BioImpacts. 9(1):45-56.
doi: 10.15171/bi.2019.06
Original Research
In silico design of a triple-negative breast cancer vaccine by targeting cancer testis antigens
Sepideh Parvizpour 1, Jafar Razmara 2, Mohammad M. Pourseif 1, Yadollah Omidi 1, 3, * 
Author information:
1 Research Center for Pharmaceutical Nanotechnology, Biomedicine Institute, Tabriz University of Medical Sciences, Tabriz, Iran
2 Department of Computer Science, Faculty of Mathematical Sciences, University of Tabriz, Tabriz, Iran
3 Department of Pharmaceutics, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
Abstract
Introduction:
Triple-negative breast cancer (TNBC) is an important subtype of breast cancer, which occurs in the absence of estrogen, progesterone and HER-2 receptors. According to the recent studies, TNBC may be a cancer testis antigen (CTA)-positive tumor, indicating that the CTA-based cancer vaccine can be a treatment option for the patients bearing such tumors. Of these antigens (Ags), the MAGE-A family and NY-ESO-1 as the most immunogenic CTAs are the potentially relevant targets for the development of an immunotherapeutic way of the breast cancer treatment.
Methods:
In the present study, immunoinformatics approach was used to design a multi-epitope peptide vaccine to combat the TNBC. The vaccine peptide was constructed by the fusion of three crucial components, including the CD8+ cytotoxic T lymphocytes (CTLs) epitopes, helper epitopes and adjuvant. The epitopes were predicted from the MAGE-A and NY-ESO-1 Ags. In addition, the granulocyte-macrophage-colony-stimulating factor (GM-CSF) was used as an adjuvant to promote the CD4+ T cells towards the T-helper for more strong induction of CTL responses. The components were conjugated by proper linkers.
Results:
The vaccine peptide was examined for different physiochemical characteristics to confirm the safety and immunogenic behavior. Furthermore, the 3D-structure of the vaccine peptide was predicted based on the homology modeling approach using the MODELLER v9.17 program. The vaccine structure was also subjected to the molecular dynamics simulation study for structure refinement. The results verified the immunogenicity and safety profile of the constructed vaccine as well as its capability for stimulating both the cellular and humoral immune responses.
Conclusion:
Based on our in-silico analyses, the proposed vaccine may be considered for the immunotherapy of TNBC.
Keywords: Multi-epitope vaccine, T-cell epitope, Triple-negative breast cancer, T-helper epitope
Copyright and License Information
© 2019 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Breast cancer is the most prevalent malignancy and the leading cause of cancer death among women around the world. According to the statistics published by the National Cancer Institute (NCI) of United States, almost 14.5 million people live beyond a cancer diagnosis in 2014, while this figure is expected to rise up to nearly 19 million by 2024. It is also estimated that around one in nine American women will develop breast cancer during their lifetimes. The main risk factors for developing breast cancer are not clear. Many women with a high risk of breast cancer incidence do not get it, while many others without any known risk factor develop breast cancer. The impact of genetic factors on the mammary carcinogenesis has been proven, in which gene mutations involved in the development of breast cancer have been observed in around 10% of these patients.
1
Exploration of efficient prophylactic and therapeutic strategies against cancer need preliminary knowledge about the role of the immune system in controlling cancer, the origin and initiation of tumor, its progression and heterogeneity. A hypothesis for tumor origination and its heterogeneity is suggested via the clonal evolution model.
2
Based on the model, several mutations appear to happen during the time and accumulated within tumor cells. Many of these mutations are identified and removed by the immune system. However, a number of such mutations may sustain in the fittest cells, leading to heterogeneity and initiation of the cancerous cells.
3
Another hypothesis refers to the concept of cancer stem cells, which can avoid the anoikis with a capability of an extensive proliferation similar to the properties of stem cells. These cells seem to play a central role in the initiation, progression and recurrence of tumors.
4
In addition to the elimination of cancerous cells, it is essential to recognize and eradicate the cancer stem cells in order to treat cancer. Perhaps, the development of effective immunotherapy approaches might be one of the best strategies.
In fact, the presence or absence of three known receptors plays a critical role in the diagnosis and treatment of the breast cancer, including estrogen receptors (ER), progesterone receptors (PR), and the epidermal growth factor receptor 2 (HER2). To propose a robust way for the treatment of breast cancer, it is important to target these receptors. In 15%-20% of all breast cancer, however, none of these receptors is expressed by the breast tumor as the ER-, PR- and HER2-negative cancer; the so-called a triple negative breast cancer (TNBC).
2
In the case of TNBC, the patients may show a high rate of inconsistency and failure in terms of the treatment, leading to recurrence, metastasis and finally death. As a result, the treatment of patients with TNBC is very difficult
4
and it is known as the most lethal form of the breast cancer.
2
Recently, cancer testis antigens (CTAs) have been reported as the cancer molecular markers, targeting of which can provide much more significant treatment strategy in cancer therapy.
5
Due to the expression of CTAs in the malignant tumors and their absence in the normal tissues except for the testicular germ cells, they can be valuable targets for cancer immunotherapy. They can also be used as potential mediators for the diagnosis of cancer cell.
6
Until now, more than 100 CTAs and their families have been recognized, which are expressed in a wide range of cancers, including breast cancer.
2
These antigens (Ags) encode immunogenic proteins, which can induce spontaneous humoral or cellular antitumor responses. Autoimmunity in the testis and placenta, however, is disallowed, in large part due to the down-regulation of the MHC and the production of immune-suppressive factors. The detailed systematic knowledge of manually curated CTAs has been collected in the Cancer Testis database (CTdatabase)
7
by the Ludwig Institute for Cancer Research, including immunogenic data, splicing variants, and the gene expression levels for each cancer type.
The expression of CTAs in large portion of TNBC has recently been reported in several studies.
8,9
Further, NY-ESO-1 looks like one of the most immunogenic CTA, known to date, which is expressed in the TNBC. Patients with tumors containing expression of NY-ESO-1 frequently show humoral and cellular immune responses against this Ag. In addition, the human melanoma-associated antigen (MAGE) that refers to a family of CTAs categorized into the type I and II based on the tissue expression pattern. The type I MAGEs in the human contains the subfamily of MAGE-A, -B, and –C which are clustered on the X chromosome.
10
The type II MAGEs (i.e., MAGE-D, -E, -F, -G, -H, and –L) are expressed in several human tissues in the X chromosome.
11
A common MAGE homology domain (MHD) exists in both types having around 170 amino acids, which is conserved about 46% within all human MAGEs.
12
The q28 region of the X chromosome carries 12 homologous genes of MAGE-A (MAGE-A1 to -A12). The pieces of evidence accuse the expression of MAGE-A of the growth and progression of tumors.
13
It is also expressed frequently in the TNBC patients.
9,14
Recently, immunotherapies have been shown to play an outstanding role in the cancer therapy since they are well-tolerated and considered as robust treatment modalities for most of the pre-invasive and invasive tumors. Of the anticancer immunotherapies, the multi-epitope peptide cancer vaccines appear as the next generation immunomedicines designed to elicit the immune system responses against tumor cells. The anti-tumor immunity action may be enhanced through either intensifying the CD4+ helper T lymphocytes (HTLs) and CD8+ cytolytic T lymphocytes (CTLs) responses or preventing the immune response suppressors.
15
In addition, various agents can be conjugated to the peptide vaccines as an adjuvant, including granulocyte/macrophage colony-stimulating factor (GM-CSF) to augment the immune responses.
16
It was shown that the magnitude of both humoral and cellular immune responses to the vaccine constructs is enhanced by GM-CSF through professional antigen-presenting cells (APCs). The efficiency of a cancer vaccine is related to its ability for the stimulation of both HTL and CTL responses. The tumor peptide segments, which are basically identified as the T-cell immunodominant epitopes and presented by the major histocompatibility complex class I (MHC-I) molecules on the antigen presenting cells (APCs), are recognized and removed by the CTLs. These epitopes are important elements for the construction of epitope-based vaccines for cancer therapy. The epitope-based techniques for the vaccine design offer several advantages, including cost-effectiveness, safety, specificity and stability under different conditions due to the definition of epitopes. Nevertheless, the vaccine development by this approach may also face with some issues, including fast destruction by tissue and serum peptidases, inability in terms of effective activation of the HTLs and possible limitations in terms of the MHC molecules.
17
However, focusing on the molecular mechanisms of the involved Ags can lead to overcome these difficulties and improve the performance of the epitope vaccine. In this way, the computational methods play a critical role to reduce the complexity of vaccine development, whereas they can be used in different stages of the design process such as selection of proper Ags, carriers, and adjuvants. The emerging bioinformatics approaches have led researchers to propose several alternative strategies for the development of new‐generation vaccines through key approaches, including (i) immunoinformatics, (ii) reverse vaccinology, and (iii) structural vaccinology.
In the current study, we intended to in-silico design of a multi-epitope polypeptide vaccine against TNBC. For the development of the vaccine, we capitalized on NY-ESO-1, MAGE-A3, MAGE-A10 and multi MAGE-A antigens as target Ags for the epitope prediction. In addition, GM-CSF was also added to the peptide as an important component in order to augment an antibody- and cell-mediated immunity. The selected components were linked together via appropriate linkers. The designed vaccine was analyzed using several immunoinformatics tools to evaluate and validate the efficacy and capability of the vaccine for the immunotherapy of TNBC.
Methods
Sequence retrieval and preparation
The UniProt database
18
was used to retrieve the amino acid sequence of the human melanoma-associated Ags, including MAGE-A3, MAGE-A10, multi-MAGE-A (MAGE-A-3,10,6,2,12,4) and NY-ESO-1. The sequences were submitted to some different bioinformatics tools for the assessment of antigenicity and prediction of epitope(s). In the sequel, proper amino acid linkers were used to link the derived peptide segments from helper Ag, main Ags and proper adjuvant. The obtained whole protein sequence was analyzed by several online servers to determine its characteristics. The sequence is also back-translated to the DNA sequence to adapt with E. coli codon using JCAT online server.
19
The resulting DNA sequence was optimized in order to clone and process its expression in the E. coli host.
Sequence analysis by immunoinformatics tools
The prediction of MHC class I binding peptides
T cells are major agents for the induction of immune response against foreign Ags. They are activated through binding of the antigenic fragment to the MHC molecules. Thus, it is a critical step in the vaccine design to predict which antigenic fragment can bind to the MHC molecule. IEDB server provides robust tools to look for the MHC class I and II binding common epitopes. The whole numbers of MHC allele interactions can be estimated using outcomes of the IEDB server. The server provides a set of options for users to choose their desired prediction technique, including artificial neural network (ANN), average relative binding (ARB), stabilized matrix method (SMM), SMM with a peptide: MHC binding energy covariance matrix (SMMPMBEC), scoring matrices derived from the combinatorial peptide libraries, consensus, and NetMHCpan.
20
Alleles were added to the list of candidates due to the lowest level of energy (E-total) in docking between each peptide and active site of their alleles. The 3D structure of these alleles has mostly been deposited to the protein data bank (PDB). In the cases lacking 3D structures of alleles in PDB, their structures were predicted by online servers. The MHC-peptide docking was done using the Hex v8.0.0 program. The final T cell epitopes have been chosen within the peptides having the most negative E-total value as a stable system. The LigPlot v1.4.5 program was used to visualize and analyze the hydrogen and hydrophobic interactions between the MHC alleles and the peptides.
The prediction of B-cell epitopes
Linear B-cell epitopes were predicted using BCPREDS online server. A novel method of a subsequence kernel with performance accuracy of 74.57% was used for the prediction of B-cell epitopes with BCPREDS specificity threshold of 0.75%.
21
Furthermore, DiscoTope 2.0 was used to designate the discontinuous B-cell epitopes from the 3D protein structure.
22
DiscoTope uses a novel epitope propensity amino acid score and surface accessibility calculation. Combination of the propensity scores of residues in spatial proximity and the contact numbers is used to calculate the final scores. The identification of epitopes was performed using the default threshold of 3.7, by which the sensitivity and specificity values of 0.47 and 0.75 were obtained, respectively.
Construction of multi-epitope vaccine sequence
The vaccine sequence was the product of the above prediction steps using immunoinformatics tools. The sequence is constructed by linking the high-scoring CTL and HTL epitopes together via appropriate linkers. In addition, GM-CSF was included at the N-terminal end of the vaccine as an adjuvant to improve its immunogenicity.
The prediction of allergenicity
Allergenicity of the constructed multi-epitope vaccine was assessed by submitting the sequence to the online AlgPred web server.
23
The prediction of allergenicity is technically done based on the similarity of the query sequence to the known protein sequences. The AlgPred server looks for MEME/MAST allergen motifs and predicts the sequence allergenicity in the case of the existence of a motif. The server uses an SVM-based module for the composition of amino acids or dipeptides, and hence, the prediction of allergen. The sequence is considered as an allergen protein if there is a hit in its BLAST search against 2890 allergen-representative peptides.
Analysis of physicochemical properties
Physicochemical properties (e.g., molecular weight, sequence length, instability index, aliphatic index, half-life, and grand average of hydropathicity) of the constructed multi-epitope vaccine were further analyzed using the ProtParam web tool.
24
The ProtParam calculates the physicochemical properties of a submitted sequence on the basis of pK values of different amino acids. Stability of a protein is revealed by the instability index, in which an index less than 40 is considered stable. It predicts the half-life based on the ‘N-end rule’, indicating that the N-terminal amino acids determine the degradation of a protein. The aliphatic index indicates the volume occupied by the aliphatic side chains, including alanine, valine, leucine, and isoleucine. In order to calculate the grand average of hydropathicity, the sum of hydropathy of all residues is divided by the total number of residues in a protein.
Engineering the fusion protein
The prediction of secondary and tertiary structure
The secondary structure of the constructed vaccine was predicted by the PSIPRED online server.
25
The server works based on incorporating two feed-forward neural networks in order to analyze the output obtained from the PSI-BLAST. Further, the tertiary structure of the multi-epitope vaccine sequence was predicted through the homology modeling technique.
26
This prediction was performed by means of the MODELLER version 9.17 software.
27
The latter software is a standalone program applying an automated strategy for comparative modeling of a protein structure based on satisfaction of spatial restraints. UCSF Chimera
28
was also used for visualizing the 3D structures of proteins.
Molecular dynamic (MD) simulation
The constructed vaccine was subjected to the GROMACS 5.0.7 software package for the energy minimization. The structure of the model was refined with a molecular dynamics simulation using the GROMOS 96 force field. To this end, the model was placed in a cubic box with suitable size, and 13 Na+ ions were added to provide a neutralized environment. The MD simulation was done at 300 K for 30 ns. Afterward, the RMSD graph was drawn for analyzing the dynamic behavior and the protein structural changes.
The tertiary structure validation
Different tools were used to assess the resulting 3D-model, including ProSA-web,
29
PRO-CHECK and ERRAT.
30
The overall quality score is calculated by analyzing atomic coordinates of the model using the ProSA-web server. The server provides a plot of the z-score for experimentally determined structures deposited in the PDB. The Ramachandran plot of residue-by-residue stereochemical qualities of models is produced by PROCHECK. The server examines the backbone conformation based on a Psi/Phi Ramachandran plot. In addition, the statistics of the non-bonded atom-atom interactions within a database of reliable high-resolution crystallography structures is calculated by ERRAT.
Codon optimization of the vaccine sequence
Due to the frequent variations of codon usage among organisms, it is necessary to optimize the protein expression within the host cells. In addition, the formation of an unstable structure of mRNA and a large volume of G/C content possibly decrease the protein synthesis due to its negative effects on the initiation and elongation of the translation. Accordingly, the vaccine sequence was submitted to JCat server
31
for the codon optimization. The program calculates the codon adaptation index (CAI) and G/C content, and also predicts the secondary structure of mRNA.
Results
Sequence retrieval
The amino acid sequence of NY-ESO-1 (UniProt ID: P78358), MAGE-A3 (UniProt ID: P43357), MAGE-A10 (UniProt ID: P43363) and multi-MAGE-A proteins were retrieved from the UniProt database. The sequences were subjected to the immunoinformatics analysis in order to select CD8+ CTL and CD4+ helper epitopes. Further, the sequence of GM-CSF (UniProt ID: P04141) was used as the adjuvant.
Epitope prediction
The alleles were submitted to the IEDB server
32
for the prediction of the T-cell epitope. Tables 1, 2 and 3 show the outcomes of the IEDB server for the MHC class I prediction for NY-ESO-1, MAGE-A3, MAGE-A10, multi MAGE-A family, respectively. The results are shown in units of the IC50 score while a lower value specifies a higher affinity. Specifically, the peptides are classified into the high-, intermediate- and low-affinity based on the IC50 scores lower than 50, 500 and 5000 nM, respectively. Additionally, another score is calculated namely percentile rank by comparing the peptide's IC50 score against those of a set of random peptides from the Swiss-Prot database. The higher affinity is indicated by a small value of percentile rank. From the values shown in Tables 1 to 3, the best peptides were chosen based on both IC50 and percentile rank scores. The T-cell epitopes were docked with the MHC alleles. The docking energy (E-total) between each peptide and active site of its allele is shown in Tables 1 to 3. The sequences with the lowest E-total value (kcal/mol) were selected as the CTL epitopes. According to the literature, we found some of the epitopes that have experimentally been confirmed. Figs. 1, 2, and 3 represent the molecular docking of the lowest E-total value fragment between the NY-ESO-1, MAGE-A3, and MAGE-A10 antigens and HLA Class I alleles. The MAGE-A family sequences were aligned as shown in Fig. 4. Based on the alignment, two commonly aligned epitopes were chosen, and they have already been published by Graff-Dubois et al.
33
Similarly, the HTL epitopes were opted from the results obtained from the IEDB MHC-II prediction module based on the higher binding affinity with the MHC-II, as shown in Table 3.
Table 1.
CD8+ T-cell epitope selection for NY-ESO-1, MAGE-A3 and MAGE-A10
|
HLA class I
|
IEDB
|
Position
|
Length
|
Method
|
ic50 score
|
Percentile rank
|
E-total (kJ/mol)
|
Published
|
| NY-ESO-1 |
|
|
|
|
|
|
|
|
A*02:01
|
SLLMWITQC
|
157-165
|
9
|
ann
|
32.98
|
0.1
|
-540.1
|
Yes
34
|
|
A*30:01
|
AARASGPGG
|
50-58
|
9
|
ann
|
21.2
|
0.1
|
-559.95
|
No
|
|
A*32:01
|
RLLEFYLAMP |
86-98 |
10 |
ann/smm |
36.08 |
0.5 |
-418.27 |
No |
|
B*07:02
|
TPMEAELARR |
98-109 |
10 |
ann |
11.90 |
0.1 |
-490.14 |
No |
|
B*40:02
|
KEFTVSGNI
|
124-132
|
9
|
ann
|
34.4
|
0.2
|
-552.45
|
No
|
|
B*40:02
|
RLLEFYLAMP |
85-98 |
10 |
ann |
17.0 |
0.2 |
-539.32 |
YES
35
|
|
B*55:01
|
MPFATPMEA |
94-102 |
9 |
ann |
23.70 |
0.2 |
-419.10 |
No |
| MAGE-A3 |
|
|
|
|
|
|
|
|
A*02:01
|
KVAELVHFL |
112-120 |
9 |
ann |
0.1 |
45.93 |
-374.92 |
Yes
36
|
|
A*02:01
|
FLWGPRALV
|
271-279
|
9
|
ann
|
0.1
|
12.02
|
-591.74
|
Yes
37
|
|
A*24:02
|
IMPKAGLLI
|
195-203
|
9
|
ann
|
0.1
|
-
|
-529.78
|
Yes
38
|
|
A*01:01
|
ELMEVDPIGHL
|
165-174
|
9
|
ann
|
0.1
|
34.74
|
-648.0
|
No
|
|
A*03:01
|
RKVAELVHFLL |
111-122 |
10 |
ann |
0.1 |
46.43 |
-457.78 |
No |
|
A*02:06
|
GSVVGNWQYF |
137-148 |
10 |
ann |
0.1 |
45.46 |
-486.05 |
No |
| MAGE-A10 |
|
|
|
|
|
|
|
| A*01:01 |
KLLTQDWVQ |
269-280 |
9 |
ann |
44.11 |
0.1 |
-489.12 |
No |
| A*01:01 |
NGSDPRSFPL |
319-330 |
10 |
ann |
54.21 |
0.1 |
-552.37 |
No |
| A*02:01 |
SLLKFLAKV
|
310-318
|
9
|
ann
|
8.90
|
0.4
|
-663.25
|
Yes
39
|
| A*02:01 |
GLYDGMEHL
|
254-262
|
9
|
ann
|
9.09
|
0.8
|
-432.16
|
Yes
39
|
| A*03:01 |
LLFKYQMKE
|
145-153
|
9
|
ann
|
47.96
|
0.1
|
-664.36
|
No
|
| A*03:01 |
RAHAEIRKMS |
301-313 |
10 |
ann |
54.20 |
0.1 |
-699.67 |
No |
| A*11:01 |
MLSDVQSMPK |
214-223 |
10 |
ann |
12.72 |
0.2 |
-533.53 |
No |
| A*31:01 |
KFLAKVNGSD |
313-324 |
10 |
ann |
39.87 |
0.1 |
-502.90 |
No |
| A*31:01 |
RSFPLWYEEA |
324-335 |
10 |
ann |
29.13 |
0.1 |
-551.39 |
No |
| A*31:01 |
VIWEALNMMG |
245-256 |
10 |
ann |
53.0 |
0.2 |
-438.42 |
No |
* Selected as the highest peptide binding affinity.
The selected sequences are shown in bold.
Table 2.
Multi-MAGE-A family epitope selection
33
|
Sequence
|
MAGE-A family
|
| CLGLSYDGLL |
1, 2, 3, 4, 6, 12 |
|
YLEYRQVPG
|
2, 3, 4, 6, 10, 12
|
|
YEFLWGPRA
|
1, 2, 3, 4, 6, 10, 12
|
The selected sequences are shown in bold.
Table 3.
CD4+ helper T-cell epitope selection for NY-ESO-1, MAGE-A3, MAGE-A10
|
|
IEDB
|
Position
|
Length
|
Percentile rank
|
ic50 score
|
E-total (kJ/mol)
|
|
NY-ESO-1
|
| HLA-DRB1*01:01 |
SRLLEFYLAMPFATP
|
85-99
|
15
|
2.05
|
13
|
-629.80
|
| HLA-DRB1*01:06 |
HRQLQLSISSCLQQL
|
142-156
|
15
|
1.66
|
5.81
|
-590.80
|
| HLA-DRB1*01:01 |
QCFLPVFLAQPPSGQ
|
164-178
|
15
|
4
|
40
|
-557.20
|
|
MAGE-A3
|
| HLA-DRB1*01:01 |
ESEFQAALSRKVAEL
|
102-116
|
15
|
1.15
|
9
|
-475.11
|
| HLA-DRB1*01:01 |
AGLLIIVLAIIAREG |
199-203 |
15 |
3.02 |
17 |
-392.30 |
| HLA-DRB1*01:01 |
ACYEFLWGPRALVET |
267-281 |
15 |
4.16 |
22 |
-480.01 |
| HLA-DRB1*01:06 |
FPVIFSKASSSLQLV
|
147-161
|
15
|
1.61
|
4.32
|
-637.50
|
| HLA-DRB1*01:05 |
PDLESEFQAALSRKV |
99-113 |
15 |
1.91 |
8.28 |
-344.27 |
| HLA-DRB1*01:04 |
ETSYVKVLHHMVKIS
|
280-294
|
15
|
2.41
|
6.55
|
-496.60
|
|
MAGE-A10
|
| HLA-DRB1*01:01 |
PKTGILILILSIVFI
|
222-236
|
15
|
3.24
|
18
|
-821.30
|
| HLA-DRB1*01:01 |
TGHSFVLVTSLGLTY
|
197-211
|
15
|
4.39
|
6
|
-620.60
|
| HLA-DRB1*01:01 |
ARYEFLWGPRAHAEI
|
292-306
|
15
|
3.37
|
33
|
-624.00
|
| HLA-DRB1*01:04 |
AEIRKMSLLKFLAKV |
304-318 |
15 |
4.01 |
8.41 |
-366.54 |
The selected sequences are shown in bold.
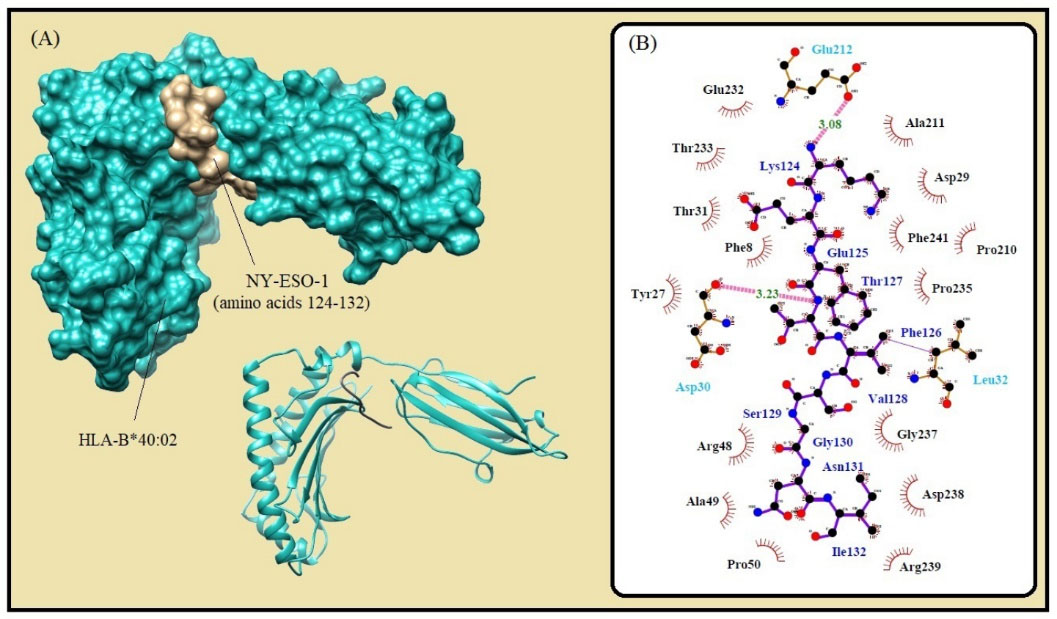
Fig. 1.
Molecular docking of the lowest E-total value fragment of NY-ESO-1 (amino acids 124-132) with the HLA-B*40:02 active site. (A) The surface and cartoon representations of the complexes HLA-B*40:02 (sky blue) and NY-ESO-1 (gold) visualized by the UCSF Chimera software. (B) The interactions between the vaccine peptide and the MHC allele prepared by Ligplot+ software. The pink dotted lines show hydrogen bonds and the red spline curves indicate hydrophobic contacts.
.
Molecular docking of the lowest E-total value fragment of NY-ESO-1 (amino acids 124-132) with the HLA-B*40:02 active site. (A) The surface and cartoon representations of the complexes HLA-B*40:02 (sky blue) and NY-ESO-1 (gold) visualized by the UCSF Chimera software. (B) The interactions between the vaccine peptide and the MHC allele prepared by Ligplot+ software. The pink dotted lines show hydrogen bonds and the red spline curves indicate hydrophobic contacts.
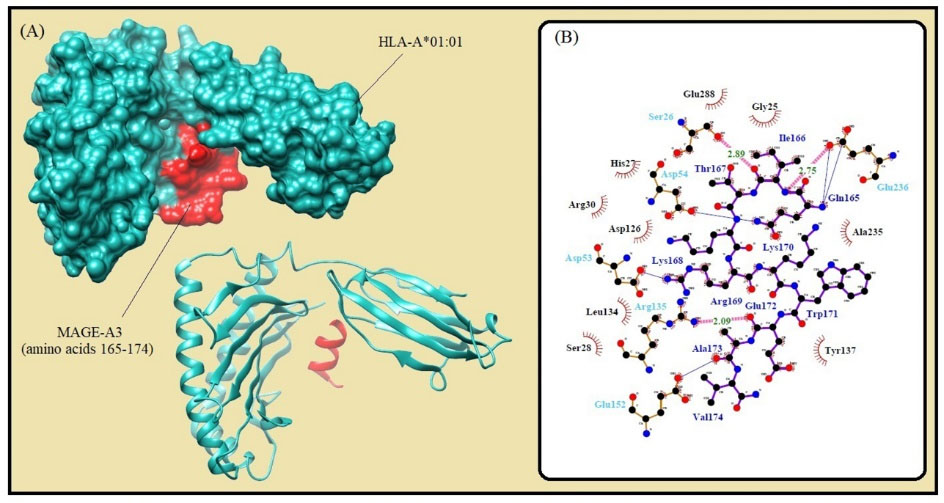
Fig. 2.
Molecular docking of the lowest E-total value fragment of MAGE-A3 (amino acids 165-174) with the HLA-A*01:01 active site. A) The surface and cartoon representation of the complexes HLA-A*01:01 (sky blue) and MAGE-A3 (red) visualised by UCSF Chimera software. B) The interactions between the vaccine peptide and the MHC allele prepared by Ligplot+ software. The pink dotted lines show hydrogen bonds and the red spline curves indicate hydrophobic contacts.
.
Molecular docking of the lowest E-total value fragment of MAGE-A3 (amino acids 165-174) with the HLA-A*01:01 active site. A) The surface and cartoon representation of the complexes HLA-A*01:01 (sky blue) and MAGE-A3 (red) visualised by UCSF Chimera software. B) The interactions between the vaccine peptide and the MHC allele prepared by Ligplot+ software. The pink dotted lines show hydrogen bonds and the red spline curves indicate hydrophobic contacts.
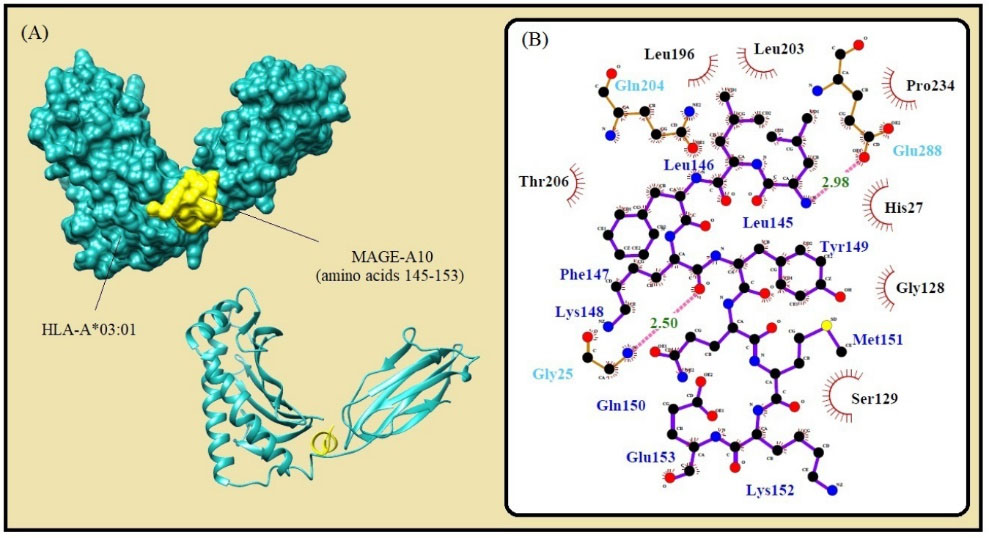
Fig. 3.
Molecular docking of the lowest E-total value fragment of MAGE-A10 (amino acids 145-153) with the HLA-A*01:01 active site. A) The surface and cartoon representation of the complexes HLA-A*01:01 (sky blue) and MAGE-A10 (yellow) visualised by UCSF Chimera software. B) The interactions between the vaccine peptide and the MHC allele prepared by Ligplot+ software. The pink dotted lines show hydrogen bonds and the red spline curves indicate hydrophobic contacts.
.
Molecular docking of the lowest E-total value fragment of MAGE-A10 (amino acids 145-153) with the HLA-A*01:01 active site. A) The surface and cartoon representation of the complexes HLA-A*01:01 (sky blue) and MAGE-A10 (yellow) visualised by UCSF Chimera software. B) The interactions between the vaccine peptide and the MHC allele prepared by Ligplot+ software. The pink dotted lines show hydrogen bonds and the red spline curves indicate hydrophobic contacts.
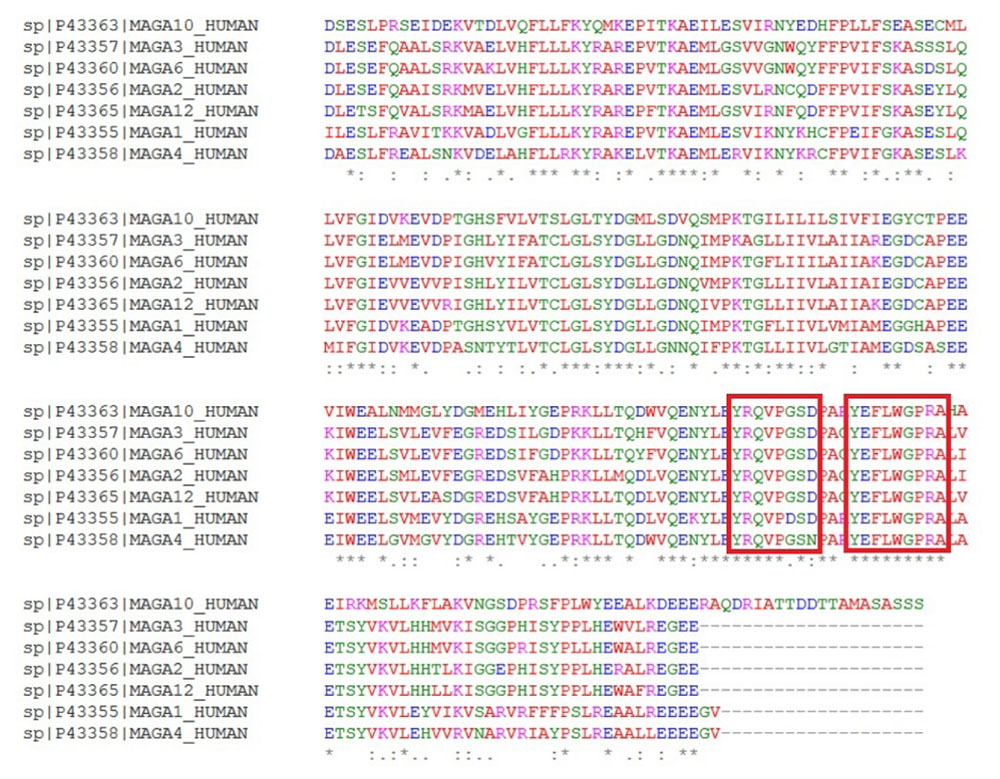
Fig. 4.
MAGE-A family sequence alignment. Two conserved epitopes are shown in red box.
.
MAGE-A family sequence alignment. Two conserved epitopes are shown in red box.
Construction of the multi-epitope peptide vaccine
Based on the results obtained from immunoinformatics analysis, three regions from each sequence of NY-ESO-1, MAGE-A3, MAGE-A10 and two regions from multi-MAGE-A (Fig. 4) were selected as the CTL epitopes. In addition, the same number of regions were selected from each sequence as the helper epitopes (Table 3). The selected CTL and helper epitopes were arranged and fused together within the vaccine peptide by proper linkers. Fig. 5 shows the schematic diagram of vaccine domains and linker's sites.
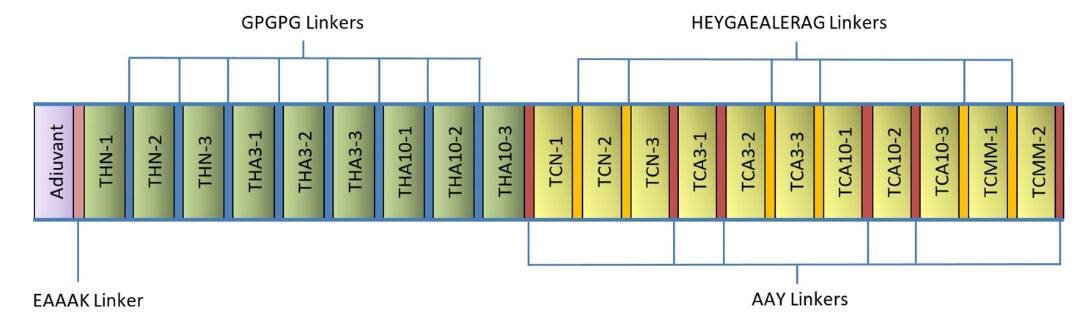
Fig. 5.
Schematic diagram of the vaccine construct consists of the adjuvant, helper epitopes and CTL epitopes, which joined together by proper linkers.
.
Schematic diagram of the vaccine construct consists of the adjuvant, helper epitopes and CTL epitopes, which joined together by proper linkers.
Allergenicity evaluation
The allergenicity of the vaccine construct was evaluated by submitting the sequence to the AlgPred server. Based on the result, the vaccine peptide was predicted to be non-allergic in the nature having a score of -1.1623. The threshold score was found to be -0.40, in which an input sequence is considered non-allergic for the scores less than this value.
Physicochemical analysis of vaccine constructs
Physicochemical characteristics of the vaccine construct were calculated by submitting the sequence to the ProtParam server. The molecular weight (MW) and theoretical isoelectric point (pI) value of the vaccine construct were 55.6 kDa and 5.70, respectively. The in vitro estimation of half-life in mammalian reticulocyte was found to be 30 hours. This value is estimated in vivo to be more than 20 and 10 hours in yeast and E. coli, respectively. The constructed vaccine obtained an instability index of 35.89 indicating stability of the protein. The aliphatic index is estimated to be 86.11 showing thermostability of the protein, in which higher value of this index indicates its more thermostability.
40
The grand average of hydropathicity (GRAVY) was found to be -0.062, while its negativity specifies its hydrophilicity in nature and ability to interact with water molecules.
The secondary structure prediction
The secondary structure of the constructed vaccine was predicted by the PSIPRED server. Fig. 6 shows the predicted secondary structure of the protein. The prediction revealed that the protein consists of 32.8% alpha helix (H), 3.8% beta strand and 63.4% coil (C). The output of the secondary structure prediction was used to refine the 3D model of the vaccine construct.
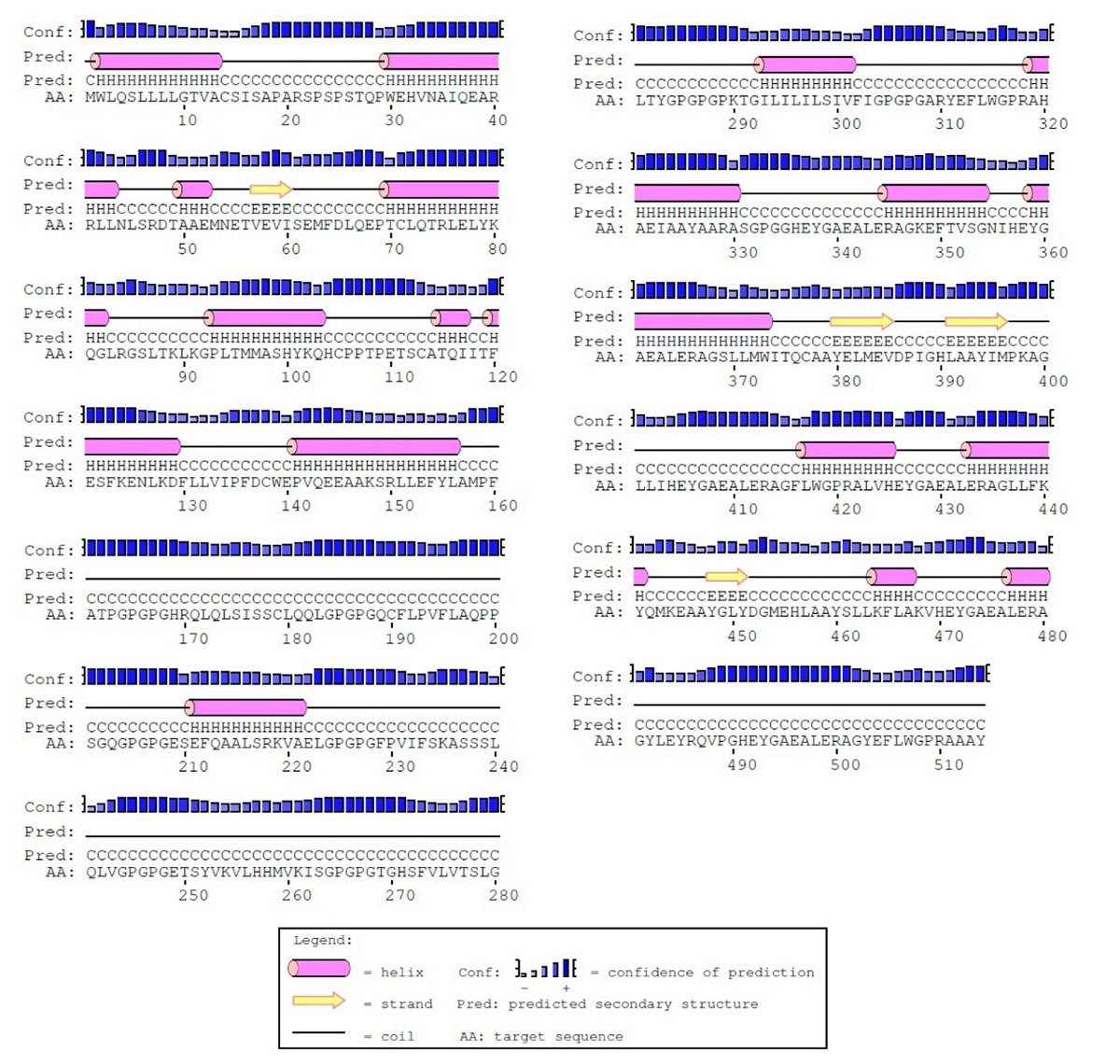
Fig. 6.
The secondary structure prediction of the constructed vaccine prepared by PSIPRED. Based on prediction, the protein consists of alpha-helix (32.8%), beta strands (3.8%) and coils (63.4%).
.
The secondary structure prediction of the constructed vaccine prepared by PSIPRED. Based on prediction, the protein consists of alpha-helix (32.8%), beta strands (3.8%) and coils (63.4%).
Homology modeling
The amino acid sequence of the constructed vaccine was submitted to the PSI-BLAST server to look for the potential structural templates of homology modeling. Based on the PSI-BLAST search result, different parts of the construct showed significant alignments with different proteins along its amino acid sequence, including PDB-IDs: 5D71, 4V0P, 2WA0, 4RS1, and 2L27. Accordingly, based on multi-template homology modeling, multiple alignments of these proteins and the vaccine sequence was calculated and used to generate a 3D-model for the construct by the MODELLER 9.17 program.
27
The MODELLER program predicts the 3D-model of an input sequence based on its alignment with sequences of known protein structures as a template. The best model was chosen based on the DOPE and GA341 objective functions, while the higher GA341 and/or the lower DOPE indicates higher quality of a generated model.
The tertiary structure prediction and refinement
The best generated 3D-model was refined in three steps. The model was first submitted to a loop refinement procedure running by MODELLER 9.17. The output structure of the loop refinement was further refined by GalaxyRefine server in order to refine the whole protein structure.
41
The GalaxyRefine method first reconstructs side chains and performs side-chain repacking and following overall structure relaxation (mild and aggressive relaxation) by molecular dynamics simulation.
Energy minimization of the constructed vaccine
A molecular dynamics simulation was done for 30ns in order to minimize the energy level of the constructed vaccine. It can be observed from the Fig. 8(A) that the RMSD value of the vaccine structure is constant and did not change significantly after 6 ns. This observation illustrates that the simulation time was long enough to achieve an equilibrium structure. Additionally, an average temperature of 30 ns simulation at 300 K for the studied system was equal to 300 ± 0.5 K as shown in Fig. 8(B) . Fig. 9 shows superimposition of the initial model and the refined model. It is clear from the figure that the quality of the 3D model was increased after efficient loop refinement and energy minimization.
Fig. 7.
The 3D structure of the constructed vaccine
.
The 3D structure of the constructed vaccine
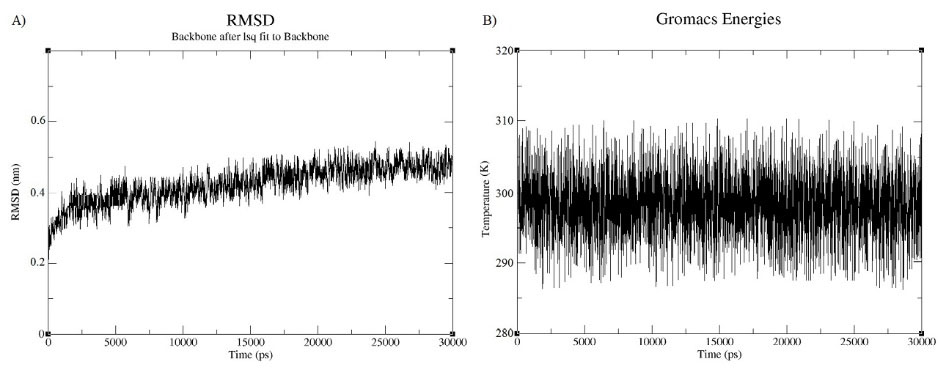
Fig. 8.
Molecular dynamic simulation analysis. A) A RMSD for the constructed vaccine. B) An average temperature for the vaccine.
.
Molecular dynamic simulation analysis. A) A RMSD for the constructed vaccine. B) An average temperature for the vaccine.
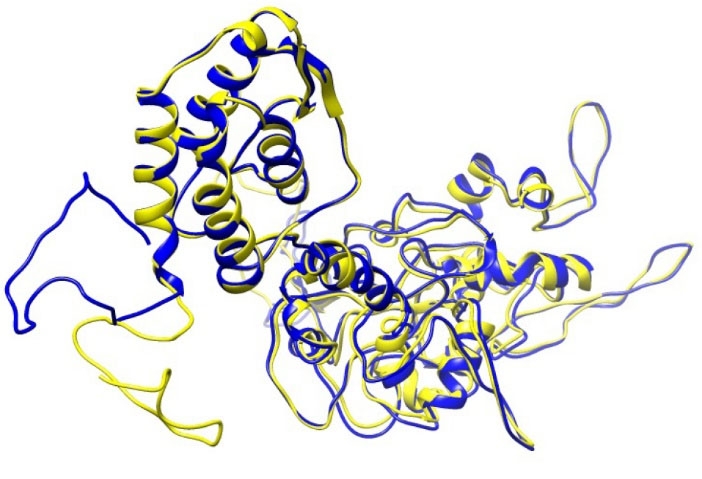
Fig. 9.
Superimposition of the initial model and the refined model
shown in yellow and blue colors, respectively. To obtain the
refined model, quality of the initial model was refined after precise
loop refinement and energy minimization.
.
Superimposition of the initial model and the refined model
shown in yellow and blue colors, respectively. To obtain the
refined model, quality of the initial model was refined after precise
loop refinement and energy minimization.
Model evaluation
The quality of the constructed 3D-model was assessed before and after the refinement using different evaluation tools. The PROCHECK web tool was used to investigate the backbone conformation of the model based on a Psi/Phi Ramachandran plot. The quality of the model after refinement was improved up to 82.4% in the most favored region and 14.2% in generally allowed region, in which only 2.9% of residues were located in the outlier region. ERRAT is another model evaluation tool to examine the overall quality of a model for non-bonded atomic interactions. Evaluation of the model by the ERRAT server indicates an improvement in the structure from 26.4% to 65.2% after the refinement. Furthermore, ProSA-web was used to calculate an overall quality score for the model. The server calculates a z-score of -4.43, which is in the normal range of scores for the native proteins. The overall scores obtained for the model are considered reasonable regarding that the vaccine structure was constructed based on multiple templates with a low sequence identity and using the threading approach.
42,43
Reverse translation
The sequence of vaccine sequence was submitted to the JCat server in order to generate a codon optimized DNA sequence. The gene sequence resulted in a codon adaptation index (CAI) of 1, where a CAI index more than 0.8 is acceptable for the high-level expression of a gene. The calculated frequency distribution in the gene was about 62%. The overall GC content of the gene was 40.89%, where the ideal range is from 30% up to 70%. The optimized gene does not contain the negative CIS elements and repeat sequences. The gene was cloned in E. coli vectors by introducing the NcoI and XhoI restriction sites to N and C- terminals of the sequence, respectively.
Identification of B-cell epitopes
The role of B-cell epitopes in cancer immunotherapy has been already proven. To predict the B-cell epitopes within the constructed vaccine, the full-length protein was submitted to BCPred and DiscoTope servers. Linear B-cell epitopes were predicted by BCPred as shown in Table S1, and discontinuous B-cell epitopes were determined by discoTope as listed in Table S2 (See online supplementary data).
Discussion
TNBC is known as one of the most difficult types of breast cancer for the treatment having a high probability of dissemination and recurrence. The rate of the survival of TNBC comparatively is lower than the other types of breast cancer, showing short overall survival, a high degree of malignancy and strong invasive potential. It is mostly diagnosed at advanced stages with a high risk of visceral metastasis.
44-46
Regarding these specific characteristics of TNBC and limited modalities of its treatment, immunotherapy can be considered as a relevant strategy for its prevention and treatment.
Cancer vaccines generally work based on the cellular immunity deduced from the CTL responses.
47
The efficiency of a constructed vaccine depends on the selection of proper CTL epitopes as the most key component, helper epitopes and adjuvant.
48
In addition, the fusion of these components needs appropriate linkers in order to generate a protein with the desired functionality. In other words, the selection of improper linkers may result in a protein having different characteristics. Accordingly, it is critically important to choose accurate components for the construction of a peptide vaccine. To achieve this goal, the tools from immunoinformatics can provide useful facilities for high accurate vaccine design. The feasibility, accuracy and speed of in silico tools for the analysis and design of multi-epitope vaccines against different infectious diseases or even cancer has been demonstrated by different computational studies.
49,50
Peptide-based vaccines generally induce antigen-specific T-cell responses against cancer-related antigens when they are administered with proper adjuvants. Essentially, the adjuvant is a key component within a peptide vaccine for optimizing the functionality of dendritic cells and generating an appropriate immune response. GM-CSF has been used in a number of tumor-associated antigen (TAA) protein-based vaccines. The capability of the GM-CSF-based vaccines to induce potent antiviral and antitumor response in preclinical experiments has been demonstrated.
16,51
It provides a critical link between dendritic cells and T cells, whereas it activates dendritic cells through the mechanism known as GM-CSF-induced antitumor immunity. Consequently, the GM-CSF can be an efficient candidate immune adjuvant to induce antitumor responses.
52
Additionally, linkers play an essential role in the structural and functional behavior of a peptide vaccine. The selected components for the vaccine construction were joined together via different linkers. Two motifs were used to link CTL epitopes, including HEYGAEALERAG and AAY. The specific cleavage site for both proteasomal and lysosomal degradation system is provided by the HEYGAEALERAG sequence. Moreover, the epitope presentation is improved by the AAY sequence whereas the generated C-terminal after cleavage properly binds to the TAP transporter or other chaperons.
53,54
Furthermore, the predicted T-helper components were linked together via the GPGPG linker. These linkers stimulate HTL responses and the conserve conformational dependent immunogenicity of helpers as well as antibody epitopes.
55
The EAAAK sequence was also used as a rigid linker for controlling the molecular distances and reducing interferences between the domains.
54,56
Ultimately, the whole vaccine structure was constructed by linking the three major sections of the vaccine, namely adjuvant, T-helper epitopes and CTL epitopes via the above appropriate linkers.
Several evaluations were carried out to investigate the immunological, physicochemical and structural features of the constructed vaccine. The protein sequence consists of 514 amino acids, which is considered a reasonable length for a cancer vaccine. A longer peptide enforces its presentation by dendritic cells increasing the induction strength of T-cell immune responses. However, sequences with a length less than 30 residues possibly bind to MHC-I or MHC-II molecules of nonqualified APCs.
57
In addition to the main goal of cancer vaccination in the induction of cellular arm of the host immune system, the maximum efficiency could be achieved by considering the HTL responses in the vaccine design. Furthermore, the critical role of CD4+ T-helper cells in the preservation of CTL responses has been demonstrated.
Conclusion
TNBC has emerged as an important subtype of breast cancer and the reason for a large number of deaths among women worldwide. In this work, we used the reverse vaccinology approach to design a multi-epitope based vaccine for the cancer immunotherapy using immunoinformatics tools. The vaccine was constructed by the fusion of multiple CTL and HTL epitopes via appropriate linkers in order to stimulate the humoral, cellular and innate immune responses. The vaccine was also included GM-CSF as an adjuvant to increase its immunogenicity. Based on several immunoinformatic analyses, the designed multi-epitope peptide can be efficiently used for the therapeutic or prophylactic purposes against TNBC. The successful immunogenicity of the proposed vaccine would be proved by the future in vitro and in vivo studies.
Acknowledgment
The authors like to acknowledge the Research Center for Pharmaceutical Nanotechnology at Tabriz University of Medical Sciences and also the Department of Computer Science at the Faculty of Mathematical Sciences at the University of Tabriz for their technical support.
Funding sources
The authors would like to thank Iranian Elite National Foundation “Bonyad Melli Nokhbegan” and the Iranian National Science Foundation (INSF) for the financial support (grant No: 95005404).
Ethical approval
None to be declared.
Competing interests
As the contributing author to this study, YO acts as the Editor of the Bioimpacts journal. It is hereby declared that his association with the journal has neither influenced the review process nor affected the acceptance of this study.
Authors contribution
SP and YO developed the concept of the study. SP, MMP and JR performed the analyses. SP and MMP drafted the manuscript. YO finalized the manuscript and the submission process. All the coauthors approved the submission of the manuscript.
Supplementary Materials
Supplementary data contains Tables S1-S2.
Research Highlights
What is the current knowledge?
simple
-
√ Triple-negative breast cancer (TNBC) is an important
subtype of breast cancer, which occurs in the absence of
estrogen, progesterone and HER-2 receptors.
-
√ Cancer testis antigens (CTAs) have been reported as one
of the promising be a more significant therapeutic way for
cancer treatment.
What is new here?
simple
-
√ In the present study, immunoinformatics approach was
used to design a multi-epitope peptide vaccine to combat the
TNBC malignancies.
-
√ The vaccine peptide was constructed by the fusion of
three crucial components, including the CD8+ cytotoxic T
lymphocytes (CTLs) epitopes, helper epitopes and adjuvant.
-
√ The homology modeling approaches were conducted for
prediction of the 3D structure of the constructed cancer
vaccine.
References
-
Vighnesh Walavalkar AKaDK. Familial Breast Cancer and Genetic Predisposition in Breast Cancer. Precision Molecular Pathology of Breast Cancer: Springer; 2015. p. 15-37.
- Nowell PC. The clonal evolution of tumor cell populations. Science 1976; 194:23-8. [ Google Scholar]
- Parvizpour S, Razmara J, Omidi Y. Breast cancer vaccination comes to age: impacts of bioinformatics. Bioimpacts 2018; 8:213-22. doi: 10.15171/bi.2018.25 [Crossref] [ Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414:105-11. doi: 10.1038/35102167 [Crossref] [ Google Scholar]
- Curigliano G, Viale G, Ghioni M, Jungbluth AA, Bagnardi V, Spagnoli GC. Cancer-testis antigen expression in triple-negative breast cancer. Ann Oncol 2011; 22:98-103. doi: 10.1093/annonc/mdq325 [Crossref] [ Google Scholar]
- Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci 2009; 100:2014-21. doi: 10.1111/j.1349-7006.2009.01303.x [Crossref] [ Google Scholar]
- Almeida LG, Sakabe NJ, deOliveira AR, Silva MC, Mundstein AS, Cohen T. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res 2009; 37:D816-9. doi: 10.1093/nar/gkn673 [Crossref] [ Google Scholar]
- Lee HJ, Kim JY, Song IH, Park IA, Yu JH, Gong G. Expression of NY-ESO-1 in Triple-Negative Breast Cancer Is Associated with Tumor-Infiltrating Lymphocytes and a Good Prognosis. Oncology 2015; 89:337-44. doi: 10.1159/000439535 [Crossref] [ Google Scholar]
- Badovinac Crnjevic T, Spagnoli G, Juretic A, Jakic-Razumovic J, Podolski P, Saric N. High expression of MAGE-A10 cancer-testis antigen in triple-negative breast cancer. Med Oncol 2012; 29:1586-91. doi: 10.1007/s12032-011-0120-9 [Crossref] [ Google Scholar]
- Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 2005; 5:615-25. doi: 10.1038/nrc1669 [Crossref] [ Google Scholar]
- Barker PA, Salehi A. The MAGE proteins: emerging roles in cell cycle progression, apoptosis, and neurogenetic disease. J Neurosci Res 2002; 67:705-12. doi: 10.1002/jnr.10160 [Crossref] [ Google Scholar]
- Weon JL, Potts PR. The MAGE protein family and cancer. Curr Opin Cell Biol 2015; 37:1-8. doi: 10.1016/j.ceb.2015.08.002 [Crossref] [ Google Scholar]
- Krishnadas DK, Bai F, Lucas KG. Cancer testis antigen and immunotherapy. Immunotargets Ther 2013; 2:11-9. doi: 10.2147/ITT.S35570 [Crossref] [ Google Scholar]
- Wang H, Sang M, Geng C, Liu F, Gu L, Shan B. MAGE-A is frequently expressed in triple negative breast cancer and associated with epithelial-mesenchymal transition. Neoplasma 2016; 63:44-56. doi: 10.4149/neo_2016_006 [Crossref] [ Google Scholar]
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011; 480:480-9. doi: 10.1038/nature10673 [Crossref] [ Google Scholar]
- Ahlers JD, Belyakov IM, Terabe M, Koka R, Donaldson DD, Thomas EK. A push-pull approach to maximize vaccine efficacy: abrogating suppression with an IL-13 inhibitor while augmenting help with granulocyte/macrophage colony-stimulating factor and CD40L. Proc Natl Acad Sci U S A 2002; 99:13020-5. doi: 10.1073/pnas.192251199 [Crossref] [ Google Scholar]
- Slingluff CL, Jr Jr. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination?. Cancer J 2011; 17:343-50. doi: 10.1097/PPO.0b013e318233e5b2 [Crossref] [ Google Scholar]
- Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S. The Universal Protein Resource (UniProt). Nucleic Acids Res 2005; 33:D154-9. doi: 10.1093/nar/gki070 [Crossref] [ Google Scholar]
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic acids research 2005; 33:W526-W31. [ Google Scholar]
- Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N. The immune epitope database 20. Nucleic acids research 2009; 38:D854-D62. [ Google Scholar]
- EL‐Manzalawy Y, Dobbs D, Honavar V. Predicting linear B‐cell epitopes using string kernels. Journal of molecular recognition 2008; 21:243-55. [ Google Scholar]
- Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS computational biology 2012; 8:e1002829. [ Google Scholar]
- Saha S, Raghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res 2006; 34:W202-9. doi: 10.1093/nar/gkl343 [Crossref] [ Google Scholar]
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 1999; 112:531-52. [ Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics 2000; 16:404-5. [ Google Scholar]
- Razmara J, Deris S, Parvizpour S. A rapid protein structure alignment algorithm based on a text modeling technique. Bioinformation 2011; 6:344-7. [ Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics 2006; Chapter 5:Unit-5 6. doi: 10.1002/0471250953.bi0506s15 [Crossref] [ Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605-12. doi: 10.1002/jcc.20084 [Crossref] [ Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 2007; 35:W407-10. doi: 10.1093/nar/gkm290 [Crossref] [ Google Scholar]
- Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 1993; 2:1511-9. doi: 10.1002/pro.5560020916 [Crossref] [ Google Scholar]
- Grote A, Hiller K, Scheer M, Munch R, Nortemann B, Hempel DC. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 2005; 33:W526-31. doi: 10.1093/nar/gki376 [Crossref] [ Google Scholar]
- Kim Y, Sette A, Peters B. Applications for T-cell epitope queries and tools in the Immune Epitope Database and Analysis Resource. J Immunol Methods 2011; 374:62-9. doi: 10.1016/j.jim.2010.10.010 [Crossref] [ Google Scholar]
- Graff-Dubois S, Faure O, Gross DA, Alves P, Scardino A, Chouaib S. Generation of CTL recognizing an HLA-A*0201-restricted epitope shared by MAGE-A1, -A2, -A3, -A4, -A6, -A10, and -A12 tumor antigens: implication in a broad-spectrum tumor immunotherapy. J Immunol 2002; 169:575-80. [ Google Scholar]
- Webb AI, Dunstone MA, Chen W, Aguilar MI, Chen Q, Jackson H. Functional and structural characteristics of NY-ESO-1-related HLA A2-restricted epitopes and the design of a novel immunogenic analogue. J Biol Chem 2004; 279:23438-46. doi: 10.1074/jbc.M314066200 [Crossref] [ Google Scholar]
- Schmidt J, Guillaume P, Dojcinovic D, Karbach J, Coukos G, Luescher I. In silico and cell-based analyses reveal strong divergence between prediction and observation of T-cell-recognized tumor antigen T-cell epitopes. J Biol Chem 2017; 292:11840-9. doi: 10.1074/jbc.M117.789511 [Crossref] [ Google Scholar]
- Zhao L, Zhang M, Cong H. Advances in the study of HLA-restricted epitope vaccines. Hum Vaccin Immunother 2013; 9:2566-77. doi: 10.4161/hv.26088 [Crossref] [ Google Scholar]
- Peng JR, Dong N, Liu HW, Leng XS. [Identification of a naturally presented MAGE-A3 epitope on the surface of HLE cell line by mass spectrometry]. Zhonghua Wai Ke Za Zhi 2007; 45:595-7. [ Google Scholar]
- Katsura F, Eura M, Chikamatsu K, Oiso M, Yumoto E, Ishikawa T. Analysis of individual specific cytotoxic T lymphocytes for two MAGE-3-derived epitopes presented by HLA-A24. Jpn J Clin Oncol 2000; 30:117-21. [ Google Scholar]
- Jia ZC, Tian Y, Huang ZM, Wang JX, Fu XL, Ni B. Identification of a new MAGE-A10 antigenic peptide presented by HLA-A*0201 on tumor cells. Cancer Biol Ther 2011; 11:395-400. [ Google Scholar]
- Ali M, Pandey RK, Khatoon N, Narula A, Mishra A, Prajapati VK. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci Rep 2017; 7:9232. doi: 10.1038/s41598-017-09199-w [Crossref] [ Google Scholar]
- Heo L, Park H, Seok C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res 2013; 41:W384-8. doi: 10.1093/nar/gkt458 [Crossref] [ Google Scholar]
- Parvizpour S, Razmara J, Shamsir MS, Illias RM, Abdul Murad AM. The role of alternative salt bridges in cold adaptation of a novel psychrophilic laminarinase. J Biomol Struct Dyn 2017; 35:1685-92. doi: 10.1080/07391102.2016.1191043 [Crossref] [ Google Scholar]
- Parvizpour S, Razmara J, Jomah AF, Shamsir MS, Illias RM. Structural prediction of a novel laminarinase from the psychrophilic Glaciozyma antarctica PI12 and its temperature adaptation analysis. J Mol Model 2015; 21:63. doi: 10.1007/s00894-015-2617-1 [Crossref] [ Google Scholar]
- Jia H, Truica CI, Wang B, Wang Y, Ren X, Harvey HA. Immunotherapy for triple-negative breast cancer: Existing challenges and exciting prospects. Drug Resist Updat 2017; 32:1-15. doi: 10.1016/j.drup.2017.07.002 [Crossref] [ Google Scholar]
- Anders CK, Deal AM, Miller CR, Khorram C, Meng H, Burrows E. The prognostic contribution of clinical breast cancer subtype, age, and race among patients with breast cancer brain metastases. Cancer 2011; 117:1602-11. doi: 10.1002/cncr.25746 [Crossref] [ Google Scholar]
- Papa A, Caruso D, Tomao S, Rossi L, Zaccarelli E, Tomao F. Triple-negative breast cancer: investigating potential molecular therapeutic target. Expert Opin Ther Targets 2015; 19:55-75. doi: 10.1517/14728222.2014.970176 [Crossref] [ Google Scholar]
- Kessler JH, Melief CJ. Identification of T-cell epitopes for cancer immunotherapy. Leukemia 2007; 21:1859-74. doi: 10.1038/sj.leu.2404787 [Crossref] [ Google Scholar]
- van der Burg SH, Bijker MS, Welters MJ, Offringa R, Melief CJ. Improved peptide vaccine strategies, creating synthetic artificial infections to maximize immune efficacy. Adv Drug Deliv Rev 2006; 58:916-30. doi: 10.1016/j.addr.2005.11.003 [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Naghili B, Saeedi N, Parvizpour S, Nematollahi A. A novel in silico minigene vaccine based on CD4(+) T-helper and B-cell epitopes of EG95 isolates for vaccination against cystic echinococcosis. Comput Biol Chem 2018; 72:150-63. doi: 10.1016/j.compbiolchem.2017.11.008 [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Daghighkia H, Nematollahi A, Omidi Y. A novel B- and helper T-cell epitopes-based prophylactic vaccine against Echinococcus granulosus. Bioimpacts 2018; 8:39-52. doi: 10.15171/bi.2018.06 [Crossref] [ Google Scholar]
- Schneble EJ, Berry JS, Trappey FA, Clifton GT, Ponniah S, Mittendorf E. The HER2 peptide nelipepimut-S (E75) vaccine (NeuVax) in breast cancer patients at risk for recurrence: correlation of immunologic data with clinical response. Immunotherapy 2014; 6:519-31. doi: 10.2217/imt.14.22 [Crossref] [ Google Scholar]
- Yan WL, Shen KY, Tien CY, Chen YA, Liu SJ. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017; 9:347-60. doi: 10.2217/imt-2016-0141 [Crossref] [ Google Scholar]
- Bergmann CC, Yao Q, Ho CK, Buckwold SL. Flanking residues alter antigenicity and immunogenicity of multi-unit CTL epitopes. J Immunol 1996; 157:3242-9. [ Google Scholar]
- Hajighahramani N, Nezafat N, Eslami M, Negahdaripour M, Rahmatabadi SS, Ghasemi Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect Genet Evol 2017; 48:83-94. doi: 10.1016/j.meegid.2016.12.010 [Crossref] [ Google Scholar]
- Livingston B, Crimi C, Newman M, Higashimoto Y, Appella E, Sidney J. A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J Immunol 2002; 168:5499-506. [ Google Scholar]
- Nezafat N, Ghasemi Y, Javadi G, Khoshnoud MJ, Omidinia E. A novel multi-epitope peptide vaccine against cancer: an in silico approach. J Theor Biol 2014; 349:121-34. doi: 10.1016/j.jtbi.2014.01.018 [Crossref] [ Google Scholar]
- Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer 2008; 8:351-60. doi: 10.1038/nrc2373 [Crossref] [ Google Scholar]