Bioimpacts. 2025;15:29993.
doi: 10.34172/bi.29993
Review
Regulation of ubiquitin-proteasome system and its relative pathways in pancreatic adenocarcinoma
Bahareh Shateri Amiri 1  , Mehrasa Naserranjbar 2, AyAna Mirhaji 3, Alireza Hejrati 2, Lina Hejrati 2, Fatemeh Aliabadi 4, *
, Mehrasa Naserranjbar 2, AyAna Mirhaji 3, Alireza Hejrati 2, Lina Hejrati 2, Fatemeh Aliabadi 4, *
Author information:
1Department of Internal Medicine, School of Medicine, Hazrat-e Rasool General Hospital, University of Medical Sciences, Tehran, Iran
2School of Medicine, Iran University of Medical Sciences, Tehran, Iran
3Tabatabaei Highschool, Shiraz, Iran
4Physiology Research Center, Faculty of Medicine, Iran University of Medical Sciences, Tehran, Iran
Abstract
Introduction:
Pancreatic cancer, which results from the uncontrolled growth of pancreatic cells, is the fourth most frequent cause of cancer-related mortality in the United States. About 90% of instances of pancreatic cancer are pancreatic adenocarcinomas, and occasionally "pancreatic cancer" is used exclusively to describe this subtype. Nab-paclitaxel, gemcitabine, and FOLFIRINOX are examples of modern chemotherapeutic drugs that have the ability to quickly confer resistance in pancreatic tumor cells. Therefore, in order to treat this dreadful condition, it is essential to develop more effective medicines. Inhibition of the ubiquitin-proteasome system (UPS) causes pancreatic cancer cells to die apoptotically. In eukaryotes, UPS is an essential mechanism for protein breakdown. Pancreatic cancer cells are more susceptible to endoplasmic reticulum stress (endoplasmic reticulum [ER] stress) and apoptosis when treated with bortezomib, a proteasome inhibitor that is the first in this group of drugs approved for the treatment of cancer, especially multiple myeloma.
Methods:
Searching through PubMed and Google Scholar and gathering data.
Results:
UPS is still a popular target for pancreatic cancer treatment among researchers. However, despite the favorable results of UPS-based therapies in vitro and in vivo, the clinical results are not as promising as expected.
Conclusion:
A deep understanding of it, is essential to achieving the maximum results. In this review, we aim to look into the UPS along with searching for the novelist therapies for pancreatic adenocarcinoma based on manipulating it.
Keywords: Pancreatic cancer, Pancreatic adenocarcinoma, Ubiquitin-proteasome system, Regulating factors
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
The authors declare that they did not receive any funds.
Introduction
The most common cancer among those with a bad prognosis in western populations is pancreatic cancer. In terms of new instances, this cancer is currently in tenth place.1 Globally, the incidence of all forms of pancreatic cancer (85% of whom are adenocarcinomas) varies from one to ten cases per 100 000 persons.2 In the Western world, pancreatic cancer ranks fourth for men and fifth for women in terms of cancer-related death.3 Europe and North America have the highest age-standardized incidence, while Africa and South Central Asia have the lowest.4 It is uncommon for anyone under the age of 40 to be diagnosed with pancreatic adenocarcinoma.2 The incidence is highest in the seventh and eighth decades of life.5
Among all pancreatic malignancies, pancreatic adenocarcinoma is the most common, accounting for 80% of them.5,6 Over the next 10 years, lung cancer is expected to be passed on by pancreatic ductal adenocarcinoma (PDAC), the most common form of pancreatic cancer, as the second-leading cause of cancer-related mortality in the US.7,8 About 10–20% of pancreatic carcinomas are resectable and potentially curable, and the five-year survival rate is only 4%; hence, the majority of pancreatic cancer treatments are palliative in nature.9,10 Gemcitabine, nab-paclitaxel, and FOLFIRINOX are examples of modern chemotherapeutic drugs that have the ability to quickly confer resistance in pancreatic tumor cells. Therefore, in order to treat this dreadful condition, it is essential to develop more effective medicines.7,11
Interestingly, subcellular localization, concentration, and conformation of proteins in cells are maintained by a highly complicated and coordinated process known as protein homeostasis or proteostasis.12 It contains multiple pathways controlling protein transport, disposal, protein synthesis, and folding.13 In general, a proper balance between normal protein production and aberrant protein breakdown is required to preserve cellular proteostasis and guarantee normal cell function.14 In fact, stressors such as diseases, UV exposure, and aging increase the misfolding and mutation of proteins.15,16 Essentially, chaperones attempt to repair misfolded proteins, but when their function fails, the proteins accumulate and may cause cytotoxicity as well as abnormal cell states. The UPS is a "highly conserved system" that causes cells to permanently remove protein substrates and is essential for cells to efficiently and immediately degrade misfolded/damaged proteins through ubiquitination.17 Unfortunately, cancer cells can disturb normal protein synthesis and protein degradation and, in this regard, up-regulate the destruction systems, which leads to an increase in tumor suppressor protein degradation, which in turn causes apoptosis avoidance.18 Therefore, UPS is still a popular target for cancer treatment among researchers.19 Significantly, in the last decades, multiple effective chemotherapy regimens have been developed, which has led to the advancement of pancreatic adenocarcinoma treatment, but the prognosis of metastatic disease still remains poor compared to other cancers, and long-term survival is exceptional.20 Interestingly, UPS is still a popular target for pancreatic cancer treatment among researchers; however, despite the favorable results of UPS-based therapies in vitro and in vivo, the clinical results are not as promising as expected. Therefore, a deep understanding of it, is essential to achieving the maximum results. In this review, we attempt to investigate the UPS along with the search for the newest treatments for pancreatic adenocarcinoma based on its manipulation.
Pathophysiology of pancreatic adenocarcinoma
Involving genes
The pancreatic adenocarcinoma genome exhibits numerous amplifications, deletions, and rearrangements, along with a variety of large-scale chromosomal alterations.2 Somatic mutations have been used as molecular clocks to evaluate the molecular evolution of pancreatic tumors. According to this research, it will take a precursor neoplastic clone around 10 years to develop into a malignant clone, and it will take another few years for metastatic subclones to form from the original malignancy.21 Comparing a patient's primary pancreatic cancer and its metastases reveals that genetic instability persists after cancer spreads, with some genetic heterogeneity developing in different metastases, as well as that almost all of the key driver genes have been altered before the onset of invasive adenocarcinoma. These estimations show that a primary tumour can remain in the pancreas for a number of years before metastasizing, affording chances for screening,22 although taking into account the substantial variation in tumor behavior in different individuals.
Pancreatic cancer is mostly driven by four genes: KRAS, TP53, CDKN2A, and SMAD4.23 Oncogenes including EGFR, AKT2, MYB, and BRAF, as well as tumor-suppressor genes like FBXW7, MAP2K4, ACVR2A, TGFBR1, TGFBR2, STK11, ACVR1B, and EP300, are among the genes altered in a small percentage of (20%) pancreatic malignancies.22 The underlying genetic propensity of the great majority of people at familial risk is yet unknown.24
Oncogenes
KRAS
Even in low-grade pancreatic intraepithelial neoplasias, KRAS mutations and telomere shortening have been identified as the early genetic abnormalities; telomere shortening is believed to contribute to chromosomal instability. In contrast, invasive carcinomas and advanced intraepithelial neoplasias exhibit inactivation of TP53, BRCA2, and SMAD4.22 A highly frequent genetic anomaly in invasive pancreatic adenocarcinomas is KRAS oncogene mutational activation.22
The KRAS mutation has a significant impact on the onset and progression of the illness.23 KRAS mutations are present in more than 90% of pancreatic tumours, and they are among the early genetic changes in pancreatic carcinogenesis.25 KRAS2 oncogene mutations led to the activation of proliferative survival signaling pathways.26 Numerous studies have demonstrated that both individuals with resectable tumors and those with unresectable malignancies who carry the KRAS mutation have a poor prognosis. Functional analyses indicate that KRAS is necessary for the advanced pancreatic cancer to continue progressing.2
The KRAS gene produces the small GTPase protein KRAS, which serves as a molecular switch for a number of cellular activities by linking cell membrane growth factor receptors to transcription factors and intracellular signaling pathways.27 It has been determined that codon G12 hotspot mutations in KRAS are found in 95% of PDACs.28 KRAS is active when attached to guanosine triphosphate (GTP), but inactive once attached to guanosine diphosphate (GDP). In physiologic contexts, the GTP-bound state switches from GTP-bound to GDP-bound once KRAS intrinsic GTPase activation is induced by GTPase-activating proteins (GAPs).29
Over 80 downstream effector proteins and signaling pathways, including mitogen-activated protein kinase (MAPK)-MAPK kinase (MEK), rapidly accelerated fibrosarcoma (RAF), phosphoinositide 3-kinase (PI3K), and the mechanistic target of rapamycin (mTOR)-AKT-MEK-extracellular signal-regulated kinase (ERK), interact with the KRAS protein once it has bound GTP.27 Additionally, nuclear transcription factors (including ELK, JUN, and MYC) are activated, which promotes cell proliferation, differentiation, migration, conversion, adherence, and survivability.27 The intrinsic and GAP-stimulated GTPase activities of various KRAS mutations are distinctive. When it comes to KRAS mutations, G12C and G12D both have the greatest rates of intrinsic and GAP-stimulated GTP hydrolysis. Due to these variations, G12C inhibitors have been developed; nevertheless, there is no specific treatment for other mutant variants of KRAS.29
The most frequent mutational event in human pancreatic cancer is an acquired missense mutation in KRAS, which changes the protein's 12th codon from glycine to aspartate or valine and irreversibly activates it. KRAS encodes a small GTPase that resides at the plasma membrane and delivers signals to a number of downstream signalling molecules when growth factor receptors, such as EGFR, are activated.30 The intrinsic GTPase activity of RAS is compromised by the KRAS point mutation, and GAPs are unable to promote the transformation of GTP (active) to GDP (inactive). As a result of its constant association with GTP, the KRAS protein persistently activates downstream signaling pathways, supporting cellular activities such as proliferation and survival. INK4a-ARF, TP53, and DPC4-SMAD4 tumor suppressor pathways are genetically inactive in the majority of pancreatic carcinomas, despite oncogenic KRAS being activated in these tumors (as well as losses of heterozygosity of 9p21, 17p, and 18q chromosomes, respectively).27
RalA/B, RAF/MEK/ERK, and PI3K are the three signaling arms that are activated as a result of the small GTPase RAS being activated. Fig. 1 depicts these three arms as having several direct connections between proteins, denoted by solid black arrows, and indirect connections, shown by dotted black arrows.30 Inhibitory interactions are shown by solid red bars, whereas established regulatory feedback loops are represented by dotted red lines. KRAS, the activated RAS protein in pancreatic cancer, is caused by acquired oncogenic point mutations like the missense mutation G12D, which prevents GAPs (GTPase Activating Proteins) from hydrolyzing GTP (guanosine triphosphate) to GDP (guanosine diphosphate) and deactivating RAS.30 On the other hand, RAS (epidermal growth factor receptor) can be activated by receptor-related tyrosine kinases like EGFR. The activation of the adaptor protein GRB2 (growth factor receptor bound) and the GEF SOS, which removes GDP from RAS and activates the molecule by allowing the binding of GTP, occurs when extracellular growth factors like EGF (epidermal growth factor) bind to these receptors. RAF serine/threonine kinases are subsequently drawn to the cell membrane by KRAS-GTP, where they are turned on. B-RAF, the primary RAF molecule implicated in pancreatic cancer, when activated, initiates a phosphorylation cascade that activates MEK (mitogen-activated protein kinase) and ERK (downstream extracellular signal-related kinases) (Fig. 1).30
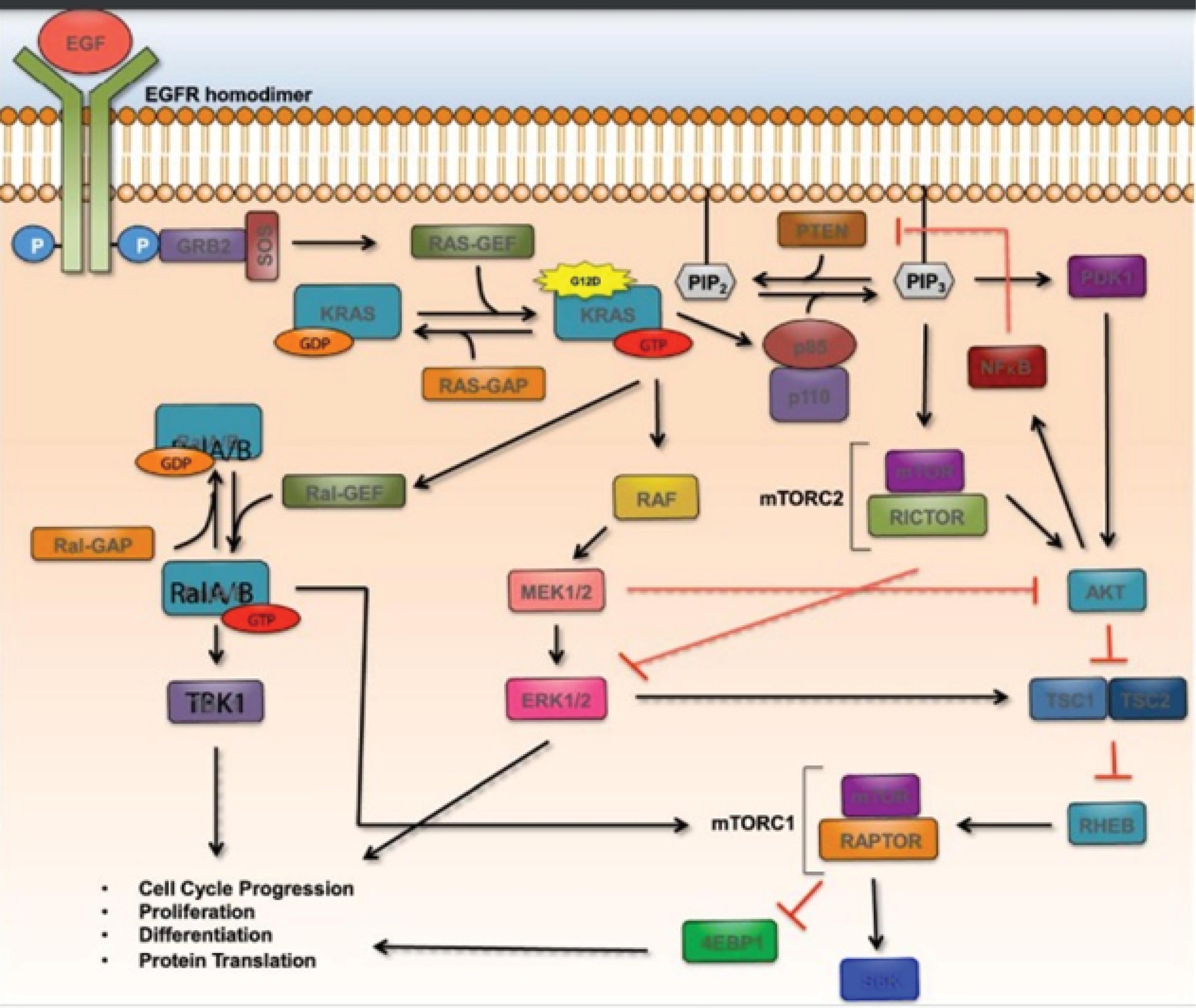
Fig. 1.
RalA/B, RAF/MEK/ERK, and PI3K are the three signaling arms that are activated as a result of the small GTPase RAS being activated. With permission from the Mann et al30 (Elsevier 2016).
.
RalA/B, RAF/MEK/ERK, and PI3K are the three signaling arms that are activated as a result of the small GTPase RAS being activated. With permission from the Mann et al30 (Elsevier 2016).
KRAS-GTP also stimulates PI3K signaling by activating the p110 subunit of PI3K (composed of p85 and p110). PIP2 (phosphatidylinositol 4,5-biphosphate) is converted to PIP3 by active PI3K (phosphatidylinositol 4,5 triphosphate). AKT (which is additionally referred to protein kinase B) is then promoted to become activated by PIP3 via activating PDK1 at the plasma membrane or the mTORC2 complex.30 AKT suppresses PTEN, a negative regulator of AKT, and stimulates NF-kB, which blocks the TSC1/TSC2 inhibitory complex of mTORC1. Last but not least, KRAS-GTP stimulates Ral-GEF, which in turn stimulates the Ral A/B small GTPases by allowing GTP to attach to it. One known downstream target of Ral A/B, which relieves NF-kB inhibition brought on by TANK, is TBK1 (TANK-binding kinase 1). Cell cycle progression, proliferation, differentiation, and protein translation are all impacted by all three signaling axes.30
By phosphorylating and then ubiquitinating FBW7, the KRAS mutation in pancreatic cancer reduced its expression. Furthermore, FBW7 (F-box and WD repeat domain-containing 7) reduced aerobic glycolysis in pancreatic cancer through activating the tumor suppressor thioredoxin-interacting protein (TXNIP), which is located in the mitochondria.31 Through the proteosomal degradation of myeloid cell leukemia-1 (MCL-1), an anti-apoptotic factor, FBW7 has been linked to anti-apoptosis and treatment resistance. The role of FBW7 in gemcitabine-resistant pancreatic cancer, however, has not been well studied. Researchers have shown that pancreatic cancer cells with overexpressed FBW7 were more sensitive to gemcitabine. In a different way than via transcription, FBW7 enhanced gemcitabine sensitivity by increasing the protein level of the equilibrative nucleoside transporter 1 (ENT1).31
KRAS activity is increased by mutations in p53 that drive sequence-specific alterations in RNA splicing. In order to encourage inclusion of cytosine-rich exons inside GTPase-activating proteins (GAPs), negative regulators of RAS family members, mutant p53 boosts production of the splicing regulator hnRNPK.29 KRAS activity is increased as a result of mutant p53-enforced GAP isoforms losing their cell membrane associations. In mutant KRAS/p53 PDACs, cytosine-rich exon insertion inhibits tumor development. Furthermore, spliceosome inhibitors make mutant p53 PDACs more susceptible to splicing suppression. These findings shed light on the treatments that target the co-enrichment of KRAS and p53 mutations in PDAC.29
Tp53
More than 50% of all malignancies in people have mutant p53 genes.3 In 50%–75% of pancreatic cancer cells that have been described, an aberrant TP53 gene has been found.26 This gene allows cells to evade DNA damage control checkpoints and, consequently, apoptotic signals. High-risk groups had considerably more KRAS and TP53 alterations, and these mutations were also strongly associated with a poor outcome.23 The tumor-suppressor activity of TP53 is rendered inactive by hotspots and truncating mutations. The development of more aggressive PDACs in mice with hotspot p53 mutations than in their heterozygous or null counterparts suggests that mutant variants of p53 have gain-of-function activities.29
The p53 pathway serves as a physical representation of the molecular link between apoptosis and the cell cycle. Nuclear phosphoprotein 53-kDa is produced by the p53 gene. Through the triggering of cell cycle arrest and death, p53 suppresses cell growth.3 More than 50% of PDAC patients have a p53 tumor suppressor gene mutation, which often involves missense changes to the DNA-binding domain.32,33 The presence of the p53 mutation in later-stage PanINs that have developed major dysplastic characteristics is consistent with a function in limiting the course of cancer.34 The selective pressure to eradicate p53 in these more developed PanINs could be partially caused by a buildup of genetic damage, such as telomere erosion and ROS, which activate p53-dependent DNA damage checkpoint responses. The widespread genomic instability in PDAC may both contribute to the spread of advanced disease and serve as a reason for it to be resistant to treatment options.34 ARF prevents the p53 protein from being targeted by MDM2 for destruction by the proteasome. In several cancer-relevant activities, p53 pathway activity would be diminished and p53 protein levels would be markedly reduced as a result of ARF deficiency. However, in 40% of instances of human PDAC, both p53 mutations and ARF deletions are present, suggesting that their roles in pancreatic cancer suppression are distinct and may not overlap.34
Smad4
The SMAD4/MADH4 gene is lost in 50% of pancreatic ductal adenocarcinoma cancer cells, which causes the TGF-β cell surface receptor to signal abnormally.26 The TGF-β signal transduction pathway contains the gene product Smad 4.35 One of the main signaling routes controlling the proliferation, differentiation, and death of distinct cell types, loss of sensitivity to transforming growth factor (TGF)-β-dependent signaling, is considered to play a significant role in the formation of tumors. Smad4, also called DPC4 (deleted in pancreatic cancer locus 4), is an important factor in TGF-β signaling's downstream regulation.36 Smad4/DPC4 was first discovered as a tumor suppressor gene for pancreatic cancer on human chromosome 18q21.1.37 Smad4/DPC4 mutations have been found in only 10% or fewer of other cancers, suggesting that Smad4 has a crucial role in TGF-β functional loss during the development of pancreatic tumors. In fact, Smad4 mutations are crucial for the development of malignant tumors. Because they are more invasive and angiogenic, tumors that lack functioning Smad4 are more likely to develop metastatic lesions.36 Patients whose pancreatic adenocarcinoma tumors expressed Smad4 protein had a considerably longer life after surgery to remove the tumor. Recent studies have demonstrated that certain Smad4 mutations target the protein for quick degradation via the ubiquitin-proteasome pathway, suggesting that Smad4 protein instability may be a factor in cancer's lack of cellular response to TGF-β.36
Through Smad4-regulated genes, TGF-β/Smad4 signaling promotes a tumor suppressive effect in normal pancreatic cells. On the other hand, TGF-β loses its tumor-suppressing action and behaves as a tumor promoter in tumor cells in some PDAC patients. Mutations in TGF- transduction and the absence of Smad4 signaling are to blame for the change in function.38 A progressive allele for PDAC has been identified as SMAD4 due to its loss in PanINs at a later stage.34 Transforming growth factor beta (TGF-β) signaling pathways are centrally mediated by Smad4, a member of the Smad family of signal transducers. Numerous physiological activities, such as cell development, differentiation, proliferation, fibrosis, and scar formation, are supported by the TGF-β signaling pathway. Through the stimulation of angiogenesis and the inhibition of the immune system, it also plays a significant part in the growth of malignancies.38
Ligand TGF-β causes transphosphorylation of the receptors by binding a complex of Type I and Type II transmembrane receptor serine/threonine kinases on the cell surface. As a result, chosen Smads are phosphorylated at C-terminal serines by the correspondingly activated receptors, and these receptor-activated Smads (R-Smads) then join forces with a common Smad4 to create a complex.38 By interacting physically and working together with DNA-binding transcription factors, activated Smad complexes go into the nucleus, where they influence the transcription of their target genes. Along with Smad4-mediated signaling, Smad2/3 and Tifl form a complex, and Smad complexes subsequently go into the nucleus to control the transcription of target genes. Smad6 or Smad7 block the activation of R-Smads by Type-I receptor kinases. Additionally activating Ras and ERK in a Smad-independent manner and causing cancer are phosphorylated TGF-β receptors.38
Genes related to apoptosis inhibition
The extrinsic, or death-receptor, pathway and the intrinsic, or mitochondrial, pathway are the two alternate intracellular methods to induce apoptosis. Apoptosis, also known as programmed cell death, is a key regulator of healthy tissue homeostasis. Apoptosis is a physiological process that helps the body get rid of extra, damaged, and diseased cells. A basic anti-neoplastic process that stops the growth of tumors in normal cells is apoptosis.39 PDAC cells, like other cancer cells, have developed resistance mechanisms that focus on the death receptor level and prevent TNFa, FasL, and TRAIL from properly initiating apoptosis.40 Furthermore, by overexpressing Bcl-family proteins (Bfl1, BCL-XL, and MCL-1) and limiting caspase activation, such as by inhibiting caspase inhibitors, PDAC cells are protected from the mitochondrial cascade of death.
By overexpressing caspase inhibitors (cIAP, XIAP1, survivin), downregulating procaspase gene expression through epigenetics, or directly inhibiting caspase by cysteine nitrosylation,39 lack of responsiveness to apoptotic stimuli is primarily responsible for tumor formation, progression, and resistance to the majority of oncologic therapy. Defects in the apoptotic pathway can lead to cancer cell resistance since chemotherapy and radiation work largely by triggering apoptosis.3 Multiple routes are used by tumor cells to down-modulate apoptosis.41
Genes related to metastasis
In fact, a number of genes, including integrins, members of the cadherin family, cell-cell adhesion molecules (CAMs), and others, have been discovered to modulate the formation of metastatic lesions. In addition, it appears that the formation of distant organ metastasis is dependent on metalloproteinases, the uPA/uPAR system, and the metastasis suppressor gene KAI1. Not to mention, the newly discovered involvement of lymphangiogenesis and angiogenesis in invasion and metastasis looked to be crucial for the spread of tumor cells.42
Angiogenesis is one method of encouraging cancer spread, as TGF-β affects endothelial cell migration and proliferation as well as capillary formation, hence boosting angiogenesis and vascular metastasis. Vascular endothelial growth factor (VEGF), which is brought on by TGF-β, is a significant contributor to this vascularization. Hypoxia, a typical milieu in a developing tumor, is the main stimulant for its expression.43 Through the activation of inflammatory cells in the tumor environment, TGF-β stimulates the secretion of proangiogenic factors like matrix-metalloproteinases 2 (MMP2) and MMP-9 and inhibits the production of anti-angiogenic factors like the protease inhibitor TIMP. In addition to VEGF, TGF-β also induces connective tissue growth factor (CTGF) to further increase angiogenesis.38
A new mechanism for the invasion and metastasis of cancer has recently been identified as the exosome secretion signaling pathway.44 Exosomes are membrane vesicles that are formed from multivesicular endosomes and range in size from 40 to 100 nm. They are discharged after fusing with the plasma membrane. Exosomes are used by cancer cells to connect with their surroundings by releasing growth factors and cytokines and delivering surface proteins.45 Exosome messaging has been shown to aid in the establishment of the metastatic niche and tumor immune evasion. The Rab family of small GTPases, which controls vesicle fusion and trafficking, includes RAB27A. A human melanoma cell line and melanocytes were used to isolate it first. Most normal tissues and tumor cell lines have been shown to express it. Cancers of the breast, lung, bladder, rectal, prostate, and liver have abnormal RAB27A expression, which has been linked to aggressive tumor behavior.46 RAB27A is expressed ectopically in glioma cell lines, where it improves cell viability, encourages proliferation and invasion, and inhibits apoptosis.47
In glioma and hepatocellular carcinoma, RAB27A expression has been demonstrated as a prognostic marker in vivo.46 In recent research, it has also been shown that TP53 plays a role in the control of exosome secretion. On the one hand, exosomes can preserve the TP53 protein to foster an environment that is favorable for tumors,48 and on the other, TP53 regulates exosome synthesis and secretion by regulating the transcription of important endosomal compartment regulators. However, it is unknown how RAB27A and TP53 work together to control exosome secretion. PDAC has been shown to accumulate TP53 protein and to have TP53 mutations.46 High levels of TP53 and RAB27A protein expression were linked to distant metastasis and tumor stage, respectively. Although researchers did not find an association between high RAB27A protein expression and TP53 expression, high RAB27A protein expression was also linked to vascular invasion. High RAB27A and high TP53 expression were shown to be substantially linked with patients' poor overall survival in both univariate and multivariate analyses.46,49
Other genes
The CDKN2A tumor suppressor gene is mutated in 95% of pancreatic ductal adenocarcinoma cells, which results in the loss of the p16 protein and, consequently, the control of the G1-S transition of the cell cycle.26 In pancreatic tumors, the oncogene MDM2, which codes for an E3 ubiquitin ligase, is also overexpressed.50
Epigenetic changes
Pancreatic tumors may have altered gene function due to epigenetic alterations.51 Alterations in DNA methylation, histone modifications, and non-coding RNAs are examples of epigenetic dysregulation. The tumor-suppressor gene CDKN2A, whose promoter methylation and gene silencing in pancreatic malignancies were originally described, only exhibits epigenetic silencing in neoplasms without genetic inactivation.52 Only a tiny number of traditional DNA repair and tumour suppressor genes, such as MLH1 and CDH1, are epigenetically silenced in pancreatic malignancies. Other genes, such as CDKN1C, RELN, SPARC, TFPI2, and others, are frequently the focus of abnormal methylation and gene suppression in pancreatic tumors. The diagnostic or biological significance of several of the most often aberrantly hypermethylated genes in pancreatic neoplasms has been assessed.22 There have also been reports of promoter hypomethylation for a number of genes, including the mucin, SFN, MSLN, and S100A4 genes.53,54 The genesis and spread of cancer appear to be influenced by changes in microRNA expression. It is believed that the overexpression of a number of microRNAs, including miR-200, miR-155, miR-34, and miR-21, in pancreatic tumors contributes to the development of the disease. Additionally, given that they are persistent and detectable in human plasma, microRNAs may be valuable diagnostic indicators. The aggressiveness and patterns of tumor growth in pancreatic tumors are likely influenced by genetic and epigenetic changes.22
Alternate RNA splicing in cancer results in altered gene expression, the production of functionally aberrant proteins, and/or nonfunctional mRNAs. Splicing factor mutations, dysregulated splicing factor expression, and oncogenic transcription are all factors in this process. It is unknown if and how PDAC may impact RNA splicing.29
Ubiquitin-proteasome system (UPS)
The UPS (ubiquitin-proteasome system) For the first time in the 1980s,55-58 Hershko and Varshavsky looked into the ubiquitin-regulated degradation of proteins. Notably, Hershko et al went into more detail on the E1, E2, and E3 enzymes to better understand ubiquitin and the accompanying proteolysis. Hershko made further findings that went beyond only this, such as the biology of UPS, the requirement of protein cleavage, and the unique physiological role of UPS in transcription, cell cycle progression, protein synthesis, DNA repair, and stress response. In reality, throughout the 1990s, such discoveries led to a major advancement in the ubiquitin area. In addition, Hershko et al's discovery of proteolysis via the UPS led to the development of UPS.57 Vershavsky's scientific research in 1980 resulted in a basic comprehension of intracellular circuits.58 Hershko, Rose, and Cichanover won the 2004 Nobel Prize in Chemistry as a result of their research, though.
The 26S proteasome, a tiny protein aggregation made up of two 19S regulatory caps (RP) and a 20S core component, is only found in mammals. It’s fascinating center, which is surrounded by two holes and suitable for extracting proteins, is empty.59 Actually, the substrates can enter the core for degradation through the holes, which are close to the 19S caps as regulatory subunits. An important function of these caps is to import proteins into the core particle (CP) by cleaving them.59 The 11S complex is another component that functions similarly to the 19S cap, is related to CP, and is expressed during immunological reactions. The 11S complex normally functions to degrade proteins created during viral infection. In general, IFN-γ expression causes the immunoproteasome to be produced, which results in the generation of MHC I (major histocompatibility I)-associated peptides in viral infections.59
Each of the four heptamer rings in the 20S complexes has two distinct α and β subunits. The β subunit is catalytic and α acts as a structural subunit. Surprisingly, these subunits are identical to one another and are also pseudoenzymes. Through their shared N-terminal domains, these subunits are connected.60 The subunit often flanks the CP, and its N-terminal domain acts as a gate to block the entry of undesirable proteins.61 Seven proteolytically active subunits that proteolyze proteins form the two inner rings. Particularly, β1, β2, and β3 perform the roles of trypsin, chymotrypsin, and caspases, respectively.62 These subunits can also exist in the i, i2, and i5i, i1 forms that are produced in response to disruptive signals like cytokines, especially interferon (IFN-γ). Immunoproteasomes, which are proteasomes, include these subunits.63 The 19 proteins that make up RP in eukaryotes are further broken down into two subunits. 10 proteins (Rpn3.5-12.15) and subunits make up the lid.64 While the 19S cap functions as a deubiquitinase, the base subunit serves as a gateway and imports proteins. Actually, protein degradation by the 20S core is triggered by the 19S cap. The 19S cap's primary job is to open the gate so that proteins can get to the CP.64
Protein ubiquitination is the first stage of the proteolytic reaction. Three enzymes are needed for the proteasome, which targets proteins for breakdown. First, ATP is hydrolyzed by the ubiquitin-activating enzyme (E1) before the ubiquitin molecule is adenylate. The cysteine-active site of E1 is then translocated by this ubiquitin.65 Ultimately, adenylated ubiquitin is transferred to the second enzyme, a ubiquitin-conjugating enzyme (E2). Then, enzymes called ubiquitin ligases (E3) recognize target proteins and catalyze the translocation of ubiquitin from E2 to proteins. Basically, at least four ubiquitin molecules should tag the target protein, and this should happen before the operculum can recognize the protein.66 Furthermore, ubiquitin molecules are linked through leucine residues to form ubiquitin chains. After ubiquitination, the target protein should be sensed by the ubiquitin receptors. These receptors feature one or more ubiquitin-associated ribs (UBA) and an N-terminal domain that resembles ubiquitin (UBL). UBA binds ubiquitin by three helical connections, while RP checks UBL. These receptors enable the transfer of polyubiquitinated proteins to the proteasome.67
Proteins are recognized by RP after being ubiquitinated, as the former described. The protease and its CP should then come into contact with this protein.59,68 It must first be deubiquitinated, and this deubiquitination creates a compartment for nucleoprotein degradation that increases the catalytic activity of the proteasome.69 The N-terminal region of the subunit fills a very small (about 13 Å) opening in CP. Consequently, the unfolding of the protein is required first.59 It is important to point out that some research has focused on altering the subunit to prevent proteasome action. Unexpectedly, in order to leave the CP, all proteins need to unfurl by 20 amino acids.59 Importantly, some characters have the ability to obstruct the mining process. Disulfide bonds, for instance, prevent this process from happening.70 Long glycine and alanine sequences can also hinder unfolding and lower the effectiveness of proteolysis.71 It is interesting to think that mutations that interfere with this mechanism could help find efficient proteasome inhibitors. The structure of tiny peptides that are produced during degradation processes is often amenable to further processing into smaller amino acid chains (Fig. 2).72
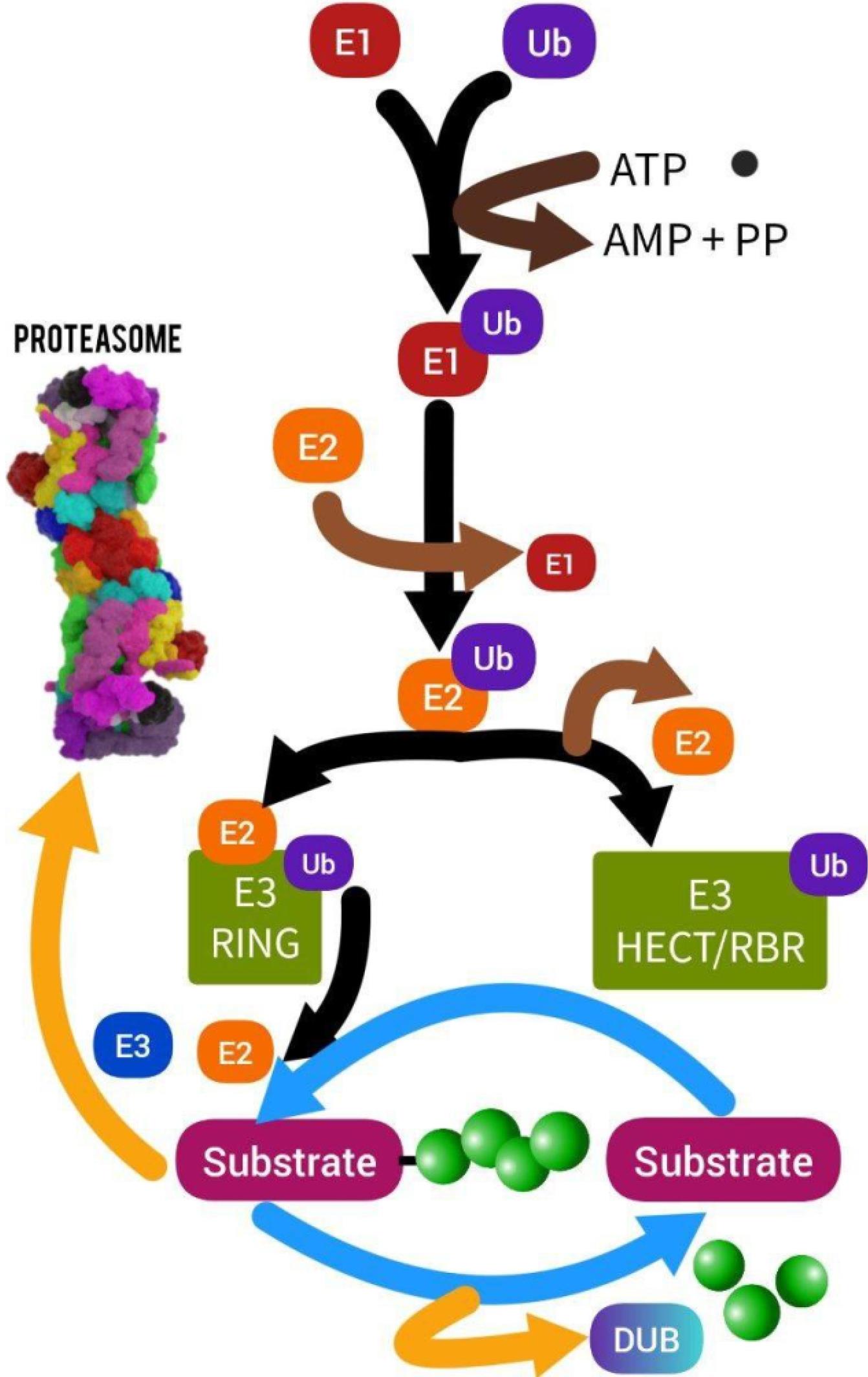
Fig. 2.
Ubiquitin-proteasome system(Designed by InShot & PicsArt).Protein ubiquitination is the first stage of the proteolytic reaction. Three enzymes are needed for the proteasome, which targets proteins for breakdown. First, ATP is hydrolyzed by the ubiquitin-activating enzyme (E1) before the ubiquitin molecule is adenylate. The cysteine-active site of E1 then receives this ubiquitin by translocation. The second enzyme, ubiquitin-conjugating enzyme (E2), receives adenylated ubiquitin in the end. The transfer of ubiquitin from E2 to proteins is next catalyzed by enzymes known as ubiquitin ligases (E3), which also detect target proteins
.
Ubiquitin-proteasome system(Designed by InShot & PicsArt).Protein ubiquitination is the first stage of the proteolytic reaction. Three enzymes are needed for the proteasome, which targets proteins for breakdown. First, ATP is hydrolyzed by the ubiquitin-activating enzyme (E1) before the ubiquitin molecule is adenylate. The cysteine-active site of E1 then receives this ubiquitin by translocation. The second enzyme, ubiquitin-conjugating enzyme (E2), receives adenylated ubiquitin in the end. The transfer of ubiquitin from E2 to proteins is next catalyzed by enzymes known as ubiquitin ligases (E3), which also detect target proteins
Regulating factors
Table 1 summarizes the regulating factors that have been targeted in PAC. Each will be explained in detail in the next sections.
Table 1.
UPS regulating factors and PAC
|
|
Name
|
Function
|
Study related to PAC
|
Ref
|
| Intrinsic Factors |
Redox Signaling |
They are regarded as crucial mediators in cellular signaling cascades and as regulators of metabolic processes |
KRAS and p53, two proteins involved in the creation and regulation of ROS, are frequently altered genetically in pancreatic ductal adenocarcinoma (PDAC). These findings prompted the suggestion that antioxidants be used to stop PDAC from developing and relapsing. Ref-1, a multifunctional DNA repair-redox signalling protein, also activates a number of transcriptional factors (TFs) including NF-B (RelA), STAT3, and AP-1 through its redox signalling activity. |
73,74
|
| Transcription Factors |
Upon treatment with proteasome inhibitors, transcriptional profiling showed a coordinated Rpn4-dependent elevation of all proteasomal subunits, indicating that Rpn4 is in charge of the cell's capacity to make up for reduced proteasome function. In order to activate Rpn4, transducing transcription factors must bind to the gene's promoter. These transcription factors include Yap1, the heat-shock transcription factor 1, and the Pdr1 and Pdr3 drug resistance-related transcription factors. |
PDAC is one of several human malignancies linked to the transcriptional coactivator yes-associated protein (YAP), a key downstream effector of the Hippo pathway. YAP is emerging as a prospective therapeutic target due to its significance in cancer. Additionally, heterozygous KrasMUT works in conjunction with oncogenic activation of YAP to promote PDAC carcinogenesis. In addition, YAP is necessary for cancer to return in the absence of KRAS. Recent research has also shown that YAP, rather than oncogenic KRAS, is a key factor in squamous subtype PDAC. These results point to a crucial realisation: YAP not only functions as a PDAC driver downstream of KRAS but also frees PDAC from KRAS reliance. |
20,75,76
|
| Heat shock factor 1 (HSF1), a highly conserved transcriptional factor that drives the normal proteotoxic stress response, is implicated in the invasion and metastasis of pancreatic cancer. |
| Extrinsic Factors |
E1 inhibitors |
The E1 enzymes' strong potential for cancer therapeutic targeting, none of these inhibitors are currently clinically effective due to their subpar pharmacokinetic features. |
In a preclinical syngeneic PDAC mouse model, Kumar et al investigated the potential therapeutic value of TAK-981, a new highly specific and powerful small molecule inhibitor of the small ubiquitin like modifier (SUMO) activating enzyme E1. They discovered that PDAC patient samples exhibit higher levels of SUMOylation than normal pancreatic tissue, a reversible post-translational modification necessary for cell cycle progression. TAK-981 caused a pause in the G2/M cell cycle, failed mitosis, and problems in chromosomal segregation in PDAC cells by reducing SUMOylation at the nanomolar level. In the KPC3 syngeneic mouse model, TAK-981 effectively reduced tumour burden without showing signs of systemic toxicity. |
77
|
| E2 inhibitors |
E2 inhibitors can function as an effective inhibitor in clinical applications because it stimulates tumour suppressors to assemble and inhibits cell growth |
IAPs perform as E3 ubiquitin ligases and aid in the development, spread, and metastasis of pancreatic cancer. Despite the fact that IAP-targeted treatments have been created and demonstrated anticancer activity in preclinical settings, none of them have yet received regulatory approval. The expression of IAPs and E2 UbcH5c are positively associated in pancreatic cancer. Overexpression of UbcH5c is linked to a bad prognosis for pancreatic cancer. A small-molecule UbcH5c inhibitor known as DHPO was discovered by Qi et al, and it directly bonded to the UbcH5c protein. In vitro, pancreatic cancer cells' migration and invasion were decreased by DHPO, which also caused apoptosis and hindered cell viability and colony formation. |
78
|
| E3 inhibitors |
The E3 enzyme, the final one in the ubiquitin cascade, controls substrate specificity. |
There is growing evidence that E3 ubiquitin ligases play critical roles in the development, spread, and response to therapy of cancer as well as in determining prognosis. The majority of the eight prognostic indicators have been associated with cancer. Further functional experiments demonstrated that RNF223 may have an oncogenic role in the development of pancreatic cancer. RNF223 was discovered by Feng et al to be an independent prognostic marker in pancreatic cancer. |
79-81
|
| The E3 ubiquitin ligase TRIM29 accelerates the growth and spread of pancreatic cancer by maintaining the stability of the Yes-associated protein 1 (YAP-1). |
| 20S core inhibitors |
They are used to inhibit the proteasome alpha and beta subunits, which are involved in the malignancy of cancers. |
Bortezomib (PS-341) is a proteasome inhibitor that has proven to be highly effective against pancreatic cancer cells. The fundamental processes, nevertheless, are not completely known. |
82
|
| 19S cap inhibitors |
ATPase inhibitors |
This inhibitor influences the development of autophagosomes and can cause the buildup of LC3-II, a common marker for autophagosomes. NMS873, a powerful and specific p97 ATPase allosteric inhibitor, is another inhibitor that can trigger the unfolded protein response and hinder autophagosome formation. In cancer cells, it can also have antiproliferative action. |
Proton Pump Blockers Selectively Targeting H + , K + -ATPases in Pancreatic Cancer and Stellate Cells Can Slow the Progression of Pancreatic Adenocarcinoma. |
83
|
| Rpn-13 inhibitors |
Rpn-13 has received more attention than Rpn-10 up to this point since it is unneeded in healthy cells and is highly expressed in a number of malignancies, including ovarian, MM, and gastric cancer. |
MM, ovarian, cervical, pancreatic, and colorectal cancers are only a few of the malignancies that have high Rpn13 expression. Anchoori et al target the ubiquitin receptor RPN13 inside the proteasome's 19S regulatory particle to create ubiquitin-proteasome system inhibitors that are effective against solid tumours. In either a 2D or 3D culture configuration, they discovered that human pancreatic cancer-derived cell lines were much less susceptible to these substances using the MTT test. |
84
|
| DUBs inhibitors |
The action of E3 ligases is counteracted by deubiquitinating enzymes (DUBs), also known as deubiquitinases, which remove ubiquitin from their target proteins. Additionally, they can participate in ubiquitin maturation, editing, and recycling. |
By targeting PD-L1 ubiquitination and stimulating the infiltrating CD8 + T cells, USP8 depletion improved immunotherapy. According to this study, USP8 deubiquitinated PD-L1 and increased the expression of this protein in pancreatic cancer. Activating cytotoxic T cells with anti-PD-L1 and a USP8 inhibitor reduced tumour development. |
81
|
| PROTACs |
PROTACs form a ternary complex with a protein of interest (POI) and an E3 ligase that encourages the target protein's lysine ubiquitination and subsequent proteosomal destruction. |
The "RC-U" "U-box-based" fusion E3 ligase successfully engaged, ubiquitinated, and encouraged KRAS oncoprotein degradation at the post-translational stage. |
85
|
Intrinsic factors
Redox signaling
Initial research focused mostly on reactive oxygen species (ROS) and reactive nitrogen species (RNS) as potentially harmful metabolic byproducts. Our knowledge of their function in metabolism has significantly altered over the past ten years, and they are now regarded as crucial mediators in cellular signaling cascades and as regulators of metabolic processes.86 The varied amounts of extracellular and intracellular free radicals have a significant impact on proteostasis, with either moderate or severe consequences. For the purpose of controlling redox signaling and preserving redox homeostasis, cells contain effective enzymatic and non-enzymatic mechanisms.87,88 The food is the primary source of antioxidants, although they can also be received via exogenous sources.89
The functioning of the cellular and molecular processes that guarantee cellular integrity is compromised by oxidative stress, as previously mentioned, which leads to the disruption of homeostasis. Proteostasis is significantly affected by oxidative stress, and the proteasome system is essential.86 Identifying and eliminating oxidatively changed or damaged proteins, proteasomes, and more especially the 20S complex, are essential for the preservation of redox equilibrium. Such substrates must be broken down by the 20S complex without the use of ATP since protein oxidation encourages protein unfolding.90,91 Exposure to H2O2 or O2 has been shown to more than tenfold accelerate proteasome-mediated degradation.92,93
It has been demonstrated that the 26S proteasome disassembles under oxidative stress to recover whole 20S and 19S particles.92 Additionally, it has been demonstrated that 20S proteasomes are more resistant to oxidative stress than 26S proteasomes, whose normal activity is severely compromised.94 It has also been demonstrated that the immunoproteasome degrades oxidatively altered proteins more effectively when the 11S regulatory particle is connected to it as opposed to operating alone.95,96 Constitutive exposure to reactive species affects the proteasome (and especially the 26S proteasome) in addition to cellular proteins.97 A variety of post-translational modifications of certain subunits, including glycoxidation, glutathionylation, lipoxidation, and carbonylation have also been demonstrated to be brought on by oxidative stress.98 In particular, it has been demonstrated that S-glutathionylation is a post-translational modification that contributes to the redox control of the proteasome.99 Additionally, under oxidative stress, 26S proteasomes are more directly impacted by modifications such as glycation, ubiquitylation, and carbonylation, as well as binding to lipid peroxidation byproducts and glutathione.100-104 The functional significance of these alterations is uncertain, though. Carbonylation is a well-known indicator of protein oxidation.105-107 yet 20S proteasomes have been shown to have a low degree of carbonylation.108 The insertion of carbonyl groups in the a2, a4, a6, and b3 subunits of pure mammalian 20S proteasomes is the primary effect of oxidative damage induction.104,109
Certain amino acid residues may interact with sugar carbonyls like glyoxal under oxidizing circumstances to produce glycation products. Damage to proteins is caused by these advanced glycation end products (AGEs), such as N-carboxymethyl-lysine.110 Proteasome subunits b1, b5i, b7, a1, and a2, as well as the 19S subunits Rpn11, Rpt1, and Rpt2 showed increased glycation with aging.100,109 The secondary protein change is production of 4-hydroxy-2-nonenal (HNE). After oxidative cell injury, HNE is a significant byproduct of lipid peroxidation.111 It is known that both DNA and proteins react with this extremely cytotoxic substance. Particularly vulnerable to HNE alteration are exposed histidine, cysteine, and lysine residues, which can result in inter- and intramolecular cross-linking and protein aggregation and inactivation.111,112
HNE addition has been connected to proteasome suppression following oxidative stress and throughout aging.103,108 HNE-modified proteasomes can be seen after causing oxidative damage for a few hours, but complete recovery occurs within 48 hours.103 This period of time fits the reported drop and rebound of proteasome activity quite well.103 In unprocessed cell extracts as well as purified proteasomes, oxidation-induced alterations in proteasome activity may be seen, suggesting that processes other than direct HNE addition to proteasomal subunits are at play. For example, oxidative damage reduces all three hydrolytic activities in rat heart extract, whereas only the trypsin-like activity was reduced in the isolated 20S proteasome from the heart that had been treated with HNE.108 According to one explanation, the formation of protein aggregates, or HNE-linked proteins, inhibits the proteasome's ability to do its job.113 However, it's likely that there are other oxidative proteasome modifications at work as well. Proteasomal function was also shown to be diminished by S-glutathionylation.101,102,114 Inhibition of chymotrypsin-like activity in mammals is correlated with sglutathionylation of the 19S complex subunits Rpn1 and Rpn2.114 However, the stability and integrity of the 26S particle are unaffected by this S-glutathionylation. S-glutathionylation of the proteasome has been demonstrated to reduce proteolytic activity in yeast.102 S-glutathionylated proteins are quite uncommon in general.115
Even in normal growth settings, minor quantities of S-glutathionylation have been seen in proteasomes,116 although it is still unclear what physiological importance this finding holds. When glutathione is taken out of the 20S protease in vitro by treatment with, say, dithiothreitol, the proteolytic activity is fully recovered.116 Deglutathionylation of the proteasome is most likely accomplished by enzymatic catalysis in a healthy cell, though. Grx2, the main glutaredoxin in yeast, may contribute to the glutathione's release from 20S proteasomes.116 The proteasome and the cytosolic yeast thioredoxins Trx1 and Trx2 interact,117 and in vitro, Trx1 and Trx2 have the potential to catalyze the deglutathionylation of the 20S proteasome.116 The 26S proteasome consistently connects with the thioredoxin Txnl1 in fission yeast and human cells.118-120 Txnl1, however, does not seem to encourage glutathione release from the 26S proteasome.121
A vicious cycle between the buildup of cross-linked protein aggregates and the highly oxidized cellular protein load results from the proteasome's function being impaired.113 More evidence for the relationship between the proteasome and oxidative stress comes from the discovery that cells treated with proteasome inhibitors have greater ROS levels.122 Data show that NADH/NAD+ is essential for the proteasome's operation during redox control. It has been demonstrated that NADH keeps the proteasome concentrations within the normal range.123
Depletion of NAD+ in myeloma cells also increases the effectiveness of the proteasome inhibitor bortezomib, indicating an active interaction between NAD+ and the proteasome.124 As previously indicated, Nrf2, which is activated in response to oxidative stress, has been demonstrated to control proteasome gene expression.125 In Drosophila melanogaster, this adaptive reaction has recently been shown to rely on both sex and age.126 It has also been demonstrated that the Nrf2/Keap-1 axis is crucial for stem cell development, where ROS levels are continually changing. More precisely, after receiving sulphoraphane therapy, Nrf2 is activated and manages the expression and upregulation of the proteasome, which controls the pluripotency of human embryonic stem cells (hESCs).127 UPS is compromised during oxidative stress following As (III) exposure to Ub-deficient N2a neuroblastoma cells. This impairment is brought on by an increase in p65-Nrf1, a type of Nrf1 that competes with p95-Nrf1 and Nrf2 in the nucleus and lowers proteasome activity and expression.128
Transcription factors
Although gene expression is required for the development of the proteasome, the control of proteasomal gene expression has long been disregarded. In eukaryotes, the yeast system provided the first example of coordinated regulation of proteasomal gene expression. The proteasome-associated control element (PACE), which is frequently found upstream of genes that make proteasomal components, is a cis-element that activates transcription in Saccharomyces cerevisiae. The transcription factor Rpn4 uses PACE as a target sequence to promote the expression of the proteasomal gene.129 The extraordinarily short-lived Rpn4 protein not only triggers transcription, but it also serves as a substrate for the 26S proteasome. A negative feedback loop is produced in yeast as a result of this regulatory mechanism when the protein that initiates the formation of the proteasomes is eliminated by the newly formed proteasomes.130 Rpn4 is crucial for yeast to have balanced amounts of proteasomal subunits and, consequently, for yeast to preserve balanced intracellular proteolysis. Rpn4 is in charge of the cell's capacity to make up for decreased proteasome function, as shown by the transcriptional profiling that showed a coordinated surge in all proteasomal subunits when proteasome inhibitors were applied.131 Over the past ten years, several groups have shown that Rpn4 is a significant stress-responsive mediator that is essential for cell survival under a variety of stressful circumstances.132-136
In order to activate Rpn4, transducing transcription factors must bind to the gene's promoter. These transcription factors include Yap1, the heat-shock transcription factor 1, and the Pdr1 and Pdr3 drug resistance-related transcription factors.132,135 As a consequence, oxidative stress indirectly increases proteasome production through Yap1-mediated activation of Rpn4. Additionally, the unfolded protein response (UPR) or proteasome inhibition can directly activate Rpn4.134 The proteasome genes, genes producing proteins associated with the proteasome, genes linked to the ubiquitylation machinery, genes involved in DNA repair, and many other cellular functions are all induced by Rpn4 in response to stress.136 No Rpn4 homologs or transcriptional regulatory elements like PACE could be found in higher eukaryotes, yet Drosophila and human proteasome abundance is likewise regulated by negative feed-back loops.137,138 The 5' untranslated sections of the mRNAs are necessary for the induction of proteasome gene expression in Drosophila in response to proteasome inhibitions.137 It is yet unknown what is mediating this induction, though.
Extrinsic factors
E1 inhibitors
Only two inhibitors, PYZD-4409 and PYR-41, have been recently introduced.139,140 PYR-41 controls the stability of the NF-kB inhibitors, which appears to prevent the nuclear factor -light-chain enhancer of activated B cells (NF-kB) from becoming active (IkB). Additionally, it prevents the tumour suppressor p53 from being destroyed, which raises p53 transcription.140 PYZD-4409 slows the development of tumors by inducing stress-induced apoptosis in leukemia mice model and cancer cells.139 Despite the fact that these results highlight the E1 enzymes' strong potential for cancer therapeutic targeting, none of these inhibitors are currently clinically effective due to their subpar pharmacokinetic features.
E2 inhibitors
CDC34, a typical E2 enzyme for cullin-ligase complexes, is an allosteric enzyme that is inhibited by CC0651. CC0651 can function as an effective inhibitor in clinical applications because it stimulates tumour suppressors to assemble and inhibits cell growth.141 However, due to pharmacokinetic issues, its development is fraught with difficulties.142 UBC13 (encoded by UEV1A), an E2 enzyme that controls the NF-kB pathway induction by generating chains dependent on ubiquitin K63, is yet another candidate for cancer treatment. It has been verified that the NSC697923 inhibitor may prevent UBC13 from forming K63 chains and can also influence the survival and proliferative capacity of the bigger B cells in lymphoma.143 IkB kinase can be inhibited by the well-known NF-kB pathway inhibitor BAY-11-7082. As a result of its ability to prevent ubiquitin from binding to UBC13, it can also limit the production of the K63 chain, acting similarly to NSC697923 (a cell-permeable and specific inhibitor of E2).144 Whereas the E2 enzyme inhibitors offer a lot of potential for treating cancer, they have only been tested in preclinical research thus far.145
E3 inhibitors
The E3 enzyme, the final one in the ubiquitin cascade, controls substrate specificity. Approximately 600 E3 ligases have been discovered so far. Since each E3 ligase is capable of binding and ubiquitinating a specific set of substrates, inhibition of one E3 is anticipated to only have an impact on the pathways that are controlled by that enzyme.146,147 In contrast to the case of proteasome or E1 inhibitors, the selectivity of ubiquitination offered by the E3s may solve, at least in part, the specificity issue raised above. Theoretically, more precise targeting of a small number of substrates might result in a focused therapy with fewer hazardous side effects.147
Three groups of E3s may be distinguished: those that include RING, U-boxes, and HECTs. Both RING and U-box operate by concurrently binding to E2, ubiquitin, and substrate, but none has intrinsic catalytic activity. Instead, they just act as a framework for the ubiquitination process. Thus, the discovery of allosteric or protein-protein inhibitors is required to target RING or U-box E3s. HECT E3s, on the other hand, have inherent enzymatic activity; therefore, inhibiting them entails blocking the catalytic site.148 Despite significant research efforts, there have only been a small number of E3 inhibitors discovered, in part because researchers have mostly concentrated on interrupting the enzyme/substrate interaction, which is thought to be more challenging to target than a catalytic site. Because of this, despite the fact that HECT E3 offers an easier and more promising druggable target than RING enzymes, they haven't drawn as much interest as prospective cancer therapies. Since RING-finger-type enzymes are the focus of the bulk of small chemical inhibitors identified so far, several are now being tested in clinical studies.148
A significant amount of research work has gone towards finding E3 inhibitors for the handful of E3s that have been identified to have a role in the growth of cancer. These E3s include MDM2, IAP, and SCF. The three primary approaches used to create inhibitors for E3 ligases are (i) to block their enzymatic activity directly, (ii) to target the substrate binding interface, and (iii) to interfere with the protein's production by blocking transcription or translation.149 Numerous E3s have an auto-ubiquitination ability that typically promotes their degradation, which is a problem when targeting the enzymatic activity. Because of this, inhibiting E3 activity frequently prevents autoubiquitination as well as substrate-directed activity, stabilizing the E3 protein and its substrates in the process.149 A stabilized pool of E3 that still binds to its substrates but does not ubiquitinate them could be detrimental to the substrate activity. For instance, the formation of E3-substrate complexes can make it more difficult for other proteins to bind to the substrate. Consideration should be given to the effects of completely halting E3 activity in light of this concern.149 Please see the reference at https://doi.org/10.1098/rsob.200390 to learn more about E3 inhibitors.18
20S core inhibitors
Although it appears that proteasome alpha subunits (PSMAs) have a role in the formation of human cancers, the expression patterns and prognostic importance of specific PSMAs in the bulk of these tumors are still unclear. Only a small number of the seven alpha subunits (PSMA1–7) have been demonstrated to be associated with cancer. Because of this, despite the fact that inhibitors of these subunits are rarely employed, PSMAs are involved in a variety of human malignancies. Cron et al150 in 2013 investigation of whole-genome RNAi screens to find knockdowns that consistently increase non-small cell lung cancer (NSCLC) cytotoxicity was the only report of their application to date. Despite receiving the best possible radiation therapy (RT), chemotherapy, and/or surgery, most patients with locally advanced NSCLC fail to respond to treatment. Significantly, they utilize doxycycline to inhibit PSMA1. It is interesting to note that Cron et al found higher proteasome inhibition following PSMA1 knockdown compared to when Bortezomib was used, which may be related to Bortezomib's poor tumor drug penetration.150 Despite this benefit, additional research is necessary before these inhibitors may be used in clinical settings.
The subunits are another target in the 20S core. Peptide aldehydes, lactacystin, epoxomicin and epoxyketones, lactone, peptide boronates, and vinyl sulfones are the five primary groups of these proteasome inhibitors. The original proteasome inhibitors were identified to be peptide aldehydes, such as MG-132.151 These inhibitors, however, have a number of drawbacks. For instance, in cell culture, MG-132 quickly oxidizes and turns into an inactive acid.152 Peptide boronates are more potent inhibitors than peptide aldehydes. Peptide boronates dissociate at a slower rate, and the process is irreversible.152 Additionally, they don't oxidize, making them significantly more stable.152 Epoxyketone is a subclass of proteasome inhibitors, and Carfilzomib is the best-known member of this subclass.153 A nanopeptide inhibitor is lactacystin.153 It's intriguing that it does not obstruct the proteasome right away. It becomes clasto-lactacystin-lactone at neutral pH, reactivating the proteasome.153 In the end, vinyl sulfones represent a different family of proteasome inhibitors with simple and affordable production.153
19S cap inhibitors
ATPase inhibitors
Six different ATPases known as Rpt1-6 make up the heterohexameric ring-like structure154 that is seen on top of the 20S CP's alpha subunits155 in the human proteasome. It was discovered that RIP-1 inhibited Rpt1-6, particularly Rpt4. However, further research is needed to demonstrate that Rpt4 targeting is a workable toxicity mechanism. A second ATPase that has an important role in the UPS in addition to the Rpt subunits is p97 (also known as VCP, or valosin-containing protein).156 Its overexpression in several illnesses and tumors is significant since it suggests it as a possible therapeutic target.157,158 P97 is known to be inhibited by four inhibitors. A selective and reversible p97 ATPase inhibitor, DBeQ can change the autophagy pathway, decrease degradation connected to the endoplasmic reticulum, and enhance cascades 3 and 7 activation in cancer cells. Similar to DBeQ, the ATP-competitive p97 ATPase inhibitor ML240 can boost caspase 3 and 7 activation and block the endoplasmic reticulum-associated degradation (ERAD) pathway in a variety of colon cancer cells. Furthermore, it affects the formation of autophagosomes and may lead to the accumulation of LC3-II, a typical marker for autophagosomes. Another inhibitor that can cause the unfolded protein response and prevent the development of autophagosomes is NMS873, a potent and precise p97 ATPase allosteric inhibitor. It may also have an antiproliferative effect on cancer cells.159 Finally, the polyphenol xanthohumol has been proposed as another inhibitor lately.160
Rpn-13 inhibitors
Two ubiquitin receptors of the 26S proteasome in the 19S RP are called Rpn-10 and Rpn-13. As was previously indicated, the precise positioning of these ubiquitin receptors at the 26S proteasome allows for the collection and degradation of polyubiquitinated substrates. Rpn-13 has received more attention than Rpn-10 up to this point 161-163 since it is unneeded in healthy cells and is highly expressed in a number of malignancies, including ovarian, MM, and gastric cancer. There are currently just two known inhibitors of Rpn-13: KDT-11, a non-covalent revocable peptoid, and RA190, a covalent irrevocable chalcone. Since KDT-11 is a well-known 20S CP 5 inhibitor, it behaves similarly to RA190 in that it causes MM cells to accumulate polyubiquitinated proteins, which in turn triggers cell death. According to a competitive fluorescence polarization (FP) experiment, KDT-11 and RA-190 differ from each other in terms of how well they bind to surfaces.164 Additionally, KDT-11 might either bind to a brand-new surface on Rpn-13 or break up the contact between Uch3 and Rpn-13. In conclusion, even though KDT-11 has an advantage over RA190 due to its selectivity for Rpn-13 in MM cells, further in vivo research is required to optimize its physical characteristics.164
DUBs inhibitors
The action of E3 ligases is counteracted by deubiquitinating enzymes (DUBs), also known as deubiquitinases, which remove ubiquitin from their target proteins. Additionally, they can participate in ubiquitin maturation, editing, and recycling.165-167 DUBs typically fall into one of six subfamilies, including t monocyte chemotactic protein-induced protein (MCPIP), he Machado-Joseph disease protein domain proteases (MJDs), ubiquitin carboxy-terminal hydrolases (UCHs), ovarian-tumor proteases (OTUs), ubiquitin-specific proteases (USPs), and JAB1/MPN/Mov34 metallo Different inhibitors have been created thus far that focus on various DUBs. For instance, ML323, (ADC-01, ADC-03, HBX41108, HBX19818, P5091, P22077), LND-57444, VLX1570, and 8-mercapto-N-((tetrahydro-3-furanyl) methyl) -4-quinoline carboxamide (1,2-b) PSMD14, 9-(ethoxyimino)-9H-indeno, USP1, USP14, UCHL5, USP9X, UCHL1, USP8, USP11, USP7, and USP20 may all be inhibited by pyrazine-2,3-dicarbonitrile, mitoxantrone, WP1130, and GSK2643943A. Except for VLX1570, which is at the clinical trial stage, all of these inhibitors are noticeably in the preclinical stage.165
PROTACs
Rapidly growing PROTACs are special dual-function molecules that can facilitate protein breakdown and are considered a preferable option to blocking small molecules for disease-specific goals. In general, PROTACs form a ternary complex with a protein of interest (POI) and an E3 ligase that encourages the target protein's lysine ubiquitination and subsequent proteosomal destruction.168
When both the POI and E3 ligase targets are involved in PROTAC, a ternary complex is created. The two proteins interact with one another through protein-protein interactions that are mediated by PROTAC as a result of this binding activity.169 The protein interface has to possess the following properties or characteristics in order to produce the ternary complex: 1) reducing PPIs that cause electrostatic or spatial barriers next to the newly formed protein interface; 2) increasing PPIs that encourage structural integration at the new protein interface; and 3) using an adequate E3 ligase in the direction of proper binding to facilitate ubiquitination of the target POI.170 The evaluation of thermodynamic parameters is also used to explain a collection of crucial energy needs for ternary set efficiency, which are collectively referred to as "cooperativity".171 According to this theory, a ternary complex whose elements "cooperate" to create an ideal free energy change stimulates well-organized protein breakdown (DG).
We can maximize PROTAC-induced PPIs, which promote structural integration at the interface and the general affinity of contacts, and raise the optimum DH for the ternary complex in order to achieve an optimal free energy change and a creative ternary complex through the PROTAC design. Additionally, entropic change (DS) for complex formation can be achieved by optimizing ligand flexibility, protein solubility, and/or ternary complex solubility.172
UPS; A novel target in pancreatic cancer
As mentioned, current chemotherapy agents such as gemcitabine, nab-paclitaxel, and FOLFIRINOX have the potential to rapidly confer resistance in pancreatic tumor cells. Therefore, it is imperative to create more potent therapies in order to cure this terrible disease. Pancreatic cancer cells undergo apoptotic cell death as a result of inhibition of the ubiquitin-proteasome system.7 Pancreatic cancer cells are more susceptible to ER stress and apoptosis when treated with bortezomib. Additionally, by triggering the action of the proteasome, proteasome activator subunit 3 (PSME3) encourages the development of pancreatic cancer. As a result, the proteasome is a desirable therapeutic target for pancreatic cancer (Table 2).7
Table 2.
Targeting UPS in PAC
|
Name of the autophagy modifying agent
|
current chemotherapy agents
|
Pathway of action
|
Effectivity
|
Method of study
|
Results
|
Reference
|
| USP21 |
None |
USP21 increases PanIN-to-tumor development of the hTERTHPNE E6/E7 xenografts in vivo and enhances colony formation and cell proliferation of immortalized human pancreatic ductal epithelial cells (hTERT-HPNE E6/E7 cells) in vitro. |
USP21 interacts with deubiquitin, and stabilizes the Wnt pathway transcription factor TCF7. Their combined research identifies USP21 as a PDAC oncogene and possible treatment target. |
In vivo & in vitro |
Nuclear USP21 up-regulates Wnt pathway signalling and enhances tumor-initiating potential in PDAC cells, as demonstrated by the findings that WT-USP21 and NLS-USP21 supported efficient tumour formation at 50%-80% with five iKPC cells, compared with 10%-20% efficiency for five iKPC cells overexpressing ED-USP21 or GFP controls. |
173
|
| USP9X |
G9 treatment (an inhibitor of USP9X) |
USP9X has been shown to control chemo-resistance in various malignancies. Due to its capacity to prevent USP9X deubiquitination, WP1130, a partly selective deubiquitinating enzyme inhibitor, has been speculated to be a possible chemosensitizer. |
In the early stages of the disease, USP9X may largely act as a tumor suppressor, but as the disease progresses, it may enhance the proliferation of tumor cells. |
Utilising well-established human pancreatic tumour cell lines (PANC1 and MIAPACA2) as well as four spontaneously immortalised human pancreatic patient-derived tumour (PDX) cell lines, we conducted gain- and loss-of-function investigations. |
A small-molecule Usp9x inhibitor was expected to have a significant influence on cancer therapy as the disease got worse. |
174,175
|
| Cell model |
Usp9x conditional deletion worked in tandem with KrasG12D to significantly speed up pancreatic carcinogenesis in mice, demonstrating the genetic connection between the two. Thus, USP9X is proposed as a significant novel tumour suppressor gene with therapeutic and prognostic significance in PDA. |
| UbcH10 |
None |
The clinical stage, level of histological differentiation, and lymph node metastasis are all correlated with the up-regulation of UbcH10. |
Compared to individuals with low UbcH10 expression, PDA patients with high UbcH10 expression have considerably lower life times. |
UbcH10 expression was examined using real-time quantitative RT-PCR in 20 pairs of PDA and surrounding non-cancerous tissues. |
For PDA patients with high UbcH10 expression, UbcH10 may act as a prognostic marker and possible biological target for tumor treatment. |
176
|
| TRIM15 |
Inhibitors of triglyceride production |
Apolipoprotein A1 (APOA1), the primary HDL component involved in lipid transport and metabolism, may be one of TRIM15's binding partners. TRIM15 promoted APOA1 polyubiquitination via its RING domain and interacted with APOA1 through its PRY/SPRY domain. In pancreatic cancer cells, APOA1 degradation accelerated lipid droplet formation and boosted lipid anabolism. |
Via controlling lipid metabolism through the APOA1-LDLR axis, TRIM15 could encourage PDAC metastasis. |
Bioinformatics analysis & cell culture |
Inhibiting triglyceride production by targeting the TRIM15-APOA1-LDLR axis may be a method to prevent PDAC metastasis. |
177
|
| KRAS |
The ornithine decarboxylase/antizyme (ODC/AZ) |
Direct interaction between an ODC and a protein substrate can facilitate the protein's breakdown by the 26S proteasome without ubiquitination, and AZ can accelerate this action. |
Both in vitro and in vivo, KRAS levels were reduced, and PANC-1 cell proliferation was inhibited. |
Cell culture |
For patients with pancreatic cancer, targeted degradation of the KRAS oncoprotein via the ODC/AZ pathway at the post-translational level may represent a promising future treatment approach |
85,178,179
|
| Engineering PROTACS |
A “U-box-based” fusion E3 ligase named “RC-U” was generated that effectively interacted with, ubiquitinated, and promoted KRAS oncoprotein degradation at the post-translational level. |
the RC-U fusion E3 ligase also successfully slowed down the proliferation of the pancreatic cancer cell lines PANC-1 and MIAPaCa-2 |
In vitro and in vivo |
Successful ubiquitination of protein substrates reduces the efficiency of “degradation of proteins by the UPS,” as well as restricts the possible applications to a certain extent. |
| Smad4 |
SCFSkp2 complex |
SMAD signaling is a key mediator of the canonical transforming growth factor β (TGFβ) pathway, with important and often contradictory roles in PDAC. Loss of SMAD4 Is Associated With Poor Tumor Immunogenicity and Reduced PD-L1 Expression in Pancreatic Cancer. |
Skp2, an F-box subunit of SCFSkp2, was discovered to physically interact with Smad4 at the physiological levels. |
Protein interaction screen with an antibody-based array method |
SCFSkp2 complex plays a significant role in causing cancer mutations of Smad4 to transition to polyubiquitination-dependent degradation |
180,181
|
| MDM2 |
MA242 |
MA242 blocks MDM2's NFAT1-mediated transcription. |
Regardless of the presence or absence of p53, MA242 reduced cell growth and caused apoptosis in pancreatic cancer cell lines. |
Employing high-throughput virtual and cell-based screening assays, In vitro analysis |
Without causing any host damage, MA242 alone or in combination with gemcitabine suppressed the development and spread of pancreatic tumours. |
182
|
USP21
In patient samples, Hou et al demonstrated a favorable correlation between USP21 (Ubiquitin Specific Peptidase 21) overexpression and its nuclear location with the development of PDAC illness. Mouse PDAC development and tumor-initiating potential are promoted by USP21.173 Additionally, USP21 increases PanIN-to-tumor development of the hTERTHPNE E6/E7 xenografts in vivo and enhances colony formation and cell proliferation of immortalized human pancreatic ductal epithelial cells (hTERT-HPNE E6/E7 cells) in vitro. In order to stimulate gene expression in the Wnt network, USP21 interacts with deubiquitin and stabilizes the Wnt pathway transcription factor TCF7 (Fig. 3). Their combined research identifies USP21 as a PDAC oncogene and possible treatment target.173
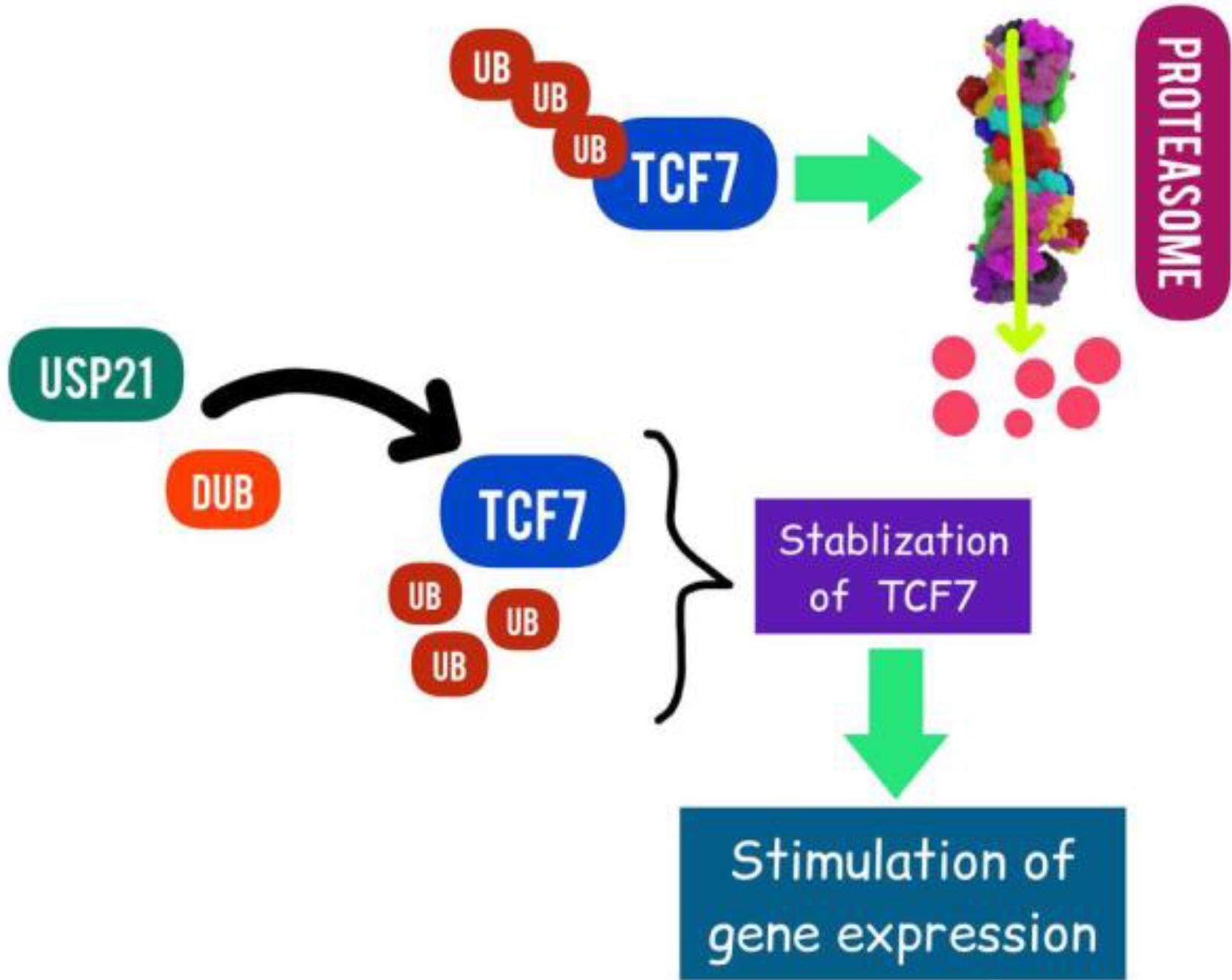
Fig. 3.
Targeting USP21 in PAC (Designed by InShot & PicsArt).In order to stimulate gene expression in the Wnt network, USP21 interacts with deubiquitin, and stabilizes the Wnt pathway transcription factor TCF7.
.
Targeting USP21 in PAC (Designed by InShot & PicsArt).In order to stimulate gene expression in the Wnt network, USP21 interacts with deubiquitin, and stabilizes the Wnt pathway transcription factor TCF7.
USP9X
Recent research has revealed that the deubiquitinating protease known as ubiquitin-specific peptidase 9X (USP9X) is essential for the growth of cancer. It's interesting to note that USP9X can function either as a tumor suppressor or an oncogene, depending on the kind of cancer.183 USP9X has been shown to control chemo-resistance in various malignancies. Due to its capacity to prevent USP9X deubiquitination, WP1130, a partly selective deubiquitinating enzyme inhibitor, has been speculated to be a possible chemosensitizer. The function of USP9X in pancreatic cancer is debatable, and nothing is known about its possible contribution to chemo-resistance. In the early stages of the disease, USP9X may largely act as a tumor suppressor, but as the disease progresses, it may enhance the proliferation of tumor cells.183
UbcH10
The UbcH10 gene, which is located on human chromosome 20q13.12, encodes UbcH10, also known as UBE2C. A crucial player in the ubiquitin-proteasome-mediated protein degradation process, UbcH10 is a member of the family of ubiquitin-conjugating enzymes (Fig. 4).184 Zhao et al show that the clinical stage, level of histological differentiation, and lymph node metastasis are all correlated with the up-regulation of UbcH10. Furthermore, compared to individuals with low UbcH10 expression, PDA patients with high UbcH10 expression have considerably shorter life expectancies. For PDA patients with high UbcH10 expression, UbcH10 may act as a prognostic marker and a possible biological target for tumor treatment.185
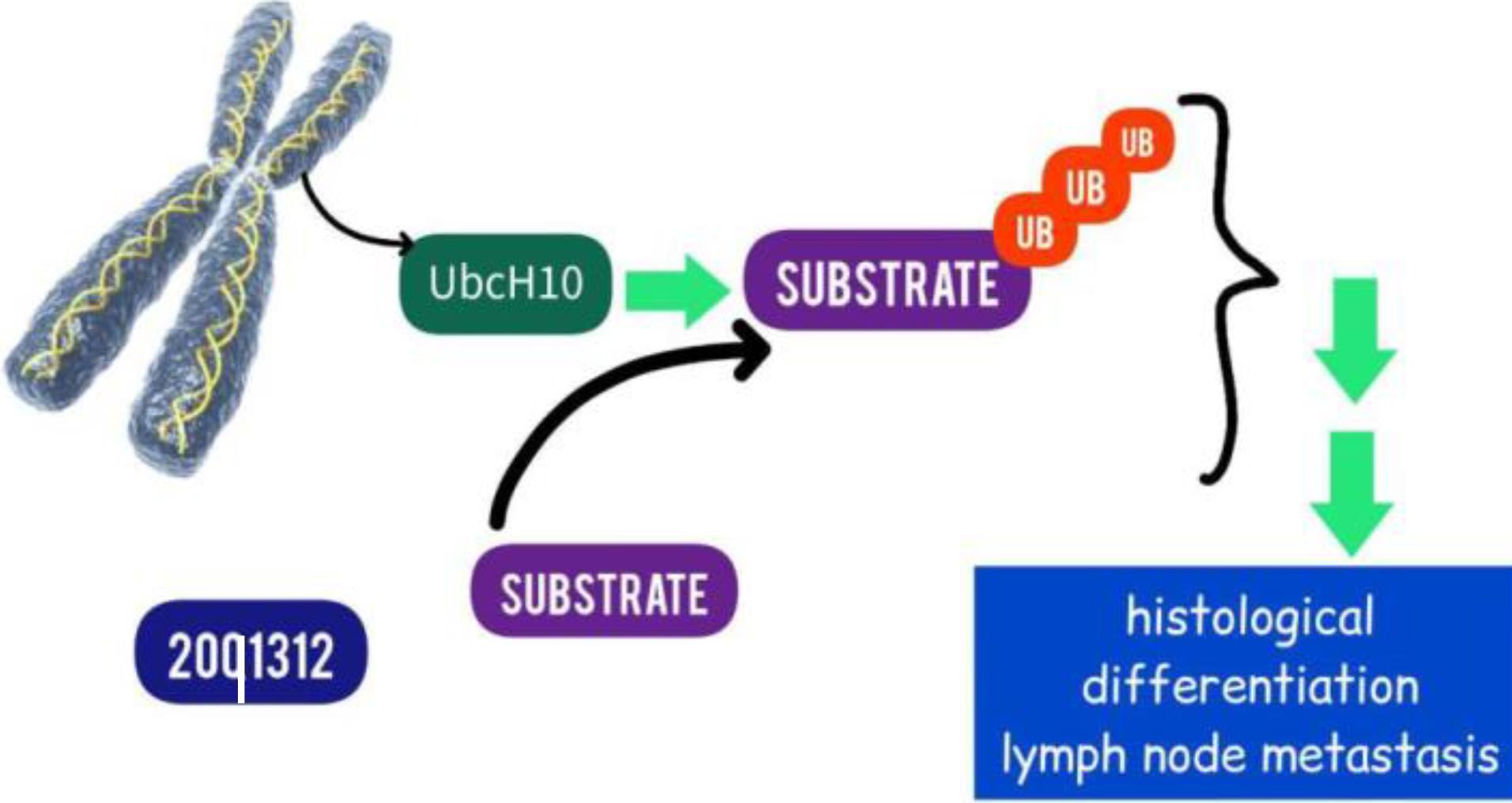
Fig. 4.
Targeting UbcH10 in PAC (Designed by InShot & PicsArt).UbcH10 is a member of the family of ubiquitin-conjugating enzymes.
.
Targeting UbcH10 in PAC (Designed by InShot & PicsArt).UbcH10 is a member of the family of ubiquitin-conjugating enzymes.
TRIM15
The TRIM15 family member, a member of the tripartite motif, has been linked to the emergence of cancer. According to a bioinformatics study, TRIM15 may have a role in controlling the spread of pancreatic cancer. Patients with PDAC frequently experience metabolic reprogramming, including dysregulated lipid synthesis. PDAC treatment plans have been presented that target lipid anabolism.177
Sun et al showed that PDAC tissues had higher TRIM15 expression and that this elevated expression is linked to a poor prognosis. Pancreatic cancer cell invasion and migration were inhibited by TRIM15 knockdown. Apolipoprotein A1 (APOA1), the primary HDL component involved in lipid transport and metabolism, may be one of TRIM15's binding partners, according to the mass spectrometry investigation, which is significant.177 Additional research revealed that TRIM15 promoted APOA1 polyubiquitination via its RING domain and interacted with APOA1 through its PRY/SPRY domain (Fig. 5). In pancreatic cancer cells, APOA1 degradation accelerated lipid droplet formation and boosted lipid anabolism. Furthermore, by controlling lipid metabolism through the APOA1-LDLR axis, TRIM15 could encourage PDAC metastasis. Therefore, inhibiting triglyceride production by targeting the TRIM15-APOA1-LDLR axis may be a method to prevent PDAC metastasis.177
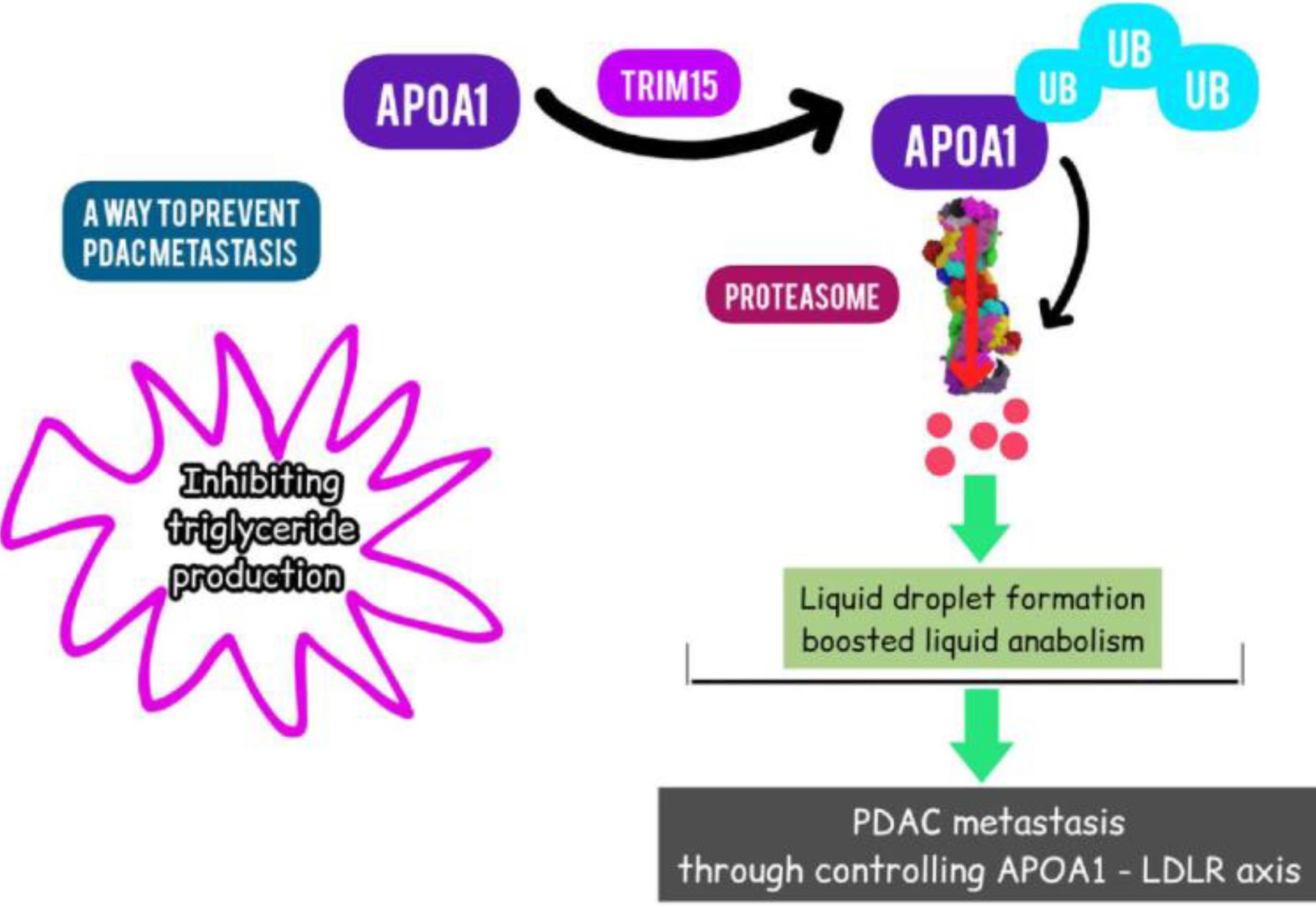
Fig. 5.
Targeting TRIM15 in PAC (Designed by InShot & PicsArt).TRIM15 promoted APOA1 polyubiquitination via its RING domain and interacted with APOA1 through its PRY/SPRY domain.
.
Targeting TRIM15 in PAC (Designed by InShot & PicsArt).TRIM15 promoted APOA1 polyubiquitination via its RING domain and interacted with APOA1 through its PRY/SPRY domain.
Targeting degradation of the KRAS
The ornithine decarboxylase/antizyme (ODC/AZ)
A process for protein breakdown that can be found in nature is the ornithine decarboxylase/antizyme (ODC/AZ) system. Direct interaction between an ODC and a protein substrate can facilitate the protein's breakdown by the 26S proteasome without ubiquitination, and AZ can accelerate this action.179 A chimeric fusion protein was created and recreated by Yihui Ma and colleagues. In a co-immunoprecipitation assay, the engineered fusion protein RC-ODC was shown to interact with the mutant KRAS oncoprotein, and in vitro, the introduction of both RC-ODC and AZ caused the exogenous and endogenous mutant KRAS oncoprotein to degrade at the posttranslational level without ubiquitination. Both in vitro and in vivo, KRAS levels were reduced, and PANC-1 cell proliferation was inhibited. At the same time, phosphorylated extracellular signal-regulated kinase 1/2 (ERK1/2) was downregulated. For patients with pancreatic cancer, targeted degradation of the KRAS oncoprotein via the ODC/AZ pathway at the post-translational level may represent a promising future treatment approach.179
Engineering PROTACS
By successfully removing their matching protein substrates from cells, a few reconstructed fusion E3 ligases and proteolysis-targeting chimeric molecules (PROTACs) have been created, which can considerably affect the malignant features of tumours in vitro and in vivo.25 A "U-box-based" fusion E3 ligase with the designation "RC-U" was developed in a study that efficiently interacted with, ubiquitinated, and promoted post-translational degradation of the KRAS oncoprotein. Importantly, the pancreatic cancer cell lines PANC-1 and MIAPaCa-2 were successfully stopped from proliferating by the RC-U fusion E3 ligase.25,186
However, proper spatial geometric structure is necessary for the development of the designed E3 ligase and protein substrate complex, which enables ubiquitin-conjugating enzymes (E2s) to catalyse the addition of ubiquitin molecules onto particular lysine residues in the protein substrate.187,188 Because of the stiffness mismatch between protein substrates and E3 ligases, proper ubiquitination of protein substrates may be challenging, according to the particular requirements for the structure.179 The fact that only RC-U efficiently interacted with, ubiquitinated, and promoted KRAS oncoprotein degradation despite attempts to design and reconstruct some chimeric fusion E3 proteins suggests that successful ubiquitination of protein substrates reduces the effectiveness of "degradation of proteins by the UPS" and limits the potential applications to some extent.179
Smad4
One method for deactivating Smad4 has been identified as the ubiquitin-proteasome pathway. An ubiquitin E3 ligase for Smad4 mutations in cancer is SCFSkp2. Skp2, an F-box subunit of SCFSkp2, was discovered by Liang et al to physically interact with Smad4 at physiological levels. Numerous unstable mutants from cancer showed markedly enhanced binding to Skp2, which expedited their proteolysis and elevated ubiquitination. These findings imply that the SCFSkp2 complex plays a significant role in causing cancer mutations in Smad4 to transition to polyubiquitination-dependent degradation.181
MDM2
An E3 ubiquitin ligase encoded by the oncogene MDM2, which is overexpressed in pancreatic tumors, is also a therapeutic target. The majority of pancreatic cancer cells do not express full-length P53; hence, the MDM2 inhibitors that are now available do not affect these cells because they do not interfere with the interaction between MDM2 and P53. Wang et al created MA242, which, in pancreatic cancer cell lines, regardless of the presence or absence of p53, reduced cell growth and promoted apoptosis. Without causing any host damage, MA242 alone or in combination with gemcitabine suppressed the development and spread of pancreatic tumours.50
Concluding remarks
Typically, unchecked pancreatic cell proliferation results in the development of pancreatic cancer, which has the potential to spread to other bodily organs. About 90% of instances of pancreatic cancer are pancreatic adenocarcinomas, and occasionally "pancreatic cancer" is used exclusively to describe this subtype. Gemcitabine, nab-paclitaxel, and FOLFIRINOX are examples of modern chemotherapeutic drugs that have the ability to quickly confer resistance in pancreatic tumor cells. Therefore, in order to treat this dreadful condition, it is essential to develop more effective medicines.
Interestingly, researcher’s achievements emphasizing the increased susceptibility of pancreatic cells to ER stress and apoptosis when treated with bortezomib have gained lots of attention as triggering proteasomes as a desirable therapeutic target for pancreatic cancer. However, despite the positive in vitro and in vivo outcomes of UPS-based therapeutics, the clinical outcomes are not as encouraging as anticipated. Therefore, a thorough comprehension of it is necessary to get the best outcomes. Up to now, many targets have been studied in this system, but there are more targets that can be triggered. For instance, in a groundbreaking finding, Rui-Hong Gong et al describe how WNT974 and ART work together to cause KRAS protein degradation in colorectal cancer. The combo therapy greatly upregulates the production of GSK-3 and the E3 ligases ANAPC2 and -TrCP, which promote the destruction of the KRAS protein through the ubiquitin-proteasome pathway.189 Additionally, they discovered that combo therapy inhibits the downstream of KRAS PI3K/Akt/mTOR signaling pathway. This research offers compelling scientific justification for the development of WNT974 and ART as KRAS-targeting therapies for the treatment of colorectal cancer.189 Because similar pathways are also involved in PDCA pathogenesis, scientists can investigate these therapies in this disease, just like colorectal cancer. There are many more examples, and therefore, further research is still needed in order to achieve the best treatment for pancreatic adenocarcinoma.
Research Highlights
What is the current knowledge?
What is new here?
Competing Interests
The authors declare that they have no competing interests.
Ethical Statement
Not applicable.
Acknowledgements
The authors gratefully acknowledge the partial support from the research council of Iran University of Medical Sciences for providing facilities.
References
- Bonacci T, Roignot J, Soubeyran P. Protein ubiquitylation in pancreatic cancer. ScientificWorldJournal 2010; 10:1462-72. doi: 10.1100/tsw.2010.133 [Crossref] [ Google Scholar]
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014; 371:1039-49. doi: 10.1056/NEJMra1404198 [Crossref] [ Google Scholar]
- Westphal S, Kalthoff H. Apoptosis: targets in pancreatic cancer. Mol Cancer 2003; 2:6. doi: 10.1186/1476-4598-2-6 [Crossref] [ Google Scholar]
- McGuigan A, Kelly P, Turkington RC, Jones C, Coleman HG, McCain RS. Pancreatic cancer: a review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol 2018; 24:4846-61. doi: 10.3748/wjg.v24.i43.4846 [Crossref] [ Google Scholar]
- Schima W, Ba-Ssalamah A, Kölblinger C, Kulinna-Cosentini C, Puespoek A, Götzinger P. Pancreatic adenocarcinoma. Eur Radiol 2007; 17:638-49. doi: 10.1007/s00330-006-0435-7 [Crossref] [ Google Scholar]
- Afghani E, Klein AP. Pancreatic adenocarcinoma: trends in epidemiology, risk factors, and outcomes. Hematol Oncol Clin North Am 2022; 36:879-95. doi: 10.1016/j.hoc.2022.07.002 [Crossref] [ Google Scholar]
- Hu S, Jin Y, Liu Y, Ljungman M, Neamati N. Synthesis and mechanistic studies of quinolin-chlorobenzothioate derivatives with proteasome inhibitory activity in pancreatic cancer cell lines. Eur J Med Chem 2018; 158:884-95. doi: 10.1016/j.ejmech.2018.09.037 [Crossref] [ Google Scholar]
- Brugel M, Carlier C, Reyes-Castellanos G, Callon S, Carrier A, Bouché O. Pesticides and pancreatic adenocarcinoma: a transversal epidemiological, environmental and mechanistic narrative review. Dig Liver Dis 2022; 54:1605-13. doi: 10.1016/j.dld.2022.08.023 [Crossref] [ Google Scholar]
- Flanders TY, Foulkes WD. Pancreatic adenocarcinoma: epidemiology and genetics. J Med Genet 1996; 33:889-98. doi: 10.1136/jmg.33.11.889 [Crossref] [ Google Scholar]
- Gupta N, Yelamanchi R. Pancreatic adenocarcinoma: a review of recent paradigms and advances in epidemiology, clinical diagnosis and management. World J Gastroenterol 2021; 27:3158-81. doi: 10.3748/wjg.v27.i23.3158 [Crossref] [ Google Scholar]
- Imaoka H, Ikeda M, Umemoto K, Sunakawa Y, Ueno M, Ueno H. Comprehensive review of undifferentiated carcinoma of the pancreas: from epidemiology to treatment. Jpn J Clin Oncol 2023; 53:764-73. doi: 10.1093/jjco/hyad062 [Crossref] [ Google Scholar]
- Anfinsen CB. Principles that govern the folding of protein chains. Science 1973; 181:223-30. doi: 10.1126/science.181.4096.223 [Crossref] [ Google Scholar]
- Hanley SE, Cooper KF. Sorting nexins in protein homeostasis. Cells 2020; 10:17. doi: 10.3390/cells10010017 [Crossref] [ Google Scholar]
- Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta 2014; 1843:182-96. doi: 10.1016/j.bbamcr.2013.06.031 [Crossref] [ Google Scholar]
- Kikis EA, Gidalevitz T, Morimoto RI. Protein homeostasis in models of aging and age-related conformational disease. Adv Exp Med Biol 2010; 694:138-59. doi: 10.1007/978-1-4419-7002-2_11 [Crossref] [ Google Scholar]
- Stefani M. Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. Biochim Biophys Acta 2004; 1739:5-25. doi: 10.1016/j.bbadis.2004.08.004 [Crossref] [ Google Scholar]
- Nargeh H, Aliabadi F, Ajami M, Pazoki-Toroudi H. Role of polyphenols on gut microbiota and the ubiquitin-proteasome system in neurodegenerative diseases. J Agric Food Chem 2021; 69:6119-44. doi: 10.1021/acs.jafc.1c00923 [Crossref] [ Google Scholar]
- Aliabadi F, Sohrabi B, Mostafavi E, Pazoki-Toroudi H, Webster TJ. Ubiquitin-proteasome system and the role of its inhibitors in cancer therapy. Open Biol 2021; 11:200390. doi: 10.1098/rsob.200390 [Crossref] [ Google Scholar]
- Kim YJ, Lee Y, Shin H, Hwang S, Park J, Song EJ. Ubiquitin-proteasome system as a target for anticancer treatment-an update. Arch Pharm Res 2023; 46:573-97. doi: 10.1007/s12272-023-01455-0 [Crossref] [ Google Scholar]
- Murugan NJ, Voutsadakis IA. Proteasome regulators in pancreatic cancer. World J Gastrointest Oncol 2022; 14:38-54. doi: 10.4251/wjgo.v14.i1.38 [Crossref] [ Google Scholar]
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010; 467:1114-7. doi: 10.1038/nature09515 [Crossref] [ Google Scholar]
- Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet 2011; 378:607-20. doi: 10.1016/s0140-6736(10)62307-0 [Crossref] [ Google Scholar]
- Yue P, Zhu C, Gao Y, Li Y, Wang Q, Zhang K. Development of an autophagy-related signature in pancreatic adenocarcinoma. Biomed Pharmacother 2020; 126:110080. doi: 10.1016/j.biopha.2020.110080 [Crossref] [ Google Scholar]
- Gnoni A, Licchetta A, Scarpa A, Azzariti A, Brunetti AE, Simone G. Carcinogenesis of pancreatic adenocarcinoma: precursor lesions. Int J Mol Sci 2013; 14:19731-62. doi: 10.3390/ijms141019731 [Crossref] [ Google Scholar]
- New M, Tooze S. The role of autophagy in pancreatic cancer—recent advances. Biology 2019; 9:7. doi: 10.3390/biology9010007 [Crossref] [ Google Scholar]
- Rossi ML, Rehman AA, Gondi CS. Therapeutic options for the management of pancreatic cancer. World J Gastroenterol 2014; 20:11142-59. doi: 10.3748/wjg.v20.i32.11142 [Crossref] [ Google Scholar]
- Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol 2020; 17:153-68. doi: 10.1038/s41575-019-0245-4 [Crossref] [ Google Scholar]
- Piffoux M, Eriau E, Cassier PA. Autophagy as a therapeutic target in pancreatic cancer. Br J Cancer 2021; 124:333-44. doi: 10.1038/s41416-020-01039-5 [Crossref] [ Google Scholar]
-
Escobar-Hoyos LF, Penson A, Kannan R, Cho H, Pan CH, Singh RK, et al. Altered RNA splicing by mutant p53 activates oncogenic RAS signaling in pancreatic cancer. Cancer Cell 2020; 38: 198-211.e8. doi: 10.1016/j.ccell.2020.05.010.
- Mann KM, Ying H, Juan J, Jenkins NA, Copeland NG. KRAS-related proteins in pancreatic cancer. Pharmacol Ther 2016; 168:29-42. doi: 10.1016/j.pharmthera.2016.09.003 [Crossref] [ Google Scholar]
- Hu Q, Qin Y, Zhang B, Liang C, Ji S, Shi S. FBW7 increases the chemosensitivity of pancreatic cancer cells to gemcitabine through upregulation of ENT1. Oncol Rep 2017; 38:2069-77. doi: 10.3892/or.2017.5856 [Crossref] [ Google Scholar]
- Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res 1997; 57:1731-4. [ Google Scholar]
- Zhang W, Qian W, Gu J, Gong M, Zhang W, Zhang S. Mutant p53 driven-LINC00857, a protein scaffold between FOXM1 and deubiquitinase OTUB1, promotes the metastasis of pancreatic cancer. Cancer Lett 2023; 552:215976. doi: 10.1016/j.canlet.2022.215976 [Crossref] [ Google Scholar]
- Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2006; 20:1218-49. doi: 10.1101/gad.1415606 [Crossref] [ Google Scholar]
- Cowgill SM, Muscarella P. The genetics of pancreatic cancer. Am J Surg 2003; 186:279-86. doi: 10.1016/s0002-9610(03)00226-5 [Crossref] [ Google Scholar]
- Wan M, Huang J, Jhala NC, Tytler EM, Yang L, Vickers SM. SCFβ-TrCP1) controls Smad4 protein stability in pancreatic cancer cells. Am J Pathol 2005; 166:1379-92. doi: 10.1016/s0002-9440(10)62356-5 [Crossref] [ Google Scholar]
- Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E. DPC4, a candidate tumor suppressor gene at human chromosome 18q211. Science 1996; 271:350-3. doi: 10.1126/science.271.5247.350 [Crossref] [ Google Scholar]
- Ahmed S, Bradshaw AD, Gera S, Dewan MZ, Xu R. The TGF-β/Smad4 signaling pathway in pancreatic carcinogenesis and its clinical significance. J Clin Med 2017; 6:5. doi: 10.3390/jcm6010005 [Crossref] [ Google Scholar]
- Arlt A, Müerköster SS, Schäfer H. Targeting apoptosis pathways in pancreatic cancer. Cancer Lett 2013; 332:346-58. doi: 10.1016/j.canlet.2010.10.015 [Crossref] [ Google Scholar]
- Hamacher R, Schmid RM, Saur D, Schneider G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol Cancer 2008; 7:64. doi: 10.1186/1476-4598-7-64 [Crossref] [ Google Scholar]
- Hager JH, Hanahan D. Tumor cells utilize multiple pathways to down-modulate apoptosis Lessons from a mouse model of islet cell carcinogenesis. Ann N Y Acad Sci 1999; 887:150-63. doi: 10.1111/j.1749-6632.1999.tb07929.x [Crossref] [ Google Scholar]
- Keleg S, Büchler P, Ludwig R, Büchler MW, Friess H. Invasion and metastasis in pancreatic cancer. Mol Cancer 2003; 2:14. doi: 10.1186/1476-4598-2-14 [Crossref] [ Google Scholar]
- Davies M, Robinson M, Smith E, Huntley S, Prime S, Paterson I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-beta1 involves MAPK, Smad and AP-1 signalling pathways. J Cell Biochem 2005; 95:918-31. doi: 10.1002/jcb.20458 [Crossref] [ Google Scholar]
- Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 2012; 18:883-91. doi: 10.1038/nm.2753 [Crossref] [ Google Scholar]
- Hoshino D, Kirkbride KC, Costello K, Clark ES, Sinha S, Grega-Larson N. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep 2013; 5:1159-68. doi: 10.1016/j.celrep.2013.10.050 [Crossref] [ Google Scholar]
- Wang Q, Ni Q, Wang X, Zhu H, Wang Z, Huang J. High expression of RAB27A and TP53 in pancreatic cancer predicts poor survival. Med Oncol 2015; 32:372. doi: 10.1007/s12032-014-0372-2 [Crossref] [ Google Scholar]
- Wu X, Hu A, Zhang M, Chen Z. Effects of Rab27a on proliferation, invasion, and anti-apoptosis in human glioma cell. Tumour Biol 2013; 34:2195-203. doi: 10.1007/s13277-013-0756-5 [Crossref] [ Google Scholar]
- Dutta S, Warshall C, Bandyopadhyay C, Dutta D, Chandran B. Interactions between exosomes from breast cancer cells and primary mammary epithelial cells leads to generation of reactive oxygen species which induce DNA damage response, stabilization of p53 and autophagy in epithelial cells. PLoS One 2014; 9:e97580. doi: 10.1371/journal.pone.0097580 [Crossref] [ Google Scholar]
- Zhang H, Zhang W, Hu B, Qin X, Yi T, Ye Y. Precise pancreatic cancer therapy through targeted degradation of mutant p53 protein by cerium oxide nanoparticles. J Nanobiotechnology 2023; 21:117. doi: 10.1186/s12951-023-01867-6 [Crossref] [ Google Scholar]
- Wang W, Qin JJ, Voruganti S, Wang MH, Sharma H, Patil S, et al. Identification of a new class of MDM2 inhibitor that inhibits growth of orthotopic pancreatic tumors in mice. Gastroenterology 2014; 147: 893-902.e2. doi: 10.1053/j.gastro.2014.07.001.
- Omura N, Goggins M. Epigenetics and epigenetic alterations in pancreatic cancer. Int J Clin Exp Pathol 2009; 2:310-26. [ Google Scholar]
- Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997; 57:3126-30. [ Google Scholar]
- Sato N, Maitra A, Fukushima N, van Heek NT, Matsubayashi H, Iacobuzio-Donahue CA. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res 2003; 63:4158-66. [ Google Scholar]
- Vincent A, Ducourouble MP, Van Seuningen I. Epigenetic regulation of the human mucin gene MUC4 in epithelial cancer cell lines involves both DNA methylation and histone modifications mediated by DNA methyltransferases and histone deacetylases. FASEB J 2008; 22:3035-45. doi: 10.1096/fj.07-103390 [Crossref] [ Google Scholar]
- Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci U S A 1980; 77:1783-6. doi: 10.1073/pnas.77.4.1783 [Crossref] [ Google Scholar]
- Hershko A, Ciechanover A, Varshavsky A. Basic Medical Research Award The ubiquitin system. Nat Med 2000; 6:1073-81. doi: 10.1038/80384 [Crossref] [ Google Scholar]
- Hershko A, Heller H, Elias S, Ciechanover A. Components of ubiquitin-protein ligase system Resolution, affinity purification, and role in protein breakdown. J Biol Chem 1983; 258:8206-14. [ Google Scholar]
- Varshavsky A. The early history of the ubiquitin field. Protein Sci 2006; 15:647-54. doi: 10.1110/ps.052012306 [Crossref] [ Google Scholar]
- Dong Y, Zhang S, Wu Z, Li X, Wang WL, Zhu Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019; 565:49-55. doi: 10.1038/s41586-018-0736-4 [Crossref] [ Google Scholar]
- Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell 2011; 41:8-19. doi: 10.1016/j.molcel.2010.12.020 [Crossref] [ Google Scholar]
- Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL. Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol Cell 2007; 27:731-44. doi: 10.1016/j.molcel.2007.06.033 [Crossref] [ Google Scholar]
- Kraus M, Rückrich T, Reich M, Gogel J, Beck A, Kammer W. Activity patterns of proteasome subunits reflect bortezomib sensitivity of hematologic malignancies and are variable in primary human leukemia cells. Leukemia 2007; 21:84-92. doi: 10.1038/sj.leu.2404414 [Crossref] [ Google Scholar]
- Oh IS, Textoris-Taube K, Sung PS, Kang W, Gorny X, Kähne T. Immunoproteasome induction is suppressed in hepatitis C virus-infected cells in a protein kinase R-dependent manner. Exp Mol Med 2016; 48:e270. doi: 10.1038/emm.2016.98 [Crossref] [ Google Scholar]
- Zwickl P, Ng D, Woo KM, Klenk HP, Goldberg AL. An archaebacterial ATPase, homologous to ATPases in the eukaryotic 26 S proteasome, activates protein breakdown by 20 S proteasomes. J Biol Chem 1999; 274:26008-14. doi: 10.1074/jbc.274.37.26008 [Crossref] [ Google Scholar]
- Deng L, Meng T, Chen L, Wei W, Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther 2020; 5:11. doi: 10.1038/s41392-020-0107-0 [Crossref] [ Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J 2000; 19:94-102. doi: 10.1093/emboj/19.1.94 [Crossref] [ Google Scholar]
- Elsasser S, Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol 2005; 7:742-9. doi: 10.1038/ncb0805-742 [Crossref] [ Google Scholar]
- Liu CW, Li X, Thompson D, Wooding K, Chang TL, Tang Z. ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol Cell 2006; 24:39-50. doi: 10.1016/j.molcel.2006.08.025 [Crossref] [ Google Scholar]
- Yasuda S, Tsuchiya H, Kaiho A, Guo Q, Ikeuchi K, Endo A. Stress- and ubiquitylation-dependent phase separation of the proteasome. Nature 2020; 578:296-300. doi: 10.1038/s41586-020-1982-9 [Crossref] [ Google Scholar]
- Wenzel T, Baumeister W. Conformational constraints in protein degradation by the 20S proteasome. Nat Struct Biol 1995; 2:199-204. doi: 10.1038/nsb0395-199 [Crossref] [ Google Scholar]
- Hoyt MA, Zich J, Takeuchi J, Zhang M, Govaerts C, Coffino P. Glycine-alanine repeats impair proper substrate unfolding by the proteasome. EMBO J 2006; 25:1720-9. doi: 10.1038/sj.emboj.7601058 [Crossref] [ Google Scholar]
- Cundiff MD, Hurley CM, Wong JD, Boscia JA 4th, Bashyal A, Rosenberg J. Ubiquitin receptors are required for substrate-mediated activation of the proteasome's unfolding ability. Sci Rep 2019; 9:14506. doi: 10.1038/s41598-019-50857-y [Crossref] [ Google Scholar]
- Mijit M, Wireman R, Armstrong L, Gampala S, Hassan Z, Schneeweis C. RelA is an essential target for enhancing cellular responses to the DNA repair/Ref-1 redox signaling protein and restoring perturbated cellular redox homeostasis in mouse PDAC cells. Front Oncol 2022; 12:826617. doi: 10.3389/fonc.2022.826617 [Crossref] [ Google Scholar]
- Abdel Hadi N, Reyes-Castellanos G, Carrier A. Targeting redox metabolism in pancreatic cancer. Int J Mol Sci 2021; 22:1534. doi: 10.3390/ijms22041534 [Crossref] [ Google Scholar]
- Mao W, Mai J, Peng H, Wan J, Sun T. YAP in pancreatic cancer: oncogenic role and therapeutic strategy. Theranostics 2021; 11:1753-62. doi: 10.7150/thno.53438 [Crossref] [ Google Scholar]
- Qin T, Chen K, Li J, Qian W, Xiao Y, Wu E. Heat shock factor 1 inhibition sensitizes pancreatic cancer to gemcitabine via the suppression of cancer stem cell-like properties. Biomed Pharmacother 2022; 148:112713. doi: 10.1016/j.biopha.2022.112713 [Crossref] [ Google Scholar]
- Kumar S, Schoonderwoerd MJA, Kroonen JS, de Graaf IJ, Sluijter M, Ruano D. Targeting pancreatic cancer by TAK-981: a SUMOylation inhibitor that activates the immune system and blocks cancer cell cycle progression in a preclinical model. Gut 2022; 71:2266-83. doi: 10.1136/gutjnl-2021-324834 [Crossref] [ Google Scholar]
- Qi S, Guan X, Zhang J, Yu D, Yu X, Li Q. Targeting E2 ubiquitin-conjugating enzyme UbcH5c by small molecule inhibitor suppresses pancreatic cancer growth and metastasis. Mol Cancer 2022; 21:70. doi: 10.1186/s12943-022-01538-4 [Crossref] [ Google Scholar]
- Feng L, Wang J, Zhang J, Diao J, He L, Fu C. Comprehensive analysis of E3 ubiquitin ligases reveals ring finger protein 223 as a novel oncogene activated by KLF4 in pancreatic cancer. Front Cell Dev Biol 2021; 9:738709. doi: 10.3389/fcell.2021.738709 [Crossref] [ Google Scholar]
- Deng X, Fu X, Teng H, Fang L, Liang B, Zeng R. E3 ubiquitin ligase TRIM29 promotes pancreatic cancer growth and progression via stabilizing Yes-associated protein 1. J Transl Med 2021; 19:332. doi: 10.1186/s12967-021-03007-w [Crossref] [ Google Scholar]
- Hou B, Chen T, Zhang H, Li J, Wang P, Shang G. The E3 ubiquitin ligases regulate PD-1/PD-L1 protein levels in tumor microenvironment to improve immunotherapy. Front Immunol 2023; 14:1123244. doi: 10.3389/fimmu.2023.1123244 [Crossref] [ Google Scholar]
- Gong L, Yang B, Xu M, Cheng B, Tang X, Zheng P. Bortezomib-induced apoptosis in cultured pancreatic cancer cells is associated with ceramide production. Cancer Chemother Pharmacol 2014; 73:69-77. doi: 10.1007/s00280-013-2318-3 [Crossref] [ Google Scholar]
- Tozzi M, Sørensen CE, Magni L, Christensen NM, Bouazzi R, Buch CM. Proton pump inhibitors reduce pancreatic adenocarcinoma progression by selectively targeting H + , K + -ATPases in pancreatic cancer and stellate cells. Cancers (Basel) 2020; 12:640. doi: 10.3390/cancers12030640 [Crossref] [ Google Scholar]
- Anchoori RK, Tan M, Tseng SH, Peng S, Soong RS, Algethami A. Structure-function analyses of candidate small molecule RPN13 inhibitors with antitumor properties. PLoS One 2020; 15:e0227727. doi: 10.1371/journal.pone.0227727 [Crossref] [ Google Scholar]
- Ma Y, Gu Y, Zhang Q, Han Y, Yu S, Lu Z. Targeted degradation of KRAS by an engineered ubiquitin ligase suppresses pancreatic cancer cell growth in vitro and in vivo. Mol Cancer Ther 2013; 12:286-94. doi: 10.1158/1535-7163.mct-12-0650 [Crossref] [ Google Scholar]
- Lefaki M, Papaevgeniou N, Chondrogianni N. Redox regulation of proteasome function. Redox Biol 2017; 13:452-8. doi: 10.1016/j.redox.2017.07.005 [Crossref] [ Google Scholar]
- Corcoran A, Cotter TG. Redox regulation of protein kinases. FEBS J 2013; 280:1944-65. doi: 10.1111/febs.12224 [Crossref] [ Google Scholar]
- Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 2014; 15:411-21. doi: 10.1038/nrm3801 [Crossref] [ Google Scholar]
- Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev 2014; 94:329-54. doi: 10.1152/physrev.00040.2012 [Crossref] [ Google Scholar]
-
Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteomics 2011; 10: R110.006924. doi: 10.1074/mcp.M110.006924.
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature 2003; 426:895-9. doi: 10.1038/nature02263 [Crossref] [ Google Scholar]
- Livnat-Levanon N, Kevei É, Kleifeld O, Krutauz D, Segref A, Rinaldi T. Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep 2014; 7:1371-80. doi: 10.1016/j.celrep.2014.04.030 [Crossref] [ Google Scholar]
- Davies KJ, Goldberg AL. Oxygen radicals stimulate intracellular proteolysis and lipid peroxidation by independent mechanisms in erythrocytes. J Biol Chem 1987; 262:8220-6. [ Google Scholar]
- Höhn A, König J, Grune T. Protein oxidation in aging and the removal of oxidized proteins. J Proteomics 2013; 92:132-59. doi: 10.1016/j.jprot.2013.01.004 [Crossref] [ Google Scholar]
- Ferrington DA, Gregerson DS. Immunoproteasomes: structure, function, and antigen presentation. Prog Mol Biol Transl Sci 2012; 109:75-112. doi: 10.1016/b978-0-12-397863-9.00003-1 [Crossref] [ Google Scholar]
- Pickering AM, Koop AL, Teoh CY, Ermak G, Grune T, Davies KJ. The immunoproteasome, the 20S proteasome and the PA28αβ proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem J 2010; 432:585-94. doi: 10.1042/bj20100878 [Crossref] [ Google Scholar]
- Reinheckel T, Ullrich O, Sitte N, Grune T. Differential impairment of 20S and 26S proteasome activities in human hematopoietic K562 cells during oxidative stress. Arch Biochem Biophys 2000; 377:65-8. doi: 10.1006/abbi.2000.1717 [Crossref] [ Google Scholar]
- Höhn TJ, Grune T. The proteasome and the degradation of oxidized proteins: part III-redox regulation of the proteasomal system. Redox Biol 2014; 2:388-94. doi: 10.1016/j.redox.2013.12.029 [Crossref] [ Google Scholar]
- Silva GM, Netto LE, Simões V, Santos LF, Gozzo FC, Demasi MA. Redox control of 20S proteasome gating. Antioxid Redox Signal 2012; 16:1183-94. doi: 10.1089/ars.2011.4210 [Crossref] [ Google Scholar]
- Carrard G, Dieu M, Raes M, Toussaint O, Friguet B. Impact of ageing on proteasome structure and function in human lymphocytes. Int J Biochem Cell Biol 2003; 35:728-39. doi: 10.1016/s1357-2725(02)00356-4 [Crossref] [ Google Scholar]
- Demasi M, Shringarpure R, Davies KJ. Glutathiolation of the proteasome is enhanced by proteolytic inhibitors. Arch Biochem Biophys 2001; 389:254-63. doi: 10.1006/abbi.2001.2332 [Crossref] [ Google Scholar]
- Demasi M, Silva GM, Netto LE. 20 S proteasome from Saccharomyces cerevisiae is responsive to redox modifications and is S-glutathionylated. J Biol Chem 2003; 278:679-85. doi: 10.1074/jbc.M209282200 [Crossref] [ Google Scholar]
- Okada K, Wangpoengtrakul C, Osawa T, Toyokuni S, Tanaka K, Uchida K. 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress Identification of proteasomes as target molecules. J Biol Chem 1999; 274:23787-93. doi: 10.1074/jbc.274.34.23787 [Crossref] [ Google Scholar]
- Zong C, Young GW, Wang Y, Lu H, Deng N, Drews O. Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics 2008; 8:5025-37. doi: 10.1002/pmic.200800387 [Crossref] [ Google Scholar]
- Levine RL, Wehr N, Williams JA, Stadtman ER, Shacter E. Determination of carbonyl groups in oxidized proteins. Methods Mol Biol 2000; 99:15-24. doi: 10.1385/1-59259-054-3:15 [Crossref] [ Google Scholar]
- Nyström T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J 2005; 24:1311-7. doi: 10.1038/sj.emboj.7600599 [Crossref] [ Google Scholar]
- Wong CM, Marcocci L, Liu L, Suzuki YJ. Cell signaling by protein carbonylation and decarbonylation. Antioxid Redox Signal 2010; 12:393-404. doi: 10.1089/ars.2009.2805 [Crossref] [ Google Scholar]
- Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem 2001; 276:30057-63. doi: 10.1074/jbc.M100142200 [Crossref] [ Google Scholar]
- Ahmed EK, Rogowska-Wrzesinska A, Roepstorff P, Bulteau AL, Friguet B. Protein modification and replicative senescence of WI-38 human embryonic fibroblasts. Aging Cell 2010; 9:252-72. doi: 10.1111/j.1474-9726.2010.00555.x [Crossref] [ Google Scholar]
- Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J 2003; 375:581-92. doi: 10.1042/bj20030763 [Crossref] [ Google Scholar]
- Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res 2003; 42:318-43. doi: 10.1016/s0163-7827(03)00014-6 [Crossref] [ Google Scholar]
- Parola M, Bellomo G, Robino G, Barrera G, Dianzani MU. 4-Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid Redox Signal 1999; 1:255-84. doi: 10.1089/ars.1999.1.3-255 [Crossref] [ Google Scholar]
- Friguet B, Szweda LI. Inhibition of the multicatalytic proteinase (proteasome) by 4-hydroxy-2-nonenal cross-linked protein. FEBS Lett 1997; 405:21-5. doi: 10.1016/s0014-5793(97)00148-8 [Crossref] [ Google Scholar]
- Zmijewski JW, Banerjee S, Abraham E. S-glutathionylation of the Rpn2 regulatory subunit inhibits 26 S proteasomal function. J Biol Chem 2009; 284:22213-21. doi: 10.1074/jbc.M109.028902 [Crossref] [ Google Scholar]
- Hansen RE, Roth D, Winther JR. Quantifying the global cellular thiol-disulfide status. Proc Natl Acad Sci U S A 2009; 106:422-7. doi: 10.1073/pnas.0812149106 [Crossref] [ Google Scholar]
- Silva GM, Netto LE, Discola KF, Piassa-Filho GM, Pimenta DC, Bárcena JA. Role of glutaredoxin 2 and cytosolic thioredoxins in cysteinyl-based redox modification of the 20S proteasome. FEBS J 2008; 275:2942-55. doi: 10.1111/j.1742-4658.2008.06441.x [Crossref] [ Google Scholar]
- Guerrero C, Milenkovic T, Przulj N, Kaiser P, Huang L. Characterization of the proteasome interaction network using a QTAX-based tag-team strategy and protein interaction network analysis. Proc Natl Acad Sci U S A 2008; 105:13333-8. doi: 10.1073/pnas.0801870105 [Crossref] [ Google Scholar]
- Andersen KM, Madsen L, Prag S, Johnsen AH, Semple CA, Hendil KB. Thioredoxin Txnl1/TRP32 is a redox-active cofactor of the 26 S proteasome. J Biol Chem 2009; 284:15246-54. doi: 10.1074/jbc.M900016200 [Crossref] [ Google Scholar]
- Wiseman RL, Chin KT, Haynes CM, Stanhill A, Xu CF, Roguev A. Thioredoxin-related protein 32 is an arsenite-regulated thiol reductase of the proteasome 19 S particle. J Biol Chem 2009; 284:15233-45. doi: 10.1074/jbc.M109.002121 [Crossref] [ Google Scholar]
- Wiseman RL, Chin KT, Haynes CM, Stanhill A, Xu CF, Roguev A. Correction: thioredoxin-related protein 32 is an arsenite-regulated thiol reductase of the proteasome 19 S particle. J Biol Chem 2020; 295:9267. doi: 10.1074/jbc.AAC120.014712 [Crossref] [ Google Scholar]
- Andersen KM, Jensen C, Kriegenburg F, Lauridsen AM, Gordon C, Hartmann-Petersen R. Txl1 and Txc1 are co-factors of the 26S proteasome in fission yeast. Antioxid Redox Signal 2011; 14:1601-8. doi: 10.1089/ars.2010.3329 [Crossref] [ Google Scholar]
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev 2002; 82:47-95. doi: 10.1152/physrev.00018.2001 [Crossref] [ Google Scholar]
- Tsvetkov P, Myers N, Eliav R, Adamovich Y, Hagai T, Adler J. NADH binds and stabilizes the 26S proteasomes independent of ATP. J Biol Chem 2014; 289:11272-81. doi: 10.1074/jbc.M113.537175 [Crossref] [ Google Scholar]
- Cagnetta A, Cea M, Calimeri T, Acharya C, Fulciniti M, Tai YT. Intracellular NAD⁺ depletion enhances bortezomib-induced anti-myeloma activity. Blood 2013; 122:1243-55. doi: 10.1182/blood-2013-02-483511 [Crossref] [ Google Scholar]
- Vriend J, Reiter RJ. The Keap1-Nrf2-antioxidant response element pathway: a review of its regulation by melatonin and the proteasome. Mol Cell Endocrinol 2015; 401:213-20. doi: 10.1016/j.mce.2014.12.013 [Crossref] [ Google Scholar]
- Pomatto LC, Wong S, Carney C, Shen B, Tower J, Davies KJA. The age- and sex-specific decline of the 20s proteasome and the Nrf2/CncC signal transduction pathway in adaption and resistance to oxidative stress in Drosophila melanogaster. Aging (Albany NY) 2017; 9:1153-85. doi: 10.18632/aging.101218 [Crossref] [ Google Scholar]
- Jang J, Wang Y, Kim HS, Lalli MA, Kosik KS. Nrf2, a regulator of the proteasome, controls self-renewal and pluripotency in human embryonic stem cells. Stem Cells 2014; 32:2616-25. doi: 10.1002/stem.1764 [Crossref] [ Google Scholar]
- Lee D, Ryu KY. Effect of cellular ubiquitin levels on the regulation of oxidative stress response and proteasome function via Nrf1. Biochem Biophys Res Commun 2017; 485:234-40. doi: 10.1016/j.bbrc.2017.02.105 [Crossref] [ Google Scholar]
- Mannhaupt G, Schnall R, Karpov V, Vetter I, Feldmann H. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett 1999; 450:27-34. doi: 10.1016/s0014-5793(99)00467-6 [Crossref] [ Google Scholar]
- Xie Y, Varshavsky A. RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: a negative feedback circuit. Proc Natl Acad Sci U S A 2001; 98:3056-61. doi: 10.1073/pnas.071022298 [Crossref] [ Google Scholar]
- Fleming JA, Lightcap ES, Sadis S, Thoroddsen V, Bulawa CE, Blackman RK. Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by PS-341. Proc Natl Acad Sci U S A 2002; 99:1461-6. doi: 10.1073/pnas.032516399 [Crossref] [ Google Scholar]
- Hahn JS, Neef DW, Thiele DJ. A stress regulatory network for co-ordinated activation of proteasome expression mediated by yeast heat shock transcription factor. Mol Microbiol 2006; 60:240-51. doi: 10.1111/j.1365-2958.2006.05097.x [Crossref] [ Google Scholar]
- Hanna J, Finley D. A proteasome for all occasions. FEBS Lett 2007; 581:2854-61. doi: 10.1016/j.febslet.2007.03.053 [Crossref] [ Google Scholar]
- Metzger MB, Michaelis S. Analysis of quality control substrates in distinct cellular compartments reveals a unique role for Rpn4p in tolerating misfolded membrane proteins. Mol Biol Cell 2009; 20:1006-19. doi: 10.1091/mbc.e08-02-0140 [Crossref] [ Google Scholar]
- Owsianik G, Balzi l L, Ghislain M. Control of 26S proteasome expression by transcription factors regulating multidrug resistance in Saccharomyces cerevisiae. Mol Microbiol 2002; 43:1295-308. doi: 10.1046/j.1365-2958.2002.02823.x [Crossref] [ Google Scholar]
- Wang X, Xu H, Ju D, Xie Y. Disruption of Rpn4-induced proteasome expression in Saccharomyces cerevisiae reduces cell viability under stressed conditions. Genetics 2008; 180:1945-53. doi: 10.1534/genetics.108.094524 [Crossref] [ Google Scholar]
- Lundgren J, Masson P, Mirzaei Z, Young P. Identification and characterization of a Drosophila proteasome regulatory network. Mol Cell Biol 2005; 25:4662-75. doi: 10.1128/mcb.25.11.4662-4675.2005 [Crossref] [ Google Scholar]
- Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel PM. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of mammalian proteasomes. J Biol Chem 2003; 278:21517-25. doi: 10.1074/jbc.M301032200 [Crossref] [ Google Scholar]
- Xu GW, Ali M, Wood TE, Wong D, Maclean N, Wang X. The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood 2010; 115:2251-9. doi: 10.1182/blood-2009-07-231191 [Crossref] [ Google Scholar]
- Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res 2007; 67:9472-81. doi: 10.1158/0008-5472.can-07-0568 [Crossref] [ Google Scholar]
- Ceccarelli DF, Tang X, Pelletier B, Orlicky S, Xie W, Plantevin V. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell 2011; 145:1075-87. doi: 10.1016/j.cell.2011.05.039 [Crossref] [ Google Scholar]
- Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res 2016; 26:484-98. doi: 10.1038/cr.2016.31 [Crossref] [ Google Scholar]
- Pulvino M, Liang Y, Oleksyn D, DeRan M, Van Pelt E, Shapiro J. Inhibition of proliferation and survival of diffuse large B-cell lymphoma cells by a small-molecule inhibitor of the ubiquitin-conjugating enzyme Ubc13-Uev1A. Blood 2012; 120:1668-77. doi: 10.1182/blood-2012-02-406074 [Crossref] [ Google Scholar]
- Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer 2005; 5:297-309. doi: 10.1038/nrc1588 [Crossref] [ Google Scholar]
- Kumari N, Anthony ML, Durgapal P, Joshi PP, Rajput D, Singh A. Ossifying fibromyxoid tumor: fine-needle aspiration cytology findings of a rare soft tissue neoplasm. Diagn Cytopathol 2020; 48:396-400. doi: 10.1002/dc.24382 [Crossref] [ Google Scholar]
- Guo ZQ, Zheng T, Chen B, Luo C, Ouyang S, Gong S. Small-molecule targeting of E3 ligase adaptor SPOP in kidney cancer. Cancer Cell 2016; 30:474-84. doi: 10.1016/j.ccell.2016.08.003 [Crossref] [ Google Scholar]
- Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell 2011; 44:304-16. doi: 10.1016/j.molcel.2011.08.029 [Crossref] [ Google Scholar]
- Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P. mTOR drives its own activation via SCF(βTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell 2011; 44:290-303. doi: 10.1016/j.molcel.2011.08.030 [Crossref] [ Google Scholar]
- Dehan E, Bassermann F, Guardavaccaro D, Vasiliver-Shamis G, Cohen M, Lowes KN. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell 2009; 33:109-16. doi: 10.1016/j.molcel.2008.12.020 [Crossref] [ Google Scholar]
- Cron KR, Zhu K, Kushwaha DS, Hsieh G, Merzon D, Rameseder J. Proteasome inhibitors block DNA repair and radiosensitize non-small cell lung cancer. PLoS One 2013; 8:e73710. doi: 10.1371/journal.pone.0073710 [Crossref] [ Google Scholar]
- Harer SL, Bhatia MS, Bhatia NM. Proteasome inhibitors mechanism; source for design of newer therapeutic agents. J Antibiot (Tokyo) 2012; 65:279-88. doi: 10.1038/ja.2011.84 [Crossref] [ Google Scholar]
- Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol 2001; 8:739-58. doi: 10.1016/s1074-5521(01)00056-4 [Crossref] [ Google Scholar]
- Thibaudeau TA, Smith DM. A practical review of proteasome pharmacology. Pharmacol Rev 2019; 71:170-97. doi: 10.1124/pr.117.015370 [Crossref] [ Google Scholar]
- Huang X, Luan B, Wu J, Shi Y. An atomic structure of the human 26S proteasome. Nat Struct Mol Biol 2016; 23:778-85. doi: 10.1038/nsmb.3273 [Crossref] [ Google Scholar]
- Confalonieri F, Duguet M. A 200-amino acid ATPase module in search of a basic function. Bioessays 1995; 17:639-50. doi: 10.1002/bies.950170710 [Crossref] [ Google Scholar]
- Jentsch S, Rumpf S. Cdc48 (p97): a "molecular gearbox" in the ubiquitin pathway?. Trends Biochem Sci 2007; 32:6-11. doi: 10.1016/j.tibs.2006.11.005 [Crossref] [ Google Scholar]
- Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 2015; 28:653-65. doi: 10.1016/j.ccell.2015.10.002 [Crossref] [ Google Scholar]
- Meyer H, Weihl CC. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 2014; 127:3877-83. doi: 10.1242/jcs.093831 [Crossref] [ Google Scholar]
- Ding R, Zhang T, Wilson DJ, Xie J, Williams J, Xu Y. Discovery of irreversible p97 inhibitors. J Med Chem 2019; 62:2814-29. doi: 10.1021/acs.jmedchem.9b00144 [Crossref] [ Google Scholar]
- Kwon M, Jang M, Kim GH, Oh T, Ryoo IJ, Ryu HW. Kushenol E inhibits autophagy and impairs lysosomal positioning via VCP/p97 inhibition. Biochem Pharmacol 2020; 175:113861. doi: 10.1016/j.bcp.2020.113861 [Crossref] [ Google Scholar]
- Fejzo MS, Anderson L, Chen HW, Anghel A, Zhuo J, Anchoori R. ADRM 1‐amplified metastasis gene in gastric cancer. Genes Chromosomes Cancer 2015; 54:506-15. doi: 10.1002/gcc.22262 [Crossref] [ Google Scholar]
- Song Y, Ray A, Li S, Das DS, Tai YT, Carrasco RD. Targeting proteasome ubiquitin receptor Rpn13 in multiple myeloma. Leukemia 2016; 30:1877-86. doi: 10.1038/leu.2016.97 [Crossref] [ Google Scholar]
- Yu GY, Wang X, Zheng SS, Gao XM, Jia QA, Zhu WW. RA190, a proteasome subunit ADRM1 inhibitor, suppresses intrahepatic cholangiocarcinoma by inducing NF-KB-mediated cell apoptosis. Cell Physiol Biochem 2018; 47:1152-66. doi: 10.1159/000490210 [Crossref] [ Google Scholar]
- Muli CS, Tian W, Trader DJ. Small-molecule inhibitors of the proteasome's regulatory particle. Chembiochem 2019; 20:1739-53. doi: 10.1002/cbic.201900017 [Crossref] [ Google Scholar]
- Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov 2018; 17:57-78. doi: 10.1038/nrd.2017.152 [Crossref] [ Google Scholar]
- Pfoh R, Lacdao IK, Saridakis V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr Relat Cancer 2015; 22:T35-54. doi: 10.1530/erc-14-0516 [Crossref] [ Google Scholar]
- Singh N, Singh AB. Deubiquitinases and cancer: a snapshot. Crit Rev Oncol Hematol 2016; 103:22-6. doi: 10.1016/j.critrevonc.2016.04.018 [Crossref] [ Google Scholar]
- Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 2017; 16:101-14. doi: 10.1038/nrd.2016.211 [Crossref] [ Google Scholar]
- Jensen SM, Potts GK, Ready DB, Patterson MJ. Specific MHC-I peptides are induced using PROTACs. Front Immunol 2018; 9:2697. doi: 10.3389/fimmu.2018.02697 [Crossref] [ Google Scholar]
- Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun 2019; 10:131. doi: 10.1038/s41467-018-08027-7 [Crossref] [ Google Scholar]
- Claveria-Gimeno R, Vega S, Abian O, Velazquez-Campoy A. A look at ligand binding thermodynamics in drug discovery. Expert Opin Drug Discov 2017; 12:363-77. doi: 10.1080/17460441.2017.1297418 [Crossref] [ Google Scholar]
- Zorba A, Nguyen C, Xu Y, Starr J, Borzilleri K, Smith J. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci U S A 2018; 115:E7285-92. doi: 10.1073/pnas.1803662115 [Crossref] [ Google Scholar]
- Hou P, Ma X, Zhang Q, Wu CJ, Liao W, Li J. USP21 deubiquitinase promotes pancreas cancer cell stemness via Wnt pathway activation. Genes Dev 2019; 33:1361-6. doi: 10.1101/gad.326314.119 [Crossref] [ Google Scholar]
- Pal A, Dziubinski M, Di Magliano MP, Simeone DM, Owens S, Thomas D. Usp9x promotes survival in human pancreatic cancer and its inhibition suppresses pancreatic ductal adenocarcinoma in vivo tumor growth. Neoplasia 2018; 20:152-64. doi: 10.1016/j.neo.2017.11.007 [Crossref] [ Google Scholar]
- Pérez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012; 486:266-70. doi: 10.1038/nature11114 [Crossref] [ Google Scholar]
- Zhao ZK, Wu WG, Chen L, Dong P, Gu J, Mu JS. Expression of UbcH10 in pancreatic ductal adenocarcinoma and its correlation with prognosis. Tumour Biol 2013; 34:1473-7. doi: 10.1007/s13277-013-0671-9 [Crossref] [ Google Scholar]
- Sun Y, Ren D, Yang C, Yang W, Zhao J, Zhou Y. TRIM15 promotes the invasion and metastasis of pancreatic cancer cells by mediating APOA1 ubiquitination and degradation. Biochim Biophys Acta Mol Basis Dis 2021; 1867:166213. doi: 10.1016/j.bbadis.2021.166213 [Crossref] [ Google Scholar]
- He Q, Liu Z, Wang J. Targeting KRAS in PDAC: a new way to cure it?. Cancers (Basel) 2022; 14:4982. doi: 10.3390/cancers14204982 [Crossref] [ Google Scholar]
- Ma Y, Xu J, Huang P, Bai X, Gao H. Ubiquitin-independent, proteasome-mediated targeted degradation of KRAS in pancreatic adenocarcinoma cells using an engineered ornithine decarboxylase/antizyme system. IUBMB Life 2019; 71:57-65. doi: 10.1002/iub.1945 [Crossref] [ Google Scholar]
- Principe DR, Underwood PW, Kumar S, Timbers KE, Koch RM, Trevino JG. Loss of SMAD4 is associated with poor tumor immunogenicity and reduced PD-L1 expression in pancreatic cancer. Front Oncol 2022; 12:806963. doi: 10.3389/fonc.2022.806963 [Crossref] [ Google Scholar]
- Liang M, Liang YY, Wrighton K, Ungermannova D, Wang XP, Brunicardi FC. Ubiquitination and proteolysis of cancer-derived Smad4 mutants by SCFSkp2. Mol Cell Biol 2004; 24:7524-37. doi: 10.1128/mcb.24.17.7524-7537.2004 [Crossref] [ Google Scholar]
- Wang W, Qin JJ, Voruganti S, Nijampatnam B, Velu SE, Ruan KH. Discovery and characterization of dual inhibitors of MDM2 and NFAT1 for pancreatic cancer therapy. Cancer Res 2018; 78:5656-67. doi: 10.1158/0008-5472.can-17-3939 [Crossref] [ Google Scholar]
- Ma T, Chen W, Zhi X, Liu H, Zhou Y, Chen BW. USP9X inhibition improves gemcitabine sensitivity in pancreatic cancer by inhibiting autophagy. Cancer Lett 2018; 436:129-38. doi: 10.1016/j.canlet.2018.08.010 [Crossref] [ Google Scholar]
- Okamoto Y, Ozaki T, Miyazaki K, Aoyama M, Miyazaki M, Nakagawara A. UbcH10 is the cancer-related E2 ubiquitin-conjugating enzyme. Cancer Res 2003; 63:4167-73. [ Google Scholar]
- Zhao ZK, Wu WG, Chen L, Dong P, Gu J, Mu JS. Expression of UbcH10 in pancreatic ductal adenocarcinoma and its correlation with prognosis. Tumour Biol 2013; 34:1473-7. doi: 10.1007/s13277-013-0671-9 [Crossref] [ Google Scholar]
- Ma Y, Gu Y, Zhang Q, Han Y, Yu S, Lu Z. Targeted degradation of KRAS by an engineered ubiquitin ligase suppresses pancreatic cancer cell growth in vitro and in vivo. Mol Cancer Ther 2013; 12:286-94. doi: 10.1158/1535-7163.mct-12-0650 [Crossref] [ Google Scholar]
- Hatakeyama S, Watanabe M, Fujii Y, Nakayama KI. Targeted destruction of c-Myc by an engineered ubiquitin ligase suppresses cell transformation and tumor formation. Cancer Res 2005; 65:7874-9. doi: 10.1158/0008-5472.can-05-1581 [Crossref] [ Google Scholar]
- Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 2008; 27:7201-11. doi: 10.1038/onc.2008.320 [Crossref] [ Google Scholar]
- Gong RH, Chen M, Huang C, Wong HLX, Kwan HY, Bian Z. Combination of artesunate and WNT974 induces KRAS protein degradation by upregulating E3 ligase ANACP2 and β-TrCP in the ubiquitin-proteasome pathway. Cell Commun Signal 2022; 20:34. doi: 10.1186/s12964-022-00834-2 [Crossref] [ Google Scholar]