Bioimpacts. 2025;15:30713.
doi: 10.34172/bi.30713
Review
The role of tumor microenvironment and self-organization in cancer progression: Key insights for therapeutic development
Milad Asadi Conceptualization, Data curation, Writing – original draft, 1 
Venus Zafari Data curation, Investigation, Writing – original draft, 1
Sanam Sadeghi-Mohammadi Formal analysis, Investigation, Writing – original draft, 2
Dariush Shanehbandi Formal analysis, Methodology, Software, 3
Ufuk Mert Methodology, Resources, Software, 4
Zahra Soleimani Data curation, Investigation, Writing – original draft, 2
Ayşe Caner Conceptualization, Supervision, Validation, Writing – review & editing, 1, *
Habib Zarredar Conceptualization, Project administration, Supervision, Visualization, Writing – review & editing, 2, *
Author information:
1Department of Basic Oncology, Ege University, Institute of Health Sciences, Izmir, Turkey
2Tuberculosis and Lung Disease Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
3Immunology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
4Institute of Health Sciences, Department of Basic Oncology, Ege University, Izmir, Turkey
Abstract
Introduction:
The tumor microenvironment (TME) plays a pivotal role in cancer progression, influencing tumor initiation, growth, invasion, metastasis, and response to therapies. This study explores the dynamic interactions within the TME, particularly focusing on self-organization—a process by which tumor cells and their microenvironment reciprocally shape one another, leading to cancer progression and resistance. Understanding these interactions can reveal new prognostic markers and therapeutic targets within the TME, such as extracellular matrix (ECM) components, immune cells, and cytokine signaling pathways.
Methods:
A comprehensive search method was employed to investigate the current academic literature on TME, particularly focusing on self-organization in the context of cancer progression and resistance across the PubMed, Google Scholar, and Science Direct databases.
Results:
Recent studies suggest that therapies that disrupt TME self-organization could improve patient outcomes by defeating drug resistance and increasing the effectiveness of conventional therapy. Additionally, this research highlights the essential of understanding the biophysical properties of the TME, like cytoskeletal alterations, in the development of more effective malignancy therapy.
Conclusion:
This review indicated that targeting the ECM and immune cells within the TME can improve therapy effectiveness. Also, by focusing on TME self-organization, we can recognize new therapeutic plans to defeat drug resistance.
Keywords: Tumor microenvironment, Self-organization, Extracellular matrix, Metastasis, Invasion
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This study was funded by the Tuberculosis and Lung Disease Research Center, Tabriz University of Medical Science, Tabriz, Iran.
Introduction
Cancer, a leading cause of mortality worldwide, manifests through aberrant cellular behaviors such as uncontrolled growth, invasion, and metastasis. Conventional treatments, like surgery, chemotherapy, and radiotherapy, though widely used, often face limitations including drug resistance and adverse effects, resulting in low curative outcomes. Recent studies have highlighted the essential role of the tumor microenvironment (TME) a complex network of extracellular matrix components (ECM), cancer cells, non-cancer cells, and soluble factors, in driving tumor development. TME not only supports cancer progression and growth but also plays a critical role in angiogenesis, tumor cell migration, immune evasion, and therapeutic resistance.1-3
The unique biochemical compositions of the TME, influenced by factors such as oxygen levels, pH, and fluid flux, creates an environment conducive to cancer cell survival, immune evasion, and resistance to anti-cancer therapies.4-7 Additionally, dysregulation of cellular pH within the tumor and the presence of an acidic microenvironment promote migration, invasion, and metastasis of cancer cells through various mechanisms.8 Notably, the TME exhibits tumor-specific variations, emphasizing the need for personalized therapeutic approaches tailored to individual tumor characteristics.9,10
The concept of self-organization within the TME, where cancer cells actively interact with their environment, leads to the formation of complex structures that promote tumor survival. In this context, neoplasms, or tumors, emerge as a result of self-organization, characterized by changes in intracellular mitotic pathways originating from asymmetric divisions of progenitor cells.11-13 Recent studies have revealed that cancer cells with specific mutations or antigens on their surface can significantly alter their TME, effectively reorganizing their surroundings to their advantage.14-16 A deeper understanding of TME characteristics and its regulation by tumor cells can elucidate cancer mechanisms and identify strategies to inhibit tumor growth.17-20
This phenomenon contributes to tumor malignancy and therapeutic resistance, underscoring the importance of understanding TME dynamics to develop effective treatment strategies. By exploring TME characteristics and their impact on cellular interactions and self-organization, this review offers a new perspective on the TME by highlighting the important role of self-organization mechanisms in tumor development and progression. The novelty of this review demonstrates in synthesizing recent findings related to the dynamic interactions between tumor cells and their microenvironment, focusing on how these interactions contribute to tumor heterogeneity and therapeutic challenges. We propose that targeting specific molecules within the TME, particularly those involved in self-organization processes, may facilitate the development of more effective personalized treatment that can overcome therapeutic resistance and improve patient outcomes.
Complexity and function of the tumor microenvironment
Understanding the TME and its interaction with tumor cells is essential for identifying potential targets for cancer therapy.21,22 Neoplastic cells utilize a diverse array of molecules and signaling pathways to manipulate cells within the TME, fostering cancer progression. This includes the secretion of cytokines and chemokines, such as tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and transforming growth factor beta (TGF-β), which recruit stromal cells like fibroblasts and immune cells. Additionally, neoplastic cells remodel the ECM, promote angiogenesis through factors like vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), and adapt to hypoxic conditions via the hypoxia-inducible factor (HIF) pathway. They also modulate immune responses, undergo epithelial-mesenchymal transition (EMT), and reprogram metabolism to support rapid proliferation. These interactions create a supportive TME that encourages tumor growth, invasion, and metastasis, highlighting their potential as therapeutic targets for cancer treatment.
The TME, which constitutes the majority of the overall tumor mass, contains consists of two key subunits, including non-cancerous cells and the ECM. Non-cancerous cells in the TME can be classified into non-immune and immune cells. The non-immune cells include mesenchymal stem cells (MSCs), adipocytes, pericytes, vascular endothelial cells, lymphatic endothelial cells, and cancer-associated fibroblasts (CAFs). Immune cells, which are a critical part of the TME include T-cells, B-cells, myeloid cells, and natural killer (NK) cells.19,23-27 This complex cellular structure creates a dynamic intercellular matrix that facilitates communication between different cell types through specific cytokines and chemokines, forming a unique environment for each solid tumor in which every cell type plays a role in supporting cancerous cells.28-31 Detailed information about cells functioning in the TME is provided below.
Non-immune cells
Human mesenchymal stem cells
Human mesenchymal stem cells (hMSCs), found in bone marrow, contribute to tumorigenesis by promoting processes like malignant transformation, tumor cell maintenance, and metastasis.32,33 Cytokines, particularly IL-6, released from tumor cells induce the migration of hMSCs to the TME.34-36 Certain members of TME, such as CAFs, adipocytes, and pericytes, are derived from hMSCs.37
Pericytes, which support blood vessel construction in the TME, influence tumor growth and metastasis.37,38 The coverage of blood vessels by pericytes depends on the tumor type, ranging from extensive to minimal or none.39 Limited pericyte coverage correlates with a higher risk of metastasis.40 In addition,adipocytes act as energy sources for malignant cells and release adipokines that contribute to the structure of the TME.37,41 Pericytes also play various roles in the immune system, such as attracting leukocytes to exit blood vessels, regulating lymphocyte activation, and triggering direct phagocytic activity.42 In a study by Kalluri et al, genetically deleting tumoral pericytes in breast cancer enhanced pulmonary carcinogenesis in an NG2-TK animal model.40 In contrast, the generation of pericytes by cancer cells was found to stimulate the development of glioma tumors.43
Cancer-associated fibroblasts
One of the most significant cells of mesenchymal origin in TME is CAFs, which are the predominant stromal cells in most malignancies.37 Although the basic mechanism of CAF formation is not fully understood, it is suggested that MSCs are a major source of CAFs. Moreover, normal fibroblasts, endothelial cells, epithelial cells, and even cancer cells can give rise to CAFs.44-47
CAFs can be classified into two subgroups based on whether their phenotype: myofibroblastic or inflammatory. CAFs activated by IL1/leukemia inhibitory factor (LIF)/Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling become inflammatory, whereas TGFβ signaling inhibits this process by downregulating the interleukin-1 receptor, leading to myofibroblast differentiation.48 Takai et al found that pirfenidone (PFD) inhibited tumor cell proliferation via CAFs, resulting in apoptosis in a 3D culture model. In vivo experiments showed that PFD, when combined with doxorubicin, reduced lung metastasis and tumor development.49
Blocking autophagy in CAFs is another strategy to reduce cancer cell growth. Drugs such as metformin and gemcitabine have been shown to induce autophagy. In in vitro and syngeneic pancreatic cancer models, chemotherapeutics such as α-cyano-4-hydroxycinnamate (CHC), either alone or in combination with metformin, have been found to inhibit autophagic flux in CAFs and limit tumor cell growth, irrespective of other chemotherapeutic drugs.50
The molecular mechanisms involved in the transformation pathway of tumor cells are varied. Initially, normal fibroblasts inhibit the effects of IL-6, preventing the expansion of cancer cells during the early stages of cancer. Subsequently, these normal cells are converted to CAFs by tumor cells, a process further accelerated by the secretion of high levels of TGF-β in malignant cells.44,51,52 Once transformed, these fibroblastic cells release molecules such as vimentin, desmin, and fibroblast activation protein (FAP).53,54 Due to their capabilities, CAFs play critical roles in supporting cancer cell and promoting carcinogenesis through variety physicochemical mechanisms, including reducing apoptosis and enhancing cancer cell proliferation, motility, and viability.47,55 Furthermore, CAFs produce ECM proteins that contribute to immunosuppression in tumor cells by attracting immunosuppressive cells.56
Endothelial cells
Endothelial cells (ECs) are crucial for the formation of blood vessels within the TME. Tumor-derived endothelial cells (TECs) differ significantly from normal ECs in both shape and function, promoting angiogenesis, tumor progression, and metastasis. TECs are influenced by pro-angiogenic factors secreted by tumor cells and CAFs, which disrupt normal vascular structures and result in leaky blood vessels, thereby hindering effective oxygen and drug delivery to the tumor core. Moreover, lymphatic endothelial cells play a pivotal role in immune evasion and metastasis, providing a pathway for cancer cells to escape from the primary tumor site.57
TECs are involved not only in in supporting initial tumor development through angiogenesis but also in facilitating tumor progression, metastasis, and resistance to therapies.58 Vascular endothelial cells in TME are affected by angiogenic factors released from cancer cells and CAFs.18,23 These factors contribute to the formation of abnormal vascular structures, primarily within the core of solid tumors. Dysregulation of vascular endothelial cells within the structural framework of blood vessels is a major reason for the formation of leaky vessels in the TME, leading to inadequate oxygen and drug delivery to the tumor core.59
Additionally, lymphatic endothelial cells are essential for the development of lymphatic vessels within the TME. In some cases, malignant cells modify their properties through factors such as VEGF-C and VEGF-D. The two primary functions of lymphatic endothelial cells in the TME are to facilitate the escape of malignant cells from the immune system and provide a favorable environment for cancer cell metastasis.26,60
Furthermore, TECs engage in bidirectional signaling with malignant cells, as cancer cells secrete factors that influence TEC function, enhancing their pro-carcinogenic properties.61 TECs possibly have a role in the progress of anti-cancer therapy resistance by helping cancer cell survival and adaptation to therapy. Controlling the impact of TECs on the TME is therefore essential for developing targeted therapies that mitigate their pro-carcinogenic functions, ultimately therapeutic outcomes.62
Immune cells
Immune cells within the TME play a crucial role in tumor surveillance, possessing both tumor-promoting and tumor-suppressing functions. Initially, the immune system recognizes transformed and damaged cells and subsequently eliminates tumor cells. This process involves various immune cells, including those of the adaptive immune system, such as T-cells. Cytotoxic T-cells directly kill malignant cells, while helper T-cells enhance the immune response by secreting cytokines that activate and stimulate other immune cells.63
Conversely, the immune system can also contribute to tumor progression by supporting chronic inflammation and suppressing antitumor immunity under certain circumstances. Chronic inflammation can lead to the recruitment of myeloid cells, such as macrophages, which may adopt pro-tumor phenotypes. These cells can secrete cytokines and growth factors that promote angiogenesis, supporting tumor survival.64 Additionally, immune cells can suppress effective antitumor responses; for example, regulatory T-cells (Tregs) inhibit the activity of effector T-cells, thereby dampening the overall immune response against the tumor. This immune evasion enables tumors to thrive and metastasize.65
The spatial distribution of immune cells within the TME can also significantly affect patient prognosis. Immune cells located near malignant cells are more likely to effectively combat tumor activity, whereas those situated farther away may be less effective.3 All major immune cell types are present in the TME, including T-cells, B-cells, myeloid cells, and natural killer cells (NK cells). A wide range of cytokines and chemokines, released by immune cells and other cells in TME, regulate the functions and subtypes of immune cells, thereby impacting cancer development in different ways.10,66-68 The interaction between immune cells and tumors are illustrated in Fig. 1.
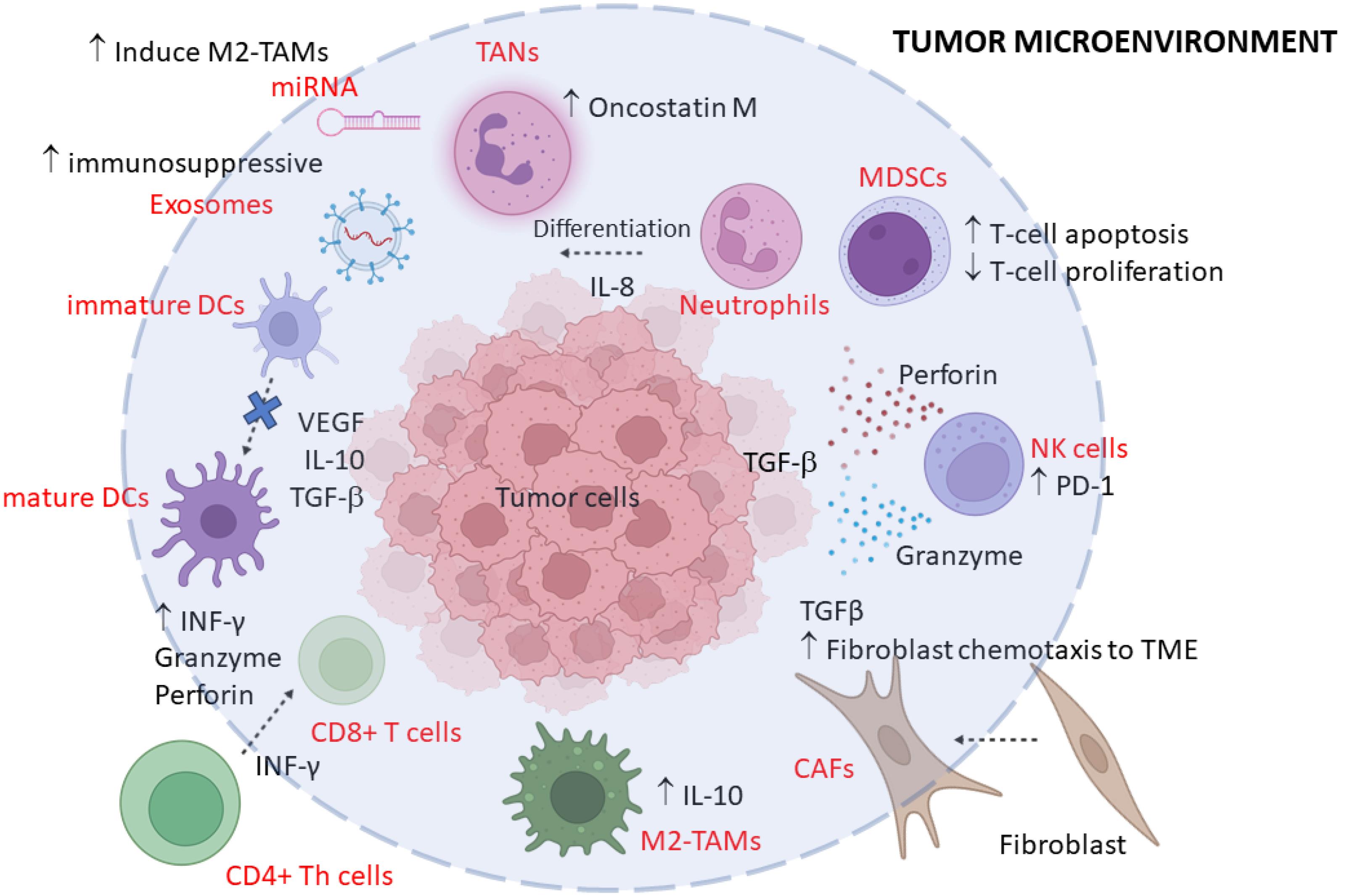
Fig. 1.
A brief schematic representation of the tumor microenvironment depicting different cell types. In this scheme, the major cells involved in the self-organization response are highlighted. (TME: tumor microenvironment; CAFs: cancer-associated fibroblasts; TAMs: Tumor-associated macrophages; RBC: red blood cell; TANs: Tumor-associated neutrophils; DCs: dendritic cells).
.
A brief schematic representation of the tumor microenvironment depicting different cell types. In this scheme, the major cells involved in the self-organization response are highlighted. (TME: tumor microenvironment; CAFs: cancer-associated fibroblasts; TAMs: Tumor-associated macrophages; RBC: red blood cell; TANs: Tumor-associated neutrophils; DCs: dendritic cells).
Myeloid cells
Myeloid cells, consisting of neutrophils, dendritic cells (DCs), monocytes/macrophages, and myeloid-derived suppressor cells (MDSCs), are essential members of TME. Tumor-associated neutrophils (TANs) are involved in TME and are less prevalent than other myeloid cells. TANs are classified as N1 or N2 according to their activation, cytokine status, and impact on tumor cell proliferation. N1-TANs fight cancer by directly or indirectly killing cells, but N2-TANs weaken the immune system, help tumors grow, create new blood vessels, and spread to other parts of the body through cytokines and chemokines.26,69 DCs, the second member of this group, play a vital role in adaptive immune responses against tumor cells. DCs in the TME are often inactive, which inhibits the adaptive immune response produced by the tumor. It is known that tumor-derived factors in the TME reduce DC function by lowering DC recruitment, activation, antigen presentation, and type 1 T helper cell (Th1) polarization.66,70
Due to their flexible nature and changing roles, macrophages can be divided into two subtypes that control tumor growth in TME in different ways. Tumor-associated macrophages (TAMs) are the most abundant population type found in TME; like TANs, they can be broadly divided into M1- and M2-TAMs. M1-TAMs produce IFN-γ, which has a powerful anti-tumor function. Furthermore, M2-TAMs are activated by IL-3 or IL-4 signals and are in charge of the anti-inflammatory response. They also significantly contribute to IL-10 production. The TME describes the remodeling of both invading and resident macrophages into TAM based on their function. TAMs have been shown to have both boosting (M1 type) and inhibiting (M2 type) effects in cancer therapies.71 TAMs enable tumor cells to migrate and metastasize by generating migration-stimulating factors.72
In addition, these cells can be divided into M2a, M2b, M2c, and M2d subtypes in response to different stimulators in TME. M2-TAMs are typically found in the hypoxic part of solid tumors with IL-10high and IL-12low phenotypes, and they promote tumor growth by releasing molecules involved in angiogenesis.27,73-76 A previous study discovered that TAMs participate in the EMT program to boost colorectal cancer (CRC) invasion, migration, and CTC-mediated metastasis. They do this by producing IL-6, which turns on the JAK2/STAT3 axis and stops miR-506-3p from stopping FoxQ1 expression.77 Targeting functional TAM molecules is a promising therapeutic approach. For instance, stopping Fc receptors on TAM stops the loss of anti-PD1 antibodies, which makes checkpoint treatment more effective.71
The last member of the myeloid system is MDSCs, which are immature myeloid cells recruited to TME to aid the establishment of an immunosuppressive TME. MDSC has functions similar to TAMs and also generates factors that stimulate the development of TAMs from M2 cells. MDSCs also stop T cells from activating by stopping the production of cytotoxic T lymphocytes (CTLs) and killing antigen-stimulated CTL clones and T regulatory cells.78 T lymphocytes (T-cells), B lymphocytes (B-cells), and NK cell types, which are members of lymphoid cells like myeloid-derived cells, play a critical role in TME.26
T-lymphocytes (T-cells)
In heterogonous TME, T cells are the most common immune cells. Due to their reaction to cancerous cells, T cells are transformed into subtypes that can exert anti-tumor and pro-tumor effects, and this cell population and characters can vary in tumor types and stages.79 T cells interact with several cells in the TME, such as macrophages, dendritic cells, and stromal cells, designing the immune response and affecting tumor development. The balance of these roles can have a significant impact on tumor development and patient outcomes, making T cells an important factor for tumor immunotherapy plans.80 Generally, CD4 + T cells, CTL, and memory T cells, which produce IL-2 and IFN-γ, and γδ T cells are acting against malignant cells. On the other hand, CD4 + Th2 and Th17 cells release IL-3, IL-4, and IL-13, as well as IL-17A, IL-17F, IL-21, and IL-22, respectively. These cells help the tumor grow in TME. Similarly, Treg cells play an anti-inflammatory and immune-suppressive role in TME, favoring tumor growth and inhibiting T cell activation and cytotoxicity. They may, however, release IL-10 and TGF-β and show CTLA-4 on their surface, which stops immune cells from attacking tumor cells. Some studies have shown that Treg cell presence and activation in TME are associated with distant metastasis and poor prognosis.81-83 Also, the type and density of immune cells have an important effect on the TME. High densities of cytotoxic T-cells, for example, are frequently associated with a favorable prognosis, whereas high levels of MDSCs are associated with a poor prognosis in patients.84 Th1 are important cells for regulating a vigorous immune response against malignancy in the TME. These characteristics make them a target for immunotherapeutic strategies to control cancer development.85 Th1 cells could change the TME by inducing macrophages and dendritic cells, leading to a more effective immune response against the malignant cells. But TME can control Th1 to help cancer progression. The TME includes many factors (MDSCs) and immunosuppressive cytokines (IL-10, TGF-β), which could suppress Th1 cell promotion and function, decreasing their capability to induce anti-tumor immunity. Also, some metabolites like adenosine and immune checkpoint molecules like PD-L1 in the TME could suppress Th1 cell activation and function, decreasing their ability to control cancer cells.86,87
B-lymphocytes (B-cells)
These cells are usually colonized at adjusted tissues to TME and, in some cases, found in the invasive margin of the tumor. B cells suppress tumor progression by secreting immunoglobulins, promoting T cell response, and killing cancer cells, but also increase tumor activity by immunosuppressive cytokines. Therefore, although infiltration of B cells into TME is associated with a good prognosis in some studies,26,67 others have revealed a controversial result in terms of the association of B cells and metastasis. Regulatory B (Breg) cells are a population of B cells that suppress the immune system through cytokine secretion and intercellular contact, like Treg cells. This regulation is mainly mediated by the cytokines produced by regulatory B cells (Breg cells), such as IL-10 IL-35, and TGF-β. These cells interact with a variety of tumor-infiltrating immune cells and tumor cells to reduce anti-tumor responses and facilitate tumor progression.88,89
Natural killer cells
These cells are usually found in the surrounding area of TME and their presence in TME is linked to good prognosis in various types of cancers. Actually, unlike other immune cells, these cells have not yet been approved for tumor progression activity. In the TME, NK cells could interact with several cell types, like TAMs and dendritic cells, which can affect the immune landscape and potentially induce or inhibit anti-tumor immunity. However, in the presence of cytokines produced by tumor cells and other kinds of immune cells, especially TGF-β, NK cells show an anergic phenotype and become inactivated.90,91
Cytokines
Cytokines are small proteins that play a vital role as a communication messenger in cell-cell communication and their effect on target cells depends on their substance, amount, and location. Certain cytokines secreted by cells in the TME play a role in organizing the surrounding region to provide better support for malignant cells. For instance, TNF-𝛼, prostaglandin E2 (PGE2), TGF-𝛽, Interferon-γ (IFN-γ), and IL-10 are involved not only in TME construction but also in the formation and progression of the malignancy.92-94
Tumor necrosis factor alfa
Chronic inflammation is one of the strongest risk factors, which can cause to initiation of cancer. Although TNF-𝛼 is one of the important mediators of primary inflammation, studies reveal the double-edged function of it in cancer. Indeed, a high concentration of TNF-𝛼 acts as a tumor-suppressive factor; on the other hand, a low concentration of it can be a stimulating factor in malignancies. For example, TNF-𝛼 can initiate defensive pathways like reactive oxygen species (ROS), which leads to the production of oxygen radicals and causes DNA damage and subsequently cancer. Moreover, in some studies, normal cells show pre-malignant features in the presence of a certain concentration of TNF-𝛼. In any case, the concentration of TNF-𝛼 is not the only factor that can determine its role in the carcinogenesis process, as some studies indicate that the expression site where this cytokine is expressed in TME is another important factor in carcinogenesis. In addition, TNF-𝛼 synergized with IFN-γ stimulates MSCs to arouse tumorigenesis and drug resistance by using the NF-κB signaling pathway.95-97
Transforming growth factor-β
TGF-β acts as a crucial mediator in cell proliferation, differentiation, apoptosis, and invasion. In the TME, M2 macrophages and MDSCs are the most important producers of TGF-β and this protein is one of the main factors, which arrest the NK-cells functions. The binding of TGF-β to its receptor leads to activity in SMAD-depended transcription factors in tumor cells, which often causes high proliferation and metastasis. Similar to TNF-α, TGF-β also has a two-sided function and, in some cases, can exhibit anti-cancer effects by activating the P21 gene. While it remains uncertain which function TGF-β will primarily exhibit, the composition of the ECM and the characteristics of host cells significantly influence whether TGF-β promotes anti-cancer effects or enhances cell proliferation pathways.94,96,98
Interleukin 10
IL-10, an anti-inflammatory cytokine, is generated by nearly all species of immune and malignant cells in a solid tumor. The presence of IL-10 in ECM reduces the antigen-presenting activity of dendritic cells, which helps malignant cells hide from immune cells. In addition, secretion of this cytokine by tumor cells causes to initiation of macrophage (TAM) infiltration to TME. On the molecular scale, IL-10 constrains the NF-𝜅B pathway and then reduces the expression of pro-inflammatory cytokines. Furthermore, IL-10 can activate of STAT-3 pathway using IL-6 and this activation leads to over-expression of BCL-2 and some other anti-apoptotic factors.30,99,100
Interferon-γ
Fibroblasts, macrophages, and T helper cells are responsible cells for the secretion of IFN-γ protein in TME. IFN-γ plays an important role in the activation of immune response by a complex mechanism. It consistently regulates both pro-tumorigenic and antitumor immunity in TME. IFN-γ acts as a cytotoxic cytokine by inducing apoptosis in tumor cells but also stimulates other immune suppressive mechanisms by providing the expression of immune checkpoint inhibitors and indoleamine-2,3-dioxygenase (IDO). In addition to its pro-apoptotic and anti-proliferative functions, IFN-γ can stimulate T-reg apoptosis, the activity of M1 pro-inflammatory, and inhibit angiogenesis in TME. IFN-γ usually achieves its effects by activation of JAK/STAT, p38/MAPK, and p16INK4a-Rb pathways in target cells and downregulating of VEGF-A, SLC7A11, and SLC3A2.6,101-103
Prostaglandin E2
PGE-2 is a type of prostaglandin, which is a group of lipid compounds derived from fatty acids. PGE-2 is produced in the body through the enzymatic conversion of arachidonic acid by the action of cyclooxygenase enzymes (COX-1 and COX-2). Like TGF-β and TNF-α, the presence of PGE-2 can have both positive and negative effects on carcinogenesis. PGE-2 prompts ROS, which generates oxygen radicals and can damage DNA. The main impact of PGE-2 on immune cells in TME is cutting the activation of NK cells down in TME, which helps tumor cells to be concealed from the immune system. As well, PGE-2 is also able to enhance tumor cell growth and angiogenesis.6,92,104
Interleukin 17
IL-17 is a pro-inflammatory factor produced by a subtype of T-helper lymphocytes called Th17. It is a family of cytokines composed of six members (IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F), and like many other TME cytokines, this group also has a double-edged influence on the tumor foundation and promotion too. This cytokine can act as the molecule to promote cell proliferation, tumor growth, and progression, as well as treatment resistance through cell IL-17R signaling activation, possibly cross-links with other receptors such as EGFR, or IGFR.104-106
Interleukin 6
Previous studies have reported that IL-6, which plays a role in tumor progression, performs this function by activating the JAK/STAT pathway. The main mediator of IL-6 in this pathway is STAT-3, and for activation of it, IL-6 must bind to its receptor and join glycoprotein gp130 to establish of IL-6/IL-6R/gp130 complex.107 Likewise, IL-6 in the presence of TGF-β induces proliferation of T Helper type 17 cells (Th-17 cells). Conversely, the secretion of IL-6 can also have some anti-cancer effects in some cases.108 For example, IL-6 is responsible for the migration of lymphocytes to high endothelial venules (HEV). The effect and resources of TME cytokines are summarized in Table 1.
Table 1.
Cytokines are highlighted in the tumor microenvironment
|
Cytokine
|
Secretor cell
|
Target cell
|
Effect in TME
|
Ref.
|
| TNF-𝛼 |
Damaged cells and several types of immune cells |
MDSCs |
DNA damage in early stages enhances tumor genesis and drug resistance by stimulating the NF-κB pathway |
97
|
| TGF-𝛽 |
M2 and MDCS in TME |
Malignant cell |
Act as a stimulator of proliferation by activating the SMAD pathway |
98
|
| IL-10 |
Various types of TME cells |
Dendritic cell |
Reduction in antigen-presenting by malignant cells and stimulation of NF𝜅B and STAT-3 |
100
|
| IFN-γ |
Fibroblasts, macrophages, and Th |
Cancer stem cell, Immune cell in TME |
Activation of JAK/STAT, p38/MAPK and PI3K pathways |
103
|
| PGE-2 |
Various types of TME cells |
NK cells, Malignant cells |
Reducing immune response in TME |
104
|
| IL-17 |
Th-17 |
Malignant cells |
Stimulation of tumor growth, and drug resistance by using IL-17R, EGFR, and IGFR |
105
|
| IL-6 |
M2 macrophages |
Malignant cells, Th-17 |
Stimulating tumor growth by affecting the JAK/STAT pathway and inducing of proliferation of Th-17, in the presence of TGF-β |
108
|
Note: TNF-α; Tumor necrosis factor- α, TGF-β; Transforming growth factor-β, IL; Interleukin, IFN-γ; Interferon-γ, PGE-2; Prostaglandin E2, MDSCs; Myeloid- derived suppressor cell, NF-κB; Nuclear Factor-κB, TME; Tumor microenvironment, Th; T helper cell, STAT-3; Signal transducer and activator of transcription-1, JAK; Janus kinase, MAPK; Mitogen-activated protein kinase, PI3K; Phosphoinositide 3-kinase, IL-17R; Interleukine-17 receptor, EGFR; Epidermal growth factor receptor, IGFR; Insulin growth factor receptor.
Non-cellular components
Extracellular matrix
The ECM is a complex framework of proteins and molecules that supports tissue structure and cellular interactions. It includes proteins like collagen, elastin, fibronectin, and laminin, as well as glycosaminoglycans (GAGs) and proteoglycans. Abnormal accumulation of ECM components, especially collagen, in conditions like desmoplasia can promote tumor growth and invasion. In low-oxygen tumor environments, cancer cells adapt by activating HIFs, which regulate genes involved in oxygen balance, angiogenesis, and glycolysis. This adaptation supports cancer cell survival and growth despite low oxygen levels, suggesting potential targets for cancer therapy.109
As a result of disturbances in the vascular system in TME, oxygen supply to the center of TME is insufficient, thus providing a lower pH in comparison to normal tissues. In this situation, tumor cells begin to use glucose via anaerobic glycolysis instead of using the Embden-Meyerhof pathway, leading to the production of lactic acid and a decrease in the pH of TME. This low pH is lethal for most normal cells, however, cancerous cells augment special types of H + and HCo3- transporters, which boost the pH inside cells and enable them to survive in such an environment.110 Understanding the TME in cancer therapy requires large patient cohort studies to predict the effect of chemotherapy on ECM and ECM cell signaling. Also, TME-based patient classification is important for evaluating the effectiveness of targeted ECM treatments and using new technologies such as artificial intelligence and machine learning to generate prognostic models for designing appropriate treatment regimens.111,112
MicroRNAs
MicroRNAs (miRNAs) are small non-coding RNAs with substantial roles in various biological pathways. As well as coding RNAs, miRNA dysregulation has been reported in various types of cancers, miRNAs are classified as oncomiR and tumor-suppressor miRNAs due to their roles and up/down-regulation in malignant cells.113 Recent studies have proven that miRNAs are not only capable of regulating neoplastic cell features but are also able to manage every feature of non-cancerous cells in TME. Secretion of specific miRNAs to ECM of TME can affect any cell, such as CAFs, and TAMs. On the other hand, the deregulation of miRNAs in host cells of TME can affect malignant cells and TME features in response to these genes.114-116 Studies have identified various miRNAs with the capability of managing TME, these miRNAs are miR-155, miR-9, miR-200-5p, miR-21, miR-141, miR-148, miR-214, miR-146a, miR-187and miR-15/16.116-118 The physiological roles of these miRNAs are summarized in Table 2.
Table 2.
Deregulated microRNAs and their targets involved in TME
|
microRNA
|
Cancer
|
Type of regulation
|
Target gene
|
Ref.
|
| miR-155 |
Ovarian cancer |
Upregulated |
- |
119
|
| miR-21 |
Various types of cancer |
Upregulated |
PDC4 |
116
|
| miR-9 |
Various types of cancer |
Upregulated |
- |
116,120
|
| miR-200 |
Breast Cancer |
Downregulated |
- |
120
|
| miR-141 |
Breast Cancer |
Downregulated |
- |
121
|
| miR-148a |
Ovarian cancer |
Downregulated |
WNT1, WNT10B |
117
|
| miR-214 |
Ovarian cancer |
Downregulated |
CCL5 |
119
|
| miR-146 |
Various types of cancer |
Upregulated |
IRAK1/ TRAF6, |
118
|
| miR-15/16 |
Prostate cancer |
Downregulated |
FGF2, FGFR1, CCND1, WNT3a, BCL2 |
118
|
Note: PDC4; Pyruvate decarboxylase 4, WNT; Wingless-related integration site, CCL5; Chemokine (C-C motif) ligand 5, IRAK1; Interleukin 1 receptor associated kinase 1, TRAF6; TNF receptor associated factor 6, FGF2; Fibroblast growth factor 2, FGFR1; Fibroblast growth factor receptor 1, CCND1; Cyclin D1, BCL2; B-cell lymphoma 2.
The non-cell-autonomous mechanism is a key factor for alteration in the miRNA profile of the neighboring cells without any genetic abnormality. Angiogenesis, metastasis, and host cell properties are three attributes in cancer TME that miRNAs can alter them. MiR-9 is one of the best-known miRNAs, which regulate the angiogenesis process by targeting E-cadherin.120 Specifically, up-regulation of miR-9 increases expression of VEGF-α by activation of the β-Catenin pathway, while IGFBP-2 is one of the targets of miR-126 and down-regulation of miR-126 increases the capacity of malignant cells to angiogenesis. On metastasis, the presence of miR-1908, miR-199a-5p, and miR-199a-3p increases the capacity of metastasis in neoplastic cells by targeting the apolipoprotein-E (Apo-E). As the third function for miRNAs in TME, they can affect the host cells and change their attributes. For example, over-expression of miR-15a, and miR-16-1 can change CAFs to better support malignant cells. MiR-135b is another miRNA that has the responsibility for determining TME characteristics and the behavior of tumor cells. It is a key mediator in the production of IL-17, which can modify immune cells in TME.122
Exosomes
Exosomes, small vesicles released by various cell types, play a significant role in TME and cancer progression.123 They can influence the TME by reprogramming the metabolism of cancer cells and their surrounding stromal cells, thereby promoting cancer progression, angiogenesis, metastasis, and drug resistance.124 Exosomes derived from cancer cells can also contribute to the formation of the TME by facilitating immune system evasion, inflammation, angiogenesis, and metastasis.125 Furthermore, they are implicated in therapy resistance, making them potential targets for cancer therapy (Fig. 2).126
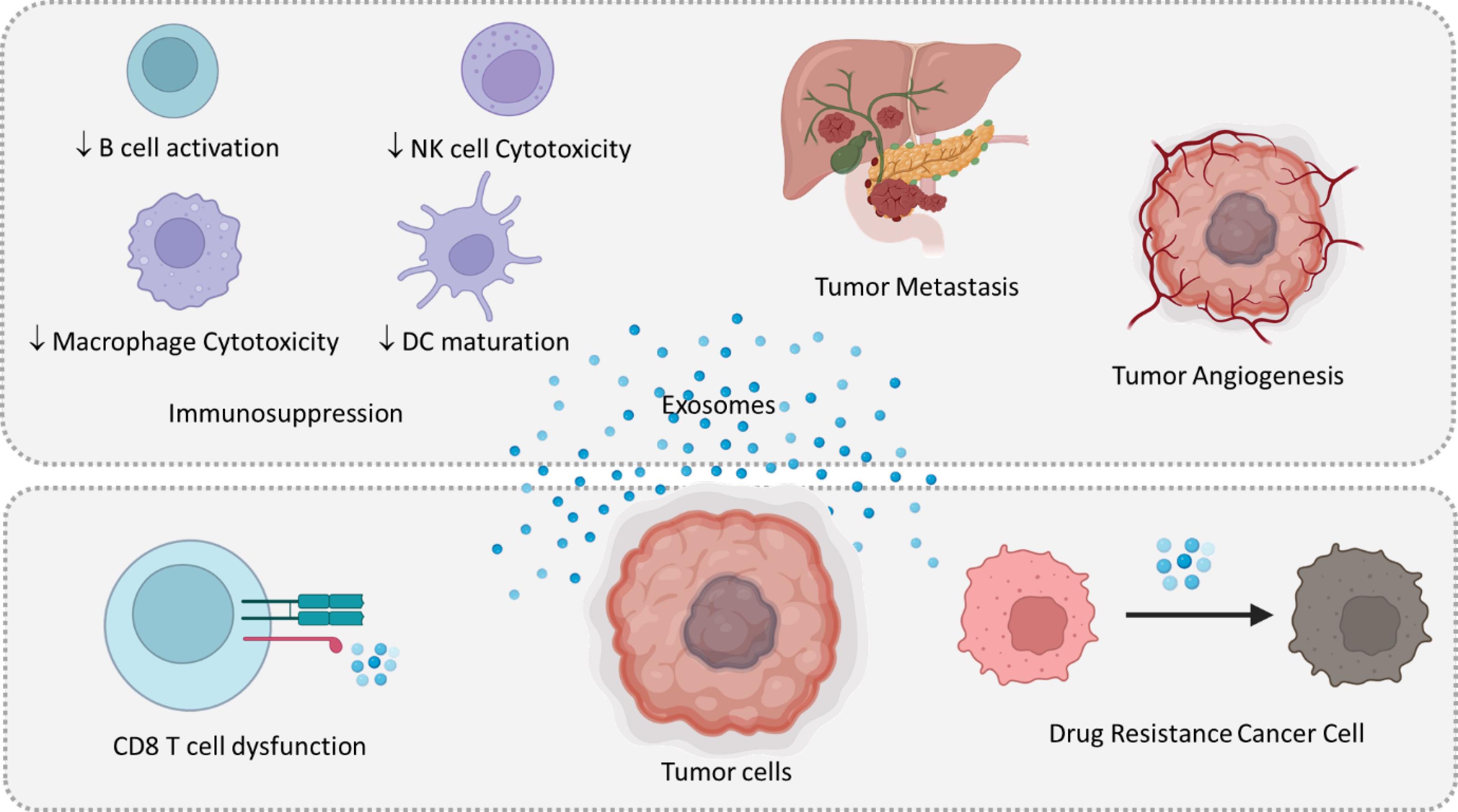
Fig. 2.
Tumor-derived Exosomes mediated immunosuppression of tumor immunity, tumor progression, metastasis, and tumor angiogenesis. Tumor-derived exosomes facilitate chemotherapeutic drug resistance in tumor cells by transporting multidrug transporters. Exosomes block the efficacy of tumor immunotherapy via the associated immunological checkpoints, particularly PD-L1.
.
Tumor-derived Exosomes mediated immunosuppression of tumor immunity, tumor progression, metastasis, and tumor angiogenesis. Tumor-derived exosomes facilitate chemotherapeutic drug resistance in tumor cells by transporting multidrug transporters. Exosomes block the efficacy of tumor immunotherapy via the associated immunological checkpoints, particularly PD-L1.
Exosomes enable communication among malignant cells and stromal cells, like endothelial cells, and fibroblasts immune cells. Exosomes released by tumor cells contain bioactive molecules like proteins, lipids, and nucleic acids, which can be transferred to nearby or distant cells, influencing their behavior and aiding tumor advancement. Exosomes change the behavior of stromal cells, to prepare an appropriate environment for cancer development and progression.127 These exosomes contribute to the formation of pre-metastatic niches in distant organs, support angiogenesis by delivering pro-angiogenic factors to endothelial cells, and modulate the immune response in the TME.128 They carry immunosuppressive molecules like programmed death-ligand 1 (PD-L1), dampening immune cell activity and facilitating tumor immune evasion. Moreover, exosomes educate immune cells to adopt a pro-tumorigenic phenotype, further bolstering tumor progression. Moreover, exosomes have been implicated in the development of drug resistance in cancer. They can transfer drug efflux pumps and other resistance-conferring molecules to sensitive cancer cells, leading to decreased sensitivity to chemotherapy and targeted therapies. Overall, the role of exosomes in the TME is multifaceted and encompasses various aspects of tumor progression. Therefore, targeting exosome-mediated communication pathways may represent a promising strategy for cancer therapy (Fig. 2).129 Also, Exosomes could impact metabolic pathways in target cells, preparing a metabolic environment in which cancer cells could survive and develop.124 One of the important features of exosomes is their ability to serve as biomarkers for prognosis and diagnosis. Exosomes have molecular signatures reflective of their cells of origin. Specific exosomal markers could be correlated with various tumor stages and types, giving the appropriate ability to exosome serve as diagnostic and prognostic biomarkers. Controlling exosomal biomarker levels during therapy could give real-time visions and data into the success of therapies and allow for personalized therapy modifications.130
Self-organization in tumor microenvironment and cancer cells
Self-organization in cancer refers to the autonomous and emergent behavior of cancer cells and their microenvironment, leading to complex and structured patterns without external direction. This process is crucial for tumor development, progression, and resistance to treatment, providing therapeutic insights.131 The TME significantly influences cancer cells, affecting their metabolic reprogramming, proliferation, and progression. The TME is dynamic, with interactions between its components and cancer cells evolving over time. Nonmalignant cells like adipocytes, fibroblasts, and immune cells within the TME play essential roles in these processes. The tumor stroma, composed of non-cancer cells, supports cancer growth and can be targeted for treatment.132-134 Non-linear interactions in neoplastic cells can cause small disorders to lead to significant changes, resulting in two outcomes: the disordered cell may die without establishing a new equilibrium, or a new pattern may form, strengthening the original cell. This phase transition often drives malignant cells towards uncontrolled proliferation, lack of differentiation, and metabolic alterations. For example, during chemotherapy, malignant cells with receptor mutations may enhance MDR gene expression, ensuring survival despite drug presence. Malignant cells act as dynamic systems, causing substantial changes in the TME.12,135-140
Hormone receptors, such as estrogen receptors (ERs) and androgen receptors (ARs), are crucial in hormone-dependent cancers like breast and prostate cancer. Mutations in these receptors drive tumor growth and influence the TME by modulating hormone-responsive pathways and stromal cell behavior. In breast cancer, ERs shape the TME and treatment response, with ER-positive and ER-negative tumors differently affecting immune cell functions. In prostate cancer, ERα promotes disease progression, while ERβ fosters an immunosuppressive environment.10 Human epidermal growth factor receptor 2 (HER-2) also impacts the TME by regulating immune cell recruitment and tumor growth through the induction of CCL2 and PD-1 ligands.141 HER-2 overexpression drives mammary carcinogenesis, tumor growth, and invasion by affecting mammary stem cells.142 In HER-2/neu-driven mammary carcinomas, CCL2/CCR2 signaling promotes the tumor-supportive microenvironment. Activation of wild-type HER-2 requires phosphorylation by EGFR-2, whereas mutant HER-2 functions independently, leading to continuous activation of the JNK/AP1 pathway and overexpression of VEGF, TGF-β, and TGF-α.143
H-RAS is another protein that can stimulate the JNK/AP1 pathway. Mutant H-RAS and HER-2 use RAC-1 to regulate the JNK/AP1 pathway. Indeed, H-RAS and HER-2 mutations not only change the intracellular pathways in malignant cells but also affect TME features by causing the secretion of a specific chemokine.144 Similar to H-RAS, also K-RAS is a member of the RAS family, which is an important GTP-ase group in cells and operates as a regulatory factor for many cancers-related cellular processes such as cell growth, proliferation, and migration. K-RAS mutations are very popular in different types of cancer and affect malignant cell environments differently. For example, in non-small cell lung cancer (NSCLC), mutant K-RAS can reduce tristetraprolin (TTP) expression level by activation of the MEK signaling pathway. TTP is a key down-regulator agent for PDL-1, which means a mutation in K-RAS directly influences the immune system in TME.145,146
It is not the only way K-RAS mutation affects immune cells in TME. In addition, the expression level of MHC-I is directly affected by K-RAS mutation, reducing the accessibility of the immune system to tumor cell antigens in some cancers. Moreover, mutant K-RAS increases the amount of CXCL-3, leading to the migration of MDSCs to TME. Mutant K-RAS can affect the CAFs population through CXCR-2 ligands, which promote invasion, angiogenesis, and migration in cancer. Due to these functions, K-RAS mutations usually induce immune suppressive microenvironment in TME.147,148
The Wnt/β-catenin signaling pathway plays a crucial role in the TME and cancer stem cells (CSCs) in various cancers, including hepatocellular carcinoma, colorectal cancer, and breast cancer. This pathway regulates interactions among different TME components, such as immune cells, stem cells, and tumor vasculature, and is involved in the maintenance of CSCs.149 In colorectal cancer, the oncogenic properties of Wnt/β-catenin signaling contribute to tumor progression and the regulation of CSCs.150 The pathway also modulates the balance between stemness and differentiation in adult stem cell niches, and its activation underlies tumorigenesis in various tissues. Pten loss and BrafV600E mutation increase activation of the Wnt-β-catenin pathway, which causes the increased expression level of inflammatory cytokines (IL-6, IL-10), and VEGF. Also, this situation is responsible for the high frequency of CD-103 + dendritic cells, which is associated with poor immune infiltration to TME.151,152
The expression of DNA mismatch repair genes (MLH-1, MSH-2, PMS-2, and MSH-6) is correlated with various clinicopathological features in colorectal cancer (CRC), including tumor location, differentiation, and lymphocytic infiltration.153 The alterations in Mismatch repair (MMR) genes can also impact TMB and PD-L1 expression in MSI-H tumors. Tumors in this category usually show high infiltration of CTL and Th1 cells, and low level of PDL-1 expression, so this type of CRC is more sensitive to immunotherapy than other classes of CRC. Thus, different types of mutations can deeply affect TME and change its total properties.152
Additionally, several known mechanisms lead to self-organization in the TME. For example, malignant cells secrete different signaling molecules, like growth factors and interleukins, that mediate paracrine signaling.154 Cancer cells release matrix metalloproteinases (MMPs) that degrade the ECM, leading to more invasion. Moreover, changes in ECM composition could increase signals that impact cell behavior to be more aggressive.155 In response to the hypoxia and nutrient gradients, malignant cells might induce HIFs, that activate metabolic and angiogenesis reprogramming.156 The physical features of the TME, like increased stiffness as a result of fibrosis, could induce cancer development.157 Tumor cells can release chemokines that attract immune cells, such as TAMs, which can shift between pro-inflammatory and anti-inflammatory states based on the signals they receive from malignant cells. This interaction often aids tumor development and growth instead of suppressing it.158
Conclusion
In conclusion, this review emphasizes the complex role of the TME and self-organization in tumor development and therapeutic resistance. By understanding the dynamic relations between tumor cells and the TME, we underline that these relationships are essential to understanding tumor heterogeneity and complexity. This vision expanded from studying the TME suggestion opportunities to recognize novel therapeutic targets, such as signaling pathways, immune cells, and the extracellular matrix, which can be used to improve more personalized and effective treatments. Additionally, the conception of self-organization offers a framework for exploring how tumors progress and resist present therapies, emphasizing the need for constant study in this area.
Future perspective
The use of artificial intelligence to model TME dynamics and predict therapy results will be an efficient method to control malignancy. Also, improving 3D organoid models to more accurately promote the TME for preclinical testing will be useful to get precise results. The potential of targeting exosome pathways as a new frontier in tumor treatment and focusing on controlling and suppressing the network communication that supports cancer proliferation and drug resistance will be useful in cancer management and therapy. In addition, personalized medicine approaches based on the patient’s TME molecular profiling lead to accurate targeted, and effective treatment.
Review Highlights
What is the current knowledge?
-
TME is composed of non-cancerous cells and various components within a tumor, including immune cells, fibroblasts, endothelial cells, the extracellular matrix (ECM), and soluble factors like chemokines and cytokines.
-
Self-organization refers to the dynamic and reciprocal interactions between tumor cells and their microenvironment, leading to the emergence of complex structures and behaviors that promote cancer progression.
-
TME and its dynamics are vital for developing new cancer therapies. Targeting specific components of the TME, such as ECM, immune cells, or signaling pathways, could improve treatment outcomes.
What is new here?
-
The manuscript currently covers a comprehensive analysis of the TME and its role in cancer progression.
-
The manuscript emphasizes that the TME is a complex and dynamic environment that plays a critical role in cancer progression. Current research is focused on unraveling these complexities to develop more effective and personalized cancer therapies.
-
The manuscript concludes by emphasizing the importance of continued research into the TME and self-organization in cancer cells, as these areas are crucial for advancing cancer treatment and developing personalized therapeutic strategies.
Competing Interests
The authors declare no conflict of interest.
Data Availability Statement
The data are available from the corresponding author upon reasonable request.
Ethical Approval
The study was approved by the Ethics Committee of Tabriz University of Medical Sciences (Ethics No.: IR.TBZMED.VCR.REC.1403.116).
Acknowledgements
The authors appreciate the researchers of the Tuberculosis and Lung Diseases Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
References
- Nallanthighal S, Heiserman JP, Cheon DJ. The role of the extracellular matrix in cancer stemness. Front Cell Dev Biol 2019; 7:86. doi: 10.3389/fcell.2019.00086 [Crossref] [ Google Scholar]
- Asadi M, Zarredar H, Zafari V, Soleimani Z, Saeedi H, Caner A. Immune features of tumor microenvironment: a genetic spotlight. Cell BiochemBiophys 2024; 82:107-18. doi: 10.1007/s12013-023-01192-7 [Crossref] [ Google Scholar]
- Wang Q, Shao X, Zhang Y, Zhu M, Wang FXC, Mu J. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med 2023; 12:11149-65. doi: 10.1002/cam4.5698 [Crossref] [ Google Scholar]
- Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 2011; 8:210-21. doi: 10.1038/nrclinonc.2011.21 [Crossref] [ Google Scholar]
- Fraisl P, Baes M, Carmeliet P. Hungry for blood vessels: linking metabolism and angiogenesis. Dev Cell 2008; 14:313-4. doi: 10.1016/j.devcel.2008.02.009 [Crossref] [ Google Scholar]
- Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci 2010; 123:4195-200. doi: 10.1242/jcs.023820 [Crossref] [ Google Scholar]
- Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE. Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res 2012; 72:2473-80. doi: 10.1158/0008-5472.Can-12-0122 [Crossref] [ Google Scholar]
- Ward C, Meehan J, Gray ME, Murray AF, Argyle DJ, Kunkler IH. The impact of tumour pH on cancer progression: strategies for clinical intervention. Explor Target Antitumor Ther 2020; 1:71-100. doi: 10.37349/etat.2020.00005 [Crossref] [ Google Scholar]
- Martin DN, Boersma BJ, Yi M, Reimers M, Howe TM, Yfantis HG. Differences in the tumor microenvironment between African-American and European-American breast cancer patients. PLoS One 2009; 4:e4531. doi: 10.1371/journal.pone.0004531 [Crossref] [ Google Scholar]
- Segovia-Mendoza M, Morales-Montor J. Immune tumor microenvironment in breast cancer and the participation of estrogen and its receptors in cancer physiopathology. Front Immunol 2019; 10:348. doi: 10.3389/fimmu.2019.00348 [Crossref] [ Google Scholar]
- Keeney S, Lange J, Mohibullah N. Self-organization of meiotic recombination initiation: general principles and molecular pathways. Annu Rev Genet 2014; 48:187-214. doi: 10.1146/annurev-genet-120213-092304 [Crossref] [ Google Scholar]
- Karsenti E. Self-organization in cell biology: a brief history. Nat Rev Mol Cell Biol 2008; 9:255-62. doi: 10.1038/nrm2357 [Crossref] [ Google Scholar]
- Kurakin A. Self-organization versus Watchmaker: ambiguity of molecular recognition and design charts of cellular circuitry. J Mol Recognit 2007; 20:205-14. doi: 10.1002/jmr.839 [Crossref] [ Google Scholar]
- Siegel PM, Shu W, Cardiff RD, Muller WJ, Massagué J. Transforming growth factor beta signaling impairs neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A 2003; 100:8430-5. doi: 10.1073/pnas.0932636100 [Crossref] [ Google Scholar]
- Sánchez-Elsner T, Botella LM, Velasco B, Corbí A, Attisano L, Bernabéu C. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J Biol Chem 2001; 276:38527-35. doi: 10.1074/jbc.M104536200 [Crossref] [ Google Scholar]
- Normanno N, Bianco C, De Luca A, Salomon DS. The role of EGF-related peptides in tumor growth. Front Biosci 2001; 6:D685-707. doi: 10.2741/normano [Crossref] [ Google Scholar]
- Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci 2019; 20:840. doi: 10.3390/ijms20040840 [Crossref] [ Google Scholar]
- Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012; 21:309-22. doi: 10.1016/j.ccr.2012.02.022 [Crossref] [ Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74. doi: 10.1016/j.cell.2011.02.013 [Crossref] [ Google Scholar]
- Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 2012; 196:395-406. doi: 10.1083/jcb.201102147 [Crossref] [ Google Scholar]
- Mbeunkui F, Johann DJ Jr. Cancer and the tumor microenvironment: a review of an essential relationship. Cancer Chemother Pharmacol 2009; 63:571-82. doi: 10.1007/s00280-008-0881-9 [Crossref] [ Google Scholar]
- Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal 2020; 18:59. doi: 10.1186/s12964-020-0530-4 [Crossref] [ Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57-70. doi: 10.1016/s0092-8674(00)81683-9 [Crossref] [ Google Scholar]
- Räsänen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res 2010; 316:2713-22. doi: 10.1016/j.yexcr.2010.04.032 [Crossref] [ Google Scholar]
- Trivanović D, Krstić J, Djordjević IO, Mojsilović S, Santibanez JF, Bugarski D. The roles of mesenchymal stromal/stem cells in tumor microenvironment associated with inflammation. Mediators Inflamm 2016; 2016:7314016. doi: 10.1155/2016/7314016 [Crossref] [ Google Scholar]
- Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci 2012; 125:5591-6. doi: 10.1242/jcs.116392 [Crossref] [ Google Scholar]
- Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol 2006; 24:5373-80. doi: 10.1200/jco.2006.05.9584 [Crossref] [ Google Scholar]
- de Looff M, de Jong S, Kruyt FA. Multiple interactions between cancer cells and the tumor microenvironment modulate TRAIL signaling: implications for TRAIL receptor targeted therapy. Front Immunol 2019; 10:1530. doi: 10.3389/fimmu.2019.01530 [Crossref] [ Google Scholar]
- Kawaguchi K, Sakurai M, Yamamoto Y, Suzuki E, Tsuda M, Kataoka TR. Alteration of specific cytokine expression patterns in patients with breast cancer. Sci Rep 2019; 9:2924. doi: 10.1038/s41598-019-39476-9 [Crossref] [ Google Scholar]
- Farajzadeh Valilou S, Keshavarz-Fathi M, Silvestris N, Argentiero A, Rezaei N. The role of inflammatory cytokines and tumor associated macrophages (TAMs) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev 2018; 39:46-61. doi: 10.1016/j.cytogfr.2018.01.007 [Crossref] [ Google Scholar]
- Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008; 27:5904-12. doi: 10.1038/onc.2008.271 [Crossref] [ Google Scholar]
- Sun Z, Wang S, Zhao RC. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J Hematol Oncol 2014; 7:14. doi: 10.1186/1756-8722-7-14 [Crossref] [ Google Scholar]
- Nwabo Kamdje AH, Kamga PT, Tagne Simo R, Vecchio L, Seke Etet PF, Muller JM. Mesenchymal stromal cells' role in tumor microenvironment: involvement of signaling pathways. Cancer Biol Med 2017; 14:129-41. doi: 10.20892/j.issn.2095-3941.2016.0033 [Crossref] [ Google Scholar]
- Han Z, Tian Z, Lv G, Zhang L, Jiang G, Sun K. Immunosuppressive effect of bone marrow-derived mesenchymal stem cells in inflammatory microenvironment favours the growth of B16 melanoma cells. J Cell Mol Med 2011; 15:2343-52. doi: 10.1111/j.1582-4934.2010.01215.x [Crossref] [ Google Scholar]
- Kabashima-Niibe A, Higuchi H, Takaishi H, Masugi Y, Matsuzaki Y, Mabuchi Y. Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer Sci 2013; 104:157-64. doi: 10.1111/cas.12059 [Crossref] [ Google Scholar]
- Liu Y, Han ZP, Zhang SS, Jing YY, Bu XX, Wang CY. Effects of inflammatory factors on mesenchymal stem cells and their role in the promotion of tumor angiogenesis in colon cancer. J Biol Chem 2011; 286:25007-15. doi: 10.1074/jbc.M110.213108 [Crossref] [ Google Scholar]
- Bergfeld SA, DeClerck YA. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev 2010; 29:249-61. doi: 10.1007/s10555-010-9222-7 [Crossref] [ Google Scholar]
- Zhang Y, Daquinag AC, Amaya-Manzanares F, Sirin O, Tseng C, Kolonin MG. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res 2012; 72:5198-208. doi: 10.1158/0008-5472.Can-12-0294 [Crossref] [ Google Scholar]
- Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 2002; 160:985-1000. doi: 10.1016/s0002-9440(10)64920-6 [Crossref] [ Google Scholar]
- Cooke VG, LeBleu VS, Keskin D, Khan Z, O'Connell JT, Teng Y. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012; 21:66-81. doi: 10.1016/j.ccr.2011.11.024 [Crossref] [ Google Scholar]
- Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA. Obesity and cancer mechanisms: tumor microenvironment and inflammation. J Clin Oncol 2016; 34:4270-6. doi: 10.1200/jco.2016.67.4283 [Crossref] [ Google Scholar]
- Birbrair A. Pericyte biology: development, homeostasis, and disease. Adv Exp Med Biol 2018; 1109:1-3. doi: 10.1007/978-3-030-02601-1_1 [Crossref] [ Google Scholar]
- Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013; 153:139-52. doi: 10.1016/j.cell.2013.02.021 [Crossref] [ Google Scholar]
- Liao Z, Tan ZW, Zhu P, Tan NS. Cancer-associated fibroblasts in tumor microenvironment - accomplices in tumor malignancy. Cell Immunol 2019; 343:103729. doi: 10.1016/j.cellimm.2017.12.003 [Crossref] [ Google Scholar]
- Stadler M, Walter S, Walzl A, Kramer N, Unger C, Scherzer M. Increased complexity in carcinomas: analyzing and modeling the interaction of human cancer cells with their microenvironment. Semin Cancer Biol 2015; 35:107-24. doi: 10.1016/j.semcancer.2015.08.007 [Crossref] [ Google Scholar]
- Mbeunkui F, Johann DJ Jr. Cancer and the tumor microenvironment: a review of an essential relationship. Cancer Chemother Pharmacol 2009; 63:571-82. doi: 10.1007/s00280-008-0881-9 [Crossref] [ Google Scholar]
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6:392-401. doi: 10.1038/nrc1877 [Crossref] [ Google Scholar]
- Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 2019; 9:282-301. doi: 10.1158/2159-8290.Cd-18-0710 [Crossref] [ Google Scholar]
- Takai K, Le A, Weaver VM, Werb Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016; 7:82889-901. doi: 10.18632/oncotarget.12658 [Crossref] [ Google Scholar]
- Zhang X, Schönrogge M, Eichberg J, Wendt EH, Kumstel S, Stenzel J. Blocking autophagy in cancer-associated fibroblasts supports chemotherapy of pancreatic cancer cells. Front Oncol 2018; 8:590. doi: 10.3389/fonc.2018.00590 [Crossref] [ Google Scholar]
- Xu K, Tian X, Oh SY, Movassaghi M, Naber SP, Kuperwasser C. The fibroblast Tiam1-osteopontin pathway modulates breast cancer invasion and metastasis. Breast Cancer Res 2016; 18:14. doi: 10.1186/s13058-016-0674-8 [Crossref] [ Google Scholar]
- Fullár A, Dudás J, Oláh L, Hollósi P, Papp Z, Sobel G. Remodeling of extracellular matrix by normal and tumor-associated fibroblasts promotes cervical cancer progression. BMC Cancer 2015; 15:256. doi: 10.1186/s12885-015-1272-3 [Crossref] [ Google Scholar]
- De Vlieghere E, Verset L, Demetter P, Bracke M, De Wever O. Cancer-associated fibroblasts as target and tool in cancer therapeutics and diagnostics. Virchows Arch 2015; 467:367-82. doi: 10.1007/s00428-015-1818-4 [Crossref] [ Google Scholar]
- Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010; 330:827-30. doi: 10.1126/science.1195300 [Crossref] [ Google Scholar]
- Lee SW, Kwak HS, Kang MH, Park YY, Jeong GS. Fibroblast-associated tumour microenvironment induces vascular structure-networked tumouroid. Sci Rep 2018; 8:2365. doi: 10.1038/s41598-018-20886-0 [Crossref] [ Google Scholar]
- Monteran L, Erez N. The dark side of fibroblasts: cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol 2019; 10:1835. doi: 10.3389/fimmu.2019.01835 [Crossref] [ Google Scholar]
- Salazar N, Zabel BA. Support of tumor endothelial cells by chemokine receptors. Front Immunol 2019; 10:147. doi: 10.3389/fimmu.2019.00147 [Crossref] [ Google Scholar]
- Dudley AC. Tumor endothelial cells. Cold Spring HarbPerspect Med 2012; 2:a006536. doi: 10.1101/cshperspect.a006536 [Crossref] [ Google Scholar]
- Hida K, Maishi N, Annan DA, Hida Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int J Mol Sci 2018; 19:1272. doi: 10.3390/ijms19051272 [Crossref] [ Google Scholar]
- Lee E, Pandey NB, Popel AS. Crosstalk between cancer cells and blood endothelial and lymphatic endothelial cells in tumour and organ microenvironment. Expert Rev Mol Med 2015; 17:e3. doi: 10.1017/erm.2015.2 [Crossref] [ Google Scholar]
- Ansardamavandi A, Tafazzoli-Shadpour M. The functional cross talk between cancer cells and cancer associated fibroblasts from a cancer mechanics perspective. BiochimBiophys Acta Mol Cell Res 2021; 1868:119103. doi: 10.1016/j.bbamcr.2021.119103 [Crossref] [ Google Scholar]
- Kazi JU. Mechanisms of anticancer therapy resistance: the role of cancer stem cells. Int J Mol Sci 2020; 21:9006. doi: 10.3390/ijms21239006 [Crossref] [ Google Scholar]
- Tesniere A, Zitvogel L, Kroemer G. The immune system: taming and unleashing cancer. Discov Med 2006; 6:211-6. [ Google Scholar]
- Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther 2021; 6:263. doi: 10.1038/s41392-021-00658-5 [Crossref] [ Google Scholar]
- Rafi MA, Omidi Y. A prospective highlight on exosomal nanoshuttles and cancer immunotherapy and vaccination. Bioimpacts 2015; 5:117-22. doi: 10.15171/bi.2015.22 [Crossref] [ Google Scholar]
- Ponomarev AV, Shubina IZ. Insights into mechanisms of tumor and immune system interaction: association with wound healing. Front Oncol 2019; 9:1115. doi: 10.3389/fonc.2019.01115 [Crossref] [ Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018; 24:541-50. doi: 10.1038/s41591-018-0014-x [Crossref] [ Google Scholar]
- Yu YR, Ho PC. Sculpting tumor microenvironment with immune system: from immunometabolism to immunoediting. Clin Exp Immunol 2019; 197:153-60. doi: 10.1111/cei.13293 [Crossref] [ Google Scholar]
- Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The emerging role of neutrophil extracellular traps (NETs) in tumor progression and metastasis. Front Immunol 2020; 11:1749. doi: 10.3389/fimmu.2020.01749 [Crossref] [ Google Scholar]
- Lee JH, Choi SY, Jung NC, Song JY, Seo HG, Lee HS. The effect of the tumor microenvironment and tumor-derived metabolites on dendritic cell function. J Cancer 2020; 11:769-75. doi: 10.7150/jca.38785 [Crossref] [ Google Scholar]
- Cassetta L, Kitamura T. Targeting tumor-associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front Cell Dev Biol 2018; 6:38. doi: 10.3389/fcell.2018.00038 [Crossref] [ Google Scholar]
- Solinas G, Schiarea S, Liguori M, Fabbri M, Pesce S, Zammataro L. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J Immunol 2010; 185:642-52. doi: 10.4049/jimmunol.1000413 [Crossref] [ Google Scholar]
- Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab 2019; 30:36-50. doi: 10.1016/j.cmet.2019.06.001 [Crossref] [ Google Scholar]
- Zhu J, Zhi Q, Zhou BP, Tao M, Liu J, Li W. The role of tumor associated macrophages in the tumor microenvironment: mechanism and functions. Anticancer Agents Med Chem 2016; 16:1133-41. doi: 10.2174/1871520616666160520112622 [Crossref] [ Google Scholar]
- Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol 2019; 12:76. doi: 10.1186/s13045-019-0760-3 [Crossref] [ Google Scholar]
- Zhou K, Cheng T, Zhan J, Peng X, Zhang Y, Wen J. Targeting tumor-associated macrophages in the tumor microenvironment. Oncol Lett 2020; 20:234. doi: 10.3892/ol.2020.12097 [Crossref] [ Google Scholar]
- Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer 2019; 18:64. doi: 10.1186/s12943-019-0976-4 [Crossref] [ Google Scholar]
- Apolloni E, Bronte V, Mazzoni A, Serafini P, Cabrelle A, Segal DM. Immortalized myeloid suppressor cells trigger apoptosis in antigen-activated T lymphocytes. J Immunol 2000; 165:6723-30. doi: 10.4049/jimmunol.165.12.6723 [Crossref] [ Google Scholar]
- Maimela NR, Liu S, Zhang Y. Fates of CD8 + T cells in tumor microenvironment. Comput Struct Biotechnol J 2019; 17:1-13. doi: 10.1016/j.csbj.2018.11.004 [Crossref] [ Google Scholar]
- Mun JY, Leem SH, Lee JH, Kim HS. Dual relationship between stromal cells and immune cells in the tumor microenvironment. Front Immunol 2022; 13:864739. doi: 10.3389/fimmu.2022.864739 [Crossref] [ Google Scholar]
- Kim HR, Park HJ, Son J, Lee JG, Chung KY, Cho NH. Tumor microenvironment dictates regulatory T cell phenotype: upregulated immune checkpoints reinforce suppressive function. J Immunother Cancer 2019; 7:339. doi: 10.1186/s40425-019-0785-8 [Crossref] [ Google Scholar]
- Leivonen SK, Pollari M, Brück O, Pellinen T, Autio M, Karjalainen-Lindsberg ML. T-cell inflamed tumor microenvironment predicts favorable prognosis in primary testicular lymphoma. Haematologica 2019; 104:338-46. doi: 10.3324/haematol.2018.200105 [Crossref] [ Google Scholar]
- Lo Presti E, Pizzolato G, Corsale AM, Caccamo N, Sireci G, Dieli F. γδ T cells and tumor microenvironment: from immunosurveillance to tumor evasion. Front Immunol 2018; 9:1395. doi: 10.3389/fimmu.2018.01395 [Crossref] [ Google Scholar]
- Kuczek DE, Larsen AM, Thorseth ML, Carretta M, Kalvisa A, Siersbæk MS. Collagen density regulates the activity of tumor-infiltrating T cells. J Immunother Cancer 2019; 7:68. doi: 10.1186/s40425-019-0556-6 [Crossref] [ Google Scholar]
- Andreu-Sanz D, Kobold S. Role and potential of different T helper cell subsets in adoptive cell therapy. Cancers (Basel) 2023; 15:1650. doi: 10.3390/cancers15061650 [Crossref] [ Google Scholar]
- Balta E, Wabnitz GH, Samstag Y. Hijacked immune cells in the tumor microenvironment: molecular mechanisms of immunosuppression and cues to improve T cell-based immunotherapy of solid tumors. Int J Mol Sci 2021; 22:5736. doi: 10.3390/ijms22115736 [Crossref] [ Google Scholar]
- Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res 2019; 79:4557-66. doi: 10.1158/0008-5472.Can-18-3962 [Crossref] [ Google Scholar]
- Wang SS, Liu W, Ly D, Xu H, Qu L, Zhang L. Tumor-infiltrating B cells: their role and application in anti-tumor immunity in lung cancer. Cell Mol Immunol 2019; 16:6-18. doi: 10.1038/s41423-018-0027-x [Crossref] [ Google Scholar]
- Yuen GJ, Demissie E, Pillai S. B lymphocytes and cancer: a love-hate relationship. Trends Cancer 2016; 2:747-57. doi: 10.1016/j.trecan.2016.10.010 [Crossref] [ Google Scholar]
- Larsen SK, Gao Y, Basse PH. NK cells in the tumor microenvironment. Crit Rev Oncog 2014; 19:91-105. doi: 10.1615/critrevoncog.2014011142 [Crossref] [ Google Scholar]
- Russo E, Laffranchi M, Tomaipitinca L, Del Prete A, Santoni A, Sozzani S. NK cell anti-tumor surveillance in a myeloid cell-shaped environment. Front Immunol 2021; 12:787116. doi: 10.3389/fimmu.2021.787116 [Crossref] [ Google Scholar]
- Chanmee T, Ontong P, Itano N. Hyaluronan: a modulator of the tumor microenvironment. Cancer Lett 2016; 375:20-30. doi: 10.1016/j.canlet.2016.02.031 [Crossref] [ Google Scholar]
- Hui L, Chen Y. Tumor microenvironment: sanctuary of the devil. Cancer Lett 2015; 368:7-13. doi: 10.1016/j.canlet.2015.07.039 [Crossref] [ Google Scholar]
- Gonda TA, Tu S, Wang TC. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle 2009; 8:2005-13. doi: 10.4161/cc.8.13.8985 [Crossref] [ Google Scholar]
- Keibel A, Singh V, Sharma MC. Inflammation, microenvironment, and the immune system in cancer progression. Curr Pharm Des 2009; 15:1949-55. doi: 10.2174/138161209788453167 [Crossref] [ Google Scholar]
- Mocellin S, Wang E, Marincola FM. Cytokines and immune response in the tumor microenvironment. J Immunother (1991) 2001; 24:392-407. [ Google Scholar]
- Wang L, Zhao Y, Liu Y, Akiyama K, Chen C, Qu C. IFN-γ and TNF-α synergistically induce mesenchymal stem cell impairment and tumorigenesis via NFκB signaling. Stem Cells 2013; 31:1383-95. doi: 10.1002/stem.1388 [Crossref] [ Google Scholar]
- Caja F, Vannucci L. TGFβ: a player on multiple fronts in the tumor microenvironment. J Immunotoxicol 2015; 12:300-7. doi: 10.3109/1547691x.2014.945667 [Crossref] [ Google Scholar]
- Becker JC, Andersen MH, Schrama D, Thor Straten P. Immune-suppressive properties of the tumor microenvironment. Cancer Immunol Immunother 2013; 62:1137-48. doi: 10.1007/s00262-013-1434-6 [Crossref] [ Google Scholar]
- Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy. Immunol Res 2011; 51:170-82. doi: 10.1007/s12026-011-8262-6 [Crossref] [ Google Scholar]
- Shen X, Li N, Li H, Zhang T, Wang F, Li Q. Increased prevalence of regulatory T cells in the tumor microenvironment and its correlation with TNM stage of hepatocellular carcinoma. J Cancer Res Clin Oncol 2010; 136:1745-54. doi: 10.1007/s00432-010-0833-8 [Crossref] [ Google Scholar]
- Mohamed MM, El-Ghonaimy EA, Nouh MA, Schneider RJ, Sloane BF, El-Shinawi M. Cytokines secreted by macrophages isolated from tumor microenvironment of inflammatory breast cancer patients possess chemotactic properties. Int J Biochem Cell Biol 2014; 46:138-47. doi: 10.1016/j.biocel.2013.11.015 [Crossref] [ Google Scholar]
- Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of IFN-γ in tumor progression and regression: a review. Biomark Res 2020; 8:49. doi: 10.1186/s40364-020-00228-x [Crossref] [ Google Scholar]
- Liu L, Ge D, Ma L, Mei J, Liu S, Zhang Q. Interleukin-17 and prostaglandin E2 are involved in formation of an M2 macrophage-dominant microenvironment in lung cancer. J Thorac Oncol 2012; 7:1091-100. doi: 10.1097/JTO.0b013e3182542752 [Crossref] [ Google Scholar]
- Iida T, Iwahashi M, Katsuda M, Ishida K, Nakamori M, Nakamura M. Tumor-infiltrating CD4 + Th17 cells produce IL-17 in tumor microenvironment and promote tumor progression in human gastric cancer. Oncol Rep 2011; 25:1271-7. doi: 10.3892/or.2011.1201 [Crossref] [ Google Scholar]
- Kryczek I, Wei S, Zou L, Altuwaijri S, Szeliga W, Kolls J. Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment. J Immunol 2007; 178:6730-3. doi: 10.4049/jimmunol.178.11.6730 [Crossref] [ Google Scholar]
- Manore SG, Doheny DL, Wong GL, Lo HW. IL-6/JAK/STAT3 signaling in breast cancer metastasis: biology and treatment. Front Oncol 2022; 12:866014. doi: 10.3389/fonc.2022.866014 [Crossref] [ Google Scholar]
- Cendrowicz E, Sas Z, Bremer E, Rygiel TP. The role of macrophages in cancer development and therapy. Cancers (Basel) 2021; 13:1946. doi: 10.3390/cancers13081946 [Crossref] [ Google Scholar]
- Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci 2019; 6:160. doi: 10.3389/fmolb.2019.00160 [Crossref] [ Google Scholar]
- Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol 2011; 226:299-308. doi: 10.1002/jcp.22400 [Crossref] [ Google Scholar]
- Gonzalez-Molina J, Moyano-Galceran L, Single A, Gultekin O, Alsalhi S, Lehti K. Chemotherapy as a regulator of extracellular matrix-cell communication: implications in therapy resistance. Semin Cancer Biol 2022; 86:224-36. doi: 10.1016/j.semcancer.2022.03.012 [Crossref] [ Google Scholar]
- Li Y, Jin G, Liu N, Guo H, Xu F. The post-chemotherapy changes of tumor physical microenvironment: targeting extracellular matrix to address chemoresistance. Cancer Lett 2024; 582:216583. doi: 10.1016/j.canlet.2023.216583 [Crossref] [ Google Scholar]
- Shanehbandi D, Asadi M, Seyedrezazadeh E, Zafari V, Shekari N, Akbari M. MicroRNA-based biomarkers in lung cancer: recent advances and potential applications. Curr Mol Med 2023; 23:648-67. doi: 10.2174/2772432817666220520085719 [Crossref] [ Google Scholar]
- Chou J, Shahi P, Werb Z. MicroRNA-mediated regulation of the tumor microenvironment. Cell Cycle 2013; 12:3262-71. doi: 10.4161/cc.26087 [Crossref] [ Google Scholar]
- Cheng Q, Yi B, Wang A, Jiang X. Exploring and exploiting the fundamental role of microRNAs in tumor pathogenesis. Onco Targets Ther 2013; 6:1675-84. doi: 10.2147/ott.S52730 [Crossref] [ Google Scholar]
- Suzuki HI, Katsura A, Matsuyama H, Miyazono K. MicroRNA regulons in tumor microenvironment. Oncogene 2015; 34:3085-94. doi: 10.1038/onc.2014.254 [Crossref] [ Google Scholar]
- Kohlhapp FJ, Mitra AK, Lengyel E, Peter ME. MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 2015; 34:5857-68. doi: 10.1038/onc.2015.89 [Crossref] [ Google Scholar]
- Yang N, Zhu S, Lv X, Qiao Y, Liu YJ, Chen J. MicroRNAs: pleiotropic regulators in the tumor microenvironment. Front Immunol 2018; 9:2491. doi: 10.3389/fimmu.2018.02491 [Crossref] [ Google Scholar]
- Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov 2012; 2:1100-8. doi: 10.1158/2159-8290.Cd-12-0206 [Crossref] [ Google Scholar]
- Ceppi P, Peter ME. MicroRNAs regulate both epithelial-to-mesenchymal transition and cancer stem cells. Oncogene 2014; 33:269-78. doi: 10.1038/onc.2013.55 [Crossref] [ Google Scholar]
- Zhao L, Sun Y, Hou Y, Peng Q, Wang L, Luo H. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int J Biochem Cell Biol 2012; 44:2051-9. doi: 10.1016/j.biocel.2012.08.005 [Crossref] [ Google Scholar]
- Matsuyama H, Suzuki HI, Nishimori H, Noguchi M, Yao T, Komatsu N. miR-135b mediates NPM-ALK-driven oncogenicity and renders IL-17-producing immunophenotype to anaplastic large cell lymphoma. Blood 2011; 118:6881-92. doi: 10.1182/blood-2011-05-354654 [Crossref] [ Google Scholar]
- Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl) 2013; 91:431-7. doi: 10.1007/s00109-013-1020-6 [Crossref] [ Google Scholar]
- Yang E, Wang X, Gong Z, Yu M, Wu H, Zhang D. Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct Target Ther 2020; 5:242. doi: 10.1038/s41392-020-00359-5 [Crossref] [ Google Scholar]
- Roma-Rodrigues C, Fernandes AR, Baptista PV. Exosome in tumour microenvironment: overview of the crosstalk between normal and cancer cells. Biomed Res Int 2014; 2014:179486. doi: 10.1155/2014/179486 [Crossref] [ Google Scholar]
- Li I, Nabet BY. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol Cancer 2019; 18:32. doi: 10.1186/s12943-019-0975-5 [Crossref] [ Google Scholar]
- Panda SS, Sahoo RK, Patra SK, Biswal S, Biswal BK. Molecular insights to therapeutic in cancer: role of exosomes in tumor microenvironment, metastatic progression and drug resistance. Drug Discov Today 2024; 29:104061. doi: 10.1016/j.drudis.2024.104061 [Crossref] [ Google Scholar]
- Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 2014; 30:255-89. doi: 10.1146/annurev-cellbio-101512-122326 [Crossref] [ Google Scholar]
- Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science 2020; 367:eaau6977. doi: 10.1126/science.aau6977 [Crossref] [ Google Scholar]
- Makler A, Asghar W. Exosomal biomarkers for cancer diagnosis and patient monitoring. Expert Rev Mol Diagn 2020; 20:387-400. doi: 10.1080/14737159.2020.1731308 [Crossref] [ Google Scholar]
- Kraus M, Wolf B. Emergence of self-organization in tumor cells: relevance for diagnosis and therapy. Tumour Biol 1993; 14:338-53. doi: 10.1159/000217849 [Crossref] [ Google Scholar]
- Gouirand V, Guillaumond F, Vasseur S. Influence of the tumor microenvironment on cancer cells metabolic reprogramming. Front Oncol 2018; 8:117. doi: 10.3389/fonc.2018.00117 [Crossref] [ Google Scholar]
- Arneth B. Tumor microenvironment. Medicina (Kaunas) 2019; 56:15. doi: 10.3390/medicina56010015 [Crossref] [ Google Scholar]
- Li H, Fan X, Houghton J. Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem 2007; 101:805-15. doi: 10.1002/jcb.21159 [Crossref] [ Google Scholar]
- Babloyantz A, Kaczmarek LK. Self-organization in biological systems with multiple cellular contacts. Bull Math Biol 1979; 41:193-201. doi: 10.1007/bf02460878 [Crossref] [ Google Scholar]
- Norman J, Sorrell EL, Hu Y, Siripurapu V, Garcia J, Bagwell J, et al. Tissue self-organization underlies morphogenesis of the notochord. Philos Trans R Soc Lond B Biol Sci 2018; 373. 10.1098/rstb.2017.0320.
- Misteli T. The concept of self-organization in cellular architecture. J Cell Biol 2001; 155:181-5. doi: 10.1083/jcb.200108110 [Crossref] [ Google Scholar]
- Tabony J. Historical and conceptual background of self-organization by reactive processes. Biol Cell 2006; 98:589-602. doi: 10.1042/bc20050086 [Crossref] [ Google Scholar]
- Camazine S, Deneubourg JL, Franks NR, Sneyd J, Theraula G, Bonabeau E. Self-Organization in Biological Systems. Princeton University Press; 2003.
- Wedlich-Söldner R, Betz T. Self-organization: the fundament of cell biology. Philos Trans R Soc Lond B Biol Sci 2018; 373. 10.1098/rstb.2017.0103.
- Triulzi T, Forte L, Regondi V, Di Modica M, Ghirelli C, Carcangiu ML. HER2 signaling regulates the tumor immune microenvironment and trastuzumab efficacy. Oncoimmunology 2019; 8:e1512942. doi: 10.1080/2162402x.2018.1512942 [Crossref] [ Google Scholar]
- Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008; 27:6120-30. doi: 10.1038/onc.2008.207 [Crossref] [ Google Scholar]
- Chen X, Wang Y, Nelson D, Tian S, Mulvey E, Patel B. CCL2/CCR2 regulates the tumor microenvironment in HER-2/neu-driven mammary carcinomas in mice. PLoS One 2016; 11:e0165595. doi: 10.1371/journal.pone.0165595 [Crossref] [ Google Scholar]
- Wang SE, Yu Y, Criswell TL, Debusk LM, Lin PC, Zent R. Oncogenic mutations regulate tumor microenvironment through induction of growth factors and angiogenic mediators. Oncogene 2010; 29:3335-48. doi: 10.1038/onc.2010.112 [Crossref] [ Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013; 503:548-51. doi: 10.1038/nature12796 [Crossref] [ Google Scholar]
- Wang H, Wang H, Ge L, Zhao Y, Zhu K, Chen Z. Betulinic acid targets drug-resistant human gastric cancer cells by inducing autophagic cell death, suppresses cell migration and invasion, and modulates the ERK/MEK signaling pathway. Acta Biochim Pol 2021; 69:25-30. doi: 10.18388/abp.2020_5530 [Crossref] [ Google Scholar]
- Dias Carvalho P, Machado AL, Martins F, Seruca R, Velho S. Targeting the tumor microenvironment: an unexplored strategy for mutant KRAS tumors. Cancers (Basel) 2019; 11:2020. doi: 10.3390/cancers11122010 [Crossref] [ Google Scholar]
- Zarredar H, Ansarin K, Baradaran B, Ahdi Khosroshahi S, Farajnia S. Potential molecular targets in the treatment of lung cancer using siRNA technology. Cancer Invest 2018; 36:37-58. doi: 10.1080/07357907.2017.1416393 [Crossref] [ Google Scholar]
- Wang K, Qiu X, Zhao Y, Wang H, Chen L. The Wnt/β-catenin signaling pathway in the tumor microenvironment of hepatocellular carcinoma. Cancer Biol Med 2021; 19:305-18. doi: 10.20892/j.issn.2095-3941.2021.0306 [Crossref] [ Google Scholar]
- Fuchs SY, Ougolkov AV, Spiegelman VS, Minamoto T. Oncogenic beta-catenin signaling networks in colorectal cancer. Cell Cycle 2005; 4:1522-39. doi: 10.4161/cc.4.11.2129 [Crossref] [ Google Scholar]
- Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE. β-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 2011; 20:741-54. doi: 10.1016/j.ccr.2011.10.030 [Crossref] [ Google Scholar]
- Zhang S, Yang X, Wang L, Zhang C. Interplay between inflammatory tumor microenvironment and cancer stem cells. Oncol Lett 2018; 16:679-86. doi: 10.3892/ol.2018.8716 [Crossref] [ Google Scholar]
- Karahan B, Argon A, Yıldırım M, Vardar E. Relationship between MLH-1, MSH-2, PMS-2, MSH-6 expression and clinicopathological features in colorectal cancer. Int J Clin Exp Pathol 2015; 8:4044-53. [ Google Scholar]
- Zhang X, Ma H, Gao Y, Liang Y, Du Y, Hao S. The tumor microenvironment: signal transduction. Biomolecules 2024; 14:438. doi: 10.3390/biom14040438 [Crossref] [ Google Scholar]
- Niland S, Riscanevo AX, Eble JA. Matrix metalloproteinases shape the tumor microenvironment in cancer progression. Int J Mol Sci 2021; 23:146. doi: 10.3390/ijms23010146 [Crossref] [ Google Scholar]
- Chen Z, Han F, Du Y, Shi H, Zhou W. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther 2023; 8:70. doi: 10.1038/s41392-023-01332-8 [Crossref] [ Google Scholar]
- Deng B, Zhao Z, Kong W, Han C, Shen X, Zhou C. Biological role of matrix stiffness in tumor growth and treatment. J Transl Med 2022; 20:540. doi: 10.1186/s12967-022-03768-y [Crossref] [ Google Scholar]
- Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol 2020; 11:583084. doi: 10.3389/fimmu.2020.583084 [Crossref] [ Google Scholar]