BioImpacts. 8(4):241-252.
doi: 10.15171/bi.2018.27
Original Research
Effect of PEGylation on assembly morphology and cellular uptake of poly ethyleneimine-cholesterol conjugates for delivery of sorafenib tosylate in hepatocellular carcinoma
Maryam Monajati 1, 2, Shirin Tavakoli 3, Samira Sadat Abolmaali 4, Gholamhossein Yousefi 4, AliMohammad Tamaddon 3, *
Author information:
1Department of Pharmaceutical Nanotechnology, School of Pharmacy, Shiraz University of Medical Sciences, Shiraz 71345, Iran
2Nanotechnology Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
3Center for Nanotechnology in Drug Delivery, Shiraz University of Medical Sciences, Shiraz 71345, Iran
4Department of Pharmaceutical Nanotechnology and Center for Nanotechnology in Drug Delivery, School of Pharmacy, Shiraz University of Medical Sciences, Shiraz 71345, Iran
Abstract
Introduction:
Sorafenib (SFB) is an FDA-approved chemotherapeutic agent with a high partition coefficient (log P = 4.34) for monotherapy of hepatocellular carcinoma (HCC). The oral bioavailability is low and variable, so it was aimed to study the application of the polymeric nanoassembly of cholesterol conjugates of branched polyethyleneimine (PEI) for micellar solubilization of SFB and to investigate the impact of the polymer PEGylation on the physicochemical and cellular characteristics of the lipopolymeric dispersions.
Methods:
Successful synthesis of cholesterol-PEI lipopolymers, either native or PEGylated, was confirmed by FTIR, 1H-NMR, pyrene assay methods. The nanoassemblies were also characterized in terms of morphology, particle size distribution and zeta-potential by TEM and dynamic light scattering (DLS). The SFB loading was optimized using general factorial design. Finally, the effect of particle characteristics on cellular uptake and specific cytotoxicity was investigated by flow cytometry and MTT assay in HepG2 cells.
Results:
Transmission electron microscopy (TEM) showed that PEGylation of the lipopolymers reduces the size and changes the morphology of the nanoassembly from rod-like to spherical shape. However, PEGylation of the lipopolymer increased critical micelle concentration (CMC) and reduced the drug loading. Moreover, the particle shape changes from large rods to small spheres promoted the cellular uptake and SFB-related cytotoxicity.
Conclusion:
The combinatory effects of enhanced cellular uptake and reduced general cytotoxicity can present PEGylated PEI-cholesterol conjugates as a potential carrier for delivery of poorly soluble chemotherapeutic agents such as SFB in HCC that certainly requires further investigations in vitro and in vivo.
Keywords: Poly ethyleneimine, Cholesterol, Lipopolymer, PEGylation, Polymeric micelles, Sorafenib tosylate, Hepatocellular carcinoma
Copyright and License Information
© 2018 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Hepatocellular carcinoma (HCC) is the fifth common cancer and the third cause of cancer-related death in the world
1,2
which is often diagnosed at later stages, thus the efficiency of the treatment methods including ablation, resection, and transplantation is limited.
1,3
Sorafenib tosylate (SFB, Nexavar®) is an anticancer orphan drug approved as a standard monotherapy for HCC. SFB is a dual action inhibitor which targets multiple kinase such as the B-Raf and C-Raf and also vascular endothelial growth factor receptor 2 (VEGFR2) and platelet-derived growth factor receptor (PDGFR) that leads to inhibition of tumor cell proliferation and invasion.
3-5
However, with respect to the Biopharmaceutical Classification Scheme, SFB belongs to class II compounds which demonstrate a poor solubility (25 ng/mL in deionized water) and high permeability from the gastrointestinal lumen. The oral bioavailability is low and variable which may restrict its therapeutic efficacy.
3
Many attempts have been made to increase the drug solubility and bioavailability such as encapsulation in poly-DL-lactide-co-glycolide (PLGA),
6
PLGA/dextran copolymer,
4
solid lipid nanoparticles
7
or liposomes,
3
complex formation with albumin,
8
cyclodextrin
9
and so on.
Among various nano-sized drug delivery systems intended to improve physicochemical (solubility) and pharmacokinetic characteristics of the drug, self-assemblies of amphiphilic polymers contain a hydrophobic inner core that provides the ability to dissolve hydrophobic drugs and a hydrophilic shell which grants the carrier stability in water.
10-13
Polymeric self-assembled nanoparticles offer several advantages such as high kinetic and thermodynamic stability, suitable size (20–100 nm), narrow size distribution, easily adjustable surface potential, modulated biocompatibility, tissue accumulation and penetration, controlled intracellular trafficking, controlled release, and reduced inherent toxicity.
14,15
Notably, there are crucial factors in the design of nanoparticles including morphology (size and shape) and surface characteristics that can influence on both pharmacokinetics and cell uptake.
16
Amphiphilic polymers have a tendency to self- assemble into a wide variety of structures including micelles, vesicles, nanotubes, nanofibers and lamellae.
17
For particles >100 nm, the highest cellular uptake happens for rod morphology followed by spheres, cylinders, and cubes while particles <100 nm spheres enters the cell to the greatest extent.
18
Polyethyleneimine (PEI) is one of the most studied cationic polymers widely used for cell transfection.
19,20
Due to the presence of primary, secondary and tertiary amines in the polymer structure, PEI can be highly protonated and therefore interact with negatively charged cell membrane
21
and can also induce osmolytic rupture and endosomal escape according to "proton sponge" effect.
22,23
However, PEI shows a variable degree of cytotoxicity depending on molecular weight and structure.
24,25
Several chemical modifications of PEI has been employed to reduce the toxicity of PEI such as chemical reaction to polyethylene glycol (PEGylation) which also leads to decreased aggregation and protein binding in physiological fluids and prolonged circulation time in blood stream.
26-30
Hydrophobic modification with cholesterol, an important part of the cell membrane, is another example of PEI modification. It has been observed that even at low modification percentages, cholesterol-containing lipopolymers show a high tendency to self-assembly.
31
Cholesterol modification of PEI facilitates endocytosis and reduces the inherent toxicity of PEI
32
; moreover, the presence of cholesterol leads to enhancement of drug loading capacity and reduction of the drug burst release.
11,33
An interesting fact about HCC is that cholesterol conjugated polymers transfect the cells through cholesterol uptake pathway, which is also utilized by low-density lipoprotein (LDL).
34
LDL binds to many cells such as vascular endothelial cells, hepatocytes and macrophages.
35
Due to the high growth rate, cancer cells overexpress LDL receptor and obtain a large amount of cholesterol from the blood. Studies have shown that lipopolymers containing a high amount of cholesterol have enhanced tumor distribution in comparison with cholesterol-free carriers.
36
Moreover, the cholesterol-bearing nanoparticles due to the relevance of their aromatic structure to SFB can make a reservoir interacting with the drug through hydrophobic and π-π mechanisms for efficient loading of SFB. Therefore, in the present study, taking the advantages of polycation (i.e. PEI)-cell interaction, PEGylation reaction and SFB solubilization in the core of hydrophobically modified PEI, SFB-incorporated nanoparticles composed of PEI-cholesterol lipopolymers were developed. The effect of polymer PEGylation on physicochemical characteristics such as drug encapsulation, particle size, zeta potential, morphology and critical micelle concentration (CMC) was investigated. Furthermore, cellular uptake and in vitro cytotoxicity against HepG2 cells were compared to the non-PEGylated carrier and the free drug.
Materials and Methods
Materials
Branched PEI 10 kDa (Mw/Mn of 1.4) and Methoxy PEG-COOH (NHS activated, 5 kDa) were purchased from Polysciences, Inc. (Warrington, PA, USA) and JenKem Technology USA (Allen, TX, USA), respectively. SFB was obtained from Hangzhou Hetd. Industry Co., Ltd. (Hangzhou, Zhejiang, China). Cholesteryl chloroformate, N,N- diisopropylethylamine (DIPEA), pyrene, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were supplied by Sigma-Aldrich (St Louis, MO, USA). Dichloromethane (DCM), dimethylsulfoxide (DMSO), and potassium bromide (KBr) were purchased from Merck KGaA (Darmstadt, Germany).
Synthesis of cholesterol conjugates
The chemical route for the synthesis of cholesterol-conjugated PEI was summarized in Scheme 1. The lipopolymers were synthesized according to the previous reports.
37,38
Different molar ratios of cholesterol to PEI, 7.5:1 (F2) and 15.5:1 (F3), were reacted with 250 µL of PEI (10% w/v) in methanol. The reactions were supplemented with 7.5 µL of DIPEA as a proton quencher. Then, 750 µL of cholesteryl chloroformate (12% w/v) in dichloromethane was added drop-wise to the reaction medium and maintained for 9 hours in 50°C while stirring. Afterward, the mixture was dropped slowly into the 3 mL water at 50°C and dispersed by probe sonication. To remove the unreacted cholesterol, the solution was filtered through a 0.22 µm nylon syringe filter and dialyzed (Float-A-Lyzer 6-8 kDa) according to the manufacturer’s instruction against 2 L of deionized water. The medium was removed and replaced by fresh deionized water for 3 consecutive days. Subsequently, the purified products solutions were lyophilized. The recovered mass of products (yields) was measured gravimetrically by a very sensitive balance. The relative yields were calculated from the ratio of actual to predicted mass of the products for different concentrations of cholesteryl chloroformate.
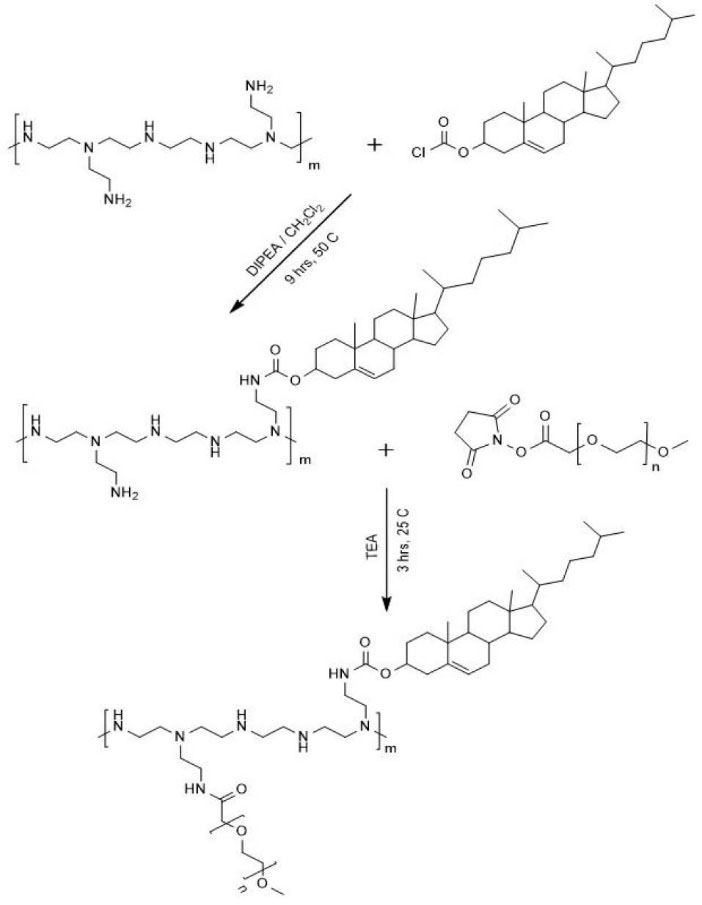
Scheme 1.
Schematic presentation of the synthesis of PEIcholesterol
and the consecutive PEGylation reaction.
.
Schematic presentation of the synthesis of PEIcholesterol
and the consecutive PEGylation reaction.
Infrared spectra were recorded on the FTIR spectrometer (Vertex, Bruker, Germany) to study spectral changes of PEI after cholesterol modification. Samples were prepared by geometric dilution of an identical amount of the lyophilized products with potassium bromide and compression of the mixtures to form discs. Twenty scans were signal averaged with a resolution of 4 cm-1 in range of 500-4000 cm.
1H-NMR spectra were recorded on Bruker-300 MHz using CDCl3 as a solvent. Proton integration method was used to calculate the average molar ratio of the conjugated cholesteryl moieties with respect to PEI based on their respective chemical shifts. The following equation was applied to calculate the average number of conjugated cholesterol per polymer chain of PEI:
where, nH is the number of a proton which exists in the chemical structure of each molecule.
PEGylation of lipopolymers
The synthesis of the PEG-PEI lipopolymers was accomplished through grafting of methoxy polyethylene glycol chains using carbodiimide chemistry.
39
Briefly, adequate volumes of 10% w/v PEG-COOH (NHS activated) solution (the respective volumes of 73 µL and 114 µL for the mole ratios of 1:7.5:1 (F4) and 2:15.5:1 (F5) PEG / cholesterol / PEI), was slowly added to 200 µL of lipopolymer solution (5% w/v in methanol). The reaction media were supplemented with 1% trimethylamine and incubated for 3 hours while mixing at 400 rpm and 25°C. The products were obtained using a rotary evaporator, reconstituted with deionized water to the final volume of 3 mL and dialyzed similarly against 2 L of deionized water. Then, the purified products were lyophilized. The recovered mass of the products (yields) was measured gravimetrically and the relative yields were calculated from their actual to expected mass ratios. Finally, the product was characterized by FTIR and 1H-NMR spectroscopy to study spectral changes and to calculate an average number of conjugated PEG per lipopolymer chain of PEI-cholesterol based on the chemical shifts according to the following equation:
where nH represents the proton number of each polymer.
Pyrene assay
To estimate CMC values of the synthesized lipopolymers, the pyrene assay was performed.
13,40
Briefly, 25 µL of pyrene solution (4 µg/µL in acetone) was transferred into microtubes and dried overnight under chemistry cabinet. One mL of each lipopolymer dispersion prepared at various concentrations (0.001, 0.005, 0.01, 0.05, 0.1, and 1 mg/mL) were added and stirred at 800 rpm for 24 hours. To remove any insoluble components, the polymer solutions were centrifuged for 15 min at 15 000 rpm. The value of the intensity ratio I339/I334 of each sample was plotted against the logarithm of the copolymer concentration and flection point of the plot was applied to determine the CMC of each lipopolymer.
HPLC assay
According to our previous report
41
with little modification, a chromatographic method was developed for SFB assay using Knauer HPLC system, equipped with P1000 LPG pump and multiple wavelengths UV P2600 detector. Separation of SFB was achieved using a reversed phase column (C18, 1250-4.6mm, Merck, Germany) at room temperature. The mobile phase composed of 80% methanol in deionized water was delivered isocratically at a flow rate of 1.3 mL/min. The HPLC method was validated in terms of specificity, linearity, accuracy, precision, and limit of quantification for an in vitro assay. (y = 56463x+156306, R² = 0.9999).
Drug loading
SFB was loaded in the lipopolymer using solid dispersion method.
42
To investigate the effect of different lipopolymer compositions and concentrations on the drug loading, an adequate amount of lipolpolymers (F2, F3, F4, or F5) and SFB were dissolved in methanol at the respective weight ratio of 0.25 and incubated at 45°C for 30 minutes. Methanol was completely removed under vacuum and each sediment was re-dispersed by distilled water at 45°C to obtain the final lipopolymer concentrations of 1, 3 and 10 mg/mL. The pH was adjusted to 7.5 and the dispersions were stirred at 45°C for 48 hours. The excess amount of SFB was removed by filtration through a 0.22 µm nylon filter. To further investigate the effect of temperature and pH on the drug loading, SFB was loaded in F3 lipopolymer as described before and the final polymer concentration was set to 3 mg/mL. The pH was adjusted to 5, 7 or 9 and the mixture was incubated for 48 hours at 30, 45 or 60°C. The amount of loaded SFB was measured by the HPLC assay method after removing the free SFB by filtration according to the following equation:
Drug loading (%) = (mass of SFB loaded / total mass of the loaded polymer) × 100.
General factorial design optimization
Various factorial experiments are generally used in analysis and optimization of drug formulations.
43,44
The effects of different factors (lipopolymer composition, concentration, pH and temperature) were investigated on the SFB loading in the lipopolymers by general factorial design. The composition was expressed in terms of cholesterol to PEI mole ratio (7.5 or 15) and the PEGylation state (PEGylated vs. non-PEGylated). The incubation temperature and pH were investigated in three levels as described in the previous section. The experiment was run in triplicate. Then, the analysis was performed using Design-Expert® software version 6.02 (Stat-Ease, Inc., Minneapolis, MN, USA).
Transmission Electron Microscopy
The suspensions of drug loaded lipopolymers, PEGylated (F5) or non-PEGylated (F3), were prepared at the concentration of 3 mg/mL for transmission electron microscopy (TEM). A drop of each specimen was deposited onto Formvar-coated, 300 mesh copper grids and dried. The specimens were photographed by Zeiss - EM10C operating at an acceleration voltage of 80 keV.
Dynamic Light Scattering
The number-, volume- and intensity-averaged hydrodynamic diameter (Z-average) and polydispersity index (PDI) of the preparations were determined by dynamic light scattering (Zeta sizer 3000HSA, Malvern Instruments, UK). Zeta-potential measurements were performed in phosphate buffer (pH 7.4) at 25°C.
Cellular uptake
To determine the effect of PEGylation on cellular uptake of the preparations, PEI-cholesterol lipopolymers were labeled with fluorescein isothiocyanate (FITC) according to our previous report.
45
Briefly, 200 μL of 10 mg/mL FITC in DMSO was added to 2 mL of the F3 or F5 at an equivalent molar concentration (respective concentrations of 1.2 and 2.1 mg/mL) in 0.1 M borate buffer (pH 9.2) and the mixture was incubated for 2 hours while stirring. The excess FITC was removed by dialysis using Float-A-Lyzer 8-10 kDa at 4°C in dark. The degree of conjugation was determined using a standard curve plotted for fluorescein absorbance at 490 nm.
Then, cellular uptake of the FITC-labeled F3 or F5 was determined by flow cytometry (FACS Calibur, Becton Dickinson, USA). Hence, HepG2 cells seeded in 6-well plates at a density of 150 000 cells /mL were treated with 50 μg/mL FITC-labeled F3 and F5 and incubated at 37°C for 3 hours. The cells were washed with PBS, detached by scraping and reconstituted in 500 μL of ice-cold PBS by pipetting. Cell-associated fluorescence intensities were determined by the flow cytometer and analyzed by FlowJo software version 7.6.2 for gated live singlet cells.
Cytotoxicity
The MTT assay was carried out to determine the lipopolymer cytotoxicity and the bioactivity of the SFB loaded particles (F3, F5) based on the published protocol.
46
HepG2 cells were plated into 96-well microtiter plates at a density of 20 000 cells/cm2. After 24 hours, the cells were treated in serum-supplemented culture medium for 24 hours. Each treatment was prepared by dilution in phosphate buffered saline (pH 7.4) and sterilization by 0.22 µm syringe filter before use in cell culture. The lipopolymer concentrations were varied from 6 to 600 μg/mL. Following treatment, the medium was aspirated and replaced by 100 µL of 1/10 diluted 5 mg/mL MTT stock solution in the culture medium. After 3 hours, the medium was aspirated again and insoluble formazan crystals were dissolved in 100 µL/well DMSO and measured spectrophotometrically in a microplate reader at λ = 570 nm minus λ = 650 nm. Cell viability was calculated relative to untreated control cells. The cytotoxicity of SFB loaded particles (F3, F5) was compared to free SFB and the lipopolymer.
Statistics
Statistical analysis was performed by Prism software version 6.0 (GraphPad, USA). The significance of each factor was determined by analysis of variance. P values less than 0.05 were considered statistically significant. Data were expressed as the mean ± standard deviation (SD).
Results and Discussion
Synthesis and characterization of PEI-cholesterol
To create hydrophobic moieties being served for interaction with SFB during the loading process, cholesteryl chloroformate was successfully conjugated to the PEI backbone to a variable extent (low, F2; high, F3) via the nucleophilic substitution reaction (Scheme 1). The recovery yield was about 76%-84%.
To confirm the cholesterol modification of PEI, FTIR and the 1H-NMR spectroscopy were used. Fig. 1 shows the FTIR spectra of the lipopolymer F3 and cholesteryl chloroformate. The appearance of new carbamate carbonyl bond at 1659 cm-1 and disappearance of acyl halide peak at 1776 cm-1, confirms the conjugation of PEI to cholesteryl chloroformate. Moreover, the 1H-NMR spectrum of the lipolymer F3 in CDCl3 ( Fig. 2) shows multiple broad peaks at 0.7-1.2 ppm that are attributed to the conjugated cholesterols. Also, 2 characteristic peaks of (-CH-COO) and (-CH=CH-) of cholesterol were observed at 4.50 ppm and 5.36 ppm, respectively. As shown in 1H-NMR spectrum, by increasing the mass of cholesterol in the lipopolymers, integrated area under the peak at 5.36 ppm was raised with respect to the PEI peaks at 2-3 ppm. The estimated molar ratios and their respective molecular weights calculated by the 1H-NMR are presented in Table 1.
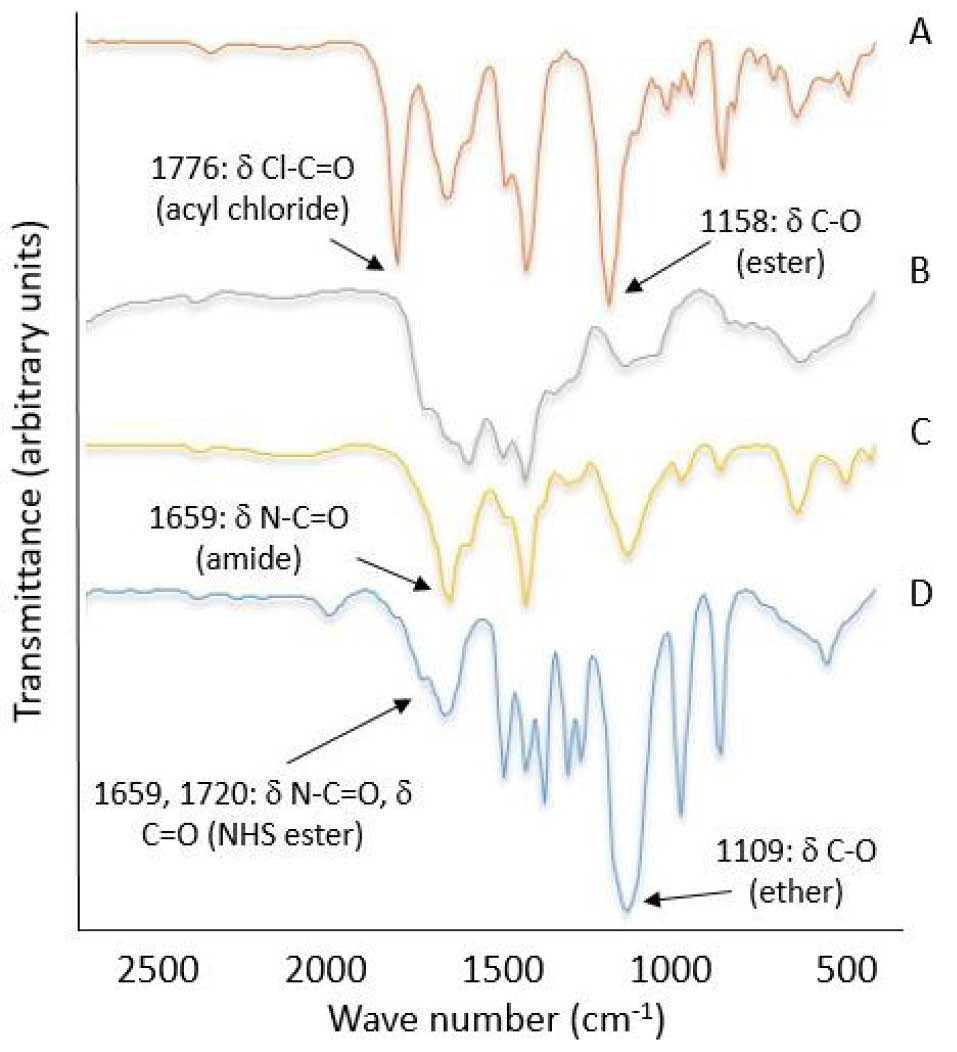
Fig. 1.
FTIR spectrum of A: cholesteryl chloroformate, B, C: F3 and F5 lipopolymer at the respective mole ratios of 1:15.5:0 and 1:15.5:2 (PEI/cholesterol/PEG), D: PEG.
.
FTIR spectrum of A: cholesteryl chloroformate, B, C: F3 and F5 lipopolymer at the respective mole ratios of 1:15.5:0 and 1:15.5:2 (PEI/cholesterol/PEG), D: PEG.
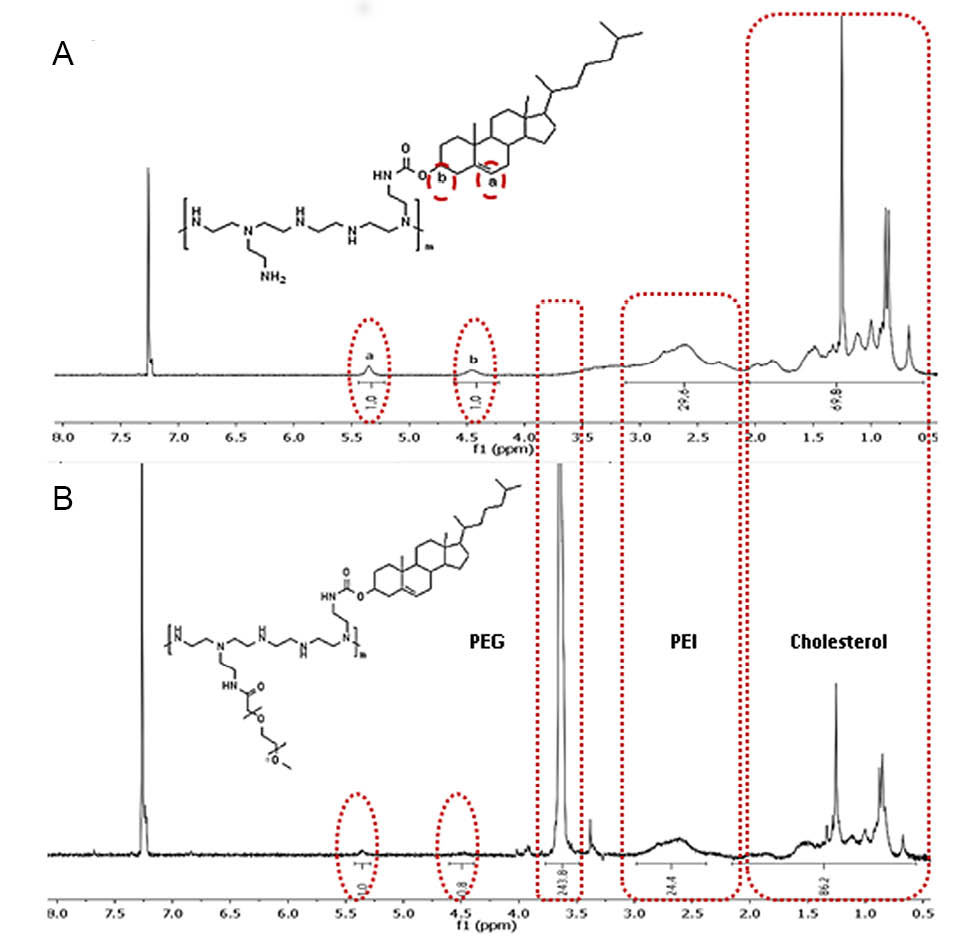
Fig. 2.
1H-NMR spectrum of F3 (A) and F5 (B) lipopolymers at the respective mole ratios of 1:15.5:0 and 1:15.5:2 (PEI/cholesterol/PEG) in CDCl3.
.
1H-NMR spectrum of F3 (A) and F5 (B) lipopolymers at the respective mole ratios of 1:15.5:0 and 1:15.5:2 (PEI/cholesterol/PEG) in CDCl3.
Table 1.
Calculated CMC, molar ratio and molecular weight of PEI-cholesterol polymer with and without PEGylating the polymer according to 1H-NMR
|
Sample ID
|
PEI/ cholesterol/PEG mole ratio
|
M
n
by
1H-NMR (kDa)
|
CMC (Mean ± SD, µg/mL)
|
| PEI |
1 : 0 : 0 |
6.7 |
- |
| F2 |
1 : 7.5 : 0 |
9.8 |
53.77 ± 4.37 |
| F3 |
1 : 15.5 : 0 |
13.1 |
45.68 ± 0.81 |
| F4 |
1 : 7.5 : 1 |
15.3 |
70.54 ± 3.84 |
| F5 |
1 : 15.5 : 2 |
23.9 |
73.43 ± 4.13 |
Synthesis and characterization of PEGylated PEI-cholesterol
The mPEG grafted PEI-cholesterol lipopolymers were synthesized using carbodiimide reaction through amide bond formation at the constant molar ratio of mEPG to cholesterol of 1: 7.5 (F4 and F5). The total recovery yields following the PEGylation reaction were similarly about 75%-80%.
The chemical structure of PEGylated lipopolymers was investigated again by FTIR and 1H-NMR spectroscopy. As shown in Fig. 1, the characteristic peak of mPEG (C-O stretching ether bonds) appears at 1109 cm-1 for the PEGylated lipopolymer (F5) while the amide bonds of NHS molecule (strong stretching N-C=O bonds) is omitted. The 1H-NMR spectrum of F5 in CDCl3 (Fig. 2) also shows multiple peaks at 2.3 - 3.1 ppm that is attributed to the ethylenimine (–N–CH2CH2) protons in PEI and the peak at 3.6 ppm refers to the ethylene glycol (-O-CH2CH2) protons in mPEG chains. The degree of PEGylation was determined by comparing the integral values obtained from the number of the (-O-CH2CH2) protons in mPEG chains and the protons of PEI. This calculation was also used as a basis for the determination of the Mn products (Table 1).
CMC determination
Cholesterol modification of PEI leads to an increase in lipophilicity of the polymer and a decrease in water solubility while PEGylation improves aqueous dispersion of the PEGylated lipopolymers. Therefore, due to the addition of hydrophilic and hydrophobic moieties to the PEI backbone, the lipopolymers can form various micelle-like structures at concentrations above CMC. To investigate the CMC of lipopolymers, the fluorescence intensity of an increasing lipopolymer concentration was determined in presence of a fixed amount of pyrene. The CMC values were determined from the concentration of the polymer that shows an abrupt increase in the pyrene fluorescence intensity. Low CMC is an important factor generally considered for self-assembled nanoparticles that guarantees the in-vivo stability of the particles.
47
As shown in Table 1, CMC values of the lipopolymers were similarly less than 75 µg/mL due to the strong hydrophobic driving force of cholesterol. It has also been demonstrated that with an increase of the degree of cholesterol conjugation, CMC of PEI lipopolymers deceases.
48
On the other hand, PEGylation with increase in the hydrophilic portion of the lipopolymers can increase CMC.
49
Similarly, in the present study, the cholesterol conjugation leads to the lower CMC, whereas, the PEGylation increases the CMC.
Drug loading
As expected, the formulation F3, with the lowest CMC, had the maximum drug loading (13.1% ± 2.65) that is substantial if compared with similar reports using heparin/chitosan (12%),
50
albumin (8.6%)
8
or galactosylated poly lactide-poly amino acid nanoparticles (3.6%).
51
Considering the SFB loading percent as a response variable, the effect of three factors including the polymer concentration, PEG/cholesterol and cholesterol/PEI mole ratios were statistically analyzed using a quadratic interaction model (P<0.0001). The model R2, adjusted-R2 and predicted R2 were determined 0.987, 0.972 and 0.929, respectively that confirms the suitability of curve fitting. Supplementary file 1 shows that the drug loading varied significantly with respect to PEG to PEI mole ratio (P=0.0005), so that the PEGylation decreased the drug loading to a modest extent, possibly due to the higher CMC values of the PEGylated lipopolymer as explained before. Inversely, increasing the cholesterol/PEI mole ratio (P=0.0558) enhanced the drug loading for the non-PEGylated lipopolymer, but no significant change happened for the PEGylated lipopolymer (P=0.2334). It seems that by increasing the number of conjugated cholesterol, more interactions can happen with SFB that may result in higher drug loading. This effect disappears following PEGylation that might be due to the well-known antifouling property of PEG molecules. Increasing the polymer concentration increased the concentration of loaded SFB (P<0.0001), however, the drug loading percent decreased by the lipopolymer concentration if non-PEGylated (P=0.0007) that can be explained by limited solubility of the lipopolymers prepared at higher degrees of the cholesterol modification. With respect to the proper stability of the drug in a wide range of temperature and pH (data not shown), the drug loading in F3 formulation was investigated in nine different environmental conditions. The data were successfully fitted in a two-factor interaction model (P=0.0012). Results revealed that unlike temperature that showed no significant effect, the drug loading increased (P=0.0004) with increasing pH in the range of 5 to 9 (Supplementary file 1). This phenomenon is possibly due to deprotonation of PEI amines in more alkaline pH that may increase the lipopolymer CMC and in turn increases the drug loading.
Transmission Electron Microscopy
According to transmission electron microscopy (TEM) image (Fig. 3), F3 lipopolymer forms rod-like micelles of 300-700 nm in length, whereas PEGylated lipopolymer (F5) assembles into smaller spherical micelles of about 30 nm in projected diameter. The morphology of copolymer micelles is determined by several variables, such as composition, chain length, hydrophilic-lipophilic balance, concentrations, as well as solvents, temperatures, additives etc.
52
Our findings is consistent with the other studies
53,54
that the higher volume fractions of hydrophilic block results in spherical micelles, whilst lower fractions favor worm-like micelles and vesicles.

Fig. 3.
TEM images of sorafenib tosylate loaded nanoparticles: A (F3) and B (F5) at the respective mole ratios of 1:15.5:0 and 1:15.5:2 (PEI/cholesterol/PEG).
.
TEM images of sorafenib tosylate loaded nanoparticles: A (F3) and B (F5) at the respective mole ratios of 1:15.5:0 and 1:15.5:2 (PEI/cholesterol/PEG).
Particle size analysis
The particle size of the polymeric micelles is generally considered as a function of different factors such as molecular weight of the amphiphilic copolymer, aggregation number, relative proportion of hydrophilic and hydrophobic chains, and the preparation process.
55
Therefore, the effects of two main determinants, i.e. cholesterol modification and PEGylation, on the particle size were investigated. As determined by DLS the particle size showed a completely different behavior if studied before and after the drug loading.
As demonstrated in Table 2 before the drug loading, by increasing the cholesterol ratio the polymer solubility decreases, so the micelle composed of uni-molecular and multi aggregates collapses and the particles become smaller.
56
Similarly, but in a different manner, conjugation of hydrophilic molecules of PEG reduced the particle size by changing the morphology from rod to spherical shape as shown in Fig. 3. Moreover, the PEGylation increases CMC of the lipopolymers (Table 1), and consequently, the aggregation number and the size may decrease.
Table 2.
Nanoparticles hydrodynamic size with and without loading of sorafenib tosylate (SFB)
|
Sample ID
|
PEI/cholesterol/PEG
|
SFB loaded
|
pH
|
Z average (nm)
|
PDI
|
N average (nm)
|
V average (nm)
|
|
F2
|
1 : 7.5 : 0 |
- |
7.5 |
132 |
0.25 |
90.0 |
155.0 |
|
F3
|
1 : 15.5 : 0 |
- |
7.5 |
142 |
0.22 |
27.9 |
89.1 |
|
F4
|
1 : 7.5 : 1 |
- |
7.5 |
120 |
0.19 |
65.0 |
112.0 |
|
F5
|
1 : 15.5 : 2 |
- |
7.5 |
115 |
0.39 |
18.1 |
15.8 |
|
F2
|
1 : 7.5 : 0 |
+ |
7.5 |
186 |
0.40 |
14.0 |
30.7 |
|
F3
|
1 : 15.5 : 0 |
+ |
5.5 |
141 |
0.43 |
18.9 |
44.3 |
|
F3
|
1 : 15.5 : 0 |
+ |
7.5 |
211 |
0.42 |
29.5 |
106.3 |
|
F3
|
1 : 15.5 : 0 |
+ |
9.5 |
175 |
0.49 |
36.4 |
374.5 |
|
F4
|
1 : 7.5 : 1 |
+ |
7.5 |
132 |
0.31 |
37.7 |
83.5 |
|
F5
|
1 : 15.5 : 2 |
+ |
7.5 |
246 |
0.24 |
185.0 |
384.0 |
The drug loading increased the particle size either by enlargement of the core of micelles or subtle particle aggregation (modest increases of PDI as noticed in Table 2). After the drug loading, the higher ratio of cholesterol modification (F3) resulted in larger micelles (Table 2) due to SFB-lipopolymer interactions and consequently increased hydrophobicity of the polymer chains that facilitates the formation of large polymer aggregates. Indeed, the polymer solubility reduction leads to the higher potential for self-assembly and larger polymer buildup in the same concentration. Possessing primary, secondary and tertiary amines, three pKa values of 5.5, 7.8 and 8.5 are reported for PEI so the aggregation process can be sensitive to pH. By increasing pH from 5.5 to 9.5, the lipopolymers become less protonated and more hydrophobic. This phenomenon would lead to larger polymer aggregates (Table 2).
The DLS size was larger than the TEM size as reported elsewhere for PEGylated micelles.
57
The discrepancy between sizes is mainly because DLS evaluates hydrodynamic size rather than projected area diameter of solid spheres by TEM.
58
Each ethylenoxide unit of PEG can interrelate with 3-5 water molecules, so the polymer hydrodynamic volume increases 5-10 times more than the estimated molecular weight of the polymer.
59
Moreover, long PEG chains decorating the surface of nanoparticles can enhance the micro-viscosity around these particles and therefore due to underestimation of viscosity in Stokes-Einstein equation, DLS might overestimate the size. While TEM analysis gives structural details of different morphology, DLS determines only the size of spherical or near-spherical nanoparticles. Asymmetrical particles result in inaccurate size determination by DLS since the amount of light they scatter attenuates depending on their orientation.
60
This phenomenon can explain the smaller size of F3 (SFB loaded, non-PEGylated lipopolymer) determined by DLS in comparison with TEM analysis.
Zeta-potential is another physicochemical characteristic of the nanoparticles that determines the particle stability, cellular and in vivo fate of the particles. Comparing zeta-potentials of the aqueous dispersions of non-PEGylated (+44.5 ± 8.9) and PEGylated (+12.4 ± 4.3 mV) lipopolymers shows the charge shielding property of the conjugated PEG chains.
61
Cellular uptake
The cellular uptake of nanoparticles is affected by various factors such as particle size,
18,45,62
surface charge,
18,45,3,64
shape
18,65,66
and structural composition.
63
It has been shown that LDL receptor saturation with free cholesterol, LDL and antibody against the LDL receptor inhibits cellular uptake of PEI-cholesterol nanoparticles. This phenomenon suggests that the cellular uptake of linear PEI-cholesterol (LPC) conjugates can occur through LDL receptor-mediated endocytosis mechanism.
34,67
As shown in Fig. 4, a significantly higher extent of cell-associated fluorescence intensity was achieved in the treated HepG2 cells with the FITC-labeled F5 (91.7%) than F3 (7.6%) formulations. Considering the first step in cellular uptake is binding to the cell membrane,
45,68
so the positive zeta-potentials of lipopolymers should play an important role in the cellular uptake,
69
however our observation emphasizes more on the effect of particle size and morphology than zeta-potential on the cellular uptake so that the spherical particles of smaller sizes (F5) were engulfed more efficiently than larger particles with rod-like shapes (F3) in HepG2 cells, though the zeta-potential of F5 was less positive than F3. This finding has also been reported in other studies that the internalization rate of spherical nanoparticles is higher than non-spherical ones such as rods, cubes and discs.
65,66
It has been shown that for nanoparticle’s invagination and wrapping, the spherical particles require the minimum energy to overcome the elastic bending if compared to other anisotropic shapes.
65
Specifically, uptake of the rod-like nanoparticles decreases with an increasing aspect ratio due to high dependency of the cellular uptake on the entry angle. The spherical shape is the only homogeneous morphology that cannot be affected from re-orientation issues that can happen for the rod-shape particles.
65,66,70
Furthermore, PEGylation stabilizes the particles and hinders the aggregate formation, which interferes with the cellular uptake. To elucidate the involvement of the endocytosis mechanisms, repetition of the experiment with various endocytosis inhibitors or specific competitive inhibitors is crucial.
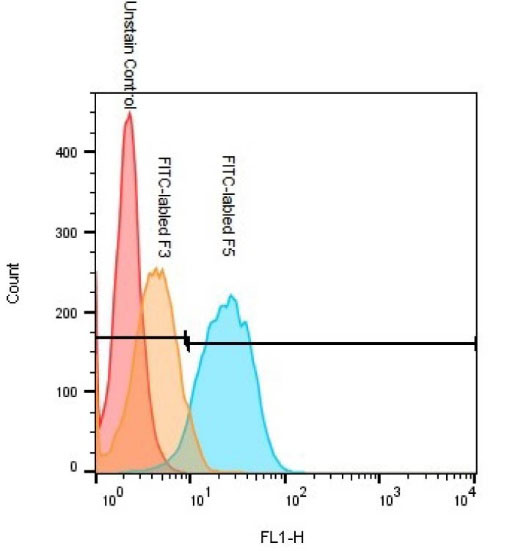
Fig. 4.
Flow cytometry analysis of HepG2 cells after incubation with FITC-labeled F3 and F5 nanoparticles prepared at the respective mole ratios of 1:15.5:0 and 1:15.5:2 (PEI/cholesterol/PEG).
.
Flow cytometry analysis of HepG2 cells after incubation with FITC-labeled F3 and F5 nanoparticles prepared at the respective mole ratios of 1:15.5:0 and 1:15.5:2 (PEI/cholesterol/PEG).
Cytotoxicity
MTT assay is a measurement of cell metabolic activity and correlates quite well with cell proliferation. Despite all promising properties of PEIs as a polymeric carrier both in vitro and in vivo, there is a major concern of cytotoxicity due to the highly positive-charge nature which imposes undesired interactions with biological matrices.
71
It has been shown that PEI conjugation with cholesterol is one of the methods to reduce the cytotoxicity on living cells by reducing the positive charge through sequestering the primary and secondary amines.
48,67,72
The cytotoxicity of cholesterol-modified lipopolymers (1:15.5, PEI to cholesterol ratio) were determined less than native PEI (Mw=10 kDa) as reported previously.
45
PEGylation has also been reported to improve the PEI biocompatibility by shielding the positive surface charges.
18,73-75
In line with the previous reports
27,39
and according to the Fig. 5, the cytotoxicity comparison of PEGylated versus non-PEGylated lipopolymers in HepG2 cells incubated for 48 hours showed that the cell viability enhanced very significantly so that F5 did not show any cytotoxic effect even at the concentration as high as 600 μg/mL. The higher cytotoxicity of F3 is also an explanation for a substantial difference in cellular uptake between F3 and F5 because the flow cytometric analysis of cellular uptake is based on the ability of particles to enter the viable cells.
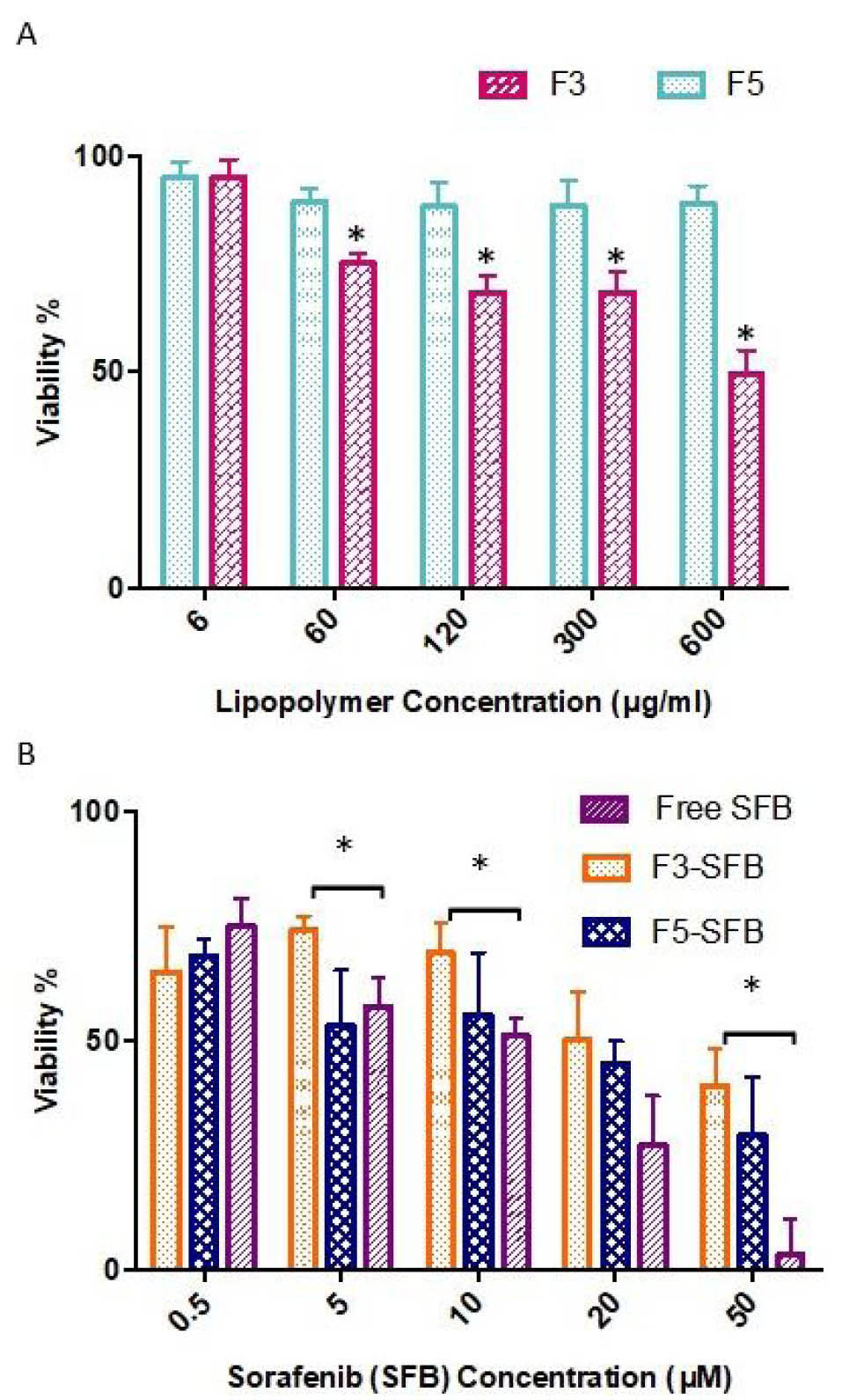
Fig. 5.
MTT assay of the cytotoxicity induced by A) lipopolymers and B) the sorafenib tosylate-loaded lipopolymeric micelles in comparison to free drug in HepG2 cells. F3 and F5: lipopolymers prepared at the respective mole ratios of 1:15.5:0 and 1:15.5:2 (PEI/cholesterol/PEG). The error bars represent SD (n=5).
.
MTT assay of the cytotoxicity induced by A) lipopolymers and B) the sorafenib tosylate-loaded lipopolymeric micelles in comparison to free drug in HepG2 cells. F3 and F5: lipopolymers prepared at the respective mole ratios of 1:15.5:0 and 1:15.5:2 (PEI/cholesterol/PEG). The error bars represent SD (n=5).
SFB inhibits tumor cell proliferation and angiogenesis via targeting numerous serine/threonine and tyrosine kinases (RAF1, BRAF, VEGFR 1, 2, 3, PDGFR, KIT, FLT3, FGFR1, and RET) in multiple oncogenic signaling pathways.
76
Coriat et al showed that SFB enhanced the production of O2ˉ, NO and H2O2 in HepG2 cells. Reactive oxygen species (ROS) can cause oxidative damages to lipids, proteins and DNA and finally to cell death.
77
Fig. 5 illustrates the cytotoxicity comparison among the drug-loaded F3 and F5 lipopolymers and free SFB (IC50 0f 5-10 μM). More cytotoxicity was found for the SFB-loaded F5 than F3 that can be explained by more cellular uptake in HepG2 cells as described in the previous section. Apart from F3 which shows the polymer-related cytotoxicity, a significant decline in cell viability was recognized for F5 at the drug concentrations above 10 µM. So, unlike the unloaded polymer (F5) that didn’t bring about significant cytotoxicity, SFB-loaded particles resulted in a concentration-dependent toxicity, indicating the drug release happens to exert the pharmacological action. Nevertheless, determination of the drug release kinetic model such as reciprocal powered time model, Weibull or Wagner’s log probability
78
has not been studied.
The cytotoxicity of free SFB was more pronounced than the loaded lipopolymeric micelles (F3 and F5). This result is in good agreement with the previous studies. Xiao et al revealed that SFB incorporated liposomes have lower cytotoxicity in comparison with SFB solution.
3
In another study, the docetaxel-loaded cholesterol polymeric micelles show less cytotoxic effect than the free drug.
11
This observation might correspond to either favorable partitioning of free lipophilic drugs in the cell membrane or the drug release property of nanoparticles that results in a sustained but lower cell exposure to effective drug concentration if compared with free drug.
33
It should also be considered that high cytotoxicity of the potent drugs if not targeted to the tumor tissue, is not favorable. Indeed, prolonged circulation of free cytotoxic agents may harm healthy normal as well as cancerous cells which lead to serious side effects. This problem could be solved by using well-designed nanoparticles that accumulate preferentially in tumors by either passive or active targeting. Here in the present study, the PEGylated nanoparticles showing enough small sizes are presumed to extravasate from the blood vessel and accumulate in the tumor.
8
Moreover, as shown in another study
79
the PEGylation through charge shielding mechanism and formation of hydrated steric barrier reduces opsonization of the nanoparticles and thus enhances blood circulation.
Conclusion
In the present study, series of PEGylated and classic PEI-cholesterol lipopolymeric micelles were successfully developed for delivery of SFB in HepG2 cells. The cholesterol-containing hydrophobic core of the micelles allows us to obtain low enough CMC values and a remarkable capacity for SFB loading. However, non-PEGylated PEI-cholesterol nanoparticles show high positive charges and general cytotoxicity. The PEGylation reaction through the charge shielding mechanism lowers the general cytotoxicity of the synthesized lipopolymers. On the other hand, the particle shape changes from large rods to small spheres that promotes to a great extent both the cellular uptake and specific SFB-related cytotoxicity. Our results confirm that the positive-charge shielded, small and spherical particles prepared from PEGylated PEI-cholesterol may have a great potential for delivery of chemotherapeutic agents such as SFB to HCC that also requires more researches on cellular internalization mechanism and in vivo fate of the particles.
Acknowledgment
The authors gratefully acknowledge use of the facilities of the Center for Nanotechnology in Drug Delivery at Shiraz University of Medical Sciences (SUMS).
Funding sources
This work is a part of Ms. Maryam Monajati Pharm.D. thesis funded by Shiraz University of Medical Sciences (Vice-Chancellery of Research and Technology).
Ethical statement
Not applicable.
Competing interests
The authors declare there is no conflict of interest.
Authors contribution
AMT, SSA and GHY conceived and planned the project, the main conceptual ideas and design of the experiments. MM and ST implemented the experiments. AMT and SSA contributed to data analysis and interpretation of the results. MM was in charge of writing the manuscript. SSA contributed to writing and also reviewing final version of the manuscript. All authors provided helpful feedback.
Supplementary materials
Supplementary file 1 contains Fig. S1.
(pdf)
Research Highlights
What is the current knowledge?
simple
-
√ Cholesterol containing lipopolymers self-assemble into
nanoparticle of different morphologies even at low degree of
modification.
-
√ High degree of cholesterol modification provides a large
hydrophobic core for an improved drug loading, but it
decreases the aqueous dispersibility.
-
√ PEGylation through the charge shielding mechanism
lowers the general cytotoxicity associated with lipopolymers.
What is new here?
simple
-
√ PEGylation changes the size and morphology of poly
ethyleneimine-cholesterol micellar nanoparticles and
increases the cellular uptake.
-
√ The cholesterol-containing hydrophobic cores allows low
CMC and a remarkable capacity for loading and delivery of
SFB tosylate to HCC cell line.
References
- Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 2006; 66:11851-8. doi: 10.1158/0008-5472 [Crossref] [ Google Scholar]
- Roberts LR. Sorafenib in liver cancer—just the beginning. N Engl J Med 2008; 359:420-2. doi: 10.1056/NEJMe0802241 [Crossref] [ Google Scholar]
- Xiao Y, Liu Y, Yang S, Zhang B, Wang T, Jiang D. Sorafenib and Gadolinium co-loaded liposomes for drug delivery and MRI-guided HCC treatment. Colloids Surf B Biointerfaces 2016. doi: 10.1016/j.colsurfb.2016.01.016 [Crossref]
- Kim DH, Kim M-D, Choi C-W, Chung C-W, Ha SH, Kim CH. Antitumor activity of sorafenib-incorporated nanoparticles of dextran/poly (dl-lactide-co-glycolide) block copolymer. Nanoscale Res Lett 2012; 7:1-6. doi: 10.1186/1556-276X-7-91 [Crossref] [ Google Scholar]
- Pawlik TM, Reyes DK, Cosgrove D, Kamel IR, Bhagat N, Geschwind J-FH. Phase II trial of sorafenib combined with concurrent transarterial chemoembolization with drug-eluting beads for hepatocellular carcinoma. J Clin Oncol 2011; 29:3960-7. doi: 10.1200/JCO.2011.37.1021 [Crossref] [ Google Scholar]
- Chen J, Sheu A, Lewandowski R, Omary R, Shea L, Larson A. Transcatheter delivery of sorafenib-eluting polylactide-co-glycolide microspheres: therapy response studies in a rat model of hepatocellular carcinoma. J Vasc Interv Radiol 2013; 24:S145. doi: 10.1016/j.jvir.2013.01.360 [Crossref] [ Google Scholar]
- Zhang H, Zhang F-M, Yan S-J. Preparation, in vitro release, and pharmacokinetics in rabbits of lyophilized injection of sorafenib solid lipid nanoparticles. Int J Nanomedicine 2012; 7:2901. doi: 10.2147/IJN.S32415 [Crossref] [ Google Scholar]
- Zhang J-Y, He B, Qu W, Cui Z, Wang Y-b, Zhang H. Preparation of the albumin nanoparticle system loaded with both paclitaxel and sorafenib and its evaluation in vitro and in vivo. J Microencapsul 2011; 28:528-36. doi: 10.3109/02652048.2011.590614 [Crossref] [ Google Scholar]
- Bondì ML, Scala A, Sortino G, Amore E, Botto C, Azzolina A. Nanoassemblies Based on Supramolecular Complexes of Nonionic Amphiphilic Cyclodextrin and Sorafenib as Effective Weapons to Kill Human HCC Cells. Biomacromolecules 2015; 16:3784-91. doi: 10.1021/acs.biomac.5b01082 [Crossref] [ Google Scholar]
- Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacology & therapeutics 2006; 112:630-48. doi: 10.1016/j.pharmthera.2006.05.006 [Crossref] [ Google Scholar]
- Varshosaz J, Taymouri S, Hassanzadeh F, Javanmard SH, Rostami M. Self-assembly micelles with lipid core of cholesterol for docetaxel delivery to B16F10 melanoma and HepG2 cells. J Liposome Res 2015; 25:157-65. doi: 10.3109/08982104.2014.961022 [Crossref] [ Google Scholar]
- Abbasi S, Yousefi G, Tamaddon A-M. Polyacrylamide–b-copolypeptide hybrid copolymer as pH-responsive carrier for delivery of paclitaxel: Effects of copolymer composition on nanomicelles properties, loading efficiency and hemocompatibility. Colloids Surf A Physicochem Eng Asp 2018; 537:217-26. doi: 10.1016/j.colsurfa.2017.09.007 [Crossref] [ Google Scholar]
- Salmanpour M, Tamaddon A, Yousefi G, Mohammadi-Samani S. " Grafting-from" synthesis and characterization of poly (2-ethyl-2-oxazoline)-b-poly (benzyl L-glutamate) micellar nanoparticles for potential biomedical applications. BioImpacts: BI 2017; 7:155. doi: 10.15171/bi.2017.19 [Crossref] [ Google Scholar]
- Li J, Li Z, Zhou T, Zhang J, Xia H, Li H. Positively charged micelles based on a triblock copolymer demonstrate enhanced corneal penetration. Int J Nanomedicine 2015; 10:6027. doi: 10.2147/IJN.S90347 [Crossref] [ Google Scholar]
- Abolmaali S, Tamaddon A, Salmanpour M, Mohammadi S, Dinarvand R. Block ionomer micellar nanoparticles from double hydrophilic copolymers, classifications and promises for delivery of cancer chemotherapeutics. Eur J Pharm Sci 2017. doi: 10.1016/j.ejps.2017.04.009 [Crossref]
- Truong NP, Whittaker MR, Mak CW, Davis TP. The importance of nanoparticle shape in cancer drug delivery. Expert Opin Drug Deliv 2015; 12:129-42. doi: 10.1517/17425247.2014.950564 [Crossref] [ Google Scholar]
- Lombardo D, Kiselev MA, Magazù S, Calandra P. Amphiphiles Self-Assembly: Basic Concepts and Future Perspectives of Supramolecular Approaches. Advances in Condensed Matter Physics 2015; 2015. doi: 10.1155/2015/151683 [Crossref]
- Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng 2012; 14:1-16. doi: 10.1146/annurev-bioeng-071811-150124 [Crossref] [ Google Scholar]
- Park TG, Jeong JH, Kim SW. Current status of polymeric gene delivery systems. Adv Drug Deliv Rev 2006; 58:467-86. doi: 10.1016/j.addr.2006.03.007 [Crossref] [ Google Scholar]
- Najafi H, Abolmaali SS, Owrangi B, Ghasemi Y, Tamaddon AM. Serum resistant and enhanced transfection of plasmid DNA by PEG-stabilized polyplex nanoparticles of L-histidine substituted polyethyleneimine. Macromol Res 2015; 23:618-27. doi: 10.1007/s13233-015-3074-5 [Crossref] [ Google Scholar]
- Tang G, Zeng J, Gao S, Ma Y, Shi L, Li Y. Polyethylene glycol modified polyethylenimine for improved CNS gene transfer: effects of PEGylation extent. Biomaterials 2003; 24:2351-62. doi: 10.1016/S0142-9612(03)00029-2 [Crossref] [ Google Scholar]
-
Huang L, Hung M-c, Wagner E. Nonviral vectors for gene therapy: Academic Press; 2005.
- Barar J, Omidi Y. Intrinsic bio-signature of gene delivery nanocarriers may impair gene therapy goals. BioImpacts: BI 2013; 3:105. doi: 10.5681/bi.2013.028 [Crossref] [ Google Scholar]
- Alshamsan A, Haddadi A, Incani V, Samuel J, Lavasanifar A, Uludag H. Formulation and delivery of siRNA by oleic acid and stearic acid modified polyethylenimine. Molecular pharmaceutics 2008; 6:121-33. doi: 10.1021/mp8000815 [Crossref] [ Google Scholar]
- Kafil V, Omidi Y. Cytotoxic impacts of linear and branched polyethylenimine nanostructures in A431 cells. BI 2011; 1:23. doi: 10.5681/bi.2011.004 [Crossref] [ Google Scholar]
- Cho KC, Jeong JH, Chung HJ, Joe CO, Kim SW, Park TG. Folate receptor-mediated intracellular delivery of recombinant caspase-3 for inducing apoptosis. J Control Release 2005; 108:121-31. doi: 10.1016/j.jconrel.2005.07.015 [Crossref] [ Google Scholar]
- Vinogradov S, Batrakova E, Kabanov A. Poly (ethylene glycol)–polyethyleneimine NanoGel™ particles: novel drug delivery systems for antisense oligonucleotides. Colloids Surf B Biointerfaces 1999; 16:291-304. doi: 10.1016/S0927-7765(99)00080-6 [Crossref] [ Google Scholar]
- Rudolph C, Sieverling N, Schillinger U, Lesina E, Plank C, Thünemann AF. Thyroid hormone (T3)-modification of polyethyleneglycol (PEG)-polyethyleneimine (PEI) graft copolymers for improved gene delivery to hepatocytes. Biomaterials 2007; 28:1900-11. doi: 10.1016/j.biomaterials.2006.12.011 [Crossref] [ Google Scholar]
- Prasannan A, Debele TA, Tsai H-C, Chao C-C, Lin C-P, Hsiue G-H. Synthesis and evaluation of the targeted binding of RGD-containing PEGylated-PEI/DNA polyplex micelles as radiotracers for a tumor-targeting imaging probe. RSC Adv 2015; 5:107455-65. doi: 10.1039/C5RA18644G [Crossref] [ Google Scholar]
- Abolmaali S, Tamaddon A, Kamali-Sarvestani E, Ashraf M, Dinarvand R. Stealth nanogels of histinylated poly ethyleneimine for sustained delivery of methotrexate in collagen-induced arthritis model. Pharmaceutical research 2015; 32:3309-23. doi: 10.1007/s11095-015-1708-0 [Crossref] [ Google Scholar]
- Xu J-P, Ji J, Chen W-D, Shen J-C. Novel biomimetic polymersomes as polymer therapeutics for drug delivery. J Control Release 2005; 107:502-12. doi: 10.1016/j.jconrel.2005.06.013 [Crossref] [ Google Scholar]
- Kim S, Choi JS, Jang HS, Suh H, Park J. Hydrophobic modification of polyethyleneimine for gene transfectants. BULLETIN-KOREAN CHEMICAL SOCIETY 2001; 22:1069-75. [ Google Scholar]
- Zhang CY, Chen Q, Wu WS, Guo XD, Cai CZ, Zhang LJ. Synthesis and evaluation of cholesterol-grafted PEGylated peptides with pH-triggered property as novel drug carriers for cancer chemotherapy. Colloids Surf B Biointerfaces 2016; 142:55-64. doi: 10.1016/j.colsurfb.2016.02.025 [Crossref] [ Google Scholar]
- Lee M, Rentz J, Han S, Bull D, Kim S. Water-soluble lipopolymer as an efficient carrier for gene delivery to myocardium. Gene therapy 2003; 10:585-93. doi: 10.1038/sj.gt.3301938 [Crossref] [ Google Scholar]
- Radwan AA, Alanazi FK. Targeting cancer using cholesterol conjugates. Saudi Pharm J 2014; 22:3-16. doi: 10.1016/j.jsps.2013.01.003 [Crossref] [ Google Scholar]
- Song Y, Tian Q, Huang Z, Fan D, She Z, Liu X. Self-assembled micelles of novel amphiphilic copolymer cholesterol-coupled F68 containing cabazitaxel as a drug delivery system. Int J Nanomedicine 2014; 9:2307. doi: 10.2147/IJN.S61220 [Crossref] [ Google Scholar]
- Dung TH, Kim JS, Juliano R, Yoo H. Preparation and evaluation of cholesteryl PAMAM dendrimers as nano delivery agents for antisense oligonucleotides. Colloids Surf A Physicochem Eng Asp 2008; 313:273-7. doi: 10.1016/j.colsurfa.2007.04.109 [Crossref] [ Google Scholar]
- Golkar N, Samani SM, Tamaddon AM. Modulated cellular delivery of anti-VEGF siRNA (bevasiranib) by incorporating supramolecular assemblies of hydrophobically modified polyamidoamine dendrimer in stealth liposomes. Int J Pharm 2016; 510:30-41. doi: 10.1016/j.ijpharm.2016.06.026 [Crossref] [ Google Scholar]
- Abolmaali SS, Tamaddon AM, Dinarvand R. Nano-hydrogels of methoxy polyethylene glycol-grafted branched polyethyleneimine via biodegradable cross-linking of Zn2+-ionomer micelle template. J Nanopart Res 2013; 15:1-21. doi: 10.1007/s11051-013-2134-z [Crossref] [ Google Scholar]
- Bahadur KR, Landry B, Aliabadi HM, Lavasanifar A, Uludağ H. Lipid substitution on low molecular weight (06–20 kDa) polyethylenimine leads to a higher zeta potential of plasmid DNA and enhances transgene expression. J Nanopart Res 2011; 7:2209-17. doi: 10.1016/j.actbio.2011.01.027 [Crossref] [ Google Scholar]
- Hashemi F, Tamaddon A, Yousefi G, Farvadi F. Development and validation of a rapid and simple HPLC-UV method for the analysis of sorafenib in the presence of polyamidoamine (PAMAM) dendrimers. J Liq Chromatogr Relat Technol 2014; 37:1427-37. doi: 10.1080/10826076.2013.794737 [Crossref] [ Google Scholar]
- Pellosi DS, Moret F, Fraix A, Marino N, Maiolino S, Gaio E. Pluronic® P123/F127 mixed micelles delivering sorafenib and its combination with verteporfin in cancer cells. Int J Nanomedicine 2016; 11:4479. doi: 10.2147/IJN.S103344 [Crossref] [ Google Scholar]
- Abolmaali SS, Tamaddon A, Yousefi G, Javidnia K, Dinarvand R. Sequential optimization of methotrexate encapsulation in micellar nano-networks of polyethyleneimine ionomer containing redox-sensitive cross-links. Int J Nanomedicine 2014; 9:2833. doi: 10.2147/IJN.S61614 [Crossref] [ Google Scholar]
- Ravanfar R, Tamaddon AM, Niakousari M, Moein MR. Preservation of anthocyanins in solid lipid nanoparticles: Optimization of a microemulsion dilution method using the Placket–Burman and Box–Behnken designs. Food Chem 2016; 199:573-80. doi: 10.1016/j.foodchem.2015.12.061 [Crossref] [ Google Scholar]
- Abolmaali SS, Tamaddon AM, Mohammadi S, Amoozgar Z, Dinarvand R. Chemically crosslinked nanogels of PEGylated poly ethyleneimine (l-histidine substituted) synthesized via metal ion coordinated self-assembly for delivery of methotrexate: Cytocompatibility, cellular delivery and antitumor activity in resistant cells. Mater Sci Eng C Mater Biol Appl 2016; 62:897-907. doi: 10.1016/j.msec.2016.02.045 [Crossref] [ Google Scholar]
- Chiou J-F, Tai C-J, Huang M-T, Wei P-L, Wang Y-H, An J. Glucose-regulated protein 78 is a novel contributor to acquisition of resistance to sorafenib in hepatocellular carcinoma. Ann Surg Oncol 2010; 17:603-12. doi: 10.1245/s10434-009-0718-8 [Crossref] [ Google Scholar]
- Kastantin M, Ananthanarayanan B, Karmali P, Ruoslahti E, Tirrell M. Effect of the lipid chain melting transition on the stability of DSPE-PEG (2000) micelles. Langmuir 2009; 25:7279-86. [ Google Scholar]
- Gusachenko O, Kravchuk Y, Konevets D, Silnikov V, Vlassov VV, Zenkova MA. Transfection efficiency of 25-kDa PEI–cholesterol conjugates with different levels of modification. J Biomater Sci Polym Ed 2009; 20:1091-110. doi: 10.1163/156856209X444448 [Crossref] [ Google Scholar]
- Ashok B, Arleth L, Hjelm RP, Rubinstein I, Önyüksel H. In vitro characterization of PEGylated phospholipid micelles for improved drug solubilization: effects of PEG chain length and PC incorporation. J Pharm Sci 2004; 93:2476-87. doi: 10.1002/jps.20150 [Crossref] [ Google Scholar]
- Yang Y-C, Cai J, Yin J, Zhang J, Wang K-L, Zhang Z-T. Heparin-functionalized Pluronic nanoparticles to enhance the antitumor efficacy of sorafenib in gastric cancers. Carbohydr Polym 2016; 136:782-90. doi: 10.1016/j.carbpol.2015.09.023 [Crossref] [ Google Scholar]
- Craparo EF, Sardo C, Serio R, Zizzo MG, Bondì ML, Giammona G. Galactosylated polymeric carriers for liver targeting of sorafenib. Int J Pharm 2014; 466:172-80. doi: 10.1016/j.ijpharm.2014.02.047 [Crossref] [ Google Scholar]
- Zhang Y, Dou X, Jin T. Synthesis and self-assembly behavior of amphiphilic diblock copolymer dextran-block-poly (epsilon-caprolactone)(DEX-b-PCL) in aqueous media. Express Polym Lett 2010; 4. doi: 10.3144/expresspolymlett.2010.75 [Crossref]
- Fujiwara S, Hashimoto M, Itoh T, Nakamura H, Tamura Y, editors editors. . Shape transition of micelles in amphiphilic solution: A molecular dynamics study 2013.
- Schuetz P, Greenall MJ, Bent J, Furzeland S, Atkins D, Butler MF. Controlling the micellar morphology of binary PEO–PCL block copolymers in water–THF through controlled blending. Soft Matter 2011; 7:749-59. doi: 10.1039/C0SM00938E [Crossref] [ Google Scholar]
- Mourya V, Inamdar N, Nawale R, Kulthe S. Polymeric micelles: general considerations and their applications. Indian J Pharm Educ Res 2011; 45:128-38. [ Google Scholar]
- Yusa S-i. Self-assembly of cholesterol-containing water-soluble polymers. Int J Polym Sci 2012:2012. doi: 10.1155/2012/609767 [Crossref]
- Stetefeld J, McKenna SA, Patel TR. Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophys Rev 2016; 8:409-27. doi: 10.1007/s12551-016-0218-6 [Crossref] [ Google Scholar]
- Cumberland SA, Lead JR. Particle size distributions of silver nanoparticles at environmentally relevant conditions. J Chromatogr A 2009; 1216:9099-105. doi: 10.1016/j.chroma.2009.07.021 [Crossref] [ Google Scholar]
-
Veronese FM. PEGylated protein drugs: basic science and clinical applications: Springer; 2009.
-
Griffiths G. Technology and applications of autonomous underwater vehicles: CRC Press; 2002.
- Luo X, Feng M, Pan S, Wen Y, Zhang W, Wu C. Charge shielding effects on gene delivery of polyethylenimine/DNA complexes: PEGylation and phospholipid coating. J Mater Sci Mater Med 2012; 23:1685-95. doi: 10.1007/s10856-012-4632-4 [Crossref] [ Google Scholar]
- Zauner W, Farrow NA, Haines AM. In vitro uptake of polystyrene microspheres: effect of particle size, cell line and cell density. J Control Release 2001; 71:39-51. doi: 10.1016/S0168-3659(00)00358-8 [Crossref] [ Google Scholar]
- Jung T, Kamm W, Breitenbach A, Kaiserling E, Xiao J, Kissel T. Biodegradable nanoparticles for oral delivery of peptides: is there a role for polymers to affect mucosal uptake?. Eur J Pharm Biopharm 2000; 50:147-60. doi: 10.1016/S0939-6411(00)00084-9 [Crossref] [ Google Scholar]
- Foster KA, Yazdanian M, Audus KL. Microparticulate uptake mechanisms of in‐vitro cell culture models of the respiratory epithelium. J Pharm Pharmacol 2001; 53:57-66. doi: 10.1211/0022357011775190 [Crossref] [ Google Scholar]
- Li Y, Yue T, Yang K, Zhang X. Molecular modeling of the relationship between nanoparticle shape anisotropy and endocytosis kinetics. Biomaterials 2012; 33:4965-73. doi: 10.1016/j.biomaterials.2012.03.044 [Crossref] [ Google Scholar]
- Li Y, Kröger M, Liu WK. Shape effect in cellular uptake of pegylated nanoparticles: comparison between sphere, rod, cube and disk. Nanoscale 2015; 7:16631-46. doi: 10.1039/C5NR02970H [Crossref] [ Google Scholar]
- Furgeson DY, Chan WS, Yockman JW, Kim SW. Modified linear polyethylenimine-cholesterol conjugates for DNA complexation. Bioconjug Chem 2003; 14:840-7. doi: 10.1021/bc0340565 [Crossref] [ Google Scholar]
- Zhao F, Zhao Y, Liu Y, Chang X, Chen C, Zhao Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small 2011; 7:1322-37. doi: 10.1002/smll.201100001 [Crossref] [ Google Scholar]
- Oh N, Park J-H. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int J Nanomedicine 2014; 9:51. doi: 10.2147/IJN.S26592 [Crossref] [ Google Scholar]
- Chithrani BD, Chan WC. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett 2007; 7:1542-50. doi: 10.1021/nl070363y [Crossref] [ Google Scholar]
- Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials 2003; 24:1121-31. doi: 10.1016/S0142-9612(02)00445-3 [Crossref] [ Google Scholar]
- Abolmaali SS, Tamaddon A, Najafi H, Dinarvand R. Effect of l-Histidine substitution on Sol–Gel of transition metal coordinated poly ethyleneimine: synthesis and biochemical characterization. J Inorg Organomet Polym Mater 2014; 24:977-87. doi: 10.1007/s10904-014-0067-3 [Crossref] [ Google Scholar]
-
Misra A. Challenges in delivery of therapeutic genomics and proteomics: Elsevier; 2010.
- Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine 2012; 7:5577-91. doi: 10.2147/IJN.S36111 [Crossref] [ Google Scholar]
- Malek A, Czubayko F, Aigner A. PEG grafting of polyethylenimine (PEI) exerts different effects on DNA transfection and siRNA-induced gene targeting efficacy. J Drug Target 2008; 16:124-39. doi: 10.1080/10611860701849058 [Crossref] [ Google Scholar]
- Cervello M, Bachvarov D, Lampiasi N, Cusimano A, Azzolina A, McCubrey JA. Molecular mechanisms of sorafenib action in liver cancer cells. Cell cycle 2012; 11:2843-55. doi: 10.4161/cc.21193 [Crossref] [ Google Scholar]
- Coriat R, Nicco C, Chéreau C, Mir O, Alexandre J, Ropert S. Sorafenib-induced hepatocellular carcinoma cell death depends on reactive oxygen species production in vitro and in vivo. Mol Cancer Ther 2012; 11:2284-93. doi: 10.1158/1535-7163.MCT-12-0093 [Crossref] [ Google Scholar]
- Barzegar-Jalali M, Adibkia K, Valizadeh H, Shadbad MRS, Nokhodchi A, Omidi Y. Kinetic analysis of drug release from nanoparticles. J Pharm Pharm Sci 2008; 11:167-77. [ Google Scholar]
- Hu Y, Xie J, Tong YW, Wang C-H. Effect of PEG conformation and particle size on the cellular uptake efficiency of nanoparticles with the HepG2 cells. J Control Release 2007; 118:7-17. doi: 10.1016/j.jconrel.2006.11.028 [Crossref] [ Google Scholar]