BioImpacts. 8(4):305-320.
doi: 10.15171/bi.2018.33
Review
Recent advances in improving oral drug bioavailability by cocrystals
Shahram Emami 1, 2, Mohammadreza Siahi-Shadbad 3, Khosro Adibkia 4  , Mohammad Barzegar-Jalali 5, *
, Mohammad Barzegar-Jalali 5, *
Author information:
1Drug Applied Research Center and Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
2Student Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran
3Department of Pharmaceutical and Food Control, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
4Research Center for Pharmaceutical Nanotechnology, Biomedicine Institute, and Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
5Biotechnology Research Center, and Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
Abstract
Introduction
: Oral drug delivery is the most favored route of drug administration. However, poor oral bioavailability is one of the leading reasons for insufficient clinical efficacy. Improving oral absorption of drugs with low water solubility and/or low intestinal membrane permeability is an active field of research. Cocrystallization of drugs with appropriate coformers is a promising approach for enhancing oral bioavailability.
Methods
: In the present review, we have focused on recent advances that have been made in improving oral absorption through cocrystallization. The covered areas include supersaturation and its importance on oral absorption of cocrystals, permeability of cocrystals through membranes, drug-coformer pharmacokinetic (PK) interactions, conducting in vivo-in vitro correlations for cocrystals. Additionally, a discussion has been made on the integration of nanocrystal technology with supramolecular design. Marketed cocrystal products and PK studies in human subjects are also reported.
Results
: Considering supersaturation and consequent precipitation properties is necessary when evaluating dissolution and bioavailability of cocrystals. Appropriate excipients should be included to control precipitation kinetics and to capture solubility advantage of cocrystals. Beside to solubility, cocrystals may modify membrane permeability of drugs. Therefore, cocrystals can find applications in improving oral bioavailability of poorly permeable drugs. It has been shown that cocrystals may interrupt cellular integrity of cellular monolayers which can raise toxicity concerns. Some of coformers may interact with intestinal absorption of drugs through changing intestinal blood flow, metabolism and inhibiting efflux pumps. Therefore, caution should be taken into account when conducting bioavailability studies. Nanosized cocrystals have shown a high potential towards improving absorption of poorly soluble drugs.
Conclusions
: Cocrystals have found their way from the proof-of-principle stage to the clinic. Up to now, at least two cocrystal products have gained approval from regulatory bodies. However, there are remaining challenges on safety, predicting in vivo behavior and revealing real potential of cocrystals in the human.
Keywords: Bioavailability, Cocrystal, Oral absorption, Permeability, Pharmacokinetic, Poorly soluble drug
Copyright and License Information
© 2018 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
The oral route is often the preferred mode of administration for chronic drug therapy since it is simple, cheap, non-invasive, and patient-friendly.
1
A successful drug delivery through the gastrointestinal tract (GI) requires that the drug exhibits sufficient bioavailability to be clinically effective. Poorly bioavailable drugs usually result in insufficient therapeutic efficacy with high inter-subject variability in plasma concentrations.
2,3
Bioavailability is defined as “the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action”.
4
The oral bioavailability of a drug is influenced by the physiological condition of GI and physicochemical properties of the drug. Suboptimal solubility and dissolution, chemical instability in GI fluids, inability to permeate the intestinal barrier, and extensive first-pass intestinal or hepatic metabolism are the main reasons for the poor oral bioavailability of drugs.
5
Biopharmaceutical classification system (BCS) is a unique tool which relates physicochemical properties of drugs to oral bioavailability.
6
BCS classifies drugs into 4 classes based on their solubility and permeability properties. The classes are class I (high solubility and permeability); class II (low solubility and high permeability); class III (high solubility and low permeability); and class IV (low solubility and permeability). About 40% of orally administered drug products currently on the market are estimated to exhibit low water solubility from which 30% are placed in class II and 10% in class IV. Furthermore, drug candidates in discovery pipelines with the poor aqueous solubility are increasing and class II (70%) and class IV (20%) comprise the major fraction of the candidates.
7
This increase in the number of water-insoluble compounds has been attributed to the recent advances in high-throughput screening tools that produce numerous compounds with high affinity to the biological targets but exhibiting poor biopharmaceutical properties.
8,9
Various strategies have been developed to improve oral absorption of poorly water-soluble drugs such as cocrystals,
10
amorphous solid dispersions,
11-13
liquisolid technology,
14
nanocrystal,
15
salts,
16
cyclodextrin complexation,
17
cosolvents,
18
and lipid formulation.
19
As a new crystal engineering strategy, cocrystals have opened a new avenue to modify the physicochemical properties of pharmaceutical solids. Cocrystals are defined as “solids that are crystalline single phase materials composed of two or more different molecular and/or ionic compounds generally in a stoichiometric ratio which are neither solvates nor simple salts”.
20
Non-covalent interactions such as hydrogen bonds (the main interaction), π-π interactions, halogen bonding, and van der Waals forces are responsible for the formation of cocrystals.
21
When components of a cocrystal are a drug and a pharmaceutically acceptable excipient (coformer) the resultant molecular complex is called a pharmaceutical cocrystal. The coformers usually are water-soluble molecules such as saccharin, caffeine, nicotinamide, and carboxylic acids.
22
The availability of numerous coformers makes it possible to prepare several cocrystals for each drug and select the most appropriate one. Fig. 1 shows an example of pharmaceutical cocrystals that formed between carbamazepine and saccharin. Nowadays, cocrystal screening is a routine part of solid form selection during development studies. Cocrystals can modify properties of drugs without any impact on the intrinsic pharmacological activity of the molecule. Cocrystallization has been studied to optimize physicochemical properties of drugs such as mechanical properties
23
, stability,
24
solubility,
25,26
permeability
27
and bioavailability.
28
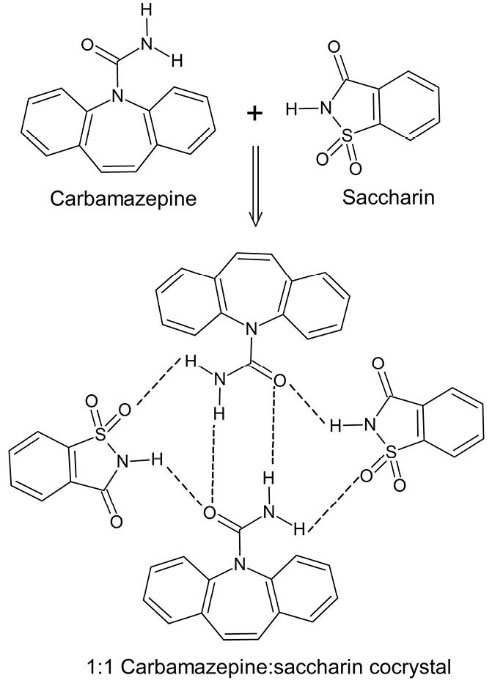
Fig. 1.
Cocrystal formation between carbamazepine and saccharin through the formation of hydrogen bonding network between the drug and coformer.
29
.
Cocrystal formation between carbamazepine and saccharin through the formation of hydrogen bonding network between the drug and coformer.
29
Improving bioavailability of poorly water-soluble drugs through cocrystallization has attracted expanding interest over recent years. In contrast to salts, the cocrystal principle can be applied to non-ionizable drugs.
30
Because of their crystalline nature, cocrystals do not exhibit physicochemical stability issues of amorphous solid dispersions. Mechanisms by which a cocrystal improves solubility is supposed to be changed lattice and solvation energies due to the presence of the coformer.
8
Improved solubility of cocrystals has been translated to higher GI absorption of cocrystals during animal studies. Shan et al
31
analyzed the correlation between solubility and pharmacokinetic (PK) parameters of cocrystals. They found that for cocrystals of class II drugs there is a strong positive correlation between in vitro solubility data and the area under the curve (AUC) of plasma drug concentration-time. Interestingly, recent studies showed that cocrystals not only can improve dissolution properties of drugs but also may modify the diffusion/permeability properties of drugs.
27,32
These results may expand the applications of cocrystals from class II drugs to class III and more importantly to class IV of BCS.
In the current manuscript, first, we present a brief information about the preparation and characterization of cocrystals then we address the different aspects of modifying GI absorption of drugs through cocrystal formation. The covered aspects include supersaturation and its effect on the oral absorption of cocrystals, the permeability of cocrystals through membranes, the effects of coformers on drug absorption, performing in vivo-in vitro correlations for cocrystals and studies conducted on human subjects. We also discuss the utility of nanococrystals for improvement of bioavailability.
Synthesis and characterization of cocrystals
Generally, cocrystals can be prepared by solution-based or solid state reactions. The solution-based techniques include slow solvent evaporation,
33
spray drying,
34
slurry crystallization,
26
and anti-solvent addition
35
while solid-state methods include neat grinding,
36
liquid assisted grinding,
37
and melt crystallization.
38
To form a cocrystal by solvent evaporation, solutions of the drug and coformer are prepared at different solvents then the solvents are removed to crystallize the intended cocrystal. Although this method is simple and easy to scale up, a major challenge for the successful production of a pure cocrystal phase by the solvent evaporation method is the separate crystallization of cocrystal components due to the difference in the solubility of components in the studied solvents.
39
Therefore, experimental conditions must be optimized carefully by conducting a mass of laboratory experiments to reach the desired product. Recent studies have shown that solid based methods are more successful in the formation of cocrystals in comparison with solution-based methods.
40
Solid-based methods represent attractive approaches for the cocrystal preparation because these methods are green, simple, highly productive, and do not exhibit the solubility problems but these methods suffer from insufficient purity, the possibility of formation of disordered crystals, and formation of polydisperse particles.
41
Taken together, most of the reported methods in the literature are difficult to translate to industrial applications and usually better suited to lab scale screening operations.
Recent trends and advances in synthesis, manufacturing, and scale-up of cocrystals have been discussed in detail by Douroumis et al.
42
The new methods should provide continuous production of high-quality cocrystals with high productivity, minimum wastage of materials, and coupled with process analytical tools for quality control. Only limited studies have been published concerning the issues of large-scale production of cocrystals. Spray drying,
34
screw extrusion,
43
and supercritical fluid technology
44
have been reported as scalable cocrystallization techniques to produce multi-kilogram batches of cocrystals.
A combination of solid-state analytical techniques is usually utilized to fully characterize the formed cocrystals. The common methods include crystallography techniques such as powder and single-crystal X-ray diffraction (PXRD and SCXRD, respectively), spectroscopic methods such as Fourier-transform infrared (FTIR) and Raman spectroscopy and solid-state nucleic magnetic resonance (solid-state NMR), and thermal methods such as polarized optical hot-stage microscopy (HSM), thermo-gravimetric analysis (TGA) and differential scanning calorimetry (DSC).
45
The crystal structure of a cocrystal can be determined by SCXRD but this technique is applicable only when a suitable single crystal can be prepared for the cocrystal of interest. The PXRD analysis is used as a fingerprint characterization technique to prove the cocrystal formation.
46
The PXRD pattern of a cocrystal exhibits unique diffraction peaks that are distinctly different in comparison to the drug and coformer. The molecular interactions between the drug and coformer, mainly as hydrogen bonding, can be evaluated by vibrational spectroscopy methods such as FTIR.
47
As the melting points of cocrystals are different from the melting points of the starting materials, therefore, thermal methods, especially DSC are of great importance in the characterization of cocrystals. In addition, DSC and TGA are valuable to evaluate the presence of solvents in the crystalline structure of cocrystals.
48
When the drug and coformer contain acidic and basic groups there is a possibility of formation of a salt instead of a cocrystal. To differentiate a cocrystal from a salt, the possibility of transfer of proton between the drug and coformer can be evaluated by solid-state NMR, SCXRD, and FTIR.
49
Effects of supersaturation/precipitation on cocrystals in vivo performance
Cocrystals of drugs with highly water-soluble coformers along with amorphous systems and salts are thermodynamically high-energy forms of drugs.
50
Higher kinetic solubility in the GI fluid is expected to achieve for drugs in these forms due to the supersaturation (Fig. 2). Supersaturation occurs when drug molecules present in solution at a concentration higher than equilibrium solubility of the thermodynamically stable form of the drug.
51
A supersaturated drug solution is thermodynamically unstable therefore it is crystallized as the stable form of the drug. The transformation leads to the rapid decrease of concentration to the solubility level of stable form (spring effect). The degree of supersaturation and its transformation rate to less soluble forms are determining the in vivo performance of supersaturable systems.
52
Furthermore, in vivo occurrence of supersaturation, precipitation, and re-dissolution of precipitate are largely dependent on physiological conditions of GI such as pH, transit time, and food effect.
53,54
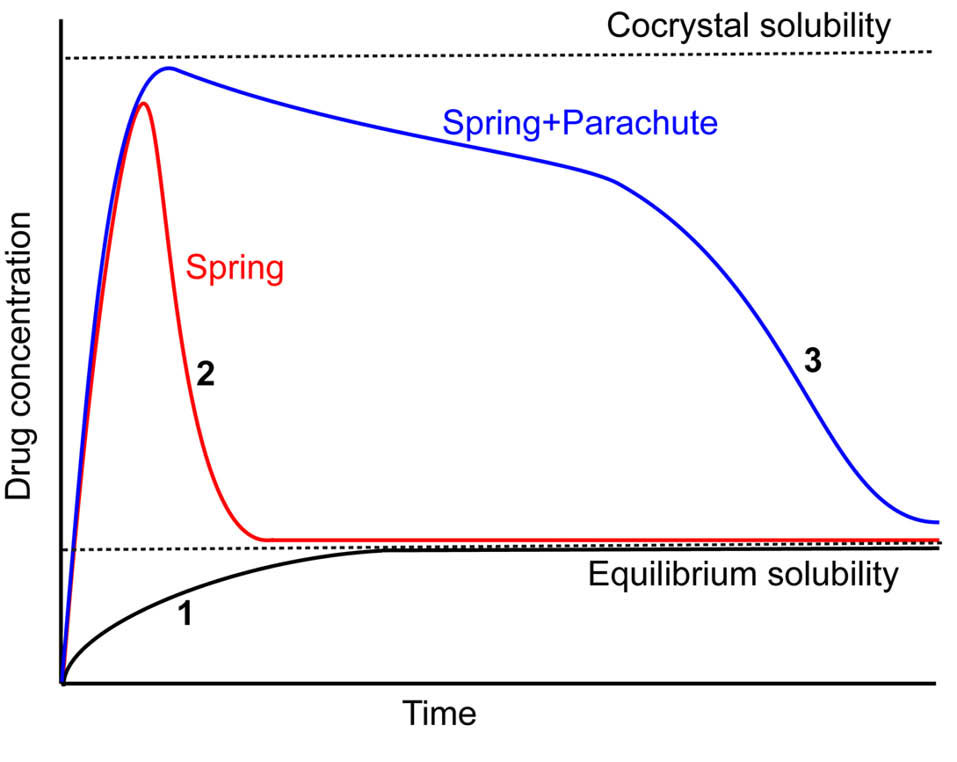
Fig. 2.
Schematic diagram of drug concentration-time profile displaying spring and spring with parachute concept. A typical dissolution profile of most stable solid phase of drug (1), a cocrystal with supersaturation/precipitation process (2), a cocrystal with crystallization inhibitor that acts as a parachute (3).
.
Schematic diagram of drug concentration-time profile displaying spring and spring with parachute concept. A typical dissolution profile of most stable solid phase of drug (1), a cocrystal with supersaturation/precipitation process (2), a cocrystal with crystallization inhibitor that acts as a parachute (3).
Box 1 summarizes important characteristics to be evaluated in the design of supersaturable cocrystals to efficiently improve oral absorption.
52,55
Ability to generate and maintain a high degree of supersaturation for sufficient period of time in vivo is the most important property. The drug plasma exposure may be significantly increased if a sustained level of supersaturated concentration has to be maintained in the GI tract for the time scale that is sufficient for absorption to take place. On the other hand, if precipitation occurs very rapidly, expected in vivo benefits may not be achieved from supersaturable formulations. Phase transformation must be evaluated and if the cocrystal exhibits rapid phase transformation, an appropriate stabilizer should be added. It is favorable that the resulted precipitate from the transformation can display higher solubility and dissolution than the stable form of the drug. From the industrial perspective, cocrystals to be selected as the final formulation option must not only provide comparable performance but also exhibit advantages over well-established solubility enabling formulation methods such as solid dispersions and nanocrystals.
Key factors to be considered in the development of supersaturable cocrystals
simple
-
► Ability to generate and maintain a high degree of supersaturation
-
► Low rate of phase transformation
-
► Higher solubility and dissolution rate of the resulted precipitate
-
► Advantages over well-established solubility enabling formulation methods
The rate of phase transformation can be controlled through the inclusion of crystallization inhibitors such as polymers and surfactants which can stabilize the supersaturated solution and reduce the rate of conversion.
56
Capturing solubility advantages of cocrystals by addition of crystallization inhibitors is called parachute effect (Fig. 2).
8
Crystallization inhibitors have widely been investigated for preventing crystallization of drug from amorphous systems
57,58
and avoiding the phase transformation of anhydrous forms to hydrate forms.
59
One of the notable differences between Cocrystals and amorphous systems is that in the case of cocrystals dissolved coformers may considerably influence the degree of supersaturation and drug transformation kinetics.
60
Only a limited number of attempts have been made to investigate the effects of different stabilizers on in vitro dissolution and in vivo performance of cocrystals.
A major fraction of cocrystals of poorly water-soluble drugs have indicated typical supersaturation/precipitation behavior during in vitro dissolution studies
61-66
however, most of the studies did not take to account this important aspect. Conventionally, PK studies of cocrystals are conducted using simple aqueous dispersions of cocrystals without conducting any additional formulation step. Although these studies reported improvements in bioavailability of drugs in cocrystal form, it seems that manipulated formulation of cocrystals is necessary to capture the full potential of cocrystals in vitro and translating the potentials to in vivo. In addition, for cocrystals that fail to attain the expected oral bioavailability improvements, performing an appropriate formulation step may lead to considerably better in vivo performance.
Dihydromyricetin (DMY) is a naturally occurring flavonoid which exhibits low solubility and permeability (class IV).
67
Cocrystals of DMY with caffeine and urea were prepared to improve the oral absorption of the compound and experiments were performed on in vitro and in vivo performances of the cocrystals.
60
In the dissolution studies (Fig. 3A), dihydromyricetin-caffeine and dihydromyricetin-urea cocrystals showed supersaturation with consequent rapid precipitation of dihydromyricetin dihydrate as the most stable form of DMY in water. The conversion rate was dependent on coformer type and precipitation rate of the dihydromyricetin-caffeine cocrystal was slower than dihydromyricetin-urea. To maintain supersaturations created by cocrystals, various crystallization inhibitors were screened, including polymers and surfactants. Among the eight excipients evaluated, polyvinyl pyrrolidone K30 (PVP K30) demonstrated the most effective inhibitory function. As it is presented in Fig. 3A, in 2.0 mg/mL of PVP K30 solution, both of the cocrystals generated approximately 6-fold higher concentrations than DMY and maintained the plateau concentration values over at least 300 minutes. Oral absorption studies in rats were conducted on cocrystals suspended in 0.5% sodium carboxymethylcellulose (NaCMC), dihydromyricetin dihydrate suspended in 0.05% NaCMC and cocrystals suspended in 2% PVP K30. Fig. 3B compares the AUC (0-6h) of different formulations. The dihydromyricetin-urea cocrystal dosed with NaCMC failed to improve the bioavailability which was attributed to a rapid solvent-mediated transformation (in less than 10 minutes) to the dihydrate form. On the other hand, when the dihydromyricetin-urea cocrystal dosed with PVP K30, a 4.9-fold enhancement in AUC(0-6h) was achieved over dihydromyricetin dihydrate. This finding was in agreement with the stabilized supersaturation state of the cocrystal in the dissolution studies. In the case of the dihydromyricetin-caffeine cocrystal, in the absence of a crystallization inhibitor, significant improvement was observed in AUC(0-6h) (3.9-fold increase). The higher bioavailability could be explained by the lower precipitation rate of this cocrystal and its ability to maintain a supersaturation to a longer period of time (about 30 min). When the supersaturation maintained by PVP K30, a 5.3-fold increase was obtained by the dihydromyricetin-caffeine cocrystal.
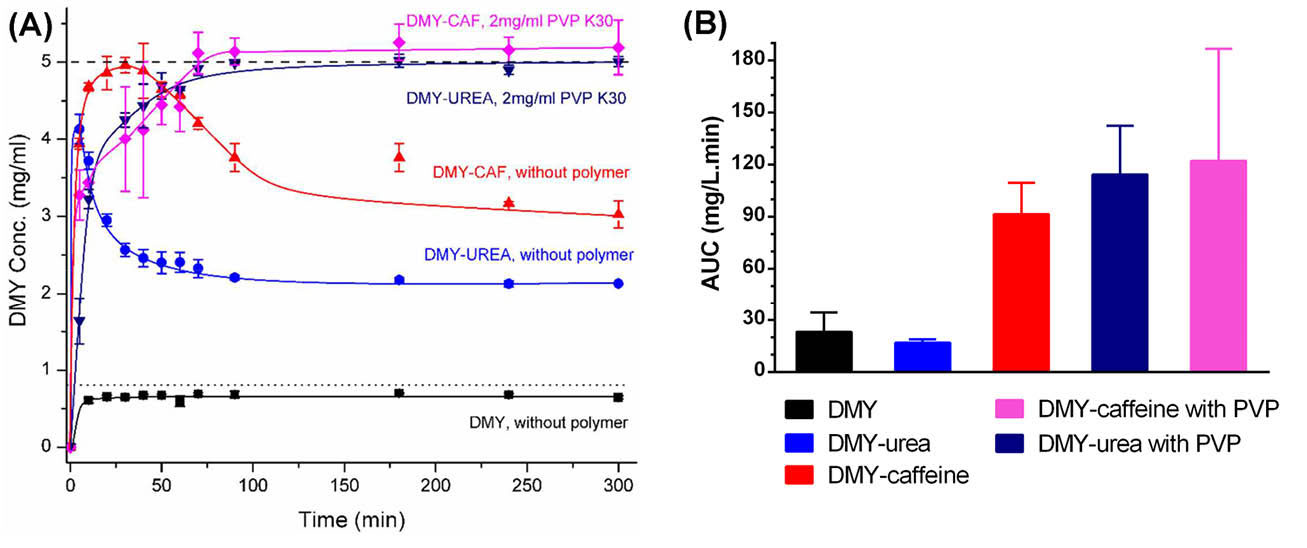
Fig. 3.
(A) In vitro powder dissolution profiles of different solid forms of DMY with or without polymer (PVP) in water at 37°C. Adapted with permission from Wang et al.
60
Copyright (2016) American Chemical Society. (B) AUC (0-6 h) of plasma concentration-time curves of different solid forms of DMY in the absence and presence of pre-dissolved PVP (2 mg/mL) administered to the rats.
.
(A) In vitro powder dissolution profiles of different solid forms of DMY with or without polymer (PVP) in water at 37°C. Adapted with permission from Wang et al.
60
Copyright (2016) American Chemical Society. (B) AUC (0-6 h) of plasma concentration-time curves of different solid forms of DMY in the absence and presence of pre-dissolved PVP (2 mg/mL) administered to the rats.
Child et al
55
have investigated the effects of crystallization inhibitors and solubilizer excipients on the in vitro dissolution properties and oral absorption of a supersaturable cocrystal of danazol. The optimized formulation consisted of the danazol-vanillin cocrystal suspended in 1% tocopheryl polyethylene glycol-1000 succinate as a solubilizer and 2% hydroxypropyl cellulose as a crystallization inhibitor. The formulated cocrystal was successful in creating a higher degree of supersaturation and maintaining supersaturation level for a longer period of time in vitro non-sink dissolution studies. The unformulated danazol-vanillin cocrystal resulted in a 1.7-fold increase in AUC (0-6h) compared to a water-insoluble solid form of danazol when administered to rats. On the other hand, the formulated cocrystal exhibited a 10-fold higher AUC (0-6h) than danazol. The results of these studies showed the importance of adding appropriate excipients to capture solubility advantages of cocrystals.
Data from several studies suggest that the physicochemical properties of drug and coformer determine the degree of supersaturation and its stability.
8
However, up to now, far too little attention has been paid to factors controlling the supersaturation level of cocrystals in vitro and in vivo. Therefore, efforts should be made to clarify the behavior of supersaturable cocrystals in the GI tract.
Membrane permeability/diffusion behavior of cocrystals
For orally administered drugs, besides solubility, sufficient permeability from membranes of the GI tract is an essential prerequisite for sufficient systemic absorption.
6
Unacceptable intestinal membrane permeability may lead to insufficient bioavailability of drugs. Permeability also affects the minimum aqueous solubility that is required for a reasonable in vivo performance.
3
The bioavailability of drugs with low rates of transfer from the intestinal membrane is likely to be impeded by limited aqueous solubility. It was anticipated that drugs with low intestinal permeability need a 20-fold higher minimum solubility than highly permeable drugs to achieve sufficient oral absorption.
68
It is estimated that about 35% of marketed drugs and 25% of drug candidates in discovery and development pipelines exhibit low membrane permeability.
7
Several strategies have been developed to tackle low permeability of drugs including structural modification or prodrug synthesis,
69
using permeation enhancers,
70
solid lipid nanoparticles,
71
and polymeric nanoparticles.
72
All of these strategies possess some obstacles. Structural modifications will necessitate preclinical efficacy and toxicology studies which are very time-consuming and costly processes. There are concerns about using permeation enhancers due to the possible membrane damaging effects. In the case of polymeric nanoparticles, their industrial uses have been restricted by difficulties in controlling manufacturing parameters, reproducibility issues and lack of regulatory guidelines. Lipid-based formulations are faced with problems related to solubility and physicochemical stability of drugs in lipidic matrix.
73
Cocrystals can improve membrane permeability/diffusion without the need for modifying the structure of drug molecule or disturbing membrane integrity. Although modifying solubility/dissolution of drugs by cocrystal formation has been extensively investigated.
8,74
Only very recent studies have reported that cocrystallization may modify membrane permeability of drugs. Table 1 summaries the representative examples of studies on the permeability of cocrystals. The studies indicated that cocrystals can improve, reduce, or do not remarkable impact on membrane permeability at all.
Table 1.
Permeability/diffusivity studies of cocrystals
|
Drug
|
Coformer
|
In vitro
Model
|
Effect on solubility (Cs)/ dissolution
|
Effect on permeability
|
Proposed mechanism of modifying permeability
|
BCS
class
|
|
TDZ
84
|
Vanillic acid |
Cellulose membrane |
4.2-fold higher Cs |
Improved flux |
Increased solubility |
- |
|
5-Fluorouracil
32
|
3-Hydroxybenzoic acid |
Silicon membrane |
No remarkable impact on Cs |
1.4-fold higher steady penetrate rate |
New supramolecular synthon formation Drug-coformer interactions Molecular packing in the crystals Lipophilicity of the coformers |
III |
|
|
4-Aminobenzoic acid |
|
No remarkable impact on Cs |
1.6-fold higher steady penetrate rate |
|
|
|
|
Cinnamic acid |
|
No remarkable impact on Cs |
1.8-fold higher steady penetrate rate |
|
|
|
Adefovir
85
|
Stearic Acid |
Caco-2 cells monolayer |
Enhanced dissolution |
No remarkable impact on the apical to basal transport rate |
Similar partition coefficient (log P) |
IV |
|
Acyclovir
76
|
Fumaric acid |
Skin of rat |
1.53-fold higher apparent Cs |
2.8-fold higher permeated amount |
Lipophilicity of the coformers (log P) Melting points of cocrystals |
IV |
|
|
Glutaric acid |
|
2.14-fold higher apparent Cs |
4-fold higher permeated amount |
|
|
|
Furosemide
77
|
Anthranilamide |
Cellulose nitrate membrane |
Higher Cs |
Higher cumulative permeated amount/flux |
Modified solubility |
IV |
|
|
Caffeine |
|
Lower apparent Cs |
Lower cumulative permeated amount/flux |
|
|
|
|
Adenine |
|
No remarkable impact on Cs |
Higher cumulative permeated amount/flux |
|
|
|
|
TMP |
|
Higher apparent Cs |
Higher cumulative permeated amount/flux |
|
|
|
Indomethacin
82
|
HMP |
NCM460 cells monolayer |
1.8-fold lower apparent Cs |
2-fold higher apparent permeability |
Drug-coformer interactions in solution Forming molecular aggregations in solution |
II |
|
|
MNA |
|
No remarkable impact on Cs |
No remarkable impact on apparent permeability |
|
|
|
|
Saccharine |
|
155-fold lower apparent Cs |
2.5-fold higher apparent permeability |
|
|
|
HCTZ
86
|
Piperazine |
Cellulose nitrate membrane |
6.6-fold higher apparent Cs |
Higher cumulative permeated amount/flux |
Drug-coformer interactions Increased solution concentration |
IV |
|
|
TMP |
|
No remarkable impact on Cs |
Lower cumulative permeated amount/flux |
|
|
|
|
Picolinamide |
|
3-fold higher apparent Cs |
Higher cumulative permeated amount/flux |
|
|
|
|
Isoniazid |
|
No remarkable impact on Cs |
Higher cumulative permeated amount/flux |
|
|
|
|
Malonamide |
|
No remarkable impact on Cs |
Higher cumulative permeated amount/flux |
|
|
|
HCTZ
27
|
Nicotinic acid |
Dialysis membrane |
1.4-fold lower apparent Cs |
2-fold higher permeability |
Drug-coformer interactions |
IV |
|
|
Nicotinamide |
|
1.3-fold higher apparent Cs |
1.8-fold higher permeability |
|
|
|
|
4-Aminobenzoic acid |
|
2.4-fold higher apparent Cs |
1.3-fold higher permeability |
|
|
|
|
Succinamide |
|
4.7-fold lower apparent Cs |
Lower permeability |
|
|
|
|
Resorcinol |
|
2.4-fold increase in Cs |
1.6-fold higher permeability |
|
|
|
Theophylline
87
|
o-Aminobenzoic acid |
Dialysis membrane |
11.9-fold higher Cs |
1.4-fold higher flux |
Drug-coformer interactions |
I |
|
|
m-Aminobenzoic acid |
|
3-fold lower Cs
|
2.4-fold lower flux |
|
|
|
|
p-Aminobenzoic acid |
|
3-fold lower Cs
|
2.4-fold lower flux |
|
|
As Table 1 indicates, most of the experiments were conducted using Franz-type diffusion cells through porous artificial membranes such as the cellulose membrane. These types of experiments assess the passive diffusion behavior of molecules through the pores filled with water instead of permeability through a biological membrane, therefore, more studies are needed to be conducted in the well-established epithelial cellular models (e.g. Caco-2, MDCK).
75
In addition, mechanisms by which cocrystals can change permeability/diffusion are still unclear. A number of researchers have proposed drug-coformer interactions in the solution phase as a possible underlying mechanism (Table 1). It has been assumed that the drug and coformer can exist as molecular aggregates in solution due to the retaining of some interactions formed in the crystalline lattice of the respective cocrystal.
By considering that BCS class IV drugs suffer from low solubility and permeability, cocrystals can be used to simultaneously increase solubility and permeability of these drugs. Some examples of drugs (class IV) with improved solubility and membrane permeability through cocrystallization strategy are acyclovir,
76
Furosemide,
77
and hydrochlorothiazide.
27
However, the potential solubility-permeability interplay should be considered when cocrystallization principle is used for bioavailability improvement of poorly water-soluble drugs.
78
The solubility-permeability has been investigated for some of the solubility enhancing approaches such as cyclodextrins,
79
amorphous solid dispersions,
80
and cosolvents.
81
The results revealed that depending on the approach used for enhancing solubility the permeability may decrease, remain unchanged, or even increase. For example, in the case of amorphous solid dispersions enhancing apparent solubility does not lead to a decrease in permeability. On the other hand, when cyclodextrin or cosolvent is used to enhance solubility, by increasing apparent solubility the permeability simultaneously is decreased.
81
Thus such studies should be performed to investigate the solubility-permeability interplay of cocrystals.
Ferretti et al
82
performed in vitro cellular permeability experiments on indomethacin, indomethacin cocrystals, and respective physical mixtures. The studied cocrystals were cocrystals of indomethacin with 2-hydroxy-4-methyl-pyridine (cocrystal 1), 2-methoxy-5-nitroaniline (cocrystal 2) and saccharine (cocrystal 3). The used cell line was the human normal colonic epithelial NCM460 cells. Fig. 4 displays a schematic representation of the obtained results. Pure indomethacin did not induce any change in the cellular integrity. Incubation of the cells monolayer with cocrystal 1 induced a considerable reduction in trans epithelial electrical resistance (TEER) showing an impaired cellular integrity. When cells were separated, the apparent permeability (Papp) of indomethacin was increased in comparison to pure indomethacin. In contrast, the physical mixture of indomethacin with 2-hydroxy-4-methyl-pyridine did not show any effect on cellular integrity and its TEER and Papp was similar to pure indomethacin. Cocrystal 2 and mixture 2 showed similar TEER and Papp with pure indomethacin. In the case of cocrystal 3 (indomethacin-saccharine), Papp improved without inducing any cellular damage. Interestingly, when cells were incubated with the indomethacin-saccharine physical mixture, tight junctions of NCM460 cells were damaged, the cells were separated and TEER was significantly reduced. The results of this study demonstrated that cocrystals and their parent physical mixtures can react very differently with biological membranes. In the subsequent work, Dalpiaz et al
83
have found that carbamazepine cocrystals and parent physical mixtures show different effects on NCM460 cells monolayer same as what has been shown for indomethacin cocrystals. The differences in behaviors of cocrystals and physical mixtures were attributed to the formation of molecular aggregates in solution.
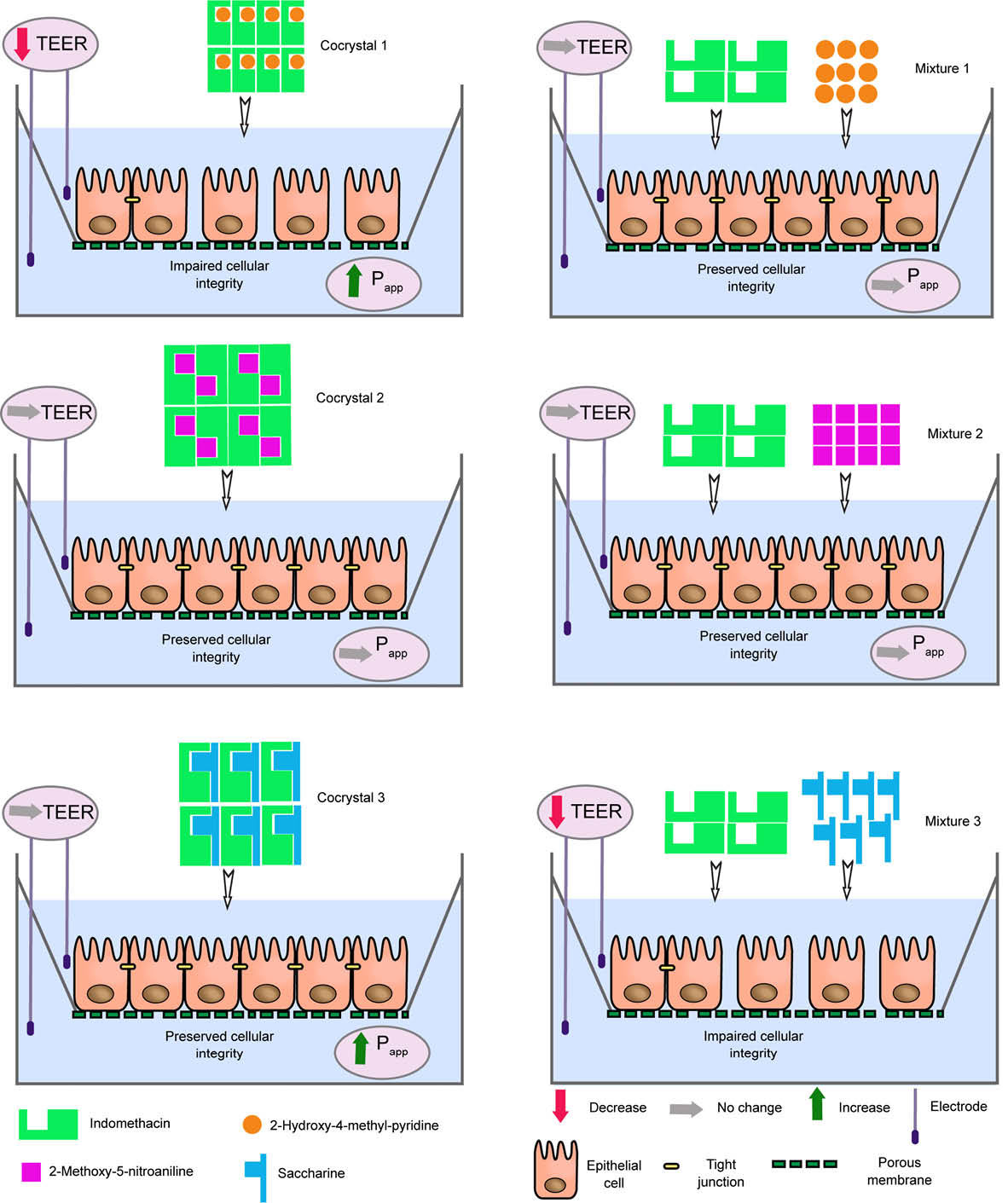
Fig. 4.
Effect of different solid forms of indomethacin on cellular integrity, apparent permeability of indomethacin (Papp), and trans epithelial electrical resistance (TEER) of in vitro cell culture monolayer (NCM460 cell monolayers). Cocrystal 1: Indomethacin-2-hydroxy-4-methylpyridine cocrystal, Cocrystal 2: Indomethacin-2-methoxy-5-nitroaniline cocrystal, Cocrystal 2: Indomethacin-saccharin cocrystal, Mixture: A physical mixture of indomethacin with the coformer. No Papp data was reported for mixture 3.
.
Effect of different solid forms of indomethacin on cellular integrity, apparent permeability of indomethacin (Papp), and trans epithelial electrical resistance (TEER) of in vitro cell culture monolayer (NCM460 cell monolayers). Cocrystal 1: Indomethacin-2-hydroxy-4-methylpyridine cocrystal, Cocrystal 2: Indomethacin-2-methoxy-5-nitroaniline cocrystal, Cocrystal 2: Indomethacin-saccharin cocrystal, Mixture: A physical mixture of indomethacin with the coformer. No Papp data was reported for mixture 3.
The effects of coformers on drug absorption
Coformers generally are considered as pharmacologically inactive materials.
88
However, recent studies have indicated that some coformers may exert biological functions. Studies on PK of cocrystals relate the changes in the PK profile of drugs mainly to the altered dissolution properties. Definitely not all, but at least some of coformers may interact with oral absorption of drugs through routes other than dissolution changes. These routes, which can be influenced by coformer presence, are metabolism, membrane transport of drugs, and physiological parameters of GI (i.e. gastric emptying time, intestinal blood flow and pH of the environment).
89
Current research on PK studies of cocrystals are focused on comparing the bioavailability of cocrystals with pure drugs and little is known about the alteration of PK of the studied drugs due to the presence of coformer molecules in the GI tract. There are only limited efforts that conducted bioavailability studies on the physical mixture of drugs with coformers to evaluate possible effects of coformers on the absorption of drugs.
A widely studied coformer in cocrystallization of pharmaceuticals is caffeine. It has been shown in the literature that co-administration of caffeine with acetaminophen
90
and ergotamine tartrate
91
lead to significantly higher plasma maximum concentration(Cmax) and AUC values for these drugs. The underlying mechanism of interaction has been attributed to improved intestinal blood flow. Therefore, caffeine can alter the PK profile of a cocrystalizing drug through the same mechanism. In this context, Zhu and coworkers
62
synthesized baicalein-caffeine cocrystal and conducted PK experiments on rats. The cocrystal, baicalein and the physical mixture were suspended in 0.1% sodium carboxymethylcellulose aqueous solution and were given to rats through gavage. The AUC24 h of baicalein-caffeine cocrystal was about 4.1 times that of pure baicalein which was attributed to the higher dissolution rate of the cocrystal. A 2-fold improvement in AUC 24 h was achieved using the physical mixture of baicalein and caffeine compared to baicalein itself (Fig. 5). This improvement was explained by the synergic effect of caffeine on baicalein absorption.
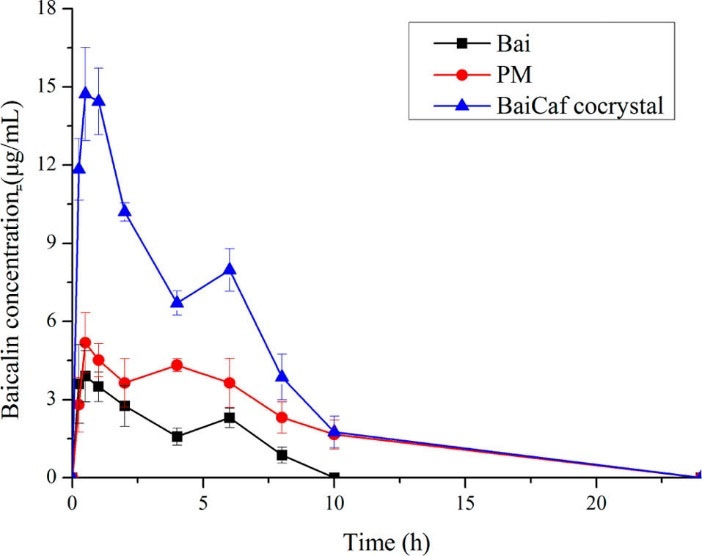
Fig. 5.
Plasma baicalin (the main active metabolite and mainly existing form of baicalein in plasma) concentrations after oral administrations of crystalline baicalein (Bai), physical mixture of baicalien with caffeine (PM) and baicalein-caffeine cocrystal (BaiCaf cocrystal) to fasted rats. Adapted with permission from Zhu et al.
62
Copyright (2017) American Chemical Society.
.
Plasma baicalin (the main active metabolite and mainly existing form of baicalein in plasma) concentrations after oral administrations of crystalline baicalein (Bai), physical mixture of baicalien with caffeine (PM) and baicalein-caffeine cocrystal (BaiCaf cocrystal) to fasted rats. Adapted with permission from Zhu et al.
62
Copyright (2017) American Chemical Society.
The first evidence of the effects of coformers on the cellular transport of drugs has been reported for adefovir dipivoxil-stearic acid cocrystal.
85
Stearic acid is a long chain fatty acid. Experiments that were conducted on Caco-2 cell line monolayers showed that stearic acid can inhibit P-glycoprotein mediated efflux of adefovir dipivoxil. However, the majority of coformers are small molecules with no known inhibitory effects on P-glycoprotein but some coformers such as polyethylene glycols and fatty acids can display inhibitory action. These examples demonstrated that some of coformers may have biopharmaceutical implications, therefore, coformers should be selected carefully. When there is a possibility of interaction, PK studies on the respective physical mixture is desirable to clarify the condition.
The PK interaction between a drug and coformer seems to be more common when dealing with drug-drug cocrystals because each of two components is a pharmacologically active agent. There is a well-known PK interaction between aspirin with meloxicam. Oral co-administration of aspirin with meloxicam to healthy volunteers increased the plasma concentration of meloxicam, by 10 and 25% higher Cmax and AUC, respectively.
92
The meloxicam-aspirin cocrystal has been synthesized and showed improved solubility and bioavailability in rats compared with pure meloxicam.
93
Unfortunately, the authors did not perform the bioavailability study on the physical mixture of meloxicam with aspirin but a portion of bioavailability improvement of cocrystal was likely originated from the PK interaction. PK interaction between components of cocrystals has also been reported for celecoxib-tramadol hydrochloride cocrystal
94
(For more details please see human PK studies section). A considerable decrease was observed in Cmax and AUC of celecoxib when celecoxib was given with tramadol hydrochloride as a combination to healthy volunteers. The mechanism of interaction was proposed to be the changing dissolution and absorption profiles of celecoxib in the presence of tramadol hydrochloride. Therefore, cautions should be taken into consideration when designing a drug-drug cocrystal as drug-drug PK interactions may occur between components of the cocrystal.
In vitro-in vivo correlation (IVIVC)
In vitro-in vivo correlation (IVIVC) refers to the establishment of a meaningful relationship between in vitro drug dissolution behavior with the in vivo PK parameters.
95
IVIVC can assign in vivo meaning to the in vitro data. It can be a useful tool for rational development and evaluation process to reduce development time and optimize the formulation.
96
Generally, IVIVC has been best suited for controlled release dosage forms where oral absorption is controlled by the release rate of the drug from the dosage form.
97
There are examples in literature for constructing IVIVC for supersaturable systems such as salts
98
and amorphous systems.
99-101
In the case of cocrystals, only a few studies have explored the correlation between the in vitro dissolution/solubility and the corresponding in vivo oral bioavailability data.
65,102
Obtaining good IVIVC for supersaturable systems is hard to achieve due to the several factors affecting supersaturation state in GI tract such as physicochemical properties of the drug, precipitation rate, solid state and particle size of precipitate, and GI physiology (pH, motility, and food effect).
103
For successful predicting of PK parameters from in vitro dissolution data, dissolution of a cocrystal in intestinal medium must be the main controlling factor of the oral absorption process. In addition, appropriate biorelevant in vitro dissolution test should be developed for the cocrystal. All in all, it is more likely to establish IVIVC for cocrystals of BCS class II drugs because these drugs are highly permeable and their absorption usually is limited by the dissolution.
AMG-517 is a candidate compound for treating chronic pain.
104
This compound has low water solubility and high permeability. Dissolution properties and PK of AMG-517 cocrystals with 12 carboxylic acid coformers were compared with AMG-517 free base.
102
Powder dissolution and intrinsic dissolution rate (IDR) studies were carried out in fasted simulated intestinal fluid (FaSIF). Cocrystals exhibited faster IDRs and different degrees of supersaturation with subsequent precipitation to free base of AMG-517 in powder dissolution studies. Bioavailability studies were conducted in rats by administering cocrystals and AMG-517 as suspensions in polyvinyl pyrrolidone (PVP) 1% aqueous solution. PK experiments indicated that higher dissolution rate of cocrystals resulted in improved absorption of the compound. Weak correlations were observed between IDRs (R2= 0.4) or apparent solubility at 15 min (R2=0.5) with plasma area under the concentration-time curves. No better correlation was found between in vitro data and other PK parameters such as Cmax. The poor IVIVC was attributed to the inherent variability within in vivo systems. Another possible reason for weak IVIVC was may be due to the fact that authors did not take into account the effect of PVP on stabilizing supersaturated condition of cocrystals at in vivo experiments.
Meloxicam, as a poorly soluble nonsteroidal anti-inflammatory drug, belongs to class II of BCS.
105
The non-sink dissolution studies of meloxicam and its 12 cocrystals with a series of aliphatic and aromatic carboxylic acid derivatives were performed in the phosphate buffer (pH 6.5).
65
The cocrystals showed the typical dissolution profile of supersaturating system with solvent-mediated phase transformation to meloxicam. In a single-dose PK study in rats,
65
most of the cocrystals displayed an improvement in the intestinal absorption of meloxicam in comparison to pure meloxicam. A linear regression analysis was performed for mean in vitro dissolution data versus mean serum concentration data at the same time point for each cocrystal and pure meloxicam. Strong linear correlations were observed between in vitro powder dissolution profiles and in vivo profile for the majority of cocrystals (R2 range 0.73–0.97). Fig. 6A indicates that serum concentration data correlated significantly with in vitro dissolution data for meloxicam and two examples of cocrystals.
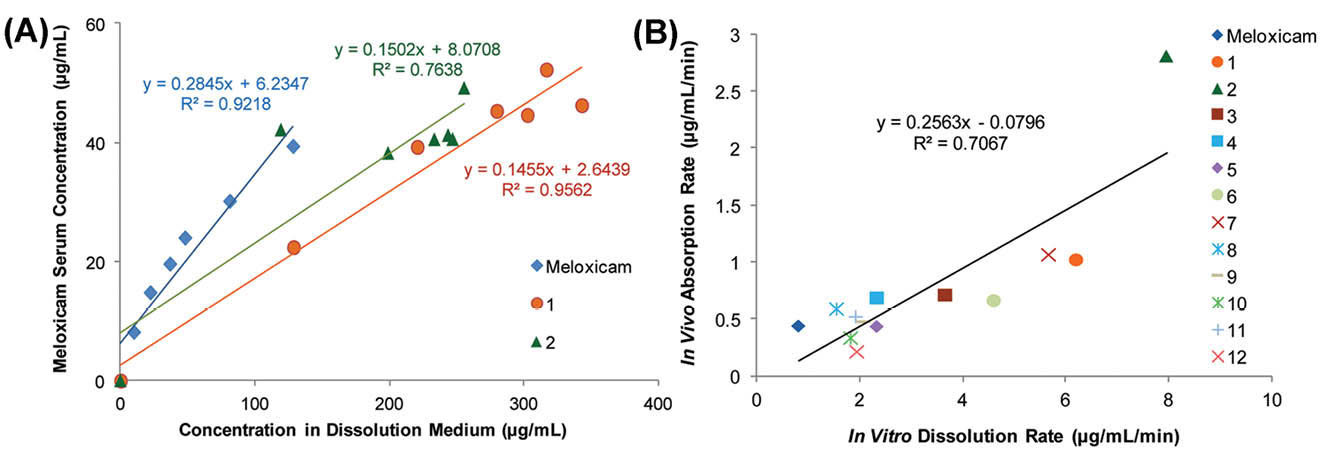
Fig. 6.
(A) Plots of mean serum concentration data versus mean in vitro dissolution data for meloxicam, meloxicam-1-hydroxy-2-naphthoic acid cocrystal (1) and meloxicam-salicylic acid cocrystal (2). (B) Plot of absorption rate versus dissolution rate of meloxicam and 12 meloxicam cocrystals (1-12). Adapted with permission from Weyna et al
65
Copyright (2012) American Chemical Society.
.
(A) Plots of mean serum concentration data versus mean in vitro dissolution data for meloxicam, meloxicam-1-hydroxy-2-naphthoic acid cocrystal (1) and meloxicam-salicylic acid cocrystal (2). (B) Plot of absorption rate versus dissolution rate of meloxicam and 12 meloxicam cocrystals (1-12). Adapted with permission from Weyna et al
65
Copyright (2012) American Chemical Society.
A correlation analysis was used to explore the relationship between dissolution and absorption rates of cocrystals (Fig. 6B). A mild linear correlation (R2= 0.71) was observed between dissolution and absorption rates. The results of the correlational analysis proposed that the meloxicam cocrystals with a higher dissolution rate would result in a better bioavailability and a reduced onset of action.
The prediction of in vivo performance of cocrystals by in vitro testing remains unclear. studies should be performed to gain knowledge about the impact of different parameters such as coformer type, composition and/or properties of the dissolution media (pH, additives) on cocrystals dissolution/solubility properties at in vitro. Various parameters such as AUC of the concentration-time curve in dissolution test, intrinsic dissolution rate, powder dissolution rate, and apparent solubility should be examined to find which parameter exhibit the best correlation with PK parameters. The contribution of the degree of supersaturation and stability of supersaturation on oral absorption of cocrystals are expected to be different for highly permeable and low permeable drugs and should be considered when constructing IVIVC.
Using nanosized cocrystals for bioavailability improvement
Nanometer-sized crystals have attracted great attention from the pharmaceutical industry. There are at least 6 pharmaceutical formulations with nanocrystal base in the market for poorly water-soluble drugs.
106
Nanocrystals improve the dissolution rate mainly by an increased surface area.
107,108
In addition, a slightly higher saturation solubility is predicted for particles with sizes lower than 1000 nm by the Ostwald-Freundlich equation.
109
While both cocrystal and nanocrystal approaches have been extensively studied for improving intestinal absorption of water-insoluble drugs, the combination of these technologies to maximize potential advantages is needed to be further investigated. A nanococrystal (NCC) can theoretically combine the dissolution rate enhancement potential of a nanocrystal with solubility improvement advantage of cocrystal which leads to improved GI absorption and efficacy of poorly water-soluble drugs. Development of NCCs is more challenging than developing nanocrystals because of differences in solubility of cocrystals components in solvents
35
and phase instability of cocrystals in the aqueous medium.
110
Generally speaking, top-down (size reduction) and bottom-up (precipitation) approaches are utilized to the synthesis of nanocrystals.
111
The majority of commercialized pharmaceutical nanocrystals have been prepared using wet milling
106
in which milling of a suspension of the drug is performed in aqueous medium containing an appropriate stabilizer. In the bottom-up techniques such as antisolvent precipitation, the precipitation of drug molecules from a solution occurred in the presence of a stabilizer.
112
The first report of preparation of pharmaceutical NCCs dates back to 2010.
113
The authors utilized caffeine-2,4-dihydroxybenzoic acid monohydrate as a model pharmaceutical cocrystal. NCCs with a mean particle size of 136 nm were prepared by a bottom-up approach. The used method was precipitation-ultrasonication using antisolvent in the presence of sorbitan oleate (Span 85) as the stabilizer. In another study,
114
NCCs of myricetin-nicotinamide have been synthesized using top-down (neat grinding) and bottom-up (antisolvent precipitation under ultrasonication) methods. The dissolution rate of NCCS prepared by precipitation method was higher than NCCs prepared by grinding.
De Smet et al
115
utilized wet milling as a typical top-down method to prepare nanosized particles of itraconazole cocrystals. Suspensions of itraconazole in combination with dicarboxylic acids were ground using milling pearls in an aqueous medium containing a surfactant (Tween 80) to form a stable nanosuspension. Among the studied coformers (maleic, adipic, glutaric and succinic acid), best results obtained when adipic acid used as coformer. Cogrinding of itraconazole with adipic acid produced a stable nanosuspension (549±51 nm) of itraconazole-adipic cocrystal with a narrow particle size distribution. The phase purity of formed NCCs was confirmed by PXRD, Raman and FTIR spectroscopy. To prevent agglomeration of NCCs during drying and facilitate re-dispersion in water, mannitol was added to the nanosuspension and then conversion of nanosuspension to a solid form was conducted by spray drying or bead layering. Nanococrystal-based formulations exhibited remarkable faster dissolution rate than commercially available amorphous formulation (Sporanox®) during in vitro dissolution studies. However, when oral bioavailability studies were conducted in dogs, different formulations did not show significant differences in PK parameters. Only, time to reach the highest plasma concentration (Tmax) of the formulated nanococrystal were half of the Sporanox® which were attributed to the higher dissolution rate of NCCs.
In a study by Karashima et al,
116
nanosuspension of carbamazepine-saccharin, indomethacin-saccharin, and furosemide-caffeine cocrystals with particle sizes lower than 300 nm were prepared using wet milling in the presence of hydroxypropyl methylcellulose and sodium dodecyl sulfate as the stabilizers. The cocrystals in the nanosuspension forms exhibited higher dissolution rates than the conventional cocrystals and pure nanocrystals of the drugs. By considering that carbamazepine, indomethacin, and furosemide have dissolution limited bioavailability, the improved dissolution rates of NCCs may result in higher bioavailability. Emami et al
117
have reported the formation of NCCs of indomethacin-saccharin by using electrospray deposition technique. NCCs with a mean size of 219 nm displayed a 3-fold higher dissolution rate than the cocrystals prepared by the solvent evaporation method.
Huang et al
118
prepared a nanosuspension of cocrystal of phenazopyridine (PAP) with phthalimide (PI) with a mean particle diameter of 21 nm by the antisolvent method under ultrasonication. The conversion of nanosuspension to solid form was performed by centrifugation. PAP as a BCS class II drug, exhibits a low water solubility (0.02 mg/mL),
119
therefore, its hydrochloride salt with a higher solubility (8.5 mg/mL) is utilized in the tablet form of this drug.
120
Phenazopyridine hydrochloride possesses some problems such as poor oral absorption due to the common ion effect of chloride in the GI tract and severe hygroscopicity under elevated humidity.
119
Formation of the PAP-PI cocrystal effectively tackled the hygroscopicity problem but the dissolution properties of cocrystal were not favorable. NCCs of PAP-PI showed higher in vitro dissolution and better bioavailability as compared to phenazopyridine hydrochloride and the PAP-PI cocrystal. When administered to rats as a suspension, the NCC showed 1.39- and 2.44-fold improvements in Cmax and oral bioavailability, respectively as compared to phenazopyridine hydrochloride.
Pharmacokinetic studies in human
Celecoxib-tramadol hydrochloride cocrystal (CTC) is a drug-drug cocrystal for treatment of acute pain that is under investigation by Esteve pharmaceutical and Mundipharma research.
121
Fig. 7 presents the molecular structure of CTC. Celecoxib and tramadol hydrochloride both are analgesic drugs with different mechanisms of action.
122,123
The purpose of development of CTC was improving pain control and providing a better safety and efficacy profile for these drugs. Tramadol hydrochloride belongs to class I of BCS and is highly soluble and highly permeable.
124
It is believed that rapid absorption of tramadol (higher peak concentrations) is related to a higher incidence of adverse effects
125
, therefore, designing a formulation with sustained release of the drug may lead to better safety profile. On the other hand, celecoxib as a member of BCS class II is a water-insoluble drug (7 µg/mL) and its bioavailability is limited because of the poor dissolution in GI fluid.
126
Almansa et al
127
identified CTC and investigated dissolution properties of the cocrystal. IDR studies depicted a 3-fold increase in celecoxib IDR and a 7-fold decrease in tramadol hydrochloride IDR in CTC than drugs alone. The modified dissolution rates of components were expected to lead to an optimized PK behavior.
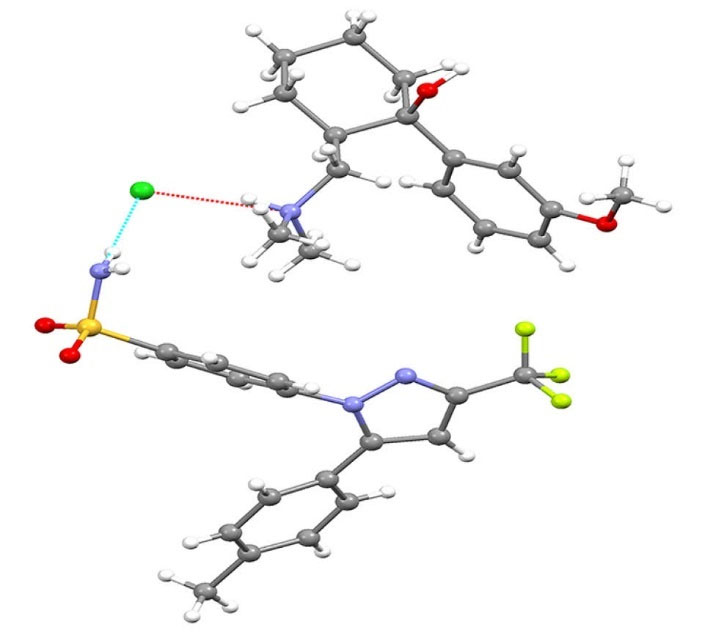
Fig. 7.
The molecular structure of celecoxib-tramadol cocrystal (CTC). The structure was created by using mercury software from the crystallographic information file of the cocrystal.
.
The molecular structure of celecoxib-tramadol cocrystal (CTC). The structure was created by using mercury software from the crystallographic information file of the cocrystal.
The oral absorbability of formulated CTC tablets in comparison to commercially available forms of tramadol hydrochloride (Adolonta® immediate-release capsules) and celecoxib (Celebrex® capsules) were evaluated and compared in beagle dogs.
121
The animal PK studies revealed that appropriately formulated CTC tablets could exhibit higher Cmax and AUC for celecoxib than Celebrex® capsules which means superior bioavailability of the cocrystal. On the other hand, in the case of tramadol hydrochloride, CTC resulted in lower peak concentration and reduced AUC than the commercial product of tramadol hydrochloride. The authors concluded that CTC exhibited a potential to simultaneously improve the bioavailability of celecoxib and safety of tramadol hydrochloride.
Videla et al
94
conducted a phase I single-dose PK study in healthy volunteers to evaluate the performance of CTC. The subjects received Celebrex® capsules (100 mg celecoxib), Adolonta® immediate-release capsules (100 mg tramadol hydrochloride), CTC formulated tablet (88 mg tramadol hydrochloride and 112 mg celecoxib) and the open combination of Celebrex® and Adolonta® (100 mg tramadol hydrochloride + 100 mg celecoxib). The Cmax values of tramadol were 263.23, 345.78, and 349.38 ng/mL from CTC, Adolonta® and tramadol plus celecoxib, respectively. The reduced Cmax of tramadol in the cocrystal form was attributed to the lower dissolution rate of tramadol from CTC which was observed at in vitro dissolution tests. After the dose correction, all three treatments showed similar AUC values for tramadol. Combining tramadol with celecoxib did not exhibit a significant effect on the concentration profile of tramadol.
The results were quite different for celecoxib. Co-administration of tramadol with celecoxib remarkably reduced oral bioavailability of celecoxib that may be due to the negative effect of tramadol on dissolution or absorption of celecoxib. While celecoxib in the form of cocrystal failed to exhibit higher Cmax or AUC than Celebrex® but the oral absorption of celecoxib from CTC was better than Celebrex® plus Adolonta®. After a single dose of the cocrystal, Tmax of celecoxib was remarkably lower compared to Celebrex® and the combination (1.50 hours versus 2.33 and 3 hours) which could be explained by improved dissolution of celecoxib in the form of cocrystal.
The results obtained for celecoxib from human PK studies were in disagreement with animal studies in beagle dogs. The animal studies indicated that CTC resulted in an improvement of celecoxib bioavailability compared to Celebrex®. Taken together, however, CTC was successful in the modifying PK profile of tramadol to access better safety profile but failed to improve oral bioavailability of celecoxib.
As another example of studies on the bioavailability of cocrystals in human, Golob and coworkers
128
reported vinpocetine-boric acid ionic cocrystal which remarkably improved in vitro powder dissolution in comparison to pure vinpocetine. The authors conducted the in vivo oral absorption studies in healthy human subjects. The cocrystal and pure vinpocetine were administered as a single dose of 10 mg of vinpocetine in hard gelatin capsule form. The cocrystal resulted in an approximately 2-fold increase in the oral bioavailability of vinpocetine in humans.
To date, most of PK studies have been conducted on animal models and only very limited studies have investigated in vivo performance of cocrystals on human subjects. However, extrapolating animal data to human has come under question recently.
129
Therefore, further studies should be conducted on humans to confirm the real potential of cocrystals.
Marketed cocrystals and cocrystals under clinical development
In less than two decades since research has been started in cocrystal field, pharmaceutical cocrystals have found their way from the bench (preclinical studies) to the bedside (approved drug products). Currently, two cocrystal formulations that are available in the market include Entresto™ (valsartan-sacubitril) by Novartis
130
and Suglat® (ipragliflozin-L-proline) by Astellas pharma and Kotobuki pharmaceutical.
131
Entresto™ tablet contains a drug-drug ionic cocrystal of monosodium sacubitril (neprilysin inhibitor), disodium valsartan (angiotensin receptor blocker) and a water molecule (Fig. 8).
132
This drug product was approved by the Food and Drug Administration and European Medicines Agency in 2015 to treat chronic heart failure.
133
PK studies demonstrated that bioavailability of valsartan in cocrystal form (Entresto™) was higher than reference valsartan.
94
The Suglat® tablet has been approved in 2014 in Japan for treatment of diabetes mellitus but has not gained approval in the United States or the EU.
134
The cocrystal systems that are under clinical development include TAK-020 Cocrystal,
135
tramadol-celecoxib
127
and ertugliflozin-L-pyroglutamic acid.
136
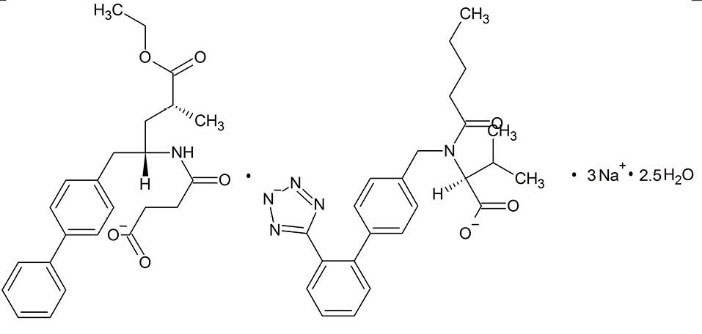
Fig. 8.
Structural formula of EntrestoTM.
.
Structural formula of EntrestoTM.
Concluding remarks
Nowadays, Cocrystals are finding their place in crystal engineering field to become a well-established part of solid screening during development stages. Significant progress has been made on cocrystal synthesis, characterization, and scale-up methods. Various studies have indicated the potentials of cocrystals to improve problematic properties of drugs. Regulatory bodies published guidelines for industry and some cocrystal products have already entered the market such as Entresto™ and Suglat®.
In this review, we have attempted to discuss findings from recent studies on different aspects of oral absorption of cocrystals. Cocrystals, as supersaturable drug delivery systems, can significantly improve solubility. However, translating in vitro improved solubility results to in vivo is difficult due to the instability of supersaturated state and its rapid precipitation and, albeit different condition of GI. Studies showed that cocrystallization may change the permeability of drugs through membranes which may be useful for low permeable drugs. The current belief is that the components of cocrystal are completely dissociated in solution and cocrystallization does not change the pharmacological profile of the parent drug. This belief has been questioned by some studies that showed cocrystals can act very differently than respective physical mixtures on biological systems which may raise toxicity concerns about utilizing cocrystals for improving bioavailability without performing appropriate evaluations. In addition, some coformers may interact with oral absorption of drugs by routes such as altering intestinal blood flow and inhibiting efflux pumps.
Integration of cocrystal principle with reducing the size to nanometer ranges may be necessary for drugs that each of this technologies is failed to achieve sufficient bioavailability. From an industrial perspective, cocrystals must show better in vivo performance than other conventional solubility enabling strategies to be selected by the industry as the final formulation option. Further studies should be performed in human subjects to reveal the advantages of cocrystals. There are remaining challenges on the prediction of in vivo behavior of cocrystals and IVIVC for cocrystals is still not yet established.
Acknowledgments
The authors acknowledge that this article is a part of the Ph.D. thesis No.103 registered in the Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran.
Funding sources
The financial support (grant No. 52/1533) from Vice Chancellor for Research of Tabriz University of Medical Sciences is gratefully acknowledged.
Ethical statement
None to be declared.
Competing interests
Authors declare no conflict of interests.
Authors contribution
MBJ conceived the original idea and supervised the project. MRS supervised the project and aided in interpretation of data. KA contributed to the conceptualization of the manuscript and to the overall writing and editing of the manuscript. SE collected the data and drafted the manuscript. All authors discussed the contents and contributed to the final manuscript.
Review Highlights
What is the current knowledge?
simple
-
√ Cocrystals can be used to modify physicochemical
properties of drugs.
-
√ PK studies indicated that Cocrystallization is a promising
approach for improvement of oral bioavailability of poorly
soluble drugs.
What is new here?
simple
-
√ Supersaturation/precipitation plays a crucial role in
bioavailability of cocrystals.
-
√ Cocrystallization may modify membrane permeability of
drugs.
-
√ Some of coformers can interact with intestinal absorption
of drugs.
-
√ Nanococrystals combine the benefits of nanocrystal
technology and supramolecular design.
References
- Sastry SV, Nyshadham JR, Fix JA. Recent technological advances in oral drug delivery–a review. Pharm Sci Technolo Today 2000; 3:138-45. doi: 10.1016/s1461-5347(00)00247-9 [Crossref] [ Google Scholar]
- Hellriegel ET, Bjornsson TD, Hauck WW. Interpatient variability in bioavailability is related to the extent of absorption: implications for bioavailability and bioequivalence studies. Clin Pharmacol Ther 1996; 60:601-7. doi: 10.1016/s0009-9236(96)90208-8 [Crossref] [ Google Scholar]
- Aungst BJ. Optimizing Oral Bioavailability in Drug Discovery: An Overview of Design and Testing Strategies and Formulation Options. J Pharm Sci 2017; 106:921-9. doi: 10.1016/j.xphs.2016.12.002 [Crossref] [ Google Scholar]
-
Food, Administration D. Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products—general considerations. Food and Drug Administration, Washington, DC 2003.
- Chan OH, Stewart BH. Physicochemical and drug-delivery considerations for oral drug bioavailability. Drug Discov Today 1996; 1:461-73. doi: 10.1016/1359-6446(96)10039-8 [Crossref] [ Google Scholar]
- Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 1995; 12:413-20. doi: 10.1208/s12248-014-9620-9 [Crossref] [ Google Scholar]
- Thayer AM. Finding solutions. Chem Eng News 2010; 88:13-8. doi: 10.1021/cen-v088n022.p013 [Crossref] [ Google Scholar]
- Babu NJ, Nangia A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst Growth Des 2011; 11:2662-79. doi: 10.1021/cg200492w [Crossref] [ Google Scholar]
- Emami S, Jouyban A, Valizadeh H, Shayanfar A. Are Crystallinity Parameters Critical for Drug Solubility Prediction?. J Solution Chem 2015; 44:2297-315. doi: 10.1007/s10953-015-0410-5 [Crossref] [ Google Scholar]
- Shayanfar A, Jouyban A. Physicochemical characterization of a new cocrystal of ketoconazole. Powder Technol 2014; 262:242-8. doi: 10.1016/j.powtec.2014.04.072 [Crossref] [ Google Scholar]
- Emami S, Valizadeh H, Islambulchilar Z, Zakeri-Milani P. Development and physicochemical characterization of sirolimus solid dispersions prepared by solvent evaporation method. Adv Pharm Bull 2014; 4:369. doi: 10.5681/apb.2014.054 [Crossref] [ Google Scholar]
- Barzegar-Jalali M, Ghanbarzadeh S, Adibkia K, Valizadeh H, Bibak S, Mohammadi G. Development and characterization of solid dispersion of piroxicam for improvement of dissolution rate using hydrophilic carriers. Bioimpacts 2014; 4:141-8. doi: 10.15171/bi.2014.007 [Crossref] [ Google Scholar]
- Barzegar-Jalali M, Valizadeh H, Shadbad M-RS, Adibkia K, Mohammadi G, Farahani A. Cogrinding as an approach to enhance dissolution rate of a poorly water-soluble drug (gliclazide). Powder Technol 2010; 197:150-8. doi: 10.1016/j.powtec.2009.09.008 [Crossref] [ Google Scholar]
- Azharshekoufeh BL, Shokri J, Adibkia K, Javadzadeh Y. Liquisolid technology: What it can do for NSAIDs delivery?. Colloids Surf B Biointerfaces 2015; 136:185-91. doi: 10.1016/j.colsurfb.2015.09.014 [Crossref] [ Google Scholar]
- Ige PP, Baria RK, Gattani SG. Fabrication of fenofibrate nanocrystals by probe sonication method for enhancement of dissolution rate and oral bioavailability. Colloids Surf B Biointerfaces 2013; 108:366-73. doi: 10.1016/j.colsurfb.2013.02.043 [Crossref] [ Google Scholar]
- Serajuddin AT. Salt formation to improve drug solubility. Adv Drug Deliv Rev 2007; 59:603-16. doi: 10.1016/j.addr.2007.05.010 [Crossref] [ Google Scholar]
- Rawat S, Jain SK. Solubility enhancement of celecoxib using β-cyclodextrin inclusion complexes. Eur J Pharm Biopharm 2004; 57:263-7. doi: 10.1016/j.ejpb.2003.10.020 [Crossref] [ Google Scholar]
- Vahdati S, Shayanfar A, Hanaee J, Martínez F, Acree Jr WE, Jouyban A. Solubility of carvedilol in ethanol+ propylene glycol mixtures at various temperatures. Ind Eng Chem Res 2013; 52:16630-6. doi: 10.1021/ie403054z [Crossref] [ Google Scholar]
- Luo Y, Chen D, Ren L, Zhao X, Qin J. Solid lipid nanoparticles for enhancing vinpocetine's oral bioavailability. J Control Release 2006; 114:53-9. doi: 10.1016/j.jconrel.2006.05.010 [Crossref] [ Google Scholar]
- Aitipamula S, Banerjee R, Bansal AK, Biradha K, Cheney ML, Choudhury AR. Polymorphs, salts, and cocrystals: What’s in a name?. Cryst Growth Des 2012; 12:2147-52. doi: 10.1021/cg3002948 [Crossref] [ Google Scholar]
- Lara-Ochoa F, Espinosa-Pérez G. Cocrystals definitions. Supramol Chem 2007; 19:553-7. doi: 10.1080/10610270701501652 [Crossref] [ Google Scholar]
- Cysewski P, Przybylek M. Selection of effective cocrystals former for dissolution rate improvement of active pharmaceutical ingredients based on lipoaffinity index. Eur J Pharm Sci 2017; 107:87-96. doi: 10.1016/j.ejps.2017.07.004 [Crossref] [ Google Scholar]
- Karki S, Friščić T, Fábián L, Laity PR, Day GM, Jones W. Improving mechanical properties of crystalline solids by cocrystal formation: new compressible forms of paracetamol. Adv Mater 2009; 21:3905-9. doi: 10.1002/adma.200900533 [Crossref] [ Google Scholar]
- Trask AV, Motherwell WS, Jones W. Physical stability enhancement of theophylline via cocrystallization. Int J Pharm 2006; 320:114-23. doi: 10.1016/j.ijpharm.2006.04.018 [Crossref] [ Google Scholar]
- Shayanfar A, Asadpour-Zeynali K, Jouyban A. Solubility and dissolution rate of a carbamazepine–cinnamic acid cocrystal. J Mol Liq 2013; 187:171-6. doi: 10.1016/j.molliq.2013.06.015 [Crossref] [ Google Scholar]
- Emami S, Adibkia K, Barzegar-Jalali M, Siahi-Shadbad M. Piroxicam cocrystals with phenolic coformers: preparation, characterization, and dissolution properties. Pharm Dev Technol 2018:1-12. doi: 10.1080/10837450.2018.1455210 [Crossref]
- Sanphui P, Devi VK, Clara D, Malviya N, Ganguly S, Desiraju GR. Cocrystals of hydrochlorothiazide: solubility and diffusion/permeability enhancements through drug–coformer interactions. Mol Pharm 2015; 12:1615-22. doi: 10.1021/acs.molpharmaceut.5b00020 [Crossref] [ Google Scholar]
- McNamara DP, Childs SL, Giordano J, Iarriccio A, Cassidy J, Shet MS. Use of a glutaric acid cocrystal to improve oral bioavailability of a low solubility API. Pharm Res 2006; 23:1888-97. doi: 10.1007/s11095-006-9032-3 [Crossref] [ Google Scholar]
- Hickey MB, Peterson ML, Scoppettuolo LA, Morrisette SL, Vetter A, Guzmán H. Performance comparison of a co-crystal of carbamazepine with marketed product. Eur J Pharm Biopharm 2007; 67:112-9. doi: 10.1016/j.ejpb.2006.12.016 [Crossref] [ Google Scholar]
- Xu J, Huang Y, Ruan S, Chi Z, Qin K, Cai B. Cocrystals of isoliquiritigenin with enhanced pharmacokinetic performance. CrystEngComm 2016; 18:8776-86. doi: 10.1039/C6CE01809B [Crossref] [ Google Scholar]
- Shan N, Perry ML, Weyna DR, Zaworotko MJ. Impact of pharmaceutical cocrystals: the effects on drug pharmacokinetics. Expert Opin Drug Metab Toxicol 2014; 10:1255-71. doi: 10.1517/17425255.2014.942281 [Crossref] [ Google Scholar]
- Dai X-L, Li S, Chen J-M, Lu T-B. Improving the membrane permeability of 5-fluorouracil via cocrystallization. Cryst Growth Des 2016; 16:4430-8. doi: 10.1021/acs.cgd.6b00552 [Crossref] [ Google Scholar]
- Weyna DR, Shattock T, Vishweshwar P, Zaworotko MJ. Synthesis and Structural Characterization of Cocrystals and Pharmaceutical Cocrystals: Mechanochemistry vs Slow Evaporation from Solution. Cryst Growth Des 2009; 9:1106-23. doi: 10.1021/cg800936d [Crossref] [ Google Scholar]
- Patil SP, Modi SR, Bansal AK. Generation of 1:1 Carbamazepine:Nicotinamide cocrystals by spray drying. Eur J Pharm Sci 2014; 62:251-7. doi: 10.1016/j.ejps.2014.06.001 [Crossref] [ Google Scholar]
- Lee M-J, Chun N-H, Wang I-C, Liu JJ, Jeong M-Y, Choi GJ. Understanding the formation of indomethacin–saccharin cocrystals by anti-solvent crystallization. Cryst Growth Des 2013; 13:2067-74. doi: 10.1021/cg400135a [Crossref] [ Google Scholar]
- Friščić T, Jones W. Recent Advances in Understanding the Mechanism of Cocrystal Formation via Grinding. Cryst Growth Des 2009; 9:1621-37. doi: 10.1021/cg800764n [Crossref] [ Google Scholar]
- Bysouth SR, Bis JA, Igo D. Cocrystallization via planetary milling: enhancing throughput of solid-state screening methods. Int J Pharm 2011; 411:169-71. doi: 10.1016/j.ijpharm.2011.03.037 [Crossref] [ Google Scholar]
- Yan Y, Chen J-M, Lu T-B. Thermodynamics and preliminary pharmaceutical characterization of a melatonin-pimelic acid cocrystal prepared by a melt crystallization method. CrystEngComm 2015; 17:612-20. doi: 10.1039/C4CE01921K [Crossref] [ Google Scholar]
- Rager T, Hilfiker R. Cocrystal Formation from Solvent Mixtures. Cryst Growth Des 2010; 10:3237-41. doi: 10.1021/cg100361y [Crossref] [ Google Scholar]
- Fucke K, Myz SA, Shakhtshneider TP, Boldyreva EV, Griesser UJ. How good are the crystallisation methods for co-crystals? A comparative study of piroxicam. New J Chem 2012; 36:1969-77. doi: 10.1039/C2NJ40093F [Crossref] [ Google Scholar]
- Duarte I, Andrade R, Pinto JF, Temtem M. Green production of cocrystals using a new solvent-free approach by spray congealing. Int J Pharm 2016; 506:68-78. doi: 10.1016/j.ijpharm.2016.04.010 [Crossref] [ Google Scholar]
- Douroumis D, Ross SA, Nokhodchi A. Advanced methodologies for cocrystal synthesis. Adv Drug Deliv Rev 2017; 117:178-95. doi: 10.1016/j.addr.2017.07.008 [Crossref] [ Google Scholar]
- Moradiya HG, Islam MT, Scoutaris N, Halsey SA, Chowdhry BZ, Douroumis D. Continuous Manufacturing of High Quality Pharmaceutical Cocrystals Integrated with Process Analytical Tools for In-Line Process Control. Cryst Growth Des 2016; 16:3425-34. doi: 10.1021/acs.cgd.6b00402 [Crossref] [ Google Scholar]
- Padrela L, Rodrigues MA, Velaga SP, Matos HA, de Azevedo EG. Formation of indomethacin-saccharin cocrystals using supercritical fluid technology. Eur J Pharm Sci 2009; 38:9-17. doi: 10.1016/j.ejps.2009.05.010 [Crossref] [ Google Scholar]
- Healy AM, Worku ZA, Kumar D, Madi AM. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv Drug Deliv Rev 2017; 117:25-46. doi: 10.1016/j.addr.2017.03.002 [Crossref] [ Google Scholar]
- Li P, Chu Y, Wang L, Wenslow RM, Yu K, Zhang H. Structure determination of the theophylline-nicotinamide cocrystal: a combined powder XRD, 1D solid-state NMR, and theoretical calculation study. CrystEngComm 2014; 16:3141-7. doi: 10.1039/C4CE00012A [Crossref] [ Google Scholar]
- Brittain HG. Brittain HGVibrational Spectroscopic Studies of Cocrystals and Salts2The Benzylamine−Benzoic Acid System. Cryst Growth Des 2009; 9:3497-503. doi: 10.1021/cg9001972 [Crossref] [ Google Scholar]
- Izutsu KI, Koide T, Takata N, Ikeda Y, Ono M, Inoue M. Characterization and Quality Control of Pharmaceutical Cocrystals. Chem Pharm Bull (Tokyo) 2016; 64:1421-30. doi: 10.1248/cpb.c16-00233 [Crossref] [ Google Scholar]
- Mohamed S, Tocher DA, Price SL. Computational prediction of salt and cocrystal structures--does a proton position matter?. Int J Pharm 2011; 418:187-98. doi: 10.1016/j.ijpharm.2011.03.063 [Crossref] [ Google Scholar]
- Kawakami K. Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. Adv Drug Deliv Rev 2012; 64:480-95. doi: 10.1016/j.addr.2011.10.009 [Crossref] [ Google Scholar]
- Kawakami K. Theory and practice of supersaturatable formulations for poorly soluble drugs. Ther Deliv 2015; 6:339-52. doi: 10.4155/tde.14.116 [Crossref] [ Google Scholar]
- Hisada N, Takano R, Takata N, Shiraki K, Ueto T, Tanida S. Characterizing the dissolution profiles of supersaturable salts, cocrystals, and solvates to enhance in vivo oral absorption. Eur J Pharm Biopharm 2016; 103:192-9. doi: 10.1016/j.ejpb.2016.04.004 [Crossref] [ Google Scholar]
- Mitra A, Fadda H. Effect of surfactants, gastric emptying, and dosage form on supersaturation of dipyridamole in an in vitro model simulating the stomach and duodenum. Mol Pharm 2014; 11:2835-44. doi: 10.1021/mp500196f [Crossref] [ Google Scholar]
- Bevernage J, Brouwers J, Clarysse S, Vertzoni M, Tack J, Annaert P. Drug supersaturation in simulated and human intestinal fluids representing different nutritional states. J Pharm Sci 2010; 99:4525-34. doi: 10.1002/jps.22154 [Crossref] [ Google Scholar]
- Childs SL, Kandi P, Lingireddy SR. Formulation of a danazol cocrystal with controlled supersaturation plays an essential role in improving bioavailability. Mol Pharm 2013; 10:3112-27. doi: 10.1021/mp400176y [Crossref] [ Google Scholar]
- Alhalaweh A, Ali HRH, Velaga SP. Effects of polymer and surfactant on the dissolution and transformation profiles of cocrystals in aqueous media. Cryst Growth Des 2013; 14:643-8. doi: 10.1021/cg4015256 [Crossref] [ Google Scholar]
- Konno H, Taylor LS. Influence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine. J Pharm Sci 2006; 95:2692-705. doi: 10.1002/jps.20697 [Crossref] [ Google Scholar]
- Wegiel LA, Mauer LJ, Edgar KJ, Taylor LS. Crystallization of amorphous solid dispersions of resveratrol during preparation and storage—Impact of different polymers. J Pharm Sci 2013; 102:171-84. doi: 10.1002/jps.23358 [Crossref] [ Google Scholar]
- Gift AD, Luner PE, Luedeman L, Taylor LS. Influence of polymeric excipients on crystal hydrate formation kinetics in aqueous slurries. J Pharm Sci 2008; 97:5198-211. doi: 10.1002/jps.21379 [Crossref] [ Google Scholar]
- Wang C, Tong Q, Hou X, Hu S, Fang J, Sun CC. Enhancing bioavailability of dihydromyricetin through inhibiting precipitation of soluble cocrystals by a crystallization inhibitor. Cryst Growth Des 2016; 16:5030-9. doi: 10.1021/acs.cgd.6b00591 [Crossref] [ Google Scholar]
- Wang J-R, Wang X, Yang Y, Chen X, Mei X. Solid-state characterization of 17β-estradiol co-crystals presenting improved dissolution and bioavailability. CrystEngComm 2016; 18:3498-505. doi: 10.1039/c6ce00433d [Crossref] [ Google Scholar]
- Zhu B, Zhang Q, Wang J-R, Mei X. Cocrystals of Baicalein with Higher Solubility and Enhanced Bioavailability. Cryst Growth Des 2017; 17:1893-901. doi: 10.1021/acs.cgd.6b01863 [Crossref] [ Google Scholar]
- Wang J-R, Yu X, Zhou C, Lin Y, Chen C, Pan G. Improving the dissolution and bioavailability of 6-mercaptopurine via co-crystallization with isonicotinamide. Bioorg Med Chem Lett 2015; 25:1036-9. doi: 10.1016/j.bmcl.2015.01.022 [Crossref] [ Google Scholar]
- Chen Y, Li L, Yao J, Ma Y-Y, Chen J-M, Lu T-B. Improving the solubility and bioavailability of apixaban via apixaban–oxalic acid cocrystal. Cryst Growth Des 2016; 16:2923-30. doi: 10.1021/acs.cgd.6b00266 [Crossref] [ Google Scholar]
- Weyna DR, Cheney ML, Shan N, Hanna M, Zaworotko MJ, Sava V. Improving solubility and pharmacokinetics of meloxicam via multiple-component crystal formation. Mol Pharm 2012; 9:2094-102. doi: 10.1021/mp300169c [Crossref] [ Google Scholar]
- He H, Zhang Q, Li M, Wang J-R, Mei X. Modulating the Dissolution and Mechanical Properties of Resveratrol by Cocrystallization. Cryst Growth Des 2017. doi: 10.1021/acs.cgd.7b00637 [Crossref]
- Hou X, Tong Q, Wang W, Xiong W, Shi C, Fang J. Dihydromyricetin protects endothelial cells from hydrogen peroxide-induced oxidative stress damage by regulating mitochondrial pathways. Life Sci 2015; 130:38-46. doi: 10.1016/j.lfs.2015.03.007 [Crossref] [ Google Scholar]
- Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 2000; 44:235-49. doi: 10.1016/s1056-8719(00)00107-6 [Crossref] [ Google Scholar]
- Varghese Gupta S, Gupta D, Sun J, Dahan A, Tsume Y, Hilfinger J. Enhancing the intestinal membrane permeability of zanamivir: a carrier mediated prodrug approach. Mol Pharm 2011; 8:2358-67. doi: 10.1021/mp200291x [Crossref] [ Google Scholar]
- Whitehead K, Karr N, Mitragotri S. Safe and effective permeation enhancers for oral drug delivery. Pharm Res 2008; 25:1782-8. doi: 10.1007/s11095-007-9488-9 [Crossref] [ Google Scholar]
- Abuasal BS, Lucas C, Peyton B, Alayoubi A, Nazzal S, Sylvester PW. Enhancement of intestinal permeability utilizing solid lipid nanoparticles increases γ-tocotrienol oral bioavailability. Lipids 2012; 47:461-9. [ Google Scholar]
- Varma MV, Panchagnula R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivo. Eur J Pharm Sci 2005; 25:445-53. doi: 10.1016/j.ejps.2005.04.003 [Crossref] [ Google Scholar]
- Ghadi R, Dand N. BCS class IV drugs: Highly notorious candidates for formulation development. J Control Release 2017. doi: 10.1016/j.jconrel.2017.01.014 [Crossref]
- Thakuria R, Delori A, Jones W, Lipert MP, Roy L, Rodríguez-Hornedo N. Pharmaceutical cocrystals and poorly soluble drugs. Int J Pharm 2013; 453:101-25. doi: 10.1016/j.ijpharm.2012.10.043 [Crossref] [ Google Scholar]
- Joubert R, Steyn JD, Heystek HJ, Steenekamp JH, Du Preez JL, Hamman JH. In vitro oral drug permeation models: the importance of taking physiological and physico-chemical factors into consideration. Expert Opin Drug Deliv 2017; 14:179-87. doi: 10.1080/17425247.2016.1211639 [Crossref] [ Google Scholar]
- Yan Y, Chen J-M, Lu T-B. Simultaneously enhancing the solubility and permeability of acyclovir by crystal engineering approach. CrystEngComm 2013; 15:6457-60. doi: 10.1039/c3ce41017j [Crossref] [ Google Scholar]
- Banik M, Gopi SP, Ganguly S, Desiraju GR. Cocrystal and salt forms of furosemide: solubility and diffusion variations. Cryst Growth Des 2016; 16:5418-28. doi: 10.1021/acs.cgd.6b00902 [Crossref] [ Google Scholar]
- Dahan A, Beig A, Lindley D, Miller JM. The solubility–permeability interplay and oral drug formulation design: Two heads are better than one. Adv Drug Deliv Rev 2016; 101:99-107. doi: 10.1016/j.addr.2016.04.018 [Crossref] [ Google Scholar]
- Beig A, Miller JM, Lindley D, Carr RA, Zocharski P, Agbaria R. Head‐To‐Head Comparison of Different Solubility‐Enabling Formulations of Etoposide and Their Consequent Solubility–Permeability Interplay. J Pharm Sci 2015; 104:2941-7. doi: 10.1002/jps.24496 [Crossref] [ Google Scholar]
- Miller JM, Beig A, Carr RA, Spence JK, Dahan A. A win–win solution in oral delivery of lipophilic drugs: supersaturation via amorphous solid dispersions increases apparent solubility without sacrifice of intestinal membrane permeability. Mol Pharm 2012; 9:2009-16. doi: 10.1021/mp300104s [Crossref] [ Google Scholar]
- Miller JM, Beig A, Carr RA, Webster GK, Dahan A. The solubility–permeability interplay when using cosolvents for solubilization: revising the way we use solubility-enabling formulations. Mol Pharm 2012; 9:581-90. doi: 10.1021/mp200460u [Crossref] [ Google Scholar]
- Ferretti V, Dalpiaz A, Bertolasi V, Ferraro L, Beggiato S, Spizzo F. Indomethacin co-crystals and their parent mixtures: does the intestinal barrier recognize them differently?. Mol Pharm 2015; 12:1501-11. doi: 10.1021/mp500826y [Crossref] [ Google Scholar]
- Dalpiaz A, Ferretti V, Bertolasi V, Pavan B, Monari A, Pastore M. From physical mixtures to co-crystals: how the coformers can modify solubility and biological activity of carbamazepine. Mol Pharm 2017. doi: 10.1021/acs.molpharmaceut.7b00899 [Crossref]
- Surov AO, Volkova TV, Churakov AV, Proshin AN, Terekhova IV, Perlovich GL. Cocrystal formation, crystal structure, solubility and permeability studies for novel 1, 2, 4-thiadiazole derivative as a potent neuroprotector. Eur J Pharm Sci 2017; 109:31-9. doi: 10.1016/j.ejps.2017.07.025 [Crossref] [ Google Scholar]
- Seo J-W, Hwang K-M, Lee S-H, Kim D-W, Park E-S. Preparation and Characterization of Adefovir Dipivoxil-Stearic Acid Cocrystal with Enhanced Physicochemical Properties. Pharm Dev Technol 2017:1-30. doi: 10.1080/10837450.2017.1334664 [Crossref]
- Gopi SP, Banik M, Desiraju GR. New Cocrystals of Hydrochlorothiazide: Optimizing Solubility and Membrane Diffusivity. Cryst Growth Des 2016; 17:308-16. doi: 10.1021/acs.cgd.6b01540 [Crossref] [ Google Scholar]
- Saikia B, Bora P, Khatioda R, Sarma B. Hydrogen bond synthons in the interplay of solubility and membrane permeability/diffusion in variable stoichiometry drug cocrystals. Cryst Growth Des 2015; 15:5593-603. doi: 10.1021/acs.cgd.5b01293 [Crossref] [ Google Scholar]
-
Food U, Administration D. Guidance for Industry: Regulatory Classification of Pharmaceutical Co-crystals. Center for Drug Evaluation and Research, Silver Spring, US 2013.
- Elder DP, Kuentz M, Holm R. Pharmaceutical excipients—quality, regulatory and biopharmaceutical considerations. Eur J Pharm Sci 2016; 87:88-99. doi: 10.1016/j.ejps.2015.12.018 [Crossref] [ Google Scholar]
- Renner B, Clarke G, Grattan T, Beisel A, Mueller C, Werner U. Caffeine accelerates absorption and enhances the analgesic effect of acetaminophen. J Clin Pharmacol 2007; 47:715-26. doi: 10.1177/0091270007299762 [Crossref] [ Google Scholar]
- Schmidt R, Fanchamps A. Effect of caffeine on intestinal absorption of ergotamine in man. Eur J Clin Pharmacol 1974; 7:213-6. doi: 10.1007/bf00560383 [Crossref] [ Google Scholar]
- Busch U, Heinzel G, Narjes H, Nehmiz G. Interaction of meloxicam with cimetidine, Maalox, or aspirin. J Clin Pharmacol 1996; 36:79-84. doi: 10.1002/j.1552-4604.1996.tb04155.x [Crossref] [ Google Scholar]
- Cheney ML, Weyna DR, Shan N, Hanna M, Wojtas L, Zaworotko MJ. Coformer selection in pharmaceutical cocrystal development: a case study of a meloxicam aspirin cocrystal that exhibits enhanced solubility and pharmacokinetics. J Pharm Sci 2011; 100:2172-81. doi: 10.1002/jps.22434 [Crossref] [ Google Scholar]
- Videla S, Lahjou M, Vaqué A, Sust M, Encabo M, Soler L. Single‐dose pharmacokinetics of co‐crystal of tramadol–celecoxib: Results of a four‐way randomized open‐label phase I clinical trial in healthy subjects. Br J Clin Pharmacol 2017. doi: 10.1111/bcp.13395 [Crossref]
- Uppoor VRS. Regulatory perspectives on in vitro (dissolution)/in vivo (bioavailability) correlations. J Control Release 2001; 72:127-32. doi: 10.1016/s0168-3659(01)00268-1 [Crossref] [ Google Scholar]
- Kostewicz ES, Abrahamsson B, Brewster M, Brouwers J, Butler J, Carlert S. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci 2014; 57:342-66. doi: 10.1016/j.ejps.2013.08.024 [Crossref] [ Google Scholar]
- Skelly JP, Amidon GL, Barr WH, Benet LZ, Carter JE, Robinson JR. In vitro and in vivo testing and correlation for oral controlled/modified-release dosage forms. Pharm Res 1990; 7:975-82. doi: 10.1002/9780470640487.ch15 [Crossref] [ Google Scholar]
- Takano R, Takata N, Saito R, Furumoto K, Higo S, Hayashi Y. Quantitative analysis of the effect of supersaturation on in vivo drug absorption. Mol Pharm 2010; 7:1431-40. doi: 10.1021/mp100109a [Crossref] [ Google Scholar]
- Guzmán HR, Tawa M, Zhang Z, Ratanabanangkoon P, Shaw P, Gardner CR. Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations. J Pharm Sci 2007; 96:2686-702. doi: 10.1002/jps.20906 [Crossref] [ Google Scholar]
- Barzegar-Jalali M, Nayebi AM, Valizadeh H, Hanaee J, Barzegar-Jalali A, Adibkia K. Evaluation of in vitro-in vivo correlation and anticonvulsive effect of carbamazepine after cogrinding with microcrystalline cellulose. J Pharm Pharm Sci 2006; 9:307-16. [ Google Scholar]
- Dastmalchi S, Garjani A, Maleki N, Sheikhee G, Baghchevan V, Jafari-Azad P. Enhancing dissolution, serum concentrations and hypoglycemic effect of glibenclamide using solvent deposition technique. J Pharm Pharm Sci 2005; 8:175-81. [ Google Scholar]
- Stanton MK, Kelly RC, Colletti A, Langley M, Munson EJ, Peterson ML. Improved pharmacokinetics of AMG 517 through co‐crystallization part 2: Analysis of 12 carboxylic acid co‐crystals. J Pharm Sci 2011; 100:2734-43. doi: 10.1002/jps.22502 [Crossref] [ Google Scholar]
- Higashino H, Hasegawa T, Yamamoto M, Matsui R, Masaoka Y, Kataoka M. In vitro–in vivo correlation of the effect of supersaturation on the intestinal absorption of BCS Class 2 drugs. Mol Pharm 2014; 11:746-54. doi: 10.1021/mp400465p [Crossref] [ Google Scholar]
- Stanton MK, Kelly RC, Colletti A, Kiang Y-H, Langley M, Munson EJ. Improved pharmacokinetics of AMG 517 through co-crystallization part 1: comparison of two acids with corresponding amide co-crystals. J Pharm Sci 2010; 99:3769-78. doi: 10.1002/jps.22181 [Crossref] [ Google Scholar]
- Han H-K, Choi H-K. Improved absorption of meloxicam via salt formation with ethanolamines. Eur J Pharm Biopharm 2007; 65:99-103. doi: 10.1016/j.ejpb.2006.07.003 [Crossref] [ Google Scholar]
- Möschwitzer JP. Drug nanocrystals in the commercial pharmaceutical development process. Int J Pharm 2013; 453:142-56. doi: 10.1016/j.ijpharm.2012.09.034 [Crossref] [ Google Scholar]
- Junghanns J-UA, Müller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomedicine 2008; 3:295. doi: 10.2147/ijn.s595 [Crossref] [ Google Scholar]
- Dizaj SM, Vazifehasl Z, Salatin S, Adibkia K, Javadzadeh Y. Nanosizing of drugs: Effect on dissolution rate. Res Pharm Sci 2015; 10:95-108. [ Google Scholar]
- Müller RH, Peters K. Nanosuspensions for the formulation of poorly soluble drugs: I Preparation by a size-reduction technique. Int J Pharm 1998; 160:229-37. doi: 10.1002/9781118444726.ch9 [Crossref] [ Google Scholar]
- Greco K, Bogner R. Solution‐mediated phase transformation: Significance during dissolution and implications for bioavailability. J Pharm Sci 2012; 101:2996-3018. doi: 10.1002/jps.23025 [Crossref] [ Google Scholar]
- Verma S, Gokhale R, Burgess DJ. A comparative study of top-down and bottom-up approaches for the preparation of micro/nanosuspensions. Int J Pharm 2009; 380:216-22. doi: 10.1016/j.powtec.2014.02.011 [Crossref] [ Google Scholar]
- de Waard H, Frijlink HW, Hinrichs WL. Bottom-up preparation techniques for nanocrystals of lipophilic drugs. Pharm Res 2011; 28:1220-3. doi: 10.1007/s11095-010-0323-3 [Crossref] [ Google Scholar]
- Sander JR, Bučar DK, Henry RF, Zhang GG, MacGillivray LR. Pharmaceutical Nano‐Cocrystals: Sonochemical Synthesis by Solvent Selection and Use of a Surfactant. Angew Chem Int Ed Engl 2010; 49:7284-8. doi: 10.1002/anie.201002588 [Crossref] [ Google Scholar]
- Liu M, Hong C, Li G, Ma P, Xie Y. The generation of myricetin–nicotinamide nanococrystals by top down and bottom up technologies. Nanotechnology 2016; 27:395601. doi: 10.1088/0957-4484/27/39/395601 [Crossref] [ Google Scholar]
- De Smet L, Saerens L, De Beer T, Carleer R, Adriaensens P, Van Bocxlaer J. Formulation of itraconazole nanococrystals and evaluation of their bioavailability in dogs. Eur J Pharm Biopharm 2014; 87:107-13. doi: 10.1016/j.ejpb.2013.12.016 [Crossref] [ Google Scholar]
- Karashima M, Kimoto K, Yamamoto K, Kojima T, Ikeda Y. A novel solubilization technique for poorly soluble drugs through the integration of nanocrystal and cocrystal technologies. Eur J Pharm Biopharm 2016; 107:142-50. doi: 10.1016/j.ejpb.2016.07.006 [Crossref] [ Google Scholar]
- Emami S, Siahi-Shadbad M, Barzegar-Jalali M, Adibkia K. Feasibility of electrospray deposition for rapid screening of the cocrystal formation and single step, continuous production of pharmaceutical nanococrystals. Drug Dev Ind Pharm 2018; 44:1034-47. doi: 10.1080/03639045.2018.1430821 [Crossref] [ Google Scholar]
- Huang Y, Li J-M, Lai Z-H, Wu J, Lu T-B, Chen J-M. Phenazopyridine-phthalimide nano-cocrystal: Release rate and oral bioavailability enhancement. Eur J Pharm Sci 2017; 109:581. doi: 10.1016/j.ejps.2017.09.020 [Crossref] [ Google Scholar]
- Tao Q, Chen J-M, Ma L, Lu T-B. Phenazopyridine cocrystal and salts that exhibit enhanced solubility and stability. Cryst Growth Des 2012; 12:3144-52. doi: 10.1021/cg300327x [Crossref] [ Google Scholar]
- Rao RN, Maurya PK, Raju AN. Isolation and characterization of a potential process related impurity of phenazopyridine HCl by preparative HPLC followed by MS–MS and 2D-NMR spectroscopy. J Pharm Biomed Anal 2009; 49:1287-91. doi: 10.1016/j.jpba.2009.03.004 [Crossref] [ Google Scholar]
-
Ranzani LS, Aldea AF. Pharmaceutical compositions of co-crystals of tramadol and coxibs. Google Patents; 2013.
- Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet 2004; 43:879-923. doi: 10.2165/00003088-200443130-00004 [Crossref] [ Google Scholar]
- Antoniou K, Malamas M, Drosos AA. Clinical pharmacology of celecoxib, a COX-2 selective inhibitor. Expert Opin Pharmacother 2007; 8:1719-32. doi: 10.1517/14656566.8.11.1719 [Crossref] [ Google Scholar]
- Bergström CA, Andersson SB, Fagerberg JH, Ragnarsson G, Lindahl A. Is the full potential of the biopharmaceutics classification system reached?. Eur J Pharm Sci 2014; 57:224-31. doi: 10.1016/j.ejps.2013.09.010 [Crossref] [ Google Scholar]
- Moore R, McQuay H. Single-patient data meta-analysis of 3453 postoperative patients: oral tramadol versus placebo, codeine and combination analgesics. Pain 1997; 69:287-94. doi: 10.1016/s0304-3959(96)03291-5 [Crossref] [ Google Scholar]
- Shono Y, Jantratid E, Janssen N, Kesisoglou F, Mao Y, Vertzoni M. Prediction of food effects on the absorption of celecoxib based on biorelevant dissolution testing coupled with physiologically based pharmacokinetic modeling. Eur J Pharm Biopharm 2009; 73:107-14. doi: 10.1016/j.ejpb.2009.05.009 [Crossref] [ Google Scholar]
- Almansa C, Mercè R, Tesson N, Farran J, Tomàs J, Plata-Salamán CR. Co-crystal of Tramadol Hydrochloride–Celecoxib (ctc): A Novel API–API Co-crystal for the Treatment of Pain. Cryst Growth Des 2017; 17:1884-92. doi: 10.1021/acs.cgd.6b01848 [Crossref] [ Google Scholar]
- Golob S, Perry M, Lusi M, Chierotti MR, Grabnar I, Lassiani L. Improving Biopharmaceutical Properties of Vinpocetine Through Cocrystallization. J Pharm Sci 2016; 105:3626-33. doi: 10.1016/j.xphs.2016.09.017 [Crossref] [ Google Scholar]
- Musther H, Olivares-Morales A, Hatley OJ, Liu B, Hodjegan AR. Animal versus human oral drug bioavailability: Do they correlate?. Eur J Pharm Sci 2014; 57:280-91. doi: 10.1016/j.ejps.2013.08.018 [Crossref] [ Google Scholar]
- Duggirala NK, Perry ML, Almarsson Ö, Zaworotko MJ. Pharmaceutical cocrystals: along the path to improved medicines. Chem Commun (Camb) 2016; 52:640-55. doi: 10.1039/c5cc08216a [Crossref] [ Google Scholar]
- Duggirala NK, Vyas A, Krzyzaniak JF, Arora KK, Suryanarayanan R. Mechanistic Insight into Caffeine–Oxalic Cocrystal Dissociation in Formulations: Role of Excipients. Mol Pharm 2017. doi: 10.1021/acs.molpharmaceut.7b00587 [Crossref]
- Feng L, Karpinski PH, Sutton P, Liu Y, Hook DF, Hu B. LCZ696: a dual-acting sodium supramolecular complex. Tetrahedron Lett 2012; 53:275-6. doi: 10.1016/j.tetlet.2011.11.029 [Crossref] [ Google Scholar]
- Fala L. Entresto (Sacubitril/Valsartan): First-in-Class Angiotensin Receptor Neprilysin Inhibitor FDA Approved for Patients with Heart Failure. Am Health Drug Benefits 2015; 8:330. [ Google Scholar]
- Madaan T, Akhtar M, Najmi AK. Sodium glucose CoTransporter 2 (SGLT2) inhibitors: Current status and future perspective. Eur J Pharm Sci 2016; 93:244-52. doi: 10.1016/j.ejps.2016.08.025 [Crossref] [ Google Scholar]
- Kale DP, Zode SS, Bansal AK. Challenges in Translational Development of Pharmaceutical Cocrystals. J Pharm Sci 2017; 106:457-70. doi: 10.1016/j.xphs.2016.10.021 [Crossref] [ Google Scholar]
- Bowles P, Brenek SJ, Caron Sp, Do NM, Drexler MT, Duan S. Commercial route research and development for SGLT2 inhibitor candidate ertugliflozin. Org Process Res Dev 2014; 18:66-81. doi: 10.1021/op4002802 [Crossref] [ Google Scholar]