Bioimpacts. 2025;15:30098.
doi: 10.34172/bi.30098
Original Article
Preparation and evaluation of lipid-based sustained release pellets of chlorpheniramine maleate by the wet extrusion-spheronization method
Mohammadreza Abbaspour Conceptualization, Funding acquisition, Methodology, Supervision, 1, 2 
Asieh Sadooghi Data curation, Formal analysis, Methodology, 1
Elham Khodaverdi Conceptualization, Formal analysis, Methodology, Supervision, 1, 2
Hossein Shahdadi Sardou Conceptualization, Formal analysis, Methodology, Writing – review & editing, 1, 2, 3, *
Ali Nokhodchi Conceptualization, Methodology, Writing – review & editing, 4, 5, 6, *
Author information:
1Department of Pharmaceutics, School of Pharmacy, Mashhad University of Medical Sciences, Mashhad, Iran
2Targeted Drug Delivery Research Center, Pharmaceutical Technology Institute, Mashhad University of Medical Sciences, Mashhad, Iran
3Health Incubator and Innovation Center, Health Science and Technology Park, Mashhad University of Medical Sciences, Mashhad, Iran
4School of Life Sciences, University of Sussex, Brighton, UK
5Lupin Research Inc, Coral Springs, Florida, USA
6Daru Vira Iranian Pharmaceutical Group, Isfahan, Iran
Abstract
Introduction:
This study aimed to investigate the feasibility of preparation of sustained-release chlorpheniramine maleate (CPM) pellets based on Compritol® as a lipid matrix and evaluation of the affecting factors on pellet properties.
Methods:
Using the D-optimal experimental design, different pellet formulations containing various amounts of CPM, Compritol® and Avicel were prepared by the wet extrusion-spheronization method. Then the pellets were cured at 40, 65 and 90 ̊C for 4 and 8 h to study the effect of the thermal process. The physicomechanical properties of the pellets were investigated in terms of particle size distribution, pelletization yield, mechanical strength, aspect ratio and sphericity. To investigate the possible interaction of CPM and Compritol®, as well as to evaluate the morphology and surface characteristics of the pellets DSC and SEM were used, respectively. Also, to investigate the drug release rate from pellets the dissolution test was carried out and mean dissolution time (MDT) was calculated to compare different formulations.
Results:
The results showed that the curing process up to 65 °C improves the strength of the pellets. However, increasing the curing temperature from 65 to 90 °C and also increasing the curing time from 4 to 8 h did not have a significant effect on the strength of the pellets but increased the drug release rate of pellets. Increasing the amount of the drug or decreasing Compritol® in the matrix of pellets leads to a larger particle size with greater mechanical strength. All formulations of the pellets had an aspect ratio and sphericity of about 1.1 and 0.9 respectively, which indicates the spherical shape of the pellets as shown by SEM. DSC thermograms indicate the reduction of the crystallinity or the change of the crystalline form of the drug to amorphous during the pelletization process.
Conclusion:
The results revealed the feasibility of preparing lipid-based sustained-release matrix pellets using the wet extrusion-spheronization method. The optimal formulation in terms of physicomechanical properties and release rate was the formulation containing 8% CPM, 67% Compritol® and 25% Avicel, which were dried at 40 ° C for 4 h and released about 90% of the drug within 12 h.
Keywords: Sustained-release systems, Chlorpheniramine maleate, Compritol®, Wet extrusion-spheronization, Matrix pellet
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This research (Grant number: 951795) was supported by the Vice-Chancellor for Research and Technology of Mashhad University of Medical Sciences, Mashhad, Iran (MUMS).
Introduction
Sustained-release systems release the drug slowly over a long period.1 These systems create a suitable therapeutic concentration of the drug over a long period with benefits such as a reduction in fluctuations of drug concentration in the blood, and a reduction in the amount of drug consumption hence less side effects.2 This class of drug delivery system is usually used orally because there is more flexibility in the design of oral dosage forms.3 Several dependent variables, including the type of disease, drug characteristics, duration of treatment and the type of drug delivery system influence the design of slow-release drug delivery systems which should be considered in designing the dosage form.4-8
Chlorpheniramine maleate (CPM) as a model drug is one of the most common anti-allergic drugs in the antihistamine drug group which reduces allergic responses by competing with histamine to bind H1 receptors.9 This drug is usually converted into mono- and Di-desmethyl metabolites in the liver, which are produced through N-dealkalization and about 20% of this substance is excreted unchanged from the urine.9-11 CPM does not have active metabolites and its metabolism is carried out with the help of cytochrome P450 (CYP) 2D6 and does not seem to involve CYP2C in the metabolism.12 Due to these characteristics, the effectiveness of the sustained-release formulations of this product is between 8 and 12 h while this amount is about 4 h in fast-release products.12,13 The high dose and rapid absorption of this drug following dose dumping in single unit sustained release dosage forms have led to serious side effects such as imbalanced body movements, anaemia, dry mouth, nausea, anorexia, diarrhea and sensitivity to light in some patients.14 To overcome this problem, multi-unit sustained-release systems can be used to adjust the release rate of the drug and prevent the sudden release of the dosage form.15 Multi-unit dosage forms are more popular than single-unit forms because they have potential benefits such as predictable gastric emptying, lack of dose dumping, flexible release patterns and increased bioavailability.
Pellets are the most popular multi-unit dosage form and their particle size varies from 0.5 to 1.5 mm.16-19 There are different methods for producing pellets, which can be mentioned as rotary fluid-bed granulators, suspension layering, powder layering and extrusion–spheronization methods.20 The extrusion-spheronization as a multi-stage pellet production process is widely used in the pharmaceutical industry, which is its main advantage over other methods such as its ability to produce pellets of high-dose drugs without creating large particles, easy mixing of particles, the production of low dust during the production process, the reduction of the surface-to-volume ratio and the use of the least amount of coated materials, solving the problems related to fluctuations in weight and dosage during packaging, suitable hardness and packaging.20-22 In some studies, the melt extrusion technique has been utilized for pellet production. However, this method presents several drawbacks including high energy consumption, limited compatibility with specific materials, and high costs. Consequently, recent research has shifted its focus towards the wet extrusion technique, which is a more straightforward and cost-effective approach than the melt extrusion technique.23,24 The pellet formulations are mainly coated with a polymer film to have a control-release effect and depending on the type of polymer film and the thickness of the coating layer affect the drug release pattern.25-27 On the other hand, it is possible to change the rate of drug release by using hydrophobic substances as excipients in the matrix of the pellet.28-30 Different types of lipids have been widely used as pharmaceutical additives due to their relatively low price, low toxicity and biodegradable properties.31 Glycerides are lipid molecules that are used as porous additives in the matrix of dosage forms.32 Drug release from these matrices occurs through erosion and diffusion from the pores.33 Consequently, the release properties of these matrices are more sensitive to the composition of gastrointestinal fluids than insoluble polymer matrixes.33
Among different glycerides, Compritol® has attracted special attention. This material has been widely used as a matrix-forming agent in the manufacture of various slow-release tablets and also as a coating agent in the melt coating method for producing powders or granules to create slow-release materials.34 Due to the complex structure and incomplete orientation of its crystals, Compritol® provides more space for drug loading and also has shown good performance in terms of drug-trapping ability.34,35
Compritol® remains solid at ambient temperature, and its inclusion in the matrix of dosage forms results in a reduction in the rate of drug release.2 Research has demonstrated that the curing process applied to drug forms containing Compritol® can effectively slow down the drug release rate. Essentially, the curing process causes Compritol® to melt and combine with other components present in the matrix of dosage forms. By removing the heating process and subjecting the dosage form to ambient temperature, the Compritol® within the matrix reverts to a solid state and in this way prevents the rapid release of the drug.36,37
In the present study, a multi-unit slow-release drug delivery system for CPM was designed and evaluated as a model drug. Compritol® was used as a lipid base drug release retardant in matrix pellet formulation. The effect of different ratios of the drug and lipid as well as the effect of temperature and curing time at different temperatures were investigated on the morphological characteristics, mechanical strength and drug release pattern of pellets.
Materials and Methods
Materials
Chlorpheniramine maleate was purchased from Merck (Germany), and Compritol® was supplied by Gattefossé (France). Microcrystalline cellulose (Avicel® PH101) was obtained from Akbarieh (Tehran, Iran). All chemical reagents used were of analytical grade.
Experimental design
Based on preliminary studies, three factors were considered including drug percentage (as a formulation variable) and two process variables (temperature and curing time) (Table 1). To achieve the optimal formulation, the response-surface method (RSM) and D-optimal design were used (Design-Expert software, Version 10, Stat-Ease, USA) and 18 different types of formulations were determined (Table 2).
Table 1.
Factors and levels used in experimental design
|
Factors
|
Levels used
|
| −1 |
0 |
+ 1 |
| Drug (%) |
1 |
8 |
15 |
| Temperature (°C) |
40 |
65 |
90 |
| Time (h) |
4 |
- |
8 |
Table 2.
Formulation components and pellet curing conditions
|
Formulations
|
Drug (%)
|
Compritol®
(%)
|
Avicel (%)
|
Temperature (C)
|
Time (h)
|
| A1 |
1 |
74 |
25 |
40 |
4 |
| A2 |
1 |
74 |
25 |
40 |
8 |
| A3 |
1 |
74 |
25 |
65 |
4 |
| A4 |
1 |
74 |
25 |
65 |
8 |
| A5 |
1 |
74 |
25 |
90 |
4 |
| A6 |
1 |
74 |
25 |
90 |
8 |
| B1 |
8 |
67 |
25 |
40 |
4 |
| B2 |
8 |
67 |
25 |
40 |
8 |
| B3 |
8 |
67 |
25 |
65 |
4 |
| B4 |
8 |
67 |
25 |
65 |
8 |
| B5 |
8 |
67 |
25 |
90 |
4 |
| B6 |
8 |
67 |
25 |
90 |
8 |
| C1 |
15 |
60 |
25 |
40 |
4 |
| C2 |
15 |
60 |
25 |
40 |
8 |
| C3 |
15 |
60 |
25 |
65 |
4 |
| C4 |
15 |
60 |
25 |
65 |
8 |
| C5 |
15 |
60 |
25 |
90 |
4 |
| C6 |
15 |
60 |
25 |
90 |
8 |
Preparation of matrix pellets
Three groups of pellets were prepared as formulations A, B and C which contained 1, 8 and 15% of CPM, respectively (Table 2). Avicel content in all formulations was fixed at 25%. Pellets were prepared using the wet extrusion-spheronization method. Briefly, the solid components of the formulations including the drug, Compritol® and Avicel were weighed according to Table 2 and mixed with a kitchen mixer (FUMA, Fu-1877 Hand Mixer, Japan) for 10 min. Then with adding distilled water as granulation liquid a uniform wet mass with a suitable consistency was obtained. The wet mass was passed through an extruder (DorsaTech, Iran) with a 1 mm pore size die at a speed of 100 rpm to obtain suitable extrudates. The resulting extrudates were placed in a spheronizer at a speed of 1000 rpm for 2 min to form spherical pellets. To evaluate the effect of thermal processing or curing on pellets, according to Table 2, wet pellets were placed at 45, 65, and 90 °C for different times (4 and 8 h).
Particle size analysis
To evaluate the pelletization yield and choose the appropriate size range for subsequent tests, the pellets were sieved by mesh 14 and 25 with sizes of 1400 and 710 μm, respectively. The pellets with particle size less than 710 μm were considered as fine particles and pellets with particle size greater than 1400 μm were considered as large particles. Also, pellets between 710 and 1400 μm were selected as pellets of appropriate particle size and their percentages were calculated as pelletization yield.
Mechanical testing of pellets
To evaluate the mechanical properties of pellets, 30 pellets from each formulation were selected and evaluated for mechanical strength using a material testing machine (Hounsfield, England) equipped with a 1 kN load cell. According to the diagram of force-displacement and stress-strain, the values of crushing strength and elastic modulus were calculated by QMat software.
Image analysis
To assess the shape and sphericity of the pellets, several pellets were randomly selected from each group of formulations A, B and C and fixed on a black background surface and imaging was conducted by a stereomicroscope (Olympus, DP25, Okura, Japan) equipped with a digital camera (Canon, Japan). The images were analyzed using ImageJ 1.52v software and the parameters of aspect ratio and sphericity were calculated as follows:
Eq. (1)
Eq. (2)
Where A is the area, Pm is the perimeter, dmax and dmin are the longest and shortest Feret’s diameters.
Differential scanning calorimetry (DSC)
To study the thermal behavior of pellets components, samples of the drug, Compritol®, their physical mixture and two selected pellet formulations (B1 and B6) were examined using differential scanning calorimetry (DSC; Metler Teldo DSC822, Switzerland). The samples were scanned by DSC at 25 to 300 °C at a rate of 10 °C/min under nitrogen gas with a flow of 20 mL /min.
Fourier transform infrared spectroscopy (FTIR)
FT-IR spectroscopy was accomplished in KBr discs over a range of 4000–450 cm-1 using IR spectroscope equipment (vortex 70, Bruker, Germany). Sample preparation involved mixing the samples with potassium bromide (KBr). Potassium bromide discs were then prepared by compressing the mixture using a hydraulic press.
Determination of drug content in pellets
To determine the drug content in pellets, 200 mg of different pellet formulations were ground with 2 mL of ethanol followed by adding distilled water to make up to 100 mL. The sample was stirred for 1 h on a magnetic stirrer to ensure the complete extraction and dissolution of the drug. The obtained solution was filtered with a paper filter followed by the reading the absorbance of the filtered solution at a wavelength of 262 nm to determine the concentration of the drug in the sample using the standard curve. To ensure the stability of the drug during the curing process at 90 °C, the content of formulation B1 was also measured before and after curing.
Scanning electron microscope (SEM)
To investigate the surface morphology of the pellets, a number of pellets were fixed on a black steel grid and covered with a thin gold coating and were examined by TESCAN MIRA3 SEM (Czech Republic) at 50, 100 and 1000 magnification.
In-vitro release test
The CPM release profile from pellets was examined using a USP dissolution apparatus I (Pharmatest, PTWS, Germany). To perform the dissolution test, 500 mg (pellet) of each formulation was placed in the basket and immersed in 900 mL of distilled water as dissolution medium at 37 ℃ and rotated at 100 rpm (suggested by US Pharmacopoeia). At several time intervals, samples were taken and the rate of CPM absorption was measured by using a UV spectrophotometer (Shimadzu, UV-1204, Japan) at wavelength 262 nm (n = 3). Finally, by using the standard curve, the amount of drug released at each time point was determined. From drug release data, mean dissolution time (MDT) was computed via the following equations.18
Eq. (3)
Where
is the midpoint of the time during which the fraction Mi of the drug is released from the dosage form. A high MDT value means that the drug delivery system has a slow in vitro drug release.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism software (version 8.4.0). Kruskal Wallis non-parametric test and other parameters were compared by a one-way ANOVA test. All results were expressed as mean ± SD. For all tests, P values less than 0.05 were considered to be statistically significant.
Results and Discussion
Particle size distribution
Sieve test results showed the highest pelleting efficiency is related to formulation A (Table 3). In other words, about 65% of the pellets were in the range of 710 to 1400 μm. As in formulation A, the amount of drug was at a lower amount and Compritol® (hydrophobic excipient)was at a higher level compared to formulations B and C, therefore the hydrophobic nature of this formulation is more than formulations B and C. It was interesting to note that the adhesion of the particles increased with the reduction of the hydrophobic nature from formulation A to C. For this reason, the contribution of particles larger than 1400 μm increased from formulation A to C, respectively. Qazi et al also showed that the lower amount of Compritol® in the pellet formulation leads to the production of larger pellets.38
Table 3.
Particle size distribution, sieve test and pelletizing efficiency results
|
Formulation
|
Weight (%)
|
Pelletizing efficiency
(%)
|
|
Smaller than 710 (µm)
|
Between 710 and 1400 (µm)
|
More than 1400 (µm)
|
| A |
6 |
64.58 |
29.42 |
64.5 |
| B |
0.75 |
45.71 |
53.54 |
45.7 |
| C |
19.8 |
36.7 |
43.5 |
36.7 |
Mechanical testing of pellets
The result of the mechanical test of different formulations showed that in uncured pellets, the crushing strength and elastic modulus increased with increasing the amount of drug and decreasing the amount of Compritol®, which attributed to the increase in the strength of the pellets. As can be seen in Table 4, the curing process led to an increase in the mechanical strength of the pellets, especially the effect was more significant in the case of the A series formulations, which have a low level of drug and a high level of lipid, which can be related to the melting of Compritol® at high temperatures and the formation of an integrated matrix structure. However, increasing the temperature from 65 to 90 ℃ does not show much effect on the strength of the pellets. Among the cured formulations compared to the uncured formulations, the change in the amount of drug from 1 to 15% showed little effect on the strength of the pellets. Also, the curing time had almost no effect on the strength of the pellets and only in the C series formulations that the amount of drug and lipid were high and low respectively, the strength of the pellets increased slightly by increasing the heating time from 4 to 8 h. Probably in these formulations, due to the high percentage of drug and Avicel among the lipid particles, the lipid particles separated from each other and as a result with the increase of time, the molten lipids have more opportunity to get closer to each other and merge. Several studies showed that very strong bonds are established between microcrystalline cellulose particles in the presence of water, which become weaker with the increase of Compritol® in the formulation.1,31,39 As a result, the lower amount of Compritol® leads to an increase in the hardness of pellets. Some studies have also used Compritol® as a lubricant alone or in combination with magnesium stearate at a rate of less than 5%.2,34,35 However, the higher percentages of drug (8-15%) used in our study create a highly lubricated formulation that covers the connection points and leads to poor connections in the pellets.
Table 4.
The results of the mechanical test of different formulations
|
Formulation
|
Crushing strength
(N)
|
Elastic modulus
(MPa)
|
| A1 |
2.28 ± 0.29 |
39.99 ± 1.93 |
| A2 |
2.19 ± 0.26 |
40.16 ± 2.31 |
| A3 |
2.66 ± 0.38 |
42.1 ± 3.5 |
| A4 |
2.5 ± 0.3 |
43.6 ± 3.8 |
| A5 |
2.65 ± 0.37 |
45.68 ± 2.57 |
| A6 |
2.81 ± 0.36 |
44.17 ± 3.39 |
| B1 |
2.36 ± 0.18 |
46.18 ± 2.98 |
| B2 |
2.8 ± 0.28 |
48.19 ± 4.35 |
| B3 |
2.95 ± 0.33 |
47.52 ± 3.65 |
| B4 |
2.72 ± 0.35 |
50.1 ± 4.26 |
| B5 |
2.45 ± 0.24 |
48.19 ± 3.36 |
| B6 |
2.3 ± 0.25 |
52.3 ± 2.59 |
| C1 |
2.63 ± 0.18 |
52.08 ± 3.89 |
| C2 |
2.84 ± 0.35 |
53.06 ± 2.93 |
| C3 |
2.75 ± 0.31 |
54.3 ± 4.47 |
| C4 |
2.88 ± 0.39 |
55.17 ± 3.67 |
| C5 |
2.58 ± 0.37 |
55.1 ± 3.31 |
| C6 |
2.75 ± 0.44 |
57.19 ± 4.36 |
As shown in Fig. 1, the crushing strength and elastic modulus increased with the increase of drug amount, which is more obvious in the pellets cured at low temperature and the difference decreased with increasing the temperature. The results showed that by increasing the curing time in the C series formulation (the series that had a higher percentage of the drug) the mechanical strength of the pellets slightly increased by increasing the time from 4 to 8 h at 40 ℃. This was not the case for other series of formulations. The relationship between temperature and curing time and drug percentage with crushing strength and elastic modulus were shown in Figs. 1A-B and 1C-D, respectively. The following equations characterized the relationship between these variables.
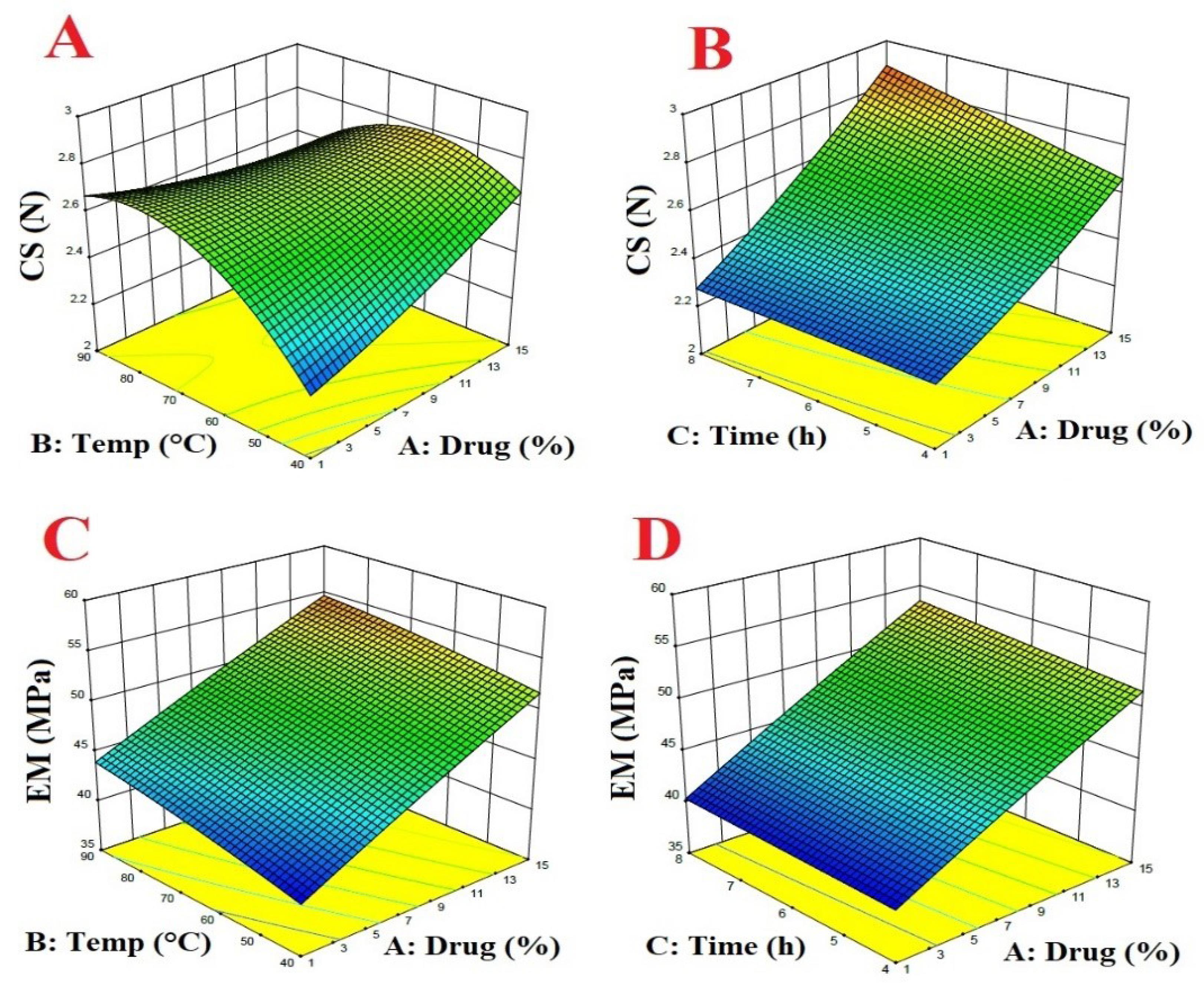
Fig. 1.
The relationship between temperature, curing time and drug percent with crushing strength and elastic modulus.
.
The relationship between temperature, curing time and drug percent with crushing strength and elastic modulus.
CS = 0.684292 + 0.0377109 × Drug + 0.0510306 × temp + 0.0271351 × time − 0.000805952 × Drug × temp + 0.00357341 × Drug × time − 0.000634333 × temp × time + 0.000571315 × Drug2 −0.000302942 × temp2
R2 = 0. 621
EM = 34.0554 + 0.89522 × Drug + 0.1328 × temp + 9.8055E-003 × time-1.8214E-003 × Drug × temp + 0.0225 × Drug × time + 2.55003E-003 × temp × time − 4.01360E-003 × Drug2 − 4.38666E-004 × temp2
R2 = 0. 98
Morphological observation
Fig. 2 shows the stereomicroscope image of C series pellets. The results of the image analysis of the pellets showed that all three series of formulations had an aspect ratio and sphericity of about 1.1 and 0.9, respectively. In the case of a completely spherical shape, both aspect ratio and sphericity values should be closer to 1. On the other hand, the increase in the aspect ratio and the decrease in sphericity indicate that the particles departed from the spherical shape. In general, no significant difference was observed in all three series of pellet formulations.

Fig. 2.
Stereomicroscope image of C series formulation pellets.
.
Stereomicroscope image of C series formulation pellets.
Differential scanning calorimetry (DSC)
The thermogram obtained from the DSC test for CPM showed a sharp endothermic peak at 134 °C, which is related to its melting point (Fig. 3). Also, a sharp endothermic peak at 70 °C can be seen for Compritol®. In the thermogram of the physical mixture of the drug and Compritol® (1:1), both peaks related to the drug and Compritol® are observed almost unchanged and shifted, which shows that these two substances did not interfere with each other. However, in the thermogram related to the pellets, only one sharp peak at 70 °C can be seen, which is probably related to Compritol®, but the peak related to the drug is not observed. The disappearance of the drug peak is probably related to the decrease in the drug crystallinity or the change in the nature of the drug from crystalline to amorphous during the pelleting process, such as dissolution of the drug in the granulation liquid and/or the change in the structure during drying at 40 °C or curing at 65 and 90 °C. Another reason could be due to the low percentage of chlorpheniramine maleate in the pellet structure compared to Compritol® which can reduce the intensity of the drug peak significantly or make it undetectable during the DSC scan.
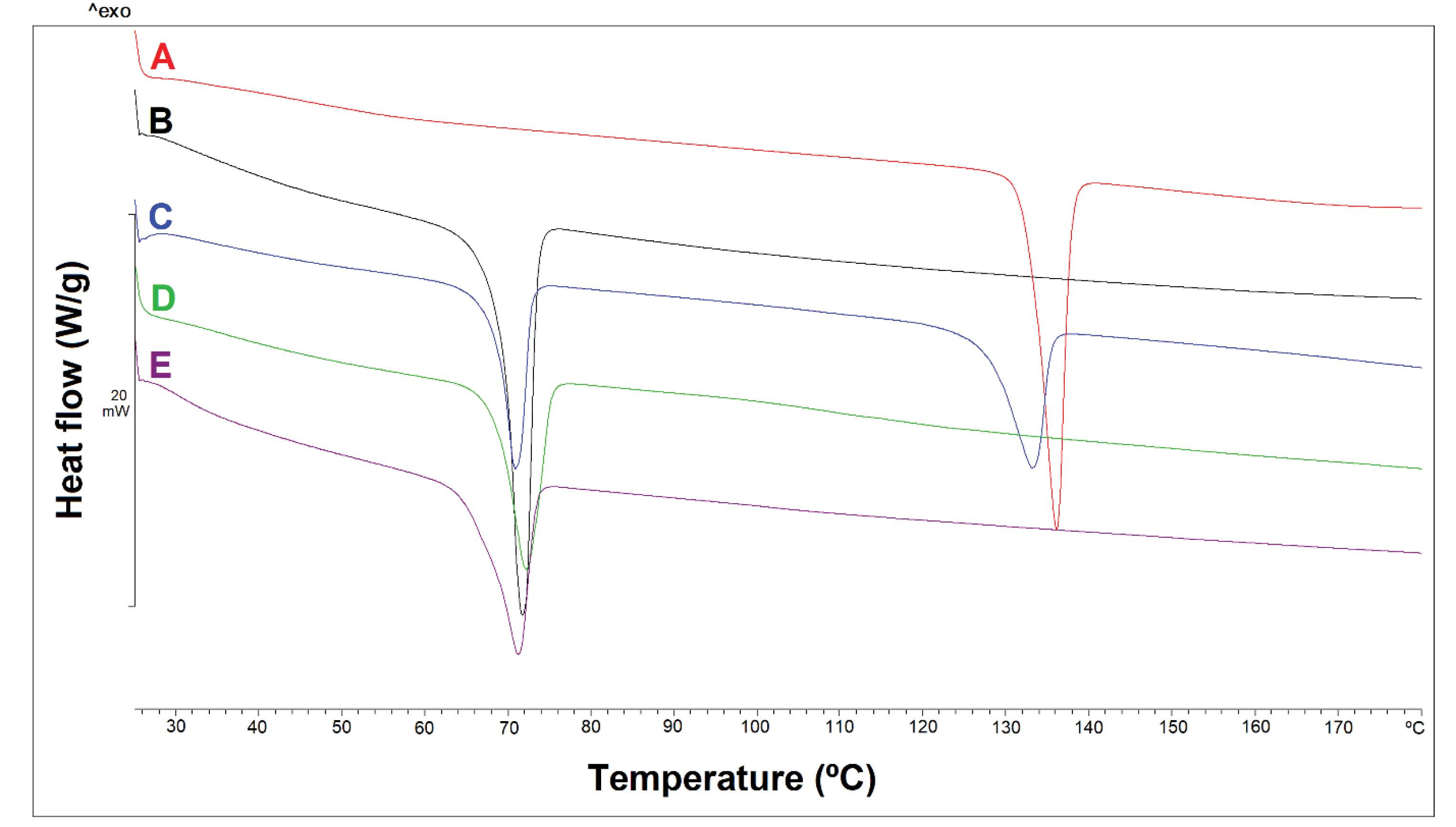
Fig. 3.
DSC diagrams of CPM (A), Compritol® (B), Physical mixture of CPM and Compritol® (C), B1 formulation pellet placed in 40 °C oven for 4 h (D), B1 formulation pellet placed in 90 °C oven for 8 h (E).
.
DSC diagrams of CPM (A), Compritol® (B), Physical mixture of CPM and Compritol® (C), B1 formulation pellet placed in 40 °C oven for 4 h (D), B1 formulation pellet placed in 90 °C oven for 8 h (E).
FTIR
The spectrum of the CPM shows an index peak in the range of 1600 cm-1 and weaker peaks in the region of 450-600 cm-1 (Fig. 4A). These peaks are also detectable in the physical mixture (Fig. 4C) and B1 formulation placed in the oven at temperatures of 40 (Fig. 4D) and 90 °C (Fig. 4E). However, the peak intensity of the CPM in the physical mixture is higher than that of the B1 formulation (Figs. 4D & 4E), which is due to the high ratio of the CPM in the physical mixture. A comparison of the spectra of formulation B1 placed in the oven at 40 °C for 4 h (Fig. 4D) with formulation B1 placed in the oven at 90 °C for 8 h (Fig. 4E) shows that both peaks match each other. In other words, the temperature of 90 °C did not change the FTIR spectrum.
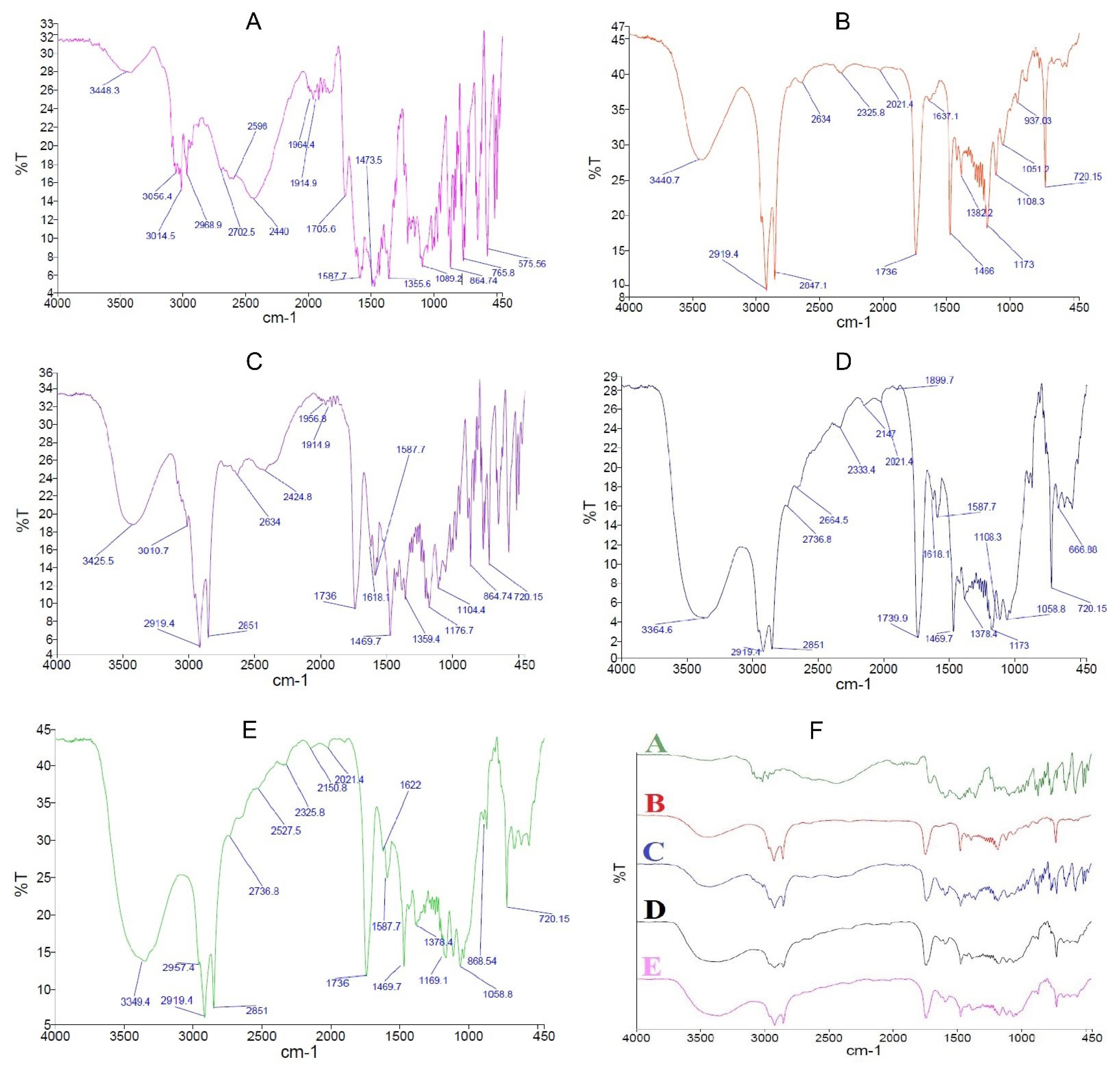
Fig. 4.
FTIR Spectra of CPM (A), Compritol® (B), Physical mixture of CPM and Compritol® with a ratio of 1:1 (C), B1 formulation pellet placed in 40 °C oven for 4 h (D), B1 formulation pellet placed in 90 °C oven for 8 h (E), and overlap of all spectra (F).
.
FTIR Spectra of CPM (A), Compritol® (B), Physical mixture of CPM and Compritol® with a ratio of 1:1 (C), B1 formulation pellet placed in 40 °C oven for 4 h (D), B1 formulation pellet placed in 90 °C oven for 8 h (E), and overlap of all spectra (F).
Determination of drug content in pellets
The drug content determination results showed that B1 formulation pellets contained 8.450 ± 0.055 and 8.540 ± 0.060 chlorpheniramine before and after the curing process, respectively.
Scanning electron microscopy (SEM)
To evaluate the surface morphology of the pellets, formulation C was examined by SEM before and after the curing and dissolution test (Fig. 5). The images of the pellets before curing show the spherical and rough appearance, but curing at high temperatures led to a smoother and more uniform surface for these pellets. This phenomenon is probably caused by the melting and fusion of Compritol® particles. As can be seen in the images of this series of pellets, some holes and pores have appeared on the surface of the pellet, which is probably due to the contraction resulting from the merging of Compritol® particles together and the creation of empty spaces in the pellet structure. It was expected that this contraction would occur in the entire pellet structure leading to denser pellets, but due to the fact that about 30% of the pellet is composed of Avicel and drug, possibly these two components of the formulation have created a strong matrix structure inside the pellet which prevent whole pellet shrinkage. SEM images of the pellets after the dissolution test show an increase in pores and voids in the pellet structure which could be related to the dissolving of the drug and its removal from the pellet structure (Figs. 5 j-l). The increase in the pores of the pellet surface after the dissolution test in the case of the cured pellets is much more than the uncured pellets, while in both pellets, almost all of the drug content of the pellets has been released to the same extent.
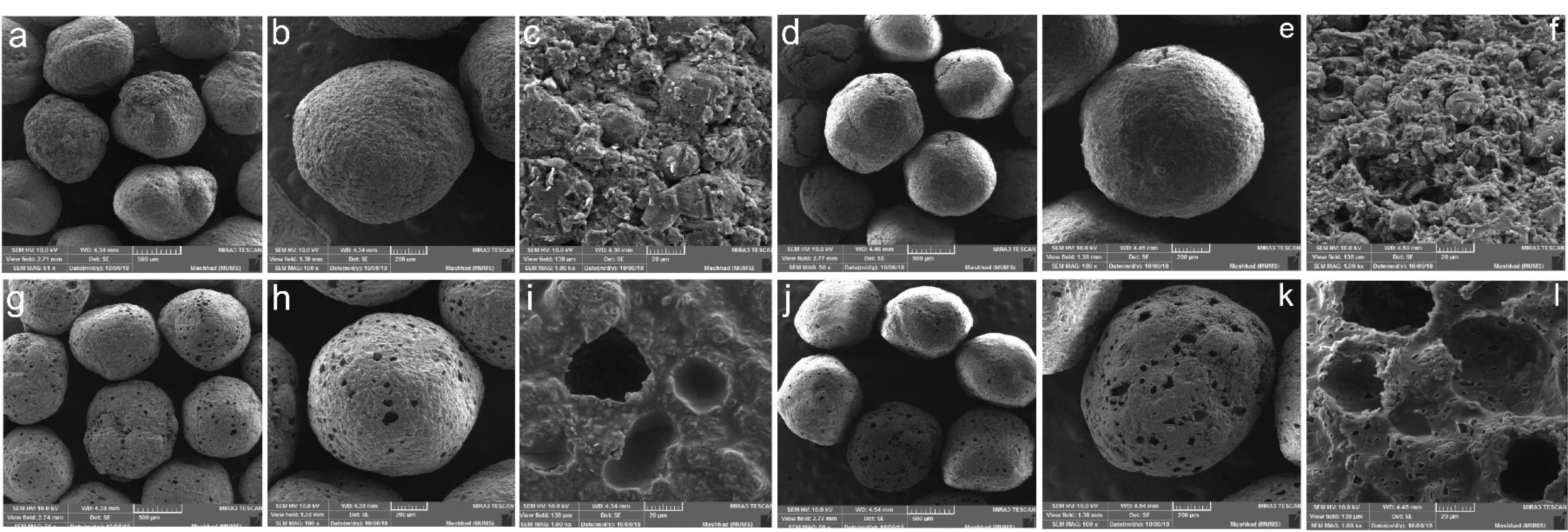
Fig. 5.
Scanning electron microscopy of the a-c) Uncured pellets of formulation C, d-f) Uncured pellets of formulation C after dissolution test, g-i) Cured pellets at 90°C for 8 h from formulation C, j-l) Cured pellets at 90°C for 8 h from formulation C after dissolution test in 50, 100 and 1000 magnifications.
.
Scanning electron microscopy of the a-c) Uncured pellets of formulation C, d-f) Uncured pellets of formulation C after dissolution test, g-i) Cured pellets at 90°C for 8 h from formulation C, j-l) Cured pellets at 90°C for 8 h from formulation C after dissolution test in 50, 100 and 1000 magnifications.
Dissolution studies
To conduct this test, at first, the uniformity of the content was initially assessed in all formulations. The evaluation indicated that the drug content of the pellets remained consistent before and after the curing process (the results are not shown). In other words, no drug was lost after the curing process. The results of the dissolution test show that the release rate of the CPM from the pellets is related to the percentage of the drug and Compritol® as well as the curing process (Fig. 5).
In Fig. 6, series A (Fig. 6A), Series B (Fig. 6B) and series C (Fig. 6C) contained 1% 8% and 15% of the drug in the matrix pellet structure. Most of the matrix structure of these pellets is composed of Compritol® and two parameters namely temperature and curing time were investigated. In Fig. 6A-C, these three series of formulations were compared with each other. In Fig. 6A, A1 and A2 showed the release profile of pellets cured at 40 °C for 4 and 8 h. A3 and A4 represented the release profile of pellets cured at 65 °C for 4 and 8 h, and finally, A5 and A6 represented the release profile of pellets cured at 90 °C for 4 and 8 h. Series B and C formulations also followed the same procedure. In Fig. 6D, the formulations of series A (A1-A2), B (B1-B2), and C (C1-C2), which contained different percentages of drugs, were compared in pairs. In Fig. 6E-F, other formulations of series A, B and C were compared in pairs.
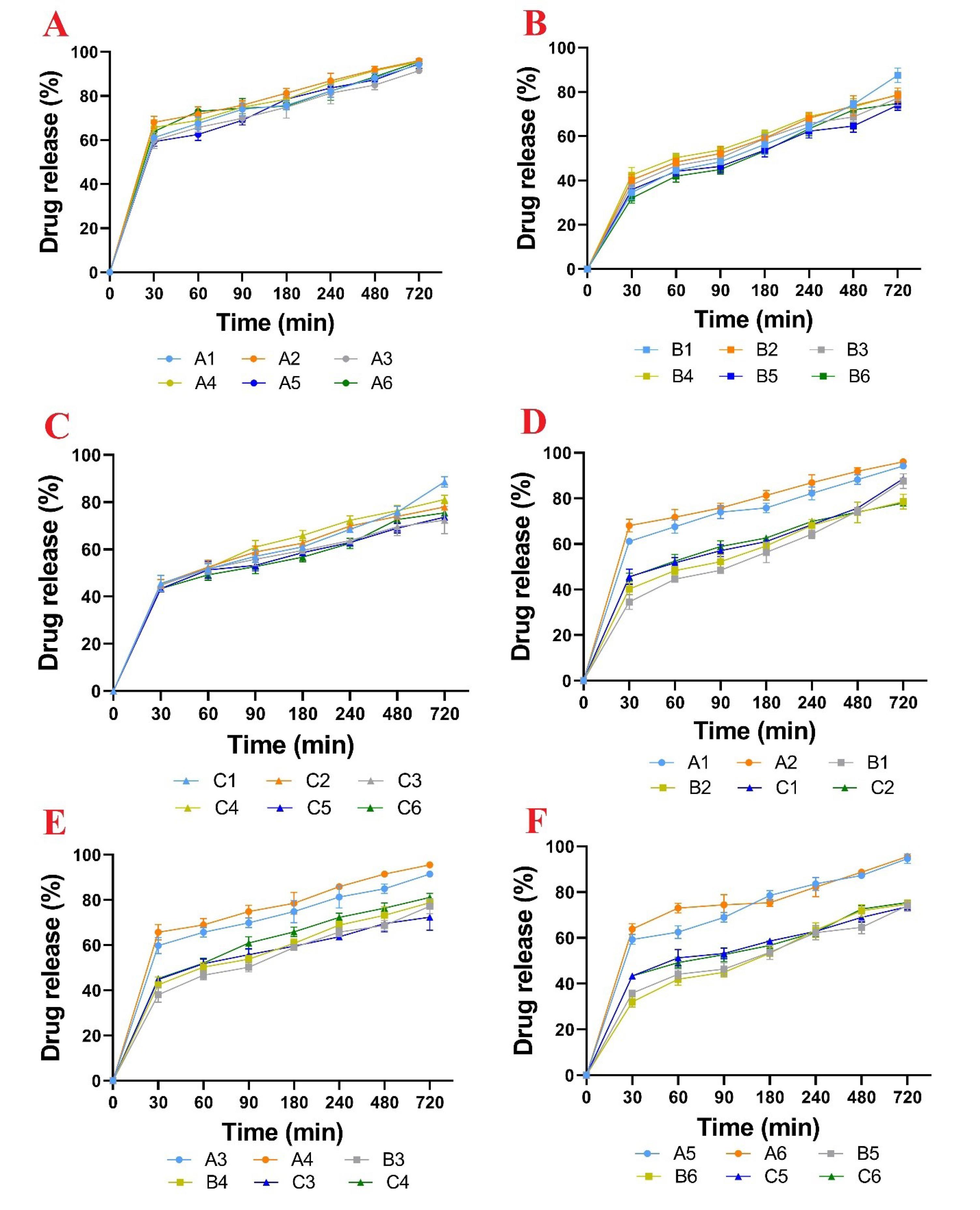
Fig. 6.
The release profile of the drug from series A formulation (A), series B formulation (B), series C formulation (C), Six formulations under curing at 40 °C (D), Six formulations under curing at 65 °C (E), Six formulations under curing at 90 °C (F). Data are shown as mean (n = 3).
.
The release profile of the drug from series A formulation (A), series B formulation (B), series C formulation (C), Six formulations under curing at 40 °C (D), Six formulations under curing at 65 °C (E), Six formulations under curing at 90 °C (F). Data are shown as mean (n = 3).
Comparing the dissolution of formulations in series A with series B showed that with an increase in drug concentration from 1 to 8% (despite the reduction of Compritol®) the MDT value increased and as a result, the drug release rate decreased. Probably, this result can be attributed to the increase in hardness of series B pellets compared to series A. Previous studies showed that the rate of drug release from pellets had an inverse relationship with increasing their mechanical strength. In general, by increasing the percentage of the drug from 8 to 15 in the pellet formulation, the drug release rate increased, which can be attributed to the increase in the drug exposed to the dissolution medium as well as the decrease in Compritol® percentage. Another point that could be seen in the drug release profiles of different formulations was that the A series formulations released more than 60% of the drug content during the first 30 min and released almost the entire drug content after 12 h, while the B and C series formulations released about 40% of the drug within the first 30 min and 75% within 12 h. In the B and C series formulations, only uncured pellets were able to release about 90% of the drug content within 12 h. However, their release percentage within 6 h did not show a significant difference when compared with the cured pellets. It seems that in formulations series B and C, rapid release occurs due to high surface porosity but in the pellets that have been subjected to the curing process, Compritol® particles melt and then merge and condense in the deeper region of the pellet. Consequently, the drug release takes place at a lower rate and much more slowly in the final stages. While in uncured pellets, due to the lack of Compritol® melting/densification, the drug is released almost completely within 12 h. In our study, the best slow-release pattern takes place by applying 67% Compritol® in combination with CPM (series B). It seems that despite the lipophilicity and retarding effect of Compritol®, the high amounts of Compritol® probably had a negative effect on the slow release of the pellets due to the increase in plasticity and hence decrease in the strength of the pellets.
In this study, MDT decreased with increasing temperature and curing time. However, it was expected that the release rate of the drug would be slower in tablet formulations with the lipid matrix.33 But in this study, contradictory results were obtained, which can be attributed to the creation of voids in the pellet structure after the curing process. As can be seen in the SEM images, these voids facilitate the infiltration of the dissolution medium into the matrix pellet and lead to faster dissolution and release of the drug. Yan et al showed that the accelerated stability test and keeping the lipid-based pellets at 40 °C for 5 months increased the release rate of the slow-release aspirin pellets. The SEM images of the pellets in their study also showed pores and holes in the surface of the pellets that it is attributed to the melting and accumulation of lipid particles.40 In another study, Amador Ríos et al investigated the effect of lipid amount on the slow-release properties of Lisinopril pellets that were pressed into tablets.39 The results of their study showed that tablets containing 30% Avicel and 30% Compritol® released about 80% of the drug content within 6 h, while the release of the drug from tablets with 30% Avicel and 60% Compritol® was completely done in the same period. They interpreted their results in such a way that the addition of Compritol® probably interferes with the formation of the Avicel matrix during the pressing process. However, their thermal curing of the tablets showed the reverse results, such that the formulation including 60% Compritol® and 30% Avicel had 85% release and the formulation including 60% Avicel and 30% Compritol® released all of the drug content. Their reasoning was that in the thermal curing process, Compritol® melted and migrated to the surface of the tablet and acts as a lipophilic coating against drug release.39 Their study also showed similar results on Ketorolac Tromethamine slow-release tablets based on Compritol®.39
The SEM images and the smooth and non-porous surfaces of the tablets in this study indicated that with increasing the thermal curing time, Compritol® melts and is placed on the surface of the tablets and the hydrophobic property of these lipids causes the slow release of the drug.39
Fouad et al designed and produced matrix tablets containing CPM in different proportions with Compritol® and Kollidone SR by direct compression method. These tablets were organoleptically tested and evaluated for CPM release. Diffusion kinetics results showed the Fickian diffusion mode for Kollidone and anomalous diffusion mechanism for Compritol® matrixes. The combination of Compritol® as a lipophilic agent and Kollidone matrix produced controlled drug release. They attributed the delay in drug release to the Compritol® to Kollidone ratio and reported that the lower the Compritol® component, the slower the drug release. However, when the ratio of Compritol® was increased, the drug release followed unusual non-Fickian kinetics due to the dehydrating effect of Compritol®. This study focuses on single-unit dosage forms (tablets) and has generally investigated the release profile of CPM by changing the ratios of Compritol® to Kollidone SR. In our study, the pellet dosage form was used due to the advantages of multi-unit dosage forms compared to single-unit dosage forms. In addition to examining the drug release profile, DSC studies were used to examine the interaction between the drug and excipients, as well as SEM studies for structural investigation and comparison of the pores formed after the curing process in the pellets have been used.41
In the present study, the pellets prepared by the wet extrusion-spheronization method have a lower density than tablets, and by performing thermal curing, Compritol® melted and probably accumulated in the center of the pellets. In general, series A formulations showed fast release due to lower hardness despite having high Compritol® and series C formulations also showed fast release due to low Compritol percentage. Finally, formulation B1, which had an appropriate Compritol® amount and hardness showed the lowest release rate.
Conclusion
The results of this study showed, all series of pellets had an aspect ratio of about 1.1 and a sphericity of about 0.9, which indicates their high sphericity. The drug release pattern from these pellets can be modulated by changing the amount of drug and excipients such as Avicel® and Compritol®. In general, by increasing the amount of the drug or decreasing the amount of Compritol® in the matrix the size of the pellets became larger, and also their mechanical strength increased. The DSC thermogram indicated the reduction of the crystal structure or the change of the crystalline form of the drug to amorphous during the pelleting process. The curing process had a negative effect on the control of the release of the CMP from pellets containing Compritol®, and the lowest release was observed in uncured pellets. Finally, the formulation containing 8% CMP, 67% Compritol® and 25% Avicel®, which were dried at 40 ℃ for 4 h showed a slower release than other formulations and was selected as the optimal formulation due to acceptable physical and mechanical properties. These pellets released about 50% of the drug in the first 2 h and released the remaining 50% slowly within 12 h according to the US Pharmacopoeia standard for CMP slow-release capsules.
Research Highlights
What is the current knowledge?
√ Single unit sustained release dosage forms with high doses show dose dumping risk hence side effects.
√ The effectiveness of fast-release CPM formulations is approximately 4 hours.
√ Sustained-release products offer a therapeutic concentration over a longer period (8-12h) with less side effects
What is new here?
√ Compritol® offers ample space for drug loading as a multi-unit sustained-release system.
√ Release profile of CPM can be modulated by the composition and curing conditions.
√ Morphological changes and mechanical strength could play a role in modulating drug release pattern.
Competing Interests
There is no conflict of interest.
Ethical Statement
Not applicable. This paper does not involve research on animals and humans.
Acknowledgements
The authors thank the Vice-Chancellor for Research and Technology of Mashhad University of Medical Sciences, Mashhad, Iran (MUMS) for supporting this research.
References
- Adepu S, Ramakrishna S. Controlled drug delivery systems: current status and future directions. Molecules 2021; 26:5905. doi: 10.3390/molecules26195905 [Crossref] [ Google Scholar]
- Rao H, Ahmad S, Madni A, Rao I, Ghazwani M, Hani U. Compritol-based alprazolam solid lipid nanoparticles for sustained release of alprazolam: preparation by hot melt encapsulation. Molecules 2022; 27:8894. doi: 10.3390/molecules27248894 [Crossref] [ Google Scholar]
- Sun S, Du X, Fu M, Khan AR, Ji J, Liu W. Galactosamine-modified PEG-PLA/TPGS micelles for the oral delivery of curcumin. Int J Pharm 2021; 595:120227. doi: 10.1016/j.ijpharm.2021.120227 [Crossref] [ Google Scholar]
- Arévalo-Pérez R, Maderuelo C, Lanao JM. Recent advances in colon drug delivery systems. J Control Release 2020; 327:703-24. doi: 10.1016/j.jconrel.2020.09.026 [Crossref] [ Google Scholar]
- Shahdadi Sardo H, Saremnejad F, Bagheri S, Akhgari A, Afrasiabi Garekani H, Sadeghi F. A review on 5-aminosalicylic acid colon-targeted oral drug delivery systems. Int J Pharm 2019; 558:367-79. doi: 10.1016/j.ijpharm.2019.01.022 [Crossref] [ Google Scholar]
- Kotla NG, Rana S, Sivaraman G, Sunnapu O, Vemula PK, Pandit A. Bioresponsive drug delivery systems in intestinal inflammation: state-of-the-art and future perspectives. Adv Drug Deliv Rev 2019; 146:248-66. doi: 10.1016/j.addr.2018.06.021 [Crossref] [ Google Scholar]
- Shahdadi Sardou H, Rahnama Vosough P, Abbaspour M, Akhgari A, Sathyapalan T, Sahebkar A. A review on curcumin colon-targeted oral drug delivery systems for the treatment of inflammatory bowel disease. Inflammopharmacology 2023; 31:1095-105. doi: 10.1007/s10787-023-01140-0 [Crossref] [ Google Scholar]
- Shahdadi Sardou H, Nazari SE, Abbaspour M, Akhgari A, Sheikh A, Kesharwani P. Nano-curcumin formulations for targeted therapy of colorectal cancer. J Drug Deliv Sci Technol 2023; 88:104943. doi: 10.1016/j.jddst.2023.104943 [Crossref] [ Google Scholar]
- Guo Q, Lin H, Lin J. Chlorpheniramine poisoning as a potential cause of rhabdomyolysis. Am J Emerg Med 2022; 57: 236.e1-236.e3. 10.1016/j.ajem.2022.04.010.
- Yen YT, Chang YJ, Lai PJ, Chang CL, Chen TY, Chyueh SC. A study of opiate, opiate metabolites and antihistamines in urine after consumption of cold syrups by LC-MS/MS. Molecules 2020; 25:972. doi: 10.3390/molecules25040972 [Crossref] [ Google Scholar]
- Xu W, Xia S, Pu J, Wang Q, Li P, Lu L. The antihistamine drugs carbinoxamine maleate and chlorpheniramine maleate exhibit potent antiviral activity against a broad spectrum of influenza viruses. Front Microbiol 2018; 9:2643. doi: 10.3389/fmicb.2018.02643 [Crossref] [ Google Scholar]
- Sychev DA, Ashraf GM, Svistunov AA, Maksimov ML, Tarasov VV, Chubarev VN. The cytochrome P450 isoenzyme and some new opportunities for the prediction of negative drug interaction in vivo. Drug Des DevelTher 2018; 12:1147-56. doi: 10.2147/dddt.s149069 [Crossref] [ Google Scholar]
- Kalaydina RV, Bajwa K, Qorri B, Decarlo A, Szewczuk MR. Recent advances in "smart" delivery systems for extended drug release in cancer therapy. Int J Nanomedicine 2018; 13:4727-45. doi: 10.2147/ijn.s168053 [Crossref] [ Google Scholar]
- Ma L, Zhou L, Xu M, Huang X, Zhang Q, Dai S. Investigation of the distributional homogeneity on chlorpheniramine maleate tablets using NIR-CI. Spectrochim Acta A Mol BiomolSpectrosc 2018; 204:783-90. doi: 10.1016/j.saa.2018.06.081 [Crossref] [ Google Scholar]
- Sufi SA, Hoda M, Pajaniradje S, Mukherjee V, Coumar SM, Rajagopalan R. Enhanced drug retention, sustained release, and anti-cancer potential of curcumin and indole-curcumin analog-loaded polysorbate 80-stabilizied PLGA nanoparticles in colon cancer cell line SW480. Int J Pharm 2020; 588:119738. doi: 10.1016/j.ijpharm.2020.119738 [Crossref] [ Google Scholar]
- Shahdadi Sardou H, Akhgari A, Mohammadpour AH, Beheshti Namdar A, Kamali H, Jafarian AH. Optimization study of combined enteric and time-dependent polymethacrylates as a coating for colon targeted delivery of 5-ASA pellets in rats with ulcerative colitis. Eur J Pharm Sci 2022; 168:106072. doi: 10.1016/j.ejps.2021.106072 [Crossref] [ Google Scholar]
- Shahdadi Sardou H, Akhgari A, Mohammadpour AH, Kamali H, Jafarian AH, Afrasiabi Garekani H. Application of inulin/Eudragit RS in 5-ASA pellet coating with tuned, sustained-release feature in an animal model of ulcerative colitis. Int J Pharm 2021; 597:120347. doi: 10.1016/j.ijpharm.2021.120347 [Crossref] [ Google Scholar]
- Kaffash E, Saremnejad F, Abbaspour M, Mohajeri SA, Afrasiabi Garekani H, Jafarian AH. Statistical optimization of alginate-based oral dosage form of 5-aminosalicylic acid aimed to colonic delivery: in vitro and in vivo evaluation. J Drug Deliv Sci Technol 2019; 52:177-88. doi: 10.1016/j.jddst.2019.04.006 [Crossref] [ Google Scholar]
- Sardou HS, Sadeghi F, Garekani HA, Akhgari A, Hossein Jafarian A, Abbaspour M. Comparison of 5-ASA layered or matrix pellets coated with a combination of ethyl cellulose and Eudragits L and S in the treatment of ulcerative colitis in rats. Int J Pharm 2023; 640:122981. doi: 10.1016/j.ijpharm.2023.122981 [Crossref] [ Google Scholar]
- Desai N, Momin M. Colon targeted bioadhesive pellets of curcumin and cyclosporine for improved management of inflammatory bowel disease. Drug DelivTransl Res 2020; 10:1288-301. doi: 10.1007/s13346-020-00756-x [Crossref] [ Google Scholar]
- Teruel AH, Gonzalez-Alvarez I, Bermejo M, Merino V, Marcos MD, Sancenon F. New insights of oral colonic drug delivery systems for inflammatory bowel disease therapy. Int J Mol Sci 2020; 21:6502. doi: 10.3390/ijms21186502 [Crossref] [ Google Scholar]
- Chen J, Li X, Chen L, Xie F. Starch film-coated microparticles for oral colon-specific drug delivery. CarbohydrPolym 2018; 191:242-54. doi: 10.1016/j.carbpol.2018.03.025 [Crossref] [ Google Scholar]
- Patil H, Tiwari RV, Repka MA. Hot-melt extrusion: from theory to application in pharmaceutical formulation. AAPS PharmSciTech 2016; 17:20-42. doi: 10.1208/s12249-015-0360-7 [Crossref] [ Google Scholar]
- Theismann EM, Keppler JK, Owen M, Schwarz K, Schlindwein W. Modelling the effect of process parameters on the wet extrusion and spheronisation of high-loaded nicotinamide pellets using a quality by design approach. Pharmaceutics 2019; 11:154. doi: 10.3390/pharmaceutics11040154 [Crossref] [ Google Scholar]
- Thakral S, Thakral NK, Majumdar DK. Eudragit: a technology evaluation. Expert Opin Drug Deliv 2013; 10:131-49. doi: 10.1517/17425247.2013.736962 [Crossref] [ Google Scholar]
- Patra CN, Priya R, Swain S, Kumar Jena G, Panigrahi KC, Ghose D. Pharmaceutical significance of Eudragit: a review. Futur J Pharm Sci 2017; 3:33-45. doi: 10.1016/j.fjps.2017.02.001 [Crossref] [ Google Scholar]
- Akhgari A, Abbaspour M, Moradkhanizadeh M. Combination of pectin and Eudargit RS and Eudragit RL in the matrix of pellets prepared by extrusion-spheronization for possible colonic delivery of 5-amino salicylic acid. Jundishapur J Nat Pharm Prod 2013; 8:86-92. [ Google Scholar]
- Sinha VR, Agrawal MK, Kumria R. Influence of formulation and excipient variables on the pellet properties prepared by extrusion spheronization. Curr Drug Deliv 2005; 2:1-8. doi: 10.2174/1567201052772898 [Crossref] [ Google Scholar]
- Muley S, Nandgude T, Poddar S. Extrusion–spheronization a promising pelletization technique: in-depth review. Asian J Pharm Sci 2016; 11:684-99. doi: 10.1016/j.ajps.2016.08.001 [Crossref] [ Google Scholar]
- Bölcskei É, Regdon G, Sovány T, Kleinebudde P, Pintye-Hódi K. Optimization of preparation of matrix pellets containing Eudragit® NE 30D. Chem Eng Res Des 2012; 90:651-7. doi: 10.1016/j.cherd.2011.09.005 [Crossref] [ Google Scholar]
- Siepmann J, Faham A, Clas SD, Boyd BJ, Jannin V, Bernkop-Schnürch A. Lipids and polymers in pharmaceutical technology: lifelong companions. Int J Pharm 2019; 558:128-42. doi: 10.1016/j.ijpharm.2018.12.080 [Crossref] [ Google Scholar]
- Nakmode D, Bhavana V, Thakor P, Madan J, Singh PK, Singh SB. Fundamental aspects of lipid-based excipients in lipid-based product development. Pharmaceutics 2022; 14:831. doi: 10.3390/pharmaceutics14040831 [Crossref] [ Google Scholar]
- Saini N, George M, Joseph L. Matrix tablets: an effective way for oral controlled release drug delivery: Controlled drug delivery using matrix tablets. Iran J Pharm Sci 2012; 8:165-70. doi: 10.22037/ijps.v8.40934 [Crossref] [ Google Scholar]
- Aburahma MH, Badr-Eldin SM. Compritol 888 ATO: a multifunctional lipid excipient in drug delivery systems and nanopharmaceuticals. Expert Opin Drug Deliv 2014; 11:1865-83. doi: 10.1517/17425247.2014.935335 [Crossref] [ Google Scholar]
- Hashem FM, Nasr M, Fathy G, Ismail A. Formulation and in vitro and in vivo evaluation of lipid-based terbutaline sulphate bi-layer tablets for once-daily administration. AAPS PharmSciTech 2016; 17:727-34. doi: 10.1208/s12249-015-0404-z [Crossref] [ Google Scholar]
- Roberts M, Vellucci D, Mostafa S, Miolane C, Marchaud D. Development and evaluation of sustained-release Compritol® 888 ATO matrix mini-tablets. Drug Dev Ind Pharm 2012; 38:1068-76. doi: 10.3109/03639045.2011.638302 [Crossref] [ Google Scholar]
- Kang C, Lee JH, Kim DW, Lee BJ, Park JB. Preparation of sustained release tablet with minimized usage of glyceryl behenate using post-heating method. AAPS PharmSciTech 2018; 19:3067-75. doi: 10.1208/s12249-018-1128-7 [Crossref] [ Google Scholar]
- Qazi F, Shoaib MH, Yousuf RI, Nasiri MI, Ahmed K, Ahmad M. Lipids bearing extruded-spheronized pellets for extended release of poorly soluble antiemetic agent-Meclizine HCl. Lipids Health Dis 2017; 16:75. doi: 10.1186/s12944-017-0466-x [Crossref] [ Google Scholar]
- Rao M, Ranpise A, Borate S, Thanki K. Mechanistic evaluation of the effect of sintering on Compritol 888 ATO matrices. AAPS PharmSciTech 2009; 10:355-60. doi: 10.1208/s12249-009-9211-8 [Crossref] [ Google Scholar]
- Yan X, He H, Meng J, Zhang C, Hong M, Tang X. Preparation of lipid aspirin sustained-release pellets by solvent-free extrusion/spheronization and an investigation of their stability. Drug Dev Ind Pharm 2012; 38:1221-9. doi: 10.3109/03639045.2011.645829 [Crossref] [ Google Scholar]
- Fouad EA, Ibrahim MA, El-Badry M. Embedment of chlorpheniramine maleate in directly compressed matrix tablets of Compritol and Kollidone SR. Trop J Pharm Res 2015; 14:371-7. doi: 10.4314/tjpr.v14i3.3 [Crossref] [ Google Scholar]