Bioimpacts. 9(2):79-88.
doi: 10.15171/bi.2019.11
Original Research
Therapeutic potency of substituted chromones as Alzheimer’s drug: Elucidation of acetylcholinesterase inhibitory activity through spectroscopic and molecular modelling investigation
Prayasee Baruah 1, Mostofa Ataur Rohman 1, Semen O. Yesylevskyy 2, Sivaprasad Mitra 1, * 
Author information:
1Centre for Advanced Studies in Chemistry, North-Eastern Hill University, Shillong – 793 022, India
2Department of Physics of Biological Systems, Institute of Physics of the National Academy of Sciences of Ukraine, Prospect Nauky 46, 03028 Kyiv, Ukraine
Abstract
Introduction:
Documentation on the potency of chromones as acetylcholinesterase (AChE) antagonists has paved the way for the design and usage of new chromone analogues as inhibitors of AChE modelled on the hypothesis based on cholinergic pathway of Alzheimer’s disease (AD). Here, 2 minimally substituted chromones, namely 3-cyanochromone (CyC) and 7-amino-3- methylchromone (AMC), were checked for their AChE inhibition efficacies and plasma protein modulation.
Methods:
Colorimetric enzymatic assay as well as fluorescence measurements were performed for obtaining the experimental results, which were further corroborated by molecular docking and simulation studies.
Results:
The investigated systems exhibited strong inhibition activities against AChE, with CyC (IC50= 85.12 ± 6.70 nM) acting as better inhibitor than AMC (IC50 = 103.09 ± 11.90 nM) and both having IC50 values in the range of FDA approved cholinergic drug Donepezil (IC50 = 74.13 ± 8.30 nM). Non-competitive inhibition was observed in both the cases with the inhibitors binding near the peripheral anionic site (PAS) of the enzyme. Having one planar nitrile group in CyC as compared to sp3 hybridised substituents in AMC facilitated stacking interactions in the former, accounting for its higher inhibitory efficacy. A significant decrease in the inhibition potency of CyC (~32%) was noted in comparison with AMC (~5%) when the experiments were performed in presence of human serum albumin (HSA) instead of pure aqueous buffer.
Conclusion:
This comparative study affirms the importance of meticulous substitution in the chromone scaffold to promote maximum inhibition potency, while considering their usage as AD drugs.
Keywords: Acetylcholinesterase inhibition, AD drug, Chromones, PAS binding, Stacking interactions, Thioflavin T fluorescence
Copyright and License Information
© 2019 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Chromones, which are 1-benzopyran-4-one ring systems, are renowned for their miscellany of therapeutic properties.
1-6
Chromones act as exceptional prototypes for modifications based on structural changes and chemical substitutions, which pave the way for synthesis of varied compounds with a multitude of different pharmacological utilities.
4
Because of this, they are considered vital in the world of medicine and deemed as an advantageous framework for drug discovery.
7,8
Along with numerous applications, they have been known to exhibit agonistic effects towards the activity of acetylcholinesterase (AChE), which is an extremely effectual enzyme and a part of the central nervous system (CNS). Experiments testing the effect of chromones on AChE peaked up with the observation of ensaculin
9,10
; a coumarin derivative which is an isomer of chromones, inhibiting AChE activity to significant proportions.
AChE causes the depletion of acetylcholine (ACh), which is a transmitter in the CNS, by rapid hydrolysis decline, which is significant in the progression of Alzheimer’s disease (AD), a neurodegenerative symptom which usually culminates in dementia and is the reason for the alarming worldwide statistics of the same.
11-14
Therefore, cholinesterase inhibitors, which retard the activity of AChE, subsequently increasing the ACh levels in the brain, have been deliberated as characteristic treatment for this rapidly escalating disease.
15,16
However, treatment options being symptomatic, an extensive search is on for developing new drugs with enhanced therapeutic effectiveness.
There exist quite a number of recent references that show the AChE inhibitory effects of coumarin isomers and their derivatives. In this context, several 3-formylchromone derivatives, with very promising AChE inhibitory activities, have been synthesized by Parveen et al.
17
Through a multi-target-directed ligand strategy, some chromone-based compounds have been synthesized which can exhibit plural biologically relevant activities.
18
Novel chromones like 2-carboxamidoalkylbenzylamines have been developed by Liu et al
19
as AChE inhibitors showing inhibitory efficiency in the sub-micromolar concentration range (the lowest with IC50 = 0.07 μM), with some compounds possessing higher selectivity for AChE over butyl cholinesterase (BuChE). These compounds also demonstrated perceptible inhibition of the aggregation of amyloid-β (Aβ) fibrils through self- as well as Cu2+-induction, another important pathway in the hypothesis of AD.
20
Recently, some chromone derivatives from Agarwood have also been developed and tested to be potent AChE inhibitors.
21
Pertinent to general observation, coumarin analogues have been identified to associate with AChE at a site different from the substrate binding site, known as the peripheral anionic site (PAS).
22
Judicious substitution with amine functional groups using appropriate spacer may amend the binding sites, in turn facilitating superior interaction with AChE and transforming them into more potent inhibitors.
23-25
Hence, identifying the site and mode of inhibition deems vital in AChE inhibition.
In the passageway of drugs in the body, many of them associate with blood constituents, mainly plasma proteins, wherein only that fraction of the total amount which is not bound remains accessible for dispersing out of the vascular structure to the targeted locations for therapeutic efficacy.
26
In fact, the role of serum proteins in modulating the inhibitory efficacies of targeted drugs and the importance of considering the behaviour of inhibitors in presence of serum matrix as a potential screening test for AD drugs was reported in earlier communications from this laboratory.
27
Therefore, a comparison on the activities of potential drugs in buffer and human serum albumin (HSA) seems important while screening potential drugs.
In connection with AD, blood plasma proteins like HSA have portrayed the ability of controlling the aggregation of Aβ by controlling the equilibrium of Aβ peptide through the blood brain barrier.
28,29
Reports on 2 groups of patients, one with higher level of serum albumin and the other with lower level suggested better cognizance and reasoning performance in the former over the latter.
30,31
Three homologous domains (named I, II, and III) form the core of HSA’s structural organisation, each comprised of 2 distinct helical subdomains (named A and B) which are joined together by a loop.
32
HSA, on account of its modular arrangement, consists of many different sites which facilitate ligand binding. It, however, encompasses 2 sites with higher affinity for binding of ligands called Sudlow’s sites I and II located in Subdomain IIA and IIIA, respectively.
33
In this report, 2 chromone derivatives, one containing a methyl group and a primary amine in its chromone skeleton (7-amino-2-methylchromone, AMC) and the other possessing a tertiary amine moiety (3-cyanochromone, CyC), the structures and other relevant parameters of which have been depicted in Table 1, have been tested for their AChE inhibition capabilities. The modulation of inhibitor potency in HSA medium has also been quantified. The affirmative experimental results of these 2 simple chromone prototypes imply the benefits of designing new drugs with the chromone scaffold as a backdrop and validating several structure activity relationship (SAR) reviews pertaining to the enzyme inhibitory capacities of chromones.
Table 1.
Structures and relevant pharmacological parameters for the tested systems
|
Name
|
Structure
|
Molecular weight (g mol-1)
|
Log p
|
TPSA (Ǻ
2
)
|
| 3-cyanochromone (CyC) |
|
171.15 |
1.50 |
54.00 |
| 7-amino-2-methylchromone (AMC) |
|
175.18 |
1.00 |
56.23 |
Materials and Methods
Chemicals
The reagents used in the present study were of highest quality analytical grade, which were used as received. AMC and CyC were procured from Sigma Aldrich, Germany (cat. No. 383481 and 30779705, respectively). AChE from Electrophorus electricus (electric eel) (cat. No. C2888), acetylthiocholine iodide (cat no. A5751), Donepezil hydrochloride monohydrate (cat no. D6821), and thioflavin T (ThT, cat. No. T3516) were all purchased from Sigma Aldrich, Germany. Dithiobis (2-nitrobenzoic acid) was received from Sisco Research Laboratories (SRL, India) (product NO. 32363) and HSA (Cat. No. 10878) was purchased from Sigma Aldrich, Germany, in a lyophilized powder form. The experiments were performed in analytical grade type II water of ~10 MΩ cm resistivity (at 298 K), obtained from Elix 10 (Millipore India Pvt. Ltd) water purification system. The pH of the buffer solutions were checked by Systronics µ-pH system 361.
Kinetic characterization of AChE activity
Ellman method with slight modification was used for assaying the activity of AChE in phosphate buffer of pH = 8.0 (0.1 M) at 298 K and for its kinetic characterization.
34
The details of the experimental procedure is given in supplementary section ST1.The scheme for the basis of the enzyme kinetics is shown in Scheme 1.
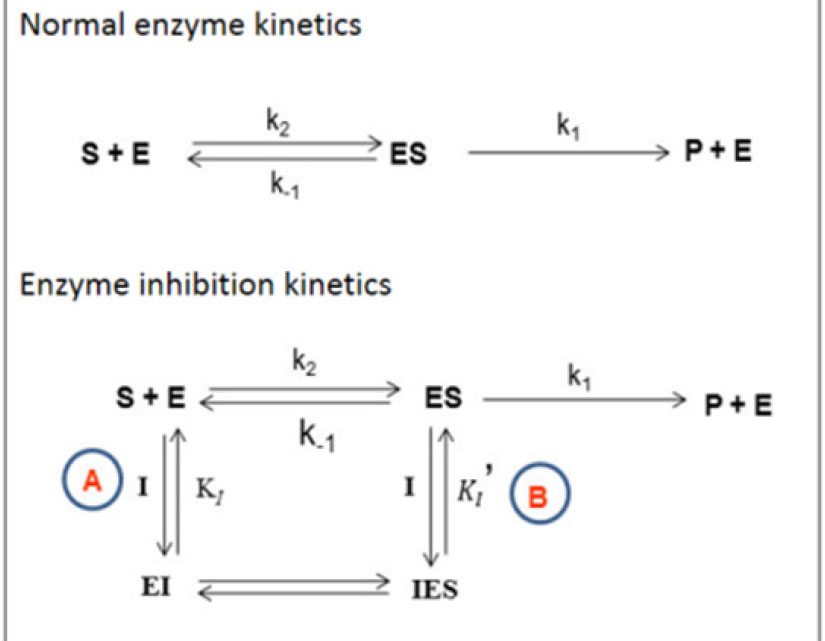
Scheme 1.
The Michaelis-Menten (MM) scheme for enzyme inhibitory kinetics. A and B represent different inhibition pathways.
.
The Michaelis-Menten (MM) scheme for enzyme inhibitory kinetics. A and B represent different inhibition pathways.
Equation 1 gives the initial rate (ν0) of the hydrolysis reaction:
Eq. (1)
where ε represents the absorption coefficient of the yellow anion, l is the path length (= 0.442 cm
35
).
Non-linear regression analysis of ν0 against initial substrate concentration (S0) gave the values of the Michaelis-Menten (MM) constant, Km and maximum hydrolysis rate, Vmax in the normal and enzyme inhibition cases using Eq. 2 and Eq. 3, respectively.
36
Eq. (2)
Eq. (3)
where,
(representing path A only for inhibition);
(representing both paths A and B).
The characteristic IC50 parameter for inhibition in both the media were obtained from the modified Hill relation (Eq. 4).
37-39
Eq. (4)
where ∆V is the initial rate decrease observed in presence of a definite concentration of inhibitor [I], ∆Vmax represents maximal initial velocity decrease, K0.5 which is pharmacologically equivalent to IC50, the inhibitor concentration to induce half-maximal change in the initial velocity. The term nH represents the Hill coefficient.
Fluorescence measurements
All steady state fluorescence studies were carried out in Quanta Master (QM-40) apparatus (Photon Technology International, PTI), the details of which have been given in supplementary section ST2. The following relation was used to obtain the corrected fluorescence intensity after removal of the inner filter effect
40
Eq. (5)
Here, the absorbance of the free ThT is given by A and Atot represents the total absorbance of the solution at λE, which is the experimental excitation wavelength.
Molecular dynamics simulation and docking calculation
Molecular docking calculations were performed for a deeper understanding of the binding of the inhibitors to the macromolecules. After retrieval of the 3D structures of AChE (PDB ID: 1C2B) and HSA (PDB ID: 1AO6) from Protein Data Bank (http://www.rcsb.org), they were made suitable for docking by eliminating all heteroatoms, water molecules, and ions. The ligand structures were fully optimized with density functional theory at B3LYP/6-311++g (d,p) levels as incorporated in Gaussian 09 platform.
Recent molecular dynamics (MD) simulation results revealed the crystal structure of AChE protein to be significantly different from its equilibrium structure in the solvent.
41
In this work, extensive MD simulations were carried out on the protein structure in aqueous medium to equilibrate it. Gromacs 5.1.2 package was used for the simulations,
42
utilizing AMBER99SB force field. The curtailed dodecahedron box used for counterbalancing the protein’s negative charge contained ~13500 TIP3P water molecules and 9 Na+ ions. Solvated system was energy minimized using the methodology of the steepest descent algorithm. Temperature and pressure were kept constant at 310 K and 1 bar, respectively, throughout the course of production simulations. Velocity rescale thermostat
43
and Berendsen barostat
44
were used for maintaining the temperature and pressure, respectively. PME method was used for long state electrostatics and 2 fs was used as the integration step
45
with Verlet cutoff.
46
The system was simulated for 465 ns. The equilibration was monitored by RMSD of protein in respect to its initial conformation.
It is to be noted that AChE exhibits large conformational changes in water in comparison to crystal structure. Therefore, the backbone RMSD is more relevant than RMSD of heavy atoms; because it reflects the stabilization of the tertiary and secondary structures reliably without any noise introduced by the motion of side chains. That is why the backbone RMSD was used in evaluating the protein equilibration (Fig. S1). It is clear that the RMSD stabilizes only after approximately 400 ns of simulation. Thus, only last 65 ns of the trajectory were considered equilibrated and used for subsequent docking simulations. One hundred thirty frames were extracted with the step of 0.5 ns from equilibrated part of the trajectory. The structures aligned by their peptide backbones, thus obtained, were taken to represent the ensemble of all solvated protein conformations. All docking took place inside a volume of 25 Å
3
which was at the middle of the center of masses of the residues 70, 72, 121, 279, and 334. The MD trajectory was evidence of the flexibility of the protein and the reason the protein was fixed for all dockings.
In the case of HSA, all-atom MD trajectory of 100 ns of pre-equilibrated protein in water was utilized as per standard protocol.
47
In this case, 200 frames were extracted from this trajectory and the docking volume of 40 Å
3
was centered at the center of masses of the residues 134, 186, 123, 117, 161, 138, 165, 138, 161, and 157 as suggested by blind docking in the case of HSA.
All structures to be docked were fashioned using MGLTools-1.5.6 software whereas the default protocol of Autodock Vina
48
was used for the actual docking. The docked poses were fashioned using Chimera
49
and Ligplot.
50
Pteros 2.0 molecular modeling library was the base of the analysis of docking results.
51
Five top-ranked poses were recorded for each docking simulation. VMD 1.9.3
52
was used for visualization purposes.
Results
Physico-chemical properties and comparative AChE inhibition activity of the investigated systems
All the relevant physico-chemical parameters for the investigated systems have been depicted in Table 1. The logP and total polar surface area (TPSA) values of the chromones were calculated using Molinspiration cheminformatics software (https://www.molinspiration.com) and found to be 1.50 and 1.00 for CyC and AMC, respectively. The corresponding TPSA values were 54.00 and 56.23 Ǻ2 respectively.
The performed kinetic experiments revealed the inhibition to be a non-competitive one. This type of inhibition falls under the category of mixed inhibition, and as per the defining characteristics of non-competitive inhibition, here only the Vmax suffered a decrease. On the other hand, Km remained practically unaltered (Table 2). Here, the values of α and αʹ were found to be identical. The MM and LB plots (Fig. 1, (ia) and (ic), respectively) show that the reduction in Vmax is higher in case of CyC, which was further verified by the estimation of IC50 values of individual systems described later. The cholinergic drug, DON, which was used as a control in our experiment showed comparable kinetic parameters that matched the previously published reports.
27
All experimental results of DON have been supplied in the supplementary section (Figs. S2-S4).
Table 2.
List of kinetic parameters for enzymatic reaction at varying concentrations of inhibitor in aqueous buffer media
|
Inhibitor
|
K
m
/µM
|
V
max
/nMs
-1
|
α
|
α'
|
IC
50
(nM)
|
n
H
|
|
Michaelis-Menten parameters
|
Hill parameters
|
|
AChE in CyC
|
| [CyC] = 0 nM |
174 ± 15
(177±20)
|
825 ± 24
(841±20)
|
1.0
(1.0)
|
1.0
(1.0)
|
85.12 ± 6.7
(113.43±4.6)
|
1.51 ± 0.1
(1.89±0.17)
|
[CyC] =5 nM
|
172 ± 20
(178±25)
|
631 ± 21
(730±17)
|
1.3
(1.1)
|
1.3
(1.1)
|
| [CyC] =10 nM |
180 ± 23
(179±19)
|
538 ± 31
(671±30)
|
1.5
(1.2)
|
1.5
(1.2)
|
|
AChE in AMC
|
| [AMC] = 0 nM |
149 ± 14
(146±35)
|
793 ± 23
(798±23)
|
1.0
(1.0)
|
1.0
(1.0)
|
110.64 ± 12.1
(115.11±12.2)
|
1.45 ± 0.1
(1.52±0.1)
|
| [AMC] =5 nM |
148 ± 18
(150±38)
|
670 ± 26
(711±47)
|
1.2
(1.2)
|
1.2
(1.2)
|
| [AMC] =10 nM |
155 ± 36
(179±27)
|
560 ± 39
(557±39)
|
1.4
(1.5)
|
1.4
(1.4)
|
|
AChE in DON
|
| [DON] = 0 nM |
146 ± 28
(140±32)
|
793 ± 47
(794±59)
|
1.0
(1.0)
|
1.0
(1.0)
|
74.13 ± 5.6
(110.64±12)
|
1.45 ± 0.1
(1.51±0.1)
|
| [DON] = 5 nM |
160 ± 36
(163±38)
|
550 ± 48
(625±42)
|
1.4
(1.4)
|
1.4
(1.4)
|
| [DON] =10 nM |
138 ± 13
(140±26)
|
484 ± 15
(571±35)
|
1.5
(1.4)
|
1.6
(1.4)
|
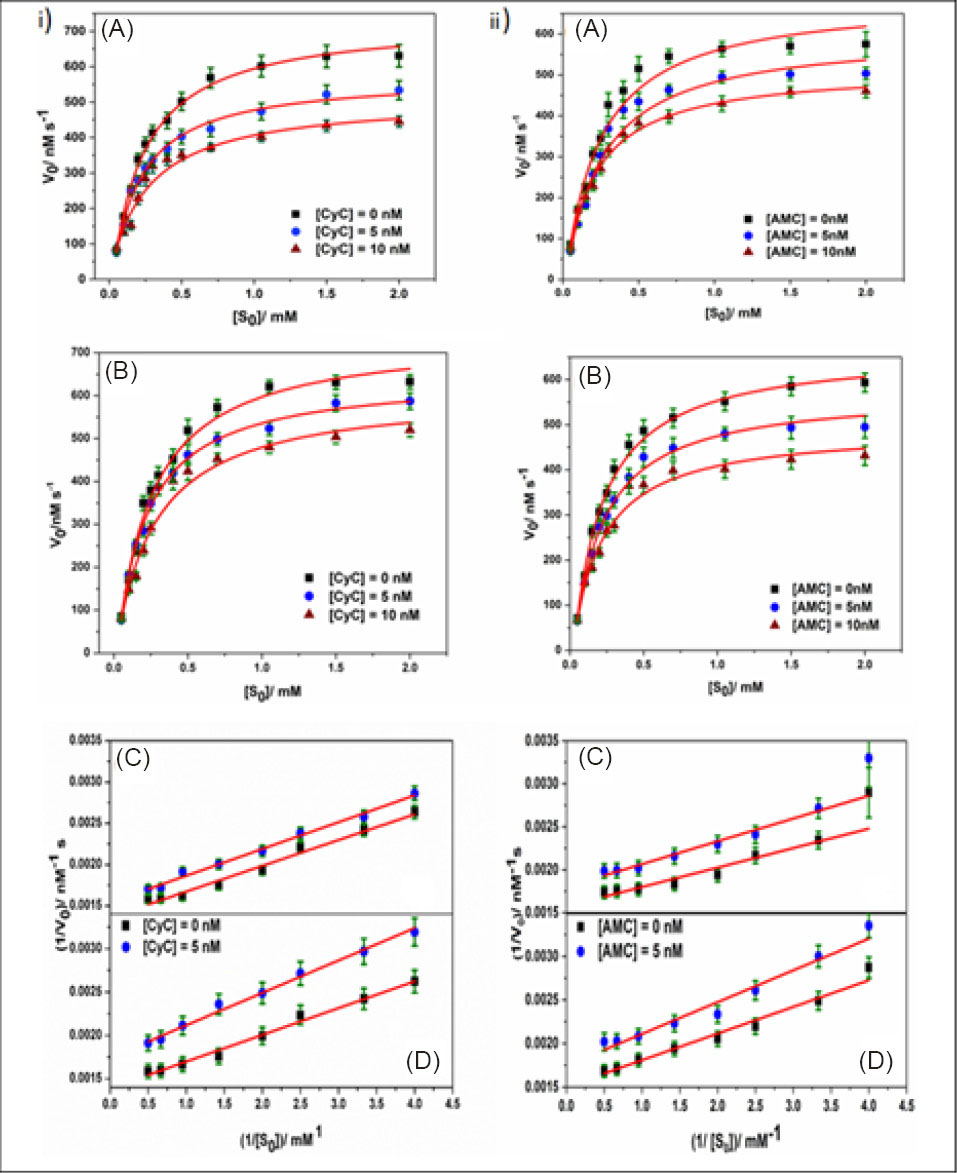
Fig. 1.
Hydrolysis curve (scattered points) (A, B) and Lineweaver-Burk (LB) plots (C, D) for the AChE hydrolysis data in aqueous buffer of pH 8.0 (A, C) and HSA medium (B, D) in absence and presence of different concentrations of CyC (1) and AMC (2). The solid line in hydrolysis curves represents non-linear regression of the experimental data points. [AChE] = 0.079 u/mL.
.
Hydrolysis curve (scattered points) (A, B) and Lineweaver-Burk (LB) plots (C, D) for the AChE hydrolysis data in aqueous buffer of pH 8.0 (A, C) and HSA medium (B, D) in absence and presence of different concentrations of CyC (1) and AMC (2). The solid line in hydrolysis curves represents non-linear regression of the experimental data points. [AChE] = 0.079 u/mL.
ThT displacement studies
The fluorescence intensities of ThT-AChE system suffer considerable hypochromic shifts on addition of gradually surging amounts of the investigated compounds. The quenching of fluorescence was quantitatively analysed using the Stern-Volmer (SV) equation. The greater binding in case of CyC is demonstrated with its higher KSV value (3.7 × 105 M-1) in comparison to that of AMC (2.3 × 105 M-1). The fluorescence emission quenching spectra as well as the SV plot for CyC are depicted in Fig. 2. The corresponding plots for AMC are shown in Fig. S5.
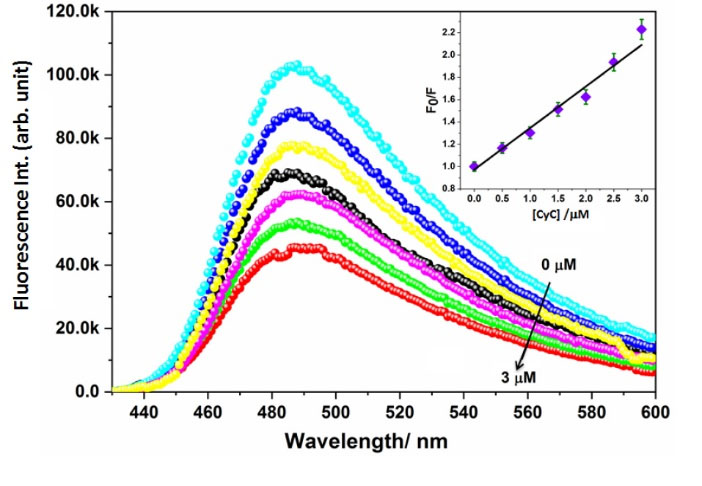
Fig. 2.
Quenching of emission intensity of ThT-AChE binary system in the presence of increasing concentrations of CyC (left) and the corresponding Stern-Volmer (SV) plot (right).
.
Quenching of emission intensity of ThT-AChE binary system in the presence of increasing concentrations of CyC (left) and the corresponding Stern-Volmer (SV) plot (right).
Modulation of inhibitory potency in HSA matrix
All experiments were carried out under fixed HSA concentration (ca. 250 mM), which is in the range of its reported functional quantity in the body.
53
Higher modulation was observed in case of CyC than AMC with a greater reduction in Vmax value, as seen in Fig. 1(ii), as well as significant alteration in IC50 values, as obtained from Hill analysis (Fig. 3). The IC50 values obtained in aqueous buffer medium proved CyC to be more potent inhibitor (IC50 = 85.12 ± 6.7 µM) and shows the value quite close to the used standard, DON (IC50 = 74.13 ± 5.6 nM); while, AMC possessed an IC50 value of 103.09 ± 11.9 nM (Table 2). In HSA medium, the IC50 changed to 113.43 ±4.5 nM and 115.11 ± 12.2 nM for CyC and AMC, respectively.
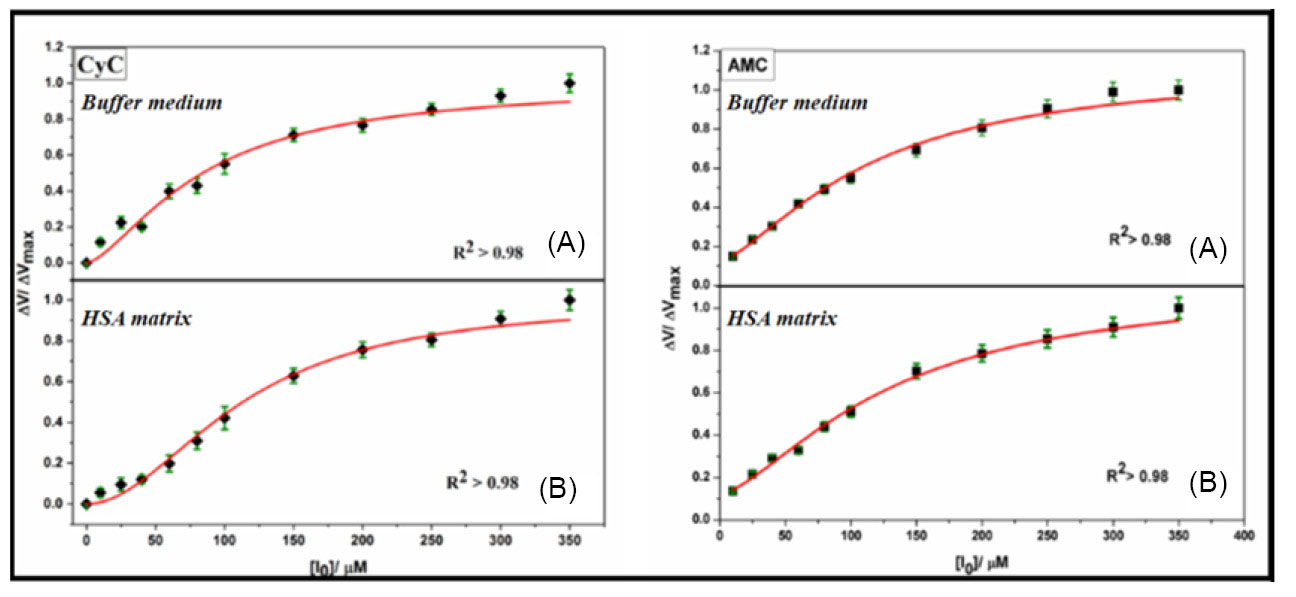
Fig. 3.
Modified Hill analysis of AChE inhibition in aqueous buffer (A) and HSA medium (B) of CyC and AMC.
.
Modified Hill analysis of AChE inhibition in aqueous buffer (A) and HSA medium (B) of CyC and AMC.
The mechanistic pathway for the enzyme inhibition was the same for AChE in both the experimental media (buffer and HSA), as apparent from the MM and LB plots depicted in Fig. 1 (iia and iic, respectively).
The following equation gives a quantitative estimation of the relative percentage of difference of inhibitor potencies on changing the experimental medium
27
:
Eq. (6)
which was found to be 32% and a mere 5% for CyC and AMC, respectively. The significant difference between the modulation of CyC and AMC inhibition activity in HSA medium is worth noting and has been discussed in detail in the later section.
To show the differences in IC50 between the various systems and measure how large a difference holds significance, the standard student t-test (details of which has been described in supplementary section ST3) was used as a measure of statistical hypothesis.
54
The t-value (given in Table S1, See Supplementary file 1) was found to be more than 0.05 in all cases, signifying a significant difference. The df value, was found to be 70 for 12 data points in each set.
Molecular modelling results
The docking scores for both ligands are distributed across a significant range due to different protein structures present in the ensemble of equilibrium conformations, as shown in Fig. 4. It is evident that CyC has a much better maximal binding score of -31.79 kJ/mol, in comparison with -29.28 kJ/mol for AMC in the case of AChE. Distribution of the maximum binding score for CyC is -25.52 kJ/mol in comparison to -24.68 kJ/mol for AMC, and the whole distribution for CyC is shifted to lower binding score values in the case of AChE. The docking results (Table 3) give the energies of both the chromones to be in the range of ThT (-31.31 kJ/mol), represented in Table S2. The best docked poses of the ligands with AChE is depicted in Fig. 5, where the Ligplots reveal the closer contact with the residues of PAS than with those of AMC. To find more about the pi-pi stacking interactions, Achilles Server was used (http://bio-hpc.eu/software/blind-docking-server/).
55
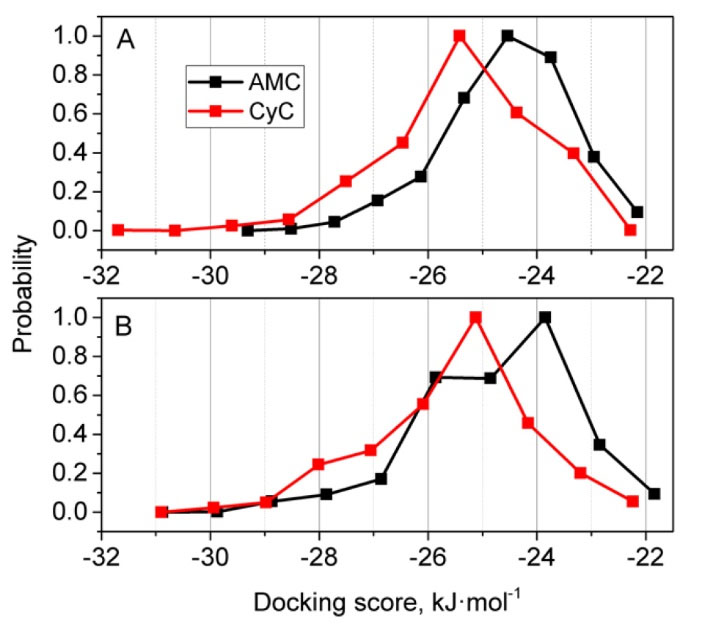
Fig. 4.
Normalized distributions of binding scores for docking of AMC and CyC to the ensembles of conformations of AChE (A) and HSA (B) proteins.
.
Normalized distributions of binding scores for docking of AMC and CyC to the ensembles of conformations of AChE (A) and HSA (B) proteins.
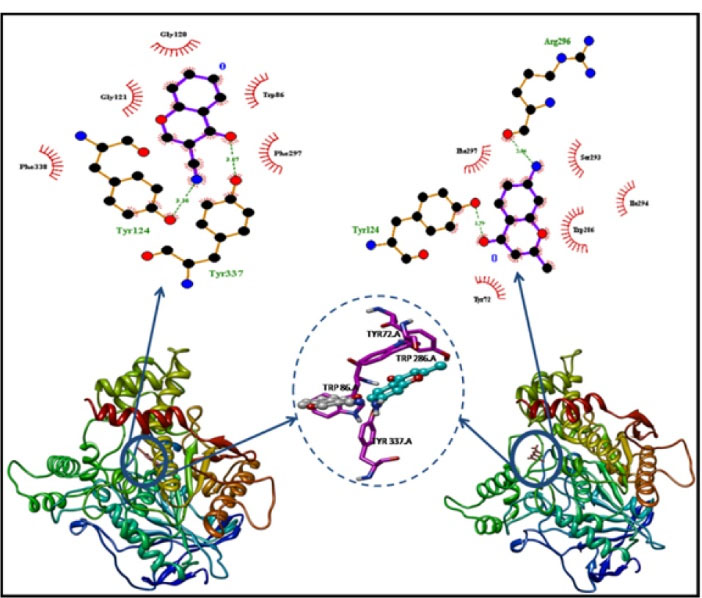
Fig. 5.
Docked poses of CyC (left) and AMC (right) with AChE. The binding site has been magnified to depict different binding poses of CyC (grey) and AMC (aqua blue) with the neighbouring residues.
.
Docked poses of CyC (left) and AMC (right) with AChE. The binding site has been magnified to depict different binding poses of CyC (grey) and AMC (aqua blue) with the neighbouring residues.
Table 3.
Affinities of the most stable docked pose of the inhibitors with AChE and HSA from molecular docking calculations
|
Receptor
|
Ligand
|
Binding affinity (kJ/mol)
|
K
i
(µM)
|
H-bonded residues
|
Hydrophobic interactions
|
| AChE |
CyC |
-35.02 |
0.9859 |
Tyr 124, Tyr 337 |
Phe 338, Gly 124, Gly 120, Trp 86, Phe 297 |
| AChE |
AMC |
-33.37 |
0.9866 |
Tyr 124, Arg 296 |
Tyr 72, Trp 286, Ser 293, Phe 297, Ile 294 |
| HSA |
CyC |
-34.60 |
0.9861 |
Nil |
Arg 186, Arg 117, Met 123, Tyr 161, Phe 165, Leu 182, Ile 142, Tyr 138, Phe 134 |
| HSA |
AMC |
-32.96 |
0.9867 |
Nil |
Phe 134, Phe 165, Met 123, Ile 142, Tyr 138, Tyr 161, Leu 135, Leu 139, Ala 158, Phe 157, Leu 154 |
In the case of HSA, the maximal binding scores for both ligands were the same (-30.96 kJ/mol).Very similar trend was also observed for HSA as with AChE but the maximum distributions were shifted to the right by approximately 0.42 kJ/mol. The best docked poses of the ligands with HSA are depicted in Fig. S6. All results corresponding to the best docked pose are given in Table 3. On the other hand, the energy of other poses is listed in Table S3.
Discussion
Several molecular properties including molecular weight, lipophilicity (logP), and TPSA are widely used as important parameters in modern drug discovery. Lipophilicity is an important physiochemical property for evaluating the capacity of a drug for passing through the blood-brain barrier (BBB). The optimal logP value for efficacious permeation of CNS was reported to be ca. 2±0.7; whereas, TPSA cut-off value was determined to be < 90 Å2 for potential drugs.
56
For FDA approved drugs, the molecular weight was determined to be ideally less than 450 g/mol.
57
The logP and TPSA values of the drugs (1.50 and 1.00 Ǻ2for CyC, and 54.00 and 56.23 Ǻ2for AMC) implied that the drugs were lipophilic enough to cross BBB and had optimum TPSA to be considered as potential drugs.
The calculated logP value of AMC was lower than the optimal value reported for CNS penetrated drugs. However, it is to be noted that although logP is an important parameter for prediction and optimisation, the suitability of a CNS drug does not depend only on this quantity. Several other factors like low molecular weight, TPSA <90 Å2, number of heteroatoms < 5, and ability to form hydrogen bonds also contribute to the drug efficacy. Except for logP being lower than the prescribed value in case of AMC, all other parameters fall within the suitable range for both the studied chromones, rendering their suitability as possible CNS drugs.
When tested via the Ellman method, both the chromones were found to exhibit inhibitory activity against AChE with the experimental assessment revealing CyC to be the more potent inhibitor (Table 2). In the non-competitive pathway, binding of the inhibitor to either the enzyme or the enzyme-substrate complex is equally plausible, meaning that the pathways A and B in Scheme 1 have equal probability of occurrence, rendering α and α' to be equal. Here, the inhibitor binds to an allosteric site, which is different from the primary substrate binding site.
37
In case of AChE, the non-competitive way is linked with PAS,
58
which is also confirmed by molecular docking results discussed later. The estimated IC50values indicate that both the chromones showed inhibitory efficiency akin to the reference drug DON. However, experiments on 2 coumarin derivatives reported elsewhere
59
revealed that replacement of the coumarin moiety with chromones results in a loss of inhibitory efficiency. Similar observation has already been reported in several SAR reviews and it is well-known that coumarin derivatives normally show better inhibition potency toward AChE activity in comparison with their chromone counterparts.
23,60
However, a noteworthy feature of this study is that both the present systems, with the slightest substitution in the chromone moiety, exhibit an inhibitory efficacy comparable and, even in some cases, greater than other generously substituted chromone derivatives reported in the literature.
17-19,21
The emission intensity of ThT, a probe that binds selectively to the PAS of AChE, has been known to increase multi-fold when bound to AChE, as seen in this case (Fig. 2 and Fig. S5).
61
The change in intensity of this ThT-bound AChE fluorescence in the presence of inhibitors can be used as an assay for quantifying the inhibitory potencies of cholinergic drugs.
62
The results obtained signifies greater inhibitory effectiveness of CyC, which is in conformity with kinetic experiments discussed above; as percentage of ThT fluorescence quenching is a direct manifestation of inhibitory efficiency.
63
The quenching of fluorescence intensity can be perceived because of displacement of ThT from the AChE gorge by the inhibitors,
64
resulting in a significant reduction of the emission intensity.
When HSA was used as a modulatory medium, the reason behind the reduction of inhibitory efficacy of the compounds could be ascribed to the confiscation of the chromones by HSA. This sequestration, in turn, results in diminution of the unbound fraction of drug to bind with the enzyme. Thus, greater affinity for HSA, as also verified by molecular docking calculation, results in greater reduction of inhibitory potency of CyC in HSA. The t values demonstrated that the difference in the IC50 values of all the systems were significant. This represents the modulation of the inhibitory potency between the 2 systems in buffer as well as while switching the medium to HSA.
Structurally, AChE consists of a narrow and deep gorge, spanning 20Å in length. The substrate binding site is the catalytic active site (CAS), which along with an allosteric site called PAS compose the 2 ends of the gorge; the CAS being near the bottom and the PAS residing at the surface.
57,65
In case of eeAChE, the catalytic site is comprised of a triad of Ser203, His447, Glu334, whereas the PAS consists of 4 amino acids: Trp86, Tyr337, Trp286, and Tyr72.
66
PAS inhibitors occlude the admittance of substrates and exodus of products from the enzyme’s active site, causing steric obstruction and allosteric variation.
25,67
Since both the chromones have an ability to form H-bonds (a nitrile group in CyC and a primary amine in AMC), the variance in their binding affinities cannot be hypothesized to be an inference of the stability caused by H-bonding alone. The role of pi-pi stacking interactions of the drugs with aromatic residues of AChE (especially Trp and Tyr) in inhibition of enzyme AChE as well as BuChE have been documented.
68
CyC that contains a linear cyano group owing to tertiary nitrogen atom, can be considered as a planar molecule and can stack parallel with aromatic residues, facilitating stacking interactions. On the other hand, AMC that contains a methyl (-CH3) as well as a primary amine (-NH2) group do not exhibit planarity due to the tetrahedral shape of the substituents. These enhanced stacking interactions in case of CyC may be the cause of its higher inhibitory efficiency towards AChE.
The docking results suggests that in average CyC binds better to the specified binding site of AChE in comparison to AMC and this result stands for different protein conformations found in equilibrium ensemble in water. This observation is concurrent with the experimental observation of superior inhibitory activity of CyC over AMC. However, CyC also binds better to the selected region of HSA in comparison to AMC, demonstrating higher degree of sequestration in case of the former. Therefore, a significant fraction of CyC becomes unavailable to demonstrate AChE inhibition in albumin matrix and causes relatively large deviation in IC50 parameter in comparison with buffer medium. The association propensities for the ligands are surprisingly similar in both AChE and HSA, which suggests that the binding of the studied ligands to these proteins might be highly concurrent.
The simulation results confirm CyC and AMC to be PAS-binding ligands as that of ThT, thereby proving the ease of displacement as discussed previously. The results obtained from Achilles Blind Docking validated our hypothesis of pi-pi stacking since CyC had stacking interactions both with Trp 86 and Phe 295, whereas AMC had stacking interactions only with Trp 86, because of structural constraints. Further justifying the concept of stacking interactions may play an important role in inhibition potency (Fig. S7).
Conclusion
The discovery and design of curative therapies for AD is an exigent task owing to the elusive understanding of its causative mechanisms. Due to the limited effectiveness of the available drugs as well as the side effects, designing new AChE inhibitors with greater effectiveness is the need of the hour. The investigated chromones used in this study, CyC and AMC, possess pertinent lipophilicity as well as TPSA values and exhibit substantial inhibitory efficiency on AChE. CyC showed higher potency of inhibition, probably due to the enhanced facility for pi-pi stacking interactions owing to the presence of a planar tertiary amine group. CyC also showed higher modulation in HSA medium. The modulation in serum albumin matrix re-instates the significance of taking the delivery medium into consideration for screening potential AD drugs. The chromones displaced Thioflavin-T from the AChE binding site and bound non-competitively with AChE at the PAS. A comparison of the anti-cholinesterase potencies of these chromones amplify the probability of the usage of chromones, even with minimal albeit prudent fabrication of its skeleton in acting as possible cholinergic drugs in the Alzheimer’s treatment.
Acknowledgement
The authors thank Prof. K. Aguan of Department of Biotechnology and Bio-informatics, NEHU for many helpful discussions.
Funding sources
The research was financially supported by the Department of Biotechnology (BT/232/NE/TBP/2011) and Department of Science and Technology (SR/FST/CSI194/2008), Govt. of India.
Ethical statement
There is none to be disclosed.
Competing interests
No conflicting interests are to be declared.
Authors’ contribution
Planned the experiments: PB, SM; executed the experiments: PB, MAR; data analysis: PB, MAR; supplied the chemicals and analysis tools: SM; manuscript writing: PB, SOY, SM; docking and MD simulation: SOY.
Supplementary Materials
Supplementary file 1 contains Figs. S1-S7 and Tables S1-S3.
(pdf)
Research Highlights
What is the current knowledge?
simple
-
√ AChE inhibitors are used as therapeutic agents in the treatment of AD.
-
√ Coumarins and chromones have been found to have AChE antagonistic property.
What is new here?
simple
-
√ Minimum albeit judicious substitution in the chromone scaffold helps to attain maximum inhibition efficiency.
√ Binding with HSA medium and replacement of ThT have been further established as standard pre-screening AD drug tests.
-
√ Validity of computational studies has been checked in the periphery of enzymatic and fluorimetric assays.
-
√ The site and mode of binding with AChE and plasma protein plays an important role in the pharmacological efficacy of potential AD drugs.
References
- Tawfik HA, Ewies EF, El-hamouly WS. Synthesis of chromones and their applications during the last ten years. Int J Res Pharm Chem 2014; 4(4):1046-1085. [ Google Scholar]
- Nawrot-Modranka J, Nawrot E, Graczyk J. In vivo antitumor, in vitro antibacterial activity and alkylating properties of phosphorohydrazine derivatives of coumarin and chromone. Eur J Med Chem 2006; 41(11):1301-1309. doi: 10.1016/j.ejmech.2006.06.004 [Crossref] [ Google Scholar]
- Marques NFLM, Marques M PM. Bioactive Chromone Derivatives – Structural Diversity. Current Bioactive Compounds 2010; 6(2):76-89. doi: 10.2174/157340710791184859 [Crossref] [ Google Scholar]
- Reis J, Gaspar A, Milhazes N, Borges F. Chromone as a Privileged Scaffold in Drug Discovery: Recent Advances. J Med Chem 2017; 60(19):7941-7957. doi: 10.1021/acs.jmedchem.6b01720 [Crossref] [ Google Scholar]
- Augstein J, Cairns H, Hunter D, Lee TB, Suschitzky J, Altounyan REC. New Orally Effective Chromone Derivatives for the Treatment of Asthma. Agents Actions 1977; 7(4):443-445. doi: 10.1007/BF01966850 [Crossref] [ Google Scholar]
- Edwards AM, Howell JBL. The chromones: history, chemistry and clinical development. Clin Exp Allergy 2000; 30(30):756-774. doi: 10.1046/j.1365-2222.2000.00879.x [Crossref] [ Google Scholar]
- Gaspar A, Matos MJ, Garrido J, Uriarte E, Borges F. Chromone: a valid scaffold in medicinal chemistry. Chem Rev 2014; 114(9):4960-4992. doi: 10.1021/cr400265z [Crossref] [ Google Scholar]
- Keri RS, Budagumpi S, Pai RK, Balakrishna RG. Chromones as a privileged scaffold in drug discovery: a review. Eur J Med Chem 2014; 78:340-374. doi: 10.1016/j.ejmech.2014.03.047 [Crossref] [ Google Scholar]
- Hilgert M, Nöldner M, Chatterjee SS, Klein J. KA-672 inhibits rat brain acetylcholinesterase in vitro but not in vivo. Neurosci Lett 1999; 263(2–3):193-196. doi: 10.1016/S0304-3940(99)00149-4 [Crossref] [ Google Scholar]
- Hoerr R, Noeldner M. Ensaculin (KA-672 HCl): a multitransmitter approach to dementia treatment. CNS Drug Rev 2002; 8(2):143-158. doi: 10.1111/j.1527-3458.2002.tb00220.x [Crossref] [ Google Scholar]
- Martyn JAJ, Fagerlund MJ, Eriksson LI. Basic Principles of neuromuscular transmission. Anaesthesia 2009; 649(suppl 1):1-9. doi: 10.1111/j.1365-2044.2008.05865.x [Crossref] [ Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK. The Cholinergic Hypothesis of Alzheimer’s Disease: A Review of Progress. J Neurol Neurosurg Psychiatry 1999; 66(2):137-147. doi: 10.1136/jnnp.66.2.137 [Crossref] [ Google Scholar]
- Craig LA, Hong NS, McDonald RJ. Revisiting the Cholinergic Hypothesis in the Development of Alzheimer’s Disease. Neurosci Biobehav Rev 2011; 35(6):1397-1409. doi: 10.1016/j.neubiorev.2011.03.001 [Crossref] [ Google Scholar]
- Wimo A, Guerchet M, Ali GC, Wu YT, Prina AM, Winblad B. The Worldwide Costs of Dementia 2015 and Comparisons with 2010. Alzheimers Dement 2017; 13(1):1-7. doi: 10.1016/j.jalz.2016.07.150 [Crossref] [ Google Scholar]
- Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D. Dementia Prevention, Intervention, and Care. Lancet 2018; 390(10113):2673-2734. doi: 10.1016/S0140-6736(17)31363-6 [Crossref] [ Google Scholar]
- Alzheimer’s Association. 2014 Alzheimer’s Disease Facts and Figures. Alzheimers Dement 2014; 10(2):1-80. [ Google Scholar]
- Parveen M, Malla AM, Yaseen Z, Ali A, Alam M. Synthesis, Characterization, DNA-Binding Studies and Acetylcholinesterase Inhibition Activity of New 3-Formyl Chromone Derivatives. J Photochem Photobiol B Biol 2014; 130:179-187. doi: 10.1016/j.jphotobiol.2013.11.019 [Crossref] [ Google Scholar]
- Lu C, Guo Y, Yan J, Luo Z, Luo HB, Yan M. Design, synthesis, and evaluation of multitarget-directed resveratrol derivatives for the treatment of Alzheimer’s disease. J Med Chem 2013; 56(14):5843-5859. doi: 10.1021/jm400567s [Crossref] [ Google Scholar]
- Liu Q, Qiang X, Li Y, Sang Z, Li Y, Tan Z. Design, Synthesis and Evaluation of Chromone-2-Carboxamido-Alkylbenzylamines as Multifunctional Agents for the Treatment of Alzheimer’s Disease. Bioorganic Med Chem 2015; 23(5):911-923. doi: 10.1016/j.bmc.2015.01.042 [Crossref] [ Google Scholar]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016; 8(6):595-608. doi: 10.15252/emmm.201606210 [Crossref] [ Google Scholar]
- Liao G, Mei WL, Kong FD, Li W, Yuan JZ, Dai HF. 5,6,7,8-Tetrahydro-2-(2-Phenylethyl)chromones from Artificial Agarwood of Aquilaria Sinensis and Their Inhibitory Activity against Acetylcholinesterase. Phytochemistry 2017; 139:98-108. doi: 10.1016/j.phytochem.2017.04.011 [Crossref] [ Google Scholar]
- Fallarero A, Oinonen P, Gupta S, Blom P, Galkin A, Mohan CG. Inhibition of acetylcholinesterase by coumarins: the case of coumarin 106. Pharmacol Res 2008; 58(3–4):215-221. doi: 10.1016/j.phrs.2008.08.001 [Crossref] [ Google Scholar]
- Anand P, Singh B, Singh N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorganic Med Chem 2012; 20(3):1175-1180. doi: 10.1016/j.bmc.2011.12.042 [Crossref] [ Google Scholar]
- Shen Q, Peng Q, Shao J, Liu X, Huang Z, Pu X. Synthesis and Biological Evaluation of Functionalized Coumarins as Acetylcholinesterase Inhibitors. Eur J Med Chem 2005; 40(12):1307-1315. doi: 10.1016/j.ejmech.2005.07.014 [Crossref] [ Google Scholar]
- Bourne Y, Taylor P, Radić Z, Marchot P. Structural Insights into Ligand Interactions at the Acetylcholinesterase Peripheral Anionic Site. EMBO J 2003; 22(1):1-12. doi: 10.1093/emboj/cdg005 [Crossref] [ Google Scholar]
- Lohman JJHM, Merkus FWHM, Rahn KH. Plasma Protein Binding of Drugs. Pharm Weekbl 1986; 8(6):302-304. [ Google Scholar]
- Islam MM, Gurung AB, Bhattacharjee A, Aguan K, Mitra Mitra, S S. Human Serum Albumin Reduces the Potency of Acetylcholinesterase Inhibitor Based Drugs for Alzheimer’s Disease. Chem Biol Interact 2016; 249:1-9. doi: 10.1016/j.cbi.2016.02.012 [Crossref] [ Google Scholar]
- Stanyon Stanyon, H H. Stanyon, HF; Viles, JHHuman Serum Albumin Can Regulate Amyloid-β Peptide Fiber Growth in the Brain Interstitium: Implications for Alzheimer Disease. J Biol Chem 2012; 287(33):28163-28168. doi: 10.1074/jbc.C112.360800 [Crossref] [ Google Scholar]
- Milojevic J, Melacini G. Stoichiometry and Affinity of the Human Serum Albumin-Alzheimer’s Aβ Peptide Interactions. Biophys J 2011; 100(1):183-192. doi: 10.1016/j.bpj.2010.11.037 [Crossref] [ Google Scholar]
- Ezra A, Rabinovich-Nikitin I, Rabinovich-Toidman P, Solomon B. Multifunctional Effect of Human Serum Albumin Reduces Alzheimer’s Disease Related Pathologies in the 3xTg Mouse Model. J Alzheimers Dis 2015; 50(1):175-188. doi: 10.3233/JAD-150694 [Crossref] [ Google Scholar]
- Llewellyn DJ, Langa KM, Friedland RP, Lang IA. Serum albumin concentration and cognitive impairment. Curr Alzheimer Res 2010; 7(1):91-96. [ Google Scholar]
- Petitpas I, Bhattacharya AA, TwineS TwineS, East M, Curry S. Crystal structure analysis of warfarin binding to human serum albumin anatomy of drug site I. J Biol Chem 2001; 276(25):22804-22809. doi: 10.1074/jbc.M100575200 [Crossref] [ Google Scholar]
- Fasano M, Curry S, Terreno E, Galliano M, Fanali G, Narciso P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005; 57(12):787-796. doi: 10.1080/15216540500404093 [Crossref] [ Google Scholar]
- Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961; 7(2):88-95. doi: 10.1016/0006-2952(61)90145-9 [Crossref] [ Google Scholar]
- Pheifer JH, Briggs DE. The estimation of thiols and disulphides in barley. J Inst Brew 1995; 101(1):5-10. doi: 10.1002/j.2050-0416.1995.tb00843.x [Crossref] [ Google Scholar]
-
Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 4th ed. W.H. Freeman; 2004. p. 1119.
-
Copeland RA. Evaluation of Enzyme Inhibitors in Drugs Discovery: A Guide for Medicinal Chemists and Pharmacologists. 2nd ed. New York: John Wiley & Sons, Inc.; 2013.
- Chen X, Wehle S, Kuzmanovic N, Merget B, Holzgrabe U, König B. Acetylcholinesterase inhibitors with photoswitchable inhibition of β- amyloid aggregation. ACS Chem Neurosci 2014; 5:377-389. doi: 10.1021/cn500016p [Crossref] [ Google Scholar]
- Pratap PR, Mikhaylyants LO, Olden-Stahl N. Fluorescence measurement of nucleotide association with the Na+/K+-ATPase. Biochim Biophys Acta 2009; 1794(11):1549-1557. doi: 10.1016/j.bbapap.2009.06.023 [Crossref] [ Google Scholar]
- Islam MM, Sonu VK, Gashnga PM, Moyon NS, Mitra S. Caffeine and sulfadiazine interact differently with human serum albumin: a combined fluorescence and molecular docking study. Spectrochim Acta A Mol Biomol Spectrosc 2016; 152:23-33. doi: 10.1016/j.saa.2015.07.051 [Crossref] [ Google Scholar]
- Cheng S, Song W, Yuan X, Xu Y. Gorge motions of acetylcholinesterase revealed by microsecond molecular dynamics simulations. Sci Rep 2017; 7(1):3219. doi: 10.1038/s41598-017-03088-y [Crossref] [ Google Scholar]
- Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015; 1(2):19-25. doi: 10.1016/j.softx.2015.06.001 [Crossref] [ Google Scholar]
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys 2007; 126(1):014101. doi: 10.1063/1.2408420 [Crossref] [ Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984; 81(8):3684-3690. doi: 10.1063/1.448118 [Crossref] [ Google Scholar]
- Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: Fast, Flexible and Free. J Comp Chem 2005; 26:1701-1718. doi: 10.1002/jcc.20291 [Crossref] [ Google Scholar]
- Páll S, Hess B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput Phys Commun 2013; 184(12):2641-2650. doi: 10.1016/j.cpc.2013.06.003 [Crossref] [ Google Scholar]
- Yesylevskyy SO, Hushcha TO. Conformational relaxations of human serum albumin studied by molecular dynamics simulations with pressure jumps. Biopolymers and Cell 2012; 28(6):486-492. [ Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comp Chem 2010; 31(2):455-461. doi: 10.1002/jcc.21334 [Crossref] [ Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC. UCSF Chimera–a visualization system for exploratory research and analysis. J Comp Chem 2015; 25:1605-1612. doi: 10.1002/jcc.20084 [Crossref] [ Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model 2011; 51:2778-2786. doi: 10.1021/ci200227u [Crossref] [ Google Scholar]
- Yesylevskyy SO. Pteros: Fast and easy to use open-source C++ library for molecular analysis. J Comp Chem 2012; 33(19):1632-1636. doi: 10.1002/jcc.22989 [Crossref] [ Google Scholar]
- Yesylevskyy SO. Pteros 20: Evolution of the fast parallel molecular analysis library for C++ and python. J Comp Chem 2015; 36(19):1480-1488. doi: 10.1002/jcc.23943 [Crossref] [ Google Scholar]
- Taverna M, Marie AL, Mira JP, Guidet B. Specific antioxidant properties of human serum albumin. Ann Intensive Care 2013; 3(1):4. doi: 10.1186/2110-5820-3-4 [Crossref] [ Google Scholar]
- Sobhani R, Pal AK, Bhattacharjee A, Mitra S, Aguan K. Screening Indigenous Medicinal Plants of Northeast India for Their Anti-Alzheimers Properties. Pharmacogn J 2017; 9(1):46-54. doi: 10.5530/pj.2017.1.9 [Crossref] [ Google Scholar]
- Sánchez-Linares I, Pérez-Sánchez H, Cecilia JM, García JM. High-throughput parallel blind virtual screening using BINDSURF. BMC Bioinformatics 2012; 13 Suppl 14:S13. doi: 10.1186/1471-2105-13-S14-S13 [Crossref] [ Google Scholar]
- Pajouhesh H, Lenz GR.
Medicinal chemical properties of successful
central nervous system drugs
. Neurotherapeutics 2005; 2:541-553. doi: 10.1602/neurorx.2.4.541 [Crossref] [ Google Scholar]
-
Cornish-Bowden A. Inhibitors and Activators BT - Fundamentals of Enzyme Kinetics. Butterworth-Heinemann; 1979. p. 73–98.
- Dvir H, Silman I, Harel M, Rosenberry TL, Sussman JL. Acetycholinesterase: From 3D Structure to Function. Chem Biol Interact 2010; 187(1–3):10-22. doi: 10.1016/j.cbi.2010.01.042 [Crossref] [ Google Scholar]
- Baruah P, Basumatary G, Yesylevskyy SO, Aguan K, Bez G, Mitra S. Novel coumarin derivatives as potent acetylcholinesterase inhibitors: insight into efficacy, mode and site of inhibition. J Biomol Struct Dyn 2018. doi: 10.1080/07391102.2018.1465853 [Crossref]
- de Souza LG, Rennã MN, Figueroa-Villar JD. Coumarins as cholinesterase inhibitors: a review. Chem Biol Interact 2016; 254:11-23. doi: 10.1016/j.cbi.2016.05.001 [Crossref] [ Google Scholar]
- De Ferrari GV, Mallender WD, Inestrosa NC, Rosenberry TL. Thioflavin T Is a fluorescent probe of the acetylcholinesterase peripheral site that reveals conformational interactions between the peripheral and acylation sites. J Biol Chem 2001; 276(26):23282-23287. doi: 10.1074/jbc.M009596200 [Crossref] [ Google Scholar]
- Islam MM, Mitra S. Cholinergic Inhibitors Replace Thioflavin-T from Acetylcholinesterase Binding Pocket: A Potential Fluorescence Based Molecular Switch. Chem Phys Lett 2016; 664:63-69. doi: 10.1016/j.cplett.2016.09.079 [Crossref] [ Google Scholar]
- Islam MM, Rohman MA, Gurung AB, Bhattacharjee A, Aguan K, Mitra S. Correlation of cholinergic drug induced quenching of acetylcholinesterase bound thioflavin-Tfluorescence with their inhibition activity. Spectrochim Acta A Mol Biomol Spectrosc 2018; 189:250-257. doi: 10.1016/j.saa.2017.08.009 [Crossref] [ Google Scholar]
- Bajda M, Wiȩckowska A, Hebda M, Guzior N, Sotriffer CA, Malawska B. Structure-Based Search for New Inhibitors of Cholinesterases. Int J Mol Sci 2013; 14(3):5608-5632. doi: 10.3390/ijms14035608 [Crossref] [ Google Scholar]
- Imramovsky A, Stepankova S, Vanco J, Pauk K, Monreal-Ferriz J, VinSova J. Acetylcholinesterase-Inhibiting Activity of Salicylanilide N-Alkylcarbamates and Their Molecular Docking. Molecules 2012; 17:10142-10158. doi: 10.3390/molecules170910142 [Crossref] [ Google Scholar]
- Colletier JP, Fournier D, Greenblatt HM, Stojan J, Sussman JL, Zaccai G. Structural Insights into Substrate Traffic and Inhibition in Acetylcholinesterase. EMBO J 2006; 25(12):2746-2756. doi: 10.1038/sj.emboj.7601175 [Crossref] [ Google Scholar]
- Nawaz SA, Ayaz M, Brandt W, Wessjohann LA, Westermann B. Cation-π and π-π Stacking Interactions Allow Selective Inhibition of Butyrylcholinesterase by Modified Quinine and Cinchonidine AlkaloidsBiochem. Biophys Res Commun 2011; 404(4):935-940. doi: 10.1016/j.bbrc.2010.12.084 [Crossref] [ Google Scholar]
- Deb PK, Sharma A, Piplani P, Akkinepally RR. Molecular Docking and receptor-specific 3D-QSAR studies of acetylcholinesterase inhibitors. Mol Divers 2012; 16(4):803-823. doi: 10.1007/s11030-012-9394-x [Crossref] [ Google Scholar]