Bioimpacts. 2025;15:30905.
doi: 10.34172/bi.30905
Review
Potential role of epidermal growth factor receptors (EGFR) signaling in the pathogenesis and management of hepatocellular carcinoma
Samah Saleh Ahmed Al-Awadhi Conceptualization, Writing – original draft, Writing – review & editing, 1 
Prakash Patil Conceptualization, Writing – review & editing, 1
Praveenkumar Shetty Validation, 1, 2
Padma K Shetty Validation, 3
Reshma A Shetty Validation, 4
Vijith V Shetty Writing – review & editing, 5, *
Author information:
1Central Research Laboratory, KS Hegde Medical Academy, NITTE (Deemed to be University), Deralakatte, Mangaluru-575018, Karnataka, India
2Department of Biochemistry, KS Hegde Medical Academy, NITTE (Deemed to be University), Deralakatte, Mangaluru-575018, Karnataka, India
3Department of Pathology, K S Hegde Medical Academy, NITTE (Deemed to be University), Deralakatte, Mangaluru-575018, Karnataka, India
4KSHEMA Centre for Genetic Services, K S Hegde Medical Academy, NITTE (Deemed to be University), Deralakatte, Mangaluru-575018, Karnataka, India
5Department of General Medicine and Oncology, Justice KS Hegde Charitable Hospital, KS Hegde Medical Academy, NITTE (Deemed to be University), Deralakatte, Mangaluru-575018, Karnataka, India
Abstract
Hepatocellular carcinoma (HCC) is the most prevalent type of primary liver cancer, accounting for roughly 90% of all liver malignancies worldwide, and remains the leading cause of cancer death worldwide. Cirrhosis, viral hepatitis, non-alcoholic fatty liver disease (NAFLD), or liver injuries caused by alcohol are all chronic diseases that have a close association with the pathogenesis of HCC. A key factor in the progression of these diseases to HCC is the activation of the epidermal growth factor receptor (EGFR) signalling pathway. The ErbB family of receptor tyrosine kinases, which includes EGFR, is essential for inflammation, cell division, and liver regeneration. In HCC, EGFR expression and hyperactivation are closely associated with tumor growth, metastasis, and patient prognosis. This review explores the structural and functional aspects of EGFR, its signalling mechanisms in hepatocellular proliferation and apoptosis, its role in liver fibrosis, and the transition from chronic liver injury to advanced HCC. Moreover, crosstalk between EGFR-mediated pathways and other signalling pathways, such as PI3K/AKT/mTOR and MAPK/ERK, contributes to resistance to targeted therapies, suggesting that molecular regulation needs to be improved in strategies targeting EGFR and its downstream pathways.
Keywords: Hepatocellular carcinoma, EGFR, Liver cancer, Signalling pathways, Fibrosis, Apoptosis, Drug resistance
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
None.
Introduction
Liver cancer is the fifth most frequent cancer worldwide, accounting for 5.6% of all human cancers, and the second most common cancer that can result in death.1 Hepatocellular carcinoma (HCC) is the most common type of liver cancer, representing 90% of all cases.2 HCC is the fourth most common cancer that causes death in the world.3 Patients with HCC typically live between six and twenty months, which is a low duration of life and probably correlates with the nearly one-to-one mortality/incidence ratio.4 HCC develops within hepatic damage, involving inflammation that causes hepatocyte necrosis, regeneration, and chronic liver disease namely cirrhosis.5,6 Numerous environmental and genetic risk factors have been linked to nonalcoholic fatty liver disease (NAFLD), chronic infection with the hepatitis B (HBV) and hepatitis C (HCV) viruses, excessive alcohol consumption, consumption of foods contaminated with aflatoxin B1, diabetes, obesity, tobacco use, and hereditary hemochromatosis.7,8 Avoiding exposure to the risk factors of HCC can reduce chronic liver disease. Early treatments with vaccination and antivirals can facilitate the establishment of cirrhosis and control cell cycle, proliferation, differentiation, cell survival, and apoptosis by diverse signalling pathways activated by abnormal molecules resulting in HCC progression.9,10 The treatment of HCC depends on the stage of diagnosis. Treatment of choice and the surgical resection, orthotropic liver transplant, is a curative treatment available for early-stage diagnosis of HCC. Treatment options for intermediate-stage illness include trans-catheter chemoembolization, percutaneous ethanol injection, and radiofrequency ablation. Systemic treatments such as bevacizumab, doxorubicin, and sorafenib are available for later stages.11
The epidermal growth factor receptor (EGFR), formerly referred to as ERBB1, is a well-known tyrosine kinase receptor within the ErbB receptor family and is abundantly expressed in the liver.12 Many cancers, including HCC, have been associated with the induction and development of the EGFR system, which is important for cell migration, survival, and proliferation.13 EGFR levels are elevated in human tissues with liver cirrhosis and HCC.14,15 EGFR is highly expressed in the liver and is crucial in regulating hepatocyte proliferation. This signalling pathway is vital for liver regeneration and is implicated in liver cancer development. Beyond these functions, EGFR is a key player throughout the liver's response to injury, influencing inflammation, cell proliferation, fibrosis, and cancerous changes.16 In cases of chronic liver injury and conditions like metabolic dysfunction-associated steatosis liver disease, EGF, EGFR, and its phosphorylated form (phospho-EGFR) increase.12 Studies suggest that EGFR may contribute to hepatic stellate cell (HSC) activation and extracellular matrix (ECM) modification, central to liver fibrosis and steatosis.17 However, some research indicates that EGFR signalling could also have protective effects during liver injury and fibrosis in certain contexts.18
Overexpression of EGFR plays an important role in the function of hepatocytes compared to other tissue cells.19 Further, EGFR has liver-protective and regeneration properties during acute and chronic liver disease. Abnormal activation of EGFR through the sequence of growth factors, receptors, and signalling pathways leads to cancerous alteration.20 Invasive tumors, inadequately differentiated HCC, metastasis, and poor patient survival are associated with overexpression of EGFR in 68% of HCC.21 Increased activation of EGFR or its downstream signalling pathways are associated with drug resistance and poor patient outcomes in HCC.22 RAS-RAF-MEK-ERK pathway is the primary pathway of EGFR which plays an important role in HCC progression and development.23 Targeting EGFR with antibodies and small molecule inhibitors can show promising outcomes in HCC therapy, but many patients develop resistance by overexpressing alternative pathways. This resistance results in a more aggressive tumor phenotype, characterized by increased cell migration and invasion compared to non-resistant tumors.24 Therefore, it is crucial to elucidate the molecular mechanisms that regulate EGFR and their downstream signalling pathways. This review provides a detailed description of EGFR and its function in HCC development, metastasis proliferation, and their potential therapeutic targets for HCC to cure patients.
EGFR structure and function
The EGFR/ErbB/HER receptor tyrosine kinase (RTK) family consists of four homologous transmembrane proteins that are ubiquitously expressed in cardiac, neuronal, and mesenchymal cells25: ErbB1 (EGFR or HER1), ErbB2 (HER2 or Neu), ErbB3 (HER3), and ErbB4 (HER4).23 The mature EGFR, consisting of 1186 residues, is generated from a precursor protein with 1210 residues through cleavage at the N-terminal region.26 The hydrophobic transmembrane domain in exon 17, the nonspecific ligand binding arm and dimerization arm spanning exons 1–16, and the C-terminal tail comprising the tyrosine kinase domain and exons 18–28 constitute the three major working structural components of EGFR from N-terminal to C-terminal (Fig. 1).27 Sixty-one amino acids make up the extracellular region of EGFR, which is separated into four domains: I (L1), II (CR1), III (L2), and IV (CR2) and characterized as leucine-rich segments, domains I and III are involved in ligand binding. When combined with the matching domain in family members, domain II can generate homo- or hetero-dimers. It is possible for domain IV to link to the transmembrane (TM) domain and form disulfide bonds with domain II. The areas known as cysteine-rich domains II and IV do not interact with the ligand. The receptor is anchored to the membrane by the TM domain, a hydrophobic structure consisting of 23 amino acids that function as a single-pass membrane.28 The N-terminal portion of the TM helices is thought to come into contact during the dimerization process, which is facilitated by the EGFR transmembrane domain.28 The 542 amino acid intracellular domain includes the tyrosine kinase domain, the flexible juxta-membrane region, and the C-terminal tail. Approximately 37 residues constitute the juxta-membrane (JM) domain of EGFR, which links the transmembrane TM and kinase domains and is crucial for receptor dimerization, activation, and regulation. It contains important motifs such as lysosomal sorting signals, nuclear localization sequences, and phosphorylation sites (such as the Thr654) which affect receptor activation.29 The ATP-binding site is located between the N-lobe, which is primarily arranged as a β-sheet, and the C-lobe, which is arranged mainly as an α-helical structure, in the domain associated with the tyrosine kinase of EGFR.30 The binding of the N-lobe to the C-lobe of various receptors facilitates trans-autophosphorylation.31 The kinase domain has lysine residues that act as the primary locations for receptor ubiquitination. Different tyrosine residues in the C-terminal tail can be phosphorylated to allow the binding of various intracellular proteins to the activated receptor and start the signal transduction relay.32
At a glance
Types of EGFR ligands:
The EGFR family includes several ligands with a common EGF-like receptor binding domain. They are classified into three main types according to their ability to bind specifically to EGFR or even to interact with other ErbB receptors:
-
High-affinity EGFR ligands: bind exclusively to EGFR, such as EGF and factor transforming growth factor alpha (TGF-a)
-
Dual specificity ligands: Ligands that bind to EGFR and other ErbB receptors, such as betacellulin (BTC) and epiregulin (EREG).
-
Heparin-binding ligands: Ligands such as HB-EGF that require heparan sulfate proteoglycans (HSPGs) to bind and can interact with EGFR and other ErbB receptors.
Role of EGFR ligands in HCC:
-
Epidermal Growth Factor (EGF): The first EGFR ligand discovered binds to the receptor with great affinity, causing dimerization and activation. EGF plays a vital role in HCC by promoting cellular proliferation, migration, and angiogenesis—all of which contribute to the formation and dissemination of tumors. Elevated EGF levels in the hepatic microenvironment further support the development of HCC.
-
Transforming Growth Factor-Alpha (TGF-α): in HCC, TGF-α is often upregulated, driving cancer cell growth and survival by strengthening EGFR signaling and activating pathways that promote proliferation and resistance to apoptosis.
-
Amphiregulin (AREG): stimulates hepatocyte proliferation and inflammatory signaling, creating a liver microenvironment conducive to HCC development. High AREG levels are linked to poor prognosis in HCC, as they amplify EGFR activation.
-
Heparin-binding EGF-like Growth Factor (HB-EGF): In HCC, HB-EGF is involved in promoting invasion and metastasis, as it supports tumor cell migration and extracellular matrix remodeling.
-
Betacellulin (BTC): BTC promotes cell proliferation and differentiation, aiding in new blood vessel formation and tumor growth in HCC. Its expression is often associated with more aggressive cancer types.
-
Epiregulin (EREG): In HCC, EREG expression is linked to greater tumor aggressiveness and poorer prognosis. It activates EGFR signaling, which promotes tumor growth, invasion, and metastasis.
-
Epigen (EPGN): While its role in HCC remains unclear, EPGN may aid tumor growth and progression by activating EGFR in tissues where it is present.
Types of EGFR dimers and their role in HCC:
-
EGFR Homodimers: They activate pathways such as MAPK/ERK and PI3K/AKT, promoting cell proliferation and survival in HCC, leading to tumor growth and resistance to cell death.
-
EGFR-ErbB2 (HER2) heterodimers: HER2 heterodimers provide stronger and more stable signaling than homodimers, promoting aggressive tumor growth and survival. They are associated with a poor prognosis in HCC and often lead to resistance to EGFR therapies.
-
EGFR-ErbB3 heterodimers: despite the altered kinase function of ErbB3, EGFR binding activates robust PI3K/AKT signaling, promoting tumor growth, survival and treatment resistance by providing alternative pathways when EGFR is inhibited.
-
EGFR-HER4 heterodimers: Less common in HCC, these dimers can support a variety of functions, including differentiation in normal tissues and cell survival and proliferation in cancer.
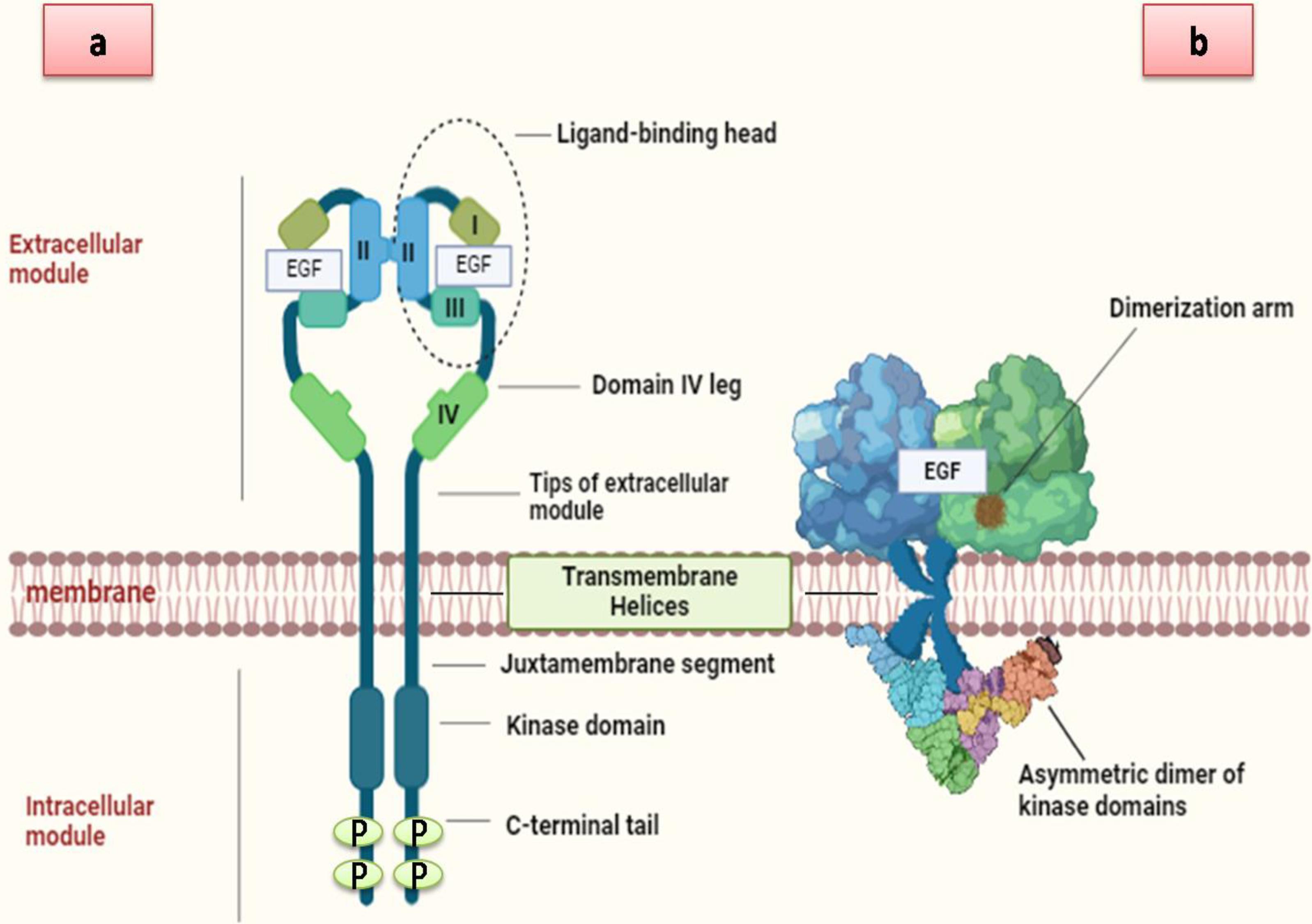
Fig. 1.
The structure of EGFR includesthree main domains: (A) The extracellular domain, which is divided into four subdomains: I, II, III, and IV. (B) The transmembrane domain, which links the extracellular and intracellular regions of EGFR. (C) The intracellular domain, consisting of: (a) the juxta membrane domain, which is critical for EGFR dimerization and activation by connecting the transmembrane domain’s C-terminus to the kinase domain, and (b) the kinase domain, which contains an NH2-terminal lobe (N-lobe). Additionally, a schematic diagram illustrates ligand-induced EGFR dimerization with EGF Created by using BioRender tool).
.
The structure of EGFR includesthree main domains: (A) The extracellular domain, which is divided into four subdomains: I, II, III, and IV. (B) The transmembrane domain, which links the extracellular and intracellular regions of EGFR. (C) The intracellular domain, consisting of: (a) the juxta membrane domain, which is critical for EGFR dimerization and activation by connecting the transmembrane domain’s C-terminus to the kinase domain, and (b) the kinase domain, which contains an NH2-terminal lobe (N-lobe). Additionally, a schematic diagram illustrates ligand-induced EGFR dimerization with EGF Created by using BioRender tool).
EGFR Pathway in Hepatocellular Carcinogenesis: Progression from Chronic Liver Damage to Advanced HCC
Development of HCC starts from exposure of the liver to injury due to long-term alcoholism, poor eating patterns that cause intrahepatic fat to accumulate, and hepatotropic infections caused by viruses that cause cirrhosis and chronic liver damage.33 The EGFR pathway regulates hepatocyte proliferation, playing important roles in liver regeneration and hepatocellular carcinogenesis.12 Overexpression of EGFR plays a crucial role in the development of HCC in all stages of liver injury, from the early stage of chronic liver injury and inflammation to hepatocellular proliferation and cirrhosis to the last stage of hepatocarcinogenesis.34 Also, the EGFR pathway is crucial in the progression and development of HCC, particularly through its interactions with key risk factors such as hepatitis C virus,35,36 hepatitis B viruses,37-39 alcohol intake,40 NAFLD,41,42 diabetes,43 and hereditary hemochromatosis44 as shown in (Box 1).
Box 1. Role of EGFR in the risk factors of HCC
-
Hepatitis C virus:
-
EGFR is required for the entry of HCV into cells and for viral endocytosis.35
-
Binding of EGFR and HCV leads to enhance invasiveness of infected liver cancer cells resulting in increased intra and extrahepatic metastatic dissemination.36
-
Hepatitis B viruses:
-
After HBV infection, the EGFR system could promote both viral amplification and immunological tolerance.37
-
The translocation of HBV from the cell surface to the endosomal network is mediated by the EGFR endocytosis mechanism.38
-
The poor prognosis of HCC patients is linked to EGFR overexpression in HBV-infected patients, which activates oncogenic MAPK and PI3K/Akt signalling pathways.39
-
Excessive alcohol consumption:
-
Non-alcoholic fatty liver disease:
-
The presence of EGFR, which is an essential tyrosine receptor factor (TKR) type, could make it a significant potential target for NASH prevention.41
-
Mutation or overexpression of EGFR leads to increased levels of cholesterol levels and accumulation of lipid in the liver resulting in induced NAFLD.42
-
Diabetes:
-
Hereditary hemochromatosis:
EGFR pathway in HCC: Mediator of inflammation, fibrosis, and tumor proliferation
Through its interactions with other inflammatory mediators and pathways, the EGFR pathway serves as a signalling hub.45 EGF-EGFR significantly increases CXCL8 and CXCL5 production in HCC cells. EGF-EGFR signalling plays a key role in HCC cell proliferation, migration, and the formation of an inflammatory microenvironment. Additionally, EGF activates EGFR downstream signalling pathways such as PI3K and ERK pathways, further promoting inflammation, proliferation, and metastasis. Inhibition of these pathways reduces HCC cell proliferation and inflammatory cytokine production.46,47 EGF-EGFR pathways play a role in the formation of liver cancer inflammatory microenvironment by regulating the production of inflammatory factors from HCC cells by stimulating PI3K and ERK pathways. Cross-talk between EGFR and CXCR2 was observed, with CXCR2 inhibition reducing EGF-induced CXCL8 production.48 CXCR2 was also found to be highly expressed in metastatic HCC cells, which may explain the differential production of CXCL8 and CXCL5. CXCL8 overexpression in HCC tissues is linked to vessel invasion and metastasis.49 Further, research is needed to explore the interactions between these pathways and mediators using systems biology and clinical bioinformatics.
EGFR plays a role in early fibrogenic pathways by activating stellate cells and modifying the ECM. This process, along with hepatocyte damage induced by EGFR, contributes to liver fibrosis. EGFR inhibition has been shown to significantly reduce hepatocyte death in chronic liver injury models like CCl4-induced injury, fast-food diet (FFD) mice, and high-fat diet (HFD) mice.50,51 Additionally, in another study, they discovered that EGFR suppression stopped the liver tissues of mice fed a high-fat diet from accumulating collagen.52 Downstream of EGFR is also implicated in liver fibrosis and injury through induction of reactive oxygen species (ROS) and oxidative stress. Numerous studies have shown that oxidative stress is a significant factor in the development of liver cirrhosis and fibrosis.53 Inhibiting ROS production has been shown to suppress liver tumor cell growth, which is accompanied by decreased EGFR phosphorylation and reduced levels of EGFR and TGF-α.54 Overexpression of EGFR can induce the expression of connective tissue growth factor (CTGF) associated with poor outcomes in fibrosis, suggesting that reducing CTGF through TGF-β inhibition may provide clinical advantages.55 Mitigating acute liver necrosis, mitochondrial dysfunction, and hepatocyte apoptosis by EGFR suppression suggests that EGFR may directly involve hepatocellular mortality during chronic liver injury.56 EGFR-associated signaling Pathway and their roles in mediator of inflammation, fibrosis, and tumor proliferation in HCC models have been listed in (Table 1).
Table 1.
Pathogenic outcomes of EGFR in hepatocellular carcinoma
|
Status
|
Model
|
Induction
|
Outcome
|
Ref.
|
| Liver fibrosis |
C57BL6/J mice |
For two months, mice were given either a regular chow diet or an FFD, with some groups also getting canertinib, an EGFR inhibitor. |
EGFR inhibition fully prevented the onset of liver fat accumulation and Liver fibrosis in this model. |
50
|
| Liver regeneration |
C57BL/6 females’ mice |
Created mice with a flexed EGFR allele to deactivate EGFR in the liver during both fetal development and adulthood. |
EGFR-deficient mice showed increased mortality and slower liver regeneration following partial hepatectomy and decreased hepatocyte proliferation. |
60
|
| Hepatocyte proliferation |
C57BL/6 WT mice |
Transgenic EGFR mice (DEGFR) were produced by integrating a truncated human EGFR into a BAC containing the albumin locus, followed by DNA microinjection into zygotes. |
EGFR catalytic activity is crucial during the early preneoplastic stages of the liver, as evidenced by DEGFR mice showing delayed tumor development, inflammation, and reduced cell proliferation. |
61
|
| [MET KO + EGFRi] mice |
Mice administration with tamoxifen for five consecutive days to create MET knockout (KO) mice.
EGFR inhibition, canertinib, an EGFR inhibitor, was incorporated into the diet of the mice at 80 mg/kg/day. |
Combined elimination of MET and EGFR signalling in mice hindered cell cycle activation and disrupted hepatocyte proliferation mechanisms. |
63
|
| Chronic liver disease |
Male AR^ + / + and AR^-/- littermates |
Liver fibrosis was induced by administering intraperitoneal injections of (CCl₄) at a dose of 0.6 mL/kg body weight, twice weekly. |
AR specifically promotes liver fibrosis by enhancing fibrogenic mediator expression and supporting fibrogenic cell survival, underscoring the EGFR system's role in hepatic fibrogenesis. |
81
|
| Therapeutic targeting |
C57BL6/J mice |
Canertinib treatment for 2 months in mice on a fast-food diet.
In mice fed the FFD for 5 months and canertinib therapy for the last 5 weeks |
EGFR inhibition completely prevented steatosis and liver injury Inhibiting
EGFR may reverse the progression of NAFLD. |
50
|
| Male Wistar rats |
Erlotinib, an EGFR inhibitor, is used to treat three cirrhosis models: rats treated with DEN, mice treated with (CCl4), and rats treated with BDL. |
Erlotinib reduced hepatocyte proliferation and liver damage, as well as EGFR phosphorylation and the quantity of activated HSC. |
76
|
| C57BL/6J mice |
Mice were injected with 25 mg/kg DEN, fed a high-fat diet after 4 weeks, and treated with 10 mg/kg honokiol (HNK). |
Inhibiting EGFR signalling by HNK significantly suppressed HCC progression in the mouse model. |
41
|
| Hep3B and Huh6 HCC cell lines |
Cells were treated with specified concentrations of HNK for designated periods. |
EGFR degradation led to significant tumour suppression. |
|
| HepG2 and HCCLM3 cell lines |
Cells were cultivated at 37°C in a humidified incubator with 5% CO2 and 95% air in DMEM or RPMI-1640 with 10% FBS. |
EGF controlled the release of CXCL5 and CXCL8 in HCC and promoted cell migration in HepG2 and HCCLM3 cells. The EGFR inhibitor prevented this manufacturing. |
100
|
| HepG2 and Huh- 7 cell lines |
Cell lines of the HepG2 (wild-type p53) and Huh-7 (mutated p53) were cultivated in RPMI 1640 media supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin. |
P53-mutated Huh-7 cells showed reduced sensitivity to cetuximab, but combining it with TKIs, fluvastatin, or doxorubicin led to significant growth reduction. |
101
|
| Human liver cancer cell lines (HuH7 and PLC/PRF/5), HEK293T cells. |
After cell cultured in DMEM, Lenvatinib-resistant (LR) cell lines were treated with increasing doses of lenvatinib, starting from 3 µmol/L and 20 µmol/L. |
EGFR inhibitors can block the EGFR–STAT3–ABCB1 signalling pathway, making lenvatinib-resistant (LR) strains more sensitive to treatment. |
102
|
| C57BL/6 mice liver cancer cell line Hep1–6 |
Hep1–6 cells were injected into the flanks of mice, with lenvatinib (10 mg/kg/day) treatment starting when the tumour reached about 100 mm³. |
The combination of Lenvatinib and erlotinib inhibited ABCB1 activity and demonstrated a strong synergistic effect in overcoming resistance, both in vitro and in vivo. |
102
|
EGFR signalling in hepatocellular proliferation and apoptosis
EGFR-mediated liver regeneration
After exposure of the liver to inflammation and liver injury, EGFR plays an important role in the regeneration of the liver and inhibition accumulation of lipids.57 In the case of liver regeneration, the expression of EGFR and its ligands (HB-EGF, TGF-α, EREG, and AR) were highly expressed in hepatocytes.58 Firstly, Lacking EGFR in embryonic mice models leads to the death of the fetus in midgestation or after birth with various defects in neural and epithelial organs.59 Likewise, in adult mice, lack of EGFR in hepatocytes showed reduced hepatocyte proliferation with defective G1-S phase entry leading to impaired liver regeneration and increased mortality after partial hepatectomy, which resulted in the development of chronic liver injury. All these findings imply that EGFR has a crucial role in controlling the proliferation of hepatocytes during the early stages of liver regeneration.60
EGFR plays an essential role in stimulating hepatocyte proliferation. In mice model stimulation of EGFR resulted in increased liver mass and increased hepatocyte proliferation.61 Besides, deletion of EGFR alone can delay only liver regeneration without a change of liver mass due to activation of the compensatory MET signalling pathway.62 In mice with hepatomegaly model, deletion of EGFR and MET signalling pathway can lead to the complete block in hepatocyte proliferation but inhibition of EGFR alone has less effect in hepatocyte proliferation.63 Also, in mice models with acute liver injury induced by using APAP overdose delay EGFR after peak injury showed substantial impairment of liver regeneration and cell-cycle activation. This led to uncontrolled liver injury development, poor recovery, and a high mortality rate (50%) from liver injury.56
EGFR-mediated apoptosis in acute liver injury
C-Met is constitutively activated by autocrine TGF-α action, leading to EGFR activation and EGFR/c-Met heterodimerization, which may amplify mitogenic signals, contributing to tumorigenesis.64 The diversity of EGFR ligands and their receptor heterodimers is associated with the intricacies of EGFR signalling during partial hepatectomy (PH). Hepatocytes have also been shown to exhibit ligand-independent EGFR activation, where hydrophobic bile acids trigger ROS-dependent EGFR tyrosine phosphorylation and MAP kinase activation. Furthermore, bile acids activate EGFR through ROS, which in turn causes Erk-dependent HSC proliferation. Bile acids cause cholangiocytes to shed TGF-α in a c-Src-dependent manner, which activates EGFR in a ligand-dependent manner. Apoptosis of liver cells mediated by CD95 also involves EGFR signalling.65,66
The role of EGFR in apoptosis cell death depends on the different harmful toxins such as hydrophobic bile acids, or hyperosmolarity cell shrinkage induce a rapid oxidative stress response, and CD95 ligands which induce activation of the EGFR in hepatocytes.67 The association of EGFR with CD95 leads to the activation of EGFR linked with FAS receptors. EGFR-FAS phosphorylation leads to the formation of the death-inducing signalling complex (DISC), resulting in apoptotic cell death. Prolonged JNK activation shifts EGFR from its usual regenerative role to a pro-apoptotic function through FAS.68 Antioxidant-sensitive Yes activation is a component of EGFR activation triggered by other proapoptotic stimuli. Hyperosmotic stress activates Src family kinase members, particularly Fyn, leading to the phosphorylation of caveolin-1 and cortactin.69 While c-Src is involved in JNK activation under oxidative stress, Fyn plays a key role in p90 ribosomal S6 kinase activation by reactive oxygen species.70 In hepatocytes, hyperosmotic exposure activates Fyn, Yes, and Lck, with Yes associating with EGFR, triggering its activation and leading to DISC formation, thereby promoting apoptosis.71 The role of EGFR in apoptosis and acute liver injury is supported by, EGFR inhibition in mice preventing APAP-induced mitochondrial dysfunction, oxidative stress, and endonuclease release, thereby protecting against DNA damage and necrosis.56
EGFR activation in chronic liver disease
Exposure of the liver to chronic hepatitis, alcoholic liver disease, NAFLD, and hemochromatosis leads to the development of chronic liver disease (CLD).72,73 CLD develops when the regenerative capacity of the liver is reduced due to the formation of fibrosis, which leads to the loss of liver function. After liver injury, the normal healing process activates various types of liver cells, including HSCs, which turn into myofibroblasts. These myofibroblasts produce collagen and ECM, contributing to fibrosis and progression of CLD.74
CLD induces a prolonged wound-healing response, leading to ECM accumulation and fibrosis. EGFR ligands contribute to the activation of ECM-producing cells, such as HSCs and hepatic myofibroblasts, which express high levels of EGFR, resulting in proliferation, survival, and collagen synthesis. EGFR expression in chronic liver injury can be induced by key inflammatory signals prevalent in acute and chronic liver injury.34,75 The role of EGFR in chronic liver disease was studied using erlotinib, which reduced hepatocyte proliferation, liver injury, and fibrosis progression in rat models of toxic fibrosis, biliary fibrosis, and cirrhosis.76 Overexpression of EGFR and its ligands such as EGF results in the stimulation of hepatocytes, Kupffer cells (KCS), and inflammatory cells, which also stimulate fibrogenic cells to produce inflammatory cytokines such as interleukin (IL)-8, CXCL8, CXCL-12, IL-6 and IL-1 and attracts tumor cells and leukocytes (monocytes, neutrophils, lymphocytes) to the tumor tissue, creating an inflammatory environment, which is a key factor for the damage of the liver and fibrosis.77,78 Recruited leukocytes and activated cancer cells then release other inflammatory mediators, promoting tumor progression.79 EGFR ligands, such as TGF-α and AR, also play a role in promoting the proliferation and migration of fibrogenic cells. Hepatocytes and ECM-producing cells produce these ligands and can act in an autocrine and paracrine manner to promote fibrosis.80 EGFR ligands such as AR and HB-EGF are upregulated in KCs by pro-inflammatory stimuli, thus contributing to the fibrogenic environment. EGFR ligands stimulate fibrogenic cells in vitro, driving their transformation by enhancing their proliferation and migration.81
Role of EGFR associated with signalling pathway in the progression of hepatocellular carcinoma
Development and progression
Hepatoprotection from the early stages of the disease has been associated with the significance of the EGFR system in the development of HCC.82 When exogenous damaging substances stimulate hepatocytes on their surface, the expression of EGFR often activates in liver fibrosis. Prolonged exposure to these damaging substances results in hyperphosphorylation of EGFR/EGF signalling, which is known to produce liver fibrosis by promoting hepatocyte EMT, HSC activation, and ECM deposition. Increased EGFR activation can speed up the various phases of the liver's response to injury, including hepatocyte proliferation, liver fibrosis, and hepatocellular carcinogenesis.82 The EGFR signalling pathway plays a role in the progression of hepatocellular carcinoma by facilitating the connection between inflammation and cancer development.46 Binding of EGFR with EGF could induce the activation of intracellular protein-tyrosine kinase, causing the initiation of a single transduction signalling pathway including the ERK-PI3K-Akt pathway and the RAS/RAF/MEK/MAPK cascade (Fig. 2).83,84 In the ERK-PI3K-Akt pathway in healthy cells, the activation of EGFR leads to the activation of tyrosine residues at the C-terminal domain of EGFR.85 This activation leads to binding PI3K to the EGFR intracellular domain. Upon activation, PI3K initiating the synthesis of PIP3 induces the activation of AKT by mTORC2 and PDK1. In turn, the activation of AKT leads to the phosphorylation of mTORC, which leads to the phosphorylation of 4EBPI and S6K1. Both proteins control the translation of proteins associated with proliferation and angiogenesis, including c-myc and cyclin-D1 in the nucleus.86 In HCC, EGFR overexpression or mutations lead to excessive activation of the PI3K/AKT pathway, promoting cell survival and tumor growth. AKT increases anti-apoptotic signals by inhibiting pro-apoptotic factors like BAD, which allows cancer cells to avoid cell death, while also activating mTOR to stimulate protein synthesis and cell cycle progression. PI3K/AKT signalling and uncontrolled cell growths are often stimulated in HCC by inhibiting PTEN, a natural inhibitor of this pathway. By targeting the PI3K/AKT pathway, the use of PI3K or mTOR inhibitors in combination with EGFR inhibitors offers a potential strategy to reduce tumor progression and improve treatment outcomes.87-89
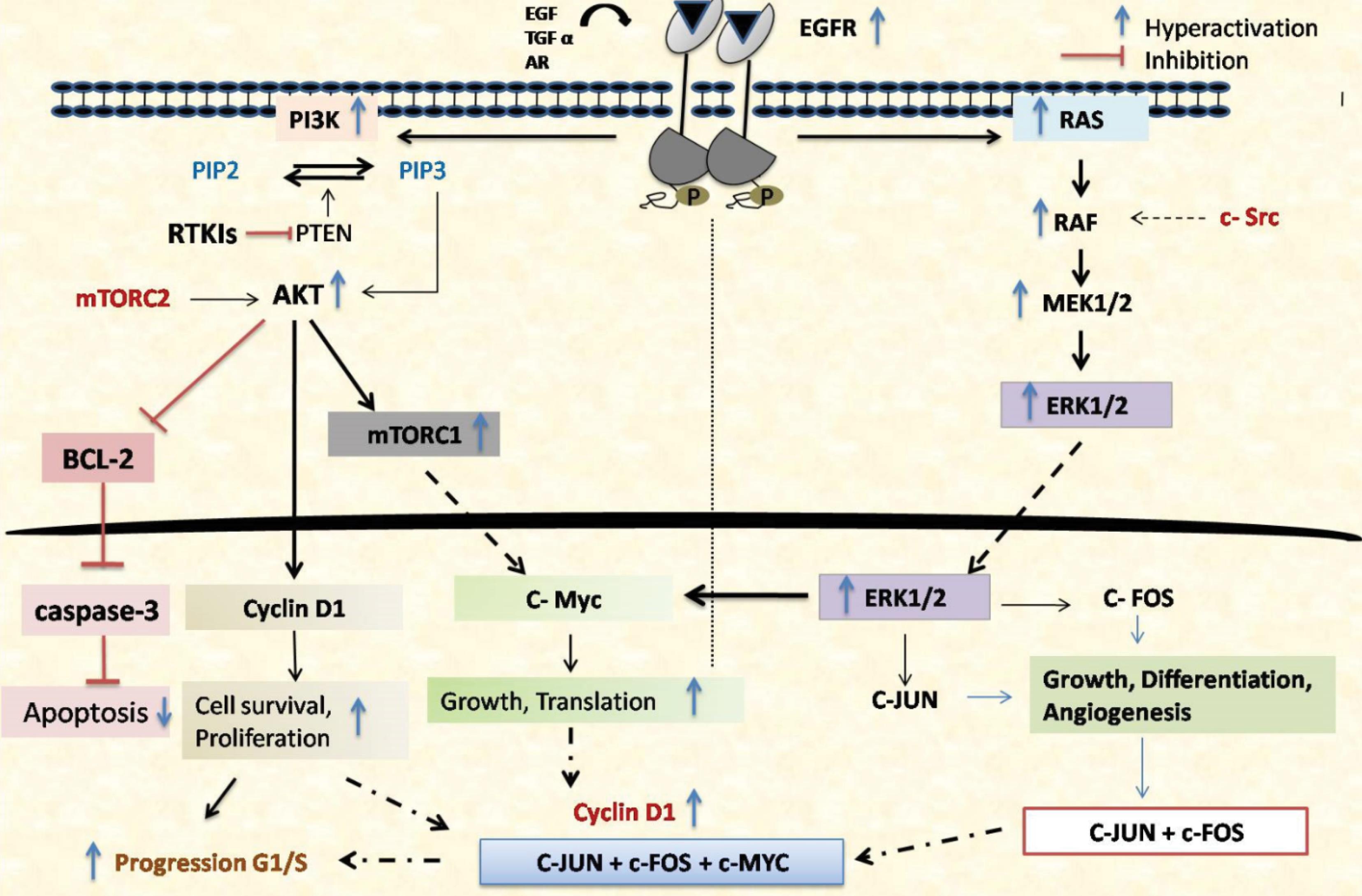
Fig. 2.
EGF activation of EGFR triggers two main pathways, RAS-RAF-MEK-ERK MAPK and PI3K-AKT-mTOR, leading to G1/S cell cycle progression. EGFR dimerization and phosphorylation recruit SHC, GRB2, and PLC-γ1. GRB2 recruits SOS or GAB1, initiating ERK MAPK or AKT signaling. SOS activates RAS, which recruits and activates RAF-1. RAF-1 activates MEK1/2, which then activates ERK1/2, leading to Cyclin D1 transcription. GRB2-GAB1 also recruits PI3K, converting PIP2 to PIP3. PIP3 recruits and activates AKT, which phosphorylates proteins that promote Cyclin D1 activity and translation. Increased Cyclin D1 boosts CDK4/6 activity, phosphorylating RB and releasing E2F, promoting Cyclin E transcription and G1/S progression. ERK, c-MYC, and AKT inhibit the CDK inhibitor p27. Both pathways also inhibit pro-apoptotic proteins, promoting cell survival.
.
EGF activation of EGFR triggers two main pathways, RAS-RAF-MEK-ERK MAPK and PI3K-AKT-mTOR, leading to G1/S cell cycle progression. EGFR dimerization and phosphorylation recruit SHC, GRB2, and PLC-γ1. GRB2 recruits SOS or GAB1, initiating ERK MAPK or AKT signaling. SOS activates RAS, which recruits and activates RAF-1. RAF-1 activates MEK1/2, which then activates ERK1/2, leading to Cyclin D1 transcription. GRB2-GAB1 also recruits PI3K, converting PIP2 to PIP3. PIP3 recruits and activates AKT, which phosphorylates proteins that promote Cyclin D1 activity and translation. Increased Cyclin D1 boosts CDK4/6 activity, phosphorylating RB and releasing E2F, promoting Cyclin E transcription and G1/S progression. ERK, c-MYC, and AKT inhibit the CDK inhibitor p27. Both pathways also inhibit pro-apoptotic proteins, promoting cell survival.
Overexpression of EGFR can lead to the increased activation of the Ras/MEK/ERK pathway by overexpression of the Ras and its downstream effectors, resulting in uncontrolled cell proliferation and tumor growth. Additionally, mutations in RAS or RAF can activate this pathway independently of EGFR signalling, causing continuous MEK/ERK activation that promotes oncogenesis. Subsequently, Raf activation begins when its SH2 domain binds to EGFR residues like GRB2.90 Extracellularly regulated kinase (ERK1/2) is subsequently activated more when Ras is activated, which in turn causes Raf to be overexpressed and phosphorylated by MEK1/2 kinases. After transferring to the nucleus, ERK1/2 regulates the expression of several genes related to the cell cycle, such as cyclin D1, facilitating the transition from the G1 to S phase and enabling unregulated cell division, resistance to apoptosis, remodeling cellular motility, angiogenesis, and medication resistance.91 Moreover, the pathway enhances angiogenesis by increasing VEGF production, thereby supporting tumor vascularization and growth. Overexpression of this pathway contributes to HCC metastasis, development, and the production of inflammatory cytokines (Fig. 2).92 HCC is characterized by the persistent activation of EGFR signalling, which supports the proliferation, apoptotic resistance, and invasive activity of HCC cells. EGFR signalling is important for maintaining the altered phenotype of HCC cells, as demonstrated by multiple in vitro experiments conducted on human HCC cell lines.20
Progression to metastasis
The first step of EGFR in HCC metastasis is starting with its ability to form a tumor-inflammatory microenvironment of HCC. EGFR binding to EGF ligands plays an initial role in developing cancer inflammation and promoting HCC cells from low metastatic potential into high metastatic potential.93 Overexpression of EGF-EGFR could be initiating the downstream PI3K and ERK signalling cascade, resulting in the production of inflammatory factors such as CXCL5 and CXCL8.46 These factors control the migration of leukocytes and the migration of cancer cells in HCC microenvironments into a certain location.94 Activation of this pathway contributes to the creation of circulating tumor cells (CTCs) (Fig. 3). The accumulation of CTCs leads to the formation of circulating tumor microemboli (CTM) of HCC, which these cells can migrate to the blood. Moreover, EGFR signalling in HCC enhances VEGF production, promoting angiogenesis to supply the tumor with oxygen and nutrients and facilitating cell spread through blood vessels. Additionally, EGFR-integrin interactions strengthen cell adhesion to the ECM, helping HCC cells anchor and survive at metastatic sites, which supports secondary tumor formation and worsens prognosis.95 Overexpression of EGFR in CTM leads to upregulation of MET.96 Patients with HCC are susceptible to extrahepatic metastases. EGFR/MET may contribute to the dissemination of cancer via circulating tumor cells. Extrahepatic metastasis is the most common type of lung metastasis.97 EGFR/MET is an appropriate CTC/CTM identification marker in theC57 xenograftmodel. The finding of CTM with high EGFR/MET in the blood vessels of lung metastasis mice demonstrates this. This level was higher than that in cancer tissues.96 In CTM, overexpression of EGFR/MET makes cells produce cytokines such as IL-8 resulting in increased liver inflammation which leads to liver injury. It also makes them resist the fatal impacts of leukocytes. Furthermore, EGFR/MET promoted phosphorylation of hetero-RTKs based on high-energy phosphorylated activity rather than direct interaction. Over-phosphorylation of RTKs is independent of RTKs' ligands and resistant to RTK inhibitors. EGFR/MET has activated the Ras/MAPK pathway more than other RTKs. Stabilization of suspended HCC cells, resistance to apoptosis, and cell proliferation are all facilitated by the Ras/MAPK pathway (Fig. 3).98,99 Inhibiting EGFR/MET could prevent HCC cells from metastasizing, particularly in CTM where EGFR/MET expression is higher.
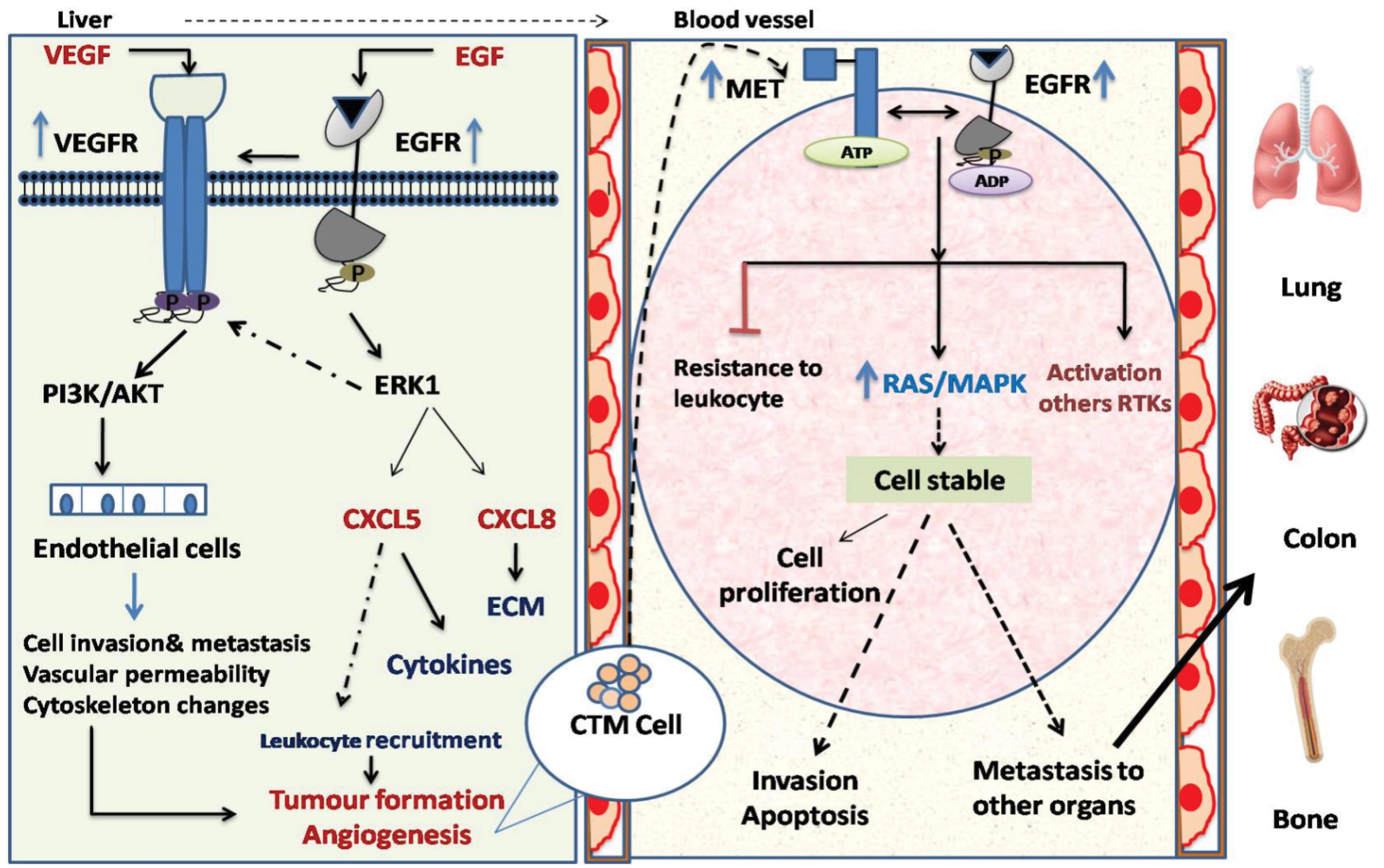
Fig. 3.
The EGF-EGFR signaling pathway plays an important function in the inflammatory microenvironment of HCC. HCC cells can produce inflammatory factors like CXCL5 and CXCL8, potentially playing a role in recruiting leukocytes and facilitating HCC metastasis. Following metastasis to the bloodstream, there is an upregulation in both the expression and phosphorylation of EGFR/MET in circulating tumor micro emboli (CTM) leading to high RTK phosphorylation resistant to RTK inhibitors and independent of ligands. EGFR/MET effectively activates the Ras/MAPK pathway, promoting cell growth, apoptosis resistance, and stability in suspended hepatocellular carcinoma (HCC) cells. Additionally, EGFR/MET helps CTM evade leukocyte-induced death which all those things l facilitating HCC metastasis to other organs such as bone, lungs and colon.
.
The EGF-EGFR signaling pathway plays an important function in the inflammatory microenvironment of HCC. HCC cells can produce inflammatory factors like CXCL5 and CXCL8, potentially playing a role in recruiting leukocytes and facilitating HCC metastasis. Following metastasis to the bloodstream, there is an upregulation in both the expression and phosphorylation of EGFR/MET in circulating tumor micro emboli (CTM) leading to high RTK phosphorylation resistant to RTK inhibitors and independent of ligands. EGFR/MET effectively activates the Ras/MAPK pathway, promoting cell growth, apoptosis resistance, and stability in suspended hepatocellular carcinoma (HCC) cells. Additionally, EGFR/MET helps CTM evade leukocyte-induced death which all those things l facilitating HCC metastasis to other organs such as bone, lungs and colon.
Therapeutic targeting of EGFR in HCC
As mentioned earlier, EGFR plays an important role in the development of HCC from the early stage to the last stage so according to this; recent studies found that, inhibition of EGFR signalling leads to the significantly reduced development of HCC. According to the study done in a mouse model, they found that inhibition of EGFR by the natural product honokiol (NHK) leads to EGFR degradation, which in turn causes strong tumor suppression.41 Also, EGFR plays a major role in the development of liver fibrosis by increased damage of hepatocytes by improving the extracellular network and activation of stellate cells. To illustrate, in a mouse with the last stage of cirrhosis induced by carbon tetrachloride (CCl4), they found that inhibition of EGFR by erlotinib leads to a decrease in the number of hepatocyte damage.76 Not only hepatocyte damage but also inhibition of EGFR leads to enhanced glucose tolerance while significantly reducing fibrosis, liver damage, and steatosis. For instance, in mice with NAFLD, inhibition of EGFR by canertinib arrested the development of steatosis and liver damage.50 Additionally, inhibition of EGFR leads to the reducing the formation of inflammatory cytokines from HCC. As a result, inhibition of EGFR leads to the inhibition of EGF EGF-regulated production of CXCL5 and CXCL8 from HCC in both HepG2 and HCCLM3 cell lines which results in preventing HCC proliferation and metastasis.100 Concludingly, EGFR inhibition is crucial for HCC treatment and prevents the development of HCC. Dysregulation of EGFR contributes to HCC development, proliferation, and migration, which provides the importance of focusing on the EGFR signalling pathway in HCC treatment.21 Also, the activity of EGFR is increased in an advanced stage of HCC, which gives importance to the targeting of EGFR. Accordingly, recent studies have attempted to use the anti-cancer EGFR inhibitors in clinical trials of HCC. Inhibitors targeting EGFR, such as the monoclonal antibody cetuximab, which interferes with the activity of ligands in the extracellular domain of the receptor, or TKIs like gefitinib, and erlotinib, have demonstrated effectiveness in inhibiting HCC proliferation and migration (Table 2).103,104 A study using a wild-type HepG2 HCC cell line treatment with cetuximab resulted in a blocked cell cycle in the G1/G0 phase, increased apoptosis, and enhanced fibrotic lesions by targeting EGFR.101 Furthermore, targeting EGFR with gefitinib or erlotinib leads to reduced EGFR phosphorylation, cell cycle arrest, apoptosis, and inhibition of growth in HCC.105 In contrast, another study found that lenvatinib activated EGFR and stimulated the EGFR–STAT3–ABCB1 axis, causing resistance in both human cell lines (HuH7 and PLC/PRF/5) and mice liver cancer cell lines (Hep1–6) which ABCB1 can strongly increase lenvatinib exocytosis and promotes resistance. In the same study, they found that erlotinib combination with lenvatinib can inhibit ABCB1 and reduce lenvatinib exocytosis. This combination showed a noteworthy beneficial effect on HCC in both vitro and in vivo settings.102 In addition, a combination of gefitinib and erlotinib in the Huh7 LR cell line mouse model showed that delays proliferation and promotes apoptosis by inhibition of EGFR-mediated MEK/ERK and PI3K/AKT pathway activation.106 The list of EGFR inhibitors together with the pathophysiological processes by which they work effectively is found in (Table 2). TKIs targeting EGFR block receptor tyrosine activity by binding to the ATP-binding domain located in the intracellular kinase domain, which prevents EGFR from activating other signalling pathways, including the PI3K/AKT/ERK pathway. TKIs significantly inhibit HCC progression by targeting EGFR (Fig. 4).105 Overexpression of EGFR results in the activation of signalling pathways such as the ERK pathway, and this activation leads to the development of HCC. In this finding, targeting EGFR by gefitinib can decrease the level of expression of EGFR and block the activation of EGFR in the ERK pathway, leading to the prevention of HCC development. In one study done on rat treatment with gefitinib, inhibition of EGFR by gefitinib leads to significantly preventing HCC metastasis, and decreased induced angiogenesis of HCC.102,107 Targeting EGFR by these agents can increase the degree of resistance in HCC cells, and be paired with other targeted compounds, such as sorafenib, erlotinib, and lenvatinib (Fig. 4), can improve the clinical outcome and block the activation of signalling pathway by EGFR.108,109
Table 2.
Therapeutic targeting of EGFR signalling
|
EGFR inhibitor
|
Target & dose
|
Outcome
|
Ref.
|
| Erlotinib |
tyrosine kinase inhibitor |
A decrease in the phosphorylation of EGFR/AKT leads to a decrease in HCC metastasis |
105
|
| Sorafenib/ erlotinib or gefitinib |
multi-kinase inhibitor |
The RAF-MEK-ERK kinase cascade activity in EGFR-positive HCC cells can be collectively controlled by sorafenib and EGFR inhibitors. |
106
|
| Gefitinib |
tyrosine kinase inhibitor |
inhibited HCC growth by blocking TNF-α-induced transactivation of EGFR by inhibiting MAPKs and Akt |
105
|
| Lenvatinib/ erlotinib |
multitarget tyrosine kinase receptor inhibitor |
It blocks the EGFR-STAT3-ABCB1 pathway and prevents lenvatinib from being exocytosed. |
102
|
| Cetuximab |
EGFR-targeted antibodies |
Blocks binding of endogenous EGFR ligands lead to strong cell cycle arrest. |
101
|
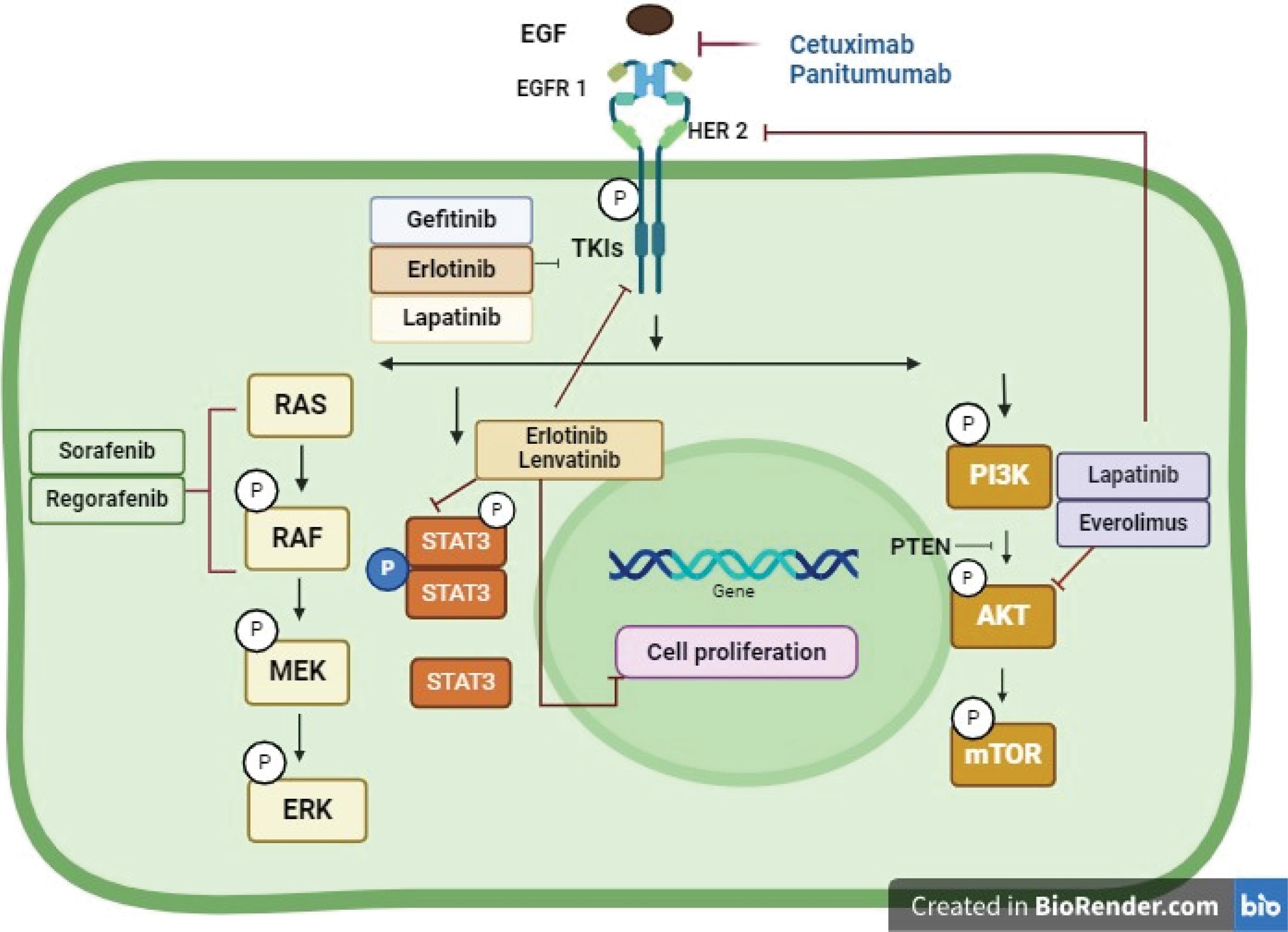
Fig. 4.
Therapeutic targeting of EGFR signaling in hepatocellular carcinoma. In HCC, EGFR is targeted by two therapies: small molecule inhibitors (gefitinib, erlotinib, lapatinib) that compete with ATP to inhibit tyrosine kinase activity, and EGFR targeted antibodies (cetuximab, panitumumab) that block ligand binding, receptor activation, and dimerization, causing receptor downregulation (Created by using BioRender software).
.
Therapeutic targeting of EGFR signaling in hepatocellular carcinoma. In HCC, EGFR is targeted by two therapies: small molecule inhibitors (gefitinib, erlotinib, lapatinib) that compete with ATP to inhibit tyrosine kinase activity, and EGFR targeted antibodies (cetuximab, panitumumab) that block ligand binding, receptor activation, and dimerization, causing receptor downregulation (Created by using BioRender software).
Conclusion
HCC is a common malignancy associated with a recurrence of metastatic disease, drug resistance, and a poor prognosis.110 HCC develops gradually after the liver injury stage; this is evidenced by the fact that more than 90% of tumors are diagnosed as chronic hepatitis or cirrhosis.111,112 HCC is characterized by its variants that exhibit different phenotypic and genetic properties, and tumorigenesis involves different molecular mechanisms.113 EGFR is pivotal in HCC pathogenesis, promoting tumor growth, repair, and metastasis. Overexpression of EGFR is associated with aggressive tumor behavior and poor outcomes, making it a key therapeutic target.114
Despite the success of EGFR inhibitors, including TKIs in preclinical and clinical trials, issues such as drug resistance and tumor heterogeneity remain significant. Therefore, combining EGFR-targeted therapies with other treatment modalities, such as immunotherapy or anti-angiogenic agents, may improve therapeutic efficacy and provide a more comprehensive approach to HCC management.
The outlook for future research on EGFR signalling in HCC should focus on overcoming challenges such as drug resistance and tumor heterogeneity. Advances in biomarker discovery could enable personalized therapies targeting EGFR, improving patient outcomes. Additionally, integrating EGFR-targeted treatments with emerging modalities like immunotherapy and anti-angiogenic agents holds promise for a more comprehensive approach to HCC management. Further exploration of EGFR-regulating miRNAs offers potential for innovative therapeutic strategies, particularly in mitigating drug resistance and addressing liver fibrosis. Combining computational and experimental approaches will be vital to unravel the complexities of EGFR signalling and its role in HCC pathogenesis. In our unpublished in-silico article, the identification of miRNAs that regulate EGFR expression may serve as potential therapeutic targets. The study provides a comprehensive framework to understand the complex interactions between miRNAs, TFs, and their target genes in HCC, highlighting the importance of integrating computational predictions with experimental data to develop effective therapies.
In conclusion, this study aims to confirm the importance of EGFR in developing HCC from the first stage of liver regeneration to the formation of chronic liver disease and then the transformation to HCC. Furthermore, this study focuses on the importance of targeting EGFR as a treatment target in HCC.
Review Highlights
What is the current knowledge?
-
EGFR overexpression in HCC is linked to tumor growth, metastasis, and resistance to targeted therapies.
-
Crosstalk between EGFR and other pathways, such as PI3K/AKT/mTOR and MAPK/ERK, exacerbates drug resistance and tumor aggressiveness.
-
EGFR inhibitors, including TKIs and monoclonal antibodies, have shown potential in reducing tumor progression but face challenges with resistance and heterogeneity.
What is new here?
-
New insights into the interaction between EGFR signalling and key inflammatory and fibrogenic mediators in HCC development.
-
Presentation of experimental models demonstrating the efficacy of EGFR inhibitors in reducing fibrosis, tumor proliferation, and metastasis.
Competing Interests
The authors declare that they have no conflict of interest.
Ethical Approval
The authors declare no ethical issues.
Acknowledgements
The authors are thankful to the Registrar of the University for providing all the support and facilities to complete this work.
References
- Pan H, Fu X, Huang W. Molecular mechanism of liver cancer. Anticancer Agents Med Chem 2011; 11:493-9. doi: 10.2174/187152011796011073 [Crossref] [ Google Scholar]
- Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, Allen C. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the Global Burden of Disease Study 2015. JAMA Oncol 2017; 3:1683-91. doi: 10.1001/jamaoncol.2017.3055 [Crossref] [ Google Scholar]
- Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol 2017; 3:524-48. doi: 10.1001/jamaoncol.2016.5688 [Crossref] [ Google Scholar]
- Motola-Kuba D, Zamora-Valdés D, Uribe M, Méndez-Sánchez N. Hepatocellular carcinoma An overview. Ann Hepatol 2006; 5:16-24. [ Google Scholar]
- Venook AP, Papandreou C, Furuse J, de Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist 2010; 15:5-13. doi: 10.1634/theoncologist.2010-S4-05 [Crossref] [ Google Scholar]
- Aravalli RN, Cressman EN, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch Toxicol 2013; 87:227-47. doi: 10.1007/s00204-012-0931-2 [Crossref] [ Google Scholar]
- Kanda T, Goto T, Hirotsu Y, Moriyama M, Omata M. Molecular mechanisms driving progression of liver cirrhosis towards hepatocellular carcinoma in chronic hepatitis B and C infections: a review. Int J Mol Sci 2019; 20:1358. doi: 10.3390/ijms20061358 [Crossref] [ Google Scholar]
- Kovalic AJ, Cholankeril G, Satapathy SK. Nonalcoholic fatty liver disease and alcoholic liver disease: metabolic diseases with systemic manifestations. Transl Gastroenterol Hepatol 2019; 4:65. doi: 10.21037/tgh.2019.08.09 [Crossref] [ Google Scholar]
- Lai CL, Yuen MF. Prevention of hepatitis B virus-related hepatocellular carcinoma with antiviral therapy. Hepatology 2013; 57:399-408. doi: 10.1002/hep.25937 [Crossref] [ Google Scholar]
- Chen C, Wang G. Mechanisms of hepatocellular carcinoma and challenges and opportunities for molecular targeted therapy. World J Hepatol 2015; 7:1964-70. doi: 10.4254/wjh.v7.i15.1964 [Crossref] [ Google Scholar]
- Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut 2014; 63:844-55. doi: 10.1136/gutjnl-2013-306627 [Crossref] [ Google Scholar]
- Bhushan B, Michalopoulos GK. Role of epidermal growth factor receptor in liver injury and lipid metabolism: emerging new roles for an old receptor. Chem Biol Interact 2020; 324:109090. doi: 10.1016/j.cbi.2020.109090 [Crossref] [ Google Scholar]
- Zandi R, Larsen AB, Andersen P, Stockhausen MT, Poulsen HS. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal 2007; 19:2013-23. doi: 10.1016/j.cellsig.2007.06.023 [Crossref] [ Google Scholar]
- Qiao Q, Zhang J, Wang W, Li Q. Over expression of transforming growth factor-alpha and epidermal growth factor receptor in human hepatic cirrhosis tissues. Hepatogastroenterology 2008; 55:169-72. [ Google Scholar]
- Harada K, Shiota G, Kawasaki H. Transforming growth factor-alpha and epidermal growth factor receptor in chronic liver disease and hepatocellular carcinoma. Liver 1999; 19:318-25. doi: 10.1111/j.1478-3231.1999.tb00056.x [Crossref] [ Google Scholar]
- Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005; 115:209-18. doi: 10.1172/jci24282 [Crossref] [ Google Scholar]
- Safadi R, Friedman SL. Hepatic fibrosis--role of hepatic stellate cell activation. MedGenMed 2002; 4:27. [ Google Scholar]
- Heldin CH. Targeting the PDGF signaling pathway in the treatment of non-malignant diseases. J Neuroimmune Pharmacol 2014; 9:69-79. doi: 10.1007/s11481-013-9484-2 [Crossref] [ Google Scholar]
- Michalopoulos GK. Liver regeneration. J Cell Physiol 2007; 213:286-300. doi: 10.1002/jcp.21172 [Crossref] [ Google Scholar]
- Berasain C, Castillo J, Prieto J, Avila MA. New molecular targets for hepatocellular carcinoma: the ErbB1 signaling system. Liver Int 2007; 27:174-85. doi: 10.1111/j.1478-3231.2006.01424.x [Crossref] [ Google Scholar]
- Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 2006; 25:3787-800. doi: 10.1038/sj.onc.1209556 [Crossref] [ Google Scholar]
- Peng S, Wang R, Zhang X, Ma Y, Zhong L, Li K. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol Cancer 2019; 18:165. doi: 10.1186/s12943-019-1073-4 [Crossref] [ Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2:127-37. doi: 10.1038/35052073 [Crossref] [ Google Scholar]
- Diamantis N, Banerji U. Antibody-drug conjugates--an emerging class of cancer treatment. Br J Cancer 2016; 114:362-7. doi: 10.1038/bjc.2015.435 [Crossref] [ Google Scholar]
- Roskoski R Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol Res 2019; 139:395-411. doi: 10.1016/j.phrs.2018.11.014 [Crossref] [ Google Scholar]
- Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel) 2017; 9:52. doi: 10.3390/cancers9050052 [Crossref] [ Google Scholar]
- Roskoski R, Jr Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res 2014; 79:34-74. doi: 10.1016/j.phrs.2013.11.002 [Crossref] [ Google Scholar]
- Cymer F, Schneider D. Transmembrane helix-helix interactions involved in ErbB receptor signaling. Cell Adh Migr 2010; 4:299-312. doi: 10.4161/cam.4.2.11191 [Crossref] [ Google Scholar]
- Red Brewer M, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell 2009; 34:641-51. doi: 10.1016/j.molcel.2009.04.034 [Crossref] [ Google Scholar]
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem 2002; 277:46265-72. doi: 10.1074/jbc.M207135200 [Crossref] [ Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006; 125:1137-49. doi: 10.1016/j.cell.2006.05.013 [Crossref] [ Google Scholar]
- Lee JC, Vivanco I, Beroukhim R, Huang JH, Feng WL, DeBiasi RM. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med 2006; 3:e485. doi: 10.1371/journal.pmed.0030485 [Crossref] [ Google Scholar]
-
White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol 2012; 10: 1342-59.e2. doi: 10.1016/j.cgh.2012.10.001.
- Berasain C, Perugorria MJ, Latasa MU, Castillo J, Goñi S, Santamaría M. The epidermal growth factor receptor: a link between inflammation and liver cancer. Exp Biol Med (Maywood) 2009; 234:713-25. doi: 10.3181/0901-mr-12 [Crossref] [ Google Scholar]
- Diao J, Pantua H, Ngu H, Komuves L, Diehl L, Schaefer G. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J Virol 2012; 86:10935-49. doi: 10.1128/jvi.00750-12 [Crossref] [ Google Scholar]
- Ninio L, Nissani A, Meirson T, Domovitz T, Genna A, Twafra S. Hepatitis C virus enhances the invasiveness of hepatocellular carcinoma via EGFR-mediated invadopodia formation and activation. Cells 2019; 8:1395. doi: 10.3390/cells8111395 [Crossref] [ Google Scholar]
- Komposch K, Sibilia M. EGFR signaling in liver diseases. Int J Mol Sci 2015; 17:30. doi: 10.3390/ijms17010030 [Crossref] [ Google Scholar]
- Iwamoto M, Saso W, Nishioka K, Ohashi H, Sugiyama R, Ryo A. The machinery for endocytosis of epidermal growth factor receptor coordinates the transport of incoming hepatitis B virus to the endosomal network. J Biol Chem 2020; 295:800-7. doi: 10.1074/jbc.AC119.010366 [Crossref] [ Google Scholar]
- Daveau M, Scotte M, François A, Coulouarn C, Ros G, Tallet Y. Hepatocyte growth factor, transforming growth factor alpha, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol Carcinog 2003; 36:130-41. doi: 10.1002/mc.10103 [Crossref] [ Google Scholar]
- Dalke DD, Sorrell MF, Casey CA, Tuma DJ. Chronic ethanol administration impairs receptor-mediated endocytosis of epidermal growth factor by rat hepatocytes. Hepatology 1990; 12:1085-91. doi: 10.1002/hep.1840120502 [Crossref] [ Google Scholar]
- Okuda K, Umemura A, Umemura S, Kataoka S, Taketani H, Seko Y. Honokiol prevents non-alcoholic steatohepatitis-induced liver cancer via EGFR degradation through the glucocorticoid receptor-MIG6 axis. Cancers (Basel) 2021; 13:1515. doi: 10.3390/cancers13071515 [Crossref] [ Google Scholar]
- Scheving LA, Zhang X, Garcia OA, Wang RF, Stevenson MC, Threadgill DW. Epidermal growth factor receptor plays a role in the regulation of liver and plasma lipid levels in adult male mice. Am J Physiol Gastrointest Liver Physiol 2014; 306:G370-81. doi: 10.1152/ajpgi.00116.2013 [Crossref] [ Google Scholar]
- Prada PO, Ropelle ER, Mourão RH, de Souza CT, Pauli JR, Cintra DE. EGFR tyrosine kinase inhibitor (PD153035) improves glucose tolerance and insulin action in high-fat diet-fed mice. Diabetes 2009; 58:2910-9. doi: 10.2337/db08-0506 [Crossref] [ Google Scholar]
- Wang B, Zhang J, Song F, Tian M, Shi B, Jiang H. EGFR regulates iron homeostasis to promote cancer growth through redistribution of transferrin receptor 1. Cancer Lett 2016; 381:331-40. doi: 10.1016/j.canlet.2016.08.006 [Crossref] [ Google Scholar]
-
Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol 2005; 1: 2005.0010. doi: 10.1038/msb4100014.
- Zhang Y, Wang L, Zhang M, Jin M, Bai C, Wang X. Potential mechanism of interleukin-8 production from lung cancer cells: an involvement of EGF-EGFR-PI3K-Akt-Erk pathway. J Cell Physiol 2012; 227:35-43. doi: 10.1002/jcp.22722 [Crossref] [ Google Scholar]
- Sun D, Sun B, Liu T, Zhao X, Che N, Gu Q. Slug promoted vasculogenic mimicry in hepatocellular carcinoma. J Cell Mol Med 2013; 17:1038-47. doi: 10.1111/jcmm.12087 [Crossref] [ Google Scholar]
- Singh JK, Farnie G, Bundred NJ, Simões BM, Shergill A, Landberg G. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin Cancer Res 2013; 19:643-56. doi: 10.1158/1078-0432.Ccr-12-1063 [Crossref] [ Google Scholar]
- Kubo F, Ueno S, Hiwatashi K, Sakoda M, Kawaida K, Nuruki K. Interleukin 8 in human hepatocellular carcinoma correlates with cancer cell invasion of vessels but not with tumor angiogenesis. Ann Surg Oncol 2005; 12:800-7. doi: 10.1245/aso.2005.07.015 [Crossref] [ Google Scholar]
- Bhushan B, Banerjee S, Paranjpe S, Koral K, Mars WM, Stoops JW. Pharmacologic inhibition of epidermal growth factor receptor suppresses nonalcoholic fatty liver disease in a murine fast-food diet model. Hepatology 2019; 70:1546-63. doi: 10.1002/hep.30696 [Crossref] [ Google Scholar]
- Choung S, Kim JM, Joung KH, Lee ES, Kim HJ, Ku BJ. Epidermal growth factor receptor inhibition attenuates non-alcoholic fatty liver disease in diet-induced obese mice. PLoS One 2019; 14:e0210828. doi: 10.1371/journal.pone.0210828 [Crossref] [ Google Scholar]
- Liang D, Chen H, Zhao L, Zhang W, Hu J, Liu Z. Inhibition of EGFR attenuates fibrosis and stellate cell activation in diet-induced model of nonalcoholic fatty liver disease. Biochim Biophys Acta Mol Basis Dis 2018; 1864:133-42. doi: 10.1016/j.bbadis.2017.10.016 [Crossref] [ Google Scholar]
- Richter K, Kietzmann T. Reactive oxygen species and fibrosis: further evidence of a significant liaison. Cell Tissue Res 2016; 365:591-605. doi: 10.1007/s00441-016-2445-3 [Crossref] [ Google Scholar]
- Mazzocca A, Fransvea E, Dituri F, Lupo L, Antonaci S, Giannelli G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 2010; 51:523-34. doi: 10.1002/hep.23285 [Crossref] [ Google Scholar]
- Urtasun R, Latasa MU, Demartis MI, Balzani S, Goñi S, Garcia-Irigoyen O. Connective tissue growth factor autocriny in human hepatocellular carcinoma: oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology 2011; 54:2149-58. doi: 10.1002/hep.24587 [Crossref] [ Google Scholar]
- Bhushan B, Chavan H, Borude P, Xie Y, Du K, McGill MR. Dual role of epidermal growth factor receptor in liver injury and regeneration after acetaminophen overdose in mice. Toxicol Sci 2017; 155:363-78. doi: 10.1093/toxsci/kfw213 [Crossref] [ Google Scholar]
- Wang T, Takikawa Y, Tabuchi T, Satoh T, Kosaka K, Suzuki K. Carnosic acid (CA) prevents lipid accumulation in hepatocytes through the EGFR/MAPK pathway. J Gastroenterol 2012; 47:805-13. doi: 10.1007/s00535-012-0546-7 [Crossref] [ Google Scholar]
- Berasain C, García-Trevijano ER, Castillo J, Erroba E, Lee DC, Prieto J. Amphiregulin: an early trigger of liver regeneration in mice. Gastroenterology 2005; 128:424-32. doi: 10.1053/j.gastro.2004.11.006 [Crossref] [ Google Scholar]
- Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation 2007; 75:770-87. doi: 10.1111/j.1432-0436.2007.00238.x [Crossref] [ Google Scholar]
- Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci U S A 2007; 104:17081-6. doi: 10.1073/pnas.0704126104 [Crossref] [ Google Scholar]
- López-Luque J, Caballero-Díaz D, Martinez-Palacián A, Roncero C, Moreno-Càceres J, García-Bravo M. Dissecting the role of epidermal growth factor receptor catalytic activity during liver regeneration and hepatocarcinogenesis. Hepatology 2016; 63:604-19. doi: 10.1002/hep.28134 [Crossref] [ Google Scholar]
- Scheving LA, Zhang X, Stevenson MC, Threadgill DW, Russell WE. Loss of hepatocyte EGFR has no effect alone but exacerbates carbon tetrachloride-induced liver injury and impairs regeneration in hepatocyte Met-deficient mice. Am J Physiol Gastrointest Liver Physiol 2015; 308:G364-77. doi: 10.1152/ajpgi.00364.2014 [Crossref] [ Google Scholar]
- Bhushan B, Stoops JW, Mars WM, Orr A, Bowen WC, Paranjpe S. TCPOBOP-induced hepatomegaly and hepatocyte proliferation are attenuated by combined disruption of MET and EGFR signaling. Hepatology 2019; 69:1702-18. doi: 10.1002/hep.30109 [Crossref] [ Google Scholar]
- Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem 2000; 275:8806-11. doi: 10.1074/jbc.275.12.8806 [Crossref] [ Google Scholar]
- Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology 2006; 43:S45-53. doi: 10.1002/hep.20969 [Crossref] [ Google Scholar]
- Carver RS, Stevenson MC, Scheving LA, Russell WE. Diverse expression of ErbB receptor proteins during rat liver development and regeneration. Gastroenterology 2002; 123:2017-27. doi: 10.1053/gast.2002.37060 [Crossref] [ Google Scholar]
- Reinehr R, Becker S, Höngen A, Haüssinger D. The Src family kinase Yes triggers hyperosmotic activation of the epidermal growth factor receptor and CD95. J Biol Chem 2004; 279:23977-87. doi: 10.1074/jbc.M401519200 [Crossref] [ Google Scholar]
- Reinehr R, Häussinger D. CD95 death receptor and epidermal growth factor receptor (EGFR) in liver cell apoptosis and regeneration. Arch Biochem Biophys 2012; 518:2-7. doi: 10.1016/j.abb.2011.12.004 [Crossref] [ Google Scholar]
- Sanguinetti AR, Cao H, Corley Mastick C. Fyn is required for oxidative- and hyperosmotic-stress-induced tyrosine phosphorylation of caveolin-1. Biochem J 2003; 376:159-68. doi: 10.1042/bj20030336 [Crossref] [ Google Scholar]
- Yoshizumi M, Abe J, Haendeler J, Huang Q, Berk BC. Src and Cas mediate JNK activation but not ERK1/2 and p38 kinases by reactive oxygen species. J Biol Chem 2000; 275:11706-12. doi: 10.1074/jbc.275.16.11706 [Crossref] [ Google Scholar]
- Abe J, Okuda M, Huang Q, Yoshizumi M, Berk BC. Reactive oxygen species activate p90 ribosomal S6 kinase via Fyn and Ras. J Biol Chem 2000; 275:1739-48. doi: 10.1074/jbc.275.3.1739 [Crossref] [ Google Scholar]
- Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008; 88:125-72. doi: 10.1152/physrev.00013.2007 [Crossref] [ Google Scholar]
- Poynard T, Mathurin P, Lai CL, Guyader D, Poupon R, Tainturier MH. A comparison of fibrosis progression in chronic liver diseases. J Hepatol 2003; 38:257-65. doi: 10.1016/s0168-8278(02)00413-0 [Crossref] [ Google Scholar]
- Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev 2017; 121:27-42. doi: 10.1016/j.addr.2017.05.007 [Crossref] [ Google Scholar]
- Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008; 134:1655-69. doi: 10.1053/j.gastro.2008.03.003 [Crossref] [ Google Scholar]
- Fuchs BC, Hoshida Y, Fujii T, Wei L, Yamada S, Lauwers GY. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 2014; 59:1577-90. doi: 10.1002/hep.26898 [Crossref] [ Google Scholar]
- Liu H, Pan Z, Li A, Fu S, Lei Y, Sun H. Roles of chemokine receptor 4 (CXCR4) and chemokine ligand 12 (CXCL12) in metastasis of hepatocellular carcinoma cells. Cell Mol Immunol 2008; 5:373-8. doi: 10.1038/cmi.2008.46 [Crossref] [ Google Scholar]
- Wong VW, Yu J, Cheng AS, Wong GL, Chan HY, Chu ES. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int J Cancer 2009; 124:2766-70. doi: 10.1002/ijc.24281 [Crossref] [ Google Scholar]
- Shirabe K, Mano Y, Muto J, Matono R, Motomura T, Toshima T. Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surg Today 2012; 42:1-7. doi: 10.1007/s00595-011-0058-8 [Crossref] [ Google Scholar]
- Gallucci RM, Simeonova PP, Toriumi W, Luster MI. TNF-alpha regulates transforming growth factor-alpha expression in regenerating murine liver and isolated hepatocytes. J Immunol 2000; 164:872-8. doi: 10.4049/jimmunol.164.2.872 [Crossref] [ Google Scholar]
- Perugorria MJ, Latasa MU, Nicou A, Cartagena-Lirola H, Castillo J, Goñi S. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology 2008; 48:1251-61. doi: 10.1002/hep.22437 [Crossref] [ Google Scholar]
- Berasain C, Avila MA. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol 2014; 49:9-23. doi: 10.1007/s00535-013-0907-x [Crossref] [ Google Scholar]
- Finot F, Masson R, Desmots F, Ribault C, Bichet N, Vericat JA. Combined stimulation with the tumor necrosis factor α and the epidermal growth factor promotes the proliferation of hepatocytes in rat liver cultured slices. Int J Hepatol 2012; 2012:785786. doi: 10.1155/2012/785786 [Crossref] [ Google Scholar]
- Citri A, Yarden Y. EGF-ErbB signalling: towards the systems level. Nat Rev Mol Cell Biol 2006; 7:505-16. doi: 10.1038/nrm1962 [Crossref] [ Google Scholar]
- Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis 2004; 9:667-76. doi: 10.1023/B:APPT.0000045801.15585.dd [Crossref] [ Google Scholar]
- Adjei AA, Hidalgo M. Intracellular signal transduction pathway proteins as targets for cancer therapy. J Clin Oncol 2005; 23:5386-403. doi: 10.1200/jco.2005.23.648 [Crossref] [ Google Scholar]
- Tian LY, Smit DJ, Jücker M. The role of PI3K/AKT/mTOR signaling in hepatocellular carcinoma metabolism. Int J Mol Sci 2023; 24:2652. doi: 10.3390/ijms24032652 [Crossref] [ Google Scholar]
- Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J. Hepatocyte-specific PTEN deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest 2004; 113:1774-83. doi: 10.1172/jci20513 [Crossref] [ Google Scholar]
- Zhou L, Huang Y, Li J, Wang Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med Oncol 2010; 27:255-61. doi: 10.1007/s12032-009-9201-4 [Crossref] [ Google Scholar]
- Jiang X, Huang F, Marusyk A, Sorkin A. Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol Biol Cell 2003; 14:858-70. doi: 10.1091/mbc.e02-08-0532 [Crossref] [ Google Scholar]
- Sridhar SS, Hedley D, Siu LL. Raf kinase as a target for anticancer therapeutics. Mol Cancer Ther 2005; 4:677-85. doi: 10.1158/1535-7163.Mct-04-0297 [Crossref] [ Google Scholar]
- Leicht DT, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M. Raf kinases: function, regulation and role in human cancer. Biochim Biophys Acta 2007; 1773:1196-212. doi: 10.1016/j.bbamcr.2007.05.001 [Crossref] [ Google Scholar]
- Song S, Yu Z, You Y, Liu C, Xie X, Lv H. EGFR/MET promotes hepatocellular carcinoma metastasis by stabilizing tumor cells and resisting to RTKs inhibitors in circulating tumor microemboli. Cell Death Dis 2022; 13:351. doi: 10.1038/s41419-022-04796-8 [Crossref] [ Google Scholar]
- Xu X, Xia J, Wang X. Potential anticancer therapies via CXCL5 and its receptors. Expert Rev Clin Pharmacol 2012; 5:347-50. doi: 10.1586/ecp.12.30 [Crossref] [ Google Scholar]
- Lorenc P, Sikorska A, Molenda S, Guzniczak N, Dams-Kozlowska H, Florczak A. Physiological and tumor-associated angiogenesis: key factors and therapy targeting VEGF/VEGFR pathway. Biomed Pharmacother 2024; 180:117585. doi: 10.1016/j.biopha.2024.117585 [Crossref] [ Google Scholar]
- Turetta M, Bulfoni M, Brisotto G, Fasola G, Zanello A, Biscontin E. Assessment of the mutational status of NSCLC using hypermetabolic circulating tumor cells. Cancers (Basel) 2018; 10:270. doi: 10.3390/cancers10080270 [Crossref] [ Google Scholar]
- Yang B, Li M, Tang W, Liu W, Zhang S, Chen L. Dynamic network biomarker indicates pulmonary metastasis at the tipping point of hepatocellular carcinoma. Nat Commun 2018; 9:678. doi: 10.1038/s41467-018-03024-2 [Crossref] [ Google Scholar]
- Chen HA, Kuo TC, Tseng CF, Ma JT, Yang ST, Yen CJ. Angiopoietin-like protein 1 antagonizes MET receptor activity to repress sorafenib resistance and cancer stemness in hepatocellular carcinoma. Hepatology 2016; 64:1637-51. doi: 10.1002/hep.28773 [Crossref] [ Google Scholar]
- Jardim DL, Tang C, De Melo Gagliato D, Falchook GS, Hess K, Janku F. Analysis of 1,115 patients tested for MET amplification and therapy response in the MD Anderson phase I clinic. Clin Cancer Res 2014; 20:6336-45. doi: 10.1158/1078-0432.Ccr-14-1293 [Crossref] [ Google Scholar]
- Huang P, Xu X, Wang L, Zhu B, Wang X, Xia J. The role of EGF-EGFR signalling pathway in hepatocellular carcinoma inflammatory microenvironment. J Cell Mol Med 2014; 18:218-30. doi: 10.1111/jcmm.12153 [Crossref] [ Google Scholar]
- Huether A, Höpfner M, Baradari V, Schuppan D, Scherübl H. EGFR blockade by cetuximab alone or as combination therapy for growth control of hepatocellular cancer. Biochem Pharmacol 2005; 70:1568-78. doi: 10.1016/j.bcp.2005.09.007 [Crossref] [ Google Scholar]
- Hu B, Zou T, Qin W, Shen X, Su Y, Li J. Inhibition of EGFR overcomes acquired lenvatinib resistance driven by STAT3-ABCB1 signaling in hepatocellular carcinoma. Cancer Res 2022; 82:3845-57. doi: 10.1158/0008-5472.Can-21-4140 [Crossref] [ Google Scholar]
- Park NH, Song IH, Chung YH. Chronic hepatitis B in hepatocarcinogenesis. Postgrad Med J 2006; 82:507-15. doi: 10.1136/pgmj.2006.047431 [Crossref] [ Google Scholar]
- Marshall J. Clinical implications of the mechanism of epidermal growth factor receptor inhibitors. Cancer 2006; 107:1207-18. doi: 10.1002/cncr.22133 [Crossref] [ Google Scholar]
- Schiffer E, Housset C, Cacheux W, Wendum D, Desbois-Mouthon C, Rey C. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology 2005; 41:307-14. doi: 10.1002/hep.20538 [Crossref] [ Google Scholar]
- Sun D, Liu J, Wang Y, Dong J. Co-administration of MDR1 and BCRP or EGFR/PI3K inhibitors overcomes lenvatinib resistance in hepatocellular carcinoma. Front Oncol 2022; 12:944537. doi: 10.3389/fonc.2022.944537 [Crossref] [ Google Scholar]
- Ueda S, Basaki Y, Yoshie M, Ogawa K, Sakisaka S, Kuwano M. PTEN/Akt signaling through epidermal growth factor receptor is prerequisite for angiogenesis by hepatocellular carcinoma cells that is susceptible to inhibition by gefitinib. Cancer Res 2006; 66:5346-53. doi: 10.1158/0008-5472.Can-05-3684 [Crossref] [ Google Scholar]
- Ezzoukhry Z, Louandre C, Trécherel E, Godin C, Chauffert B, Dupont S. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int J Cancer 2012; 131:2961-9. doi: 10.1002/ijc.27604 [Crossref] [ Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021; 71:209-49. doi: 10.3322/caac.21660 [Crossref] [ Google Scholar]
- Lupberger J, Hildt E. Hepatitis B virus-induced oncogenesis. World J Gastroenterol 2007; 13:74-81. doi: 10.3748/wjg.v13.i1.74 [Crossref] [ Google Scholar]
- Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008; 48:1312-27. doi: 10.1002/hep.22506 [Crossref] [ Google Scholar]
- Dimri M, Satyanarayana A. Molecular signaling pathways and therapeutic targets in hepatocellular carcinoma. Cancers (Basel) 2020; 12:491. doi: 10.3390/cancers12020491 [Crossref] [ Google Scholar]
- Ito Y, Takeda T, Sakon M, Tsujimoto M, Higashiyama S, Noda K. Expression and clinical significance of ErbB receptor family in hepatocellular carcinoma. Br J Cancer 2001; 84:1377-83. doi: 10.1054/bjoc.2000.1580 [Crossref] [ Google Scholar]
- Blivet-Van Eggelpoël MJ, Chettouh H, Fartoux L, Aoudjehane L, Barbu V, Rey C. Epidermal growth factor receptor and HER-3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J Hepatol 2012; 57:108-15. doi: 10.1016/j.jhep.2012.02.019 [Crossref] [ Google Scholar]