Bioimpacts. 2025;15:30795.
doi: 10.34172/bi.30795
Review
The promise of gene therapy in common types of dementia
Mojtaba Ghobadi Conceptualization, Funding acquisition, Investigation, Visualization, Writing – original draft, 1 
Mohammad Foad Heidari Writing – review & editing, 2
Arezoo Farhadi Investigation, 3
Ali Shakerimoghaddam Visualization, 4
Mahdi Ghorbani Visualization, 1, 2
Zahra Hami Writing – review & editing, 5
Naeim Ehtesham Supervision, 6
Javad Behroozi Conceptualization, Project administration, Resources, Writing – review & editing, 1, *
Author information:
1Cancer Epidemiology Research Center, Aja University of Medical Sciences, Tehran, Iran
2Department of Medical Laboratory Sciences, School of Allied Health Medicine, AJA Univers
3Department of Genetics and Molecular Medicine, School of Medicine, Zanjan University of Medical Sciences, Zanjan, Iran
4Infectious Diseases Research Center, AJA University of Medical Sciences, Tehran, Iran
5Toxicology Research Center, AJA University of Medical Sciences, Tehran, Iran
6Department of Meical Genetics, School of Medicine, Iranshahr University of Medical Sciences, Iranshahr, Iran
Abstract
Dementia is an umbrella term describing different types of diseases that lead to cognitive impairment and memory dysfunction, predominantly affecting older adults. The most common forms include Alzheimer's disease (AD), vascular dementia (VaD), dementia with Lewy bodies (DLB), and frontotemporal dementia (FTD). Despite extensive research, there is no definitive cure for dementia, primarily due to its complex and multifactorial nature, particularly the role of genetic abnormalities. Gene therapy, a novel therapeutic approach, aims to correct defective genes or introduce functional gene products by delivering specific DNA sequences to patients, and is often considered for individuals unresponsive to conventional treatments, such as those with dementia. Over the past decade, significant research has explored the potential of gene therapy in dementia, offering new hope for more effective treatments. However, several challenges remain in its practical application. One key challenge is developing safe and efficient gene delivery methods, as the brain's intricate structure and protective barriers present significant obstacles. Furthermore, ensuring the long-term expression and stability of therapeutic genes is crucial for sustained benefit. Future studies should focus on identifying genes implicated in different types of dementia, optimizing gene delivery systems, improving gene-targeting specificity, and conducting comprehensive clinical trials to assess the safety and efficacy of these therapies. Addressing these challenges could pave the way for novel treatment strategies, ultimately improving the quality of life for individuals with dementia.
Keywords: Gene therapy, Alzheimer's disease, Vascular dementia, Parkinson's disease, Frontotemporal dementias, CRISPR
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This study was financially supported by Aja University of Medical Sciences (Grant number 97002493).
Introduction
Definition of dementia
Dementia is an umbrella term used to describe neurodegenerative syndromes with similar characteristics. The common feature of these conditions is chronic cognitive and emotional impairment, interfering with daily activities and significantly reducing quality of life. Currently, about 55 progressive and non-progressive diseases have been identified as dementia-related syndromes in clinical setting. Common types of dementia include Alzheimer's disease (AD, the most common type of dementia), vascular dementia (VaD), dementia with Lewy body (DLB), and frontotemporal dementias (FTD) (Fig. 1).1 The clinical manifestations of dementia very widely among patients and may present as memory loss, communication and language disorders, agnosia (inability to object recognition), apraxia (inability to perform learned tasks), and impaired executive function (reasoning, judgment, and planning). Cognitive impairment is mainly due to brain inflammation, changes in the brain metabolism, and synaptic dysfunction.2,3
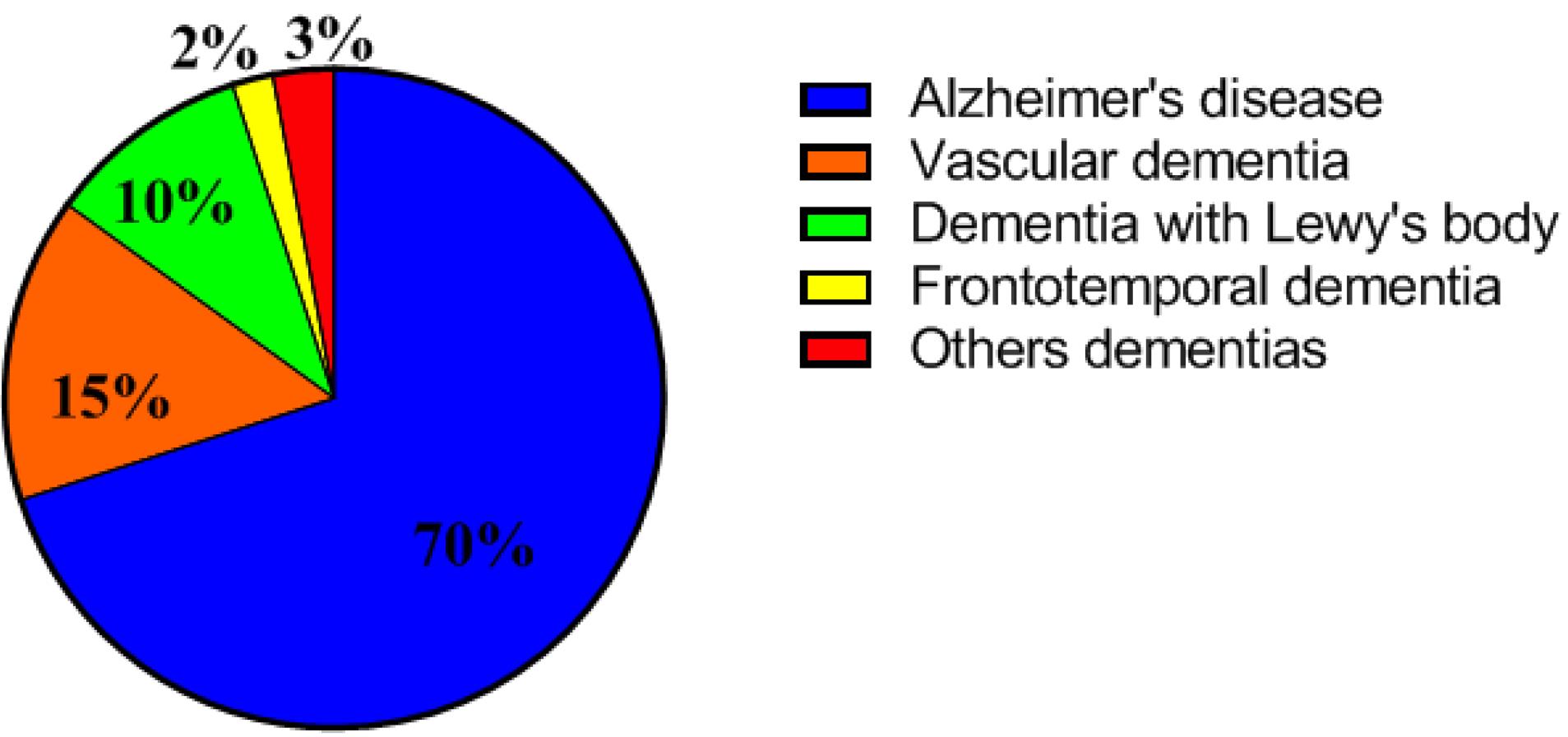
Fig. 1.
Prevalence percentage of common types of dementia.
.
Prevalence percentage of common types of dementia.
Epidemiology
The prevalence of dementia increases with age. The risk at the age 60 is about 1%, doubling every five years, reaching 30-50% at the age of 85.4 Typically, patients who develop dementia before the age of 65 have early dementia (at working age), while those diagnosed after age 65 have late-onset dementia.5 According to Prince et al, about 24 million people were affected by dementia in 2001, which increased to approximately 35 million in 2010. This number is expected to rise to 65 million by 2030 and 115 million in 2050, with nearly two-thirds of affected individuals living in developing and low-income countries.6
Risk factors
Cognitive decline is influenced by several risk factors, which can be categorized into two groups: those specific to particular types of dementia and general risk factors. Age is the most prominent risk factor, with the probability of developing cognitive impairment escalating as individuals advance in age.7 Neuroanatomical changes associated with aging include a progressive reduction in brain volume, primarily attributable to the loss of neurons and synaptic connections.8 Furthermore, the accumulation of neurotoxic protein aggregates, such as tau and amyloid beta, is known to increase with age, potentially leading to neuronal dysfunction and subsequent cell death.9 Additionally, the incidence of chronic inflammatory conditions tends to rise with age, contributing to a neuroinflammatory milieu which exacerbates oxidative stress and neuronal degeneration. Thus, aging is regarded as a critical risk factor for cognitive decline.10
Another significant contributor to cognitive impairment is the reduction in cerebral perfusion resulting from various pathological conditions. Cardiovascular diseases, including myocardial infarction, coronary artery disease, and atherosclerosis, are notable in this context. These conditions are associated with diminished cerebral blood flow and ischemia, leading to vascular damage, hypoxia, and impaired delivery of essential nutrients to neurons, ultimately resulting in oxidative injury to neuronal cells.11,12
Diabetes mellitus is also a substantial risk factor for cognitive decline. Numerous studies have demonstrated that vascular complications associated with diabetes lead to impaired perfusion of neural tissue, coupled with the induction of inflammatory pathways and oxidative stress, both of which contribute to neuronal apoptosis.13 Furthermore, a deficiency or downregulation of the insulin-degrading enzyme in the brain, responsible for the catabolism of amyloid beta, has been implicated in the accumulation of these neurotoxic aggregates within neurons.14 Alterations in the insulin signaling cascade may also facilitate pathological interactions between receptors for advanced glycation end products and Aβ peptides, further complicating the neurodegenerative process.15,16
Obesity constitutes another critical risk factor, as it can adversely affect cerebral hemodynamics. An increase in adipose tissue is associated with white matter integrity compromise and a reduction in neuronal populations, thereby heightening the susceptibility to cognitive decline.1,17
Additionally, previous studies have found that individuals with a positive family history of dementia are at double the risk compared to others. Although, Mendelian forms of dementia are rare, some dementia cases are genetically complex. Various genetic mutation, such as those in the amyloid precursor protein (APP), presenilin 1 (PSEN1), presenilin 2 (PSEN2), microtubule-associated protein tau (MAPT), granulin (GRN), and chromosome 9 open reading frame 72 (C9ORF72) genes, increase the likelihood of developing dementia, making genetic predisposition an important risk factor as described.18
Gene therapy
Gene therapy is an innovative approach to treating diseases by correcting defective genes or using gene products to alter gene expression.19,20 The U.S. Food and Drug Administration (FDA) defines gene therapy as a treatment that utilizes genetic material, such as nucleic acids (DNA or RNA), viruses, modified microorganisms, or a newly developed gene-editing method known as Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated protein 9 (CRISPR/Cas9). These modifications can occur by integrating new genetic material into existing genes or adding functional copies of genes.21,22 Genetic material may be used to alter cells inside the body or transferred to cells outside the body before being reintroduced into the patient. Gene therapy can be categorized into two types: Somatic gene therapy, were genes are inserted into specific cells to treat an individual’s condition without heritable changes, and germ-line gene therapy, where modifications are pass down to future generations. Current regulations only permit gene therapy in non-reproductive cells.23,24
Advances in gene delivery systems, such as viral vectors and CRISPR/Cas9, have made it possible to target the brain, overcoming one of the primary challenges in treating neurological disorders.25 Further, growing body of research has identified promising gene targets for therapeutic intervention in common forms of dementia. For instance, in AD, genes involved in amyloid metabolism, tau protein regulation, and neuroinflammation have been the focus of gene therapy studies. Similarly, specific gene mutations associated with familial forms of dementia have been highlighted as potential targets for gene editing or gene silencing strategies.18,26 As a result, numerous gene therapy studies have been conducted in the field of dementia treatment. In this review, we will evaluate the progress made in targeting specific genes, the challenges encountered, and the potential for future advancements in dementia therapy.
Gene therapy in common types of dementias
AD and gene therapy
AD is a multifactorial progressive dementia characterized by the accumulation of extracellular plaques composed of amyloid beta (Aβ) and intracellular neurofibrillary tangles (NFTs) containing tau protein (tauopathy). The most common manifestation of AD is short-term memory impairment.27 Additionally, AD affects other cognitive functions, such as visual-spatial processing; and executive functions. Symptoms of AD related to synaptic dysfunction include the destruction of cortical neural circuits and neuronal degeneration, particularly in the brain's cognitive cortex. This leads to a loss of synaptic stability (homeostasis), neuronal loss, and disruption of the neural network's integrity.28
AD is classified into two categories: early-onset Alzheimer’s disease (EOAD) and late-onset Alzheimer’s disease (LOAD). The early-onset type manifests in approximately 5% of cases before the age of 65. This form can be further divided into two subtypes: those exhibiting an autosomal inheritance pattern (5-10% of EOAD cases) and sporadic early-onset Alzheimer’s disease (sEOAD).29 The primary factors contributing to this type of Alzheimer’s are disruptions in several genes, including the A673V variant of the amyloid precursor protein (APP) for the autosomal recessive form, as well as mutations in the APP gene, presenilin 1 (PSEN1), and presenilin 2 (PSEN2), which are recognized as major causes of the autosomal dominant form of AD.30
In contrast, late-onset Alzheimer’s disease is primarily associated with neurodegeneration resulting from the accumulation of Aβ. Aβ is a protein derived from the APP, which is located at the presynaptic terminals of neurons in vertebrates. Enzymes such as α-secretase, γ-secretase, and β-site APP cleaving enzyme 1 (BACE-1) cleave APP, resulting in the production of amyloid beta fragments. When the activity of α-secretase is diminished, abnormal cleavage of APP outside the appropriate zone by β-secretase leads to the production and aggregation of Aβ, ultimately resulting in neuronal death.28,31
Aβ tends to aggregate and form plaques due to its biochemical properties, primarily existing in two main forms: Aβ40 and Aβ42, which differ in their β-sheet lengths. Aβ polymerization occurs through a nucleation-dependent pathway, resulting in the formation of fibrils and intermediate structures, such as paranuclei and protofibrils. Both Aβ fibrils and smaller oligomers deposit intracellularly and extracellularly, contributing to neuronal dysfunction.32 This dysfunction arises from the formation of irregular aggregates, while beta-oligomer amyloids create insoluble fibrillar structures. These aggregates resist hydrolytic degradation, leading to plaque formation within neurons, disrupting their function and contributing to the pathophysiology of Alzheimer’s disease.33
Mutations in the APP gene can result in the production of longer Aβ peptides owing to the overactivation of β-secretase, leading to an overproduction of neurotoxic forms of Aβ.34 Additionally, loss-of-function mutations in the PSEN1 and PSEN2 genes reduce the production of Aβ40 relative to Aβ42 due to altered γ-secretase activity, resulting in an increased Aβ42/Aβ40 ratio, which further contributes to the formation of amyloid plaques (Fig. 2).35
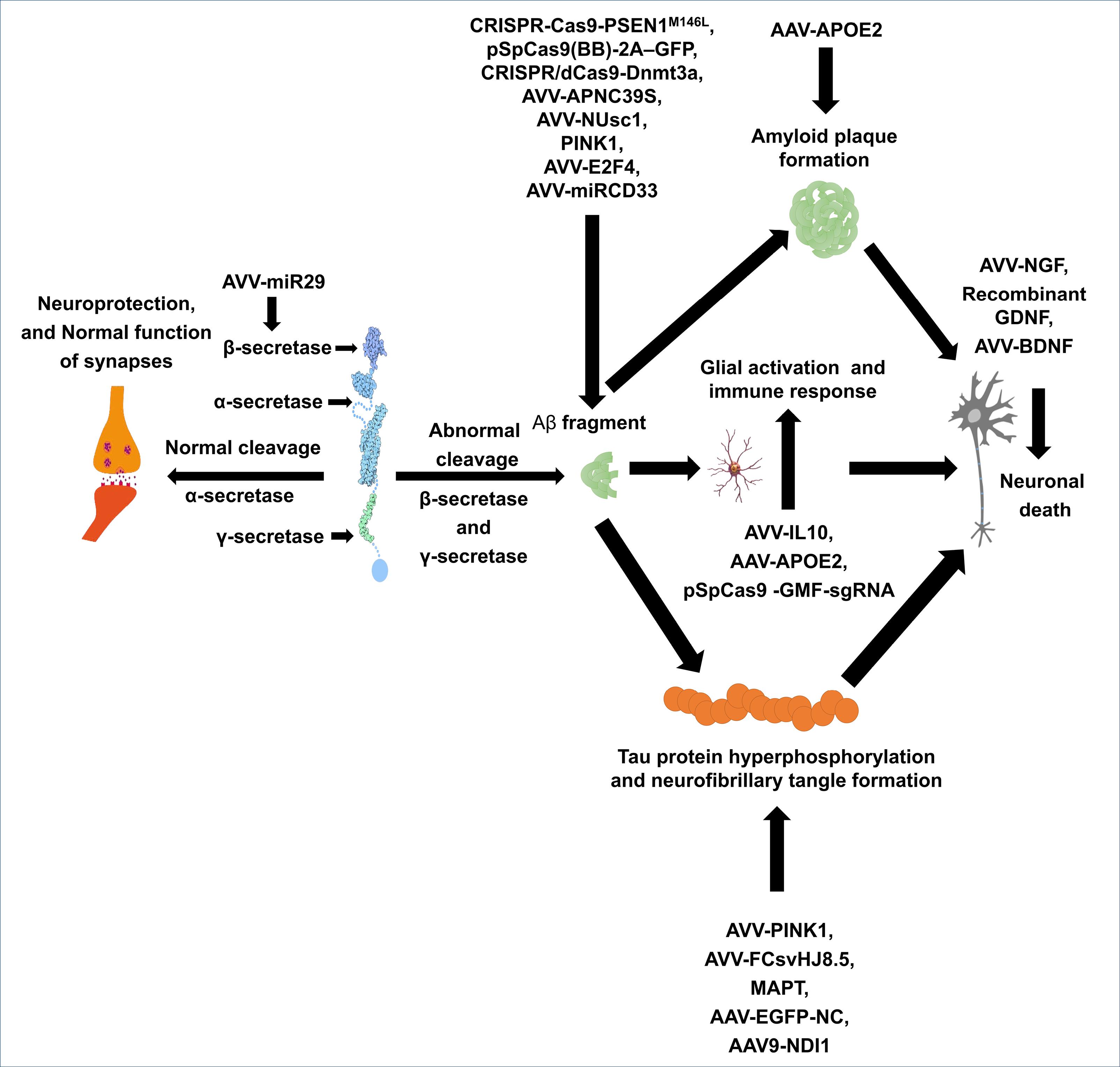
Fig. 2.
Pathophysiology and gene therapy targets in AD. AAV: Adenovirus vector vaccine, NGF: Nerve growth factor, BDNF: Brain derived neurotrophic factor, GDNF: Glial-derived neurotrophic factor, IL10: Interleukin-10, MAPT: Microtubule-associated protein tau, EGFP-NC: Enhanced green fluorescent protein-nuclear localization signal, Aβ: Amyloid beta, pSpCas9: plasmid Staphylococcus pyogenes cas9, GMF-sgRNA: Gene modification framework- single guide RNA, 2A-GFP: 2A peptide-green fluorescent protein, PINK1: Phosphatase and tensin homolog-induced putative kinase 1, NUse1: Nuclear envelope spanning protein 1.
.
Pathophysiology and gene therapy targets in AD. AAV: Adenovirus vector vaccine, NGF: Nerve growth factor, BDNF: Brain derived neurotrophic factor, GDNF: Glial-derived neurotrophic factor, IL10: Interleukin-10, MAPT: Microtubule-associated protein tau, EGFP-NC: Enhanced green fluorescent protein-nuclear localization signal, Aβ: Amyloid beta, pSpCas9: plasmid Staphylococcus pyogenes cas9, GMF-sgRNA: Gene modification framework- single guide RNA, 2A-GFP: 2A peptide-green fluorescent protein, PINK1: Phosphatase and tensin homolog-induced putative kinase 1, NUse1: Nuclear envelope spanning protein 1.
Over 200 mutations in these genes have been associated with early-onset AD. Despite its genetic basis, early-onset AD is relatively rare compared to late-onset AD. Late-onset AD typically begins after age 65 and is influenced by multiple factors. It has a genetic link to the ApoE4 allele, which increases susceptibility to the disease. ApoE4 is connected to increased neuroinflammatory responses. This allele can alter how microglia, the brain’s immune cells, are activated, resulting in a pro-inflammatory environment which may aggravate neuronal injury and lead to cognitive decline.36 In addition to its role in amyloid pathology, ApoE4 might also affect the phosphorylation and aggregation of tau proteins. Tau tangles are a critical pathological feature of AD, and those with the ApoE4 variant may show more severe tau-related pathology (Fig. 2).37 Recent research, including studies from the Genetic and Environmental Risk Factors for Alzheimer's Disease (GAAWS), has identified more than 270 genetic loci associated with an increased risk of developing late-onset AD.38
Human studies on gene therapy for AD are limited. One clinical trial study examined the bilateral stereotactic administration of adeno-associated virus carrying nerve growth factor (AAV2-NGF) into the basal forebrain region. This approach was associated with a reduction in cognitive abnormalities and progression of AD compared to baseline. However, improvements in cognitive function were not observed among the participants. This study demonstrated that long-term gene-mediated NGF expression triggered a growth response in the nucleus basalis of Meynert (NBM) neurons when exposed to the NGF protein.39 In another study, Tuszynski et al reported that all patients showed nerve fiber growth towards the NGF source and observed cholinergic neuronal hypertrophy in the treated group. They concluded that neurons in the AD brain retain the capacity to respond to growth factors with axonal sprouting, cell hypertrophy, and activation of functional markers, indicating that these neurons can still react to specific growth factors to promote growth and activity.40 However, Castle et al highlighted that the lack of cognitive improvement may be attributed to the vector delivery method. They found that AAV2-NGF did not effectively engage the target cholinergic neurons. They recommended using advanced imaging techniques and convection-enhanced delivery for future clinical trials to enhance the delivery of growth factors to the brain.41 Additionally, Mummery et al conducted a phase 1b, randomized, placebo-controlled trial to evaluate the safety and pharmacokinetics of MAPTRx, a tau-targeting antisense oligonucleotide, in 46 patients with mild AD. Participants received doses of 10 mg, 30 mg, and 60 mg or two quarterly doses of 115 mg. The results revealed that 94% experienced mild to moderate adverse events, primarily headaches following lumbar puncture, with no severe events reported. A significant reduction in tau protein concentration in cerebrospinal fluid (CSF) was also observed.42 These results suggest that while growth factor gene therapy shows promise as a safe and potentially valuable approach to AD management, further investigation is needed to optimize delivery methods and assess long-term efficacy.
Aβ and its role in AD represent another critical target for gene therapy. An experimental study utilized a vector carrying miR-29 to reduce the expression of β-site amyloid precursor protein cleaving enzyme 1 (BACE1). This approach mitigated the degenerative effects of Aβ peptide and attenuated cognitive disturbances in a rat model of AD.43 In another study, Du et al explored gene therapy-mediated overexpression of PTEN-induced putative kinase 1 (PINK1), which is typically reduced in AD. This intervention improved mitochondrial function through the activation of autophagy, leading to a reduction in Aβ-induced synaptic degeneration and cognitive decline in AD mice.44 Additionally, polymorphisms in the E2F4 transcription factor, a regulator of cell cycle genes, are associated with stress response, apoptosis, synaptic function, and memory formation, and have been linked to AD.45 Sanchez et al demonstrated that systemic injection of an AAV.E2F4 vector improved memory and cognitive function while reducing Aβ production and aggregation in the hippocampus of mice.46 In another animal study, Griciuc et al assessed the effects of the miRCD33 vector in an APP/PS1 mouse model of AD. Their findings revealed significant reductions in soluble Aβ and amyloid plaque load in both 8-month-old and 2-month-old AD mice. Additionally, this treatment reduced neuroinflammation and microglia activation in both age groups.47
Inhibition of tau protein hyperphosphorylation through gene therapy has emerged as a promising approach for Alzheimer’s treatment. Wang and colleagues demonstrated that increasing the expression of PTEN-induced PINK1 via the tail vein injection of the adeno-associated viral vector pAAV-CMV-PINK1-P2A-EGFP-3FLAG (AAV-PINK1) six weeks prior to intracerebral injection of forskolin, a tau hyperphosphorylating agent, led to the preservation of the PI3K/Akt/GSK3β signaling pathway. This increase in PINK1 expression resulted in reduced mitochondrial damage through elevated antioxidant levels, thereby maintaining mitochondrial structure and subsequently reducing neuronal loss as well as synaptic damage in the studied rats.48 The Fc domain of the anti-tau antibody HJ8.5, delivered via the AAV2/8 viral vector, significantly diminished hyperphosphorylated tau accumulation in the hippocampus of transgenic tauopathy mice through ICV injection, without affecting extracellular tau levels in the CSF. This suggests that microglial tau clearance may not be essential for the protective effects of anti-tau antibodies, indicating a potential novel therapeutic strategy.49 The use of brain-derived neurotrophic factor (BDNF) as a gene-based therapeutic approach was investigated by Jiao and colleagues in an Alzheimer’s animal model, motivated by the observed reduction of serum BDNF levels in Alzheimer’s patients. In this study, no significant changes in hyperphosphorylated tau levels were noted among the experimental groups. However, the restoration of BDNF through alternative pathways led to improvements in behavioral deficits, reduced neuronal death, and enhanced synaptic status in the studied animals, indicating an independent neuroprotective effect of BDNF in tau-related neuronal damage.50 In the study by Asih and colleagues using an Ab Alzheimer’s mouse model, it was found that wild-type human tau or phosphorylation-deficient tau T205A demonstrated that T205 modification is critical for the downstream effects of p38γ, which prevent memory impairment in APP transgenic mice. Furthermore, genome editing of the T205 codon in the MAPT gene of mice revealed that this single side chain in endogenous tau critically modulates memory deficits in APP transgenic Alzheimer’s mice. The ablation of the protective effect of p38γ activity through genetic deletion of p38γ in a tau transgenic mouse model expressing non-pathogenic tau rendered tau toxic and resulted in impaired memory function in the absence of human Aβ. These findings indicate that modulating neuronal p38γ activity serves as an intrinsic tau-dependent therapeutic approach to enhance compromised cognition in advanced dementia.51,52
Other experimental studies have demonstrated the beneficial effects of gene therapy on various aspects of AD and have developed new gene therapy methods. For instance, the study by Koller et al examined the relationship between APOE4 and tau by using bilateral ICV injection of AAV-APOE4 in mice to assess whether altering tau expression could prevent AD. The results revealed no significant changes in tau protein expression, indicating a lack of direct correlation between APOE4 and tau in this Alzheimer’s model. This finding suggests that tau may follow a distinct pathway, prompting a shift in Alzheimer’s treatment strategies towards other gene therapy approaches that address various aspects of the disease.53 Rodrigues et al examined the impact of a single dose of specially designed liposomes containing ApoE2 DNA on mouse brains. They observed a significant increase in ApoE levels following transfection, suggesting that this method could enhance the ability to target the brain and deliver genes more effectively.54 Similarly, Jackson et al reported that a single intracerebroventricular (ICV) injection of AAV-APOE2 led to a rapid reduction in Aβ plaque deposition, neuronal loss, and microglial activation in both human tissue and an APP/PS1 mouse model of AD.55 These findings indicate that gene therapy using APOE2 holds promise as a novel approach for AD treatment. Other studies are listed in Table 1 (Fig. 2).
Table 1.
articles in AD gene therapy
|
Author
|
Model
|
Method
|
Findings
|
| Selles et al56 |
|
|
-
Inhibition the AβO binding to neurons
-
Inhibition the dendritic spine loss
-
Reversed memory defect in aged mice
-
Memory impairment prevention
|
| Arora et al57 |
|
|
-
Aβ plaque reduction
-
Increase in synaptic protein
-
Increase in cell proliferation
-
Improve in nesting test score behavior
|
| Elmer et al58 |
|
|
|
| Murphy et al59 |
|
|
|
| Kiyota et al60 |
-
Tg2576 mice
-
M146 L PS1 mutant mice
|
|
|
| Revilla et al61 |
|
|
|
| Li et al62 |
|
|
-
Reduction in Tau phosphorylation and neuroinflammatory response
-
Reduction in mitochondrial dysfunction
-
Improvement in learning and memory
|
| Ng et al63 |
|
|
-
Overexpression of circulatory adiponectin and decreased both the soluble and fibrillar Aβ
-
Reduction in Aβ-induced IL-1β and IL-18 secretion by suppressing microglial NLRP3-infammasome activation
-
Improve in memory function
|
| Zheng et al64 |
|
|
|
| Jackson et al65 |
|
|
|
CRISPR-Cas9 studies represent another aspect of gene therapy for AD. This method has shown that that genome editing could be beneficial for AD treatment; however, there is no clinical study in this field due to its novelty. Konstantinidis et al utilized the CRISPR-Cas9 system from Streptococcus pyogenes (Sp) to selectively disrupt the PSEN1M146L allele in fibroblasts from individuals carrying the PSEN1 mutation, which is associated with early-onset AD. The results indicated that disruption of more than 50% of mutant alleles was achieved in all CRISPR-Cas9-treated samples, leading to a partial restoration of the extracellular Aβ42/40 ratios; however, the system was not efficient enough, necessitating further research.66 In an experimental study, Raikwar et al employed an AAV co-expressing Staphylococcus aureus (Sa) Cas9 and a glia maturation factor-specific guide RNA (GMF-sgRNA) as well as lentiviral vectors (LVs) expressing either Sp Cas9 or GMF-sgRNAs for microglia inhibition in the murine BV2 microglial cell line. They reported that microglial suppression was successfully induced by reducing p38 MAPK phosphorylation, which is beneficial for AD treatment.67 In another experimental study, Park et al used stereotaxic injection of 10 µL catalytically inactivated Cas9 fused with DNA methyltransferase3a (CRISPR/dCas9-Dnmt3a) for targeted DNA methylation of APP in the APP-KI mouse brain. This study found that the APP mRNA level, Aβ peptide level and Aβ42/40 ratio were significantly reduced in treated APP-KI mouse neurons, which also attenuated cognitive and behavioral impairments in this mice.68 Overall, these results indicate the need to explore different therapeutic strategies in CRISPR/Cas9 studies and to expand this technology for greater efficiency (Table 2, Fig. 2).
Table 2.
Therapeutic usage of CRISPR/Cas9 in AD treatment
|
Author
|
Model
|
Method
|
Findings
|
| Gyorgy et al69 |
|
|
|
| Ortiz-Virumbrales et al70 |
|
|
|
| Duan et al71 |
|
|
-
Decrease in amyloid-plaque in the different region of 5XFAD mice brain
-
Improves cognitive performance in 5XFAD mice treated group
-
Improves neuronal functions in 5XFAD mice
-
Decrease in Aβ pathology of APP/PS1 mice brain
|
| Yang et al72 |
|
|
|
Gene therapy in VaD
VaD is one of the most common forms of dementia after AD, accounting for approximately 15% of cases. This condition arises from disturbances in blood supply to the brain due to vascular issues, leading to nerve damage, brain injury, and resulting cognitive as well as memory problems. However, the relationship between cerebrovascular pathology and cognitive impairment is complex and not fully understood, largely due to variations in disease classification and diagnostic criteria.73 Nevertheless, atherosclerosis seems to be a major cause of VaD, and other causative factors include aging, coronary artery disease, hypercholesterolemia in middle age, hypertension, obesity, metabolic syndrome, arteriosclerosis, smoking, and poor educational attainment.74
The genetic basis of VaD is categorized into two main types: monogenic VaD and sporadic VaD. Monogenic VaD include cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D), and Fabry disease. These are often caused by mutations in specific genes, such as the NOTCH3 gene. The NOTCH3 gene encodes a protein receptor essential for the function and survival of vascular smooth muscle cells. Mutations in this gene are cause of cerebral small vessel disease (SVD) leading to impairment in vessels integrity, atherosclerosis, strokes, and reduction in blood perfusion.75 In sporadic VaD, genetic abnormalities exacerbate tissue responses to hypoxia, inflammation, and neuronal death following reduced blood flow due to risk factors such as arterial plaque or infarction. In both situations, reduction in blood supply impairs chemical environment and oxygenation levels of neurons. Subsequently, hypoxia, oxidative stress, and inflammation would occur, causing mitochondrial dysfunction, blood brain barrier impairment, and microvascular dysfunction, and finally neuronal death, brain atrophy, and dementia (Fig. 3).76
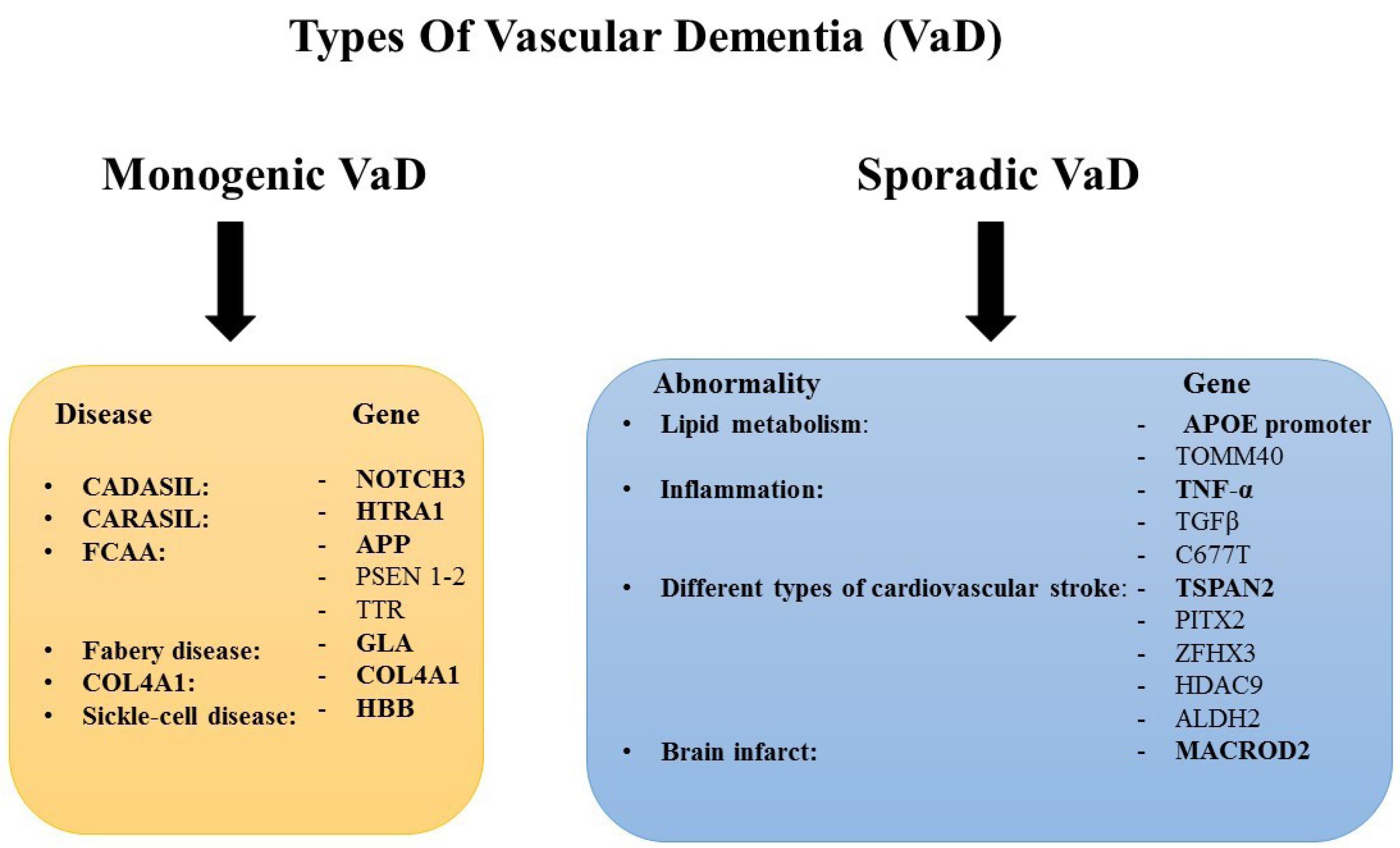
Fig. 3.
Genes causing vascular dementia in the monogenic and sporadic VaD. CADASIL: Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, CARASIL: Cerebral Autosomal-Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, FCAA: Familial Cerebral Amyloid Angiopathy.
.
Genes causing vascular dementia in the monogenic and sporadic VaD. CADASIL: Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, CARASIL: Cerebral Autosomal-Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, FCAA: Familial Cerebral Amyloid Angiopathy.
Gene therapy approaches for VaD target genetic abnormalities. For example, in cases of monogenic VaD, Rutten et al investigated the correction of a cysteine residue in the epidermal growth factor-like repeat (EGFr) domain of the NOTCH3 receptor. Using pre-mRNA antisense-mediated skipping of specific exons, their in silico and in vitro studies demonstrated that the mutation could be eliminated, restoring normal NOTCH3 receptor function.77 Additionally, the dual AAV split-ABEmax system has been employed to correct NOTCH3 gene mutations. This approach has been shown to recover deposition and correct abnormalities in the actin cytoskeleton structure of vascular smooth muscle cells (VSMCs) derived from CADASIL patient-derived induced pluripotent stem cells (hiPSCs). Furthermore, organoids generated from edited CADASIL blood vessels exhibited enhanced cell adhesion and improved vessel structure.78 In addition, CRISPR/Cas studies revealed that NOTCH3 gene edition could be useful for CADASIL treatment. Gravesteijn et al reported that exon 9 deletion from genomic DNA in HEK293 cells by CRISPR/Cas9-mediated genome editing targeting introns 8 and 9 would reduce the NOTCH3Δexon9 protein aggregation and is associated with relatively mild later-onset phenotype.79 However, this study could not provide a promising therapeutic approach for CADASIL treatment due to small number of samples but it is a good guide for further research.
According to the majority of literature, gene therapy for the sporadic form of VaD is very limited. However, existing studies suggest that gene therapy targeting HIF-1-dependent genes such as VEGF,80 or increase in neurotrophin levels may improve the signs and symptoms of VaD.81,82 For instance, the transfection of bone marrow-derived mesenchymal stem cells (BMSCs) with a recombinant NGF adenoviral vector followed by transplantation into the hippocampus of VaD-induced rats, demonstrated improvement in the animals learning and memory.83 In another experimental study, VEGF-based gene therapy improved hemodynamics and boosted parenchymal microvascular density.84 Similar findings were reported by Hiramatsu et al, who observed a significant increase in the total number of blood vessels following VEGF gene therapy in a rat model of VaD.85 Despite these promising results, due to the variability in risk factors associated with VaD, significant challenges remain before gene therapy can become a suitable treatment option. Furthermore, especially in clinical trials, several issues still need to be addressed, such as the safety and efficacy of vectors and delivery systems, therapy duration, and cell-specific targeting strategies.86
DLB or alpha-synucleinopathies
Lewy body dementia (LBD) and Parkinson's disease dementia (PDD) are often grouped together due to their shared pathological processes, collectively known as DLB. The distinguishing feature of this type of dementia is the presence of α-synuclein (Lewy bodies and Lewy neurites) in neurons, which leads to neuronal death. Aβ also plays a significant role in DLB.87 However, even in dementia associated with PDD, neurofibrillary tangles containing tau protein and Aβ contribute to more advanced dementia, suggesting that these pathologies interact.88 The genetic basis of DLB can be categorized into two groups based on subtypes. In the case of PDD, around 3-5% of cases result from monogenic mutations, while approximately 90 gene variants have been associated with a 16-36% increased risk of PDD.89 Mutations in synuclein alpha (SNCA), glucocerebrosidase (GBA), leucine-rich repeat kinase 2 (LRRK2), PTEN induced kinase 1 (PIKN1), and parkin RBR E3 ubiquitin protein ligase (PRKN) genes are the most common genetic susceptibilities reported.90 For instance, the SNCA mutation is linked to more rapid progression of motor defects and cognition decline in PDD, while LRRK2 mutations often occur in familial PDD cases.91 Conversely, LBD is associated with mutations in the APOE ε4 allele, GBA, and SNCA genes92 (Fig. 4).
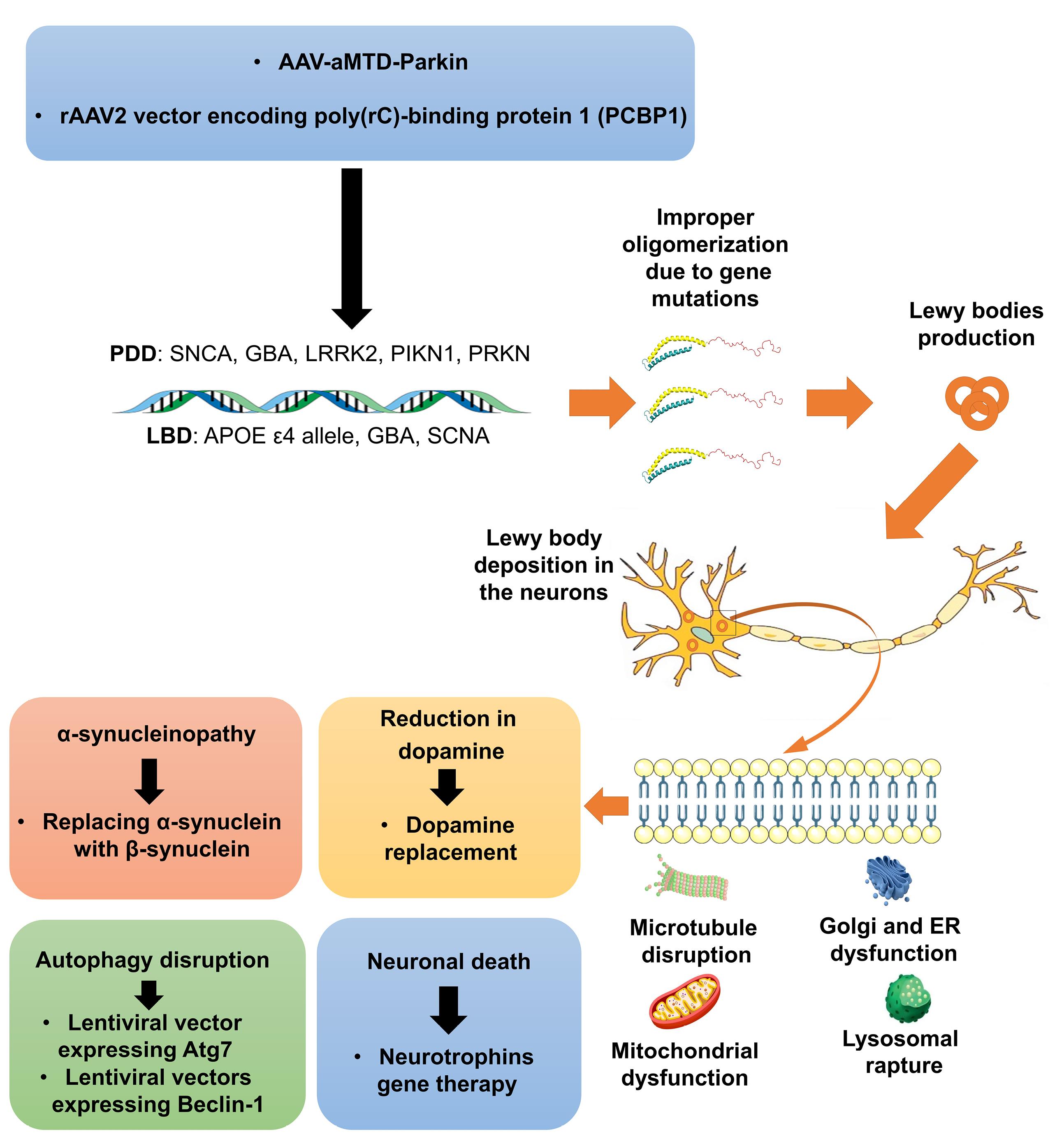
Fig. 4.
Gene therapy in PDD is primarily designed to mitigate neuronal death through the modulation of autophagic pathways, restoration of dopaminergic function, and intervention in alpha-synucleinopathy. This multifaceted approach seeks to enhance neuronal survival and function by addressing the underlying pathophysiological mechanisms associated with PDD. PDD: Parkinson’s disease dementia, LBD: Lewy body disease, ER: Endoplasmic reticulum, SNCA: Synuclein Alpha, GBA: Glucocerebrosidase, LRRK2: Leucine-Rich Repeat Kinase 2, PINK1: Phosphatase and tensin homolog-induced putative kinase 1, PRKN: Parkin RBR E3 Ubiquitin Protein Ligase, AAV: Adeno-associated Virus, aMTD: Amino-terminal modulator of drosophila.
.
Gene therapy in PDD is primarily designed to mitigate neuronal death through the modulation of autophagic pathways, restoration of dopaminergic function, and intervention in alpha-synucleinopathy. This multifaceted approach seeks to enhance neuronal survival and function by addressing the underlying pathophysiological mechanisms associated with PDD. PDD: Parkinson’s disease dementia, LBD: Lewy body disease, ER: Endoplasmic reticulum, SNCA: Synuclein Alpha, GBA: Glucocerebrosidase, LRRK2: Leucine-Rich Repeat Kinase 2, PINK1: Phosphatase and tensin homolog-induced putative kinase 1, PRKN: Parkin RBR E3 Ubiquitin Protein Ligase, AAV: Adeno-associated Virus, aMTD: Amino-terminal modulator of drosophila.
PDD arises from the progressive degeneration of dopaminergic neuron in substantia nigra pars compacta (SNpc) due to α-synuclein accumulation lowering dopamine levels leading to sensorimotor dysfunction and cognitive decline.93 Motor symptoms such as bradykinesia, tremors, gait dysfunction, and rigidity are hallmarks of PDD, while non-motor symptoms such as dementia, depression, and anxiety emerge as the disease progresses.94 The pathology and symptoms of DLB significantly overlap with those of PDD, with cognitive decline, and motor dysfunction, typically becoming apparent within the first year of disease onset.95 The hallmark symptoms of DLB include cognitive impairment, executive dysfunction, visuospatial deficits, visual hallucinations, and memory loss.96
Gene therapy for PDD primarily focuses on dopamine replacement, targeting motor symptoms more than cognitive deficits. This emphasis stems from the fact that dopamine reduction in the striatum initially causes motor dysfunction, with cognitive impairments developing later.97 For example, Jarraya et al injected a viral vector encoding dopamine synthesis genes, such as tyrosine hydroxylase, into the striatum of a macaque model of PDD. They reported increased dopamine expression and extracellular dopamine concentrations, which corrected motor deficits without inducing dyskinesia, unlike traditional L-dopa treatment.98 Similar results were observed in clinical trials conducted by Christine et al99 and in a Phase 1 trial by Chadwick et al on putaminal AADC gene therapy.100 Despite these advances, a gap remains in understanding the effects of dopamine gene therapy on cognitive symptoms in PDD. While medications such as levodopa and pramipexole have been shown to improve working memory and alleviate depression in PDD patients, there is no clear evidence linking gene therapy to cognitive improvement.101,102 Other studies have focused on neurotrophins such as GDNF and BDNF, revealing that serum levels and gene polymorphisms of these factors are related to PD. However, gene therapy involving neurotrophins has not yet yielded significant results for PDD, despite improvements in motor symptoms.103-105 These findings indicate that while neurotrophin and dopamine gene therapies hold promise for addressing motor deficits, the cognitive aspects of PDD remain underexplored.106,107 Further research is required to evaluate the impact of gene therapy on learning and memory in PDD.
Gene therapy targeting different proteins represents another promising avenue for PDD treatment. In an experimental study, Lee et al demonstrated that injecting a novel AAV-Parkin construct, termed AAV-aMTD-Parkin, which combines a hydrophobic cell-penetrating peptide sequence with AAV and Parkin DNA, into the brains of 6-OHDA- and AAV-α-Syn-induced PDD mice enhanced both motor and cognitive functions. This gene therapy also activated tyrosine hydroxylase (TH), a key enzyme in dopamine production, and significantly improved cell and tissue permeability.108 Similarly, Ma et al injected a rAAV2 vector encoding poly(rC)-binding protein 1 (PCBP1), a protein that regulates gene expression and plays a role in neurodegenerative diseases, into a 6-OHDA cell and rat model of PDD. The treated groups presented notable improvements in behavioral abnormalities, reduced anxiety, and enhanced cell protection against death in culture.109 However, further research is needed to evaluate the broader effects of gene therapy on PDD, particularly regarding movement and memory.
Another promising approach in gene therapy for LBD is the prevention of α-synucleinopathy. Research in this area primarily focuses on experimental studies with limited cognitive assessment. For instance, Spencer et al employed lentiviral vectors expressing Beclin-1, an autophagy regulator, in α-synuclein transgenic mice. Their results indicated reduced α-synuclein accumulation in the limbic system, along with diminished synaptic and dendritic dysfunction. Coexpression of Beclin-1 and α-synuclein in neuronal cell lines also led to reduced autophagy.110 However, studies by Crews et al found no significant difference in Beclin-1 levels between DLB patients and controls. Their work showed that intracerebral injection of a lentiviral vector expressing Atg7, another autophagy pathway component, in transgenic mice reduced α-synuclein accumulation, mitigated neuronal damage, and elevated levels of MAP2, a dendritic marker, in the hippocampus.111 These findings highlight the crucial role of lysosomal dysfunction and autophagy in α-synucleinopathies.
Further gene therapy research has focused on replacing α-synuclein with β-synuclein, a non-amyloidogenic homolog. Unilateral intracerebral injection of lentiviral β-synuclein in transgenic models of α-synucleinopathies has shown promise by reducing α-synuclein accumulation in synapses and promoting Akt activation, which enhances neurite development and protects neurons from the toxic effects of α-synuclein.112 These studies suggest that gene therapy has potential for addressing α-synucleinopathies by targeting various molecular pathways and mechanisms involved in the disease. Further research is necessary to refine these approaches and assess their efficacy in clinical settings.
The CRISPR/Cas9 method represents another potential gene therapy option for α-synucleinopathies, particularly in PDD. A key target in these studies is the SNCA gene. Chen et al used CRISPR/Cas9n to delete the endogenous SNCA gene in midbrain dopaminergic (mDA) neuronal precursors differentiated from human embryonic stem cells (hESCs). They reported that the transgenic mDA cells were resistant to α-synucleinopathy.113 Similarly, Sastre et al developed a CRISPR interference (CRISPRi) system targeting α-synuclein expression at the transcriptional level in iPSC-derived neuronal cultures from a patient with an SNCA genomic triplication. Their study demonstrated a variable downregulation of α-synuclein, which led to a reduction in oxidative stress and mitochondrial DNA damage, ultimately rescuing the cells from death.114
In another approach, some studies explored reprogramming striatal glial cells into induced GABAergic neurons using CRISPR/Cas gene-editing as a potential treatment for Parkinson's disease (PD).115,116 For example, Giehrl-Schwab et al showed that astrocyte-derived induced GABAergic neurons could integrate into the neural circuits of the striatum and improve motor function in a PD model. However, despite the important role of the striatum in cognition-related behaviors,117 this study did not investigate cognitive outcomes (Fig. 4).
Gene therapy in Frontotemporal dementia
Frontotemporal dementia (FTD) is an enigmatic neurodegenerative clinical syndrome characterized by progressive deficits in behavior, executive function, and language. It is one of the most common types of dementia across all age groups and is considered an early-onset form of dementia. FTD includes different subtypes, such as the behavioral variant (bvFTD), which is mainly associated with personality changes and social-emotional functioning, and primary progressive aphasia (PPA), which further divides into semantic variant PPA (svPPA) and nonfluent variant PPA (nfvPPA).118 FTD is characterized by atrophy of the frontal and temporal lobes, resulting from protein misfolding and their abnormal deposition in central nervous system cells. Protein accumulation, primarily TDP-43, MAPT, and FUS, has been observed in 50%, 40%, and 10% of cases, respectively. The abnormal protein folding is often due to genetic mutations (25%), including mutations in the chromosome 9 open reading frame 72 (C9orf72, lead to TDP-43 accumulation) gene, progranulin (GRN, has role in inflammation and neuronal survival), and tau microtubule-associated protein (MAPT, has role in tau coding).119 Elevated levels of Aβ in the cerebrospinal fluid of young individuals with progressive dementia are also linked to the occurrence of FTD, and widespread white matter damage is another hallmark of the disease.120 The hyperphosphorylated tau protein forms neurofibrillary tangles that interfere with axonal transport and disrupt neuronal communication, while TDP-43 mislocalizes from the nucleus to the cytoplasm, forming cytoplasmic inclusions that impair RNA processing and gene expression. The resulting inflammatory response, combined with the direct toxic effects of these protein aggregates, leads to progressive neuronal death and synaptic dysfunction. Ultimately, these processes contribute to the cognitive decline and behavioral changes characteristic of FTD.
GRN is a major target of gene therapy in FTD, explored in both clinical and experimental studies. For instance, sub-occipital intrathecal injection of PR006, a recombinant AAV9 vector targeting GRN, was administered to 15 participants with GRN-mutant FTD to evaluate safety and tolerability.121 Preclinical studies demonstrated that a single injection of PR006 lowered neuroinflammatory marker expression, reduced ubiquitin accumulation, and increased GRN expression in FTD mouse models, all while being well-tolerated.122 In a first-in-human phase 1/2 open-label trial, Sevigny et al reported that low-dose (n = 6) and mid-dose (n = 7) PR006 administered intrathecally was generally safe and well-tolerated, with cerebrospinal fluid (CSF) pleocytosis being the most common adverse event related to PR006. Although blood progranulin levels transiently increased in most patients, cognitive and behavioral performance, assessed by Clinical Dementia Rating (CDR), showed progressive decline within the broad range reported for FTD patients in both groups.123 Similarly, AAV9-mediated human GRN delivery via stereotaxic microinjection into the right posterior lateral ventricle of GRN knockout mice led to sustained progranulin expression in the brain, particularly in the ipsilateral striatum and frontal cortex, in a cell- and region-specific manner.124 Lysosomal dysfunction is another critical aspect of FTD pathology, especially in cases of complete GRN deficiency, which results in neuronal ceroid lipofuscinosis (NCL), a lysosomal storage disorder.125 Arrant et al demonstrated that AAV-mediated GRN delivery in a transgenic mouse model of FTD improved lysosomal function, reduced microgliosis, and corrected abnormal lysosomal enzyme activity, such as cathepsin D.126 Other studies found that inhibiting the GRN mutation RNA pathway could elevate GRN mRNA levels in transgenic FTD mouse models.127 Collectively, these data suggest that GRN gene therapy is a valuable approach for managing FTD, though further research, particularly into cognitive outcomes, is still needed (Fig. 5).
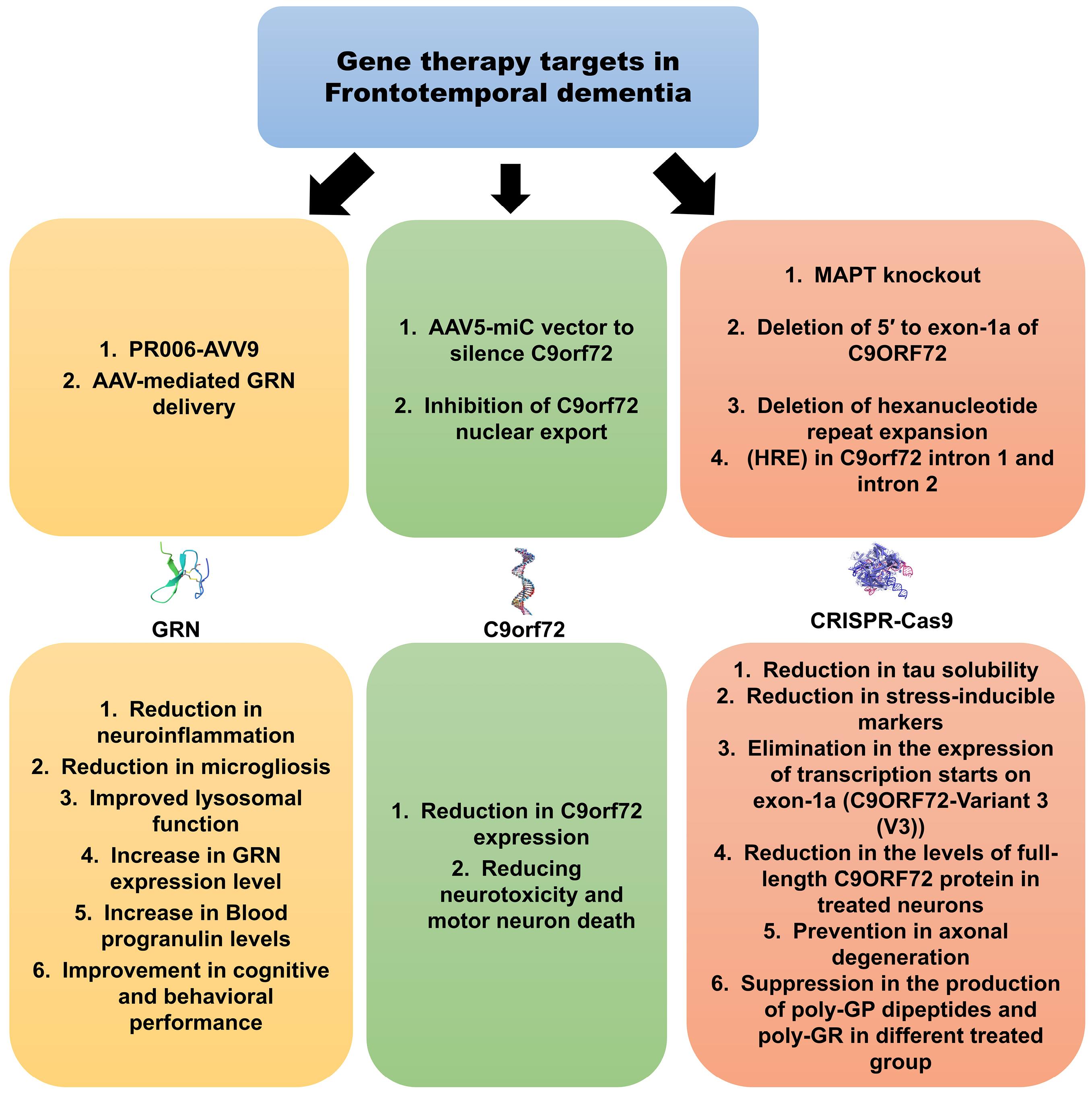
Fig. 5.
Gene therapy in frontotemporal dementia aims to address the pathophysiological effects of this disease, particularly by targeting GRN and C9orf72, to preserve neurons and reduce the incidence of cognitive impairments. GRN: Granulin, C9orf72: Chromosome 9 open reading frame 72, AAV: Adeno-associated virus, MAPT: Microtubule-associated protein tau.
.
Gene therapy in frontotemporal dementia aims to address the pathophysiological effects of this disease, particularly by targeting GRN and C9orf72, to preserve neurons and reduce the incidence of cognitive impairments. GRN: Granulin, C9orf72: Chromosome 9 open reading frame 72, AAV: Adeno-associated virus, MAPT: Microtubule-associated protein tau.
Another gene therapy target in FTD is C9orf72, though sparse studies exist on this target. Mutations in the C9orf72 gene are common in both amyotrophic lateral sclerosis (ALS) and FTD. Martier et al conducted one of the most comprehensive studies, using an AAV5-miC vector to silence C9orf72 in human-derived induced pluripotent stem cell (iPSC) neurons and an ALS mouse model. They reported reduced C9orf72 expression and diminished accumulation of repeat-containing C9orf72 transcripts in iPSCs, as well as a 20%-40% reduction in C9orf72 mRNA in the cortex and striatum of mice.128 Inhibition of C9orf72 nuclear export through depletion of SRSF1 or blocking its interaction with NXF1 also showed promise in reducing neurotoxicity and motor neuron death in vitro and in vivo ALS models, with potential implications for FTD.129 CRISPR/Cas9 gene editing has emerged as a promising therapeutic approach for FTD, particularly targeting C9orf72 and MAPT. Although most studies have been conducted in cell lines, in vivo research remains rare. Consequently, further studies, particularly focusing on cognitive symptoms, are crucial to determining the full therapeutic potential of CRISPR/Cas9 in FTD (Table 3).
Table 3.
Therapeutic usage of CRISPR/Cas9 in FTD treatment
|
Author
|
Model
|
Method
|
Findings
|
| Silva et al130 |
|
|
|
| Krishnan et al131 |
|
|
-
Elimination in the expression of transcription starts on exon-1a (C9ORF72-Variant 3 (V3))
-
Reduction in the levels of full-length C9ORF72 protein in treated neurons
-
Prevention in axonal degeneration
|
| Meijboom et al132 |
|
|
|
Concluding remarks
Gene therapy offers a novel approach to addressing the underlying genetic and molecular mechanisms of dementia. Its potential to target specific pathological processes in common forms of dementia, including AD, VaD, DLB, and FTD, provides a promising avenue for more effective treatments. However, despite the potential, several challenges remain, including concerns over long-term safety and the complexity of gene regulatory pathways. Additionally, the full genetic role in various forms of dementia is not yet fully understood, which limits the current effectiveness of gene therapy. One significant challenge is finding safe and effective methods for gene delivery, though advancements in vector technologies continue. Most research has focused primarily on the genetic basis of dementia, with less emphasis on other factors affecting cognition, learning, and memory—the primary clinical manifestations of the disease. As the field advances, collaboration among scientists, healthcare providers, and regulatory bodies will be essential for the responsible and effective application of gene therapy in dementia. The future holds great potential, and gene therapy could ultimately lead to significant advances in treatment for dementia.
Review Highlights
-
Gene therapy offers new hope for patients with various forms of dementia, as researchers investigate its potential to slow or halt disease progression.
-
Recent studies suggest that gene therapy may have a positive impact on dementia by targeting specific genetic mechanisms underlying the condition.
Competing Interests
None to be declared.
Ethical Approval
Not applicable.
Acknowledgements
The authors would like to express their gratitude to the Central Research Laboratory, Aja University of Medical Sciences, for providing the necessary resources and support for this research.
References
- World Health Organization (WHO). Towards a Dementia Plan: A WHO Guide. WHO; 2018.
- Hildreth KL, Church S. Evaluation and management of the elderly patient presenting with cognitive complaints. Med Clin North Am 2015; 99:311-35. doi: 10.1016/j.mcna.2014.11.006 [Crossref] [ Google Scholar]
- Mohseni M, Behzad G, Farhadi A, Behroozi J, Mohseni H, Valipour B. MicroRNAs regulating autophagy: opportunities in treating neurodegenerative diseases. Front Neurosci 2024; 18:1397106. doi: 10.3389/fnins.2024.1397106 [Crossref] [ Google Scholar]
- Geldmacher DS, Whitehouse PJ. Evaluation of dementia. N Engl J Med 1996; 335:330-6. doi: 10.1056/nejm199608013350507 [Crossref] [ Google Scholar]
- Dening T, Sandilyan MB. Dementia: definitions and types. Nurs Stand 2015; 29:37-42. doi: 10.7748/ns.29.37.37.e9405 [Crossref] [ Google Scholar]
- Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 2013; 9: 63-75.e2. doi: 10.1016/j.jalz.2012.11.007.
- Dintica CS, Yaffe K. Epidemiology and risk factors for dementia. Psychiatr Clin North Am 2022; 45:677-89. doi: 10.1016/j.psc.2022.07.011 [Crossref] [ Google Scholar]
- Erten-Lyons D, Howieson D, Moore MM, Quinn J, Sexton G, Silbert L. Brain volume loss in MCI predicts dementia. Neurology 2006; 66:233-5. doi: 10.1212/01.wnl.0000194213.50222.1a [Crossref] [ Google Scholar]
- Power MC, Mormino E, Soldan A, James BD, Yu L, Armstrong NM. Combined neuropathological pathways account for age-related risk of dementia. Ann Neurol 2018; 84:10-22. doi: 10.1002/ana.25246 [Crossref] [ Google Scholar]
- Mao P. Oxidative stress and its clinical applications in dementia. J Neurodegener Dis 2013; 2013:319898. doi: 10.1155/2013/319898 [Crossref] [ Google Scholar]
- Wolters FJ, Zonneveld HI, Hofman A, van der Lugt A, Koudstaal PJ, Vernooij MW. Cerebral perfusion and the risk of dementia: a population-based study. Circulation 2017; 136:719-28. doi: 10.1161/circulationaha.117.027448 [Crossref] [ Google Scholar]
- Austin BP, Nair VA, Meier TB, Xu G, Rowley HA, Carlsson CM. Effects of hypoperfusion in Alzheimer's disease. J Alzheimers Dis 2011; 26 Suppl 3:123-33. doi: 10.3233/jad-2011-0010 [Crossref] [ Google Scholar]
- Ninomiya T. Diabetes mellitus and dementia. Curr Diab Rep 2014; 14:487. doi: 10.1007/s11892-014-0487-z [Crossref] [ Google Scholar]
- Morelli L, Llovera R, Gonzalez SA, Affranchino JL, Prelli F, Frangione B. Differential degradation of amyloid beta genetic variants associated with hereditary dementia or stroke by insulin-degrading enzyme. J Biol Chem 2003; 278:23221-6. doi: 10.1074/jbc.M300276200 [Crossref] [ Google Scholar]
- Yang L, Wang H, Liu L, Xie A. The role of insulin/IGF-1/PI3K/Akt/GSK3β signaling in Parkinson's disease dementia. Front Neurosci 2018; 12:73. doi: 10.3389/fnins.2018.00073 [Crossref] [ Google Scholar]
- Cholerton B, Baker LD, Craft S. Insulin, cognition, and dementia. Eur J Pharmacol 2013; 719:170-9. doi: 10.1016/j.ejphar.2013.08.008 [Crossref] [ Google Scholar]
- Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020; 396:413-46. doi: 10.1016/s0140-6736(20)30367-6 [Crossref] [ Google Scholar]
- Loy CT, Schofield PR, Turner AM, Kwok JB. Genetics of dementia. Lancet 2014; 383:828-40. doi: 10.1016/s0140-6736(13)60630-3 [Crossref] [ Google Scholar]
- Gonçalves GAR, de Melo Alves Paiva R. Gene therapy: advances, challenges and perspectives. Einstein (Sao Paulo) 2017; 15:369-75. doi: 10.1590/s1679-45082017rb4024 [Crossref] [ Google Scholar]
- Rastgar A, Kheyrandish S, Vahidi M, Heidari R, Ghorbani M. Advancements in small interfering RNAs therapy for acute lymphoblastic leukemia: promising results and future perspectives. Mol Biol Rep 2024; 51:737. doi: 10.1007/s11033-024-09650-y [Crossref] [ Google Scholar]
- Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science 2018; 359:eaan4672. doi: 10.1126/science.aan4672 [Crossref] [ Google Scholar]
- Behroozi J, Shahbazi S, Bakhtiarizadeh MR, Mahmoodzadeh H. Genome-wide characterization of RNA editing sites in primary gastric adenocarcinoma through RNA-seq data analysis. Int J Genomics 2020; 2020:6493963. doi: 10.1155/2020/6493963 [Crossref] [ Google Scholar]
- Isazadeh H, Oruji F, Shabani S, Behroozi J, Nasiri H, Isazadeh A. Advances in siRNA delivery approaches in cancer therapy: challenges and opportunities. Mol Biol Rep 2023; 50:9529-43. doi: 10.1007/s11033-023-08749-y [Crossref] [ Google Scholar]
- Moaveni AK, Amiri M, Shademan B, Farhadi A, Behroozi J, Nourazarian A. Advances and challenges in gene therapy strategies for pediatric cancer: a comprehensive update. Front Mol Biosci 2024; 11:1382190. doi: 10.3389/fmolb.2024.1382190 [Crossref] [ Google Scholar]
- Conniot J, Talebian S, Simões S, Ferreira L, Conde J. Revisiting gene delivery to the brain: silencing and editing. Biomater Sci 2021; 9:1065-87. doi: 10.1039/d0bm01278e [Crossref] [ Google Scholar]
- Loera-Valencia R, Piras A, Ismail MA, Manchanda S, Eyjolfsdottir H, Saido TC. Targeting Alzheimer's disease with gene and cell therapies. J Intern Med 2018; 284:2-36. doi: 10.1111/joim.12759 [Crossref] [ Google Scholar]
- Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT. Alzheimer disease. Nat Rev Dis Primers 2021; 7:33. doi: 10.1038/s41572-021-00269-y [Crossref] [ Google Scholar]
- Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE. Alzheimer's disease. Lancet 2021; 397:1577-90. doi: 10.1016/s0140-6736(20)32205-4 [Crossref] [ Google Scholar]
- Hammers DB, Eloyan A, Thangarajah M, Taurone A, Beckett L, Gao S. Differences in baseline cognitive performance between participants with early-onset and late-onset Alzheimer's disease: comparison of LEADS and ADNI. Alzheimers Dement 2025; 21:e14218. doi: 10.1002/alz.14218 [Crossref] [ Google Scholar]
- Shulman JM, Chen K, Keenan BT, Chibnik LB, Fleisher A, Thiyyagura P. Genetic susceptibility for Alzheimer disease neuritic plaque pathology. JAMA Neurol 2013; 70:1150-7. doi: 10.1001/jamaneurol.2013.2815 [Crossref] [ Google Scholar]
- Trejo-Lopez JA, Yachnis AT, Prokop S. Neuropathology of Alzheimer's disease. Neurotherapeutics 2022; 19:173-85. doi: 10.1007/s13311-021-01146-y [Crossref] [ Google Scholar]
-
Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta: a crucial factor in Alzheimer's disease. Med Princ Pract 2015. 24: 1-10. doi: 10.1159/000369101.
- Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM. Amyloid-β oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol 2013; 73:104-19. doi: 10.1002/ana.23748 [Crossref] [ Google Scholar]
- Hunter S, Brayne C. Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol Psychiatry 2018; 23:81-93. doi: 10.1038/mp.2017.218 [Crossref] [ Google Scholar]
- Perrone F, Bjerke M, Hens E, Sieben A, Timmers M, De Roeck A. Amyloid-β(1-43) cerebrospinal fluid levels and the interpretation of APP, PSEN1 and PSEN2 mutations. Alzheimers Res Ther 2020; 12:108. doi: 10.1186/s13195-020-00676-5 [Crossref] [ Google Scholar]
- Uddin MS, Kabir MT, Al Mamun A, Abdel-Daim MM, Barreto GE, Ashraf GM. APOE and Alzheimer's disease: evidence mounts that targeting APOE4 may combat Alzheimer's pathogenesis. Mol Neurobiol 2019; 56:2450-65. doi: 10.1007/s12035-018-1237-z [Crossref] [ Google Scholar]
- Steward A, Biel D, Dewenter A, Roemer S, Wagner F, Dehsarvi A. ApoE4 and connectivity-mediated spreading of tau pathology at lower amyloid levels. JAMA Neurol 2023; 80:1295-306. doi: 10.1001/jamaneurol.2023.4038 [Crossref] [ Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC. Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 2019; 51:414-30. doi: 10.1038/s41588-019-0358-2 [Crossref] [ Google Scholar]
- Tuszynski MH, Thal L, Pay M, Salmon DP, Hoi-Sang U, Bakay R. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 2005; 11:551-5. doi: 10.1038/nm1239 [Crossref] [ Google Scholar]
- Tuszynski MH, Yang JH, Barba D, Hoi-Sang U, Bakay RA, Pay MM. Nerve growth factor gene therapy: activation of neuronal responses in Alzheimer disease. JAMA Neurol 2015; 72:1139-47. doi: 10.1001/jamaneurol.2015.1807 [Crossref] [ Google Scholar]
- Castle MJ, Baltanás FC, Kovacs I, Nagahara AH, Barba D, Tuszynski MH. Postmortem analysis in a clinical trial of AAV2-NGF gene therapy for Alzheimer's disease identifies a need for improved vector delivery. Hum Gene Ther 2020; 31:415-22. doi: 10.1089/hum.2019.367 [Crossref] [ Google Scholar]
- Mummery CJ, Börjesson-Hanson A, Blackburn DJ, Vijverberg EG, De Deyn PP, Ducharme S. Tau-targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer's disease: a phase 1b, randomized, placebo-controlled trial. Nat Med 2023; 29:1437-47. doi: 10.1038/s41591-023-02326-3 [Crossref] [ Google Scholar]
- Jahangard Y, Monfared H, Moradi A, Zare M, Mirnajafi-Zadeh J, Mowla SJ. Therapeutic effects of transplanted exosomes containing miR-29b to a rat model of Alzheimer's disease. Front Neurosci 2020; 14:564. doi: 10.3389/fnins.2020.00564 [Crossref] [ Google Scholar]
- Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer's disease. Brain 2017; 140:3233-51. doi: 10.1093/brain/awx258 [Crossref] [ Google Scholar]
- Karch CM, Ezerskiy LA, Bertelsen S, Goate AM. Alzheimer's disease risk polymorphisms regulate gene expression in the ZCWPW1 and the CELF1 Loci. PLoS One 2016; 11:e0148717. doi: 10.1371/journal.pone.0148717 [Crossref] [ Google Scholar]
- López-Sánchez N, Garrido-García A, Ramón-Landreau M, Cano-Daganzo V, Frade JM. E2F4-based gene therapy mitigates the phenotype of the Alzheimer's disease mouse model 5xFAD. Neurotherapeutics 2021; 18:2484-503. doi: 10.1007/s13311-021-01151-1 [Crossref] [ Google Scholar]
- Griciuc A, Federico AN, Natasan J, Forte AM, McGinty D, Nguyen H. Gene therapy for Alzheimer's disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum Mol Genet 2020; 29:2920-35. doi: 10.1093/hmg/ddaa179 [Crossref] [ Google Scholar]
- Wang XJ, Qi L, Cheng YF, Ji XF, Chi TY, Liu P. PINK1 overexpression prevents forskolin-induced tau hyperphosphorylation and oxidative stress in a rat model of Alzheimer's disease. Acta Pharmacol Sin 2022; 43:1916-27. doi: 10.1038/s41401-021-00810-5 [Crossref] [ Google Scholar]
- Ising C, Gallardo G, Leyns CE, Wong CH, Jiang H, Stewart F. AAV-mediated expression of anti-tau scFvs decreases tau accumulation in a mouse model of tauopathy. J Exp Med 2017; 214:1227-38. doi: 10.1084/jem.20162125 [Crossref] [ Google Scholar]
- Jiao SS, Shen LL, Zhu C, Bu XL, Liu YH, Liu CH. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer's disease. Transl Psychiatry 2016; 6:e907. doi: 10.1038/tp.2016.186 [Crossref] [ Google Scholar]
- Ittner A, Asih PR, Tan AR, Prikas E, Bertz J, Stefanoska K. Reduction of advanced tau-mediated memory deficits by the MAP kinase p38γ. Acta Neuropathol 2020; 140:279-94. doi: 10.1007/s00401-020-02191-1 [Crossref] [ Google Scholar]
- Asih PR, Stefanoska K, Prikas E, Ittner A. High level forebrain expression of active tau kinase p38γ exacerbates cognitive dysfunction in aged APP-transgenic Alzheimer's mice. Neuroscience 2022; 484:53-65. doi: 10.1016/j.neuroscience.2022.01.005 [Crossref] [ Google Scholar]
- Koller EJ, Gonzalez De La Cruz E, Weinrich M, Williams T, Cruz PE, Ryu D. Intracerebral expression of AAV-APOE4 is not sufficient to alter tau burden in two distinct models of tauopathy. Mol Neurobiol 2020; 57:1986-2001. doi: 10.1007/s12035-019-01859-4 [Crossref] [ Google Scholar]
- dos Santos Rodrigues B, Kanekiyo T, Singh J. ApoE-2 brain-targeted gene therapy through transferrin and penetratin tagged liposomal nanoparticles. Pharm Res 2019; 36:161. doi: 10.1007/s11095-019-2691-7 [Crossref] [ Google Scholar]
- Jackson RJ, Keiser MS, Meltzer JC, Fykstra DP, Dierksmeier SE, Melloni A, et al. APOE2 gene therapy reduces amyloid deposition, and improves markers of neuroinflammation and neurodegeneration in a mouse model of Alzheimer disease. bioRxiv [Preprint]. August 16, 2023. Available from: https://www.biorxiv.org/content/10.1101/2023.08.14.552850v1.
- Selles MC, Fortuna JT, Cercato MC, Santos LE, Domett L, Bitencourt AL. AAV-mediated neuronal expression of an scFv antibody selective for Aβ oligomers protects synapses and rescues memory in Alzheimer models. Mol Ther 2023; 31:409-19. doi: 10.1016/j.ymthe.2022.11.002 [Crossref] [ Google Scholar]
- Arora S, Kanekiyo T, Singh J. Functionalized nanoparticles for brain targeted BDNF gene therapy to rescue Alzheimer's disease pathology in transgenic mouse model. Int J Biol Macromol 2022; 208:901-11. doi: 10.1016/j.ijbiomac.2022.03.203 [Crossref] [ Google Scholar]
- Elmer BM, Swanson KA, Bangari DS, Piepenhagen PA, Roberts E, Taksir T. Gene delivery of a modified antibody to Aβ reduces progression of murine Alzheimer's disease. PLoS One 2019; 14:e0226245. doi: 10.1371/journal.pone.0226245 [Crossref] [ Google Scholar]
- Murphy SR, Chang CC, Dogbevia G, Bryleva EY, Bowen Z, Hasan MT. Acat1 knockdown gene therapy decreases amyloid-β in a mouse model of Alzheimer's disease. Mol Ther 2013; 21:1497-506. doi: 10.1038/mt.2013.118 [Crossref] [ Google Scholar]
- Kiyota T, Ingraham KL, Swan RJ, Jacobsen MT, Andrews SJ, Ikezu T. AAV serotype 2/1-mediated gene delivery of anti-inflammatory interleukin-10 enhances neurogenesis and cognitive function in APP + PS1 mice. Gene Ther 2012; 19:724-33. doi: 10.1038/gt.2011.126 [Crossref] [ Google Scholar]
- Revilla S, Ursulet S, Álvarez-López MJ, Castro-Freire M, Perpiñá U, García-Mesa Y. Lenti-GDNF gene therapy protects against Alzheimer's disease-like neuropathology in 3xTg-AD mice and MC65 cells. CNS Neurosci Ther 2014; 20:961-72. doi: 10.1111/cns.12312 [Crossref] [ Google Scholar]
- Li H, Chen Z, Shen Y, Xiong T, Chen A, Chen L. Gene therapy in Aβ-induced cell and mouse models of Alzheimer's disease through compensating defective mitochondrial complex I function. J Transl Med 2024; 22:760. doi: 10.1186/s12967-024-05571-3 [Crossref] [ Google Scholar]
- Ng RC, Jian M, Ma OK, Xiang AW, Bunting M, Kwan JS. Liver-specific adiponectin gene therapy suppresses microglial NLRP3-inflammasome activation for treating Alzheimer's disease. J Neuroinflammation 2024; 21:77. doi: 10.1186/s12974-024-03066-y [Crossref] [ Google Scholar]
- Zheng N, Li K, Cao J, Wang Z, Zhang L, Zhao Z. Electrophysiology-based screening identifies neuronal HtrA serine peptidase 2 (HTRA2) as a synaptic plasticity regulator participating in tauopathy. Transl Psychiatry 2025; 15:5. doi: 10.1038/s41398-025-03227-4 [Crossref] [ Google Scholar]
- Jackson RJ, Keiser MS, Meltzer JC, Fykstra DP, Dierksmeier SE, Hajizadeh S. APOE2 gene therapy reduces amyloid deposition and improves markers of neuroinflammation and neurodegeneration in a mouse model of Alzheimer disease. Mol Ther 2024; 32:1373-86. doi: 10.1016/j.ymthe.2024.03.024 [Crossref] [ Google Scholar]
- Konstantinidis E, Molisak A, Perrin F, Streubel-Gallasch L, Fayad S, Kim DY. CRISPR-Cas9 treatment partially restores amyloid-β 42/40 in human fibroblasts with the Alzheimer's disease PSEN 1 M146L mutation. Mol Ther Nucleic Acids 2022; 28:450-61. doi: 10.1016/j.omtn.2022.03.022 [Crossref] [ Google Scholar]
- Raikwar SP, Thangavel R, Dubova I, Selvakumar GP, Ahmed ME, Kempuraj D. Targeted gene editing of glia maturation factor in microglia: a novel Alzheimer's disease therapeutic target. Mol Neurobiol 2019; 56:378-93. doi: 10.1007/s12035-018-1068-y [Crossref] [ Google Scholar]
- Park H, Shin J, Kim Y, Saito T, Saido TC, Kim J. CRISPR/dCas9-Dnmt3a-mediated targeted DNA methylation of APP rescues brain pathology in a mouse model of Alzheimer's disease. Transl Neurodegener 2022; 11:41. doi: 10.1186/s40035-022-00314-0 [Crossref] [ Google Scholar]
- Gyorgy B, Ingelsson M, Loov C, Takeda S, Lannfelt L, Hyman BT. 567 CRISPR-Cas9 mediated gene editing in a monogenic form of Alzheimer's disease. Mol Ther 2016; 24:S226-7. doi: 10.1016/s1525-0016(16)33375-5 [Crossref] [ Google Scholar]
- Ortiz-Virumbrales M, Moreno CL, Kruglikov I, Marazuela P, Sproul A, Jacob S. CRISPR/Cas9-Correctable mutation-related molecular and physiological phenotypes in iPSC-derived Alzheimer's PSEN2 (N141I) neurons. Acta Neuropathol Commun 2017; 5:77. doi: 10.1186/s40478-017-0475-z [Crossref] [ Google Scholar]
- Duan Y, Ye T, Qu Z, Chen Y, Miranda A, Zhou X. Brain-wide Cas9-mediated cleavage of a gene causing familial Alzheimer's disease alleviates amyloid-related pathologies in mice. Nat Biomed Eng 2022; 6:168-80. doi: 10.1038/s41551-021-00759-0 [Crossref] [ Google Scholar]
- Yang J, Qin G, Liu Z, Zhang H, Du X, Ren J. A nanozyme-boosted MOF-CRISPR platform for treatment of Alzheimer's disease. Nano Lett 2024; 24:9906-15. doi: 10.1021/acs.nanolett.4c02272 [Crossref] [ Google Scholar]
- Iadecola C. The pathobiology of vascular dementia. Neuron 2013; 80:844-66. doi: 10.1016/j.neuron.2013.10.008 [Crossref] [ Google Scholar]
- Román GC. Vascular dementia prevention: a risk factor analysis. Cerebrovasc Dis 2005; 20:91-100. doi: 10.1159/000089361 [Crossref] [ Google Scholar]
- Manso-Calderón R, González-Sarmiento R. Genetic susceptibility to vascular cognitive impairment: a pathophysiological view. Future Neurol 2016; 11:119-34. doi: 10.2217/fnl-2016-0002 [Crossref] [ Google Scholar]
- Mossanen Parsi M, Duval C, Ariëns RA. Vascular dementia and crosstalk between the complement and coagulation systems. Front Cardiovasc Med 2021; 8:803169. doi: 10.3389/fcvm.2021.803169 [Crossref] [ Google Scholar]
- Rutten JW, Dauwerse HG, Peters DJ, Goldfarb A, Venselaar H, Haffner C. Therapeutic NOTCH3 cysteine correction in CADASIL using exon skipping: in vitro proof of concept. Brain 2016; 139:1123-35. doi: 10.1093/brain/aww011 [Crossref] [ Google Scholar]
- Wang J, Zhang L, Wu G, Wu J, Zhou X, Chen X. Correction of a CADASIL point mutation using adenine base editors in hiPSCs and blood vessel organoids. J Genet Genomics 2024; 51:197-207. doi: 10.1016/j.jgg.2023.04.013 [Crossref] [ Google Scholar]
- Gravesteijn G, Dauwerse JG, Overzier M, Brouwer G, Hegeman I, Mulder AA. Naturally occurring NOTCH3 exon skipping attenuates NOTCH3 protein aggregation and disease severity in CADASIL patients. Hum Mol Genet 2020; 29:1853-63. doi: 10.1093/hmg/ddz285 [Crossref] [ Google Scholar]
- Ke XJ, Zhang JJ. Changes in HIF-1α, VEGF, NGF and BDNF levels in cerebrospinal fluid and their relationship with cognitive impairment in patients with cerebral infarction. J Huazhong Univ Sci Technolog Med Sci 2013; 33:433-7. doi: 10.1007/s11596-013-1137-4 [Crossref] [ Google Scholar]
- Kim Y, Kim YJ. Effect of obesity on cognitive impairment in vascular dementia rat model via BDNF-ERK-CREB pathway. Biol Res Nurs 2021; 23:248-57. doi: 10.1177/1099800420951633 [Crossref] [ Google Scholar]
- Zhu T, Zhu M, Qiu Y, Wu Z, Huang N, Wan G. Puerarin alleviates vascular cognitive impairment in vascular dementia rats. Front Behav Neurosci 2021; 15:717008. doi: 10.3389/fnbeh.2021.717008 [Crossref] [ Google Scholar]
- Wang F, Chang G, Geng X. NGF and TERT co-transfected BMSCs improve the restoration of cognitive impairment in vascular dementia rats. PLoS One 2014; 9:e98774. doi: 10.1371/journal.pone.0098774 [Crossref] [ Google Scholar]
- Hecht N, Marushima A, Nieminen M, Kremenetskaia I, von Degenfeld G, Woitzik J. Myoblast-mediated gene therapy improves functional collateralization in chronic cerebral hypoperfusion. Stroke 2015; 46:203-11. doi: 10.1161/strokeaha.114.006712 [Crossref] [ Google Scholar]
- Hiramatsu M, Hishikawa T, Tokunaga K, Kidoya H, Nishihiro S, Haruma J. Combined gene therapy with vascular endothelial growth factor plus apelin in a chronic cerebral hypoperfusion model in rats. J Neurosurg 2017; 127:679-86. doi: 10.3171/2016.8.Jns16366 [Crossref] [ Google Scholar]
- Sato N, Shimamura M, Takeuchi D, Kurinami H, Ogihara T, Morishita R. Gene therapy for ischemic brain disease with special reference to vascular dementia. Geriatr Gerontol Int 2007; 7:1-14. doi: 10.1111/j.1447-0594.2007.00373.x [Crossref] [ Google Scholar]
- Walker L, Stefanis L, Attems J. Clinical and neuropathological differences between Parkinson's disease, Parkinson's disease dementia and dementia with Lewy bodies - current issues and future directions. J Neurochem 2019; 150:467-74. doi: 10.1111/jnc.14698 [Crossref] [ Google Scholar]
- Outeiro TF, Koss DJ, Erskine D, Walker L, Kurzawa-Akanbi M, Burn D. Dementia with Lewy bodies: an update and outlook. Mol Neurodegener 2019; 14:5. doi: 10.1186/s13024-019-0306-8 [Crossref] [ Google Scholar]
- Bloem BR, Okun MS, Klein C. Parkinson's disease. Lancet 2021; 397:2284-303. doi: 10.1016/s0140-6736(21)00218-x [Crossref] [ Google Scholar]
- Domingo A, Klein C. Genetics of Parkinson disease. Handb Clin Neurol 2018; 147:211-27. doi: 10.1016/b978-0-444-63233-3.00014-2 [Crossref] [ Google Scholar]
- Trinh J, Zeldenrust FM, Huang J, Kasten M, Schaake S, Petkovic S. Genotype-phenotype relations for the Parkinson's disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord 2018; 33:1857-70. doi: 10.1002/mds.27527 [Crossref] [ Google Scholar]
- Sanghvi H, Singh R, Morrin H, Rajkumar AP. Systematic review of genetic association studies in people with Lewy body dementia. Int J Geriatr Psychiatry 2020; 35:436-48. doi: 10.1002/gps.5260 [Crossref] [ Google Scholar]
- Balestrino R, Schapira AH. Parkinson disease. Eur J Neurol 2020; 27:27-42. doi: 10.1111/ene.14108 [Crossref] [ Google Scholar]
- Simon DK, Tanner CM, Brundin P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med 2020; 36:1-12. doi: 10.1016/j.cger.2019.08.002 [Crossref] [ Google Scholar]
- Outeiro TF, Koss DJ, Erskine D, Walker L, Kurzawa-Akanbi M, Burn D. Dementia with Lewy bodies: an update and outlook. Mol Neurodegener 2019; 14:5. doi: 10.1186/s13024-019-0306-8 [Crossref] [ Google Scholar]
- Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 2007; 68:812-9. doi: 10.1212/01.wnl.0000256715.13907.d3 [Crossref] [ Google Scholar]
- Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J. Parkinson disease. Nat Rev Dis Primers 2017; 3:17013. doi: 10.1038/nrdp.2017.13 [Crossref] [ Google Scholar]
- Jarraya B, Boulet S, Ralph GS, Jan C, Bonvento G, Azzouz M. Dopamine gene therapy for Parkinson's disease in a nonhuman primate without associated dyskinesia. Sci Transl Med 2009; 1:2ra4. doi: 10.1126/scitranslmed.3000130 [Crossref] [ Google Scholar]
- Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, Hawkins RA. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 2009; 73:1662-9. doi: 10.1212/WNL.0b013e3181c29356 [Crossref] [ Google Scholar]
- Christine CW, Bankiewicz KS, Van Laar AD, Richardson RM, Ravina B, Kells AP. Magnetic resonance imaging-guided phase 1 trial of putaminal AADC gene therapy for Parkinson's disease. Ann Neurol 2019; 85:704-14. doi: 10.1002/ana.25450 [Crossref] [ Google Scholar]
- Lewis SJ, Slabosz A, Robbins TW, Barker RA, Owen AM. Dopaminergic basis for deficits in working memory but not attentional set-shifting in Parkinson's disease. Neuropsychologia 2005; 43:823-32. doi: 10.1016/j.neuropsychologia.2004.10.001 [Crossref] [ Google Scholar]
- Barone P, Poewe W, Albrecht S, Debieuvre C, Massey D, Rascol O. Pramipexole for the treatment of depressive symptoms in patients with Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010; 9:573-80. doi: 10.1016/s1474-4422(10)70106-x [Crossref] [ Google Scholar]
- Wang Y, Liu H, Zhang BS, Soares JC, Zhang XY. Low BDNF is associated with cognitive impairments in patients with Parkinson's disease. Parkinsonism Relat Disord 2016; 29:66-71. doi: 10.1016/j.parkreldis.2016.05.023 [Crossref] [ Google Scholar]
- Guerini FR, Beghi E, Riboldazzi G, Zangaglia R, Pianezzola C, Bono G. BDNF Val66Met polymorphism is associated with cognitive impairment in Italian patients with Parkinson's disease. Eur J Neurol 2009; 16:1240-5. doi: 10.1111/j.1468-1331.2009.02706.x [Crossref] [ Google Scholar]
- Liu Y, Tong S, Ding L, Liu N, Gao D. Serum levels of glial cell line-derived neurotrophic factor and multiple neurotransmitters: In relation to cognitive performance in Parkinson's disease with mild cognitive impairment. Int J Geriatr Psychiatry 2020; 35:153-62. doi: 10.1002/gps.5222 [Crossref] [ Google Scholar]
- Choi-Lundberg DL, Lin Q, Chang YN, Chiang YL, Hay CM, Mohajeri H. Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science 1997; 275:838-41. doi: 10.1126/science.275.5301.838 [Crossref] [ Google Scholar]
- Tereshchenko J, Maddalena A, Bähr M, Kügler S. Pharmacologically controlled, discontinuous GDNF gene therapy restores motor function in a rat model of Parkinson's disease. Neurobiol Dis 2014; 65:35-42. doi: 10.1016/j.nbd.2014.01.009 [Crossref] [ Google Scholar]
- Lee S, Kang M, Lee S, Yoon S, Cho Y, Min D. AAV-aMTD-Parkin, a therapeutic gene delivery cargo, enhances motor and cognitive functions in Parkinson's and Alzheimer's diseases. Pharmacol Res 2024; 208:107326. doi: 10.1016/j.phrs.2024.107326 [Crossref] [ Google Scholar]
- Ma LY, Wang L, Liang J, Huo L. Investigating the neuroprotective potential of rAAV2-PCBP1-EGFP gene therapy against a 6-OHDA-induced model of Parkinson's disease. Brain Behav 2024; 14:e3376. doi: 10.1002/brb3.3376 [Crossref] [ Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci 2009; 29:13578-88. doi: 10.1523/jneurosci.4390-09.2009 [Crossref] [ Google Scholar]
- Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010; 5:e9313. doi: 10.1371/journal.pone.0009313 [Crossref] [ Google Scholar]
- Hashimoto M, Rockenstein E, Mante M, Crews L, Bar-On P, Gage FH. An antiaggregation gene therapy strategy for Lewy body disease utilizing beta-synuclein lentivirus in a transgenic model. Gene Ther 2004; 11:1713-23. doi: 10.1038/sj.gt.3302349 [Crossref] [ Google Scholar]
- Chen Y, Dolt KS, Kriek M, Baker T, Downey P, Drummond NJ. Engineering synucleinopathy-resistant human dopaminergic neurons by CRISPR-mediated deletion of the SNCA gene. Eur J Neurosci 2019; 49:510-24. doi: 10.1111/ejn.14286 [Crossref] [ Google Scholar]
- Sastre D, Zafar F, Torres CAM, Piper D, Kirik D, Sanders LH. Inactive S aureus Cas9 downregulates alpha-synuclein and reduces mtDNA damage and oxidative stress levels in human stem cell model of Parkinson's disease. Sci Rep 2023; 13:17796. doi: 10.1038/s41598-023-45078-3 [Crossref] [ Google Scholar]
-
Zhou H, Su J, Hu X, Zhou C, Li H, Chen Z, et al. Glia-to-neuron conversion by CRISPR-CasRx alleviates symptoms of neurological disease in mice. Cell 2020; 181: 590-603.e16. doi: 10.1016/j.cell.2020.03.024.
- Giehrl-Schwab J, Giesert F, Rauser B, Lao CL, Hembach S, Lefort S. Parkinson's disease motor symptoms rescue by CRISPRa-reprogramming astrocytes into GABAergic neurons. EMBO Mol Med 2022; 14:e14797. doi: 10.15252/emmm.202114797 [Crossref] [ Google Scholar]
- Martins GJ, Shahrokh M, Powell EM. Genetic disruption of Met signaling impairs GABAergic striatal development and cognition. Neuroscience 2011; 176:199-209. doi: 10.1016/j.neuroscience.2010.12.058 [Crossref] [ Google Scholar]
- Bright F, Werry EL, Dobson-Stone C, Piguet O, Ittner LM, Halliday GM. Neuroinflammation in frontotemporal dementia. Nat Rev Neurol 2019; 15:540-55. doi: 10.1038/s41582-019-0231-z [Crossref] [ Google Scholar]
- Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol 2019; 266:2075-86. doi: 10.1007/s00415-019-09363-4 [Crossref] [ Google Scholar]
- Tan RH, Kril JJ, Yang Y, Tom N, Hodges JR, Villemagne VL. Assessment of amyloid β in pathologically confirmed frontotemporal dementia syndromes. Alzheimers Dement (Amst) 2017; 9:10-20. doi: 10.1016/j.dadm.2017.05.005 [Crossref] [ Google Scholar]
- Uspenskaya-Cadoz O, Benoit J, Mahoney E, Velaga J, Sondergaard P, Lowrey M. Design of a phase 1/2 study of an AAV9-based gene therapy for fronto-temporal dementia patients with pathogenic GRN mutations (PROCLAIM trial). Alzheimers Dement 2020; 16:e040916. doi: 10.1002/alz.040916 [Crossref] [ Google Scholar]
- Heckman LD, Burstein SR, Brandes A, Wong LC, Yang Z, Lin HN. Preclinical development of PR006, a gene therapy for the treatment of frontotemporal dementia with progranulin mutations. Alzheimers Dement 2020; 16:e043632. doi: 10.1002/alz.043632 [Crossref] [ Google Scholar]
- Sevigny J, Uspenskaya O, Heckman LD, Wong LC, Hatch DA, Tewari A. Progranulin AAV gene therapy for frontotemporal dementia: translational studies and phase 1/2 trial interim results. Nat Med 2024; 30:1406-15. doi: 10.1038/s41591-024-02973-0 [Crossref] [ Google Scholar]
- Amado DA, Rieders JM, Diatta F, Hernandez-Con P, Singer A, Mak JT. AAV-mediated progranulin delivery to a mouse model of progranulin deficiency causes T cell-mediated toxicity. Mol Ther 2019; 27:465-78. doi: 10.1016/j.ymthe.2018.11.013 [Crossref] [ Google Scholar]
- Canafoglia L, Morbin M, Scaioli V, Pareyson D, D'Incerti L, Fugnanesi V. Recurrent generalized seizures, visual loss, and palinopsia as phenotypic features of neuronal ceroid lipofuscinosis due to progranulin gene mutation. Epilepsia 2014; 55:e56-9. doi: 10.1111/epi.12632 [Crossref] [ Google Scholar]
- Arrant AE, Onyilo VC, Unger DE, Roberson ED. Progranulin gene therapy improves lysosomal dysfunction and microglial pathology associated with frontotemporal dementia and neuronal ceroid lipofuscinosis. J Neurosci 2018; 38:2341-58. doi: 10.1523/jneurosci.3081-17.2018 [Crossref] [ Google Scholar]
- Smith DM, Niehoff ML, Ling K, Jafar-Nejad P, Rigo F, Farr SA. Targeting nonsense-mediated RNA decay does not increase progranulin levels in the Grn R493X mouse model of frontotemporal dementia. PLoS One 2023; 18:e0282822. doi: 10.1371/journal.pone.0282822 [Crossref] [ Google Scholar]
- Martier R, Liefhebber JM, García-Osta A, Miniarikova J, Cuadrado-Tejedor M, Espelosin M. Targeting RNA-mediated toxicity in C9orf72 ALS and/or FTD by RNAi-based gene therapy. Mol Ther Nucleic Acids 2019; 16:26-37. doi: 10.1016/j.omtn.2019.02.001 [Crossref] [ Google Scholar]
- Hautbergue GM, Castelli LM, Ferraiuolo L, Sanchez-Martinez A, Cooper-Knock J, Higginbottom A. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat Commun 2017; 8:16063. doi: 10.1038/ncomms16063 [Crossref] [ Google Scholar]
-
Silva MC, Watson LA, Sheridan SD, Lucente DE, Brandon NJ, Tsai LH, et al. Rescue of tau phenotypes in human iPSC-derived neuronal models of frontotemporal degeneration with experimental therapeutics. Neurology 2017; 88: P2.082. doi: 10.1212/WNL.88.16_supplement.P2.082.
- Krishnan G, Zhang Y, Gu Y, Kankel MW, Gao FB, Almeida S. CRISPR deletion of the C9ORF72 promoter in ALS/FTD patient motor neurons abolishes production of dipeptide repeat proteins and rescues neurodegeneration. Acta Neuropathol 2020; 140:81-4. doi: 10.1007/s00401-020-02154-6 [Crossref] [ Google Scholar]
- Meijboom KE, Abdallah A, Fordham NP, Nagase H, Rodriguez T, Kraus C. CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro. Nat Commun 2022; 13:6286. doi: 10.1038/s41467-022-33332-7 [Crossref] [ Google Scholar]