Bioimpacts. 2025;15:30778.
doi: 10.34172/bi.30778
Original Article
Immunoinformatics design of an mRNA vaccine against classical swine fever virus using conserved E2 protein and NS3 T-lymphocyte epitopes
Edward C. Banico Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, 1 
Ella Mae Joy S. Sira Methodology, Writing – review & editing, 1
Lauren Emily Fajardo Methodology, Writing – review & editing, 1
Fredmoore L. Orosco Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing, 1, 2, 3, *
Author information:
1irology and Vaccine Research Program, Industrial Technology Development Institute, Department of Science and Technology, Taguig City, 1634, Philippines
2S&T Fellows Program, Department of Science and Technology, Taguig City, 1634, Philippines
3Department of Biology, College of Arts and Sciences, University of the Philippines Manila, Manila City, 1000, Philippines
Abstract
Introduction:
The classical swine fever virus (CSFV) causes significant economic losses in the livestock industry. While the existing E2 marker vaccine offers protection against infections, it is characterized by delayed immunity and reduced effectiveness over time. Optimizing the existing vaccine is crucial to better control CSFV outbreaks worldwide. This study aimed to improve the existing E2 marker vaccine for CSFV by integrating NS3 T lymphocyte-inducing epitopes into the conserved E2 protein sequence and using mRNA technology for vaccine delivery.
Methods:
The design and evaluation of the vaccine were carried out exclusively through in silico methods. T lymphocyte epitopes were identified from the CSFV NS3 protein using multiple epitope prediction tools. A vaccine construct was formed after linking the predicted NS3 epitopes, E2 protein, and an immunogenic adjuvant. Molecular docking and dynamics simulations were performed to analyze the interaction between the adjuvant used and its immune receptor. Signal peptides were incorporated into the design, and mRNA sequences with varying codon usage biases were generated using LinearDesign. The mRNA sequence with minimum free energy (MFE) and codon adaptation index (CAI) closest to the controls was selected as the final design.
Results:
Twenty epitopes with high binding affinity to major histocompatibility complexes (MHCs) were identified from the CSFV NS3 protein. The vaccine construct with swine CD154 adjuvant demonstrated high antigenicity, making it the optimal choice for the final vaccine design. Molecular docking and dynamics simulations confirmed the adjuvant's strong affinity and stable interaction with its canonical receptor, swine CD40. Moreover, the final vaccine design exhibited higher populations of lymphocytes and antibodies compared to the components of the commercialized E2 marker vaccine in immune simulation. The final mRNA vaccine sequence exhibited a higher MFE and CAI than the two licensed mRNA vaccine controls.
Conclusion:
The mRNA vaccine designed in this study serves as a potential CSFV vaccine candidate. In vivo and in vitro validation is needed to confirm its efficacy.
Keywords: Classical swine fever virus, E2 marker vaccine, Epitopes, mRNA vaccine, Immunoinformatics
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
Philippine Council for Agriculture, Aquatic and Natural Resources Research and Development (PCAARRD).
Introduction
The classical swine fever virus (CSFV) causes one of the most significant viral diseases among domestic pigs and wild boars. The severe economic losses attributed to CSFV have led to the World Organization for Animal Health's classification of it as a notifiable disease.1 Controlling CSFV outbreaks typically involves stamping-out methods, vaccination, or a combination of both.2 Two types of CSFV vaccines are available: the classical live-attenuated vaccine,3 and the recently developed E2 subunit vaccine.4 The classical live-attenuated (Chinese strain) vaccine offers robust immunity within a few days that appears to persist lifelong,5 but lacks a serological marker that would allow the DIVA or Differentiation of field virus Infected from Vaccinated Animals.1 In contrast, the E2 subunit vaccine, a marker vaccine, solves the issue of differentiation and provides adequate protection in pigs.6 However, it fails to provide early protection,7 and the immunity conferred by this type of vaccine diminishes over time.5 These shortcomings were attributed to the inefficiency of the E2 protein in stimulating T lymphocyte responses.8 B and T lymphocyte responses are crucial in clearing CSFV, a virus causing non-cytopathic infections.1 The E2 protein is the major antigenic component of the E2 marker vaccine and only effectively induces B lymphocyte response.9 Thus, components that effectively activate the T lymphocyte response must be included in the design to enhance the vaccine's protective efficacy.
NS3 protein is the most potent T lymphocyte antigen.10 It induces lymphoproliferation, IFN-γ production, and effector cytotoxic T lymphocyte (CTL) responses.10 The overall induction of T lymphocyte responses by NS3 was comparable to that of the whole virus, indicating that this nonstructural protein contains immunodominant T lymphocyte epitopes.10 However, the translation of this ability in vivo was not straightforward. A study that added the full-length NS3 to the E2 subunit vaccine protected pigs against lethal CSFV challenge did not significantly improve detectable T or B lymphocyte populations compared to the NS3-lacking vaccine.10 Nevertheless, the study concluded that effectively translating the in vitro antigenic potential of NS3 to in vivo requires an appropriate delivery of NS3 to antigen-presenting cells (APCs) and, subsequently, efficient processing of NS3 peptides for cross-presentation to CD8 + T lymphocytes. Thus, this study aims to improve the immunogenicity of the existing E2 marker vaccine for CSFV by incorporating only the T lymphocyte epitopes from the NS3 protein into the conserved E2 protein of the virus and adapting an mRNA delivery approach.
In this study, the design and evaluation of the vaccine construct were performed in silico using immunoinformatics tools and servers. Tests were conducted to assess the designed vaccine construct's physicochemical properties and immunogenicity potential.
Materials and Methods
The design and evaluation of the vaccine construct for CSFV were conducted exclusively through in silico methodologies. Fig. 1 presents an overview of the study’s methodology.
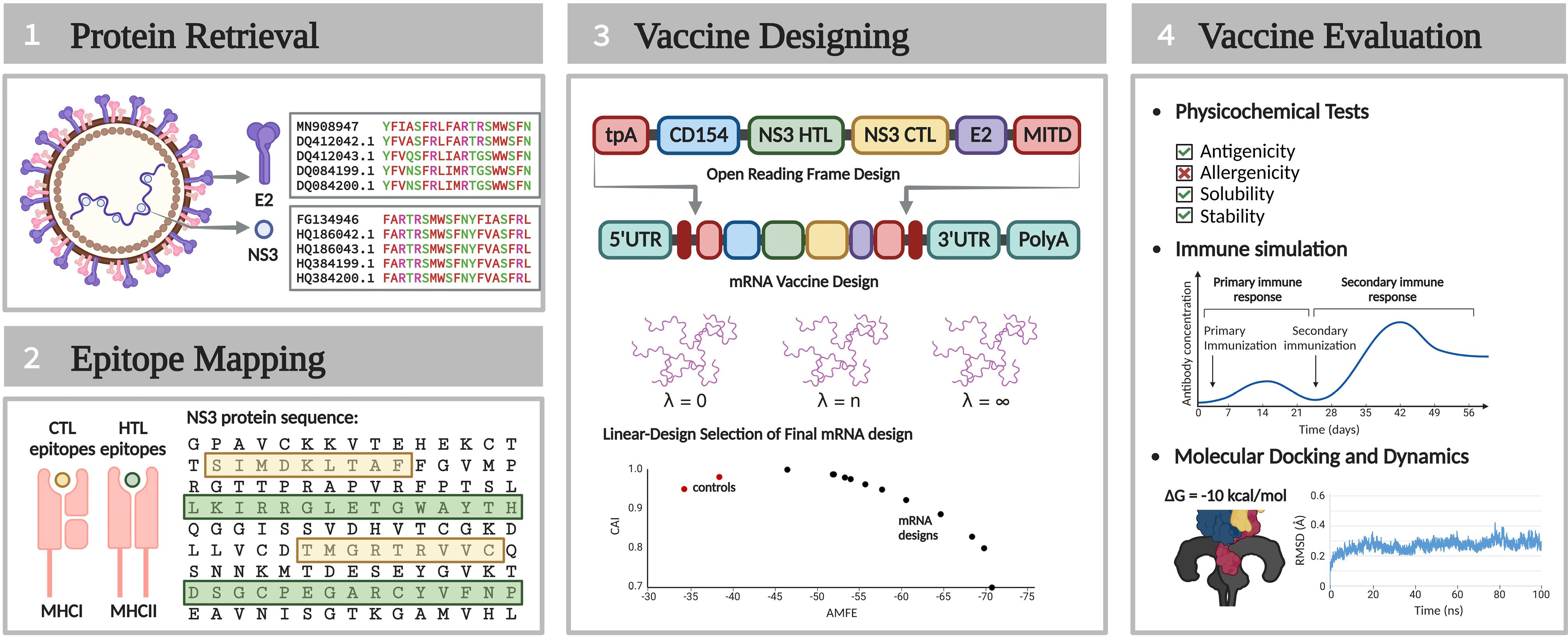
Fig. 1.
Summarized methodology of the study.
.
Summarized methodology of the study.
Protein retrieval
Reference sequences for the E2 and NS3 proteins of the CSFV were obtained from the National Center for Biotechnology Information (NCBI) database11 (https://www.ncbi.nlm.nih.gov/) in May 2024. Reference sequences are well-characterized and curated representations of protein sequences,12 hence were used as baselines for analyzing variability. The BLASTP suite13 (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to align the reference sequences against their variants in the database. The alignment was restricted to sequences derived from CSFV, ensuring a query coverage of > 90% while excluding models and uncultured or environmental sample sequences. The resulting alignment files were analyzed using the Protein Variability Server (PVS)14 (http://imed.med.ucm.es/PVS/). From this analysis, the consensus sequence of the E2 protein was derived. Variable positions within the E2 protein were identified based on Shannon entropy, with the highest variability value observed at H = 4.3. These positions were substituted with the most frequently occurring residues, resulting in a highly conserved E2 consensus sequence. This consensus sequence was incorporated into the vaccine construct to ensure it represents the most prevalent residues, enhancing its broad applicability. Additionally, highly conserved fragments within the NS3 protein were identified in PVS based on the Shannon entropy value observed at H = 1. Only these conserved fragments were used in the NS3 epitope mapping, excluding highly variable residues in the prediction.
Epitope mapping
This study focused on the major histocompatibility complex (MHC) binding to predict potential vaccine epitope components. MHC binding is the most critical characteristic of T lymphocyte epitopes and the most restrictive parameter in epitope prediction.15 This study predicted two T lymphocyte epitopes: cytotoxic T lymphocyte (CTL) and helper T lymphocyte (HTL) epitopes. CTL epitopes bind to Class I MHCs, while HTL epitopes bind to Class II MHCs. This study used swine leukocyte antigen (SLA) for swine-specific MHCs.
Potential HTL epitopes were identified using NetMHCIIpan 4.316 (https://services.healthtech.dtu.dk/services/NetMHCIIpan-4.3/). This server identified 15-mer peptides with high binding affinity to 43 human leukocyte antigen II molecules (HLA; human-specific MHC). HLAs were used because SLAs were not available for the prediction and since these HLAs are also considered homologs of SLAs.17 A percentile rank (PR) cutoff of ≤ 1 was applied to ensure high binding affinity. Further screening of the predicted epitopes was performed using MixMHC2pred18 (http://mixmhc2pred.gfellerlab.org), which integrates predictions of SLA binding affinity with antigen processing and CD4 + T cell recognition and outputs a PR for every epitope-SLA complex. This study screened ten epitopes with the lowest average PR as the best HTL epitope components for the vaccine design.
NetMHCcons 1.119 (https://services.healthtech.dtu.dk/services/NetMHCcons-1.1/) was employed to identify CTL epitopes. This server predicted 9-mer peptides with high binding affinity to standard SLA molecules. A cut-off value of PR ≤ 0.5 or half maximal inhibitory constant (IC50) ≤ 50 was set to ensure the screening of epitopes with high binding affinity to the SLAs. MHCflurry 2.020 was used to screen the predicted epitopes further. This server combines the prediction of affinity to SLAs with antigen processing, including proteasomal cleavage and TAP transport, and outputs a PR for every epitope-SLA complex. This study screened ten epitopes with the lowest average PR as the best CTL epitopes for the vaccine design.
Vaccine designing
Open reading frame design
To construct the vaccine's open reading frame (ORF), HTL epitopes were linked using the GPGPG linker, while CTL epitopes were connected using the AAY linker. The linker HEYGAEALERAG joined the HTL and CTL epitope groups, with this sequence also appended to the N- and C-terminal ends of the HTL-CTL sequence. Six distinct adjuvants, commonly used in swine immunization, were included at the N-terminal end of the HTL-CTL sequence, generating six unique vaccine constructs. These adjuvants included Sus scrofa CD154 (ID: NP_999291.1),21 Clostridium difficile flagellin (FliC) (ID: WP_009888062.1),21 Mycobacterium 50S ribosomal protein L7/L12 (ID: WP_003403353.1),22 bacterial phenol-soluble modulin α4 (ID: WP_014532416.1),23 S. scrofa β-defensin-1 (ID: NP_999607.1),24 and Mycobacterium heparin-binding hemagglutinin adhesin (HBHA) (ID: WP_003402339.1).25 These sequences were downloaded in NCBI and linked to the HTL-CTL sequence's N-terminus using the EAAAK linker.
The vaccine constructs incorporated a 192-amino acid region truncated from the N-terminus of the E2 consensus sequence. This region represented the protein's neutralizing domain.26 The resulting adjuvant-HTL-CTL-E2 polyprotein formed the vaccine's immunogenic core. The physicochemical properties of these immunogenic cores with different adjuvants were analyzed using the ExPASy ProtParam tool27 (https://web.expasy.org/protparam). This server assessed molecular weight, theoretical pI, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) scores. Additional features, such as antigenicity in VaxiJen v2.028 (https://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html), allergenicity in AllerTOP v2.029 (https://www.ddg-pharmfac.net/AllerTOP/feedback.py), and solubility upon overexpression in Escherichia coli using SCRATCH SolPro30 (https://scratch.proteomics.ics.uci.edu) were also evaluated.
Immune response profiles of the six immunogenic cores were simulated using C-Immsim31 (https://kraken.iac.rm.cnr.it/C-IMMSIM/). Three immunizations were administered at time steps 1, 84, and 168 with 1000 particles per dose, and the simulations were run until the 300th time step. Comparative graphs of T and B lymphocyte populations and antibody titers were generated. The final design was chosen as the design exhibiting the most optimal physicochemical properties and immune simulation profile.
Additional sequences were incorporated into the final design to facilitate delivery to appropriate destinations. A tissue plasminogen activator (tPA) signal peptide32 was added to the N-terminus, and an MHC I-trafficking domain (MITD) sequence33 was added to the C-terminus. The tPA peptide was previously incorporated into a vaccine against porcine epidemic diarrhea virus (PEDV), where it enhanced antibody production,34 highlighting its potential use in swine vaccine designs. While the use of MITD in swine vaccines has not been reported, its role in improving antigen presentation is well-documented in other systems, such as murine.33 Combined with tpA, it significantly enhanced the presentation of CTL and HTL epitopes.33 This study's immunogenic core with the translocation peptides (tPA and MITD) constitutes the vaccine's open reading frame (ORF).
Optimization of the vaccine’s mRNA sequence
LinearDesign software35 (https://github.com/LinearDesignSoftware/LinearDesign.git) generated mRNA sequences for the ORF, employing a swine-specific codon usage frequency available in the Codon Statistics Database36 (http://codonstatsdb.unr.edu/). The sequences' codon adaptation indices (CAI) were calculated using DAMBE.37 The human α-globin (hαg) 5'-untranslated region (UTR) and Kozak sequence were added to the N-terminal end of the ORF. The C-terminal region was designed with a double translation termination signal (UGAUGA), an hαg 3' UTR, and a 120-nucleotide poly(A) tail. Hαg UTRs were selected as it has been shown to result in higher protein amounts in different mammalian cellular systems.38,39
The optimal secondary structures of the mRNA sequences and their minimum free energies (MFE) were visualized and calculated using the RNAfold webserver40 (https://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi). Two positive controls to validate the design were positive mRNA sequences from approved COVID-19 vaccines—BioNTech/Pfizer BNT162b2 and Moderna mRNA-1273. Their sequences were obtained from Jeong and colleagues,41 and their CAI and MFE values were also determined. Given the significant difference in the ORF length between the mRNA sequences of the designed vaccine and the controls, adjusted minimum free energies (AMFE)42,43 were calculated, normalizing the MFE values. The mRNA sequence with CAI and AMFE values most comparable to those of the control vaccines was selected as the final ORF sequence for the mRNA vaccine.
Vaccine evaluation
Structure prediction
The tertiary structure of the immunogenic core of the final vaccine design was predicted using the AlphaFold 3 server44 (https://golgi.sandbox.google.com/). The initial structure underwent energy minimization in Chimera 1.17.1,45 employing the steepest descent algorithm over 5000 steps. To enhance structural accuracy, further refinement was conducted using the GalaxyWEB Refine service46 (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE). The quality of the refined structures was evaluated with the PROCHECK tool47 available in PDBsum48 (https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/Generate.html). The overall G-factor scores and the percentage of residues within the most favored regions of the Ramachandran plot were noted. The structure demonstrating the highest quality on these metrics was selected as the representative structure of the immunogenic core.
Molecular docking and dynamics
Molecular docking was conducted to ensure the favorable binding characteristic of the vaccine to its target immune receptor. The identity of the receptor used in the docking analysis depends on the identity of the final adjuvant of the vaccine. Toll-like receptors (TLR) 2 for phenol-soluble modulin α4,49 TLR 4 for 50S L7/L12 ribosomal proteinand HBHA,22,25 TLR 5 for FliC,21 TLR 9 for S. scrofa β-defensin-1,24 and CD40 for CD15421 were considered. The sequence of the final receptor was downloaded from the NCBI, and its structure was modeled using the AlphaFold 3 server. Energy minimization and refinement of the generated structure were also performed using Chimera 1.17.1 and GalaxyWEB Refine service, respectively. The parameters applied were the same as discussed above. The qualities of the refined structures were also assessed using the PROCHECK tool, and the structure with the highest overall G-factor and the highest percentage of residues in the most favored regions of the Ramachandran plot was selected as the representative structure of the receptor.
The adjuvant component was truncated from the immunogenic core’s representative structure using ChimeraX and docked to the receptor's representative structure using ClusPro 2.050 (https://cluspro.bu.edu/login.php). In the docking, an attraction parameter defines specific residues of the receptor to which the ligand (or, in this case, the vaccine adjuvant) is directed to bind. This parameter ensures that the docking focuses on biologically relevant sites rather than allowing the ligand to bind arbitrarily across the receptor surface. To define the attraction parameter, the receptor's structure bound to its natural ligand was initially retrieved in the RCSB Protein Data Bank51 (https://www.rcsb.org/). Residues involved in the binding interaction of the natural ligand were determined using Chimera 1.17.1. Residues with van der Waals (VDW) interaction to the ligand at ≥ -0.40 Å were treated as key residues and were specified as an attraction site in the docking server.
After docking, the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) service of the HawkDock server52 (http://cadd.zju.edu.cn/hawkdock/) was used to determine the overall binding free energy (∆G) of the complex. Moreover, an MD simulation was performed using the Groningen Machine for Chemical Simulations (GROMACS) 2023.253 to assess the stability of the complexes. The force field parameter of the complex was assigned using the Chemistry at Harvard Macromolecular Mechanics (CHARMM) 36 force field.9 The system was prepared by placing the complex within a cubic simulation box with periodic boundary conditions. A minimum distance of 1 Å was maintained between the edges of the complex and the box boundary to avoid interactions between periodic images. The box was then solvated using the TIP3P water model,54 and added with sodium and chloride ions to achieve a concentration of 0.15 M. Energy minimization was performed using 50,000 steps of the steepest descent algorithm, eliminating atomic clashes and optimizing the system’s configuration. Following minimization, the system underwent two equilibration phases. The first phase involved NVT equilibration, during which the temperature was gradually stabilized at 312 K using a modified Berendsen thermostat. The second phase, NPT equilibration, was conducted to stabilize pressure at 1 bar, controlled by a Berendsen barostat. After equilibration, a 200 ns production simulation was carried out with a timestep of 2 fs, generating 100 million simulation steps. Snapshots of the system were saved at 10 ps intervals, producing a trajectory of 20000 frames. Structural properties, including root mean square deviation (RMSD), root mean square fluctuation (RMSF), the radius of gyration (Rg), number of hydrogen bonds, and solvent-accessible surface area (SASA), were computed to characterize the behavior of the complex throughout the simulation.
As a positive control, the receptor structure bound to its natural ligand was also subjected to MM/GBSA and MD simulations, applying the same parameters. The results, including the binding free energy from MM/GBSA and structural properties throughout the MD simulation (RMSD, RMSF, Rg, H-bonds, and SASA), were compared with those of the vaccine construct-receptor complex to assess its binding efficiency.
Immune simulation
The host immune response profile of the immunogenic core of the final vaccine design was compared to the components of the commercialized Tian Wen Jing (TWJ)-E2® CSFV vaccine (positive control). The TWJ-E2® vaccine consists of a CD154-adjuvanted E2 protein from subgenotype 1.1 C-strain.2 The sequences of CD154 (NCBI ID: NP_999291.1) and E2 subgenotype 1.1 C-strain protein (HM175885.1) were retrieved from NCBI. A C-ImmSim immune simulation was performed using these sequences, applying parameters identical to those of the immune simulation of the immunogenic cores of the vaccine design. The graphs of lymphocyte populations and antibody titers induced by this control were generated and overlayed with the final immunogenic core design for comparison.
Results
Protein retrieval
The E2 reference sequence (NP_777498.1) and the NS3 reference sequence (NP_777502.1) were retrieved from NCBI and aligned using BLASTP. Each sequence was compared against variants found in the database, which, in this case, consisted of 99 isolates. Analysis with the PVS revealed 13 highly variable residues in the E2 protein, specifically at positions E24, K31, N34, D36, V49, E72, M165, A168, E192, L200, N212, T197, T250. In contrast, only one highly variable residue, P481, was identified in the NS3 protein.
Epitope mapping
NetMHCIIpan 4.3 identified 65 HTL epitopes from the NS3 protein, each demonstrating a PR ≤ 1 for binding affinity to at least one of the 43 Class II SLAs. From these 65 epitopes, ten with the lowest average PR in MixMHC2pred were selected. The results from MixMHC2pred reflect binding affinity across all Class II SLAs in the database and their potential for antigen processing and CD4 + T cell recognition. Table 1 lists the final ten HTL epitopes, including the Class II SLAs to which they exhibited the highest binding affinity.
Table 1.
HTL epitopes predicted from the CSFV NS3 protein
|
Epitopes
|
Position
|
Best binding Class II SLA(s)
a
|
| CYVFNPEAVNISGTK |
115-129 |
DQA*02-DQB1*04 |
| AVNISGTKGAMVHLQ |
122-136 |
DQA*04-DQB1*04 |
| GLPIFEASSGRVVGR |
164-178 |
DRA*01-DQB1*02 |
| PQPKLRAAMVEYSFI |
304-318 |
DQA*01-DQB1*05 |
| NLRVVAMTATPAGTV |
343-357 |
DRA*01-DRB1*03 |
| KHPIEEFIAPEVMKG |
363-376 |
DRA*02-DRB1*13, DRA*03-DRB1*13, DRA*04-B1*13 |
| SQSPYVVVATNAIES |
442-456 |
DRA*01-DRB1*01, DRA*03-DRB1*01, DRA*01-DRB1*01, DRA*02-DRB1*01, DRA*03-DRB1*01, DRA*04-DRB1*01, DRA*01-DRB1*05, DRA*02-DRB1*05, DRA*03-DRB1*05, DRA*01-DRB1*06, DRA*02-DRB1*06, DRA*03-DRB1*06, DRA*04-DRB1*06, DRA*01-DRB1*12, DRA*02-DRB1*12, DRA*03-DRB1*12, DRA*04-DRB1*12, DRA*02-DRB1*17, DRA*03-DRB1*17, DRA*04-DRB1*17 |
| QSPYVVVATNAIESG |
443-457 |
DRA*01-DRB1*01, DRA*02-DRB1*01, DRA*03-DRB1*01, DRA*04-DRB1*01, DRA*03-DRB1*05, DRA*01-DRB1*06, DRA*02-DRB1*06, DRA*03-DRB1*06, DRA*04-DRB1*06, DRA*01-DRB1*12, DRA*02-DRB1*12, DRA*03-DRB1*12, DRA*04-DRB1*12, DRA*03-DRB1*17 |
| YDNYTFLNARKLGDD |
626-640 |
DRA*01-DRB1*02, DRA*02-DRB1*02, DRA*03-DRB1*02, DRA*04-DRB1*02 |
| QGTVEAGRALKQVVG |
668-682 |
DQA*01-DRB1*05 |
a SLA notation (e.g., DQA*02-DQB1*04) specifies heterodimeric MHC molecules, where DQA/DRA and DQB1/DRB1 denote the alpha and beta chains, respectively, followed by specific identifiers or variants (*XX).
Identifying SLAs with strong epitope binding is essential for assessing epitope coverage. Notably, three epitopes demonstrated high binding affinity to the Class II SLA DRA*01-DRB1*01 and DRA*03-DRB1*01, suggesting that populations with a higher prevalence of these antigens may respond more effectively to the vaccine. Although data on the prevalence of DRA genes in swine populations remain unavailable, DRB1*01 is reported in approximately 10% of European-farmed pigs and 7% of pigs in Thailand.17,55 Moreover, none of the ten HTL epitopes contained highly variable residues, ensuring their potential as conserved vaccine components.
NetMHCcons 1.1 identified 57 CTL epitopes from the NS3 protein, each demonstrating a PR ≤ 1 or IC50 ≤ 50 for binding affinity to all Class I SLAs with standard designations. From these 57 epitopes, ten with the lowest average PR in MHCflurry 2.0 were selected. The results from the MHCflurry reflect strong binding affinity across all Class I SLA molecules and efficient proteasomal cleavage and TAP transport, ensuring that the predicted epitopes are effectively processed and presented to T lymphocytes. Table 2 lists the final ten CTL epitopes, including the Class I SLAs to which they exhibited the highest binding affinity. None of the 10 CTL epitopes contained highly variable residues, ensuring their potential as conserved vaccine components.
Table 2.
CTL epitopes predicted from the CSFV NS3 protein
|
Epitopes
|
Position
|
Best binding Class I SLA(s)a
|
| SIMDKLTAF |
17-25 |
1*16:01 |
| VTASGTPAF |
145-153 |
1*16:01 |
| KLMSGIQTV |
191-199 |
2*10:06 |
| AAAESVYQY |
258-266 |
1*16:01 |
| AMVEYSFIF |
311-319 |
1*16:01 |
| AIMGKIHRF |
332-340 |
1*16:01 |
| SENLRVVAM |
341-349 |
2*06:07 |
| SEYLDIAGL |
382-390 |
2*06:07 |
| REMNYDWSL |
551-559 |
2*06:07 |
| TQLEILNNL |
568-576 |
2*06:07 |
a SLA notation (e.g., 1*16:01) represents SLA loci (1 or 2) followed by specific identifiers or variants (XX*XX).
The 10 CTL epitopes exhibited strong binding affinity to three types of Class I SLAs: 1*16:01, 2*06:07, and 2*10:06. While SLA 1*16:XX is relatively rare, with a prevalence of only > 1% in European farmed pigs and no reported occurrence in Thailand, SLA 2*06:XX and 2*10:XX are more prominent, with ~2-10% and 8-15% occurrences in European and Thailand pigs, respectively.17,55
Vaccine designing
Open reading frame design
Six vaccine constructs were designed by linking the NS3 epitopes to the conserved E2 protein using peptide linkers and adding six adjuvants. These constructs were subsequently evaluated on their physicochemical properties and immune response profile (Fig. 2). All constructs successfully passed the antigenicity, allergenicity, and stability assessments, with their immune simulation graphs indicating almost similar profiles on all parameters. Therefore, the final vaccine design selection was prioritized based on the antigenicity score, where the construct with CD154 adjuvant exhibited the highest score (0.62), making it the most promising candidate. Other relevant physicochemical properties of the final design with CD154 adjuvant are listed in Table 3. The vaccine had a 684-residue immunogenic core, and the addition of the tPA signal peptide and the MITD sequence extended the construct to a 764-residue ORF.
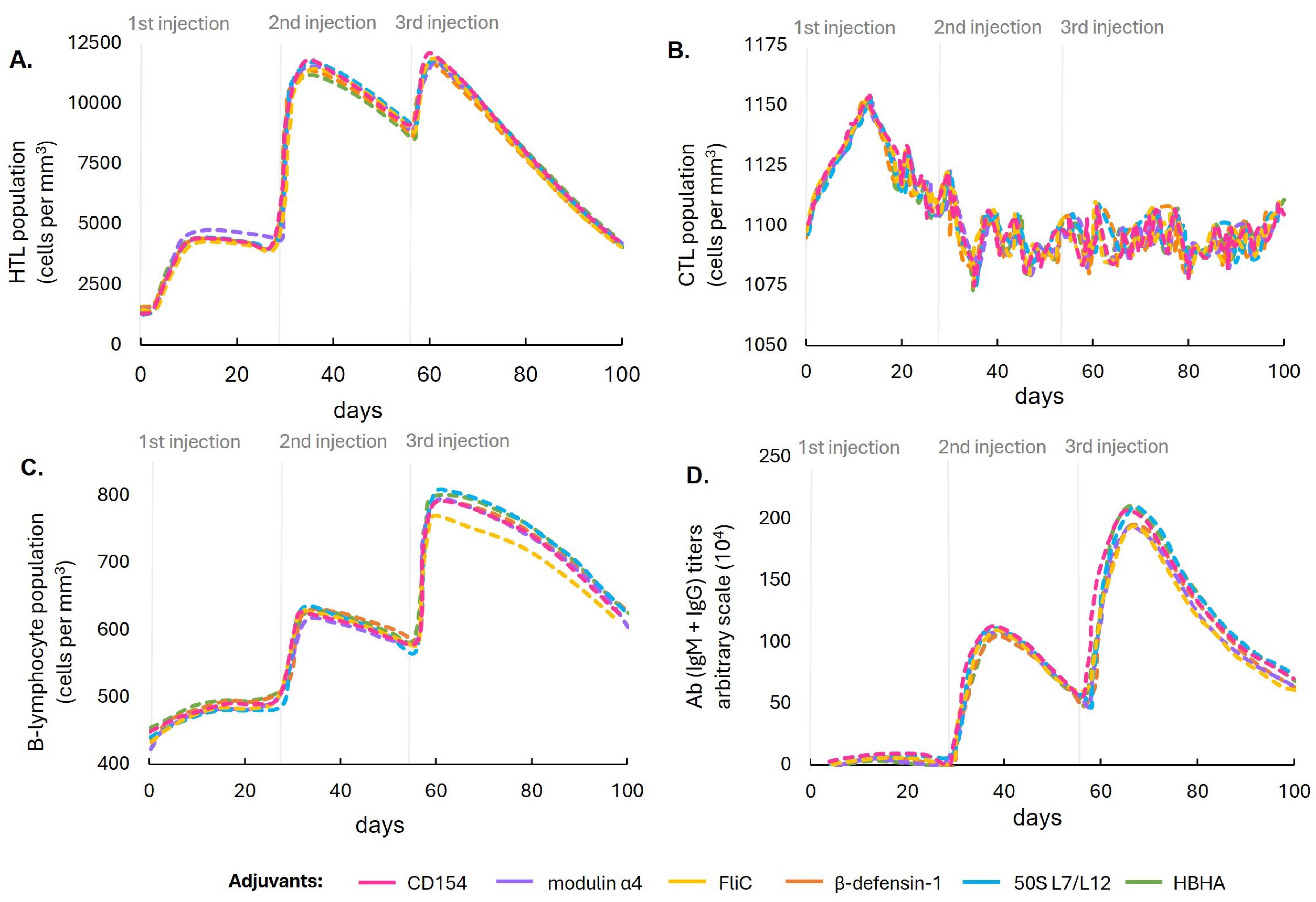
Fig. 2.
Computational simulation comparing the populations of (A) HTLs, (B) CTLs, (C) B lymphocytes, and (D) antibody titers in response to immunizations with the six designed vaccine constructs with different adjuvant formulations.
.
Computational simulation comparing the populations of (A) HTLs, (B) CTLs, (C) B lymphocytes, and (D) antibody titers in response to immunizations with the six designed vaccine constructs with different adjuvant formulations.
Table 3.
Other physicochemical properties of the designed CD154-adjuvanted vaccine for CSFV
|
Properties
|
Results
|
| Molecular weight |
72 336.09 g/mol |
| Theoretical pI |
pH 6.12 |
| Estimated half-life (mammalian reticulocytes, in vitro) |
5.5 hours |
| Instability index (stability) |
36.62 (stable) |
| Aliphatic index |
77.68 |
| Grand average of hydropathicity |
-0.077 |
Optimization of the vaccine’s mRNA sequence
The mRNA sequences of the ORF were generated using LinearDesign, applying a range of codon bias values (λ), from the most optimal codons (λ = ∞) to the least optimal (λ = 0), along with intermediate values (λ = 1–10). The CAIs of these sequences were calculated, and after incorporating UTRs, initiation and termination signals, and a poly(A) tail, the AMFEs were determined. Fig. 3 illustrates the relationship between CAI and AMFE across these mRNA sequences. A negative correlation was observed, wherein sequences with higher CAI values—indicating greater codon optimality—demonstrated lower AMFE values, reflecting reduced structural stability (Fig. 3A).
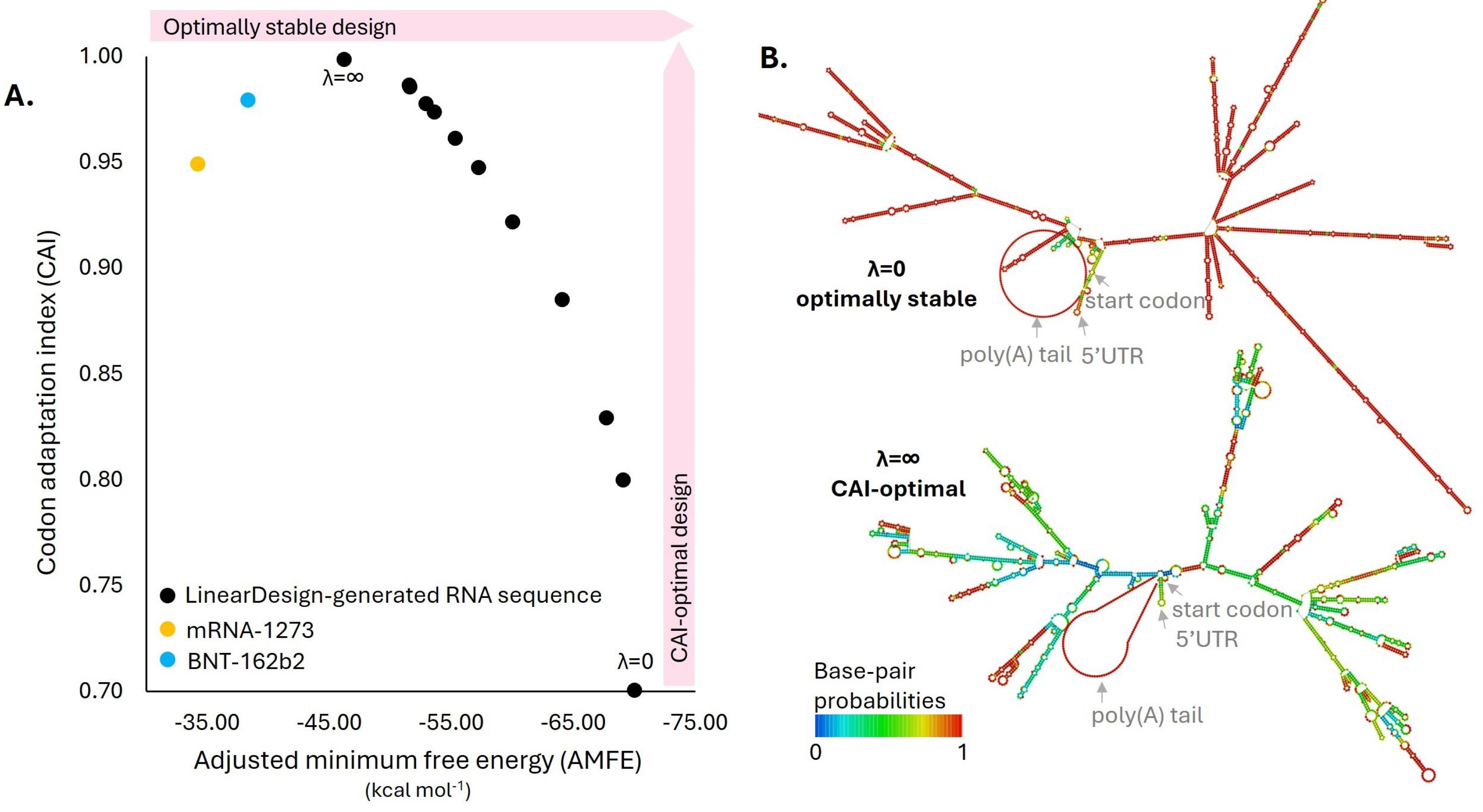
Fig. 3.
Optimization of the mRNA sequence for the CSFV vaccine design using LinearDesign. (A) A two-dimensional scatter plot illustrating the stability and codon optimality of the mRNA sequences generated for the CSFV vaccine. Stability is represented by adjusted minimum free energy (AMFE) values on the x-axis. At the same time, codon adaptation index (CAI) scores, indicative of codon usage bias with the swine host, are plotted on the y-axis. (B) Secondary structure representations of the mRNA designs were identified as optimally stable and CAI-optimal. These structures highlight regions with high base-pairing probabilities, emphasizing areas contributing to the stability.
.
Optimization of the mRNA sequence for the CSFV vaccine design using LinearDesign. (A) A two-dimensional scatter plot illustrating the stability and codon optimality of the mRNA sequences generated for the CSFV vaccine. Stability is represented by adjusted minimum free energy (AMFE) values on the x-axis. At the same time, codon adaptation index (CAI) scores, indicative of codon usage bias with the swine host, are plotted on the y-axis. (B) Secondary structure representations of the mRNA designs were identified as optimally stable and CAI-optimal. These structures highlight regions with high base-pairing probabilities, emphasizing areas contributing to the stability.
This study used two approved COVID-19 mRNA vaccines, the BNT-162b2 and mRNA-1273, as positive controls. As shown in Fig. 3A, the mRNA sequences with λ > 8 exhibited higher CAI values than those of the two controls, indicating better codon optimality. Notably, the sequence with λ = ∞, which had the lowest MFE among the LinearDesign-generated sequences, still had a higher MFE value than the control. This was selected as the final sequence for the CSFV mRNA vaccine due to its superior codon optimality and greater stability than the controls.
Vaccine evaluation
Structure prediction
From the AlphaFold-predicted and Chimera-energy-minimized tertiary structures of the immunogenic core, five refined models were generated using GalaxyRefine. All refined models have z-scores within the plot containing z-scores of all experimentally determined protein groups of similar size. However, among these refined models, Refined model 2 was selected as the representative structure of the vaccine construct. This selection was based on its overall G-factor and percentage of residues in the favored regions of the Ramachandran plot, which is the highest among the refined structures. The G-factor assesses stereochemical properties, with higher values indicating a more favorable structure,47 The percentage of residues in favored regions of the Ramachandran plot reflects proper backbone dihedral angles, which are critical for stable and accurate protein folding.47 Choosing the structure with the highest scores on these metrics ensures that the structure for subsequent docking and simulation analyses represents the closest form to the immunogenic core’s native conformation. Table 4 displays the scores of the refined structures in the quality assessment servers. Fig. 4 shows the tertiary structure model of the representative structure of the immunogenic core.
Table 4.
Scores of the raw, energy-minimized, and refined structures of the designed immunogenic core of the CD154-adjuvanted vaccine in tertiary structure quality assessment servers
|
Models
|
ProSA (Z-score)
|
PROCHECK (Ramachandran)
|
PROCHECK (G-Factor)
|
| Raw from Alphafold |
-6.47 |
79.5 |
-0.32 |
| Energy minimized by Chimera |
-6.65 |
84.8 |
-0.27 |
| Refined model 1 by GalaxyRefine |
-7.00 |
91.8 |
0.08 |
| Refined model 2 by GalaxyRefine |
-6.96 |
93.4 |
0.08 |
| Refined model 3 by GalaxyRefine |
-7.00 |
93.0 |
0.07 |
| Refined model 4 by GalaxyRefine |
-7.04 |
92.5 |
0.03 |
| Refined model 5 by GalaxyRefine |
-6.83 |
93.0 |
0.08 |
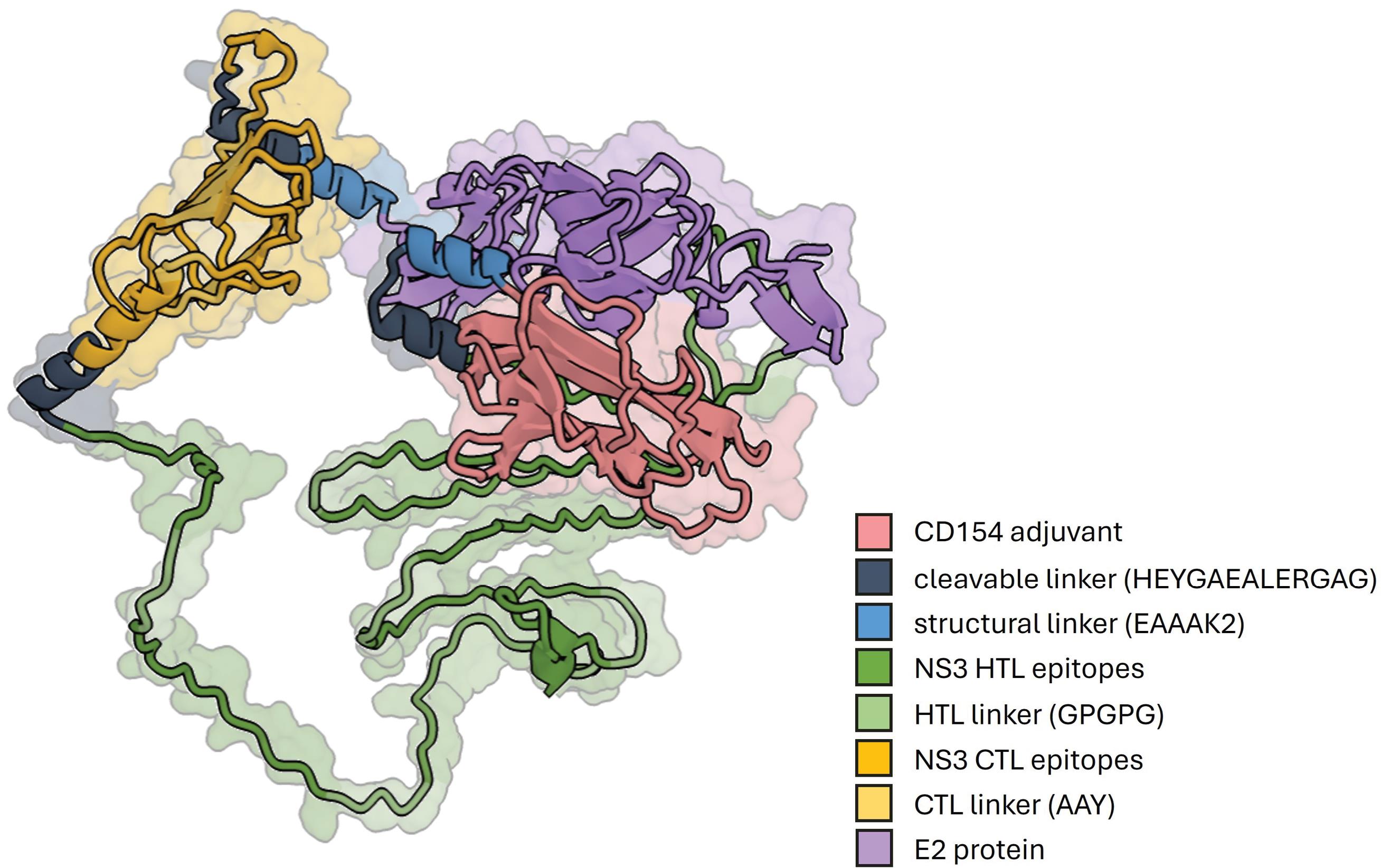
Fig. 4.
The tertiary structure of the designed immunogenic core of the CD154-adjuvanted vaccine construct. The construct consisted of four primary domains: the CD154 adjuvant, NS3 CTL epitopes, NS3 HTL epitopes, and the E2 protein. GPGPG linkers joined the HTL epitopes, while the CTL epitopes were joined using AAY linkers. The cleavable HEYGAEALERAG linker joined the CTL and HTL domains. The structural EAAAK2 linker is used to space the CD154 adjuvant, the combined NS3 CTL-HTL epitopes, and the E2 protein.
.
The tertiary structure of the designed immunogenic core of the CD154-adjuvanted vaccine construct. The construct consisted of four primary domains: the CD154 adjuvant, NS3 CTL epitopes, NS3 HTL epitopes, and the E2 protein. GPGPG linkers joined the HTL epitopes, while the CTL epitopes were joined using AAY linkers. The cleavable HEYGAEALERAG linker joined the CTL and HTL domains. The structural EAAAK2 linker is used to space the CD154 adjuvant, the combined NS3 CTL-HTL epitopes, and the E2 protein.
Molecular docking and dynamics
The immunogenic core of the vaccine construct is expected to be cleaved intracellularly by proteasomes at the HEYGAEALERAG linker, resulting in the formation of four structures: tPA-CD154, NS3 HTL epitopes, NS3 CTL epitopes, and the E2 protein. The tPA-CD154 is expected to be expressed extracellularly, enabling it to interact with the swine CD40 structure, its natural receptor.
CD154 has demonstrated efficient binding to CD40 in vitro and was already documented in the crystal structure available in the RCSB PDB (ID: 3QD6).56 However, this structure is from humans, and no corresponding structure for swine is available. In this study, swine CD154, the adjuvant component of the vaccine construct, was docked to swine CD40. The binding affinity and structural properties of the swine CD154-CD40 complex were then compared to the human CD154-CD40 complex using MM/GBSA and dynamic simulations (See Fig. 5).
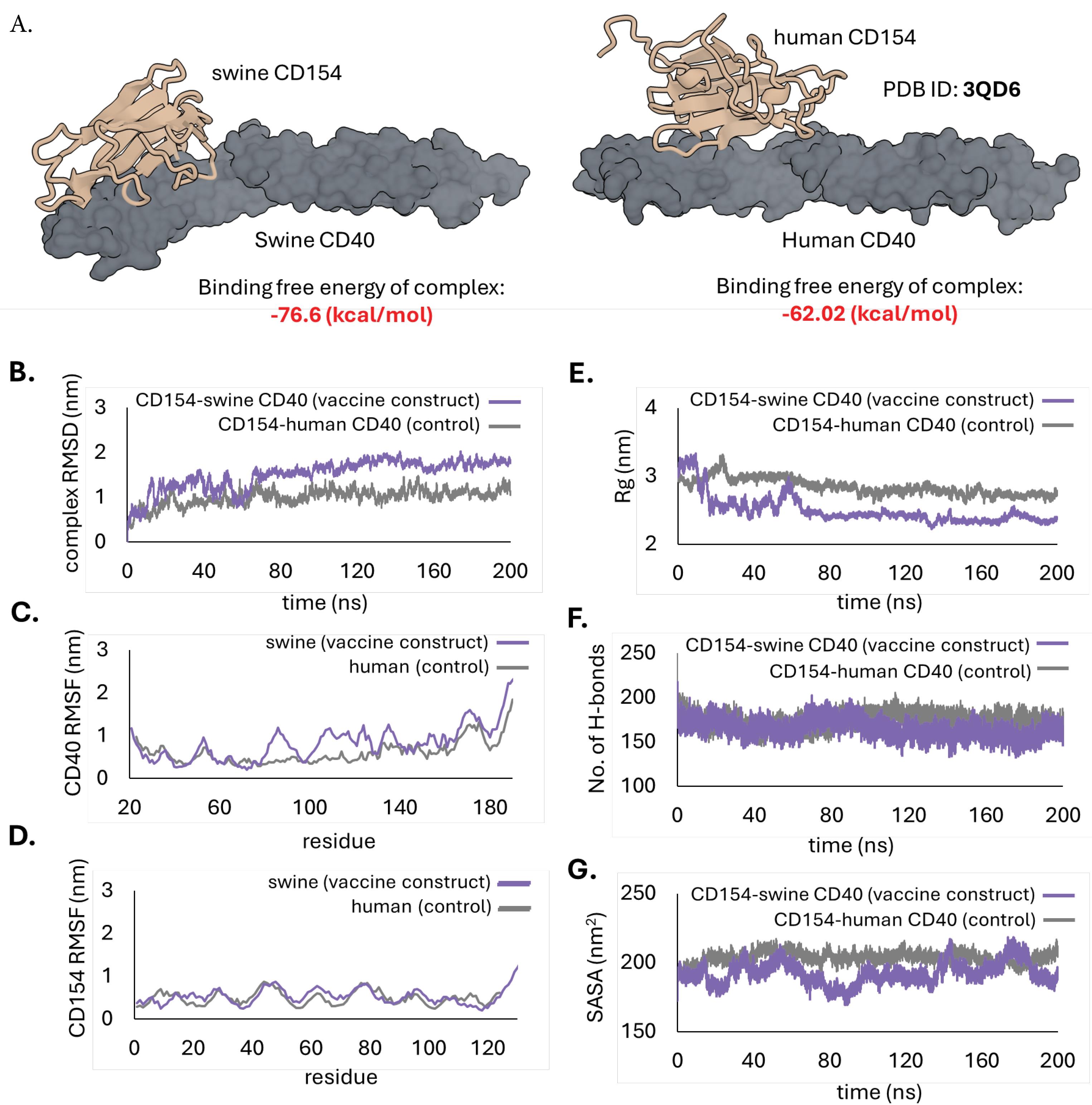
Fig. 5.
Docking of the CD154 to CD40 in swine and humans. The swine CD40 (left) served as the receptor for CD154 when used as an adjuvant in swine immunizations, while the human CD40 (right) was used as the control (from PDB ID: 3QD6). (A) Model of the docked complexes and their estimated binding free energies determined using MM/GBSA. (B) Root mean square deviations (RMSD) graph, (C) Root mean square fluctuations (RMSF) of the CD40 receptor, (D) RMSF of the CD154 ligand, (E) Radius of gyration (Rg) graph, (F) number of hydrogen bonds, and (G) Solvent accessible surface area (SASA) plot of the complexes following 200 ns MD simulations.
.
Docking of the CD154 to CD40 in swine and humans. The swine CD40 (left) served as the receptor for CD154 when used as an adjuvant in swine immunizations, while the human CD40 (right) was used as the control (from PDB ID: 3QD6). (A) Model of the docked complexes and their estimated binding free energies determined using MM/GBSA. (B) Root mean square deviations (RMSD) graph, (C) Root mean square fluctuations (RMSF) of the CD40 receptor, (D) RMSF of the CD154 ligand, (E) Radius of gyration (Rg) graph, (F) number of hydrogen bonds, and (G) Solvent accessible surface area (SASA) plot of the complexes following 200 ns MD simulations.
The overall binding free energy of the swine CD154-CD40 complex was -76.6 kcal/mol. This is relatively lower than the control at -62.02 kcal/mol, suggesting CD154 may have a better binding affinity to CD40 in swine than humans. A low (negative) binding energy indicates the interaction is energetically favored, hence the greater possibility of binding.57
The MD simulation of the swine CD154-CD40 complex demonstrated stability throughout the 200 ns simulation. The RMSD trajectory reached a steady state at approximately 70 ns and remained constant for the remaining time of the simulation (Fig. 5B). However, a deviation of ~1.5 nm from its original pose was recorded, slightly higher than that of the human complex (~1.0 nm). The RMSF analysis revealed no significant difference in the fluctuation of swine CD154 compared to human CD154. Six regions of the swine CD40 (Q79-G95, T99-L129, K132-T141, P147-P163, and S166-D183) showed higher fluctuations compared to human CD40, but these are loops outside the key interaction residues, which are irrelevant to the binding. The Rg graph indicated that the swine CD154-CD40 complex initially had a larger Rg than the human complex but eventually declined after ~15 ns. Low Rg suggests a more stable, compact, and well-packed structure. Meanwhile, the number of hydrogen bonds of the swine CD154-CD40 complex remained constant throughout the simulation, similar to the human complex. The SASA plot also showed lower values for the swine CD154-CD40 complex, indicating an energetically favorable interface compared to the human complex. Overall, the results of the MM/GBSA and MD simulations suggest that the swine CD154-CD40 interaction was stable and had favorable binding characteristics that were better than the human complex (which already has an established binding efficiency). These results support the potential use of swine CD40 as an effective component in vaccine designs for swine populations.
Immune simulation
Immune simulations were performed to compare the potential of the designed vaccine to elicit T and B lymphocytes, as well as antibody responses relative to the components of the E2 CSFV vaccine (positive control) (Fig. 6). The designed vaccine showed higher levels of HTLs, B lymphocytes, and antibodies relative to the control only after the second and third injections. Moreover, it exhibited a relatively higher CTL population following the first injection. While this CTL response gradually decreased, it was effectively maintained after subsequent doses, unlike the control vaccine, which showed a continuous decline in CTL levels. The study achieved its primary objective of initially enhancing the T lymphocyte population, particularly CTLs, after the first immunization and sustaining it over time.
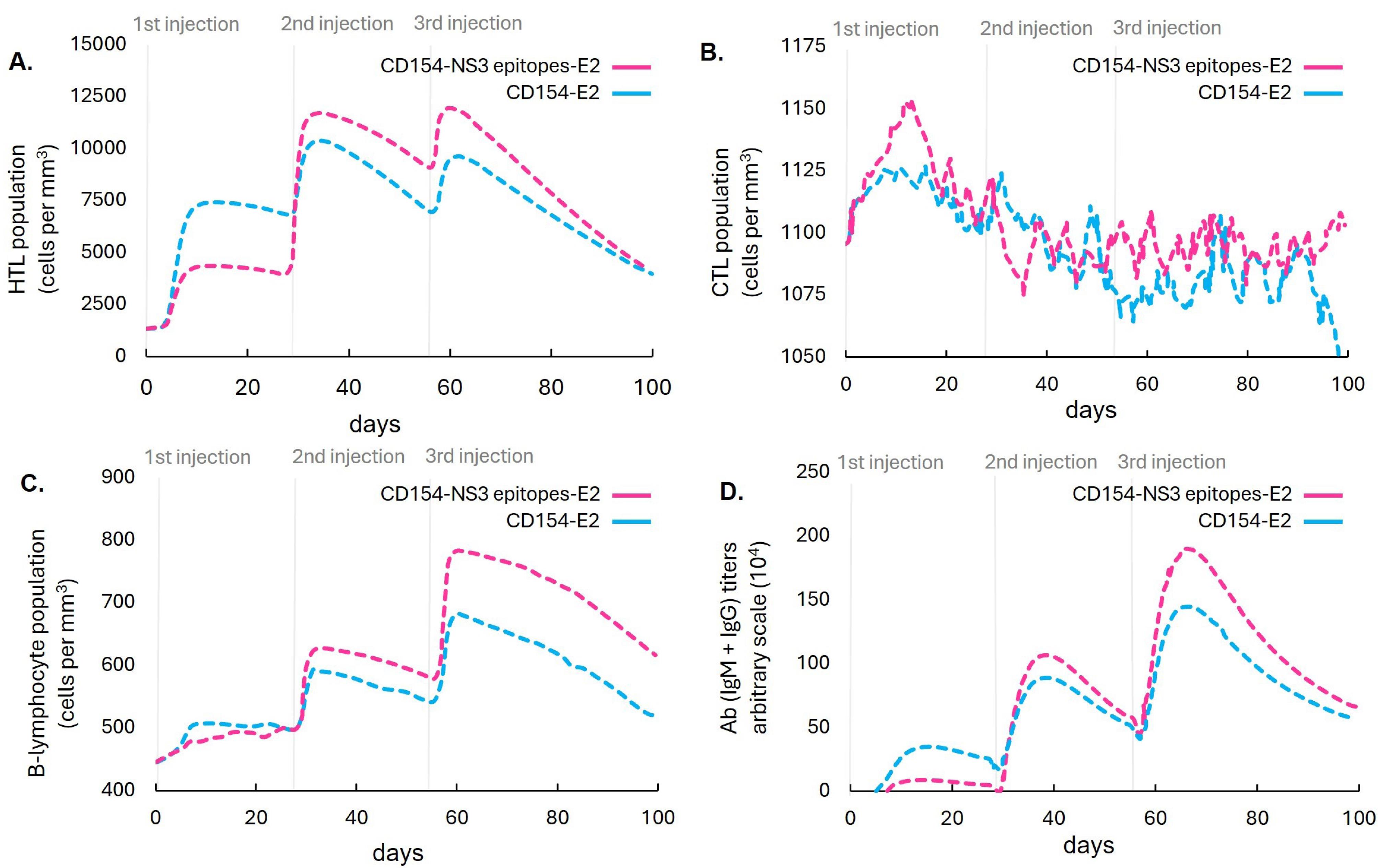
Fig. 6.
Computational simulation comparing the populations of (A) HTLs, (B) CTLs, (C) B lymphocytes, and (D) antibody titers in response to immunizations with the designed vaccine and the components of the E2 marker vaccine in a mammalian host.
.
Computational simulation comparing the populations of (A) HTLs, (B) CTLs, (C) B lymphocytes, and (D) antibody titers in response to immunizations with the designed vaccine and the components of the E2 marker vaccine in a mammalian host.
Discussion
This study aimed to improve the immunogenicity of the existing E2 marker vaccine for CSFV, potentially addressing its limitation in stimulating T lymphocyte responses.8 The first modification introduced was using an mRNA delivery approach to direct the translation of the immunogenic core to the cytoplasm. mRNA delivery has demonstrated significant success in eliciting robust immune responses, as evidenced by its application in numerous COVID-19 vaccines.58-60 mRNA delivery introduces genetic instructions directly into host cells, enabling the in situ production of antigens. This approach is more effective than the exogenous antigens utilized by protein subunit vaccines, as it more efficiently engages MHC I molecules,61,62 which are critical for activating CD8 + T lymphocytes and inducing a cytotoxic immune response. As a result, mRNA vaccines are known to induce more potent CTL responses than protein-subunit vaccines.63,64
The second modification introduced was using conserved viral protein sequences instead of the subgenotype 1.1 sequence used in E2-CD154 vaccines.2 In addition to subgenotype 1.1, this study considered other subgenotypes available in the NCBI database, such as 1.3, 1.4, 2.1, and 2.5. Notably, genotype 2 strains are genetically distant from the current vaccine strain,65 raising concerns about the efficacy of existing vaccines against these divergent strains. By including genotype 2 strains in the vaccine formulation, the study sought to design a vaccine with broader immunogenic coverage, potentially enhancing the T lymphocyte response and the overall immunity conferred by the vaccine.
The third modification introduced was the addition of T lymphocyte-inducing epitopes from the NS3 protein to the vaccine. Epitope components are specifically designed to include only the key regions necessary for inducing an immune response. In comparison to full-length protein, this focused approach ensures that only the most relevant peptides for cross-presentation to CD8 + T lymphocytes are included, improving antigen processing and presentation efficiency.66
Despite the ability of the CSFV NS3 protein to elicit T lymphocyte responses,10 no study has identified the specific epitopes responsible for this effect. This study represents the first effort to map these epitopes using immunoinformatics. Immunoinformatics accelerates vaccine development by leveraging computational tools to predict and analyze potential epitopes and vaccine constructs before laboratory experiments, streamlining the experimental phase and focusing resources on the most promising leads. Immunoinformatics has already demonstrated remarkable success in developing vaccines against numerous pathogens, including Neisseria meningitidis serogroup B,67 and antibiotic-resistant Staphylococcus aureus and Streptococcus pneumonia, to name a few. This study incorporated insights from multiple benchmark analyses conducted across diverse server platforms to optimize the selection of immunoinformatics tools for specific applications and reduce the risk of pursuing suboptimal vaccine candidates.68,69
This study identified ten epitopes capable of inducing CTL responses and ten capable of inducing HTL responses from the CSFV NS3 protein. The predicted epitopes were selected based on their strong binding affinity to swine major histocompatibility complexes, applying strict thresholds to ensure accuracy. These epitopes must still be tested in vitro to confirm their ability to induce specific T lymphocyte responses.
Six vaccine constructs were designed by combining the predicted epitopes with the E2 protein and six adjuvants. The vaccine construct with CD154 adjuvant had the most favorable physicochemical properties, specifically antigenicity. Moreover, the immune simulation revealed a relative increase in CTL, HTL, B lymphocyte, and antibody populations of the designed vaccine compared to the components of the E2 subunit vaccine design. The increased B lymphocyte population and antibody titer observed were likely due to additional B lymphocyte epitopes within the added NS3 sequences. However, further in vitro tests are also required to verify this finding. Moreover, the relatively higher CTL population following the first injection and maintained CTL population following subsequent injections is evidence of the potential ability of the designed construct to address the delayed and diminishing immunity associated with the original vaccine. However, it is essential to emphasize that the immune simulation analysis conducted by the study is limited to the protein component of the E2 vaccine and does not encompass the complete formulation of the vaccine (e.g., delivery systems). Additional components may significantly influence the vaccine's efficacy and were not accounted for by the analysis.
CD154, acting as an adjuvant in the vaccine construct, demonstrated a stable interaction with swine CD40. This is the first study to elucidate this potential binding using molecular docking and dynamics simulations. Successful interactions between CD154 and CD40 can mediate CD4 + T lymphocyte priming by dendritic cells (DCs), subsequently aiding CD4 + T lymphocyte assistance to B lymphocytes and classical macrophage activation.70 CD154 effectively binds to human CD40.56 However, in this study, verifying whether CD154 would also effectively bind to swine CD40 is crucial because the vaccine's intended host is swine. This study provided evidence supporting this effective binding, reinforcing the potential of CD154 as an effective adjuvant for vaccines used in swine immunizations.
This study also generated a codon-optimized mRNA sequence for the vaccine construct using the LinearDesign tool. LinearDesign has successfully improved mRNA vaccines' protein expression and half-life against SARS-CoV-2 and varicella-zoster viruses.35 This tool integrates CAI and folding dynamics considerations in the mRNA vaccine design. In this study, the LinearDesign-generated mRNA sequence of the ORF exhibited a higher CAI and AMFE compared to two licensed mRNA vaccines against SARS-CoV2 (BioNTech/Pfizer BNT162b2 and Moderna mRNA-1273). These results indicate that the generated mRNA vaccine can match the performance of the established vaccines in terms of stability and translation efficiency, thereby making it a promising candidate for further development and potential clinical use.
Despite the promise of the methodology used in this study, it is crucial to acknowledge the limitations of in silico approaches to vaccine development, specifically their predictive limitations. The study's methods relied heavily on existing data and assumptions that may not always capture the full complexity of biological systems. Computational models may not fully account for the intricate interactions and environmental factors in living organisms, potentially leading to oversimplified conclusions. Moreover, some servers used in this study relied on generalized mammalian models as hosts, which do not specifically account for the unique aspects of swine immunology. This limitation could affect the precision of the predictions regarding immune responses and vaccine efficacy in swine, potentially overlooking species-specific factors that influence the performance of the designed vaccine construct.
To advance future research, it is essential to customize immunoinformatics tools and databases to align with the unique immunological characteristics of target animal species. Such tailored approaches can significantly enhance the precision and effectiveness of veterinary vaccine design. Given that computational models in immunoinformatics are based on evolving scientific knowledge, these models must be periodically updated or re-evaluated to ensure their continued accuracy and applicability.
The mRNA vaccine framework presented in this study serves as an initial design for validation experiments. Should the vaccine candidate fail in these tests, alternative strategies can be pursued. One approach is to revisit the pool of epitopes that were initially shortlisted but excluded due to stringent selection thresholds. These previously omitted epitopes may hold the potential for improving vaccine efficacy. Similarly, alternative mRNA sequences and design parameters can be explored. For instance, while the current design employed a threshold of λ = ∞, experimenting with thresholds such as λ > 8 might improve mRNA stability and expression.
Furthermore, a systematic re-evaluation of structural elements, including UTRs and poly(A) tails, is critical. Testing various combinations and modifications of these components can optimize vaccine design and enhance performance. These iterative refinements in epitope selection and mRNA structural elements will facilitate the development of improved vaccine candidates, ultimately guiding the formulation chosen for further development and eventual deployment.
Conclusion
Overall, this study successfully mapped epitopes from the CSFV NS3 protein using immunoinformatics and designed and evaluated a vaccine construct against the pathogen. This provides a comprehensive methodological blueprint for using immunoinformatics tools to map epitopes, design vaccine constructs, and evaluate their potential efficacy, which future researchers can adapt and refine for different pathogens and host species.
Research Highlights
What is the current knowledge?
-
Current E2 marker vaccine for CSFV had delayed and diminishing immunity.
-
Improving T lymphocyte response can address the limitations of the CSFV E2 vaccine.
-
Epitope-based mRNA vaccines boost antigen presentation and T lymphocyte response.
-
Immunoinformatics can streamline the mRNA vaccine development process.
What is new here?
This vaccine is a promising candidate for further development and potential clinical use.
Data Availability Statement
The raw data is available from the corresponding author (Fredmoore L. Orosco) on reasonable request.
Competing Interests
None declared.
Ethical Approval
Not applicable. This study did not involve human participants or animal subjects.
Acknowledgements
To the Department of Science and Technology - Advanced Science and Technology Institute (DOST-ASTI), Philippines for their high-performance computing units used to run the MD analyses.
References
- Blome S, Staubach C, Henke J, Carlson J, Beer M. Classical swine fever-an updated review. Viruses 2017; 9:86. doi: 10.3390/v9040086 [Crossref] [ Google Scholar]
- Coronado L, Perera CL, Rios L, Frías MT, Pérez LJ. A critical review about different vaccines against classical swine fever virus and their repercussions in endemic regions. Vaccines (Basel) 2021; 9:154. doi: 10.3390/vaccines9020154 [Crossref] [ Google Scholar]
- Zhao JJ, Cheng D, Li N, Sun Y, Shi Z, Zhu QH. Evaluation of a multiplex real-time RT-PCR for quantitative and differential detection of wild-type viruses and C-strain vaccine of classical swine fever virus. Vet Microbiol 2008; 126:1-10. doi: 10.1016/j.vetmic.2007.04.046 [Crossref] [ Google Scholar]
- Gong W, Li J, Wang Z, Sun J, Mi S, Xu J. Commercial E2 subunit vaccine provides full protection to pigs against lethal challenge with 4 strains of classical swine fever virus genotype 2. Vet Microbiol 2019; 237:108403. doi: 10.1016/j.vetmic.2019.108403 [Crossref] [ Google Scholar]
- van Oirschot JT. Vaccinology of classical swine fever: from lab to field. Vet Microbiol 2003; 96:367-84. doi: 10.1016/j.vetmic.2003.09.008 [Crossref] [ Google Scholar]
- Li F, Li B, Niu X, Chen W, Li Y, Wu K. The development of classical swine fever marker vaccines in recent years. Vaccines (Basel) 2022; 10:603. doi: 10.3390/vaccines10040603 [Crossref] [ Google Scholar]
- Beer M, Reimann I, Hoffmann B, Depner K. Novel marker vaccines against classical swine fever. Vaccine 2007; 25:5665-70. doi: 10.1016/j.vaccine.2006.12.036 [Crossref] [ Google Scholar]
- Sordo Y, Suárez M, Caraballo R, Sardina T, Brown E, Duarte C. Humoral and cellular immune response in mice induced by the classical swine fever virus E2 protein fused to the porcine CD154 antigen. Biologicals 2018; 52:67-71. doi: 10.1016/j.biologicals.2017.12.004 [Crossref] [ Google Scholar]
- Huang YL, Deng MC, Wang FI, Huang CC, Chang CY. The challenges of classical swine fever control: modified live and E2 subunit vaccines. Virus Res 2014; 179:1-11. doi: 10.1016/j.virusres.2013.10.025 [Crossref] [ Google Scholar]
- Rau H, Revets H, Balmelli C, McCullough KC, Summerfield A. Immunological properties of recombinant classical swine fever virus NS3 protein in vitro and in vivo. Vet Res 2006; 37:155-68. doi: 10.1051/vetres:2005049 [Crossref] [ Google Scholar]
- Sayers EW, Bolton EE, Brister JR, Canese K, Chan J, Comeau DC. Database resources of the national center for biotechnology information. Nucleic Acids Res 2022; 50:D20-d6. doi: 10.1093/nar/gkab1112 [Crossref] [ Google Scholar]
- O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 2016; 44:D733-45. doi: 10.1093/nar/gkv1189 [Crossref] [ Google Scholar]
- Boratyn GM, Camacho C, Cooper PS, Coulouris G, Fong A, Ma N. BLAST: a more efficient report with usability improvements. Nucleic Acids Res 2013; 41:W29-33. doi: 10.1093/nar/gkt282 [Crossref] [ Google Scholar]
- Garcia-Boronat M, Diez-Rivero CM, Reinherz EL, Reche PA. PVS: a web server for protein sequence variability analysis tuned to facilitate conserved epitope discovery. Nucleic Acids Res 2008; 36:W35-41. doi: 10.1093/nar/gkn211 [Crossref] [ Google Scholar]
- Stranzl T, Larsen MV, Lundegaard C, Nielsen M. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics 2010; 62:357-68. doi: 10.1007/s00251-010-0441-4 [Crossref] [ Google Scholar]
- Nilsson JB, Kaabinejadian S, Yari H, Kester MG, van Balen P, Hildebrand WH. Accurate prediction of HLA class II antigen presentation across all loci using tailored data acquisition and refined machine learning. Sci Adv 2023; 9:eadj6367. doi: 10.1126/sciadv.adj6367 [Crossref] [ Google Scholar]
- Techakriengkrai N, Nedumpun T, Golde WT, Suradhat S. Diversity of the swine leukocyte antigen class I and II in commercial pig populations. Front Vet Sci 2021; 8:637682. doi: 10.3389/fvets.2021.637682 [Crossref] [ Google Scholar]
-
Racle J, Guillaume P, Schmidt J, Michaux J, Larabi A, Lau K, et al. Machine learning predictions of MHC-II specificities reveal alternative binding mode of class II epitopes. Immunity 2023; 56: 1359-75.e13. doi: 10.1016/j.immuni.2023.03.009.
- Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 2012; 64:177-86. doi: 10.1007/s00251-011-0579-8 [Crossref] [ Google Scholar]
-
Donnell V, Holinka LG, Krug PW, Gladue DP, Carlson J, Sanford B, et al. African swine fever virus Georgia 2007 with a deletion of virulence-associated gene 9GL (B119L), when administered at low doses, leads to virus attenuation in swine and induces an effective protection against homologous challenge. J Virol 2015. 89: 8556-66. doi: 10.1128/jvi.00969-15.
- Charerntantanakul W. Adjuvants for swine vaccines: mechanisms of actions and adjuvant effects. Vaccine 2020; 38:6659-81. doi: 10.1016/j.vaccine.2020.08.054 [Crossref] [ Google Scholar]
- Lee SJ, Shin SJ, Lee MH, Lee MG, Kang TH, Park WS. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS One 2014; 9:e104351. doi: 10.1371/journal.pone.0104351 [Crossref] [ Google Scholar]
- Li L, Pian Y, Chen S, Hao H, Zheng Y, Zhu L. Phenol-soluble modulin α4 mediates Staphylococcus aureus-associated vascular leakage by stimulating heparin-binding protein release from neutrophils. Sci Rep 2016; 6:29373. doi: 10.1038/srep29373 [Crossref] [ Google Scholar]
- Fagbohun OA, Aiki-Raji CO, Omotosho OO. Contriving a multi-epitope vaccine against African swine fever utilizing immunoinformatics. Res Sq [Preprint]. September 6, 2022. Available from: https://www.researchsquare.com/article/rs-1978238/v1.
- Lei Y, Shao J, Ma F, Lei C, Chang H, Zhang Y. Enhanced efficacy of a multi-epitope vaccine for type A and O foot-and-mouth disease virus by fusing multiple epitopes with Mycobacterium tuberculosis heparin-binding hemagglutinin (HBHA), a novel TLR4 agonist. Mol Immunol 2020; 121:118-26. doi: 10.1016/j.molimm.2020.02.018 [Crossref] [ Google Scholar]
- Fox JM, Long F, Edeling MA, Lin H, van Duijl-Richter MKS, Fong RH. Broadly neutralizing alphavirus antibodies bind an epitope on E2 and inhibit entry and egress. Cell 2015; 163:1095-107. doi: 10.1016/j.cell.2015.10.050 [Crossref] [ Google Scholar]
-
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, ed. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005. p. 571-607. doi: 10.1385/1-59259-890-0:571.
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 2007; 8:4. doi: 10.1186/1471-2105-8-4 [Crossref] [ Google Scholar]
- Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP v2--a server for in silico prediction of allergens. J Mol Model 2014; 20:2278. doi: 10.1007/s00894-014-2278-5 [Crossref] [ Google Scholar]
- Magnan CN, Randall A, Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics 2009; 25:2200-7. doi: 10.1093/bioinformatics/btp386 [Crossref] [ Google Scholar]
- Rapin N, Lund O, Bernaschi M, Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One 2010; 5:e9862. doi: 10.1371/journal.pone.0009862 [Crossref] [ Google Scholar]
- Wang JY, Song WT, Li Y, Chen WJ, Yang D, Zhong GC. Improved expression of secretory and trimeric proteins in mammalian cells via the introduction of a new trimer motif and a mutant of the tPA signal sequence. Appl Microbiol Biotechnol 2011; 91:731-40. doi: 10.1007/s00253-011-3297-0 [Crossref] [ Google Scholar]
- Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol 2008; 180:309-18. doi: 10.4049/jimmunol.180.1.309 [Crossref] [ Google Scholar]
- Wang H, Yi W, Qin H, Wang Q, Guo R, Pan Z. A genetically engineered bivalent vaccine coexpressing a molecular adjuvant against classical swine fever and porcine epidemic diarrhea. Int J Mol Sci 2023; 24:11954. doi: 10.3390/ijms241511954 [Crossref] [ Google Scholar]
- Zhang H, Zhang L, Lin A, Xu C, Li Z, Liu K. Algorithm for optimized mRNA design improves stability and immunogenicity. Nature 2023; 621:396-403. doi: 10.1038/s41586-023-06127-z [Crossref] [ Google Scholar]
- Subramanian K, Payne B, Feyertag F, Alvarez-Ponce D. The codon statistics database: a database of codon usage bias. Mol Biol Evol 2022; 39:msac157. doi: 10.1093/molbev/msac157 [Crossref] [ Google Scholar]
-
Xia X. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol 2013. 30: 1720-8. doi: 10.1093/molbev/mst064.
- Babendure JR, Babendure JL, Ding JH, Tsien RY. Control of mammalian translation by mRNA structure near caps. RNA 2006; 12:851-61. doi: 10.1261/rna.2309906 [Crossref] [ Google Scholar]
- Trepotec Z, Aneja MK, Geiger J, Hasenpusch G, Plank C, Rudolph C. Maximizing the translational yield of mRNA therapeutics by minimizing 5'-UTRs. Tissue Eng Part A 2019; 25:69-79. doi: 10.1089/ten.TEA.2017.0485 [Crossref] [ Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res 2008; 36:W70-4. doi: 10.1093/nar/gkn188 [Crossref] [ Google Scholar]
- Jeong DE, McCoy M, Artiles K, Ilbay O, Fire A, Nadeau K, et al. Assemblies-of-putative-SARS-CoV2-spike-encoding-mRNA-sequences-for-vaccines-BNT-162b2-and-mRNA-1273. 2021. Available from: https://www.semanticscholar.org/paper/Assemblies-of-putative-SARS-CoV2-spike-encoding-for-Jeong-McCoy/150b70589516b969ce20fe83b9808478dd6f0e72. Accessed May 9, 2024.
- Zhang BH, Pan XP, Cox SB, Cobb GP, Anderson TA. Evidence that miRNAs are different from other RNAs. Cell Mol Life Sci 2006; 63:246-54. doi: 10.1007/s00018-005-5467-7 [Crossref] [ Google Scholar]
- Trotta E. On the normalization of the minimum free energy of RNAs by sequence length. PLoS One 2014; 9:e113380. doi: 10.1371/journal.pone.0113380 [Crossref] [ Google Scholar]
- Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024; 630:493-500. doi: 10.1038/s41586-024-07487-w [Crossref] [ Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605-12. doi: 10.1002/jcc.20084 [Crossref] [ Google Scholar]
- Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res 2013; 41:W384-8. doi: 10.1093/nar/gkt458 [Crossref] [ Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 1993; 26:283-91. doi: 10.1107/s0021889892009944 [Crossref] [ Google Scholar]
- Laskowski RA, Jabłońska J, Pravda L, Vařeková RS, Thornton JM. PDBsum: structural summaries of PDB entries. Protein Sci 2018; 27:129-34. doi: 10.1002/pro.3289 [Crossref] [ Google Scholar]
- Pang M, Tu T, Wang Y, Zhang P, Ren M, Yao X. Design of a multi-epitope vaccine against Haemophilus parasuis based on pan-genome and immunoinformatics approaches. Front Vet Sci 2022; 9:1053198. doi: 10.3389/fvets.2022.1053198 [Crossref] [ Google Scholar]
- Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C. The ClusPro web server for protein-protein docking. Nat Protoc 2017; 12:255-78. doi: 10.1038/nprot.2016.169 [Crossref] [ Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H. The protein data bank. Nucleic Acids Res 2000; 28:235-42. doi: 10.1093/nar/28.1.235 [Crossref] [ Google Scholar]
- Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods 1 The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 2011; 51:69-82. doi: 10.1021/ci100275a [Crossref] [ Google Scholar]
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015; 1-2:19-25. doi: 10.1016/j.softx.2015.06.001 [Crossref] [ Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983; 79:926-35. doi: 10.1063/1.445869 [Crossref] [ Google Scholar]
- Hammer SE, Duckova T, Groiss S, Stadler M, Jensen-Waern M, Golde WT. Comparative analysis of swine leukocyte antigen gene diversity in European farmed pigs. Anim Genet 2021; 52:523-31. doi: 10.1111/age.13090 [Crossref] [ Google Scholar]
- An HJ, Kim YJ, Song DH, Park BS, Kim HM, Lee JD. Crystallographic and mutational analysis of the CD40-CD154 complex and its implications for receptor activation. J Biol Chem 2011; 286:11226-35. doi: 10.1074/jbc.M110.208215 [Crossref] [ Google Scholar]
- Zhuang Q, Chen H, Xiong F. First-principles study of monolayer GeTe and the effect of external strain and electric field. J Electron Mater 2025; 54:718-26. doi: 10.1007/s11664-024-11530-x [Crossref] [ Google Scholar]
- Pourseif MM, Masoudi-Sobhanzadeh Y, Azari E, Parvizpour S, Barar J, Ansari R. Self-amplifying mRNA vaccines: mode of action, design, development and optimization. Drug Discov Today 2022; 27:103341. doi: 10.1016/j.drudis.2022.103341 [Crossref] [ Google Scholar]
- Omidi Y, Pourseif MM, Ansari RA, Barar J. Design and development of mRNA and self-amplifying mRNA vaccine nanoformulations. Nanomedicine (Lond) 2024; 19:2699-725. doi: 10.1080/17435889.2024.2419815 [Crossref] [ Google Scholar]
- Salemi A, Pourseif MM, Omidi Y. Next-generation vaccines and the impacts of state-of-the-art in-silico technologies. Biologicals 2021; 69:83-5. doi: 10.1016/j.biologicals.2020.10.002 [Crossref] [ Google Scholar]
- Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol 2010; 10:403-14. doi: 10.1038/nri2780 [Crossref] [ Google Scholar]
- Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 2011; 11:823-36. doi: 10.1038/nri3084 [Crossref] [ Google Scholar]
- Hoerr I, Obst R, Rammensee HG, Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol 2000; 30:1-7. doi: 10.1002/1521-4141(200001)30:1<1::Aid-immu1>3.0.Co;2-# [Crossref] [ Google Scholar]
- Racanelli V, Behrens SE, Aliberti J, Rehermann B. Dendritic cells transfected with cytopathic self-replicating RNA induce crosspriming of CD8 + T cells and antiviral immunity. Immunity 2004; 20:47-58. doi: 10.1016/s1074-7613(03)00353-4 [Crossref] [ Google Scholar]
- Fatima M, Luo Y, Zhang L, Wang PY, Song H, Fu Y. Genotyping and molecular characterization of classical swine fever virus isolated in China during 2016-2018. Viruses 2021; 13:664. doi: 10.3390/v13040664 [Crossref] [ Google Scholar]
- Heegaard PM, Boas U, Sorensen NS. Dendrimers for vaccine and immunostimulatory uses A review. Bioconjug Chem 2010; 21:405-18. doi: 10.1021/bc900290d [Crossref] [ Google Scholar]
- Rappuoli R. Reverse vaccinology. Curr Opin Microbiol 2000; 3:445-50. doi: 10.1016/s1369-5274(00)00119-3 [Crossref] [ Google Scholar]
- Andreatta M, Trolle T, Yan Z, Greenbaum JA, Peters B, Nielsen M. An automated benchmarking platform for MHC class II binding prediction methods. Bioinformatics 2018; 34:1522-8. doi: 10.1093/bioinformatics/btx820 [Crossref] [ Google Scholar]
- Trevizani R, Yan Z, Greenbaum JA, Sette A, Nielsen M, Peters B. A comprehensive analysis of the IEDB MHC class-I automated benchmark. Brief Bioinform 2022; 23:bbac259. doi: 10.1093/bib/bbac259 [Crossref] [ Google Scholar]
- Díaz Á, González-Alayón I, Pérez-Torrado V, Suárez-Martins M. CD40-CD154: a perspective from type 2 immunity. Semin Immunol 2021; 53:101528. doi: 10.1016/j.smim.2021.101528 [Crossref] [ Google Scholar]