Bioimpacts. 14(4):29951.
doi: 10.34172/bi.2023.29951
Original Article
Thiazolo-pyridopyrimidines: An in silico evaluation as a lead for CDK4/6 inhibition, synthesis and cytotoxicity screening against breast cancer cell lines
Chaithra R Shetty Conceptualization, Formal analysis, Funding acquisition, Methodology, Validation, Visualization, Writing – original draft, 1 
C. S. Shastry Conceptualization, Funding acquisition, Project administration, Supervision, Visualization, Writing – review & editing, 2, *
Parasuraman P Data curation, Methodology, Software, 3
Srinivas Hebbar Investigation, Resources, 4
Author information:
1Nitte Deemed to be University, NGSM Institute of Pharmaceutical Sciences, Department of Pharmaceutical Chemistry, Deralakatte, Mangaluru, Karnataka, India, 575018
2Nitte Deemed to be University, NGSM Institute of Pharmaceutical Sciences, Department of Pharmacology, Deralakatte, Mangaluru, Karnataka, India, 575018
3Department of Pharmaceutical Chemistry, Faculty of Pharmacy, M S Ramaiah University of Applied Sciences, Bengaluru, Karnataka, India, 560054
4Pharmaceutics Department, Manipal College of Pharmaceutical Sciences, MAHE, Manipal, Karnataka, India, 576104
Abstract
Introduction:
Pyridopyrimidines belong to a class of compounds characterized by the presence of nitrogen as heteroatoms. These compounds exhibit diverse biological effects, particularly showing promise as anticancer agents, including actions that inhibit CDK4/6.
Methods:
We designed and synthesized a range of substituted thiazolo-pyridopyrimidines (4a-p). Computational ADME/T analysis and molecular docking were performed using the crystal structure of CDK4/6. Subsequently, we synthesized the top-scoring compounds, characterized them using IR, NMR, and Mass spectroscopy, and assessed their impact on MCF-7 and MDAMB-231 cell lines using the SRB assay. To further evaluate stability, molecular dynamics simulations were conducted for the two most promising compounds within the binding site.
Results:
The docking scores indicated stronger interactions for compounds 4a, 4c, 4d, and 4g. As a result, these specific compounds (4a, 4c, 4d, and 4g) were chosen for synthesis and subsequent screening to assess their cytotoxic effects. Remarkably, compounds 4c and 4a exhibited the most promising activity in terms of their IC50 values across both tested cell lines. Furthermore, molecular dynamics simulation studies uncovered an elevated level of stability within the 4c-6OQO complex.
Conclusion:
By integrating insights from computational, in vitro, and molecular dynamics simulation findings, compound 4c emerges as a leading candidate for future investigations. The presence of a polar hydroxyl group at the C2 position of the 8-phenyl substitution on the pyridopyrimidine rings appears to contribute to the heightened activity of the compound. Further enhancements to cytotoxic potential could be achieved through structural refinements.
Keywords: Thiazolo-pyridopyrimidines, CDK4/6, Molecular docking, Cytotoxicity, MD simulation
Copyright and License Information
© 2024 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Pyridopyrimidines belong to the category of heterocyclic compounds that showcase a diverse range of biological and pharmacological effects. These effects encompass antimicrobial, antiparasitic, anticoagulant, anti-inflammatory, antiallergic, potassium-suppressing, analgesic, diuretic, anti-aggressive, antihypertensive, antifolate, anticonvulsant, and anticancer activities. Pyrido[2,3-d]pyrimidines have been documented to exhibit antitumor characteristics, potentially stemming from their ability to inhibit various enzymes implicated in carcinogenesis pathways. Most of the pyrido[2,3-d]pyrimidine series demonstrated robust inhibitory activity against a range of kinases, especially CDK4/6.1–10
Heterocyclic structures based on thiazole have demonstrated a broad spectrum of biological activities, establishing them as a prominent class of heterocycles widely employed in drug design and synthetic chemistry. The subgroup of 1,3-thiazoles is not only found in animal cells but also constitutes a fundamental framework in numerous natural products, including vitamins, alkaloids, and pigments. Additionally, they play a role in the structure of 18 clinically approved drugs sanctioned by the FDA, among which are two antitumor drugs—epothilone and tiazofurin.11
Many diseases are prevented by inhibiting many of the existing enzymes in living organisms. Kinases are the enzymes, which transfer phosphate groups to proteins, whereas phosphatases pull them out. In a cell, the above enzymes work together to regulate the activity of different proteins, often in response to an external stimuli/factor. The human genome contains approximately 538 identified kinases, which is responsible for maintaining the cellular activity by turning protein function on, whereas related phosphatases reverse the same.12–14
Significant gains in understanding the fundamental molecular mechanisms regulating cancer cell signalling have revealed that kinases play an important role in cancer carcinogenesis and metastasis including breast cancer.15,16 The cell cycle consists of G0/G1, S, G2, and M phases. G1 and G2 are key checkpoints regulated by cyclins and CDKs. Cyclins D1, D2, and D3 activate CDK4/6 in mid-G1. They phosphorylate and inactivate pRb, releasing E2F factors. This allows transcription of CDK2, E-cyclins, promoting S phase entry. D-type cyclin transcription and CDK4/6 activity link signaling pathways to cell cycle initiation. This pathway is often disrupted in cancers like breast cancer, where CDK4/6 drive proliferation in HR-positive breast cancer. The CDK (cyclin D-cyclin-dependent kinase) 4/6 i.e., CDK4/6 (INK4)-retinoblastoma (Rb) pathway regulates cell proliferation by monitoring the cell cycle checkpoint between the G1 (pre-DNA synthesis) and S (DNA synthesis) phases as shown in Fig. 1. Cyclin D-CDK4/6-INK4-Rb pathway is typically dysregulated in most of the cancer including breast cancer, and it contribute in cell cycle progression and proliferation. The shift from G1 to S phase by interacting with D-type cyclins and status of Rb phosphorylation is regulated by CDK4/6. Rb binds to E2 family (E2F) transcription factors and inhibits their function while unphosphorylated; when phosphorylated, Rb gets dissociate from transcription factors of E2F allowing them to engage in DNA replication and cell division. Increased expression of D-type cyclins, amplification or mutation of CDK4/6, or loss of negative regulators of cyclinD-CDK4/6 like p16INK4A can all lead to increased action of cyclinD-CDK4/6, which enhances Rb phosphorylation and eventually proliferation of breast cancer cells. The coordinated role of cyclin D and CDK4/6 in cell cycle regulation has made this axis an appealing target for therapeutic strategies. In this context, the discovery of CDK4/6 inhibitors stands out as particularly significant. These inhibitors have been remarkably intriguing due to their ability to exploit the vulnerability of cancer cells while also preserving a manageable level of toxicity.17,18
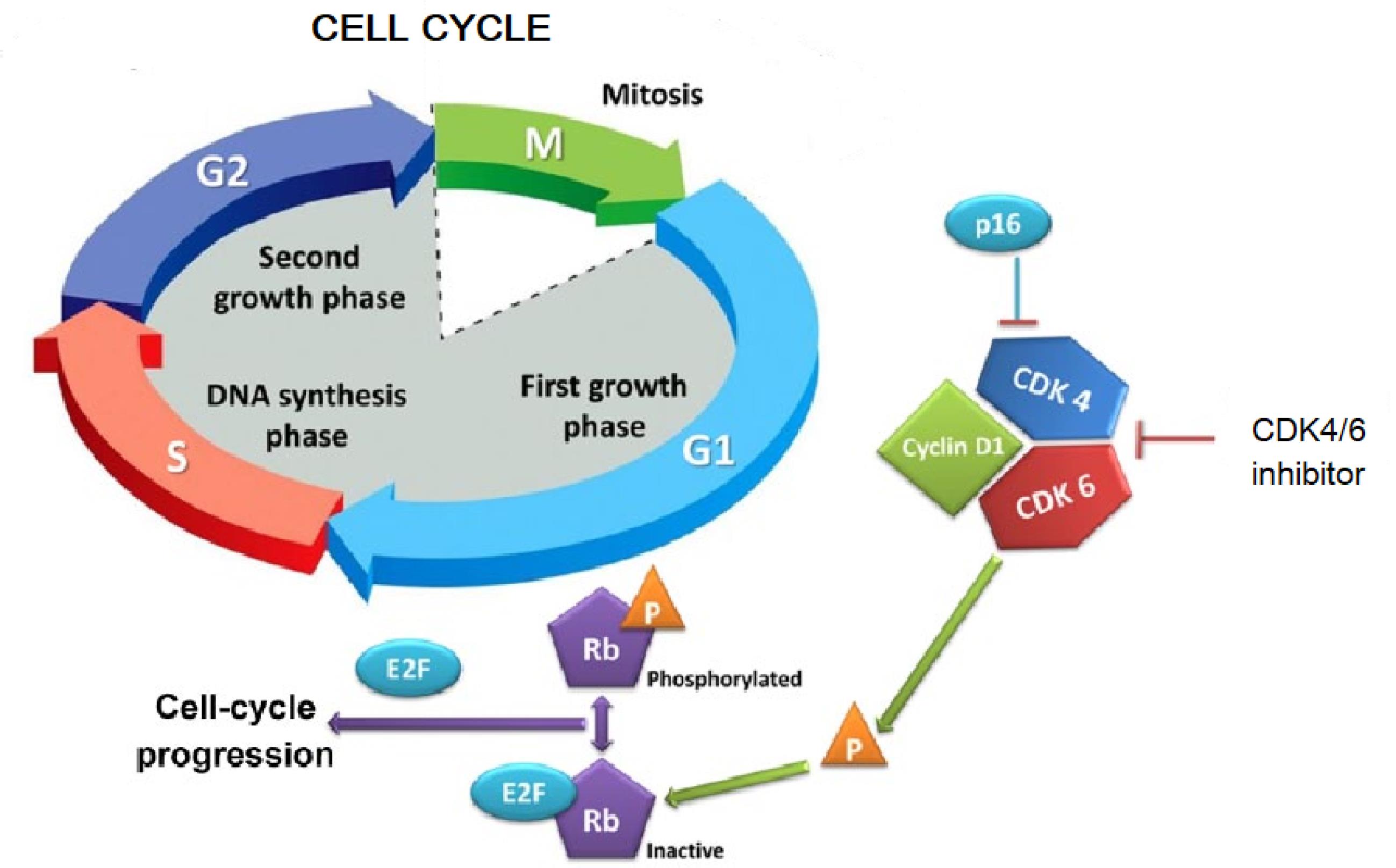
Fig. 1.
Cell cycle and action of CDK4/6 inhibitor.19
.
Cell cycle and action of CDK4/6 inhibitor.19
FDA granted the first CDK4/6 inhibitor i.e., Palbociclib (Fig. 2) which is a pyridopyrimidine derivative mainly to increase progression-free survival rate in patients with breast cancer when used along with fulvestrant or letrozole. Most of the pyrido[2,3-d] pyrimidine series demonstrated robust inhibitory activity against a range of kinases, especially CDK4/6. The Cyclin D-CDK4/6-INK4-Rb pathway dysregulation is common in breast cancer. Targeting this pathway holds promise for breast cancer treatment. Furthermore, existing literature substantiates the potential of CDK4/6 inhibition using diverse pyridopyrimidines. Hence, we wanted to investigate the interaction of our designed pyridopyrimidine compounds with CDK4/6 and explore their anticancer properties.20,21
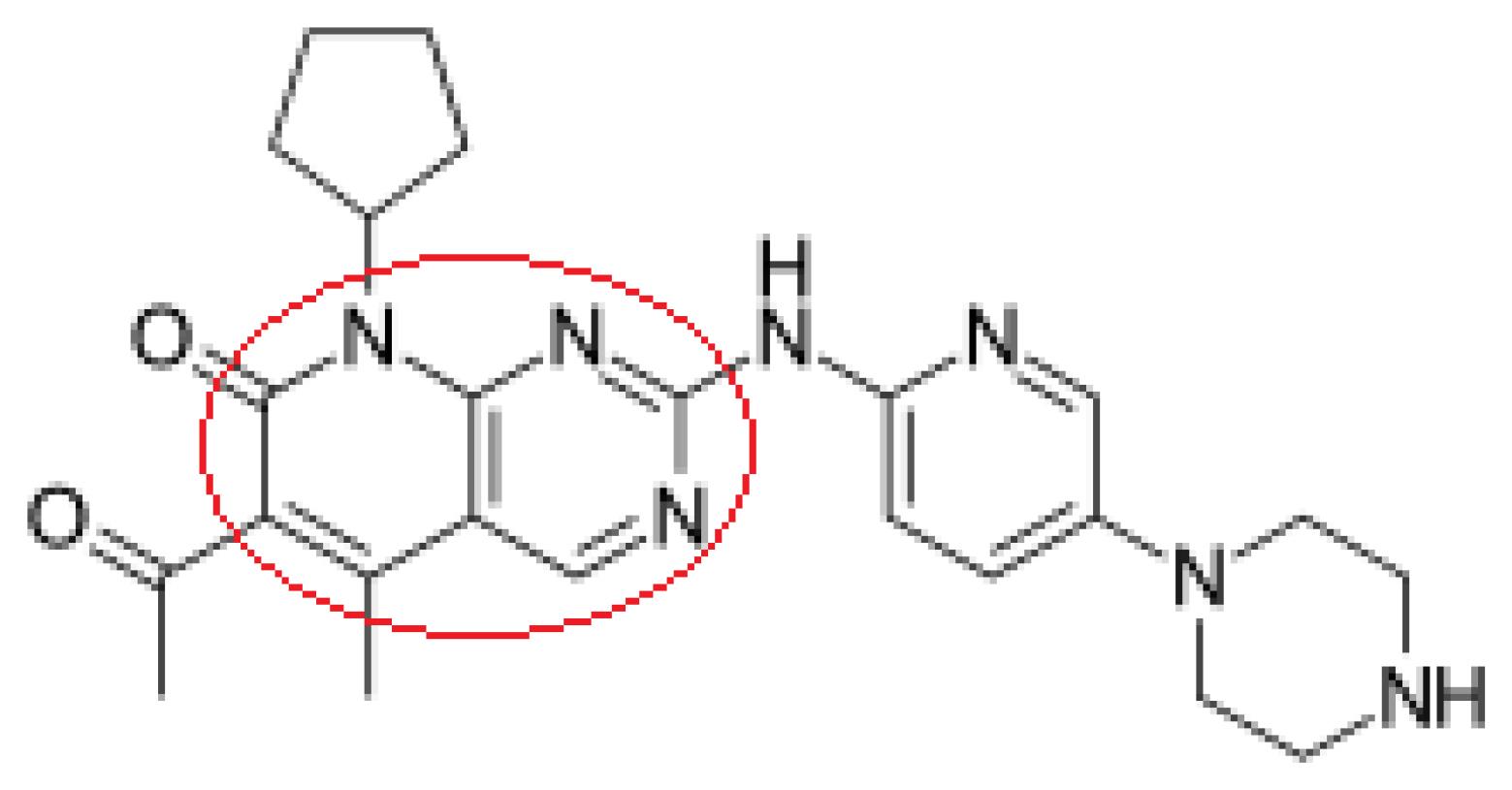
Fig. 2.
Structure of palbociclib.
.
Structure of palbociclib.
The variety of compounds synthesized based on multiple investigations with the aim of designing and developing new CDK4/6 inhibitors, such as 4-thiazol-N-(pyridine-2-yl) pyrimidin-2-amine, 7-azabenzimidazoles, piperidine sulfonamide derivatives and N-(pyridin-2-yl)-4-(thiazol-5-yl) pyrimidine-2-amines.22–25
In the process of discovery and development of a drug molecule, computational methods are often used to analyze and design new drug candidates for different targets. In silico screening is carried out to find a hit compounds sequence from a database using a precise and organized process which performs a vital function in boosting the drug development efficiency. Combining structure-based and ligand-based techniques will almost certainly improve screening success.26,27
Drawing inspiration from the structural and pharmacological characteristics of the prior pyrido[2,3-d]pyrimidines and thiazoles, our objective was to synthesize a fresh series of pyrido[2,3-d]pyrimidinone derivatives integrated with a thiazole ring. This endeavor was undertaken with the goal of obtaining a compound of greater potency by mainly targeting CDK4/6. This study encompasses an initial screening of compounds using computational tools to assess their in silico ADME properties. Subsequently, molecular docking techniques were employed to identify potential novel compounds with CDK4/6 inhibitory potential. Compounds with the highest scores were selected for synthesis, characterization, and evaluation against breast cancer cell lines through in vitro cytotoxicity tests such as the SRB assay. To gain insights into the stability of proteins and ligand-protein complexes, molecular dynamics (MD) simulation studies were conducted on the two most promising compounds.28,29 The workflow's schematic representation is depicted in Fig. 3.
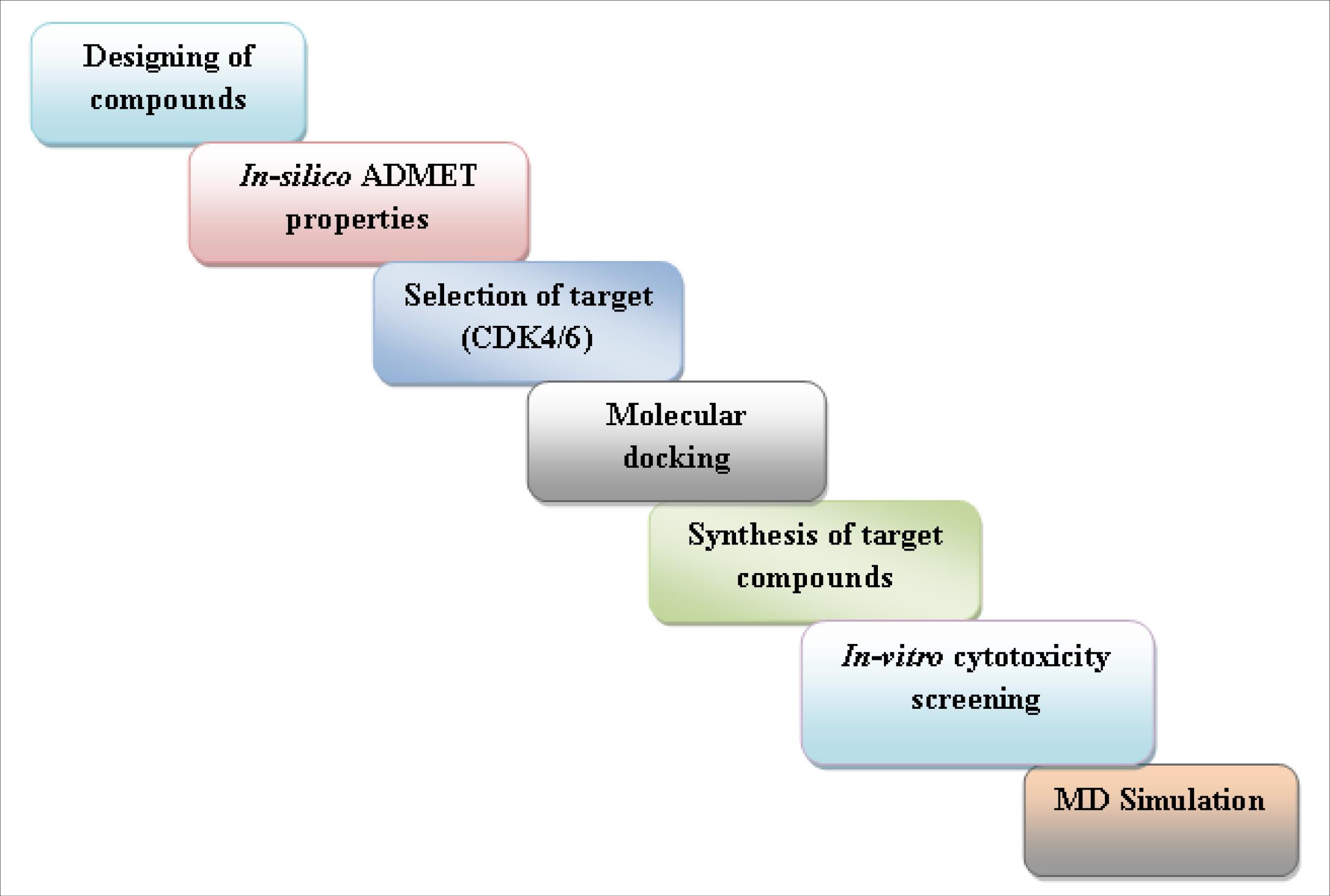
Fig. 3.
Schematic representation of workflow.
.
Schematic representation of workflow.
Methods
Designing of compounds
Lipinski’s rule of 5 determination for 4a-p
Lipinski's "Rule of 5" is a thumb rule for determining a chemical compound's drug likeness. It defines whether a compound has the chemical and physical properties that might render it a potentially effective drug in humans which is active orally. It states that majority of "drug-like" molecules have molecular weight <= 500, logP<= 5, number of hydrogen bond donors <= 5 and number of hydrogen bond acceptors <= 10. Bioavailability issues could be seen if compounds violate more than one of these rules.30 Molinspiration, a free online tool, is used to determine it.
Determination of physicochemical characteristics of 4a-p
Molecular polar surface Area tPSA
Properties of drug transport are explained by an extremely helpful parameter like tPSA. PSA is defined as the sum of the surfaces of polar atoms in a molecule (typically nitrogens, oxygens and attached hydrogens). tPSA has showed strong correlation with blood-brain barrier penetration, Caco-2 monolayer permeability and human intestinal absorption. It is determined as the sum of contributions of each fragment. O and N centered polar fragments are taken into consideration. PSA has been shown to be a highly accurate predictor of bioavailability, drug absorption including intestinal absorption, blood-brain barrier penetration and Caco-2 permeability.31
Number of rotatable bonds –nrotb
Molecular flexibility is quantified by the presence of number of rotatable bonds which is a simple and clear topological parameter. It has been demonstrated to be an excellent predictor of drug oral bioavailability. According to Veber's rule, as the number of rotatable bonds decreased (10 or fewer), we can see the greater oral bioavailability.31
Determination of ADME properties of 4a-p
The in silico ADME properties were evaluated by Qikprop (Schrodinger 2018-3 suite device Maestro 11.7.012).32,33
QPPCaco
Caco-2 cell apparent permeability was predicted in nanometers per second. Caco-2 cells are used to simulate the blood-gut barrier. The results obtained by QikProp are for passive transportation.
QPlogB
The brain/blood partition coefficient has been predicted.
QPPMDCK
Permeability of MDCK cells predicted in nanometers per second. MDCK cells are thought to be an excellent blood-brain barrier mimic. The predictions made by QikProp are for passive transportation.32,33
QPlogKp
Skin permeability predicted, log Kp.
QPlogKhsa
Prediction of binding to human serum albumin.
Human oral absorption
Qualitative human oral absorption predicted: 1, 2, or 3 for low, medium, or high. The evaluation employs a set of knowledge-based rules that include checking for appropriate values of percent human oral absorption.
Evaluation of toxicity of 4a-p
The free online tool admetSAR was used to evaluate in silico toxicity parameters.34
Molecular docking studies of 4a-p
The crystal structures of CDK4 (PDB ID: 2W96) and CDK6 (PDB ID: 6OQO) from the RCSB PDB were used for molecular docking studies. 2W96 is a CDK4 crystal structure with no native ligand (Resolution 2.30), and 6OQO is a CDK4 crystal structure with an active ligand (Resolution 1.98). All ligands and selected proteins were prepared using the Schrodinger 2018-3 suite device Maestro 11.7.012's ligprep and protein preparation wizard, respectively. Grid box was created by generating receptor grids. The same software was used for extra precision molecular docking.35-37
MM-GBSA free energy calculation
Estimation of binding energy of complex formed by the ligand and receptor using Molecular Mechanics-Generalised Born Surface Area (MM-GBSA) method developed by Miller and colleagues. Calculation of binding energy from MD simulation trajectories using the following formula:
∆Gbinding = RT ln Ki= Gcomplex- (Greceptor+Gligand)
The following is how the energy term is calculated:
G= Eele + Evdw + ESA+ EGB
The Electrostatic, Vander Waals, Surface area and Seneral Born solvation energies are denoted by Eele, Evdw, ESA and EGB respectively.38
Synthesis of substituted thiazolo -pyridopyrimidines
Synthesis of hydrazinylpyridpyrimidines (3a-p)
In 20 mL of dry DMF, 6-amino-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (0.01 mol) and α, β- unsaturated ketones (0.01 mol) were refluxed for 12-15 hours (completion of reaction was ensured by TLC). Once the reaction was completed, it was added to crushed ice. Filtered, washed with water and dried. Synthesized compounds were purified by recrystallization using DMF.39
Synthesis of thiazolo-pyridopyrimidines (4a-p)
Ten mmol of 2-thioxo derivatives (3a-p) was mixed with 10 mmol of chloroacetic acid and 20 mmol of sodium acetate in 20ml of glacial acetic acid. For 2-4 hours, the mixture was gently heated with stirring on a water bath (60 °C). After completion of the reaction, flask containing reaction mixture was cooled to room temperature and added to the beaker containing crushed ice, stirred well and resultant precipitate was filtered, washed and purified (Fig. 4).40
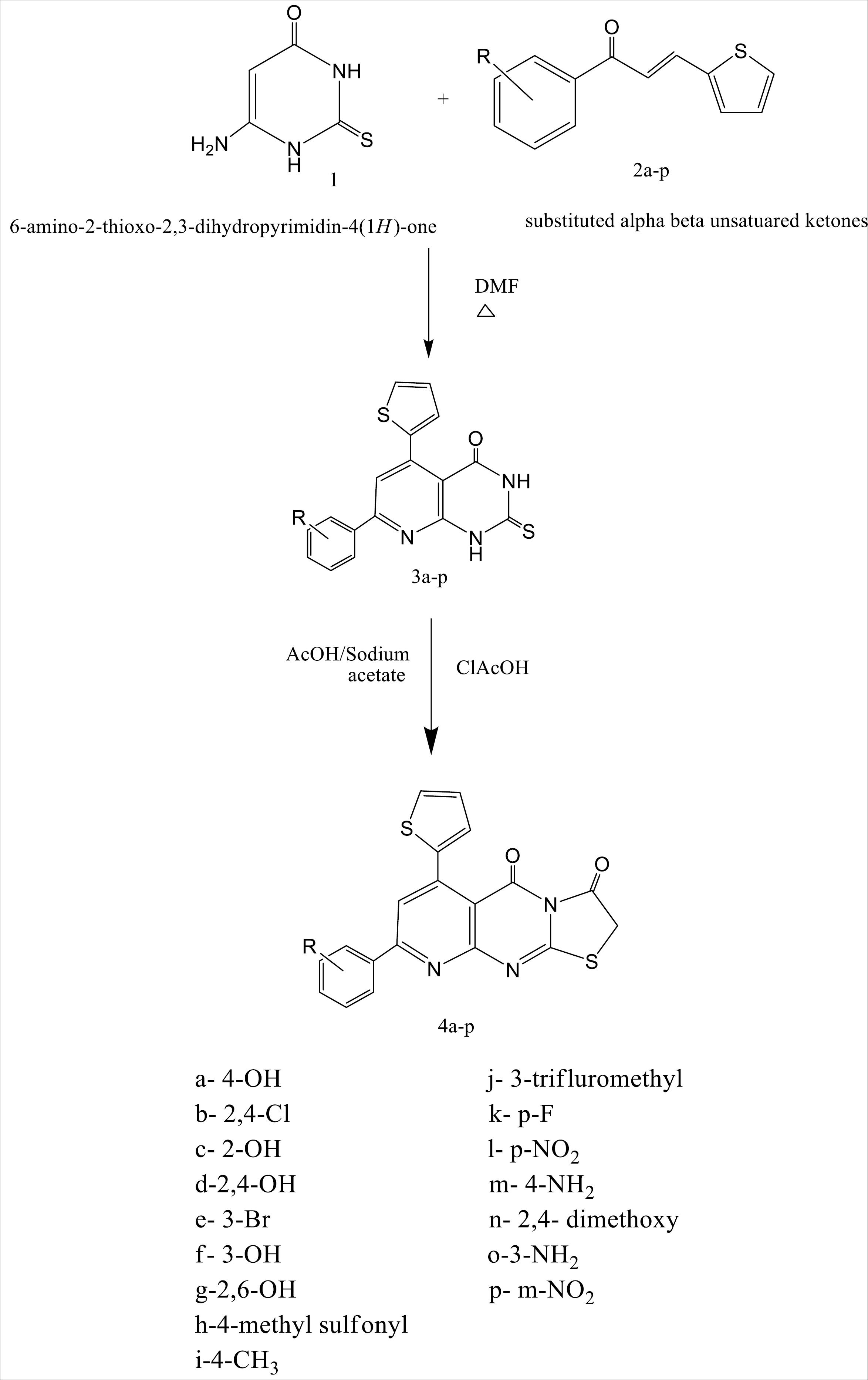
Fig. 4.
Schematic representation of route of synthesis of target thiazolo pyridopyrimidines.
.
Schematic representation of route of synthesis of target thiazolo pyridopyrimidines.
In vitro anticancer studies
Semi-automated in vitro assays using sulforhodamine-B were used to determine the cytotoxicity of the synthesized compounds. The human breast cancer cell lines (MCF-7 and MDAMB-231) have been grown in culture flasks at 37 °C in RPMI-1640 medium which is a complete growth medium with 2 mM glutamine, pH 7.4, with added 10% foetal bovine serum, streptomycin (100 g/mL), and penicillin (100 units/mL) in an incubator with 5% carbon dioxide and temperature maintained at 37 °C at 90% relative humidity. Flasks at the subconfluent growth stage were chosen, and cells were recovered using trypsin-EDTA. Cytotoxicity was evaluated using cells with more than 98% viability as found by trypan blue exclusion assay. A haemocytometer was used to count the number of cells per millilitre of suspension.41,42
SRB assay
Sulforhodamine B (SRB) assay is used to evaluate the antineoplastic properties of test compoundThis method is based on SRB's ability to attach to proteins stoichiometrically under slight acidic environment which can be then extracted in basic conditions; As a result, the quantity of bound dye is useful as an indicator for cell population, from which cell growth can be extrapolated. A haemocytometer was used to determine the number of cells per millilitre of suspension. The cell count was set to 10 000 cells per 100 μL. Each well of 96-well plates are placed with 100 μL of cell suspension. For 24 hours, the plates were left to incubate at 37 °C in a 5% CO2 atmosphere. Following that, 100 μL of test compounds at various concentrations was added to each of the wells containing cells.43,44
The plates were kept for incubation at 37 °C in 5% CO2 for 24 hours. To each well of 96 well plates 30 μL of chilled 30% TCA was added, and in order to fix the cells attached to the bottom of the wells plates were kept at 4°C for 1 hour incubation. After that, wells were washed with distilled water and air dried. 100 μL of SRB dye (0.4% w/v in 1% acetic acid) was added to each well and kept it at room temperature for 1h at dark. Thereafter, SRB was removed and wells were washed using 1% acetic acid and air dried. Further, each well was treated with 100 μL of Tris buffer (10 mM; pH 10.5) and kept for incubation for 30 min at room temperature. By using a microplate reader optical density (OD) of the plate wells was recorded at 540 nm to measure the OD of protein bound dye.45
The following formula is employed to calculate the percentage inhibition/ cytotoxicity
% cell cytotoxicity= Control OD- Sample OD/ Control OD *100
The concentration which can kill 50% of the cells (IC50) was determined using a plot of percentage cytotoxicity against concentration of sample.
Molecular dynamics (MD) simulations
This method is mainly based on the particle motion and distribution, and also on the dynamical response of the system, which is estimated using statistical mechanics and Newton's motion equations. Specific behavior of the macromolecules can be evaluated by this method in under various conditions.10
In MD simulation studies, the best configuration of the ligand-protein complexes obtained from molecular docking were used in order to investigate the stability, compactness and fluctuations of the protein-ligand complexes as demonstrated by molecular docking studies. For minimization and position restraint, each protein-ligand complex was embedded in an orthorhombic box using an aqueous model with a simple point charge (SPC) and an OPLS3e force field. A suitable number of counter-ions (Na+/Cl-) was added to neutralize the complexes. The simulation duration was set to 100 ns for each complex in the Normal pressure and temperature (NPT) ensemble.46,47
Results and Discussion
Based on the estimated values, the conclusion drawn was that the majority of the compounds conform to all five characteristics outlined in Lipinski's rule of five, as detailed in Table 1. The primary factors of significance are the molecular weight and log P, which exhibit a close association with passive intestinal absorption. The compounds' molecular weights were below 500 Daltons, and their Log P values ranged from 3.05 to 4.99, indicating that the majority of the compounds, with the exception of 4b and 4j (which recorded values of 5.49 and 5.08, respectively), are likely to exhibit favorable intestinal absorption and enhanced permeability across cell membranes. The consideration of absorption, distribution, metabolism, and excretion (ADME) characteristics is crucial, as lipophilicity predominantly determines the capacity to traverse critical biological barriers and membranes. Notably, substances with high lipophilicity often display poor solubility, affecting their bioavailability, or can accumulate in adipose tissues, potentially hindering excretion and thereby influencing systemic substance toxicity. A pivotal parameter indicating oral bioavailability is the total count of hydrogen bond donors and acceptors. The findings revealed that all screened compounds adhered to the permissible range of 5 hydrogen bond donors and 10 acceptors. The computed parameters of the tested compounds are in line with established standards.
Table 1.
In-silico evaluation of Lipinski’s rule of 5 and physicochemical characteristics of 4a-p
|
Compound
|
Mol. Wt
|
Log P
|
nON
|
nOHNH
|
nviolation
|
tPSA (<1402 Ǻ) |
nrotb (<10) |
| 4a |
393.434 |
3.73 |
6 |
1 |
0 |
85.09 |
2 |
| 4b |
446.325 |
5.49 |
5 |
0 |
1 |
64.86 |
2 |
| 4c |
393.434 |
3.94 |
6 |
1 |
0 |
85.09 |
2 |
| 4d |
409.434 |
3.44 |
7 |
2 |
0 |
105.32 |
2 |
| 4e |
456.331 |
4.99 |
5 |
0 |
0 |
64.86 |
2 |
| 4f |
393.434 |
3.71 |
6 |
1 |
0 |
85.09 |
2 |
| 4g |
409.434 |
3.67 |
7 |
2 |
0 |
105.32 |
2 |
| 4h |
455.52 |
3.05 |
7 |
0 |
0 |
99.00 |
3 |
| 4i |
391.462 |
4.66 |
5 |
0 |
0 |
64.86 |
2 |
| 4j |
445.433 |
5.08 |
5 |
0 |
1 |
64.86 |
3 |
| 4k |
395.425 |
4.37 |
5 |
0 |
0 |
64.86 |
2 |
| 4l |
422.432 |
4.17 |
8 |
0 |
0 |
110.68 |
3 |
| 4m |
392.449 |
3.28 |
6 |
2 |
0 |
90.88 |
2 |
| 4n |
437.487 |
4.25 |
7 |
0 |
0 |
83.33 |
4 |
| 4o |
392.449 |
3.26 |
6 |
2 |
0 |
90.88 |
2 |
| 4p |
422.432 |
4.14 |
8 |
0 |
0 |
110.68 |
3 |
| Palbociclib |
447.4 |
2.96 |
9 |
2 |
0 |
105.04 |
5 |
tPSA serves as a robust descriptor for factors encompassing bioavailability, BBB penetration, drug absorption, and Caco-2 permeability. Additionally, the propensity for hydrogen bonding can also be correlated with tPSA. The investigation revealed that the tPSA values spanned from 64.86 to 110.68 across all examined compounds, falling within the allowable limit of 140Å. The calculated values for all tested compounds align with established standards. Evaluation of the number of rotatable bonds within the screened compounds indicated a range of 2 to 4, rendering them moderately flexible compared to the reference standard, palbociclib, which features 5 rotatable bonds.
The estimated apparent Caco-2 cell permeability exhibited a spectrum ranging from excellent to satisfactory among the compounds. Notably, compounds 4b, 4e, 4i, 4j, 4k, and 4n displayed a likelihood of superior intestinal permeability compared to the standard. The remaining molecules were predicted to possess intestinal permeability equivalent to the reference standard. Forecasts for brain/blood partition coefficient values of all evaluated compounds adhered to the recommended range. With the exception of 4b and 4j, the majority of compounds demonstrated comparability to the standard. A substantial proportion of the tested compounds are anticipated to exhibit outstanding permeability across MDCK cells, suggesting their potential to accurately mimic BBB permeability. Notably, 4b, 4e, 4i, 4j, and 4k outperformed the standard drug in terms of BBB permeability. Forecasts regarding skin permeability and anticipated protein binding to human serum albumin indicated favorable outcomes, with values closely resembling those of the standard palbociclib. Most derivatives were projected to share a heightened oral absorption profile akin to the standard palbociclib in terms of human oral absorption. However, compounds 4b, 4e, and 4j are anticipated to exhibit low oral absorption. These findings are succinctly summarized in Table 2.
Table 2.
In-silico evaluation of ADME characteristics of 4a-p
|
Compound
|
QPPCaco (nm/sec)
|
QPlogBB
|
QPPMDCK
|
QPlogKp
|
QPlogKhsa
|
Human oral absorption
|
|
Range
|
<25 poor, >500 great
|
–3.0 – 1.2
|
<25 poor,
>500 great
|
–8.0 – –1.0
|
–1.5 – 1.5
|
1, 2 or 3 for low, medium, or high
|
| 4a |
306.129 |
-0.926 |
437.827 |
-3.119 |
0.401 |
3 |
| 4b |
958.793 |
-0.064 |
6764.376 |
-2.391 |
0.609 |
1 |
| 4c |
412.448 |
-0.78 |
606.289 |
-2.828 |
0.399 |
3 |
| 4d |
124.777 |
-1.41 |
166.788 |
-3.89 |
0.262 |
3 |
| 4e |
1016.265 |
-0.143 |
4257.021 |
-2.222 |
0.522 |
1 |
| 4f |
308.05 |
-0.924 |
441.752 |
-3.112 |
0.401 |
3 |
| 4g |
147.145 |
-1.335 |
198.543 |
-3.72 |
0.268 |
3 |
| 4h |
177.849 |
-1.269 |
247.333 |
-3.698 |
-0.259 |
3 |
| 4i |
1024.083 |
-0.33 |
1618.162 |
-2.243 |
0.542 |
3 |
| 4j |
1004.925 |
-0.069 |
6958.059 |
-2.294 |
0.656 |
1 |
| 4k |
995.954 |
-0.206 |
2851.856 |
-2.208 |
0.415 |
3 |
| 4l |
124.812 |
-1.375 |
166.722 |
-3.936 |
0.264 |
3 |
| 4m |
258.642 |
-1.009 |
366.835 |
-3.276 |
0.369 |
3 |
| 4n |
910.204 |
-0.559 |
1431.028 |
-2.309 |
0.354 |
3 |
| 4o |
260.907 |
-1.004 |
370.641 |
-3.269 |
0.368 |
3 |
| 4p |
118.18 |
-1.41 |
157.635 |
-3.983 |
0.271 |
3 |
| Palbociclib |
178.394 |
-0.554 |
84.93 |
-4.93 |
0.148 |
3 |
The Ames toxicity assessment serves the purpose of determining the mutagenic potential of a compound. This widely utilized technique helps evaluate whether a given compound can trigger DNA mutations in bacteria. Compounds 4l and 4p have exhibited Ames toxicity, with a probability of 79.58%, which might be attributed to the presence of the -NO2 group. The remaining compounds, along with the standard samples, demonstrated non-toxicity in the Ames test with nearly equivalent probabilities. The assessed compounds are predicted to be non-carcinogenic with a higher probability, aligning their values with those of standard drugs. In the domain of in silico acute oral toxicity assessment, all tested compounds fall within category III, indicating favorable LD50 values exceeding 500 mg and 5000 mg, comparable to the standard drug. The toxicity profiles for rats and fish mirror those of the standard palbociclib. A consolidated summary of the in silico toxicity screening results for the compounds is provided in Table 3.
Table 3.
In-silico evaluation of toxicity of 4a-p
|
Compound code
|
AMES toxicity
|
Carcinogenicity
|
Acute oral toxicity
|
Rat acute toxicity LD50
mol/kg
|
Fish toxicity pLC50
mg/L
|
|
Result
|
Probability
|
Result
|
Probability
|
Result
|
Probability
|
| 4a |
- |
0.6451 |
- |
0.8129 |
III |
0.5733 |
2.2704 |
1.4532 |
| 4b |
- |
0.6433 |
- |
0.8190 |
III |
0.6085 |
2.2446 |
1.3431 |
| 4c |
- |
0.6828 |
- |
0.8021 |
III |
0.5584 |
2.2795 |
1.4100 |
| 4d |
- |
0.7270 |
- |
0.8003 |
III |
0.5641 |
2.2473 |
1.3885 |
| 4e |
- |
0.6227 |
- |
0.8545 |
III |
0.5942 |
2.3101 |
1.4166 |
| 4f |
- |
0.6451 |
- |
0.8129 |
III |
0.5733 |
2.2473 |
1.3885 |
| 4g |
- |
0.7270 |
- |
0.8003 |
III |
0.5641 |
2.4071 |
1.8511 |
| 4h |
- |
0.6372 |
- |
0.6313 |
III |
0.6164 |
2.2447 |
1.6136 |
| 4i |
- |
0.6268 |
- |
0.8709 |
III |
0.5114 |
2.4642 |
1.4286 |
| 4j |
- |
0.5798 |
- |
0.8339 |
III |
0.5573 |
2.3738 |
1.3983 |
| 4k |
- |
0.6031 |
- |
0.8454 |
III |
0.5876 |
2.3894 |
1.3929 |
| 4l |
+ |
0.7958 |
- |
0.7239 |
III |
0.6410 |
2.2786 |
1.6183 |
| 4m |
- |
0.5091 |
- |
0.8605 |
III |
0.5655 |
2.2786 |
1.6183 |
| 4n |
- |
0.6680 |
- |
0.8360 |
III |
0.5965 |
2.2786 |
1.6183 |
| 4o |
- |
0.5091 |
- |
0.8605 |
III |
0.5655 |
2.2786 |
1.6183 |
| 4p |
+ |
0.7958 |
- |
0.7239 |
III |
0.6410 |
2.3894 |
1.3929 |
| Palbociclib |
- |
0.6543 |
- |
0.8632 |
III |
0.6438 |
2.390 |
1.5620 |
Molecular docking investigations were conducted by subjecting compounds to docking against the respective crystal structures of CDK4 (2W96) and CDK6 (6OQO), yielding favorable outcomes for the majority of compounds in comparison to the standard. The docking scores, provided in Table 4, indicated that compounds 4a, 4c, 4d, and 4g are poised to exhibit superior interactions with both CDKs, marking them as potential hit molecules for subsequent assessment. The interaction pattern of 4a with 2W96 was illustrated in Fig. 5, revealing that the nitrogen atom of the pyrimidine ring (N1) engaged in hydrogen bonding with LYS35, the ketone moiety at C3 of the thiazole ring established a hydrogen bond with VAL96, and the 4-OH group of the phenyl ring located on C8 of the pyridopyrimidine ring formed a hydrogen bond with LYS142. Fig. 6 showcased the interaction of 4c with 2W96, where the ketone group at C4 and the thiophene ring at C6 of the pyridopyrimidine ring participated in hydrogen bonding with LYS35 and engaged in a pi-cation interaction, respectively. Furthermore, the 2-OH group of the phenyl ring situated at C8 of the pyridopyrimidine ring formed a hydrogen bond with ILE12. Interaction of 4c with 6OQO was depicted in Fig. 7, revealing the hydrogen bonding between the ketone group at C3 of the thiazole ring and LYS43, while the 2-OH group of the phenyl ring at C8 of the pyridopyrimidine ring established a hydrogen bond with a water molecule, forming a water bridge. Fig. 8 demonstrated the interaction of 4g with 6OQO, indicating a water-bridged hydrogen bonding between THR107 and the 2-OH group of the phenyl ring located at C8 of the pyridopyrimidine ring.
Table 4.
Molecular docking studies of 4a-p
|
Compound
|
2W96
|
6OQO
|
|
Docking score
|
MMGBSA energy
|
Docking score
|
MMGBSA energy
|
| 4a |
-7.711 |
-76.21 |
-6.765 |
-69.53 |
| 4b |
-3.512 |
-66.31 |
-4.484 |
-63.98 |
| 4c |
-6.461 |
-62.43 |
-7.137 |
-60.36 |
| 4d |
-6.247 |
-64.99 |
-6.747 |
-68.89 |
| 4e |
-6.424 |
-76.82 |
-5.112 |
-58.28 |
| 4f |
-6.356 |
-66.74 |
-5.029 |
-73.35 |
| 4g |
-6.383 |
-64.12 |
-6.838 |
-62.32 |
| 4h |
-5.003 |
-38.52 |
--6.527 |
-58.47 |
| 4i |
-3.971 |
-63.22 |
-4.186 |
-71.8 |
| 4j |
-4.959 |
-74.91 |
-5.174 |
-61.74 |
| 4k |
-5.597 |
-59.29 |
-5.528 |
-57.02 |
| 4l |
-3.113 |
-73.2 |
-5.792 |
-69.96 |
| 4m |
-4.293 |
-60.08 |
-6.5 |
-71.18 |
| 4n |
-6.295 |
-75.76 |
-5.914 |
-68.04 |
| 4o |
-5.87 |
-81.82 |
-6.373 |
-72.83 |
| 4p |
-3.896 |
-67.11 |
-4.651 |
-72.15 |
| Palbociclib |
-5.685 |
-59.91 |
-8.076 |
-73.28 |
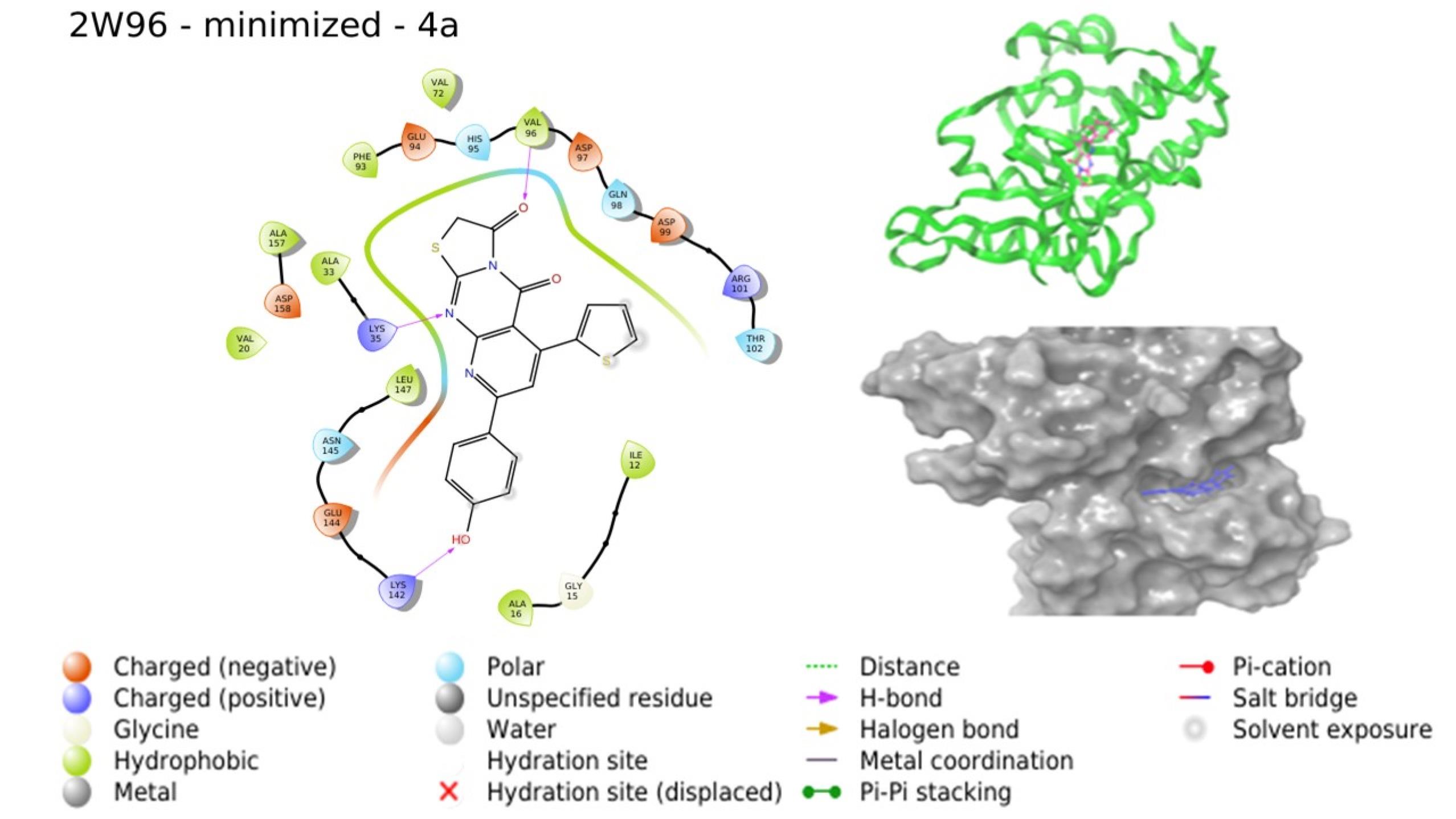
Fig. 5.
Interaction of 4a with 2W96 (Image extracted from Schrödinger software).
.
Interaction of 4a with 2W96 (Image extracted from Schrödinger software).
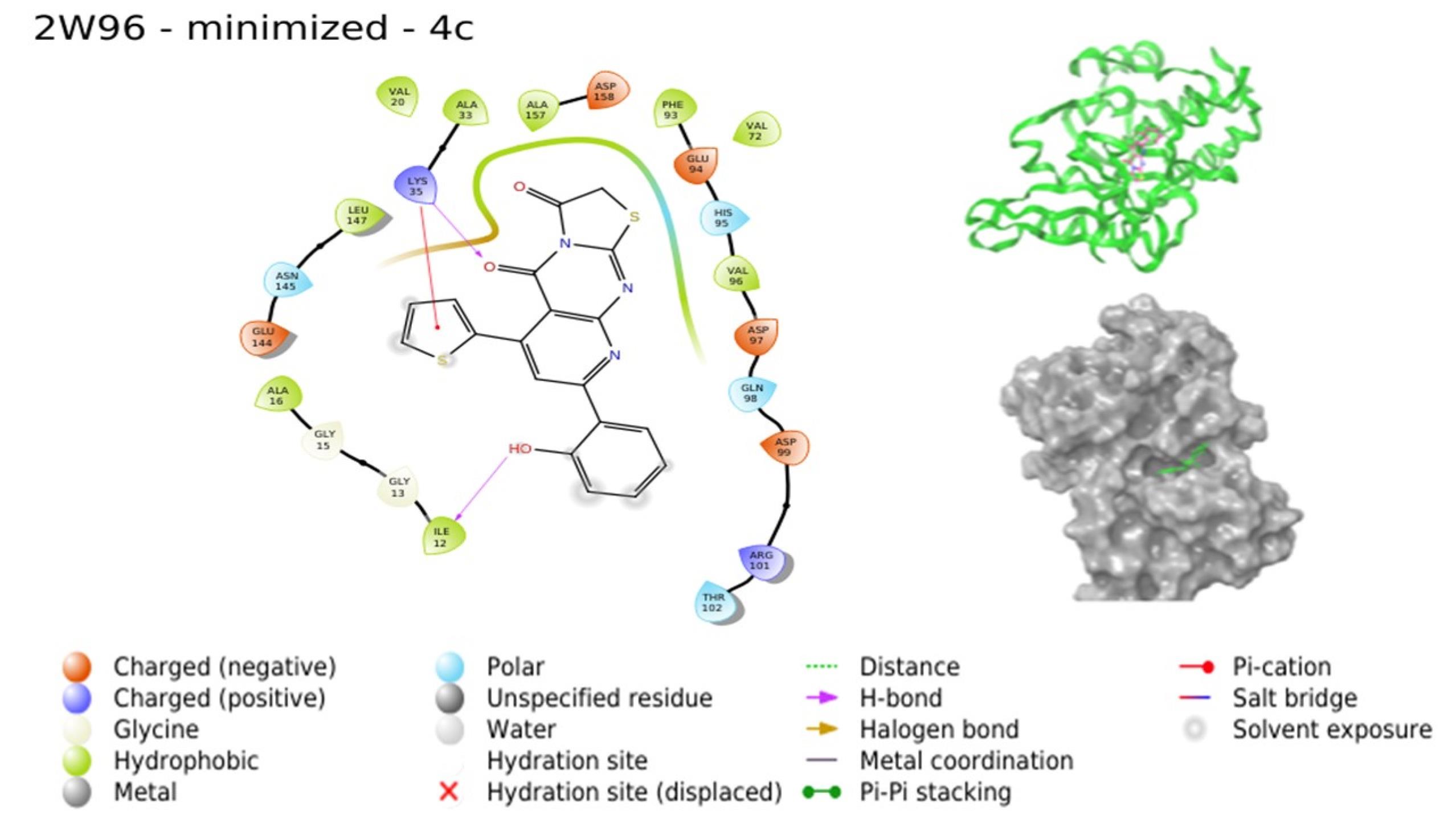
Fig. 6.
Interaction of 4c with 2W96 (Image extracted from Schrödinger software).
.
Interaction of 4c with 2W96 (Image extracted from Schrödinger software).
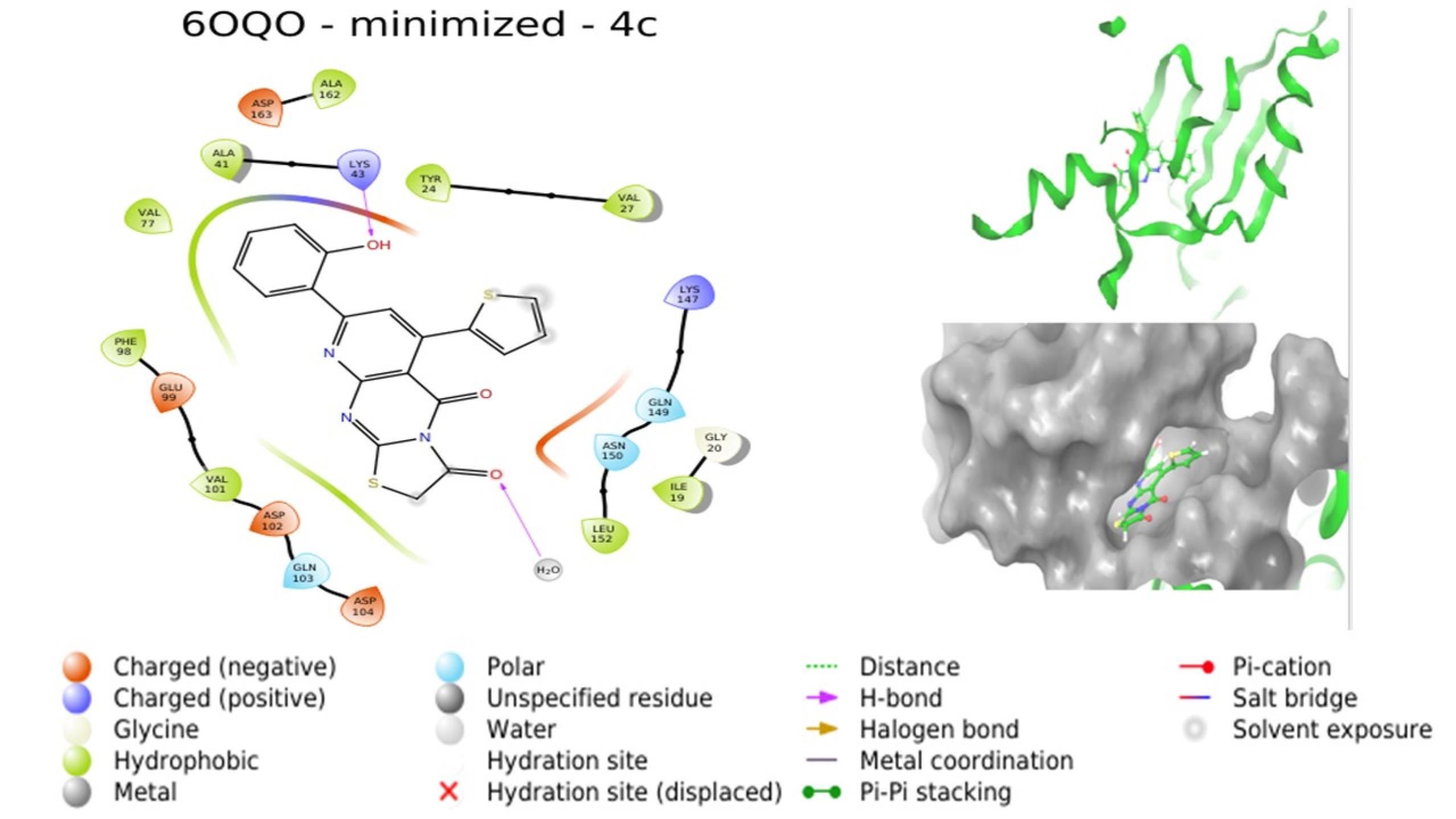
Fig. 7.
Interaction of 4c with 6OQO (Image extracted from Schrödinger software).
.
Interaction of 4c with 6OQO (Image extracted from Schrödinger software).
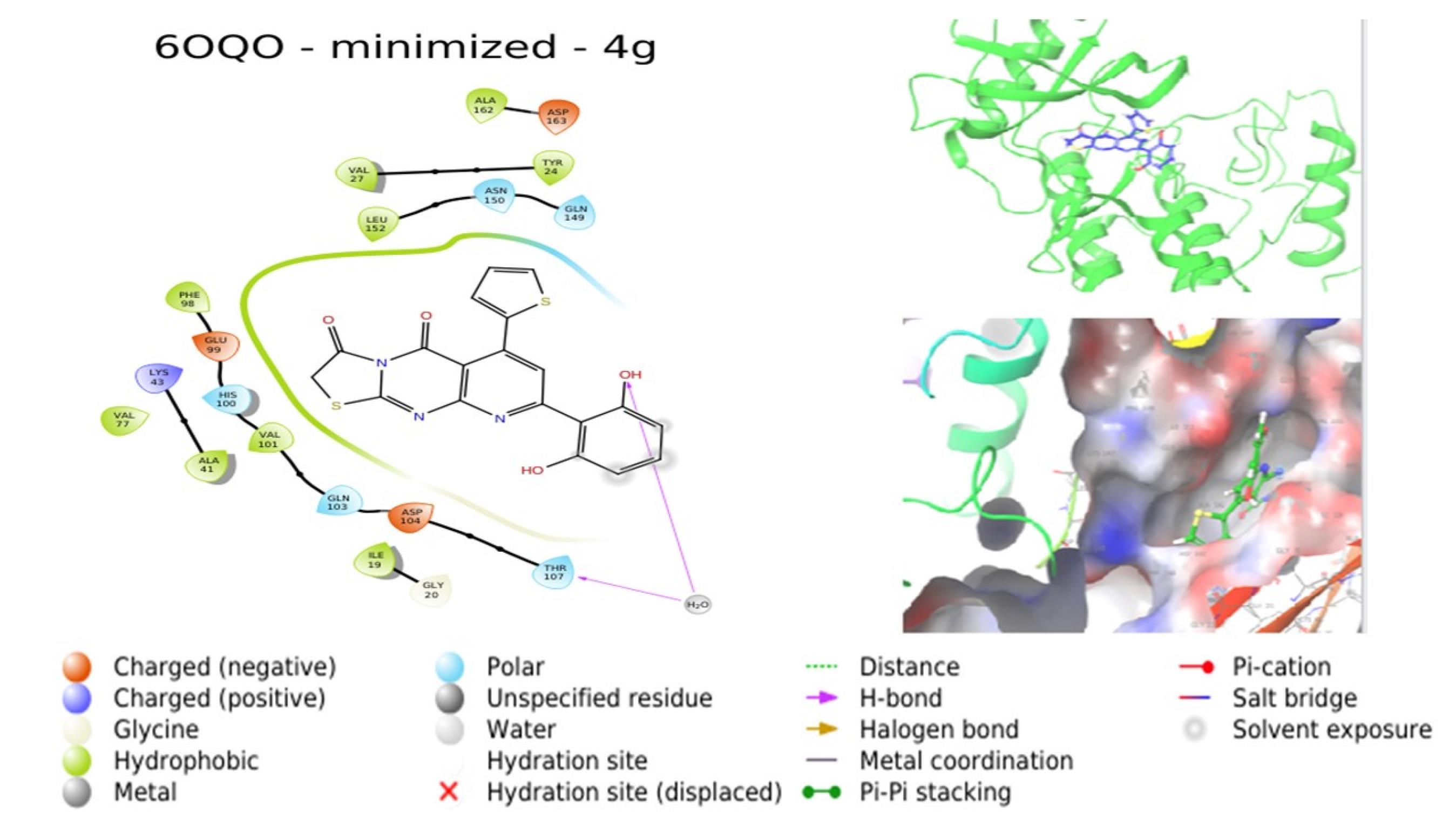
Fig. 8.
Interaction of 4g with 6OQO (Image extracted from Schrödinger software).
.
Interaction of 4g with 6OQO (Image extracted from Schrödinger software).
The compounds with the highest docking scores underwent subsequent synthesis, followed by determination of their IC50 values in breast cancer cell lines through cytotoxicity studies such as the SRB assay.
The initial step involved the synthesis of the first intermediate pyridopyrimidines (3a, 3c, 3d, 3g) by subjecting α, β-unsaturated ketones to reflux with 6-amino-2,3-dihydro-2-thioxopyrimidin-4(1H)-one for a duration of 12-15 hours in 20 ml of anhydrous DMF. The reaction completion was verified via TLC. Subsequently, these intermediate pyridopyrimidines (3a, 3c, 3d, 3g) were mixed with 10 mmol of chloroacetic acid and 20 mmol of sodium acetate in an acidic medium, employing glacial acetic acid as the solvent. The mixture was then subjected to reflux with stirring for 2-4 hours, leading to the synthesis of thiazolo pyridopyrimidines (4a, 4c, 4d, and 4g). The physicochemical attributes of the synthesized compounds are detailed in Table S1 (See Supplementary file 1). Characterization of all four synthesized compounds encompassed IR, NMR, and mass spectral analyses. The IR spectra of the compounds displayed discernible peaks corresponding to the various functional groups inherent in each compound. Notably, significant peaks were observed for the hydroxyl group on the phenyl ring at C8 of the pyridopyrimidine ring, the two distinct ketone peaks at C3 of the thiazole ring and C4 of the pyrimidine ring, as well as prominent features denoting C=C, C=N, C-N, and C-S-C bonds. In the NMR spectra, a singlet was noted for the hydrogen at C6 of the pyridine ring, while multiplets appeared for the four hydrogen atoms present in the aromatic rings at C8 of the pyridopyrimidine ring. The thiophene ring at C6 of the pyridopyrimidine ring exhibited two doublets and a triplet corresponding to the hydrogen at C3, C5, and C4 positions of the thiophene ring. A peak indicative of the CH2 group of the thiazole ring was also observed. The hydroxyl group at C4 of the phenyl ring yielded a singlet. The mass spectra of the compounds exhibited a molecular ion peak.
Characterization of synthesized compounds (4a,4c,4d and 4g)
8-(4-hydroxyphenyl)-6-(thiophen-2-yl)-2H-pyrido[2,3-d]thiazolo[3,2-a]pyrimidine-3,5-dione (4a): FT-IR (KBr, cm-1): 3603(-OH), 1706, 1643 (2 C=O), 1558(C=N), 1414(C=C), 1127(C-N), 638(C-S-C). 1H -NMR (DMSO- d6, 400 MHz):7.622(s, 1H, C-6 pyridine), 7.160-6.884 (m, 4H, Ar-H), 8.122 (d, 1H, thiophene H3, J=3 Hz), 7.757(d, 1H, thiophene H5, J=6 Hz), 7.444(t, 1H, thiophene H4, J=9 Hz), 4.081(s, 2H, CH2), 7.706(s, 1H, Ar-OH). MS (m/z): 393.5371(M+ peak).
8-(2-hydroxyphenyl)-6-(thiophen-2-yl)-2H-pyrido[2,3-d]thiazolo[3,2-a]pyrimidine-3,5-dione (4c): FT-IR (KBr, cm-1): 3577 (-OH), 1680, 1647(2 C=O), 1540 (C=N), 1396(C=C), 1229(C-N), 648(C-S-C). 1H -NMR (DMSO- d6, 400 MHz): 7.629(s, 1H, C-6 pyridine), 6.961-6.936(m, 4H, Ar-H), 7.778(d, 1H, thiophene H3, J=6 Hz), 7.483(d, 1H, thiophene H5, J=5 Hz), 7.171(t, 1H, thiophene H4, J=9 Hz), 3.342(s, 2H, CH2), 7.616(s, 1H, Ar-OH). MS (m/z): 393.4406(M+ peak).
8-(2,4-dihydroxyphenyl)-6-(thiophen-2-yl)-2H-pyrido[2,3-d]thiazolo[3,2-a]pyrimidine-3,5-dione (4d): FT-IR (KBr, cm-1): 1736, 1623 (2 C=O), 1575(C=N), 1470 (C=C), 1116(C-N), 614(C-S-C), 1344(-CF3). 7.923 (s, 1H, C-6 pyridine), 8.645-7.790 (m, 4H, Ar-H), 8.571 (d, 1H, thiophene H3, J=3 Hz), 7.516 (d, 1H, thiophene H5, J=6 Hz), 7.188 (t, 1H, thiophene H4, J=9 Hz), 3.523(s, 2H, CH2).MS (m/z): 445.4612 (M+ peak).
8-(2,6-dihydroxyphenyl)-6-(thiophen-2-yl)-2H-pyrido[2,3-d]thiazolo[3,2-a]pyrimidine-3,5-dione (4g): FT-IR (KBr, cm-1): 3694, 3608 (2 OH), 1730, 1627(2 C=O), 1539 (C=N), 1471(C=C), 1198(C-N), 622(C-S-C). 1H -NMR (DMSO- d6, 400 MHz): 7.034 (s, 1H, C-6 pyridine), 6.630-6.298 (m, 4H, Ar-H), 7.255 (d, 1H, thiophene H3, J=6 Hz), 6.832 (d, 1H, thiophene H5, J=5 Hz), 6.642 (t, 1H, thiophene H4, J=9 Hz), 3.450(s, 2H, CH2), 6.714, 6.125 (s, 1H, Ar-OH). MS (m/z): 409.4383 (M+ peak).
The synthesized compounds were screened against breast cancer cell lines, namely MCF-7 and MDAMB-231, using the SRB assay. All four tested compounds demonstrated concentration-dependent inhibitory effects in both MCF-7 and MDAMB-231 cell lines, as depicted in Figs. S1 and S2 (See Supplementary file 2). Among these compounds, 4c displayed the highest activity, followed by 4a, in both cell lines. The IC50 values for 4c and 4a in MCF-7 were determined as 27.086 µM and 32.597 µM, respectively. Similarly, for MDAMB-231, the IC50 values were 29.133 µM for 4c and 34.468 µM for 4a, as outlined in Tables S2 and S3. The enhanced cytotoxic activity could potentially be attributed to the presence of a polar, electron-donating hydroxyl group at C2 (4c) and C4 (4a) of the phenyl ring situated at C8 of the pyridopyrimidine ring.
Hence, considering these findings, 4c and 4a emerge as promising lead molecules, with potential for further augmentation through structural modifications. The visually evident cell death is demonstrated in Figs. S3 and S3, corresponding to MCF-7 and MDAMB-231 cell lines, respectively. Notably, compounds 4c and 4a demonstrated encouraging cytotoxic effects. Due to their promising outcomes in both in silico and in vitro models, we opted to subject 4a and 4c to MD simulation with the target to which they displayed the highest docking score. This simulation aims to assess their stability within the binding site.
Root mean square deviation (RMSD) analysis
RMSD analysis of target proteins was conducted to assess the stability and dynamic characteristics of the protein structure by measuring its deviation from the initial shape and arrangement. In terms of all backbone Cα atoms, the protein RMSD values were 4 and 3.2 for the simulated 4a-2W96 and 4c-6OQO protein complexes, respectively, when compared to their respective original structures over the course of each trajectory, illustrated in Figs. S5 and S6. In the context of the simulation run, RMSD values within the range of 1 to 3 Å are indicative of stability, while higher values suggest substantial conformational changes in the proteins. After 35 ns, the RMSD values within the simulation model of 2W96 reached a stable state and maintained this level throughout the simulation, exhibiting minimal variations in conformation. This stability indicates that the simulated system achieved equilibrium. In contrast, the simulated system of 6OQO displayed ongoing conformational changes throughout the simulation period.46
Ligand RMSD analysis
The ligand's RMSD (depicted on the right-hand Y-axis in Figs. S5 and S6) reflects the stability of the ligand within the protein and its binding pocket. Significantly higher values compared to the protein's RMSD (on the left-hand Y-axis of Figs. S5 and S6) suggest that the ligand has moved away from its initial binding site. The RMSD value for 4a was recorded at 6.4 Å, while for 4c it was 3.2 Å. These findings indicate that ligand 4c remained stable within the binding pocket of 6OQO, as both the ligand and protein exhibited identical RMSD values. Conversely, there was a divergence and instability observed between ligand 4a and 2W96, as the ligand's alignment with the protein's original structure led to a variation in RMSD of less than 2 Å.47,48
RMSF analysis
Root mean square fluctuation (RMSF) is useful for defining slight differences along the protein sequence and thus tracing out the flexible area in the protein structure. The peaks present in the plot represent the most fluctuating region of the protein during simulations. When compared to more rigid secondary structure elements (SSE) such as beta sheets and alpha-helices, residues/amino acids in loops, non-structured space, or protein termini (N- and C-terminal) will show greater fluctuation. Loops shows greater RMSF value is merely due to the flexibility of the residue in contrast to its average position.48 Figs. S7 and S7 provide an overview of the RMSF plots for the target proteins. The RMSF values for 2W96 and 6OQO were determined to be 5.4 and 4.5, respectively. These values suggest minimal variation in residues and their backbone atoms, signifying limited conformational deviations throughout the simulation. In Fig. S7, noticeable fluctuations were observed within the loops of residues 35-40, 75-80, 95-100, 170-180, and 230-235 in the N and C-terminal regions of 2W96. These fluctuations are attributed to the inherent flexibility of these areas. However, our primary focus is directed towards the relatively modest fluctuations within segments 52-60, 140-150, and 200-210, which contain SSE. These collective fluctuations contribute to the opening or closing of the substrate-binding groove positioned between these two segments. Elevated RMSF values signify potential communication with the binding sites. In contrast, regions 125-135, 190-200, and 235-245 of the 2W96 protein exhibited the lowest RMSF values. Within 6OQO, minor fluctuations were observed across the range of residues 150-250. Conversely, pronounced fluctuations were noted in regions encompassing residues 20-25, 35-45, 65-80, and 145-150. In Fig. S8, regions 110-130 and 170-180 of 6OQO exhibited notably subdued RMSF values.
Protein-ligand contacts
Following the simulation, each complex was evaluated individually for the ligand interaction with each residues, which is based on the mean value of the interaction occupancies of the binding site residues.49-51 The outcomes were presented in histogram format, as depicted in Figs. S9 and S10. For the 4a-2W96 complex, the most significant interaction occupancy was observed with VAL96, LYS35, and TYR17. VAL96 exhibited hydrogen bond interactions for approximately 75% of the simulation duration and consistently formed water bridges with the ligand throughout the simulation. These water bridge interactions represent hydrogen bond-based protein-ligand interactions facilitated by a water molecule, characterized by slightly different hydrogen bond geometry compared to conventional definitions. LYS35 displayed robust hydrogen bond interactions for the entire simulation period, accompanied by 20% hydrophobic interaction with the ligand. TYR17 demonstrated hydrogen bond interactions for 90% of the simulation time. Minor interactions were observed with ILE12, ALA16, GLY18, VAL20, ALA33, HIS95, ARG101, GLU144, and LEU147. Fig. S11 illustrates the interaction between the ligand and the 2W96 target after simulation. In the 4c-6OQO complex, the most substantial interaction occupancy was attributed to ASP163, which engaged in consistent hydrogen bonding throughout the simulation. VAL101 and HIS100 maintained water bridge interactions throughout the simulation. PHE98 demonstrated 55% hydrophobic interaction, while ILE19 displayed 45% hydrophobic interaction along with water bridge interactions. Ionic and water bridge interactions were observed with ASP104 and ALA162 for 35% of the simulation duration. ALA162 engaged in hydrophobic interaction for around 38% of the simulation time. Minor interactions were noted with GLU21, TYR27, ALA41, GLN149, and LEU152. Interaction between 4a and the 6OQO target after simulation is depicted in Fig. S12. The insights gleaned from the molecular dynamics study suggest that the binding of 4c-6OQO is more stable, positioning 4c as a promising lead molecule for further investigations.
Conclusion
Utilizing in silico ADME/T assessments and molecular docking analyses, a quartet of thiazolo pyridopyrimidines were deliberately chosen for synthesis, followed by their assessment against MCF-7 and MDAMB-231 via the SRB assay. Among the four compounds subjected to screening, 4c exhibited a notably promising level of activity, while 4a demonstrated a comparatively satisfactory level of activity, as evident by their minimal IC50 values. It is plausible that the enhanced activity of these compounds is attributed to the presence of polar hydroxyl groups at C2 and C4 positions within the 8-phenyl substitution of the pyridopyrimidine rings. Nonetheless, it is conjectured that the cytotoxicity of these compounds could be further enhanced through strategic structural modifications. Subsequent to these analyses, the selected compounds underwent molecular dynamics simulations, employing the crystal structure of CDK4/6 and the 4c/4a complex. The outcomes of the molecular dynamics study unveiled that the binding of 4c-6OQO exhibited heightened stability. Accordingly, compound 4c emerges as a promising candidate for CDK6 inhibition, positioning it as a potential lead molecule for further investigation whose mechanism of action needs to be confirmed by further in vitro and in vivo models.
Research Highlights
What is the current knowledge?
√ Pyridopyrimidines exhibit diverse biological effects, particularly showing promise as anticancer agents, including actions that inhibit CDK4/6.
√ FDA granted the first CDK4/6 inhibitor i.e., palbociclib which is a pyridopyrimidine derivative mainly used in patients with breast cancer along with fulvestrant or letrozole.
What is new here?
√ A range of novel substituted thiazolo-pyridopyrimidines are designed and synthesized.
√ Selected molecules are evaluated for their anticancer activity against breast cancer by in silico, in vitro models.
√ Most potent compounds were reported and chemistry of those compounds are discussed.
Acknowledgements
Authors are thankful to: (1) Nitte deemed to be university and NGSM Institute of pharmaceutical sciences, Deralakatte, Mangalore, for providing CADD lab and cell culture lab facilities to conduct this research; (2)Department of Pharmaceutical Chemistry, Faculty of Pharmacy, M S Ramaiah University of Applied Sciences, Bengaluru, Karnataka for MD simulation studies; and (3) NMR instrument centre and DST-purse laboratory, Mangalore university, Mangalagangothri for the spectral studies.
Competing Interests
The authors declare no competing interests.
Ethical Statement
There is none to be stated.
Funding
This research was funded byNitte Deemed to be University, Deralakatte, Mangalore.
Supplementary files
Supplementary file 1 contains Tables S1-S3.
(pdf)
Supplementary file 2 contains Figs. S1-S12
(pdf)
References
- Cordeu L, Cubedo E, Bandrés E, Rebollo A, Sáenz X, Chozas H. Biological profile of new apoptotic agents based on 2,4-pyrido[2,3-d]pyrimidine derivatives. Bioorg Med Chem 2007; 15:1659-69. doi: 10.1016/j.bmc.2006.12.010 [Crossref] [ Google Scholar]
- Farag B, Agili F, El‐Kalyoubi S, Said SA, Youssif S, Shehta W. Synthesis, Molecular Docking and Anticancer Activity of Some 5‐Aryl‐5, 10‐dihydropyrido [2, 3‐d: 6, 5‐d′] dipyrimidine‐2, 4, 6, 8‐tetraone Derivatives and Pyrido [2, 3‐d] pyrimidines. ChemistrySelect 2022; 7:e202103834. doi: 10.1002/slct.202103834 [Crossref] [ Google Scholar]
- Abbas SE, George RF, Samir EM, Aref MM, Abdel-Aziz HA. Synthesis and anticancer activity of some pyrido [2, 3-d] pyrimidine derivatives as apoptosis inducers and cyclin-dependent kinase inhibitors. Future Med Chem 2019; 11:2395-2414. doi: 10.4155/fmc-2019-0050 [Crossref] [ Google Scholar]
- Dorsey JF, Jove R, Kraker AJ, Wu J. The pyrido[2,3-d]pyrimidine derivative PD180970 inhibits p210Bcr-Abl tyrosine kinase and induces apoptosis of K562 leukemic cells. Cancer Res 2000;60:3127–31..
- Font M, González Á, Palop JA, Sanmartín C. New insights into the structural requirements for pro-apoptotic agents based on 2,4-diaminoquinazoline, 2,4-diaminopyrido[2,3-d]pyrimidine and 2,4-diaminopyrimidine derivatives. Eur J Med Chem 2011; 46:3887-99. doi: 10.1016/j.ejmech.2011.05.060 [Crossref] [ Google Scholar]
- Abdel-Aziem A, El-Gendy MS, Abdelhamid AO. Synthesis and antimicrobial activities of pyrido [2, 3-d] pyrimidine, pyridotriazolopyrimidine, triazolopyrimidine, and pyrido [2, 3-d: 6, 5d'] dipyrimidine derivatives. Eur J Chem 2012; 3:455-460. doi: 10.5155/eurjchem.3.4.455-460.683 [Crossref] [ Google Scholar]
- Pastor del Castillo A, Alajarín Ferrández LR, Vaquero López JJ, Álvarez-Builla Gómez J, Casa-Juana Muñoz MF, Sunkel Letelier C. Synthesis and structure of new pyrido[2,3-D]pyrimidine derivatives with calcium-channel antagonist activity. Tetrahedron 1994; 50:8085-98. doi: 10.1016/S0040-4020(01)85291-1 [Crossref] [ Google Scholar]
- Suresh M, Lavanya P, Vasu K, Sudhakar D, Rao CV. Synthesis and bioassay studies of 7-substituted pyrido[2,3-d]pyrimidines. J Chem Pharm Res 2010; 2:82-9..
- Tadayon M, Garkani-Nejad Z. In silico study combining QSAR, docking and molecular dynamics simulation on 2,4-disubstituted pyridopyrimidine derivatives. J Recept Signal Transduct Res 2019; 39:167-174. doi: 10.1080/10799893.2019.1641821 [Crossref] [ Google Scholar]
- Elzahabi HS, Nossier ES, Alasfoury RA, El-Manawaty M, Sayed SM, Elkaeed EB. Design, synthesis, and anti-cancer evaluation of new pyrido [2, 3-d] pyrimidin-4 (3H)-one derivatives as potential EGFRWT and EGFRT790M inhibitors and apoptosis inducers. J Enzyme Inhib Med Chem 2022; 37:1053-76. doi: 10.1080/14756366.2022.2062752 [Crossref] [ Google Scholar]
- Salem MG, El-Maaty DM, El-Deen YI, Elesawy BH, Askary AE, Saleh A, et al. Novel 1,3-Thiazole Analogues with Potent Activity against Breast Cancer: A Design, Synthesis, In Vitro, and In Silico Study. Molecules 2022; 27: 4898. .
- Khajouei MR, Khodarahmi G, Ghaderi A. Synthesis and cytotoxic evaluation of some novel 3-[2-(2-phenyl-thiazol-4-yl)-ethyl]-3H-pyrido [2, 3-d] pyrimidin-4-one derivatives. Res Pharm Sci 2021; 16: 455. .
- Fabbro D, Cowan‐Jacob SW, Moebitz H. Ten things you should know about protein kinases: IUPHAR Review 14. Br J Pharmacol 2015; 172:2675-700. doi: 10.1111/bph.13096 [Crossref] [ Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002; 298:1912-1934. doi: 10.1126/science.1075762 [Crossref] [ Google Scholar]
- Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, Hubbard BP. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 2018; 17:1-20. doi: 10.1186/s12943-018-0804-2 [Crossref] [ Google Scholar]
- Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 2006; 24:1770-83. doi: 10.1200/JCO.2005.03.7689 [Crossref] [ Google Scholar]
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9:153-66. doi: 10.1038/nrc2602 [Crossref] [ Google Scholar]
- Ortega S, Malumbres M, Barbacid M. Biochim Biophys Acta 2002; 1602: 73–87. 10.1016/s0304-419x(02)00037-9.
- Battisti NM, De Glas N, Sedrak MS, Loh KP, Liposits G, Soto-Perez-de-Celis E. Use of cyclin-dependent kinase 4/6 (CDK4/6) inhibitors in older patients with ER-positive HER2-negative breast cancer: Young International Society of Geriatric Oncology review paper. Ther Adv Med Oncol 2018; 10:1758835918809610. doi: 10.1177/1758835918809610 [Crossref] [ Google Scholar]
- Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 2016; 375:1925-36. doi: 10.1056/NEJMoa1607303 [Crossref] [ Google Scholar]
- Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015; 16:25-35. doi: 10.1016/S1470-2045(14)71159-3 [Crossref] [ Google Scholar]
- Cho YS, Angove H, Brain C, Chen CH, Cheng H, Cheng R. Fragment-Based Discovery of 7-Azabenzimidazoles as Potent, Highly Selective, and Orally Active CDK4/6 Inhibitors. ACS Med Chem Lett 2012; 3:445-49. doi: 10.1021/ml200241a [Crossref] [ Google Scholar]
- Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol 2016; 17:425-39. doi: 10.1016/S1470-2045(15)00613-0 [Crossref] [ Google Scholar]
- Freeman-Cook KD, Hoffman RL, Behenna DC, Boras B, Carelli J, Diehl W. Discovery of PF-06873600, a CDK2/4/6 Inhibitor for the Treatment of Cancer. J Med Chem 2021; 64:9056-77. doi: 10.1021/acs.jmedchem.1c00159 [Crossref] [ Google Scholar]
- Tadesse S, Yu M, Mekonnen LB, Lam F, Islam S, Tomusange K. Highly Potent, Selective, and Orally Bioavailable 4-Thiazol-N-(pyridin-2-yl) pyrimidin-2-amine Cyclin-Dependent Kinases 4 and 6 Inhibitors as Anticancer Drug Candidates: Design, Synthesis, and Evaluation. J Med Chem 2017; 60:1892-915. doi: 10.1021/acs.jmedchem.6b01670 [Crossref] [ Google Scholar]
- Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr Med Chem 2013; 20:2839-60. doi: 10.2174/09298673113209990001 [Crossref] [ Google Scholar]
- Tadesse S, Zhu G, Mekonnen LB, Lenjisa JL, Yu M, Brown MP. A novel series of N-(pyridin-2-yl)-4-(thiazol-5-yl)pyrimidin-2-amines as highly potent CDK4/6 inhibitors. Future Med Chem 2017; 9:1495-506. doi: 10.4155/fmc-2017-0076 [Crossref] [ Google Scholar]
- Lionta E, Spyrou G, K Vassilatis D, Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem 2014; 14:1923-38. doi: 10.2174/1568026614666140929124445 [Crossref] [ Google Scholar]
- Svensson F, Karlén A, Sköld C. Virtual screening data fusion using both structure- and ligand-based methods. J Chem Inf Model 2012; 52:225-32. doi: 10.1021/ci2004835 [Crossref] [ Google Scholar]
- Shetty CR, Mendonca V, Shastry CS, Murugeshwaria V, Lamare W. In-silico evaluation of pyrazole and triazine containing pyridopyrimidines. Indian Drugs 2023; 60:19-32. doi: 10.53879/id.60.01.13285 [Crossref] [ Google Scholar]
- Shetty CR, Bhat KI, Kumar A, Kumar P, Krishnamurthy PT, Merugumolu VK. Synthesis, in-silico Studies and Evaluation of Anticancer Activity of Some Novel Benzothiazole Substituted 4-Thiazolidinones. Indian J Pharmaceut Educ Res 2020; 54:1121-32. doi: 10.5530/ijper.54.4.208 [Crossref] [ Google Scholar]
- Domínguez-Villa FX, Durán-Iturbide NA, Ávila-Zárraga JG. Synthesis, molecular docking, and in silico ADME/Tox profiling studies of new 1-aryl-5-(3-azidopropyl)indol-4-ones: Potential inhibitors of SARS CoV-2 main protease. Bioorg Chem 2021; 106:104497. doi: 10.1016/j.bioorg.2020.104497 [Crossref] [ Google Scholar]
- Kovačević SZ, Jevrić LR, Kuzmanović SO, Lončar ES. Prediction of In-silico ADME Properties of 1,2-O-Isopropylidene Aldohexose Derivatives. Iran J Pharm Res 2014; 13: 899–907..
- Ammar O. In silico pharmacodynamics, toxicity profile and biological activities of the Saharan medicinal plant Limoniastrum feei. Braz J Pharm Sci 2017; 53:e00061. doi: 10.1590/s2175-97902017000300061 [Crossref] [ Google Scholar]
- Ali A, Abdellattif MH, Ali A, AbuAli O, Shahbaaz M, Ahsan MJ. Computational Approaches for the Design of Novel Anticancer Compounds Based on Pyrazolo[3,4-d]pyrimidine Derivatives as TRAP1 Inhibitor. Molecules 2021; 26(19):5932. doi: 10.3390/molecules26195932 [Crossref] [ Google Scholar]
- Panchabhai VB, Butle SR, Ingole PG. Synthesis, characterization and molecular docking studies on some new N-substituted 2-phenylpyrido[2,3-d]pyrimidine derivatives. Research Journal of Pharmacy and Technology 2021; 14:3846-54. [ Google Scholar]
- Verma VA, Halu B, Saundane AR, Meti RS. Synthesis, Biological Validation, and Docking Studies of Novel Purine Derivatives Containing Pyridopyrimidine, Pyrazolopyridine, and Pyranonapthyridine Rings. Polycyclic Aromatic Compounds 2021; 42:1-23. doi: 10.1080/10406638.2020.1871384 [Crossref] [ Google Scholar]
- Arwansyah A, Arif AR, Syahputra G, Sukarti S, Kurniawan I. Theoretical studies of Thiazolyl-Pyrazoline derivatives as promising drugs against malaria by QSAR modelling combined with molecular docking and molecular dynamics simulation. Molecular Simulation 2021; 47:988-1001. doi: 10.1080/08927022.2021.1935926 [Crossref] [ Google Scholar]
- Elzahabi HS, Nossier ES, Alasfoury RA, El-Manawaty M, Sayed SM, Elkaeed EB. Design, synthesis, and anti-cancer evaluation of new pyrido[2,3-d]pyrimidin-4(3H)-one derivatives as potential EGFRWT and EGFRT790M inhibitors and apoptosis inducers. J Enzyme Inhib Med Chem 2022; 37:1053-76. doi: 10.1080/14756366.2022.2062752 [Crossref] [ Google Scholar]
- Youssef A, Fouda AM, Faty RM. Microwave assisted synthesis of some new thiazolopyrimidine and pyrimidothiazolopyrimidopyrimidine derivatives with potential antimicrobial activity. Chem Cent J 2018; 12:1-4. doi: 10.1186/s13065-018-0419-0 [Crossref] [ Google Scholar]
- Razali S, Firus Khan AA, Khatib A, Ahmed QU, Abdul Wahab R, Zakaria ZA. An In Vitro Anticancer Activity Evaluation of Neolamarckia cadamba (Roxb) Bosser Leaves’ Extract and its Metabolite Profile. Front Pharmacol 2021; 12:741683. doi: 10.3389/fphar.2021.741683 [Crossref] [ Google Scholar]
- Yadav B, Bajaj A, Saxena M, Saxena AK. In Vitro Anticancer Activity of the Root, Stem and Leaves of Withania Somnifera against Various Human Cancer Cell Lines. Indian J Pharm Sci 2010; 72:659-63. doi: 10.4103/0250-474X.78543 [Crossref] [ Google Scholar]
- Abou-Seri SM, Eissa AA, Behery MG, Omar FA. Synthesis, in vitro anticancer activity and in silico studies of certain isoxazole-based carboxamides, ureates, and hydrazones as potential inhibitors of VEGFR2. Bioorg Chem 2021; 116:105334. doi: 10.1016/j.bioorg.2021.105334 [Crossref] [ Google Scholar]
- Lu Y, Wang Z, Li CM, Chen J, Dalton JT, Li W. Synthesis, in vitro structure-activity relationship, and in vivo studies of 2-arylthiazolidine-4-carboxylic acid amides as anticancer agents. Bioorg Med Chem 2010; 18:477-95. doi: 10.1016/j.bmc.2009.12.020. [Crossref] [ Google Scholar]
- Khan N, Kumar N, Ballal A, Datta D, Belle VS. Unveiling antioxidant and anti-cancer potentials of characterized Annona reticulata leaf extract in 1,2-dimethylhydrazine-induced colorectal cancer in Wistar rats. J Ayurveda Integr Med 2021; 12:579-89. doi: 10.1016/j.jaim.2021.05.010 [Crossref] [ Google Scholar]
- Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol 2011; 9:1-9. doi: 10.1186/1741-7007-9-71 [Crossref] [ Google Scholar]
- Navyashree V, Kant K, Kumar A. Natural chemical entities from Arisaema genus might be a promising break-through against Japanese encephalitis virus infection: a molecular docking and dynamics approach. J Biomol Struct Dyn 2021; 39:1404-16. doi: 10.1080/07391102.2020.1731603 [Crossref] [ Google Scholar]
- Gopinath P, Kathiravan MK. Docking studies and molecular dynamics simulation of triazole benzene sulfonamide derivatives with human carbonic anhydrase IX inhibition activity. RSC Adv 2012; 1:38079-93. doi: 10.1039/d1ra07377j [Crossref] [ Google Scholar]
- Radwan A, Mahrous GM. Docking studies and molecular dynamics simulations of the binding characteristics of waldiomycin and its methyl ester analog to Staphylococcus aureus histidine kinase. PLoS One 2020; 15:e0234215. doi: 10.1371/journal.pone.0234215 [Crossref] [ Google Scholar]
- Pashaei H, Rouhani A, Nejabat M, Hadizadeh F, Mirzaei S, Nadri H. Synthesis and molecular dynamic simulation studies of novel N-(1-benzylpiperidin-4-yl) quinoline-4-carboxamides as potential acetylcholinesterase inhibitors. Journal of Molecular Structure 2021; 1244:130919. doi: 10.1016/j.molstruc.2021.130919 [Crossref] [ Google Scholar]
- Thakral S, Narang R, Kumar M, Singh V. Synthesis, molecular docking and molecular dynamic simulation studies of 2-chloro-5-[(4-chlorophenyl) sulfamoyl]-N-(alkyl/aryl)-4-nitrobenzamide derivatives as antidiabetic agents. BMC Chemistry 2020; 14:1-6. doi: 10.1186/s13065-020-00703-4 [Crossref] [ Google Scholar]