BioImpacts. 7(1):59-71.
doi: 10.15171/bi.2017.08
Review
Pleiotropic cytotoxicity of VacA toxin in host cells and its impact on immunotherapy
Farnaz Fahimi 1, †, Mohammad Reza Tohidkia 1, †, Mehdi Fouladi 1, Reza Aghabeygi 2, Naser Samadi 2, Yadollah Omidi 1, 2, 3, *
Author information:
1Research Center for Pharmaceutical Nanotechnology, Biomedicine Institute, Tabriz University of Medical Sciences, Tabriz, Iran
2School of Advanced Biomedical Sciences, Tabriz University of Medical Sciences, Tabriz, Iran
3Department of Pharmaceutics, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
† These authors have an equal contribution.
Abstract
Introduction:
In the recent decades, a number of studies have highlighted the importance of Helicobacter pylori in the initiation and development of peptic ulcer and gastric cancer. Some potential virulence factors (e.g., urease, CagA, VacA, BabA) are exploited by this microorganism, facilitating its persistence through evading human defense mechanisms. Among these toxins and enzymes, vacuolating toxin A (VacA) is of a great importance in the pathogenesis of H. pylori. VacA toxin shows different pattern of cytotoxicity through binding to different cell surface receptors in various cells.
Methods:
To highlight attempts in treatment for H. pylori infection, here, we discussed the VacA potential as a candidate for development of vaccine and targeted immunotherapy. Furthermore, we reviewed the related literature to provide key insights on association of the genetic variants of VacA with the toxicity of the toxin in cells.
Results:
A number of investigations on the receptor(s) binding of VacA toxin confirmed the pleiotropic nature of VacA that uses a unique mechanism for internalization through some membrane components such as lipid rafts and glycophosphatidylinositol (GPI)-anchored proteins (GPI-AP). Considering the high potency of VacA toxin in the clinical presentations in infection and assisting persistence and colonization of H. pylori, it is considered as one of the pivotal components in production vaccines and monoclonal antibodies (mAbs).
Conclusion:
It is possible to generate mAbs with a considerable potential to convert into secretory immunoglobulins that could penetrate into the niche of H. pylori and inhibit its normal functionalities. Further, conjugation of H. pylori targeting Ab fragments with the toxic agents or drug delivery systems (DDSs) offers new generation of H. pylori treatments.
Keywords: Helicobacter pylori, VacA, Cell receptor, Vaccine, Immunotherapy
Copyright and License Information
© 2017 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Long-term colonization of Helicobacter pylori in the human stomach is often associated with the high incidence of development of peptic ulcers, gastric lymphoma, and gastric adenocarcinoma. Gastric cancer is considered as one of the most prevalent malignancies worldwide, especially in the developing countries. It is largely related to the high colonization of H. pylori, which shows a high rate of morality, in large part because of diagnosis at the late stages of the disease.
1-3
H. pylori is classified as type 1 carcinogenic bacteria because more than 60% of the non-cardiac gastric cancers are related to H. pylori infection.
4-6
Although the presence of H. pylori is not enough for the initiation of cancer, it is necessary for such phenomenon. H. pylori-associated gastric cancer seems to initiate by the loss of acidic juice secretion due to the persistent atrophic gastritis.
7
H. pylori can survive and colonize in the human stomach through the functional expression of VacA that is produced by all strains, even though with different levels of expression. VacA toxin creates a selective ion channel in order to utilize the electrolyte and metabolites of the host cells for the survival of H. pylori in the mucosal layer.
8
The effects of VacA toxin on the epithelial cells are mediated by diverse mechanisms. Endocytosis of VacA associates with the pattern alteration of intracellular endocytic pathways, resulting in formation of vacuoles, and the impairment of the mitochondrial membrane potential and hence induction of apoptosis.
9,10
Besides, VacA binds to variable cell surface receptors and triggers several intracellular signaling pathways, in particular, mitogen-activated protein kinase (MAPK)/p38 and extracellular signal-regulated kinases 1 and 2 (ERK1/2), which lead to recruitment of cytokines initiating inflammatory response.
11-13
The active VacA toxin is made up of two functional (p33) and binding (p55) domains. Based on sequence diversities in these domains, H. pylori is classified into 4 strains, including: s1m1, s1m2, s2m2 and s2m1. These strains were shown to pose different extent of vacuolation in various cells.
13,14
Depending on the cell type and H. pylori strain, the VacA interaction with cells occur through various receptors such as receptor protein tyrosine phosphatase beta (RPTPβ), sphingomyelin, epidermal growth factor receptor (EGFR) and heparin/heparan sulfate. Of these, the RPTPβ is the main receptor for VacA binding, which guaranties survival of H. pylori within the host cells.
15,16
The influence of cell surface receptors, in particular RPTPβ, on the VacA-induced cell vacuolation in vitro is still controversial. Collectively, the VacA toxin is considered as a multi-receptor toxin that imposes cellular vacuolization.
17
In addition to the effect of VacA on long-term persistence of H. pylori, VacA is deemed to be a marker for gastric cancer diagnosis, which is also a determinant for the initiation of metastasis in infectious patients.
18,19
Former reviews on this subject have not covered the exact roles and effects of multiple receptors on binding and in vitro cytotoxicity of VacA and its influence on immunotherapeutical approaches. Hence, the foremost goal of the current review is to discuss the genotype of VacA toxin and articulate the cell specific toxicity on epithelial cells elicited by VacA and its potential as a candidate for development of vaccine against H. pylori.
Structure and expression of VacA
The VacA gene is a monosistronic gene with the length of 3860-3940 base pairs. All strains of H. pylori possess the VacA gene, but with different levels of expression leading to different degrees of cytotoxicity. Expression level of the VacA toxin can be affected by several factors, including: (a) promoter strength, (b) stem-loop structures at the 3´ end of mRNA resulting in mRNA endurance, (c) presence of activators or inhibitor sequences, and (d) amino acid sequence differences found between H. pylori strains leading to distinct secretion pattern.
20,21
The expression of VacA gene produces a 1287 amino acids (aa), a 140 KDa length pre-protoxin, that is processed to a mature toxin through two cleavages at the N- and C-terminal ends of the toxin by membrane-associated proteases. Similar to autotransporter proteins (ATs), the first proteolysis is occurred by LepB peptidase at the N-terminal signal sequence (33 aa) responsible for transporting toxin to the bacterial periplasm.
22-24
The second proteolysis at the C-terminal domain, which happens simultaneously with the secretion of toxin to the outer membrane, produces a 88 KDa mature toxin, a 12 KDa secreted peptide (linker peptide), and a 33 KDa C-Ter autotransporter domain remaining associated with the bacterial membrane.
22,25,26
The complete processing of VacA produces the mature VacA toxin that is considered as an A-B type toxin (Fig. 1).
27,28
The mature VacA toxin is consisted of p33 and p55 domains, which are essential for its activity and binding to the host cells and is performed by various residues confirmed by several studies (Table 1).
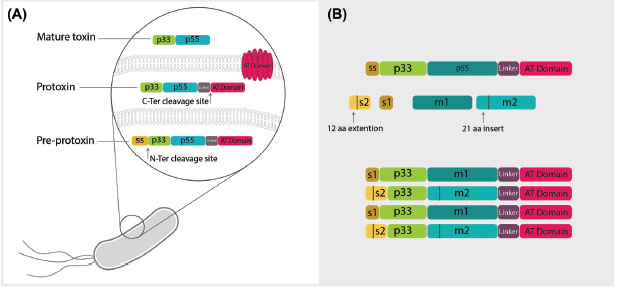
Fig. 1.
The molecular structure of vacuolating toxin of Helicobacter pylori. A) The vacuolating toxin of Helicobacter pylori is expressed
as pre-protoxin inside bacteria containing p33 and p55 domains, a linker domain and autotransporter (AT) domain responsible for toxin
secretion from bacteria. Maturation of the toxin occurs through two enzymatic cleavages. First, spontaneous N-ter cleavage site leads to
breakage of signal sequence (ss), which is a determinant of directing of toxin to periplasmic space. Next, cleavage of the AT domain occurs
simultaneously with the secretion of mature toxin to external environment of bacteria. The linker domain degrades by exposure to the
environment and mature toxin is produced including two major regions known as p33 and p55. B) There are two diverse regions inside the
mature toxin called s in p33 domain and m in p55 domain. The s region is located in the signal sequence of toxin in p33 with two types of
s1 and s2 and an extra 21 amino acid extension sequence. The other divergent portion in p55 is also classified in two types, m1 and m2,
with 21 amino acid insertion and shows different cell binding capacity to various receptors. Based on these two portions, VacA is classified
into four groups of s1m1, s1m2, s2m2 and s2m1, all of which determine various pathogenicity based on their genotype.
.
The molecular structure of vacuolating toxin of Helicobacter pylori. A) The vacuolating toxin of Helicobacter pylori is expressed
as pre-protoxin inside bacteria containing p33 and p55 domains, a linker domain and autotransporter (AT) domain responsible for toxin
secretion from bacteria. Maturation of the toxin occurs through two enzymatic cleavages. First, spontaneous N-ter cleavage site leads to
breakage of signal sequence (ss), which is a determinant of directing of toxin to periplasmic space. Next, cleavage of the AT domain occurs
simultaneously with the secretion of mature toxin to external environment of bacteria. The linker domain degrades by exposure to the
environment and mature toxin is produced including two major regions known as p33 and p55. B) There are two diverse regions inside the
mature toxin called s in p33 domain and m in p55 domain. The s region is located in the signal sequence of toxin in p33 with two types of
s1 and s2 and an extra 21 amino acid extension sequence. The other divergent portion in p55 is also classified in two types, m1 and m2,
with 21 amino acid insertion and shows different cell binding capacity to various receptors. Based on these two portions, VacA is classified
into four groups of s1m1, s1m2, s2m2 and s2m1, all of which determine various pathogenicity based on their genotype.
Table 1.
Function of various domains of the VacA toxin
|
Domain
|
Amino acid
sequence
|
Role of sequence in toxin activity
|
Ref.
|
|
P33
|
6-27 |
Vacuolation-apoptosisoligomerization |
27
,29
|
|
|
1-32
|
Ion channel formations |
30
|
|
|
49-57
|
Oligomerization |
30
|
|
P55
|
346-347 |
Vacuolating-depolarizationoligomerization |
29
|
|
|
599-628
|
Folding and secretion of protein |
31
|
|
|
312-478
|
Interaction with p33 |
29
|
|
|
342-361
|
Cleavage site |
32
|
|
|
313-478
|
P33/p55 interaction |
32
|
The function of VacA is not fully restricted to the domains introduced
for, and each domain- p33 and p55- is participating in the role of the
other domain.
P55 domain
The P55 domain shows different binding specificity to various cells, which is related to a divergent region known as "m region" with the least recombination between strains. Based on the m region, VacA toxin is classified into two types of m1 and m2, which are different in receptor binding and interaction with the host cells as well as the expression level.
33,34
Unlike the m1 VacA, the m2 VacA has an additional 23 aa insert located at residues of 460-496, a region that has no effect on the toxin cytotoxicity and/or cell specificity.
5,22
Differences in the m region contributes to both different degrees of cytotoxicity in various cells and discrete incidence of gastritis and gastric cancer, even though no relation has been observed between the m region and incidence of peptic ulcer in patients.
5,35,36
P33 domain
The p33 domain (i.e., residues 1 to 311) is a functional domain of the VacA toxin that causes cell vacuolation through formation of anion selective channels.
37,38
A hydrophobic 32 aa residual region with a-helix structure is located at the vicinity of the N-terminus of the p33 domain which assumes to be critical for the toxicity of VacA and is designated as s region.
39
Further, some researches have confirmed the importance of the transmembrane dimerization sequence consisted of three tandem repeats of GXXXG motifs for the channel and vacuole formation.
40
Based upon the sequence diversity in the "s region", VacA toxin has been classified into two types of s1 and s2. These two types -like m types- impose the toxicity potential of the toxin via (a) increased expression of the BAbA2 and cagPAI and VacA, (b) enhanced efficiency of signal sequence that facilitates toxin transformation, and (c) elevated capacity of anion selective channels. All these result in phenotypic manifestation of the toxin like peptic ulcer and gastric inflammation.
41
The s1 region shows hydrophobic characteristics that mediates dimerization of toxin leading to channel formation. The s2 region possesses a hydrophilic sequence extension of 12 aa providing a distinct cleavage site from s1, which results in inhibition of cell vacuolation.
42,43
Other polymorphic regions inside VacA
Besides two mentioned divergent regions inside VacA toxin, two additional regions are identified known as intermediate (i) and deletion (d). The i region, which is located between s and m is classified in 3 diverse types known as i1, i2 and i3. This region is considered to be responsible for increased potential of carcinogenesis and expression of CagA,
44
though one study confirmed that the presence of i is not as efficient as s and m regions on the incidence of disease.
44-46
The type of i region is somehow dependent on the presence of types of s and m, so that presence of s1m1 increases the incidence of i1 to be expressed and result in an increased outcome in severity of the disease.
47
The deletion of 81 residues between i and m regions is another polymorphic region inside VacA toxin, which is located at the N-terminus of p55 and is considered to be related to the vacuolating activity and binding to host cells, gastric mucusal atrophy and neutrophil infiltration. Despite the influence of s, m and i regions diversity on toxicity of VacA, it seems that different d regions provide the same function(s).
45
In general, it is believed that various types of VacA toxin are classified based on their polymorphic regions and distributed around the world. They are responsible for various types of diseases with different severity due to their phenotypes expressed and also the effect of host genetic background and environmental effects.
45
Cell surface receptors for VacA toxin
Receptor-like protein tyrosine phosphatase β (RPTPβ)
RPTPβ is considered as the major cell receptor for binding of VacA toxin. Similar to inhibitory ligand of RPTPβ, pleiotrophin, the VacA prohibits the phosphatase activity of RPTPβ via interacting with QTTQP motif located at 747-751 containing two O-glycosylation sites that is distinct from the binding site of pleiotrophin.
48
Inhibitory effect of VacA on RPTPβ leads to vacuolation-independent cytotoxicity through phosphorylation of Git-1 that triggers several intracellular signal transductions related to pro-inflammation and ulcerogenesis.
48
Phosphorylation of Git-1 initiates signaling cascades, leading to actin cytoskeleton impairment and focal adhesion segregation known as the first steps in ulcerogenesis.
49,50
Further, VacA induces the activation of MAPK pathways through the activation of p38 and ERK1/2 messengers, resulting in expression of various chemokines and inflammatory agents specially interleukins.
12
Several studies have been carried out to investigate whether RPTPβ is related to the VacA induced cellular vacuolation. There is somewhat controversy about relation between the presence of RPTPβ on the cell surface and the toxicity of VacA. It has been shown that the VacA-induced vacuolation in the RPTPβ-positive human promyelocytic leukemia HL-60 cells can be prohibited by antisense oligonucleotide against RPTPβ receptor.
51,52
Adversely, the vacuolation, induced by VacA on RPTPβ expressing rabbit kidney epithelial RK13 cells, was not inhibited by phorbol 12-myristate 13-acetate (PMA). In addition, in vivo study with RPTPβ-deficient mice (RPTP-/- strains) indicated that the vacuolation is not dependent on the presence of RPTPβ.
53-55
Consequently, RPTPβ is not necessary for the vacuolating activity of VacA, but rather for signal induction. Dependency of the vacuolation to RPTP receptors in various cells is shown in Table 2. These findings highlight the necessity of further justification of a pattern for the receptor-dependent vacuolation in different cells.
Table 2.
Phenotype specificity of VacA in relation with RPTPs
|
Cell line
|
Type of receptor
|
Toxic strain of VacA
|
| RK13 |
RPTPβ |
m1 and m2 |
|
Az-521
|
RPTPβ and RPTPα |
m1 and m2
|
|
AGS
|
RPTPβ and RPTPα |
m1
|
|
HeLa
|
RPTPα |
m1
|
|
G401
|
RPTPα |
m1 and m2
|
|
HL60
|
- |
No toxicity |
Phenotype specificity caused by the m region of VacA toxin is related
to the functional expression of RPTPs.
54
HeLa and G401 cell lines
expressing one type of receptor (RPTPα) demonstrate different toxicity
responses. Difference in the post-transcriptional processing of RPTPα
causes formation of smaller RPTPα in HeLa cell line and reacting solely
to the m1, while RPTPα in G401 responds to both m1 and m2.
55
In some
cell lines, lack of RPTP is the reason of VacA untoxic behavior. HL-60
cell line with no RPTP receptor and no impression by VacA intoxication
responded to VacA m1 when treated with PMA, but still unaffected
by m2.
51,55
Also two AGS and AZ521 cell lines expressing both RPTP
receptors show various reaction to VacA treatment. Additionally, in
spite of expressing just RPTPβ in RK13 cell line both m1 and m2 types
are considered as toxic strains. As a result, although m1 and m2 both
respond to RPTPβ, it seems that in all cell lines presence of RPTPβ does
not guarantee the vacuolation potential of VacA.
Receptor-like protein tyrosine phosphatase α (RPTPα)
RPTPα is a 140-KDa protein receptor (p140) recognized on Wilm’s tumor G-401 cell line for the first time.
56
RPTPα, unlike RPTPβ, has shorter glycosylated extracellular domain - necessary for the binding to VacA. In fact, attenuating VacA-induced vacuolation on G401 cells through neuraminidase-α glycosidase treatment indicated the necessity of sialic acid and glycosylation for the binding of VacA.
49
Early studies showed that the function of RPTPα in signaling cascades related to pathogenesis of VacA is still unclear.
48
Epidermal Growth Factor Receptor (EGFR)
VacA was shown to be internalized by HeLa cells through EGFR receptor inducing vacuolation. This finding was confirmed through inhibiting the vacuolation by anti-EGFR antibody as well as immunoprecipitaion of two protein fragments (p33 and p55) equivalent to the VacA domains with anti-EGFR antibody.
57
Furthermore, interfering VacA with EGFR-mediated signaling pathways has been shown by ERK1/2 signaling path, actin stress fiber formation, and wound healing. These findings suggest that VacA imposes its impact(s) on HeLa cell through EGFR-mediated signaling pathways leading to inhibition of wound re-epithelialization, renewal of the gastric mucosa and cell proliferation.
58,59
Lipid raft/glycosylphosphatidylinositol-anchored proteins (GPI-AP)
Lipid raft as a cholesterol-enriched area acts as an accumulation center in the plasma membrane. It appears to be the main pathway, which enables VacA toxin to form oligomerized structure necessary for the flow of electrolytes and ions.
39,60
Experiments about low-affinity binding of VacA toxin to the membranes containing low-cholesterol has confirmed the cholesterol-dependent nature of the VacA association to lipid rafts.
61
Furthermore, GPI-AP was shown to be possibly involved with the VacA intoxication through recruitment of GPI-AP-associated VacA by lipid rafts.
62
Incubation of Hep-2 cells with phosphatidylinositol-specific phospholipase C (PI-PLC) was shown to diminish the sensitivity to VacA, supporting the GPI-AP involvement. Also removal of GPI-AP from HeLa cells has confirmed that the localization of VacA into lipid rafts is not a GPI dependent process, but the flux of chloride ions through the membrane is affected.
63,64
There are evidences that the translocation of VacA to lipid raft is important for the vacuolation-dependent cytotoxicity. Further, addition of methyl-β-cyclodextrin (MCD), a –cholesterol depleting drug, to HeLa cells and human gastric carcinoma AZ-521 cells was shown to interfere with the VacA internalization and vacuolation, but not with its binding to cells. Thus, it can be conquered that lipid rafts are necessary for the intoxication process. Also, treatment of HeLa and AZ-521 cells by PI-PLC was shown to inhibit the internalization and vacuolation supporting the importance of GPI-AP in the VacA-induced cytotoxicity.
65
Conversely, another study using GPI-AP-deficient mutant Chinese hamster ovarian CHO-LA1 cells has revealed that lipid rafts might play an alternative role of GPI-AP for the VacA toxin.
66
In addition to intoxication, lipid raft influences signaling cascades induced through the binding of VacA toxin to the host surface receptors as well as GPI-APs. Treatment of AZ-521 cells with PI-PLC and MCD was shown to inhibit the p38/MAPK signaling pathway, in large part due to the inhibition of RPTPβ/VacA translocation to lipid raft assisting GPI-AP. As a result, VacA and RPTPβ complex can be translocated to lipid rafts, which is the most important step in triggering the RPTPβ-mediated signaling cascade while the presence of GPI-AP is contributory for the accumulation of VacA in lipid rafts.
65
Sphingomyelin (SM)
SM serves as another receptor of the VacA-mediating interaction of toxin with the plasma membrane. The importance of SM in vacuolation was shown by the inhibition of the vacuolation in multiple cell lines (e.g., HeLa, human Caucasian gastric adenocarcinoma AGS cells as well as AZ-521 cells) treated with sphingomyelinase C. Enhanced VacA-induced vacuolation has been shown through exogenously overexpressed SM onto the plasma membrane of HeLa cells, which further indicates the role of the SM in cell vacuolation. In addition, the fate of intracellular pathway in which VacA is involved could be strictly affected by the length of SM carbon chain. In cells with long chain SM (C16 and C18), the VacA intoxication occurs via a cdc42-dependent pinocytic pathway/pinocytosis trafficking to Rab7-containing late endosomal compartment essential for the vacuolating activity of the VacA toxin. However, in the short acyl chains (C2 and C4), the VacA is internalized through a cdc42-independent pathway directing to Rab11-containing compartments of the eukaryotic cells. Interestingly, the length of acyl chain affects the distribution of VacA into lipid or non-lipid raft and also it’s uptake by GPI-AP in the plasma membrane.
67,68
Fibronectin (FN)
FN, a 440 KDa extracellular matrix (ECM) glycoprotein, functions as a receptor for the VacA toxin that binds through its RGD (Arg-Gly-Asp) motif in a dose-dependent manner.
69
Adhesion of the VacA to FN or any other ECM proteins can initiate several mechanisms in favor of bacteria survival, in large part through retarding the clearance of H. pylori and preserving the infection via invasion and penetration into the intracellular junction.
70,71
In addition, the VacA binding to FN was shown to impose cell disruption and changes in cell morphology through cytoskeletal reorganization.
72
Heparin (H) and heparan sulfate (HS)
The H and HS as two types of receptors are cellular surface and extra cellular matrix-associated proteoglycans and present different binding sites for m1 and m2 strain of VacA in favor of H. pylori survival.
73
It should be also stated that H. pylori evades immune response of the host by binding of VacA to H/HS that, in return, recruits and inactivates complement components, C3b and C4b, known as one of the first barriers of the native immune system.
74
Further, attachment of H/HS onto the surface of microorganism enables H. pylori to avoid phagocytosis.
75
Low-density Lipoprotein Receptor-related Protein-1 (LRP1)
LRP1, an endocytic receptor from LDL receptor family, is thought to be the receptor mediating VacA-dependent autophagy. The study investigating the influence of LRP1 was carried out on AZ-521 cell. LRP1 gene silencing by siRNA and confocal microscopy results in the AZ-521 cells determined dependency of binding and internalization of toxin to LRP1. Furthermore, VacA-induced apoptosis occurs through autophagy and caspase independent via binding to LRP1. Interestingly, formation of small vacuoles via binding to LRP1 is exhibited by formation of phagosomes, which is different from the RPTPβ-mediated vacuolation. Overall, LRP1 is considered as one of the main receptors for binding and internalization leading to autophagy dependent apoptosis and vacuolation.
76
CD18
In addition of various receptors on epithelial cells for binding of VacA, one receptor has been identified as a ligand for VacA on T cells named CD18 and known as PKC-associated internalization. Similar to the epithelial cells internalization, the mechanism of intoxication after concentrating within the lipid raft is clathrin-independent due to phosphorylation of cytoplasmic domain of CD18, PKC, by the regulation of Rac1 and cdc-42.
77
As a result, T-cell proliferation and IL-2 signaling pathway is prohibited by inhibition of nuclear factor of activated T-cell (NFAT) making it an immunomodulator toxin.
78
Another immunosuppressive effect of VacA has been observed on B cells, causing interference with antigens (Ags) presenting cells (APCs).
79
Internalization and trafficking of VacA toxin
Generally, VacA is released from bacteria in two types, including: (a) VacA associated with vesicles known as outer membrane vesicles (OMV), and (b) free VacA. The biological activity of free VacA is triggered immediately after the internalization (known as the early internalization), while the VacA-containing OMVs remain intact for about 72 hours after the internalization (known as the late internalization).
80
Both types of VacA are internalized through clathrin-independent pathway that is believed to be regulated by the small GTPase proteins, Rack1 and Cdc42. Upon internalization, the VacA toxin is located beneath the membrane in GPI-AP early endosome compartment (GEECs). The tubulovesicular compartments containing GPI-AP are part of the GPI recycling system. Once in the early and late endosomes, the toxin either is directed to mitochondria and induces apoptosis or is oligomerized on the vacuole membranes and causes vacuolation.
65,81,82
Similar to the diphtheria and tetanus toxins, VacA intoxication through lipid rafts is a pH- dependent process. An increased interaction of VacA with lipid raft at an acidic pH is attributed to the structural alteration of p55 domain. Although the effect of low pH on the p33 domain has been shown, its impact(s) on the p55 domain appears to be higher on the toxin binding because the interaction of p33 with lipid raft is weaker/lower than that of the p55. The low pH-induced conformational changes were shown to increase the surface exposure of the hydrophobic domain of toxin in favor of its binding to the lipid rafts, which facilitates the membrane internalization.
80
Pathogenesis of VacA and signaling pathways
VacA can trigger intracellular signal transductions related to pro-inflammation, vacuolation and apoptosis. In general, the behaviour of the host cells in relation with VacA toxin is relevant to the type of toxin-cell interaction. Binding of the VacA toxin to the host cell membrane through its receptors can activate several signaling pathways leading to pro-inflammatory responses. On the other hand, the uptake of VacA toxin by cells can direct the toxin to the mitochondria and induce apoptosis or vacuolation (Fig. 2).
23
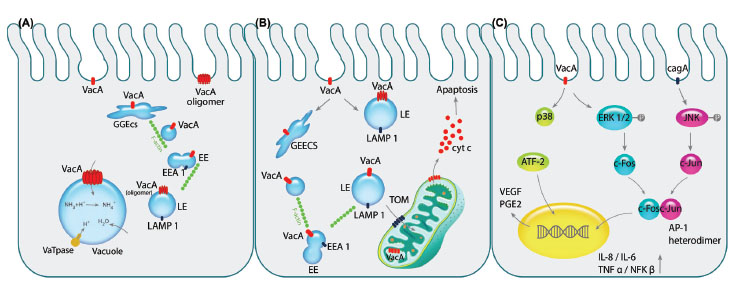
Fig. 2.
Pathogenicity of VacA toxin is exhibited on epithelial cells. A) After the pinocytosis of VacA into intracellular environment, it is
directed towards early endosomal compartment GEEC, EE and LE respectively by the aid of F-actin filaments. Next, VacA is oligomerized
on the LE membrane acting as the channel discharging chloride ions that results in LE swelling since water molecules are imported
due to overpopulation of ammonium resulted from the change in chloride ions. Consequently, over-hydration results in vacuolation of
the cell. B) Following aforementioned trafficking of the vacA to the endosomal compartments, its transportation to the mitochondria by
N-ter signal sequence of the toxin can commence the programmed cell death (apoptosis). Importing of VacA through TOM complex and
oligomerizing alters membrane potential of the mitochondria which recruit pro-apoptotic Bax and Bak complexes indirectly leading to
release of cytochrome c. C) Binding of the VacA to cell surface receptor RPTPβ activates the MAPK signaling cascades independently
or in cooperation with CagA. The activation of MAPK/p38 pathway induces stimulation of transcription factor ATF-2 that increases the
expression of PGE2, known as the angiogenic factor and mitotic signal, via binding to COX-2 promoter. Additionally, VacA phosphorylates
ERK1/2 that activates the downstream component c-fos, while CagA activates the other components of AP-1 heterodimer, c-jun, by
phosphorylation of JNK. Interaction of c-fos and c-jun together would produce the AP-1 heterodimer which is counted as the transcription
factor for several cytokines such as IL-1, IL-6 and TNFα leading to inflammation of the gastric mucosa.
.
Pathogenicity of VacA toxin is exhibited on epithelial cells. A) After the pinocytosis of VacA into intracellular environment, it is
directed towards early endosomal compartment GEEC, EE and LE respectively by the aid of F-actin filaments. Next, VacA is oligomerized
on the LE membrane acting as the channel discharging chloride ions that results in LE swelling since water molecules are imported
due to overpopulation of ammonium resulted from the change in chloride ions. Consequently, over-hydration results in vacuolation of
the cell. B) Following aforementioned trafficking of the vacA to the endosomal compartments, its transportation to the mitochondria by
N-ter signal sequence of the toxin can commence the programmed cell death (apoptosis). Importing of VacA through TOM complex and
oligomerizing alters membrane potential of the mitochondria which recruit pro-apoptotic Bax and Bak complexes indirectly leading to
release of cytochrome c. C) Binding of the VacA to cell surface receptor RPTPβ activates the MAPK signaling cascades independently
or in cooperation with CagA. The activation of MAPK/p38 pathway induces stimulation of transcription factor ATF-2 that increases the
expression of PGE2, known as the angiogenic factor and mitotic signal, via binding to COX-2 promoter. Additionally, VacA phosphorylates
ERK1/2 that activates the downstream component c-fos, while CagA activates the other components of AP-1 heterodimer, c-jun, by
phosphorylation of JNK. Interaction of c-fos and c-jun together would produce the AP-1 heterodimer which is counted as the transcription
factor for several cytokines such as IL-1, IL-6 and TNFα leading to inflammation of the gastric mucosa.
A- Vacuolation
After pinocytic-dependent and clathrin-independent endocytosis of the VacA toxin through GEECs, they are directed to the early endosomal (EE) compartments containing EEA1 as a marker with assistance of the F-actin and CD2AP as VacA-F-actin interaction mediator.
15
Vesicular trafficking of early endosomes to the LAMP1-containing late endosomes (LE) is the next step that initiates the oligomerization of VacA on the late endosomal compartment membrane. The oligomerized VacA acts as an ion selective channel altering chloride concentration and enhancing proton pump activity that results in LE swelling and vacuole formation.
24,83
B- Programmed Cell Death (apoptosis)
In addition to the vacuolation of the host cells caused by the VacA, the internalization through endocytosis can elicit apoptosis via mitochondrial-dependent cell death mechanism. Internalization to the mitochondria occurs via TOM complex through the recognition of N-terminus of VacA, which is believed to be futile for the channel formation but important for directing toxin to the mitochondrial membrane. VacA, containing LE compartment and mitochondrial membrane interaction, is also able to initiate the programmed cell death through membrane potential fluctuations.
84,85
The release of cytochrome C through translocation of the pro-apoptotic compounds- Bax and Bak caused by the alterations of membrane potential is the basic mechanism for leading cells towards apoptosis.
86,87
C- MAPK signaling pathways
Furthermore, VacA can impose its effects on the host cell through binding to certain membrane receptors and inducing p38 and ERK1/2 signaling cascades.
84
Activation of p38/MAPK pathway through binding of the VacA initiates the activating transcription factor-2 (ATF-2), which functions as a regulator on the Cox-2 by binding to its promoter via TLR2/9. Induction of Cox-2 expression was shown to enhance the expression of PGE2 - known as a mitogenic signal - and contributes to angiogenesis through up-regulation of the vascular endothelial growth factor (VEGF).
16,88
VacA also contributes to the activation of ERK1/2 via its phosphorylation and formation of AP-1 that contains heterodimer of c-fos and c-jun. While the c-fos expression is regulated by the phosphorylated ERK1/2, the other part of the AP-1 complex is activated via JNK phosphorylation through binding of cagA that is another toxin of H. pylori-. As a result, AP-1 regulates the expression and recruitment of cytokines such as IL-8, IL-6, TNF-α and NF-κB through binding to their promoters.
12,89
D- Toxin oligomerization on plasma membrane
In the host cells, the VacA toxin is functionalized in two forms of monomer and oligomer. The latter can reshape as “rosettes” in the solution and form low-conductance, anion-selective and voltage-dependent bio-structures that are water-soluble 30 nm-diameter channels as two single-layered hexameric and heptameric flowers and bilayered shapes consisting of 6-9 and 12-14 monomers, respectively.
40,90,91
A study predicting structure of oligomerized VacA by deep-etch electron microscopy (EM) viewed 3 possible forms for hexameric structure, including: (i) the flower shape, a dodecamer constructed of two hexameric flat forms with two symmetrical six-subunit arrays, (ii) the flat form composed of an individual six-petal structure known as half of a dodecamer with central ring and counterclockwise chirality, and (iii) the other flat form with no central ring and clockwise chirality with a more abundance than the former and limited accessibility in the environment (Fig. 3).
91
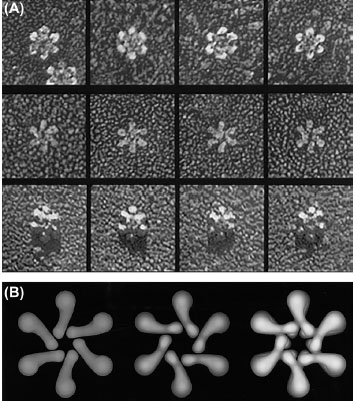
Fig. 3.
Freeze-dried transmission electron microscopy
micrographs of H. pylori VacA toxin. A) Rotary replicas of
oligomerized structures of VacA toxin. The first, second and third
rows respectively show the flower shaped dodecamer form of
VacA toxin, the flat form of VacA in oligomerized form, and the
seven-membered structure of toxin. B) Three structures predicted
based on the studies on the oligomerized forms of VacA with
deep-etch electron microscopy analysis. The right, middle and
left images respectively show the symmetrical pair of hexameric
VacA flower placed face-to-face that is a complete dodecamer
constructed, the minor flat form included half of a dodecamer with
the central ring that resembles the complete VacA structure, and
the more common flat form of oligomerized VacA with no central
ring that is sandwiched in the center of dodecamer. The data
were adopted with permission from a published work conducted
by Cover et al.
91
.
Freeze-dried transmission electron microscopy
micrographs of H. pylori VacA toxin. A) Rotary replicas of
oligomerized structures of VacA toxin. The first, second and third
rows respectively show the flower shaped dodecamer form of
VacA toxin, the flat form of VacA in oligomerized form, and the
seven-membered structure of toxin. B) Three structures predicted
based on the studies on the oligomerized forms of VacA with
deep-etch electron microscopy analysis. The right, middle and
left images respectively show the symmetrical pair of hexameric
VacA flower placed face-to-face that is a complete dodecamer
constructed, the minor flat form included half of a dodecamer with
the central ring that resembles the complete VacA structure, and
the more common flat form of oligomerized VacA with no central
ring that is sandwiched in the center of dodecamer. The data
were adopted with permission from a published work conducted
by Cover et al.
91
Affluence of the heptameric structure seems to be dependent on the length of the loop region located between p33 and p55. Since the m1 type possesses longer residue than the m2 type, the heptameric structure is more abundant. These multi-shaped structures are generated in the presence of two interacting sites and environmental effect of pH, which aids the assembly of monomeric structures in the neutral pH.
92,93
The oligomerized VacA on the host cells are able to permeabilize the membrane through rosette form channels that can efflux/transfer the metabolites and ions of the host cell to the extracellular environment in favor of survival of bacteria.
40
VacA and CagA interaction
Besides VacA-induced pathogenesis on the host cell in gastric mucusa, it is proved that the VacA association with CagA is a key regulator for the disease severity. Phosphorylated and unphosphorylated CagAs are able to function as the inhibitor of intracellular trafficking of VacA and the simulator of anti-apoptotic BCL2 in order to prevent initiation of apoptosis induced by VacA, respectively.
94,95
Besides this manifestation of CagA, it affects the internalization of the VacA to the host cells to which bacteria is attached in order to assist the survival of the bacteria. It is determined that the binding of VacA to distant cells out of access of H. pylori induces vacuolation and apoptosis, thus the intracellular nutrients become accessible.
96,97
VacA as a candidate for molecular therapy
For decades, the first line treatment of H. pylori infection is a triple drug combination, including: proton pump inhibitor (PPI), clarithromycin and amoxicillin. However, this traditional regimen often associate with some drawbacks, including: an increased resistance to antibiotic, low patients compliance against the long course of treatment with significant side effects, and relapse of the infection.
98,99
Thus, new generation molecular therapies need to be alternatively implemented since they impose less detrimental impacts on the healthy tissues with lower side effects, thereby improving the efficacy of treatment.
In several experiments, few vaccines and immunotherapies have successfully been developed against various H. pylori Ags related to infection and colonization of the bacteria and incidence of peptic ulcer and gastric cancer. The candidate Ags for molecular therapy include flagella, hsp60, urease, adhesions, cagA and VacA.
100
Of these, VacA plays a pivotal role in pathogenesis of H. pylori. It contributes to the persistent colonization of bacteria, leading to the longevity of infection through escaping from the autophagy and immunosurveillance of host.
101
VacA is considered as one of the main virulence factors involving in ulcerogenesis and gastric cancer. Further, the severity of disease symptoms among patients suffering from the gastritis and peptic ulcer disease appears to be VacA strain-dependent.
102
Vaccine development
Generally, based on multiple studies on developing an effective vaccine against H. pylori, there are two types of vaccines, including: (a) the prophylactic vaccine that prevents the infection by reducing or clearing bacteria in the healthy or previously infected individuals, and (b) the therapeutic vaccine that treats the infection in patients via stimulating immune system different from the infection-induced immunity.
103,104
To control this formidable diseases, however, multi-component vaccines need to be developed. To achieve an effective vaccine against H. pylori, multiple virulence factors mediating various clinical manifestations should be targeted. Although multivalent/polyvalent vaccines would be helpful, development of multi-antigen vaccines against H. pylori seems to be inevitable. For this purpose, the vaccine could be designed based on the H. pylori whole cell (HWC) or antigen cocktail which is composite of two or more major virulence factors of bacterium.
105
Although approaches exploiting HWC in mouse models or different clinical trials confirmed the efficacy of these vaccines, in some cases, these types of vaccines show low quality of Ags and possible cross-reactivity and immunologic reactions.
106,107
Therefore, an efficient vaccine against H. pylori should contain a series of virulent Ags presented in all strains that are involved in bacterial adhesion and intoxication. Owing to its function in pathogenicity of H. pylori, VacA has a potential ability of being a suitable candidate for vaccine development. This notion has further been verified because of the high amount of IgGs detected in patient’s serum, and the high expression rate in most H. pylori strains specially those causing the peptic ulcer. Furthermore, establishing protection with no need to native conformation by VacA toxin makes it an appropriate Ag for development of vaccine and immunotherapy.
104,108
A couple of studies have reported the efficacy of the VacA-containing vaccine in vitro and animal models. For example, formaldehyde-inactivated VacA vaccine was shown to elicit immune response(s) in mice as compared to that of the native toxin.
109
It should be noted that formaldehyde is a detoxifying agent via interacting with L-lysinin residues of the toxin, and hence can be exploited as an adjuvant in vaccination. Since toxin inactivation by formaldehyde also causes distortion of some epitopes that do not present in the native Ags, the mutant type of heat-labile enterotoxin of E. coli, LTK63, was utilized as a suitable adjuvant for the vaccine development against H. pylori.
110
In addition, VacA has been exploited for the production of multi-component vaccines. Intragastric immunization of mice infected with H. pylori SPM326 by the therapeutic vaccine consisting of recombinant VacA and CagA along with LTK63 adjuvant was shown to eradicate H. pylori for 3 months and also provide tenacity of eradication for 2 months after infection rehearsal.
111
Immunization of Beagle dog suffering from gastritis with multi-component vaccine, VacA-CagA- neutrophil-activating protein (NAP), supplemented with adjuvant Al(OH)3, resulted in an increased level of antibody (Ab) and hence protection for 4 months post-vaccination. Despite such therapeutic impacts, the gastritis was recurred after 29 weeks indicating the partial periodic effect of vaccine.
112
In contrary to mice, Beagle dogs, as animal model for the human H. pylori infection, provide researchers with high ability of tandem biopsy with no need to sacrifice the animal, which helps to gain a better understanding of the overall process of infection and immunity.
Further, a prophylactic vaccine containing three Ags (VacA, CagA and NAP) with adjuvant Al(OH)3 has been developed and examined in the non-infected individuals through a phase 1 clinical trial. This intramuscular booster vaccination demonstrated satisfactory safety and immunogenicity with anamnestic antibody and cellular responses 18–24 months post-vaccination. Long memory immunity was originated from VacA- and CagA-driven interferon ɤ production, which lasted 4.5 months post-immunization. After the third immunization, all volunteers responded to one or two Ags (mostly VacA and NAP), while 86% responded to all three Ags. VacA induced Ab response after two immunizations, whereas CagA showed cell-immune response after three immunizations.
113
Yet, there exist some debates to figure out what portion of the VacA toxin is more efficient for the vaccine development. A study using different recombinant fragments of VacA confirmed that the highest titer of Ab was produced against 297-317 residues including both p33 and p55 regions of the toxin.
114
Nonetheless, another effort was carried out to study the VacA neutralizing activity of 10 monoclonal antibodies (mAbs) produced in mice via epitope mapping using a panel of VacA deletion mutants and VacA chimera. Among these, two mAbs were able to neutralize the cytotoxic activity of VacA through recognition of amino acids 685 to 821 located at the receptor binding region.
115
In the following context, mAbs against H. pylori infection will be discussed.
Monoclonal antibody
Development of mAbs has provided new insights in the diagnosis and therapy of various diseases, in particular infections and malignancies. As a result, commercial therapeutic mAbs have made their way to the therapeutic markets in the early 1980s.
116
Passive administration of mAb directed at protective Ags such as VacA and urease might be particularly relevant as a substitute to the current therapies and diagnosis methods. Thus, VacA toxin has become a target for the production of therapeutic mAbs to neutralize the cytotoxin activity. Accordingly, various studies reported that some variants of VacA are highly associated with an increased risk of symptomatic gastroduodenal disease. Two anti-VacA mAbs, known as V36E and V41, were shown to be able to neutralize the vacuolation of rabbit kidney cell line RK13 through binding to the native structure of VacA.
117
Another experiment was conducted to produce mAbs against small and large subunits of urease. As a result, two Abs designated as S2 and L2 were determined based on their strong interactions respectively with small and large subunits, and showed significant blockage of the enzyme activity.
118
Besides application of mAbs in the field of therapy, it seems that they are promising tools for increasing accuracy of diagnosis. Common diagnostic methods of H. pylori infection have been developed on the basis of biopsy, which is painful. Further, various diagnostic methods such as Urea Breath Test (UBT) and serological techniques may provide false negative and positive results specially in asymptomatic patients, thus leading to failure of accurate diagnosis and efficient treatments.
119
Gamma-glutamyl transpeptidase (GGT) antigen is a virulence factor, necessary for colonization and cell apoptosis of H. pylori, which appears to be a promising Ag for development of mAb. In 2014, a phase 1 clinical trial (ID: NCT02123771) was successfully carried out to detect the H. pylori infection in the stool of patients using ELISA and anti-GGT mAb. Given that several toxins of H. pylori participate in the pathogenesis and survival of bacteria, a multi-component Ab cocktail may provide the next generation immunotherapy of H. pylori infection, similar to Ab cocktail used for treatment of Antrax.
120
Ab cocktail is a combination of several Abs that can recognize different virulence factors, through which the treatment outcome can be maximized by expanding the spectrum of protection and enhancing the protective efficacy.
Production of Ab cocktail to recognize the major virulence factors H. pylori (i.e., VacA, urease, CagA, GGT and flagellin) appears to be very desirable for the treatment of H. pylori infection. Use of class immunoglobulin A (IgA) in Ab cocktail against H. pylori seems to provide an improved immunotherapy modality, in large part because IgA secreting cells often react with H. pylori membrane proteins (e.g., flagellin and urease) in H. pylori-infected patients.
121
Given the high stability and avidity of IgA in the gastric environment and habituating H. pylori as a noninvasive organism in gastric mucosa, passive immunotherapeutic approaches using secretory IgA (sIgA) might provide a robust treatment modality. For example, in a study, a scFv polypeptide specific to H. pylori urease subunit A was compared with its reformatted polymeric IgA (IgAp/d) and sIgA for the degree of enzyme activity inhibition. Due to increased avidity, the sIgA and IgAp/d were found to be able to efficiently block the enzymatic activity of free and membrane-associated ureases.
122,123
Although several antibody fragments have been designed against the main virulence factor of H. pylori, urease,
124
no antibody fragment has been developed against domain of VacA by phage display ever. New approaches for production of antibodies, such as phage display technique,
125,126
have been developed in the recent decades that provide a convenient and economic procedure for production of monoclonal antibodies. To the best of our knowledge, we are among the first teams who work on the development of mAb fragments by phage display against VacA toxin.
127,128
Concluding remarks
VacA is considered as one of the main virulence factors in pathogenesis of H. pylori in the stomach with variety of cytotoxicity, in which the toxicity is assumed to be dependent on different variants of the toxin and various types of receptors and cells. Despite some attempts to justify the binding pattern of VacA, the exact receptor(s) of host cells responsible for the binding of VacA toxin is yet to be fully addressed. In general, it is believed that, regardless of the receptor involved (e.g., RPTPβ or EGFR), the process of VacA internalization occurs in the regions enriched with lipid rafts with the aid of cholesterol, GPI-AP and sphingomyelin. Notwithstanding unprecedented volume of investigations on the H. pylori infection worldwide, its effective eradication remains as a dilemma. Failure of the currently applied therapies, consisting of antibiotic regimens, is largely attributed to inaccurate treatment causing point mutations in H. pylori. New molecular therapies such as vaccines and mAbs seem to offer much more effective treatment modalities with much higher patient compliance and minimal side effects. Although successful vaccination in animal models has been reported, attempts for translation of such therapies to human cases are still under investigation. Among various types of mAbs produced against H. pylori, IgA class of Abs may be considered as one of the best candidates for passive immunotherapies since the infection of gastric mucosa by H. pylori recruit cells that are responsible for the production of IgA specific for a variety of virulence factors. Besides, sIgA was shown to prevent the cellular attachment of H. pylori and its infection. In this review, we discussed genetic diversity and receptors of VacA toxin on epithelial cells and tried to justify whether VacA cytotoxicity is dependent on the type of receptor and genetic diversity for the first time. It is obvious that receptor and genetic diversity are regulators of H. pylori pathogenesis regardless of the genetic background and environmental issues affecting the severity of the disease worldwide and also the effect of cagA on the potential of VacA. Overall, the cytotoxicity of the VacA in vivo is exhibited based on the position/condition of the host cells. Non vacuolating toxicity (e.g., peptic ulcer and gastritis) is resultant from the pathogenicity for adjacent cells with attached H. pylori and vacuolation is observed in distant cells in order to feed on cellular nutrients. Collectively, due to growing trend of mAb applications, recombinant Ab fragments (e.g., scFv, Fab, single domain Abs and bispecific Abs), multivalent Abs and Ab-conjugates with higher avidity as a cocktail of IgG and sIgA Abs as well as multivallent vaccines may be our best shot to tackle this formidable disease.
Ethical approval
Not applicable.
Competing interests
The authors declare no conflict of interests.
Acknowledgment
The authors are grateful for the financial support (Grant No: RCPN-93010) provided by Tabriz University of Medical Sciences.
Review Highlights
What is current knowledge?
simple
-
√ Application of VacA in production of a vaccine for H. pylori
treatment has been confirmed, and used as a part of multicomponent
vaccine including several virulence factors of the
bacterium.
-
√ Efficiency of therapeutic vaccines has been investigated in
various in vivo and in vitro experiments, as well as clinical
trials.
-
√ Despite success in the treatment of infection in various
studies, relapse of infection after several months appears as
a challenging issue.
What is new here?
simple
-
√ Besides production of systemic immunoglobulins
confronting H. pylori infection, local antibody secreting cells
(ASCs) appear to be the major class of Abs representing the
state of infection in patients.
-
√ New multivalent vaccines may provide novel treatment
modalities against H. pylori.
-
√ New immunotherapies could be designed based on the IgG
and IgA Ab cocktail.
-
√ Novel methods of producing recombinant Ab fragments
(scFv and Fab) such as display technologies are able to
produce mAbs with higher abilities of multimerization into
secretory forms in comparison to traditional full length Abs.
References
- Hagymasi K, Tulassay Z. Helicobacter pylori infection: new pathogenetic and clinical aspects. World J Gastroenterol 2014; 20:6386-99. doi: 10.3748/wjg.v20.i21.6386 [Crossref] [ Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90. doi: 10.3322/caac.20107 [Crossref] [ Google Scholar]
-
Tan P, Yeoh KG. Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology 2015; 149: 1153-62.e3.
- de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol 2012; 13:607-15. doi: 10.1016/s1470-2045(12)70137-7 [Crossref] [ Google Scholar]
- Bridge DR, Merrell DS. Polymorphism in the Helicobacter pylori CagA and VacA toxins and disease. Gut Microbes 2013; 4:101-17. doi: 10.4161/gmic.23797 [Crossref] [ Google Scholar]
- Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F. A review of human carcinogens--Part B: biological agents. Lancet Oncol 2009; 10:321-2. [ Google Scholar]
-
Graham DY. Helicobacter pylori update: gastric cancer, reliable therapy, and possible benefits. Gastroenterology 2015; 148: 719-31.e3. doi:10.1053/j.gastro.2015.01.040.
- Rassow J, Meinecke M. Helicobacter pylori VacA: a new perspective on an invasive chloride channel. Microbes Infect 2012; 14:1026-33. doi: 10.1016/j.micinf.2012.07.002 [Crossref] [ Google Scholar]
- Calore F, Genisset C, Casellato A, Rossato M, Codolo G, Esposti MD. Endosome-mitochondria juxtaposition during apoptosis induced by H pylori VacA. Cell Death Differ 2010; 17:1707-16. doi: 10.1038/cdd.2010.42 [Crossref] [ Google Scholar]
- Yamasaki E, Isomoto H, Hirayama T. [Pleiotropic effects of VacA: its role in the pathogenesis of Helicobacter pylori infection]. Tanpakushitsu Kakusan Koso 2009; 54:607-13. [ Google Scholar]
- Hisatsune J, Nakayama M, Isomoto H, Kurazono H, Mukaida N, Mukhopadhyay AK. Molecular characterization of Helicobacter pylori VacA induction of IL-8 in U937 cells reveals a prominent role for p38MAPK in activating transcription factor-2, cAMP response element binding protein, and NF-kappaB activation. J Immunol 2008; 180:5017-27. [ Google Scholar]
- Ki MR, Lee HR, Goo MJ, Hong IH, Do SH, Jeong DH. Differential regulation of ERK1/2 and p38 MAP kinases in VacA-induced apoptosis of gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol 2008; 294:G635-47. doi: 10.1152/ajpgi.00281.2007 [Crossref] [ Google Scholar]
- Palframan SL, Kwok T, Gabriel K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front Cell Infect Microbiol 2012; 2:92. doi: 10.3389/fcimb.2012.00092 [Crossref] [ Google Scholar]
-
Nakano M, Hirayama T, Moss J, Yahiro K. Helicobacter pylori VacA Exhibits Pleiotropic Actions in Host Cells. In: Suzuki H, R Warren, B Marshall, editors. Helicobacter pylori. Tokyo: Springer Japan; 2016. p. 49-66.
- Gauthier NC, Monzo P, Gonzalez T, Doye A, Oldani A, Gounon P. Early endosomes associated with dynamic F-actin structures are required for late trafficking of H pylori VacA toxin. J Cell Biol 2007; 177:343-54. doi: 10.1083/jcb.200609061 [Crossref] [ Google Scholar]
- Caputo R, Tuccillo C, Manzo BA, Zarrilli R, Tortora G, Blanco Cdel V. Helicobacter pylori VacA toxin up-regulates vascular endothelial growth factor expression in MKN 28 gastric cells through an epidermal growth factor receptor-, cyclooxygenase-2-dependent mechanism. Clin Cancer Res 2003; 9:2015-21. [ Google Scholar]
- Backert S, Tegtmeyer N. the versatility of the Helicobacter pylori vacuolating cytotoxin vacA in signal transduction and molecular crosstalk. Toxins (Basel) 2010; 2:69-92. doi: 10.3390/toxins2010069 [Crossref] [ Google Scholar]
- Memon AA, Atherton JC. Reply to "Helicobacter pylori vacA as marker for gastric cancer and gastroduodenal diseases: one but not the only factor". J Clin Microbiol 2014; 52:4452. doi: 10.1128/JCM.02667-14 [Crossref] [ Google Scholar]
- Abadi AT, Lee YY. Helicobacter pylori vacA as marker for gastric cancer and gastroduodenal diseases: one but not the only factor. J Clin Microbiol 2014; 52:4451. doi: 10.1128/JCM.02640-14 [Crossref] [ Google Scholar]
-
Sinnett CG, Letley DP, Narayanan GL, Patel SR, Hussein NR, Zaitoun AM, et al. Helicobacter pylori vacA transcription is genetically-determined and stratifies the level of human gastric inflammation and atrophy. J Clin Pathol 2016.
- Amilon KR, Letley DP, Winter JA, Robinson K, Atherton JC. Expression of the Helicobacter pylori virulence factor vacuolating cytotoxin A (vacA) is influenced by a potential stem-loop structure in the 5' untranslated region of the transcript. Mol Microbiol 2015; 98:831-46. doi: 10.1111/mmi.13160 [Crossref] [ Google Scholar]
- Gangwer KA, Mushrush DJ, Stauff DL, Spiller B, McClain MS, Cover TL. Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc Natl Acad Sci U S A 2007; 104:16293-8. doi: 10.1073/pnas.0707447104 [Crossref] [ Google Scholar]
- Junaid M, Linn AK, Javadi MB, Al-Gubare S, Ali N, Katzenmeier G. Vacuolating cytotoxin A (VacA) - A multi-talented pore-forming toxin from Helicobacter pylori. Toxicon 2016; 118:27-35. doi: 10.1016/j.toxicon.2016.04.037 [Crossref] [ Google Scholar]
-
Foegeding NJ, Caston RR, McClain MS, Ohi MD, Cover TL. An Overview of Helicobacter pylori VacA Toxin Biology. Toxins (Basel) 2016; 8.
- Fischer W, Buhrdorf R, Gerland E, Haas R. Outer membrane targeting of passenger proteins by the vacuolating cytotoxin autotransporter of Helicobacter pylori. Infect Immun 2001; 69:6769-75. doi: 10.1128/iai.69.11.6769-6775.2001 [Crossref] [ Google Scholar]
- Nguyen VQ, Caprioli RM, Cover TL. Carboxy-terminal proteolytic processing of Helicobacter pylori vacuolating toxin. Infect Immun 2001; 69:543-6. doi: 10.1128/iai.69.1.543-546.2001 [Crossref] [ Google Scholar]
- Torres VJ, McClain MS, Cover TL. Mapping of a domain required for protein-protein interactions and inhibitory activity of a Helicobacter pylori dominant-negative VacA mutant protein. Infect Immun 2006; 74:2093-101. doi: 10.1128/iai.74.4.2093-2101.2006 [Crossref] [ Google Scholar]
- Backert S, Neddermann M, Maubach G, Naumann M. Pathogenesis of Helicobacter pylori infection. Helicobacter 2016; 21 Suppl 1:19-25. doi: 10.1111/hel.12335 [Crossref] [ Google Scholar]
- Torres VJ, McClain MS, Cover TL. Interactions between p-33 and p-55 domains of the Helicobacter pylori vacuolating cytotoxin (VacA). J Biol Chem 2004; 279:2324-31. doi: 10.1074/jbc.M310159200 [Crossref] [ Google Scholar]
- Lupetti P, Heuser JE, Manetti R, Massari P, Lanzavecchia S, Bellon PL. Oligomeric and subunit structure of the Helicobacter pylori vacuolating cytotoxin. J Cell Biol 1996; 133:801-7. [ Google Scholar]
- Ivie SE, McClain MS, Algood HM, Lacy DB, Cover TL. Analysis of a beta-helical region in the p55 domain of Helicobacter pylori vacuolating toxin. BMC Microbiol 2010; 10:60. doi: 10.1186/1471-2180-10-60 [Crossref] [ Google Scholar]
- Burroni D, Lupetti P, Pagliaccia C, Reyrat JM, Dallai R, Rappuoli R. Deletion of the major proteolytic site of the Helicobacter pylori cytotoxin does not influence toxin activity but favors assembly of the toxin into hexameric structures. Infect Immun 1998; 66:5547-50. [ Google Scholar]
- Tombola F, Pagliaccia C, Campello S, Telford JL, Montecucco C, Papini E. How the loop and middle regions influence the properties of Helicobacter pylori VacA channels. Biophys J 2001; 81:3204-15. doi: 10.1016/s0006-3495(01)75956-5 [Crossref] [ Google Scholar]
- Matteo MJ, Armitano RI, Granados G, Wonaga AD, Sanches C, Olmos M. Helicobacter pylori oipA, vacA and dupA genetic diversity in individual hosts. J Med Microbiol 2010; 59:89-95. doi: 10.1099/jmm.0.011684-0 [Crossref] [ Google Scholar]
- Figueiredo C, Machado JC, Pharoah P, Seruca R, Sousa S, Carvalho R. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst 2002; 94:1680-7. [ Google Scholar]
- Figura N, Valassina M, Moretti E, Vindigni C, Collodel G, Iacoponi F. Histological variety of gastric carcinoma and Helicobacter pylori cagA and vacA polymorphism. Eur J Gastroenterol Hepatol 2015; 27:1017-21. doi: 10.1097/MEG.0000000000000414 [Crossref] [ Google Scholar]
- McClain MS, Czajkowsky DM, Torres VJ, Szabo G, Shao Z, Cover TL. Random mutagenesis of Helicobacter pylori vacA to identify amino acids essential for vacuolating cytotoxic activity. Infect Immun 2006; 74:6188-95. doi: 10.1128/iai.00915-06 [Crossref] [ Google Scholar]
- Gonzalez-Rivera C, Campbell AM, Rutherford SA, Pyburn TM, Foegeding NJ, Barke TL. A Nonoligomerizing Mutant Form of Helicobacter pylori VacA Allows Structural Analysis of the p33 Domain. Infect Immun 2016; 84:2662-70. doi: 10.1128/IAI.00254-16 [Crossref] [ Google Scholar]
- Domanska G, Motz C, Meinecke M, Harsman A, Papatheodorou P, Reljic B. Helicobacter pylori VacA toxin/subunit p34: targeting of an anion channel to the inner mitochondrial membrane. PLoS Pathog 2010; 6:e1000878. doi: 10.1371/journal.ppat.1000878 [Crossref] [ Google Scholar]
- Pyburn TM, Foegeding NJ, Gonzalez-Rivera C, McDonald NA, Gould KL, Cover TL. Structural organization of membrane-inserted hexamers formed by Helicobacter pylori VacA toxin. Mol Microbiol 2016; 102:22-36. doi: 10.1111/mmi.13443 [Crossref] [ Google Scholar]
- Han SR, Schreiber HJ, Bhakdi S, Loos M, Maeurer MJ. vacA genotypes and genetic diversity in clinical isolates of Helicobacter pylori. Clin Diagn Lab Immunol 1998; 5:139-45. [ Google Scholar]
- Sugimoto M, Zali MR, Yamaoka Y. The association of vacA genotypes and Helicobacter pylori-related gastroduodenal diseases in the Middle East. Eur J Clin Microbiol Infect Dis 2009; 28:1227-36. doi: 10.1007/s10096-009-0772-y [Crossref] [ Google Scholar]
-
Mendoza-Elizalde S, Arteaga-Resendiz NK, Valencia-Mayoral P, Luna RC, Moreno-Espinosa S, Arenas-Huertero F, et al. Diversification of the vacAs1m1 and vacAs2m2 Strains of Helicobacter pylori in Meriones unguiculatus. Front Microbiol 2016; 7.
- Chung C, Olivares A, Torres E, Yilmaz O, Cohen H, Perez-Perez G. Diversity of VacA intermediate region among Helicobacter pylori strains from several regions of the world. J Clin Microbiol 2010; 48:690-6. doi: 10.1128/jcm.01815-09 [Crossref] [ Google Scholar]
- Ogiwara H, Sugimoto M, Ohno T, Vilaichone RK, Mahachai V, Graham DY. Role of deletion located between the intermediate and middle regions of the Helicobacter pylori vacA gene in cases of gastroduodenal diseases. J Clin Microbiol 2009; 47:3493-500. doi: 10.1128/jcm.00887-09 [Crossref] [ Google Scholar]
- Ferreira RM, Figueiredo C, Bonet C, Pardo ML, Liso JMR, Alonso P. Helicobacter pylori vacA intermediate region genotyping and progression of gastric preneoplastic lesions. The American journal of gastroenterology 2012; 107:145. [ Google Scholar]
- Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh Hosseini M. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 2007; 133:926-36. doi: 10.1053/j.gastro.2007.06.056 [Crossref] [ Google Scholar]
- Yahiro K, Wada A, Yamasaki E, Nakayama M, Nishi Y, Hisatsune J. Essential domain of receptor tyrosine phosphatase beta (RPTPbeta) for interaction with Helicobacter pylori vacuolating cytotoxin. J Biol Chem 2004; 279:51013-21. doi: 10.1074/jbc.M406473200 [Crossref] [ Google Scholar]
- Yahiro K, Wada A, Nakayama M, Kimura T, Ogushi K, Niidome T. Protein-tyrosine phosphatase alpha, RPTP alpha, is a Helicobacter pylori VacA receptor. J Biol Chem 2003; 278:19183-9. doi: 10.1074/jbc.M300117200 [Crossref] [ Google Scholar]
- Xu Y, Fisher GJ. Receptor type protein tyrosine phosphatases (RPTPs) - roles in signal transduction and human disease. J Cell Commun Signal 2012; 6:125-38. doi: 10.1007/s12079-012-0171-5 [Crossref] [ Google Scholar]
- Padilla PI, Wada A, Yahiro K, Kimura M, Niidome T, Aoyagi H. Morphologic differentiation of HL-60 cells is associated with appearance of RPTPbeta and induction of Helicobacter pylori VacA sensitivity. J Biol Chem 2000; 275:15200-6. [ Google Scholar]
- Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 2005; 15:11-8. doi: 10.1038/sj.cr.7290257 [Crossref] [ Google Scholar]
- Fujikawa A, Shirasaka D, Yamamoto S, Ota H, Yahiro K, Fukada M. Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat Genet 2003; 33:375-81. doi: 10.1038/ng1112 [Crossref] [ Google Scholar]
- Skibinski DA, Genisset C, Barone S, Telford JL. The cell-specific phenotype of the polymorphic vacA midregion is independent of the appearance of the cell surface receptor protein tyrosine phosphatase beta. Infect Immun 2006; 74:49-55. doi: 10.1128/iai.74.1.49-55.2006 [Crossref] [ Google Scholar]
- De Guzman BB, Hisatsune J, Nakayama M, Yahiro K, Wada A, Yamasaki E. Cytotoxicity and recognition of receptor-like protein tyrosine phosphatases, RPTPalpha and RPTPbeta, by Helicobacter pylori m2VacA. Cell Microbiol 2005; 7:1285-93. doi: 10.1111/j.1462-5822.2005.00556.x [Crossref] [ Google Scholar]
- Mohebiany AN, Nikolaienko RM, Bouyain S, Harroch S. Receptor-type tyrosine phosphatase ligands: looking for the needle in the haystack. Febs j 2013; 280:388-400. doi: 10.1111/j.1742-4658.2012.08653.x [Crossref] [ Google Scholar]
- Seto K, Hayashi-Kuwabara Y, Yoneta T, Suda H, Tamaki H. Vacuolation induced by cytotoxin from Helicobacter pylori is mediated by the EGF receptor in HeLa cells. FEBS Lett 1998; 431:347-50. [ Google Scholar]
- Tegtmeyer N, Zabler D, Schmidt D, Hartig R, Brandt S, Backert S. Importance of EGF receptor, HER2/Neu and Erk1/2 kinase signalling for host cell elongation and scattering induced by the Helicobacter pylori CagA protein: antagonistic effects of the vacuolating cytotoxin VacA. Cell Microbiol 2009; 11:488-505. doi: 10.1111/j.1462-5822.2008.01269.x [Crossref] [ Google Scholar]
- Yan F, Cao H, Chaturvedi R, Krishna U, Hobbs SS, Dempsey PJ. Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology 2009; 136:1297-307, e1. doi: 10.1053/j.gastro.2008.12.059 [Crossref] [ Google Scholar]
-
Yahiro K, Hirayama T, Moss J, Noda M. New Insights into VacA Intoxication Mediated through Its Cell Surface Receptors. Toxins (Basel) 2016; 8.
- Geisse NA, Cover TL, Henderson RM, Edwardson JM. Targeting of Helicobacter pylori vacuolating toxin to lipid raft membrane domains analysed by atomic force microscopy. Biochem J 2004; 381:911-7. doi: 10.1042/bj20031719 [Crossref] [ Google Scholar]
- Kuo CH, Wang WC. Binding and internalization of Helicobacter pylori VacA via cellular lipid rafts in epithelial cells. Biochem Biophys Res Commun 2003; 303:640-4. [ Google Scholar]
- Ricci V, Galmiche A, Doye A, Necchi V, Solcia E, Boquet P. High cell sensitivity to Helicobacter pylori VacA toxin depends on a GPI-anchored protein and is not blocked by inhibition of the clathrin-mediated pathway of endocytosis. Mol Biol Cell 2000; 11:3897-909. [ Google Scholar]
- Gauthier NC, Ricci V, Gounon P, Doye A, Tauc M, Poujeol P. Glycosylphosphatidylinositol-anchored proteins and actin cytoskeleton modulate chloride transport by channels formed by the Helicobacter pylori vacuolating cytotoxin VacA in HeLa cells. J Biol Chem 2004; 279:9481-9. doi: 10.1074/jbc.M312040200 [Crossref] [ Google Scholar]
- Nakayama M, Hisatsune J, Yamasaki E, Nishi Y, Wada A, Kurazono H. Clustering of Helicobacter pylori VacA in lipid rafts, mediated by its receptor, receptor-like protein tyrosine phosphatase beta, is required for intoxication in AZ-521 Cells. Infect Immun 2006; 74:6571-80. doi: 10.1128/iai.00356-06 [Crossref] [ Google Scholar]
- Schraw W, Li Y, McClain MS, van der Goot FG, Cover TL. Association of Helicobacter pylori vacuolating toxin (VacA) with lipid rafts. J Biol Chem 2002; 277:34642-50. doi: 10.1074/jbc.M203466200 [Crossref] [ Google Scholar]
- Gupta VR, Patel HK, Kostolansky SS, Ballivian RA, Eichberg J, Blanke SR. Sphingomyelin functions as a novel receptor for Helicobacter pylori VacA. PLoS Pathog 2008; 4:e1000073. doi: 10.1371/journal.ppat.1000073 [Crossref] [ Google Scholar]
- Gupta VR, Wilson BA, Blanke SR. Sphingomyelin is important for the cellular entry and intracellular localization of Helicobacter pylori VacA. Cell Microbiol 2010; 12:1517-33. doi: 10.1111/j.1462-5822.2010.01487.x [Crossref] [ Google Scholar]
- Dubreuil JD, Giudice GD, Rappuoli R. Helicobacter pylori interactions with host serum and extracellular matrix proteins: potential role in the infectious process. Microbiol Mol Biol Rev 2002; 66:617-29, table of contents. [ Google Scholar]
- Schwarz-Linek U, Werner JM, Pickford AR, Gurusiddappa S, Kim JH, Pilka ES. Pathogenic bacteria attach to human fibronectin through a tandem beta-zipper. Nature 2003; 423:177-81. doi: 10.1038/nature01589 [Crossref] [ Google Scholar]
- Gruenheid S, Finlay BB. Microbial pathogenesis and cytoskeletal function. Nature 2003; 422:775-81. doi: 10.1038/nature01603 [Crossref] [ Google Scholar]
- Hennig EE, Godlewski MM, Butruk E, Ostrowski J. Helicobacter pylori VacA cytotoxin interacts with fibronectin and alters HeLa cell adhesion and cytoskeletal organization in vitro. FEMS Immunol Med Microbiol 2005; 44:143-50. doi: 10.1016/j.femsim.2004.10.020 [Crossref] [ Google Scholar]
- Dubreuil JD, Ruggiero P, Rappuoli R, Del Giudice G. Effect of heparin binding on Helicobacter pylori resistance to serum. J Med Microbiol 2004; 53:9-12. doi: 10.1099/jmm.0.05389-0 [Crossref] [ Google Scholar]
- Utt M, Danielsson B, Wadstrom T. Helicobacter pylori vacuolating cytotoxin binding to a putative cell surface receptor, heparan sulfate, studied by surface plasmon resonance. FEMS Immunol Med Microbiol 2001; 30:109-13. [ Google Scholar]
- Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol 2005; 3:320-32. doi: 10.1038/nrmicro1095 [Crossref] [ Google Scholar]
- Yahiro K, Satoh M, Nakano M, Hisatsune J, Isomoto H, Sap J. Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J Biol Chem 2012; 287:31104-15. doi: 10.1074/jbc.M112.387498 [Crossref] [ Google Scholar]
- Sewald X, Jimenez-Soto L, Haas R. PKC-dependent endocytosis of the Helicobacter pylori vacuolating cytotoxin in primary T lymphocytes. Cell Microbiol 2011; 13:482-96. doi: 10.1111/j.1462-5822.2010.01551.x [Crossref] [ Google Scholar]
- Sewald X, Gebert-Vogl B, Prassl S, Barwig I, Weiss E, Fabbri M. Integrin subunit CD18 Is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe 2008; 3:20-9. doi: 10.1016/j.chom.2007.11.003 [Crossref] [ Google Scholar]
-
Ricci V, Sommi P. Interaction of Helicobacter pylori VacA Toxin with Its Target Cells. In: Gopalakrishnakone P, B Stiles, A Alape-Girón, JD Dubreuil, M Mandal, editors. Microbial Toxins. Dordrecht: Springer Netherlands; 2016. p. 1-28.
- Boquet P, Ricci V. Intoxication strategy of Helicobacter pylori VacA toxin. Trends Microbiol 2012; 20:165-74. doi: 10.1016/j.tim.2012.01.008 [Crossref] [ Google Scholar]
- Howes MT, Kirkham M, Riches J, Cortese K, Walser PJ, Simpson F. Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J Cell Biol 2010; 190:675-91. doi: 10.1083/jcb.201002119 [Crossref] [ Google Scholar]
-
Bitsikas V, Corrêa IR, Nichols BJ. Clathrin-independent pathways do not contribute significantly to endocytic flux. eLife 2014; 3.
-
Kim IJ, Blanke SR. Remodeling the host environment: modulation of the gastric epithelium by the Helicobacter pylori vacuolating toxin (VacA). Front Cell Infect Microbiol 2012; 2.
- Isomoto H, Moss J, Hirayama T. Pleiotropic actions of Helicobacter pylori vacuolating cytotoxin, VacA. Tohoku J Exp Med 2010; 220:3-14. [ Google Scholar]
- Willhite DC, Blanke SR. Helicobacter pylori vacuolating cytotoxin enters cells, localizes to the mitochondria, and induces mitochondrial membrane permeability changes correlated to toxin channel activity. Cell Microbiol 2004; 6:143-54. [ Google Scholar]
- Chang H, Chen D, Ni B, Zuo Q, Wang C, Han R. Cortactin Mediates Apoptosis of Gastric Epithelial Cells Induced by VacA Protein of Helicobacter pylori. Dig Dis Sci 2016; 61:80-90. doi: 10.1007/s10620-015-3836-0 [Crossref] [ Google Scholar]
- Akazawa Y, Isomoto H, Matsushima K, Kanda T, Minami H, Yamaghchi N. Endoplasmic reticulum stress contributes to Helicobacter pylori VacA-induced apoptosis. PLoS One 2013; 8:e82322. doi: 10.1371/journal.pone.0082322 [Crossref] [ Google Scholar]
- Hisatsune J, Yamasaki E, Nakayama M, Shirasaka D, Kurazono H, Katagata Y. Helicobacter pylori VacA enhances prostaglandin E2 production through induction of cyclooxygenase 2 expression via a p38 mitogen-activated protein kinase/activating transcription factor 2 cascade in AZ-521 cells. Infect Immun 2007; 75:4472-81. doi: 10.1128/iai.00500-07 [Crossref] [ Google Scholar]
- Mitsuno Y, Yoshida H, Maeda S, Ogura K, Hirata Y, Kawabe T. Helicobacter pylori induced transactivation of SRE and AP-1 through the ERK signalling pathway in gastric cancer cells. Gut 2001; 49:18-22. [ Google Scholar]
- Wang X, Wattiez R, Paggliacia C, Telford JL, Ruysschaert J, Cabiaux V. Membrane topology of VacA cytotoxin from H pylori. FEBS Lett 2000; 481:96-100. [ Google Scholar]
- Cover TL, Hanson PI, Heuser JE. Acid-induced dissociation of VacA, the Helicobacter pylori vacuolating cytotoxin, reveals its pattern of assembly. J Cell Biol 1997; 138:759-69. [ Google Scholar]
- Chambers MG, Pyburn TM, González-Rivera C, Collier SE, Eli I, Yip CK. Structural Analysis of the Oligomeric States of Helicobacter pylori VacA Toxin. J Mol Biol 2013; 425:524-35. doi: 10.1016/j.jmb.2012.11.020 [Crossref] [ Google Scholar]
- Ivie SE, McClain MS, Torres VJ, Algood HM, Lacy DB, Yang R. Helicobacter pylori VacA subdomain required for intracellular toxin activity and assembly of functional oligomeric complexes. Infect Immun 2008; 76:2843-51. doi: 10.1128/iai.01664-07 [Crossref] [ Google Scholar]
- Oldani A, Cormont M, Hofman V, Chiozzi V, Oregioni O, Canonici A. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog 2009; 5:e1000603. doi: 10.1371/journal.ppat.1000603 [Crossref] [ Google Scholar]
- Nakano M, Yahiro K, Yamasaki E, Kurazono H, Akada J, Yamaoka Y. Helicobacter pylori VacA, acting through receptor protein tyrosine phosphatase alpha, is crucial for CagA phosphorylation in human duodenum carcinoma cell line AZ-521. Dis Model Mech 2016; 9:1473-81. doi: 10.1242/dmm.025361 [Crossref] [ Google Scholar]
- Basso D, Zambon CF, Letley DP, Stranges A, Marchet A, Rhead JL. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology 2008; 135:91-9. doi: 10.1053/j.gastro.2008.03.041 [Crossref] [ Google Scholar]
- da Costa DM, Pereira Edos S, Rabenhorst SH. What exists beyond cagA and vacA? Helicobacter pylori genes in gastric diseases. World J Gastroenterol 2015; 21:10563-72. doi: 10.3748/wjg.v21.i37.10563 [Crossref] [ Google Scholar]
- Ayala G, Escobedo-Hinojosa WI, de la Cruz-Herrera CF, Romero I. Exploring alternative treatments for Helicobacter pylori infection. World J Gastroenterol 2014; 20:1450-69. doi: 10.3748/wjg.v20.i6.1450 [Crossref] [ Google Scholar]
- Kim SY, Choi DJ, Chung JW. Antibiotic treatment for Helicobacter pylori: Is the end coming?. World J Gastrointest Pharmacol Ther 2015; 6:183-98. doi: 10.4292/wjgpt.v6.i4.183 [Crossref] [ Google Scholar]
- Sun P, Wang JQ, Zhang YT, Zhao SG. Evaluating the immune responses of mice to subcutaneous immunization with Helicobacter pylori urease B subunit. J Anim Sci Biotechnol 2014; 5:14. doi: 10.1186/2049-1891-5-14 [Crossref] [ Google Scholar]
- Tsugawa H, Suzuki H, Saya H, Hatakeyama M, Hirayama T, Hirata K. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe 2012; 12:764-77. doi: 10.1016/j.chom.2012.10.014 [Crossref] [ Google Scholar]
- Shimizu T, Marusawa H, Watanabe N, Chiba T. Molecular Pathogenesis of Helicobacter pylori-Related Gastric Cancer. Gastroenterol Clin North Am 2015; 44:625-38. doi: 10.1016/j.gtc.2015.05.011 [Crossref] [ Google Scholar]
- Garhart CA, Redline RW, Nedrud JG, Czinn SJ. Clearance of Helicobacter pylori Infection and Resolution of Postimmunization Gastritis in a Kinetic Study of Prophylactically Immunized Mice. Infect Immun 2002; 70:3529-38. [ Google Scholar]
-
Moyat M, Velin D. Use of VacA as a Vaccine Antigen. Toxins (Basel) 2016; 8.
- Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology 2007; 133:288-308. doi: 10.1053/j.gastro.2007.05.008 [Crossref] [ Google Scholar]
- Kotloff KL, Sztein MB, Wasserman SS, Losonsky GA, DiLorenzo SC, Walker RI. Safety and immunogenicity of oral inactivated whole-cell Helicobacter pylori vaccine with adjuvant among volunteers with or without subclinical infection. Infect Immun 2001; 69:3581-90. doi: 10.1128/iai.69.6.3581-3590.2001 [Crossref] [ Google Scholar]
- Del Giudice G, Covacci A, Telford JL, Montecucco C, Rappuoli R. The design of vaccines against Helicobacter pylori and their development. Annu Rev Immunol 2001; 19:523-63. doi: 10.1146/annurev.immunol.19.1.523 [Crossref] [ Google Scholar]
- Tang RX, Luo DJ, Sun AH, Yan J. Diversity of Helicobacter pylori isolates in expression of antigens and induction of antibodies. World J Gastroenterol 2008; 14:4816-22. [ Google Scholar]
- Manetti R, Massari P, Marchetti M, Magagnoli C, Nuti S, Lupetti P. Detoxification of the Helicobacter pylori cytotoxin. Infect Immun 1997; 65:4615-9. [ Google Scholar]
- Marchetti M, Rossi M, Giannelli V, Giuliani MM, Pizza M, Censini S. Marchetti M, Rossi M, Giannelli V, Giuliani MM, Pizza M, Censini S, et alProtection against Helicobacter pylori infection in mice by intragastric vaccination with Hpylori antigens is achieved using a non-toxic mutant of Ecoli heat-labile enterotoxin (LT) as adjuvant. Vaccine 1998; 16:33-7. [ Google Scholar]
- Ghiara P, Rossi M, Marchetti M, Di Tommaso A, Vindigni C, Ciampolini F. Therapeutic intragastric vaccination against Helicobacter pylori in mice eradicates an otherwise chronic infection and confers protection against reinfection. Infect Immun 1997; 65:4996-5002. [ Google Scholar]
- Rossi G, Ruggiero P, Peppoloni S, Pancotto L, Fortuna D, Lauretti L. Therapeutic vaccination against Helicobacter pylori in the beagle dog experimental model: safety, immunogenicity, and efficacy. Infect Immun 2004; 72:3252-9. doi: 10.1128/iai.72.6.3252-3259.2004 [Crossref] [ Google Scholar]
- Merchant JL. VacA pores as portable portals for urea. J Clin Invest 2001; 108:803-4. doi: 10.1172/jci14004 [Crossref] [ Google Scholar]
- Liu XL, Li SQ, Liu CJ, Tao HX, Zhang ZS. Antigen epitope of Helicobacter pylori vacuolating cytotoxin A. World J Gastroenterol 2004; 10:2340-3. [ Google Scholar]
- Ruggiero P, Peppoloni S, Rappuoli R, Del Giudice G. The quest for a vaccine against Helicobacter pylori: how to move from mouse to man?. Microbes Infect 2003; 5:749-56. [ Google Scholar]
- Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs 2015; 7:9-14. doi: 10.4161/19420862.2015.989042 [Crossref] [ Google Scholar]
- Yamaguchi H, Osaki T, Taguchi H, Kamiya S. Production of monoclonal antibodies neutralizing vacuolation of cultured cells by Helicobacter pylori cytotoxin. FEMS Microbiol Lett 1998; 168:277-82. [ Google Scholar]
- Nagata K, Mizuta T, Tonokatu Y, Fukuda Y, Okamura H, Hayashi T. Monoclonal antibodies against the native urease of Helicobacter pylori: synergistic inhibition of urease activity by monoclonal antibody combinations. Infect Immun 1992; 60:4826-31. [ Google Scholar]
- Pourakbari B, Ghazi M, Mahmoudi S, Mamishi S, Azhdarkosh H, Najafi M. Diagnosis of Helicobacter pylori infection by invasive and noninvasive tests. Braz J Microbiol 2013; 44:795-8. doi: 10.1590/s1517-83822013005000052 [Crossref] [ Google Scholar]
- Chen Z, Moayeri M, Purcell R. Monoclonal antibody therapies against anthrax. Toxins (Basel) 2011; 3:1004-19. doi: 10.3390/toxins3081004 [Crossref] [ Google Scholar]
- Mattsson A, Quiding-Jarbrink M, Lonroth H, Hamlet A, Ahlstedt I, Svennerholm A. Antibody-secreting cells in the stomachs of symptomatic and asymptomatic Helicobacter pylori-infected subjects. Infect Immun 1998; 66:2705-12. [ Google Scholar]
- Berdoz J, Corthesy B. Human polymeric IgA is superior to IgG and single-chain Fv of the same monoclonal specificity to inhibit urease activity associated with Helicobacter pylori. Mol Immunol 2004; 41:1013-22. doi: 10.1016/j.molimm.2004.05.006 [Crossref] [ Google Scholar]
- Begue RE, Moll A. Immunogenicity of Recombinant Helicobacter pylori Urease B Administered by Various Routes and with Different Adjuvants. Open Vaccine J 2009; 2:28-32. doi: 10.2174/1875035400902010028 [Crossref] [ Google Scholar]
- Ardekani LS, Gargari SLM, Rasooli I, Bazl MR, Mohammadi M, Ebrahimizadeh W. A novel nanobody against urease activity of Helicobacter pylori. International Journal of Infectious Diseases 2013; 17:e723-e8. [ Google Scholar]
- Zhao A, Tohidkia MR, Siegel DL, Coukos G, Omidi Y. Phage antibody display libraries: a powerful antibody discovery platform for immunotherapy. Crit Rev Biotechnol 2016; 36:276-89. doi: 10.3109/07388551.2014.958978 [Crossref] [ Google Scholar]
- Tohidkia MR, Barar J, Asadi F, Omidi Y. Molecular considerations for development of phage antibody libraries. J Drug Target 2012; 20:195-208. doi: 10.3109/1061186X.2011.611517 [Crossref] [ Google Scholar]
- Tohidkia MR, Asadi F, Barar J, Omidi Y. Selection of potential therapeutic human single-chain Fv antibodies against cholecystokinin-B/gastrin receptor by phage display technology. BioDrugs 2013; 27:55-67. doi: 10.1007/s40259-012-0007-0 [Crossref] [ Google Scholar]
-
Tohidkia MR, Sepehri M, Khajeh S, Barar J, Omidi Y. Improved Soluble ScFv ELISA Screening Approach for Antibody Discovery Using Phage Display Technology. SLAS Discov 2017; in press.