained 23 hits, two hits -isoquercitrin, and honokiol (Fig. S3) showed the desired interaction exactly like anastrozole.
Isoquercetin is a flavonoid compound found commonly in Mangifera indica (mango) and Rheum nobile, which possessed a lowest docking score of −71.55 and showed π-stacking interaction with the PHE134 and TRP 224 with a distance of 5.220 and 5.254 Å, respectively. Also, the compound showed hydrogen bond interactions with amino acid residues ARG115 and MET374 with a distance of 1.710 and 2.299 Å, respectively. These findings are supported by the results of Hamed et al, who isolated isoquercitrin from Carica papaya and compared the aromatase inhibitory activity with letrozole, as well as cytotoxicity against hormone-dependent MCF-7 breast cancer cell lines.31
Honokiol—a lignan abundantly present in the bark of the trees belonging to the Magnolia genus showed the lowest docking score of -74.13, which was very close to the docking score of anastrozole (-71.44). A study on lignans including honokiol also reports their role in aromatase inhibition.10,32
Likewise, the other five hits, viz., 8-prenylnaringenin, cimicifugic acid G, epigallocatechin 3-gallate, physcion, and rhein showed π-stacking interaction with TRP224 and hydrogen bond interactions with ARG115 and MET374, which was exactly observed in the clinically used imidazole aromatase inhibitor-fadrozole. The previous findings of researchers also showed the potential of lignans and flavonoids as aromatase inhibitors, which supports our findings.5,6,8,10,33,34
8-Prenylnaringenin - a prenylflavonoid phytoestrogen abundantly found in hops (Humulus lupulus) exhibited a docking score of -61.95.35 Cimicifugic acid showed the lowest docking score of -72.22. Epigallocatechin-3-gallate, physcion, and rhein showed docking scores of -76.63, -54.04, and -62.62, respectively.
Natural product as hit compounds belonging to chemical structure modification class
In the screening of natural actives as AIs, we found certain hits possessed anticancer activity, yet showed similar interactions and docking scores to that of anastrozole. Eleven hits belonged to benzo-gamma-pyrone (flavonoid) class, viz., 7, 8-dihydroxy flavone and 7-hydroxy flavone, including naringenin, centaureidin, naringenin, taxifolin, nobiletin, epicatechin, 3,5,7,3',4'-glucopyranoside, delphinidin 3-glucoside, denbinobin and phyllanthin that showed docking scores in the range between -55.19 to -80.77.
Three quinone derivatives 1-acetoxy, 9, 10 anthraquinones, and emodin showed docking scores of -54.07, -63.57, and -52.50, respectively. The other two alcoholic compounds Flemingin B and 6-gingerol exhibited docking scores of -63.13 and -64.76, respectively. All the compounds showed π-stacking interaction with PHE134 and hydrogen bond interaction with ARG115 and MET374.
QSAR studies
Among the 23 obtained hits by virtual screening, sixteen hits belonged to the flavonoid class; hence, flavonoids with aromatase inhibitory activity were subjected to QSAR studies from previous literature. In this regard, a total of 33 flavonoids were employed for QSAR studies5,10,36-39 (Table S2). Fig. 2 represents the template of the flavonoid nucleus used for QSAR studies.
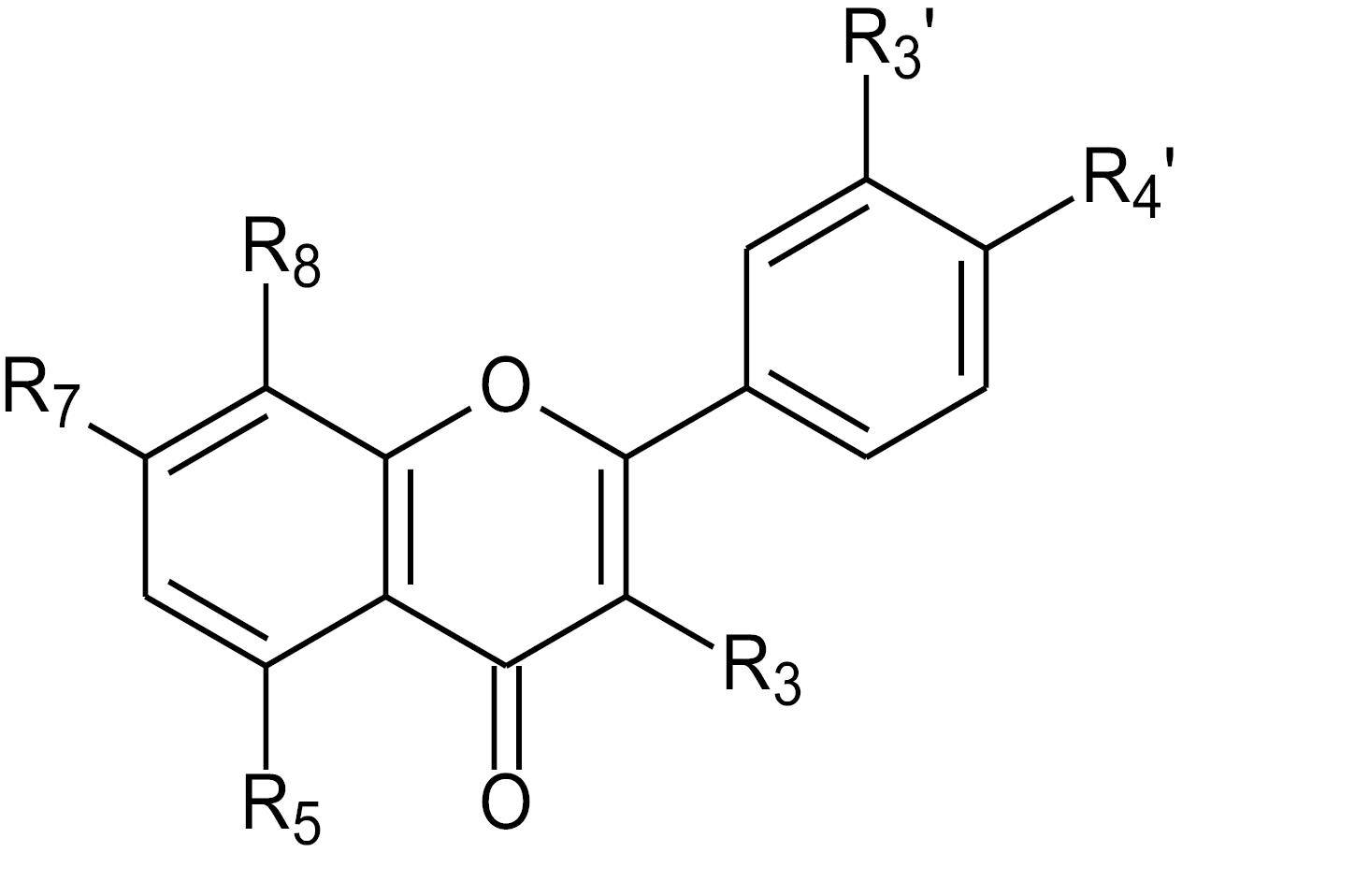
Fig. 2.
Flavonoid template used for QSAR studies.
.
Flavonoid template used for QSAR studies.
2D QSAR studies
To get insights of the effect of various molecular descriptors on the biological activity of flavonoid series, 2D QSAR studies were performed. Using the MLR method, various 2D QSAR models were generated, where the relationship between the biological activity (dependent variable) and independent variables (different molecular descriptors) was identified with the help of linear equations. Table 3 represents the best regression equation of the flavonoid series for the contribution of descriptors for obtained biological activity. A total variance of 95% (r2 = 0.95) was observed in the training set for the selected 2D QSAR model. In addition, the model showed predictive ability of 85% internal (q2) and 62% external (pred_r2). The two-tailed p-value was less than 0.0001, indicating the probability of model failure was 1 in 100 000.
Table 3.
Statistical parameters of the 2D and 3D QSAR models
|
Parameter
|
2D QSAR
|
3D QSAR
|
| Equation |
PIC50 = 14.6329 (SAMostHydrophobic) + 9.0434 (SAMostHydrophilic) -1.0545 (DistTopo) + 0.0228 (Quadrupole3) + 0.0754 (YcompDipole)+ 3.3837 |
PIC50= -0.0409 (S_540) -0.0567 (S_1156)+0.1706(E_429)+ 0.1945 (E_1149) -0.0285 (E_658) -0.1463 |
| N |
15 |
17 |
| Degree of freedom |
9 |
11 |
| r2 |
0.95 |
0.90 |
| r2_se |
0.15 |
0.19 |
| q2 |
0.85 |
0.82 |
| q2_se |
0.25 |
0.26 |
| pred_r2 |
0.62 |
0.67 |
| pred_r2se |
0.42 |
0.35 |
| F - test |
30.90 |
20.14 |
| Contributing descriptors |
SAMostHydrophobic
SAMostHydrophilic
DistTopo
Quadrupole3
YcompDipole |
S_540
S_1156
E_429
E_1149
E_658 |
Model 1 presents the positive contribution of the descriptors SAMostHydrophobic, SAMostHydrophilic, Quadrupole3, and YcompDipole (Table 3) as semi-empirical descriptors. SAMostHydrophobic and SAMostHydrophilic represent the most hydrophobic and hydrophilic values, respectively, on the vdW surface area of a molecule using the Audry method. Quadrupole3 descriptor represents the importance of magnitude, and is an individual descriptor that signifies the magnitude of the third tensor of the quadrupole moments that positively contribute to the biological activity (16%).
Furthermore, model 1 shows the negative contribution of DistTopo, a topological descriptor, which indicates the distance-based topological index (Fig. 3).
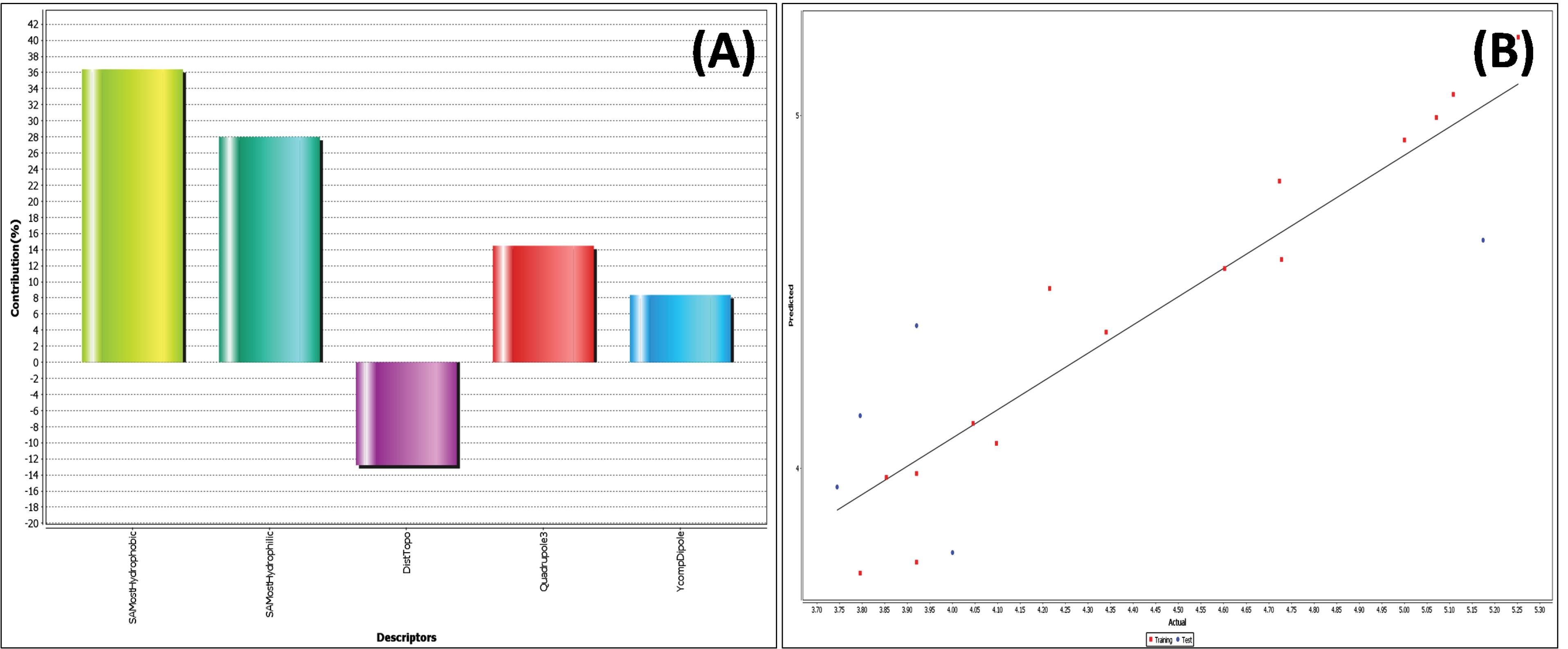
Fig. 3.
2D QSAR model of flavonoids: (A) Contribution plot of contributing descriptor in 2D QSAR (B) Fitness plot of actual vs predicted activity.
.
2D QSAR model of flavonoids: (A) Contribution plot of contributing descriptor in 2D QSAR (B) Fitness plot of actual vs predicted activity.
3D QSAR studies
3D-QSAR studies of the flavonoids were performed using the MLR method using electrostatic, hydrophobic & steric fields. Numerous 3D-QSAR models were obtained using the stepwise variable selection method from which the best model - model 2 is presented herein (Table 3). Considering the aromatase inhibitory activity, we employed the template-based 3D-QSAR model with 17 compounds in the training set, and was found to be highly statistically significant with respect to the internal predictive ability of the model (q2=0.82) and regression coefficient (r2 = 0.90). q2 is the cross-validated correlation coefficient that measures the reliability of prediction. The results indicate that the developed model was reliable and accurate. Furthermore, the residual values for the actual and predicted activity were found close to zero (Table S2), thereby indicating a good predictivity of the generated models.
The 3D data points generated in the vicinity of the pharmacophore were used to optimize the steric & electrostatic requirements of the flavonoid nucleus, in order to study the anticancer activity against hormone-dependent breast cancer. The generated data points consisted a range of property values that can assist the design of the new chemical entities. The points generated in 3D-QSAR models were, E_429, E_1149, and E_658, which represented the electrostatic energy of interactions, while S_540 and S_1156 described the interactions of the steric field between the compounds in the dataset and the probe (CH3+1). These data points suggest the importance and requirements of the electrostatic & steric properties, whose range is mentioned in the parenthesis (Table 3) for understanding the relation between the structure-activity relationship and the biological activity of the flavonoid analogues.
Model 2 shows that the steric descriptors presented negative values, indicating a low steric tolerance (Table 3 and Fig. S4). As a result, presence of steric substituent at the generated points S_540, and S_1156 was not favourable for aromatase inhibitory activity (Fig. S4).
Similarly, the two data points implied that the presence of bulky substituent at these data points could lead to a reduction in the biological activity. Additionally, the positive values of electrostatic descriptors E_429, and E_1149 suggested that electropositive (electron-donating) groups in the flavonoid pharmacophore in the vicinity of R7 or R8 and R3’ or R4’ were optimum for aromatase inhibitory activity. E_658 contributed negatively, indicating that an electron-donating group in the basic flavonoid skeleton was not favorable for the aromatase inhibitory activity.
Molecular dynamics simulation
The docking study highlighted honokiol, epicatechin, and isoquercitrin as ligands that established tight and potentially significant binding interactions with human placental aromatase (PDB 3S7S). The docking study, while offering an essential initial validation of these interactions, inherently operates under static conditions. However, it falls short in delineating the impact of dynamic molecular mobility on the complex, a crucial aspect in understanding the true nature of protein-ligand engagement. Molecular Dynamics (MD) simulations of 100 ns were executed on 3S7S-honokiol, 3S7S-epicatechin, and 3S7S-isoquercitine to bridge this gap and delve deeper into the intricacies of these interactions. This computational technique provided a dynamic outlook by simulating the temporal behavior of molecular systems, affording insights into the complex's evolution over time. In the present study, MD simulations were employed to unravel the multifaceted dynamics governing protein-ligand interactions, thereby elevating our understanding beyond mere static binding modes. A comprehensive panorama of the complex behavior was studied through an array of visual and statistical parameters scrutinized during the simulation.
The RMSD analysis was conducted to investigate the structural stability of the 3S7S-honokiol, 3S7S-epicatechin, and 3S7S-isoquercitine complexes during the course of the MD simulations. In the case of 3S7S-honokiol, the RMSD plot revealed substantial fluctuations in both protein and ligand RMSD values. Notably, the RMSD of honokiol exhibited an ascending trend prior to 20 ns, after which it converged with the RMSD of the docked protein. Between 60 ns and 100 ns, both protein and ligand RMSD values exhibited alignment with reduced scattering. However, it is worth noting that the honokiol consistently exhibited higher RMSD values compared to the docked protein structure. Conversely, the RMSD analysis for the 3S7S-epicatechin complex depicted distinctive behaviors. The protein structure of this complex showcased minimal scattering in RMSD values, underscoring its stability over the simulation period. However, the RMSD values of the ligand, epicatechin, displayed sporadic deviations between 45 ns and 55 ns. Apart from this interval, epicatechin demonstrated remarkable consistency, with negligible deviations in its RMSD values, implying a robust binding interaction. Remarkably, the simulated complex of 3S7S-isoquercitine exhibited a distinct pattern in its RMSD behavior. Both the protein and ligand RMSD values evinced minimal deviation, highlighting the structural stability of the complex throughout the simulation. While the RMSD of isoquercitrin experienced a slight increase and deviation after 60 ns, this shift was relatively minor compared to the overall stability exhibited by the complex. The RMSD plot of simulated complexes are represented in Fig. 4A-C. In Fig. 4A-C, the blue colour represents calculated RMSD values for alpha carbon (Ca) atoms of target aromatase and red colour represents the ligand fit with protein. RMSF provided insights into the flexibility of individual protein residues, shedding light on regions undergoing pronounced conformational changes. Across all three simulated complexes, the RMSF values were observed to be within appropriate ranges, with the RMSF plots displaying minimal high peaks. Remarkably, the 3S7S-isoquercitine complex exhibited a specific amino acid residue segment spanning from 180 to 200, characterized by the highest RMSF value of 4 Å. This observation signifies the potential occurrence of conformational alterations within this region of the protein structure. In contrast, the other two complexes, 3S7S-honokiol and 3S7S-epicatechin, displayed a distinct pattern characterized by minimal fluctuations in the docked protein structure. The RMSF analysis provides a lens through which the dynamic behaviour of protein-ligand interactions can be comprehended. The relatively subdued RMSF values across all complexes suggest stable binding interactions. The exceptional flexibility exhibited by the 3S7S-isoquercitine complex within the 180 to 200 amino acid residue range underscores the potential for localized structural adjustments in response to ligand binding. The RMSF plots are represented in Fig. 4D-F.
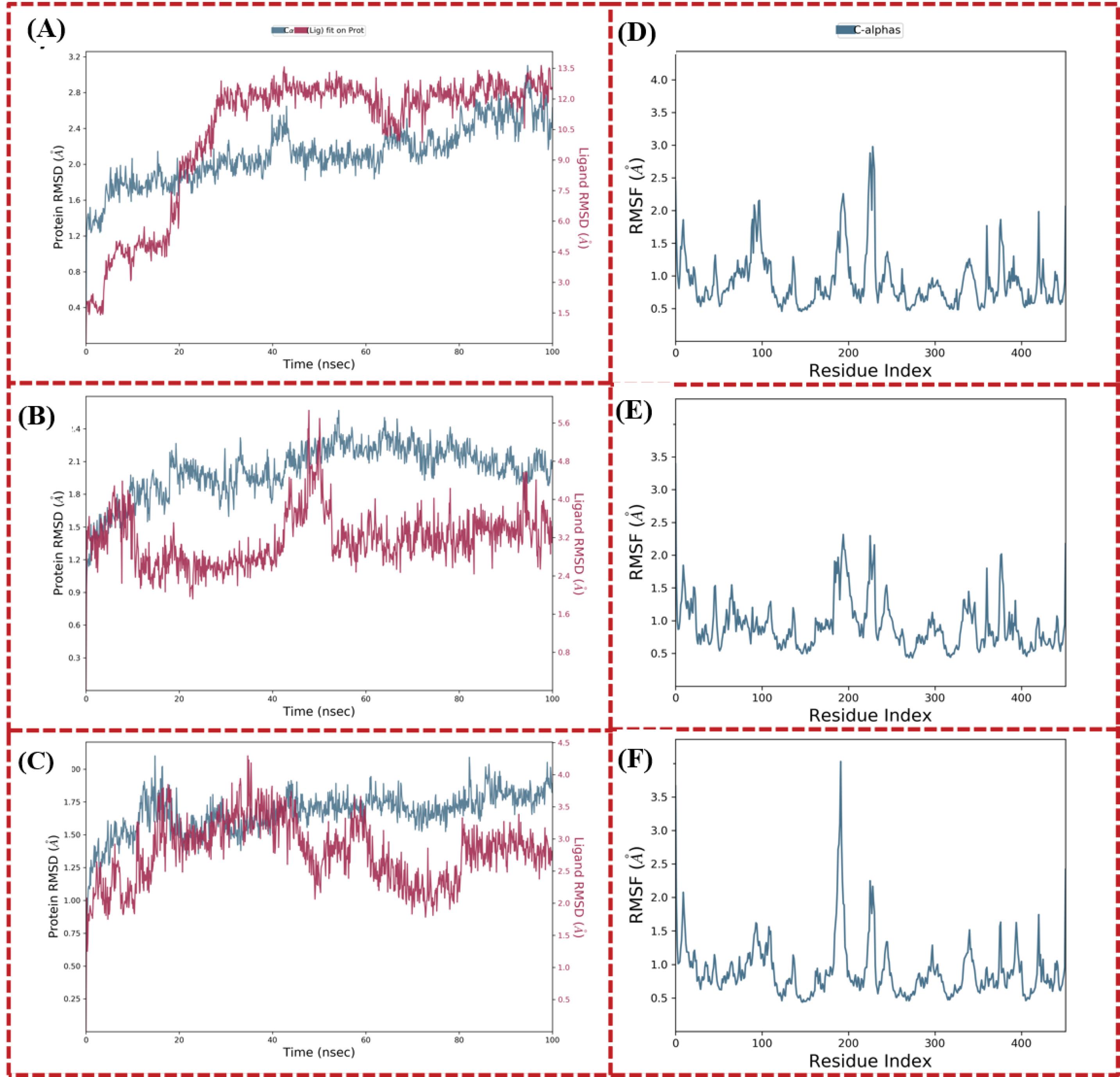
Fig. 4.
(A) to (C) Calculated RMSD values for alpha carbon atoms (blue colour) of aromatse 3S7S and protein fit ligands (red color) and (D) to (F) Line representation of the evolution of RMSF of aromtase alpha carbon atoms during the MD simulation. (A) and (D) 3S7S-honokiol, (B) and (E) 3S7S-epicatechin, and (C) and (F) 3S7S-isoquercitine.
.
(A) to (C) Calculated RMSD values for alpha carbon atoms (blue colour) of aromatse 3S7S and protein fit ligands (red color) and (D) to (F) Line representation of the evolution of RMSF of aromtase alpha carbon atoms during the MD simulation. (A) and (D) 3S7S-honokiol, (B) and (E) 3S7S-epicatechin, and (C) and (F) 3S7S-isoquercitine.
Probing the stability of protein-ligand interactions, a pivotal gauge of binding robustness, was facilitated through the exploration of protein-ligand contacts. These insights elucidate the persistent associations forged during molecular dynamics simulations, thereby corroborating the steadfastness of the binding interactions (depicted in Fig. 5). In the context of the 3S7S-honokiol complex, key amino acid residues, including ARG115, ILE133, TRP141, ALA306, ASP309, LEU372, MET374, and CYS437, exhibited hydrogen bond interactions with the docked ligand (Fig. 5A). Notably, ILE133 emerged as the central locus of interactions, displaying maximum contacts over the 100 ns simulation duration (Fig. 5D). Similarly, in the 3S7S-epicatechin complex, MET303, ALA306, THR310, VAL370, PRO429, and CYS437 were implicated in hydrogen interactions with the ligand (Fig. 5B). Further investigation revealed a consistent interaction pattern between ALA306, PRO429, and PHE430 throughout the 100 ns simulation, reinforcing the stability of these interactions (Fig. 5E). Intriguingly, the 3S7S-isoquercitine complex exhibited a network of hydrogen interactions involving amino acid residues such as ARG115, ILE132, ILE133, TRP141, GLN225, ALA306, THR310, LEU372, MET374, ARG435, GLY436, CYS437, GLY439, LEU477, and SER478 (Fig. 5C). Notably, ARG115 and THR310 showcased consistent and enduring binding interactions over the simulation, indicating the resilience of ligand binding with the targeted protein (Fig. 5F). These findings underscore the role of specific amino acid residues in facilitating and sustaining binding interactions between the ligands and the protein. The recurrent hydrogen interactions observed across complexes over extended simulation periods reinforce the stability of these complexes. Notably, residues such as ALA306 and CYS437 appear to be recurrently involved in interactions, indicating their crucial role in ligand binding stability.
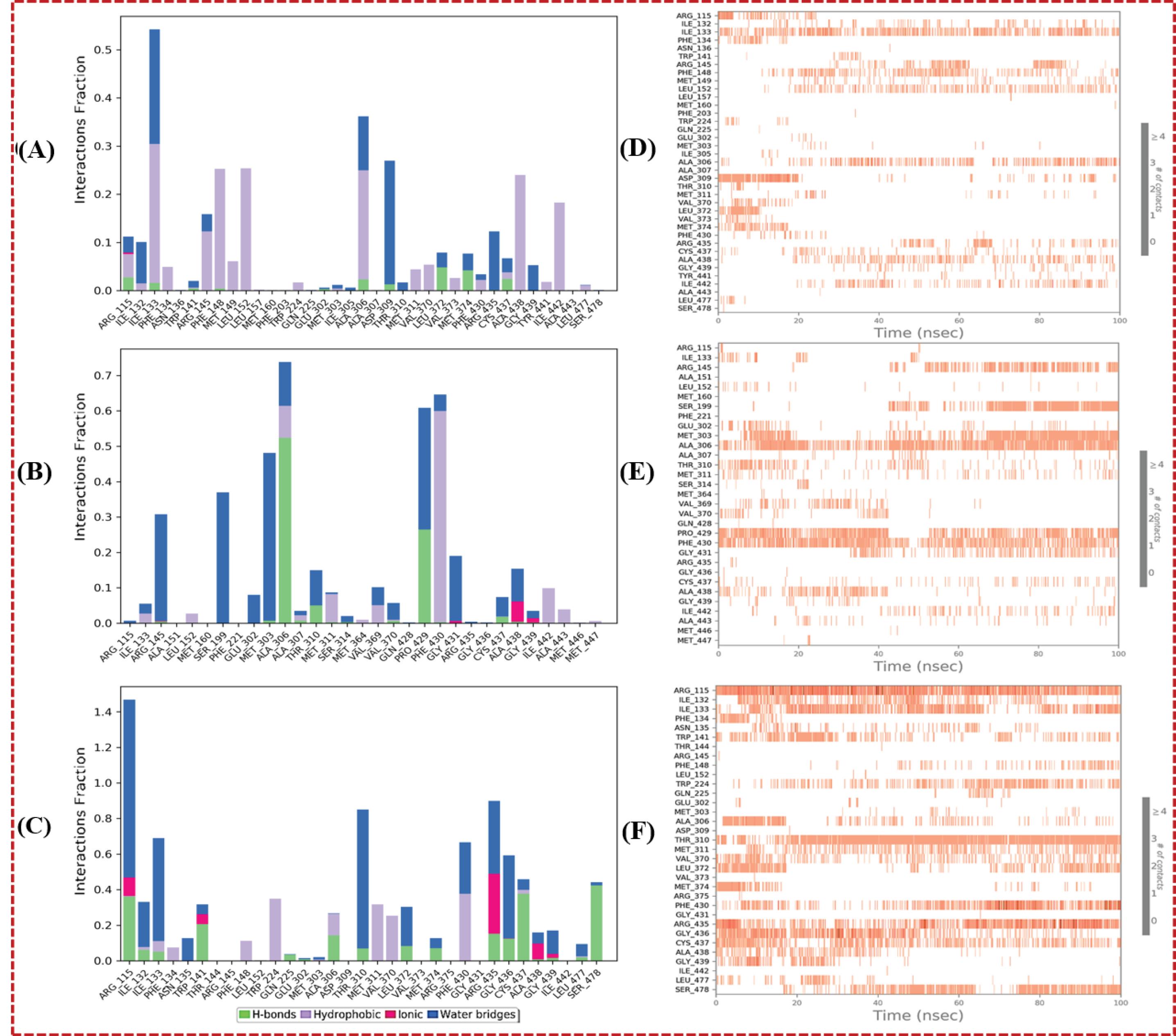
Fig. 5.
(A) to (C) Protein-ligand contacts and (D)-(F) interactions over time plot for the simulated complexes. (A) and (D) 3S7S-honokiol, (B) and (E) 3S7S-epicatechin, (C) and (F) 3S7S-isoquercitine.
.
(A) to (C) Protein-ligand contacts and (D)-(F) interactions over time plot for the simulated complexes. (A) and (D) 3S7S-honokiol, (B) and (E) 3S7S-epicatechin, (C) and (F) 3S7S-isoquercitine.
Binding free energy (MMGBSA) calculations
The MMGBSA calculations were employed to assess the binding energies of simulated complexes involving 3S7S-honokiol, 3S7S-epicatechin, and 3S7S-isoquercitine. The MMGBSA method was employed to calculate the binding free energies including ΔGbind, ΔGbindCoulomb, ΔGbindCovalent, ΔGbindHbond, ΔGbindLipo, ΔGbindSolvGB, and ΔGbindvdW was calculated. The last 50 frames of the simulation trajectory were selected for analysis, with a 1-step sampling size. The calculated binding free energy (ΔGbind) was determined as -46.9 kcal/mol for the 3S7S-honokiol complex. This significant negative value suggests a robust binding between honokiol and the target protein. The energy contributions were delineated further, revealing a substantial lipophilic interaction energy (ΔGbindLipo) of -23.2 kcal/mol, emphasizing the substantial role of hydrophobic forces in stabilizing the complex. The van der Waals interaction energy (ΔGbindvdW) was calculated as -37.0 kcal/mol, indicating a notable contribution from dispersion forces in promoting complex formation. Electrostatic interactions also play a role, with Coulombic energy (ΔGbindCoulomb) of -6.8 kcal/mol and hydrogen bonding energy (ΔGbindHbond) of -0.2 kcal/mol, highlighting the importance of both electrostatic and hydrogen bonding contributions. Similarly, the 3S7S-epicatechin complex exhibited a ΔGbind of -32.9 kcal/mol, indicating a favorable binding affinity. The energy components elucidated a ΔGbindLipo interaction energy of -13.5 kcal/mol, underscoring the hydrophobic interactions stabilizing the complex. The ΔGbindvdW interaction energy of -33.6 kcal/mol and ΔGbindCoulomb interaction energy of -14.5 kcal/mol substantiate the role of these forces in complex stability. ΔGbindHbond energy at -0.8 kcal/mol and ΔGbindSolvGB energy of 28.4 kcal/mol further contribute to the overall binding free energy. In contrast, the 3S7S-isoquercitine complex exhibited the highest binding affinity, with ΔGbind at -53.9 kcal/mol. This result highlights a more robust interaction between isoquercitine and the target protein. The individual energy terms revealed a ΔGbindLipo interaction energy of -14.2 kcal/mol and a ΔGbindvdW interaction energy of -52.5 kcal/mol. The ΔGbindCoulomb interaction energy and ΔGbindHbond energy were calculated to be -21.1 kcal/mol and -1.3 kcal/mol, respectively. Notably, the ΔGbindSolvGB energy was positive at 36.6 kcal/mol, indicating a compensatory role of solvent effects in the binding process. Comparing the three complexes, it is evident that 3S7S-isoquercitine exhibits the strongest binding energies, attributed to a synergistic interplay of various interactions including lipophilic, van der Waals, electrostatic, and hydrogen bonding contributions. The solvent effect also contributes significantly to its binding free energy. On the other hand, while both 3S7S-honokiol and 3S7S-epicatechin show favorable binding affinities, their energy profiles suggest relatively stronger hydrophobic interactions and a less pronounced contribution from solvent effects. Fig. 6 graphically represents calculated binding free energies.
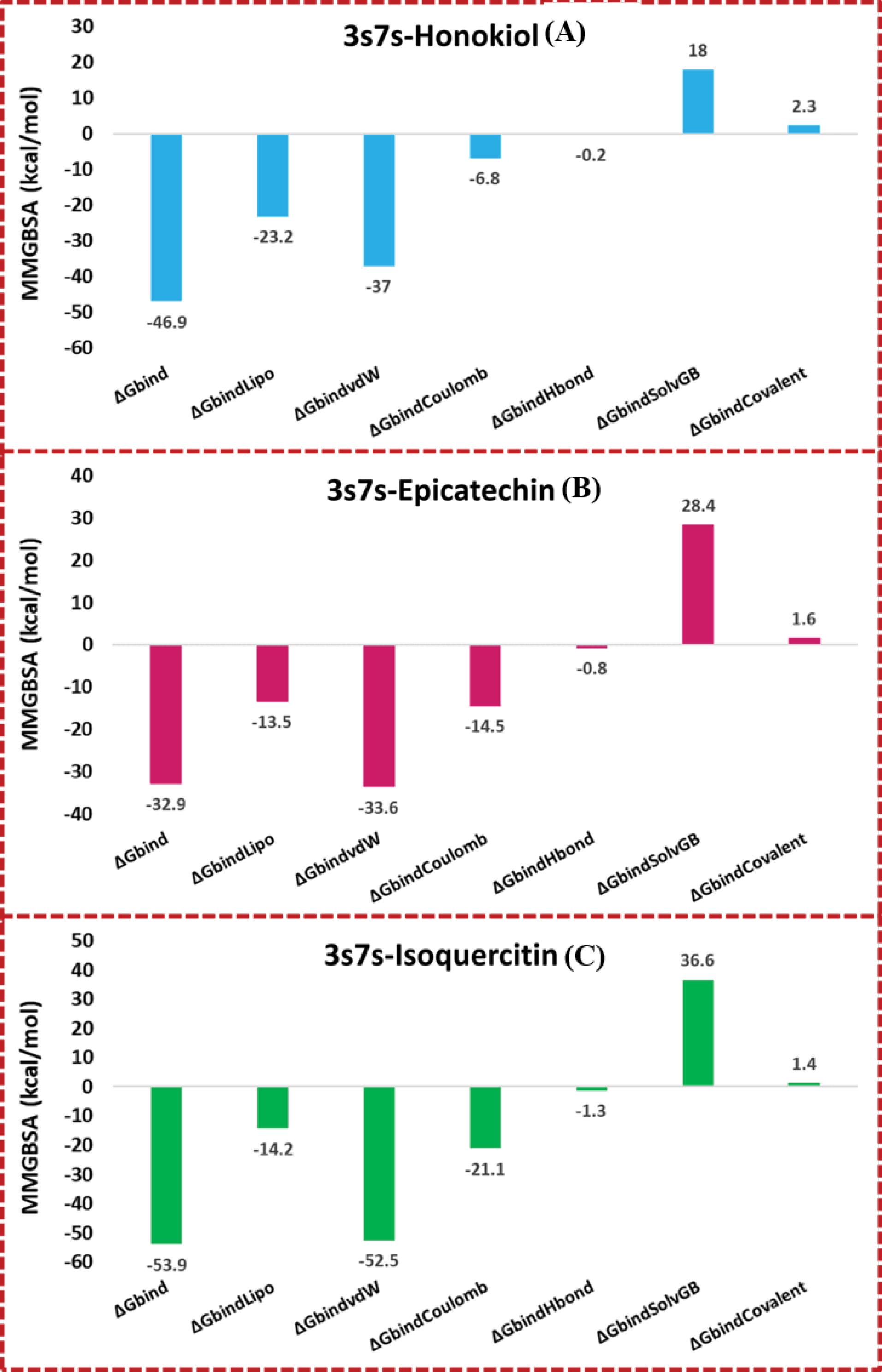
Fig. 6.
Graphical representations binding energies calculated from MMGBSA trajectories of (A) 3S7S-honokiol, (B) 3S7S-epicatechin, (C) 3S7S-isoquercitine.
.
Graphical representations binding energies calculated from MMGBSA trajectories of (A) 3S7S-honokiol, (B) 3S7S-epicatechin, (C) 3S7S-isoquercitine.
Pharmacophore modelling
In order to screen the hits with aromatase inhibitory potential, a pharmacophore model was developed using letrozole – an AI employed in clinical regimen, to get insights of the structural features required for a lead molecule to act as an aromatase inhibitor. The hits obtained after virtual screening were aligned on letrozole to develop the pharmacophoric model, which suggested the requirements to generate leads with a good aromatase inhibitory potential.
Fig. 7A shows the pharmacophore model for letrozole, and provides insights into the structural features present as an aromatase inhibitor. The developed pharmacophore model shows that the molecular structure of letrozole comprises two aromatic centres (large yellow sphere), one aromatic centre (large buff-colored sphere), one aliphatic centre (large orange sphere), and an additional aliphatic carbon centre (small orange sphere). Further, the bond length of the pharmacophoric groups aligned on active molecules is presented in Fig. 7B. Due to the similitude of the pharmacophoric features, the screened natural product hits may act as potential aromatase inhibitors.
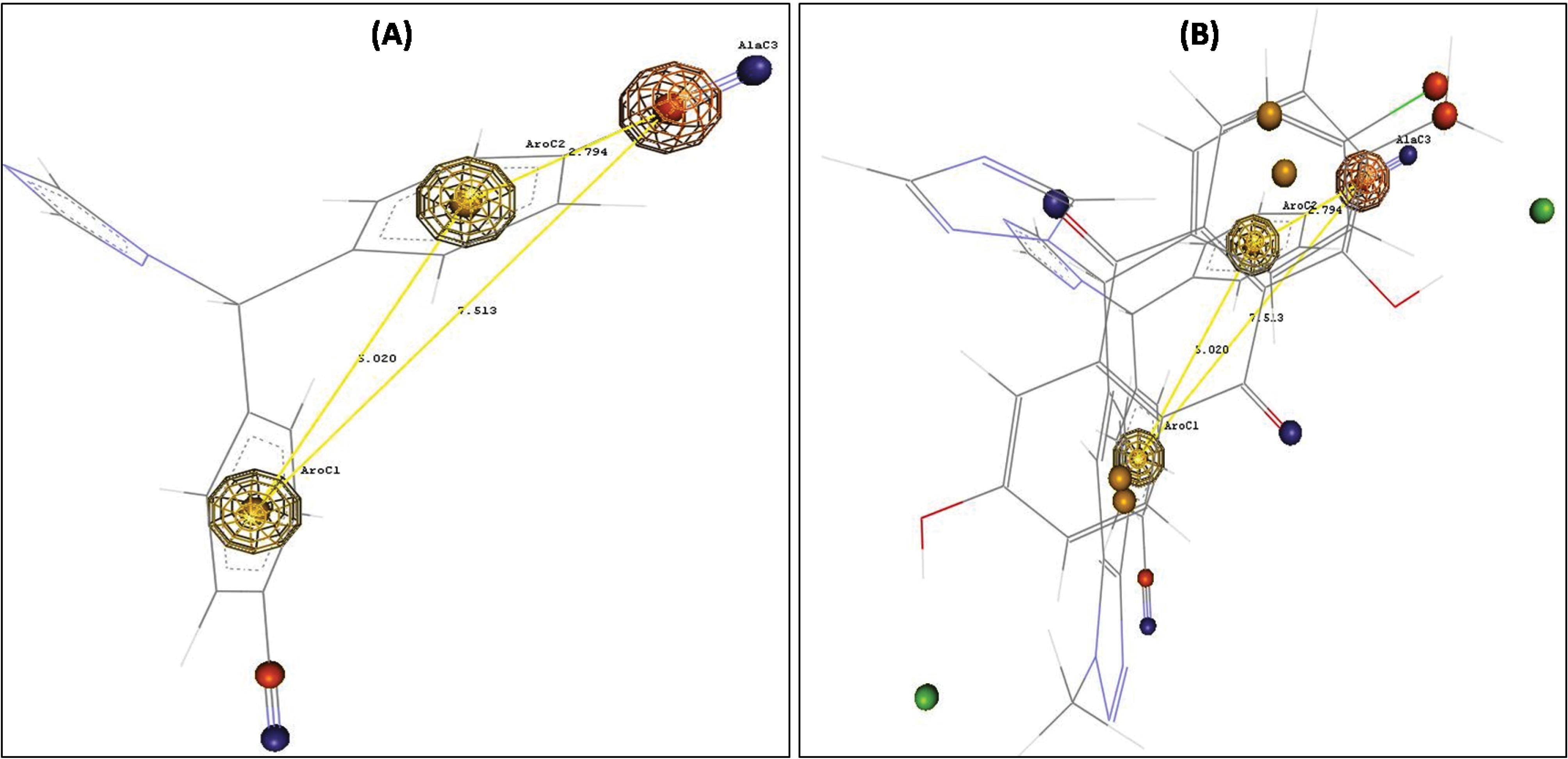
Fig. 7.
Pharmcophore model with three pharmacophoric features: (A) Reference compound –Letrozole (B) Flavonoids aligned on the reference compound
.
Pharmcophore model with three pharmacophoric features: (A) Reference compound –Letrozole (B) Flavonoids aligned on the reference compound
Discussion
Using the BioPredicta module of VLifeMDS, 6 cavities were found to be present in the receptor domain, among which due to larger hydrophobicity surface area (13054.52) and presence of amino acid residues of reference ligand, cavity 1 was selected as binding cavity for the study.
In the aromatase active site (PDB id: 3S7S), PHE134, PHE221, and TRP224 amino acid residues were found to be responsible for aromatic or π-stacking interactions, and hydrogen bonding interactions were observed prominently with ARG115 and MET374 amino acid residues. It should be noted that the amino acids present in the 3S7S active site, those interacting in hydrogen bond interactions and π-stacking usually serve as important for aromatase inhibitory activity.
In the present study, the leads were obtained from a chemically diverse set of natural actives targeting aromatase for ER+ breast cancer, by comparing the binding energy scores and the interactions with clinically used AIs in breast cancer therapy. In this pursuit, 23 hit compounds were obtained from virtual screening which had passed the selection criteria of a cut-off docking score of -40.
To validate the obtained virtual screening results, the amino acid residues involved in interaction with clinically used aromatase inhibitors, viz., letrozole, anastrozole, vorozole and fadrozole were studied and the hydrogen bond and π-stacking interactions was observed with amino acid residues in the binding cavity. This difference in the interactions among the clinically used AIs may be due to the presence of imidazole as a base nucleus in fadrozole, while others contained the triazole as a base nucleus. Furthermore, two triazole AIs letrozole and anastrozole showed aromatic interaction with TRP224 with a distance of 5.247 and 5.179 Å, respectively. But fadrozole showed a distance of 5.328 Å which is slightly longer than those with triazole nuclei and an absence of interaction with PHE134 or PHE224. On the other hand, exemestane exhibited a hydrogen bond interaction with ARG115 with a distance of 1.719 Å. The most notable interaction observed with non-steroidal AIs was the hydrogen bond interaction with amino acid residue MET374, which was absent in case of exemestane, a steroidal aromatase inhibitor.
The amino acid interactions of obtained 23 hits were compared with the interactions of standard aromatase inhibitors and the natural product hit compounds are categorized into the two different classes, viz., natural product as a hit compounds fit into aromatase inhibitors class and natural product hit compounds belongs to chemical structure modification class. Isoquercetin and honokiol showed similarity of interactions with anastrozole and thus was found to putatively target aromatase enzyme like anastrozole. On the other hand, 8-Prenylnaringenin, cimicifugic acid G, epigallocatechin 3-gallate, physcion, and rhein showed exactly similar interactions with the clinically used imidazole aromatase inhibitor-fadrozole. Among these five hits, 8-prenylnaringenin and epigallocatechin-3-gallate belong to the flavonoid class while physcion and rhein belong to the anthraquinone class.35,40-42 The results indicate that the structural requirements needed to be an AI include a six-membered ring with a carbonyl group and hydroxyl group, although this needs wet-lab experimental confirmation.
The hit compounds that showed the interactions with the aromatase-binding cavity, but not with exact similarity to the interactions of standard drugs were categorized in the natural product hit compounds belonging to the chemical structure modification class. These included 11 hits with benzo-gamma-pyrone (flavonoid), 03 with quinone nucleus and 2 hydroxy-derivative compounds. Park et al isolated anthraquinones from Ventilago denticulate and found that that emodin showed a good aromatase inhibitory activity, but chrysophanol and physcion were devoid of the activity.43,44 In line with these reports, our findings suggest that 1, 8,-dihdroxy-anthraquinones45,46 with minor modifications to their structure would be potential AIs. Thus, the natural products containing flavonoid nucleus, anthraquinone, or the benzopyran or chromone nucleus with hydroxyl and acidic groups in their structures may putatively act as potent AIs with some structural modification.
QSAR study provided the structural insights required for aromatase inhibitory activity in flavonoid nucleus. In the 2D QSAR studies, the negative values of SAMostHydrophobic and SAMostHydrophilic descriptors implies that a lower electrostatic potential in the vdW surface area of the molecule and an optimum hydrophilic and hydrophobic value on the surface of the molecule are important for aromatase inhibitory activity. The positive coefficient of Quadrupole3 showed that an increase in the values of this descriptor was beneficial for aromatase inhibitory activity. The 3D QSAR showed that presence of steric descriptors was not favourable for aromatase inhibitory activity, while the presence of an electrostatic descriptor could increase the aromatase inhibitory activity.
The performed MD simulation study unveiled a dynamic tapestry of molecular interactions, elucidating the temporal dimensions of protein-ligand engagement and illuminating the potential for functional modulation. These insights reverberate across diverse scientific disciplines, transcending the realm of ligand-receptor interactions. By influencing drug discovery strategies, aiding protein-engineering endeavours, and enhancing the understanding of biophysical phenomena, MD simulations have solidified their status as invaluable tools in unravelling the intricate fabric of the molecular landscape. The docking study's outcomes served as a stepping-stone, highlighting the promising interactions of honokiol, isoquercitrin, and epicatechin with human placental aromatase. However, the subsequent MD simulations unearthed the dynamic orchestration of these interactions, transcending the static docking paradigm and affording a dynamic comprehension. Through the lens of RMSD, RMSF, protein-ligand contacts, post-MD binding interactions, ligand torsion profiles, and ligand properties, a comprehensive view emerged, solidifying the potency of MD simulations in deciphering the nuanced dynamics underlying ligand-receptor interactions. This study underscores the pivotal role of computational methodologies in unveiling the intricacies of molecular behavior, accentuating their transformative influence across scientific domains.
The MMGBSA calculations provided comprehensive insights into the binding energetics of the studied complexes. The results suggest that 3S7S-isoquercitine forms the most energetically favorable complex, highlighting its potential as a strong ligand candidate. These findings have implications for drug design efforts, guiding the selection and optimization of ligands with enhanced binding affinities and tailored interactions for targeted therapeutic applications. Pharmacophore modeling was performed to get pharmacophoric features present in molecule for aromatase inhibitory activity. The results indicate that the minimum structural features for the aromatase inhibitory activity included three aromatic centres, one aliphatic centre and an additional aliphatic carbon centre. Using this approach, the modifications of the functional groups suggested from the pharmacophoric model can pave a way to increase the aromatase inhibitory potential of the generated leads.
Conclusion
The present study was aimed to develop new leads as aromatase enzyme inhibitors using a computational approach from a set of structurally diverse natural products reported to inhibit breast cancer. This was done by assessment of the docking scores, followed by analysing the interactions with the drugs that are clinically used in the treatment of breast cancer. Using virtual screening, 23 natural products were obtained as hits, and the comparative study of clinically used drugs revealed the putative classification of the obtained hits into aromatase inhibitors and inhibitors that require a structural modification. The results indicate that molecular docking could be reliably used to predict and understand the binding modes of the chemically diverse compounds. In addition, validation of docked complex using the MD simulation indicated that all stimulated complex systems exhibited a conformational stability under the mobile condition with minimum deviations. To supplement this data, we established the pharmacophore modelling for letrozole to obtain the insights required for the generation of new leads targeting aromatase. Besides, the hits obtained after a prior screening from traditional systems of medicine shows that these alternative systems of medicine could be potential contributors as lead compounds in the drug discovery process. In conclusion, we successfully employed a virtual screening protocol along with a structure-based drug design approach for the process of generating new aromatase-modulating natural products in the treatment of breast cancer.
Research Highlights
What is the current knowledge?
√ More than 70% of women suffer from breast cancer mainly due to over-expression of hormone estrogen.
√ Targeting the aromatase enzyme can help to block estrogen production and thereby can be used as therapy in breast cancer.
What is new here?
√ Computational studies assist the identification of natural actives from the structurally diverse set of natural products targeting the aromatase enzyme.
√ QSAR studies provided the structural functionalities needed to improve the aromatase inhibitory potential of the screened natural products.
√ Pharmacophore modelling highlighted the pharmacophoric features of the natural product scaffolds needed to develop new drugs with potential aromatase inhibitory activity.
Acknowledgements
The authors are thankful to Dr. H. N. More, Principal, Bharati Vidyapeeth College of Pharmacy for providing the necessary laboratory facilities.
Competing Interests
There are no competing interests to declare.
Ethical Statement
No animal studies involved in this research.
Supplementary files
Supplementary file 1 contains Tables S1-S2 and Figs. S1-S4.
(pdf)
References
- Khan SI, Zhao J, Khan IA, Walker LA, Dasmahapatra AK. Potential utility of natural products as regulators.pdf. Reprod Biol Endocrinol 2011; 9:91. doi: 10.1186/1477-7827-9-91 [Crossref] [ Google Scholar]
- Aggarwal S, Verma SS, Aggarwal S, Gupta SC. Drug repurposing for breast cancer therapy: Old weapon for new battle. Semin Cancer Biol 2021; 68:8-20. doi: 10.1016/j.semcancer.2019.09.012 [Crossref] [ Google Scholar]
- Awasthi M, Singh S, Pandey VP, Dwivedi UN. Molecular docking and 3D-QSAR-based virtual screening of flavonoids as potential aromatase inhibitors against estrogen-dependent breast cancer. J Biomol Struct Dyn 2015. 33: 804–819. 10.1080/07391102.2014.912152.
- Ashtekar SS, Bhatia NM, Bhatia MS. Development of leads targeting ER-α in breast cancer: An in silico exploration from natural domain. Steroids 2018; 131:14-22. doi: 10.1016/j.steroids.2017.12.016 [Crossref] [ Google Scholar]
- Suvannang N, Nantasenamat C, Isarankura-Na-Ayudhya C, Prachayasittikul V. Molecular docking of aromatase inhibitors. Molecules 2011; 16:3597-617. doi: 10.3390/molecules16053597 [Crossref] [ Google Scholar]
- Campbell DR, Kurzer MS. Flavonoid inhibition of aromatase enzyme activity in human preadipocytes. J Steroid Biochem Mol Biol 1993; 46:381-8. doi: 10.1016/0960-0760(93)90228-O [Crossref] [ Google Scholar]
- Enriori CL, Reforzo-Membrives J. Peripheral aromatization as a risk factor for breast and endometrial cancer in postmenopausal women: A review. Gynecol Oncol 1984; 17:1-21. doi: 10.1016/0090-8258(84)90055-6 [Crossref] [ Google Scholar]
- Ibrahim AR, Abul-Hajj YJ. Aromatase inhibition by flavonoids. J Steroid Biochem Mol Biol 1990; 49:257-60. doi: 10.1016/0960-0760(90)90335-I [Crossref] [ Google Scholar]
- Ashtekar SS, Bhatia NM. Synthesis of benzopyrans and evaluation of cytotoxicity against ER-MCF-7 cell lines. J Mol Struct 2022; 1268:133687. doi: 10.1016/j.molstruc.2022.133687 [Crossref] [ Google Scholar]
- Wang C, Mäkelä T, Hase T, Adlercreutz H, Kurzer MS. Lignans and flavonoids inhibit aromatase enzyme in human preadipocytes. J Steroid Biochem Mol Biol 1994; 50:205-212. doi: 10.1016/0960-0760(94)90030-2 [Crossref] [ Google Scholar]
- Khan SI, Zhao J, Khan IA, Walker LA, Dasmahapatra AK. Potential utility of natural products as regulators of breast cancer-associated aromatase promoters. Reprod Biol Endocrinol 2011; 9:91. doi: 10.1186/1477-7827-9-91 [Crossref] [ Google Scholar]
- Younas M, Hano C, Giglioli-Guivarc'h N, Abbasi BH. Mechanistic evaluation of phytochemicals in breast cancer remedy: current understanding and future perspectives. RSC Adv 2018; 8:29714. doi: 10.1039/c8ra04879g [Crossref] [ Google Scholar]
- Islam MR, Islam F, Nafady MH, Akter M, Mitra S, Das R. Natural Small Molecules in Breast Cancer Treatment: Understandings from a Therapeutic Viewpoint. Molecules 2022; 27:2165. [ Google Scholar]
- Peng B, Zhang S, Chan K, Zhong ZF, Wang YT. Novel Anti-Cancer Products Targeting AMPK: Natural Herbal Medicine against Breast Cancer. Molecules 2023; 28:740. doi: 10.3390/molecules28020740 [Crossref] [ Google Scholar]
- Balunas MJ, Su B, Brueggemeier RW, Kinghorn AD, Natural Products as Aromatase Inhibitors. Anticancer Agents Med Chem 2008; 8: 646-82.
- Duan N, Hu X, Zhou R, Li Y, Wu W, Liu N. A Review on Dietary Flavonoids as Modulators of the Tumor Microenvironment. Mol Nutr Food Res 2023; 25:e2200435. doi: 10.1002/mnfr.202200435 [Crossref] [ Google Scholar]
- Ateba SB, Mvondo MA, Ngeu ST, Tchoumtchoua J, Awounfack CF, Njamen D, Krenn L. Natural Terpenoids Against Female Breast Cancer: A 5-year Recent Research. Curr Med Chem 2018; 25:3162-213. doi: 10.2174/0929867325666180214110932 [Crossref] [ Google Scholar]
- Hu Z, Pan J, Wang J, Pei Y, Zhou R. Current research status of alkaloids against breast cancer. Chin J Physiol 2022; 65:12-20. doi: 10.4103/cjp.cjp_89_21 [Crossref] [ Google Scholar]
- Luo H, Vong CT, Chen H, Gao Y, Lyu P, Qiu L, et al. Naturally occurring anti-cancer compounds: shining from Chinese herbal medicine. Chin Med 2019;14: 48. Https://doi.org/10.1186/s13020-019-0270-9.
- Ashtekar SS, Bhatia NM, Bhatia MS. Exploration of Leads from Natural Domain Targeting HER2 in Breast Cancer: An In-Silico Approach. Int J Pept Res Ther 2019; 25:659-667. doi: 10.1007/s10989-018-9712-y [Crossref] [ Google Scholar]
- Dey S, Pratibha M, Singh Dagur H, Rajakumara E. Characterization of host receptor interaction with envelop protein of Kyasanur forest disease virus and predicting suitable epitopes for vaccine candidate. J Biomol Struct Dyn 2023; 5:1-11. doi: 10.1080/07391102.2023.2218924 [Crossref] [ Google Scholar]
- Gopinath P, Kathiravan MK. Docking studies and molecular dynamics simulation of triazole benzene sulfonamide derivatives with human carbonic anhydrase IX inhibition activity. RSC Adv 2021; 11:38079-93. doi: 10.1039/d1ra07377j [Crossref] [ Google Scholar]
- Al-Karmalawy AA, Alnajjar R, Dahabd MM, Metwaly AM, Eissa IH. Molecular docking and dynamics simulations reveal the potential of Anti-HCV drugs to inhibit COVID-19 main protease. Pharm Sci 2021; 27:S109-21. doi: 10.34172/PS.2021.3 [Crossref] [ Google Scholar]
- Thangavel M, Chandramohan V, Shankaraiah LH, Jayaraj RL, Poomani K, Magudeeswaran S. Design and Molecular dynamic Investigations of 7,8-Dihydroxyflavone Derivatives as Potential Neuroprotective Agents Against Alpha-synuclein. Sci Rep 2020; 10:559. doi: 10.1038/s41598-020-57417-9 [Crossref] [ Google Scholar]
- Rodrigues FC, Hari G, Pai KSR, Suresh A, Nayak UY, Anilkumar NV. Molecular modeling piloted analysis for semicarbazone derivative of curcumin as a potent Abl-kinase inhibitor targeting colon cancer. Biotech 2021; 11:506. doi: 10.1007/s13205-021-03051-9 [Crossref] [ Google Scholar]
- Jiang Z, You L, Dou W, Sun T, Xu P. Effects of an electric field on the conformational 602 transition of the protein: A molecular dynamics simulation study. Polymers (Basel) 2019; 11:282. doi: 10.3390/polym11020282 [Crossref] [ Google Scholar]
- Shivanika C, Deepak Kumar S, Ragunathan V, Tiwari P, Sumitha A, Brindha Devi P. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J Biomol Struct Dyn 2022; 40:585-611. doi: 10.1080/07391102.2020.1815584 [Crossref] [ Google Scholar]
- Hussein ME, Mohamed OG, El-Fishawy AM, El-Askary HI, Hamed AA, Abdel-Aziz MM. Anticholinesterase Activity of Budmunchiamine Alkaloids Revealed by Comparative Chemical Profiling of Two Albizia spp, Molecular Docking and Dynamic Studies. Plants 2022; 11:3286. doi: 10.3390/plants11233286 [Crossref] [ Google Scholar]
- Chaudhari AM, Kumar D, Joshi M, Patel A, Joshi C. E156G and Arg158, Phe-157/del mutation in NTD of spike protein in B16172 lineage of SARS-CoV-2 leads to immune evasion through antibody escape. bioRxiv 2021; 21:447321. doi: 10.1101/2021.06.07.447321 [Crossref] [ Google Scholar]
- Alnajjar R, Mostafa A, Kandeil A, Al-Karmalawy AA. Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease. Heliyon 2020; 12:E05641. doi: 10.1016/j.heliyon.2020.e05641 [Crossref] [ Google Scholar]
- Hamed ANE, Abouelela ME, El Zowalaty AE, Badr MM, Abdelkader MSA. Chemical constituents from Carica papaya Linnleaves as potential cytotoxic, EGFRwt and aromatase (CYP19A) inhibitors; a study supported by molecular docking. RSC Adv 2022; 12:9154-62. doi: 10.1039/d1ra07000b [Crossref] [ Google Scholar]
- Rauf A, Patel S, Imran M, Aneela Maalik, Arshad MU, Saeed F, Mabkhot YN, Al-Showiman SS, Ahmad N, Elsharkawy E. Honokiol: An anticancer lignan. Biomed Pharmacother 2018; 107:555-62. doi: 10.1016/j.biopha.2018.08.054 [Crossref] [ Google Scholar]
- Hong Y, Chen S. Aromatase inhibitors: Structural features and biochemical characterization. Ann N Y Acad Sci 2006; 1089:237-51. doi: 10.1196/annals.1386.022 [Crossref] [ Google Scholar]
- Le Bail JC, Varnat F, Nicolas JC, Habrioux G. Estrogenic and antiproliferative activities on MCF-7 human breast cancer cells by flavonoids. Cancer Lett 1998; 130:209-16. doi: 10.1016/S0304-3835(98)00141-4 [Crossref] [ Google Scholar]
- Monteiro R, Faria A, Azevedo I, Calhau C. Modulation of breast cancer cell survival by aromatase inhibiting hop (Humulus lupulus L) flavonoids. J Steroid Biochem Mol Biol 2007; 105:124-30. doi: 10.1016/j.jsbmb.2006.11.026 [Crossref] [ Google Scholar]
- Jeong HJ, Shin YG, Kim IH, Pezzuto JM. Inhibition of aromatase activity by flavonoids. Arch Pharm Res 1999; 22:309-12. doi: 10.1007/BF02976369 [Crossref] [ Google Scholar]
- Lee D, Bhat KPL, Fong HHS, Farnsworth NR, Pezzuto JM, Kinghorn AD. Aromatase Inhibitors from Broussonetia papyrifera. J Nat Prod 2001; 64:1286-93. doi: 10.1021/np010288l [Crossref] [ Google Scholar]
- Pouget C, Fagnere C, Basly JP, Besson AE, Champavier Y, Habrioux G, Chulia AJ. Synthesis and aromatase inhibitory activity of flavanones. Pharm Res 2002; 19:286-291. doi: 10.1023/A:1014490817731 [Crossref] [ Google Scholar]
- Khatabi K, Aanouz I, Alaqarbeh M, Ajana M, Lakhlifi1 T, Bouachrine M. Molecular docking, molecular dynamics simulation, and ADMET analysis of levamisole derivatives against the SARS-CoV-2 main protease (MPro). Bioimpacts 2022; 12:107-113. doi: 10.34172/bi.2021.22143 [Crossref] [ Google Scholar]
- Mojaddami A, Sakhteman A, Fereidoonnezhad M, Faghih Z, Najdian A, Khabnadideh S, Sadeghpour H, Rezaei Z. Binding mode of triazole derivatives as aromatase inhibitors based on docking, protein ligand interaction fingerprinting, and molecular dynamics simulation studies. Res Pharm Sci 2017; 12:21-30. doi: 10.4103/1735-5362.199043 [Crossref] [ Google Scholar]
- Bolton JL, Dunlap TL, Hajirahimkhan A, Mbachu O, Chen SN, Chadwick L. The Multiple Biological Targets of Hops and Bioactive Compounds. Chem Res Toxicol 2019; 32:222-233. doi: 10.1021/acs.chemrestox.8b00345 [Crossref] [ Google Scholar]
- Satoh K, Sakamoto Y, Ogata A, Nagai F, Mikuriya H, Numazawa M. Inhibition of aromatase activity by green tea extract catechins and their endocrinological effects of oral administration in rats. Food Chem Toxicol 2002; 40:925-33. [ Google Scholar]
- Kim YG, Park YH, Yang EY, Park WS, Park KS. Inhibition of tamoxifen’s therapeutic effects by emodin in estrogen receptor-positive breast cancer cell lines. Ann Surg Treat Res 2019; 97:230-8. doi: 10.4174/astr.2019.97.5.230 [Crossref] [ Google Scholar]
- Deborah R. Campbell, Mindy S. Kurzer, Flavonoid inhibition of aromatase enzyme activity in human preadipocytes, The Journal of Steroid Biochemistry and Molecular Biology 1993; 46:381-8. doi: 10.1016/0960-0760(93)90228-O [Crossref] [ Google Scholar]
- Alhadrami HA, Abdulaal WH, Hassan HM, Alhakamy NA, Sayed AM. In Silico-Based Discovery of Natural Anthraquinones with Potential against Multidrug-Resistant Ecoli. Pharmaceuticals (Basel) 2022; 15:86. doi: 10.3390/ph15010086 [Crossref] [ Google Scholar]
- Balunas MJ, Su B, Brueggemeier RW, Kinghorn AD. Natural products as aromatase inhibitors. Anticancer Agents Med Chem 2008; 8:646-82. [ Google Scholar]