Bioimpacts. 12(1):65-86.
doi: 10.34172/bi.2021.23511
Original Research
TEM1-targeting PEGylated PLGA shikonin nanoformulation for immunomodulation and eradication of ovarian cancer
Efthymia-Iliana Matthaiou 1, 2  , Yi Guo 1, 3, Jaleh Barar 1, 4 , Raphael Sandaltzopoulos 2, Lana E. Kandalaft 5, Chunsheng Li 1, *, George Coukos 1, 5, Yadollah Omidi 1, 6, *
, Yi Guo 1, 3, Jaleh Barar 1, 4 , Raphael Sandaltzopoulos 2, Lana E. Kandalaft 5, Chunsheng Li 1, *, George Coukos 1, 5, Yadollah Omidi 1, 6, *
Author information:
1Ovarian Cancer Research Center, Perelman School of Medicine, University of Pennsylvania, Philadelphia, USA
2Department of Molecular Biology and Genetics, Democritus University of Thrace, Alexandroupolis, Greece
3University of Shanghai, Shanghai, China
4Research Center for Pharmaceutical Nanotechnology, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
5Ludwig Institute for Cancer Research, Lausanne and University of Lausanne, Lausanne, Switzerland
6Department of Pharmaceutical Sciences, College of Pharmacy, Nova Southeastern University, Fort Lauderdale, FL 33328, USA
Abstract
Introduction:
Tumor endothelial marker 1 (TEM1) is expressed by tumor vascular endothelial cells in various cancers.
Methods:
Here, we developed poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) PEGylated with polyethylene glycol (PEG) and functionalized with anti-TEM1 antibody fragment (78Fc) and loaded them with necroptosis-inducing agent shikonin (SHK) (78Fc-PLGA-SHK NPs).
Results:
The nanoformulation showed a smooth spherical shape (~120 nm; the ζ potential of –30 mV) with high drug entrapment and bioconjugation efficiencies (~92% and ~90%, respectively) and a sustained-release profile in serum. Having significant toxicity in vitro (e.g., MS1 and TC1 cells), the nanoformulation dramatically increased the cytotoxicity in the TC1 murine lung carcinoma subcutaneous and intravenous/metastatic models as aggressive tumor models. The injection of the 78Fc-PLGA-SHK NPs to the MS1-xenograft mice resulted in significantly higher accumulation and effects in the TEM1-positive tumor targets, while they were excreted via urine track without retaining in the liver/spleen. In the TC1 subcutaneous model, C57/BL6 mice treated with the 78Fc-PLGA-SHK NPs revealed a significant therapeutic effect. The mice, which were tumor-free after receiving the nanoformulation, were re-challenged with the TC1 cells to investigate the immune response. These animals became tumor-free a week after the injection of TC1 cells.
Conclusion:
Based on these findings, we propose the 78Fc-PLGA-SHK NPs as a highly effective immunostimulating nanomedicine against the TEM1-expressing cells for targeted therapy of solid tumors including ovarian cancer.
Keywords: Tumor endothelial marker 1, endosialin, CD248, Shikonin, Targeted therapy, Nanomedicine, Tumor vasculature
Copyright and License Information
© 2022 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
As a leading cause of death worldwide, cancer continues to be the global burden. Various types of malignancies, in particular solid tumors, continue to increase with the growth and aging of the world’s population.
1
In solid tumors, the resistance to cytotoxic agents cell death and the reprogramming of metabolic pathways are the main hallmarks of cancer cells, leading to chemotherapy inefficacy.
2
Despite a rigorous chemotherapy course, many patients might inevitably induce marked toxicity mainly due to non-specific accumulation of anticancer drugs in the normal cells/tissue and drug resistance.
3-5
The occurrence of such side effects may lead to the failure of treatment modalities. To avoid non-specific inadvertent toxic impacts of the conventional chemotherapy modalities, advanced multifunctional nanomedicines with active and/or passive targeting potential have successfully been used.
6,7
Smart targeted nanoparticles (NPs)/ nanosystems (NSs) have been reported to accumulate highly in the tumor microenvironment (TME) via both the passive targeting by the enhanced permeation and retention (EPR) effect and the active targeting via interaction with unique cancer antigens/molecular markers, resulting in maximal therapeutic effects in cancerous cells with minimal side effects.
8-14
Of anticancer NSs, biodegradable polymeric NPs have widely been studied in terms of their potential in the safe delivery of loaded anticancer agents to the target cells while they elicit negligible side effects.
9
Among biodegradable polymers, poly(lactic-co-glycolic acid) (PLGA) is one of the most widely investigated biocompatible polymers,
15-17
and has been considered to serve as delivery systems.
9,18,19
For active targeting of cancer cells, NSs need to be decorated with a desired homing agent specific to cognate cancer molecular marker. In addition to epithelial cells, tumor microvasculature endothelial cells might display markers that differ from that of the normal stromal cells.
20,21
The oncomarkers expressed are called tumor vascular markers (TVMs), which might be exploited as targets for antibody-based tumor diagnosis and therapy,
22-25
in large part because of the relative stability of TVM-expressing cells within the tumor neovasculature that play a key role in the maintenance and progression of the tumor.
26
Further, the leaky capillary of tumor neovasculature facilitates the circulation of Ab-conjugated nanomedicines, resulting in their easy access to TVMs. Among the TVMs, tumor endothelial marker 1 (TEM1), known as CD248/endosialin, is a very promising candidate for the targeted therapy of tumor microvasculature.
27,28
The functional expression of the TEM1 has been shown to associate with tumor microvasculature (TMV), including pericytes and myofibroblasts,
21,25,29
and TMV endothelial cells in various human cancers.
20,30-32
However, it is not expressed by other adult normal cells/tissue.
29,33
TEM1 plays a central role in the progression of the tumor via various mechanisms, including the induction of proliferation and angiogenesis,
34
as well as migration and metastasis. Such impacts occur through the interaction of TEM1 with matrix proteins such as fibronectin, collagen type I and IV,
35
and Mac-2 BP/90K.
32
For instance, TEM1-deficient mice transplanted with the tumor in the abdominal sites were shown to present a striking reduction in the growth, invasiveness, and metastasis of the tumor.
36
All these findings support the concept that the TEM1-mediated targeted therapy might offer a great tool for the diagnostic and/or therapy of solid tumors such as ovarian cancer.
It should be noted that the battle with cancer can substantially be improved through a combination of anticancer cytotoxicity and immunosurveillance, while very few anticancer agents display such properties. Of various products assessed for anticancer and immunomodulatory effects, shikonin (SHK) appears to provide unique pharmacological effects such as wound-healing potential together with antitumor, anti-inflammatory, antioxidant, antithrombotic, and antimicrobial effects.
37-39
Given that the SHK functions on different molecular targets and exhibits a broad range of activities, it is considered a highly interesting substance.
40-43
Further, as a highly lipophilic naphthoquinone pigment, SHK inhibits (i) the activity of pyruvate kinase M2,
44,45
(ii) upregulates the activity of p53 and downregulates the functional expression of cyclin-dependent protein kinase 4,
46
(iii) upregulates BCL2-associated X protein and downregulates B-cell lymphoma 2,
37
and (iv) inhibits the pathways of extracellular signal-regulated kinases and protein kinase B.
47
SHK was shown to incite significant cellular toxicity in vitro and in vivo,
48
which is not specific to the cancer cells only. Like any other cytotoxic agent, to be effective on the target cells, SHK molecules must be delivered efficiently and specifically to the cancer cells that can be attained employing nanoscale targeted drug delivery systems (DDSs). In this current work, we developed the SHK-loaded, anti-TEM1-armed PLGA NPs as described previously,
28
and investigated their biological effects both in vitro and in vivo. Furthermore, we studied the cell death mechanism(s) induced by SHK and looked into the immunostimulatory properties of this innovative nanoformulation in vivo.
Materials and Methods
Acid terminated PLGA (lactide:glycolide 50:50, Mw 38 000-54 000), ethyl acetate (EtAc), Pluronic® F68 (PF68), N-hydroxysulfosuccinimide sodium salt (NHS), 4-morpholineethanesulfonic acid (MES hydrate), D-(+)-trehalose dehydrate, phalloidin–tetramethylrhodamine B isothiocyanate, goat serum, cardiogreen, bovine serum albumin (BSA), 0-(2-aminoethyl)polyethyleneglycol 3000 (PEG -OH), N-(3-dimethylaminopropyl)-N’-ethyl carbodiimide hydrochloride) (EDC), sulforhodamine B (SRB), and tissue culture dishes with vents CELLSTAR were purchased from Sigma Aldrich Co. (St. Louis, MO, USA). Shikonin, crystal violet, JC-1, RIP3 antibody (H-43), RIP antibody (H-207), HPV16 E6 (N-17) P, and HPV16 E7 (C-20) P were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). BD Bioscience TranswellTM; Annexin V: PE apoptosis detection kit I, anti-IFN-γ Rat IgG1 κ Mouse Ab, 3 mL BDTM slip-tip disposable syringe, rat IgG1 κ anti-mouse IFN-γ biotinylated Ab, streptavidin-alkaline phosphatase, ham IgG1 κ anti-mouse CD3e Ab clone 145-2C11, Syr Ham IgG2 λ1 anti-mouse CD28 clone, and anti-CD31 rat anti-mouse Ab were obtained from BD Biosciences (San Jose, CA, USA). Fisherbrand Cover Glasses Circles (No. 1 to 0.13 to 0.17 mm thick; Size: 12 mm), and 1-Step NBT/BCIP Solution PierceTM were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The XTT cell proliferation kit II, and the in-situ cell death detection kit AP were purchased from Roche Applied Science (Penzberg, Germany). Necrostatin-1 and 2.7-dichlorodihydrofluorescein diacetate were obtained from Cayman Chemical Co. (Ann Arbor, MI, USA). NucBlue® fixed Cell ReadyProbes® reagent, Alexa Fluor® 594 goat anti-hamster IgG (H+L), and PloLong diamond antifade mountant with DAPI were purchased from Molecular Probes Biotechnology Co. (Eugene, OR, USA). Wheat germ agglutinin, Alexa Fluor® 488 conjugate, and ACK lysing buffer were obtained from Life Technologies/Invitrogen Corporation (Carlsbad, CA, USA). DNase, RNase, protease-free ddH2O and Falcon® 70µm cell strainer white sterile individually packaged 50/Case were purchased from Mediatech Inc. (Manassas, VA, USA). Annexin V-FITC early apoptosis detection kit was obtained from Cell Signaling Technology (Danvers, MA, USA). The recombinant human IL-2 was obtained from Peprotech (Rocky Hill, NJ, USA). Acridine orange staining solution was purchased from ImmunoChemistry Technologies LLC (Bloomington, MN, USA). TNF-α (Q178) polyclonal antibody was purchased from Bioworld Technology Inc. (Louis Park, MN, USA). Rabbit anti-NFkB polyclonal antibody was purchased from Bioss Antibodies Inc. (Woburn, MA, USA). Formvar/Carbon 400 Mesh, Cu, 50/bx was purchased from Electron Microscopy Sciences (Hatfield, PA, USA). 5-Bromo-4-chloro-3-indolyl β-D-galactosidase was purchased from PanReac AppliChem (Darmstadt, Germany). MultiScreen-IP Filter Plate was purchased from EMD Millipore (Billerica, MA, USA). DAPI fluoromount G was purchased from SouthernBiotech (Birmingham, AL, USA). Rapamycin was purchased from InvivoGen (San Diego, CA, USA). 78Fc scFv_4H9 (2.435 µg/µL; 40 µL) was provided by Dr. Chunsheng Li (University of Pennsylvania). The cell lines were obtained from American Type Culture Collection (Manassas, VA, USA) and MS1 cells were engineered in-house to express the HuTem1 marker.
Nanoparticle formulation and physicochemical evaluation
The preparation and evaluation of NPs were performed employing a single emulsion-solvent evaporation technique, as described previously.
28
Briefly, SHK (15 mg/mL) was dissolved in 30 μL DMSO. About 100 mg of PLGA was dissolved in a 3 mL oil phase (chloroform: ethyl acetate 2:1). Then, the SHK solution was slowly added to the oil phase. About, 3% of surfactants (i.e., 1:1 ratio of Pluronic F68 and polyvinyl alcohol) were dissolved in 10 mL double distilled water. To this end, the oil phase was added and mixed with the water phase (10 mL) over an ice bath. The mixture was then sonicated at 40 W output for 4 minutes (2×2 minutes) employing a probe sonicator (VCX 130; Sonics and Materials, Inc, Newtown, CT, USA). Afterward, to remove the oil phase, the mixture was evaporated under a reduced pressure of 150 mbar, employing a rotary evaporator (30 rpm) (Hei-VAP Advantage ML/G5B; Heidolph North America, Grove Village, IL, USA). Further, the same procedure was employed for the formulation of the drug-free PLGA NPs. The influence of variables on the NP formulations was further evaluated. For the lyophilization of the NPs, they were resuspended in 1 mL trehalose (250 mM) as a cryoprotectant supplemented with 0.5% Tween 80, which was applied to prevent the lyophilization-induced aggregation and keep it safe for later use.
Transmission electron microscopy (TEM) analysis
To morphologically characterize NPs, TEM analysis was conducted. Briefly, 10 μL of NPs sample was suspended 1 mL distilled water, and then, one drop of the suspension was placed over a carbon 400 mesh TEM grid and allowed to dry. The NPs on the mesh were visualized on a Tecnai 12 microscope at 120 KeV at indicated magnifications (FEI, Hillsboro, OR, USA), equipped using Gatan's US1000 CCD camera (Gatan, Inc., Pleasanton, CA, USA).
Particle size and zeta potential analyses
For the analysis of the size and zeta potential of NPs, each sample (20 μL) was diluted to 1 mL of distilled water. A Zetasizer, Nano-ZS (Malvern Instruments, Malvern, UK) was recruited to measure the size and zeta potential of the NPs.
Drug content (DC%), drug entrapment (DE%), drug loading (DL%), and yielding (YE%) efficiencies
The amount of SHK in NPs was analyzed using the ultraviolet (UV) spectrophotometric method, where opted amounts of SHK-loaded NPs were dissolved in acetonitrile (1 mg/mL). To complete the release of the entrapped drug, the samples were kept on a shaker at 37°C for 72 hours. Then, to extract the drug, the samples were removed by centrifugation at 10 000 ×g, 4°C for 5 minutes. After collecting the supernatant, a UV spectrophotometer (Genesys 6; Thermo Fisher Scientific) was employed to measure the absorbance at a wavelength of 520 nm. The DC%, DE%, DL%, and yielding efficiency (YE%) values were calculated for all the formulations (Table S1).
Release kinetics analysis
To explore the kinetics profile, the release of SHK from PLGA NPs were evaluated at various pHs (pH 4.4, 5.4, 6.4, and 7.4) utilizing phosphate-buffered saline (PBS) release buffer (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4) supplemented with fetal bovine serum (FBS). All the experiments were performed at 37°C. In practice, NPs (25 mg) were distributed in 5 mL of PBS buffer, and the samples were divided into equal aliquots (1 mL each). These tubes were kept on a shaker at 37°C and 150 rpm. At designated time intervals (i.e., 1, 2, 4, 8, 12, 24, 48, and 72 hours), the samples were centrifuged at 10 000× g at 4°C for 5 minutes. Then, the extracted supernatant was read exploiting the Genesys 6 UV spectrophotometer, at a wavelength of 520 nm (SHK) for PBS and 600 nm for FBS samples. Finally, various kinetics models (Table S2) were recruited to analyze the data, as described previously.
49
PEGylation and 78Fc conjugation
A one-step approach was used for the PEGylation and Ab conjugation of NPs (Fig. 1A), as described previously.
28
Briefly, 10 mg of acid-terminated PLGA NPs was re-suspended in 2-4 mL MES (pH 6). It was then activated with 250 μL EDC (1 mg/mL) and 250 μL NHS (1 mg/mL) at room temperature under stirring at 35 rpm for 2 hours. The samples were washed (×2) with 50 mM sucrose by centrifugation at 10 000× g (4°C) for 5 minutes. The activated NPs were resuspended in PBS and incubated with 250 μL NH2-PEG3000-OH (1 mg/mL) and 100 μL 78Fc (at a final molar ratio of 10:1) at room temperature under stirring at 35 rpm for 2 hours. The NPs were washed (×2) using 50 mM sucrose (pH 8) by centrifugation at 10 000 × g (4°C) for 5 minutes, to remove the unconjugated molecules, and the supernatant was analyzed to determine the conjugation efficiency through UV spectrophotometry.
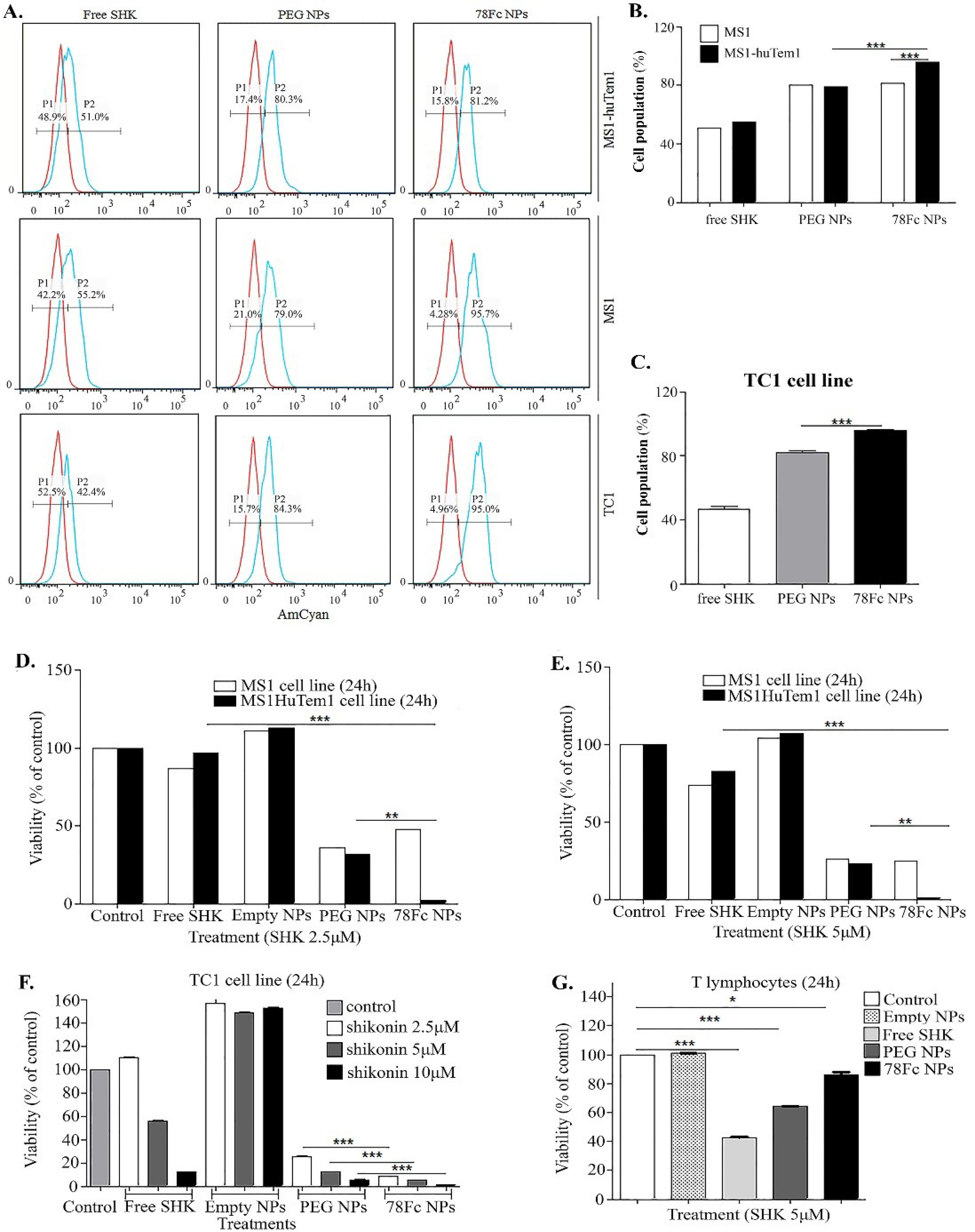
Fig. 1.
The internalization and cytotoxicity of shikonin (SHK)-loaded nanoparticles (NPs) in different cell lines. A) Flow cytometry analysis of SHK and SHK-loaded NPs uptake (P1: the untreated control cells, P2: the cells treated with SHK in TEM1-positive, MS1HuTem1 endothelial cells, and TEM1-negative, MS1 endothelial cells, as well as TC1 cells. B) The uptake of NPs by MS1 and MS1HuTem1 cells. C) The uptake of NPs by TC1 cells. D) Treated cells with 2.5µM SHK. E) Treated cells with 5 µM SHK. F) TC1 cells treated with 2.5µM, 5µM, and 10µM SHK. G) T cells treated with 5µM SHK. The cultured cells were exposed to treatments for 2 h then the treatments were removed and cells were subjected to SRB cytotoxicity assay 24h after the addition of the treatment.
.
The internalization and cytotoxicity of shikonin (SHK)-loaded nanoparticles (NPs) in different cell lines. A) Flow cytometry analysis of SHK and SHK-loaded NPs uptake (P1: the untreated control cells, P2: the cells treated with SHK in TEM1-positive, MS1HuTem1 endothelial cells, and TEM1-negative, MS1 endothelial cells, as well as TC1 cells. B) The uptake of NPs by MS1 and MS1HuTem1 cells. C) The uptake of NPs by TC1 cells. D) Treated cells with 2.5µM SHK. E) Treated cells with 5 µM SHK. F) TC1 cells treated with 2.5µM, 5µM, and 10µM SHK. G) T cells treated with 5µM SHK. The cultured cells were exposed to treatments for 2 h then the treatments were removed and cells were subjected to SRB cytotoxicity assay 24h after the addition of the treatment.
Determination of Ab conjugation efficiency
To analyze the conjugation efficiency, we capitalized on the Bradford method using Coomassie Blue dye. Briefly, Coomassie plus reagent (300 μL) was introduced to the Ab-conjugated NPs (i.e., 10 mg dispersed NPs in 200 μL PBS pH 7.4) and incubated for 10–15 minutes. Then, a microplate reader (EL800; BioTek, Winooski, VT, USA) was used to read the absorbance at 595 nm, and the readings were compared to a standard plot of BSA solution at different concentrations (ranging from 10 to 1000 μg/mL).
Cell culture
Cell lines including MS1, MS1HuTem1, TC1, and T cells were cultured in well-plates using RPMI 1640 (GlutaMAXTM) supplemented with 10% fetal bovine serum, 100 units/mL penicillin G, and 100 μg/mL streptomycin. T cells were cultivated in 24-well plates at a seeding density of 5.0×105 cells/well, which were exposed to 20 U/mL of IL-2 to help their proliferation before the assay. The cultivated cells were kept in a humidified incubator with 5% CO2 at 37°C during the culture and experimentation. The isolation of T cells was done using the service provided by the Human Immunology Core (HIC) facility at the University of Pennsylvania (Philadelphia, PA, USA).
Internalization of NPs
To analyze the internalization of 78Fc-armed NPs by the TEM1-positive/-negative MS1 cells, the flow cytometry technique was employed.
28
To this end, the cells were treated with free SHK or SHK-loaded NPs for 2 hours, and then, the plates were kept on the ice, and the internalization analysis was performed utilizing BD Canto II flow cytometer using the PE filter.
Cytotoxicity evaluation
The cytotoxic impacts of free SHK and SHK-loaded PLGA NPs were analyzed using sulforhodamine B (SRB) assay, as described previously.
28
The cells were cultivated at a seeding density of 3.0×104 cells/cm2 in 96-well plates. About 24 hours post-cultivation, they were treated with different concentrations of SHK or equivalent SHK-loaded NPs. The treatments were then removed after 2 hours of initial exposure, and the cells were incubated for 24 hours. After fixing the cells with 10% trichloroacetic acid at 4°C for 60 minutes, they were washed (×4) with distilled water. Then, the samples were stained with SRB-staining solution (0.057% w/v in 1.0% v/v acetic acid solution) at room temperature for 30 minutes. Once washed (×4) with 1.0% acetic acid solution, the samples were added with 200 μL of 10 mM Tris base solution (pH 10.5) and kept under shaking for 30 minutes. Finally, the absorbance was read by the EL800 microplate reader at 570 nm, and the viability of cells was calculated.
Cell migration and cell invasion assay
TC1 and MS1 cells were treated with SHK (0.5, 1.5, 2.5, 5 µM), doxorubicin (DOX) 100 nM as the positive control, or full media as the negative control for 6 h before the assays. For migration assay, transwell migration chambers were placed in 24-well plates. The serum-free medium (100 µL) was added into the chamber, followed by the addition of 200 µL pretreated cells in suspension in serum-free medium (2.5 × 105 cells/mL). In the lower chamber, 750 µL of full media were added. The cells were incubated at 37°C for 12 h. They were then washed with PBS (×2). The cells were fixed by the addition of formaldehyde (3.7% in PBS) (1 mL in the chamber, 1 mL in the lower chamber) at room temperature for 2 minutes. Formaldehyde was discarded and chambers were washed x2 with PBS. Cells were permeabilized by the addition of 100% ethanol at room temperature for 20 minutes. Ethanol was discarded and chambers were washed x2 with PBS. Giemsa stain was added (1 mL in the chamber, 1 mL in the lower chamber) at room temperature for 15 minutes. Giemsa was discarded and chambers were washed x2 with PBS. The non-migrated cells were scraped off with cotton swabs, while the migrated cells were counted under a light microscope.
For cell invasion assay, cells were detached using 0.25% Trypsin-EDTA solution, washed with DPBS, and re-suspended in serum-free cell culture media containing 0.1% BSA (1.0 × 106 cells/mL). About 50 µL of Matrigel was added to a 24-well transwell insert and solidified in a 37°C incubator for 15-30 minutes, to form a thin gel layer. About 100 μL of cell suspension was added on top of the Matrigel coating and incubated at 37°C and 5% CO2 for 10 minutes, to allow the cells to settle down. Using a pipette, 600 μL of full media was included to the bottom of the lower chamber in a 24-well plate. Then, the cells were incubated at 37°C and 5% CO2 for 12 hours. After the 12 h incubation, the transwell insert was removed and a cotton-tipped applicator was used to remove the media and remaining cells that have not migrated from the top of the membrane without damaging it. The transwell insert was dipped into 70% ethanol for 10 minutes, to allow cell fixation. Afterward, a cotton-tipped applicator was utilized to eliminate the remaining ethanol from the top of the membrane. Then, the transwell membrane was kept to dry for 15 minutes. The crystal violet solution (0.2%) was used to stain the cells. Then, the transwell insert was gently washed several times with distilled water to remove the crystal violet excess and the transwell membrane was allowed to air-dry. Finally, an inverted microscope was employed to count the cells and get an average sum of the cells migrated through the membrane towards the chemo-attractant and attached to the membrane.
Colony formation assay
The cells were cultivated at a seeding density of 2.5 × 105/flask in 25-T flasks for 4 hours to allow them to attach but not to multiply. Then, the cells were received the following treatments for 5 hours full media (i.e., the negative control), doxorubicin 100 nm (i.e., the positive control), SHK 0.3, 1.5, 2.5, and 5 μM. About 5 hours after initial treatment addition, samples were harvested. The number of cells in the resulting cell suspension was counted and diluted in sterile tubes so that approximately 5.0 × 103 cells could be pipetted into the 6-well plates. Samples were pipetted in 6-well plates in triplicates and placed in an incubator. They were kept until the cells in the control dishes have formed sufficiently large clones (8 days). Once clones were formed samples were washed twice and 2 mL of fixation/staining solution (6.0% glutaraldehyde and 0.5% crystal violet) were added to each well at room temperature for 30 minutes. Then, the glutaraldehyde crystal violet mixture was removed and carefully rinsed with tap water. The samples were air-dried at RT. The plating efficiency (PE) and surviving factor (SF) values were calculated as follows.
PE = (number of colonies formed/ number of cells seeded) × 100%
SF = number of colonies formed after treatment/ number of cells seeded × PE
Fluorescence microscopy
Cells at a seeding density of 1.0×104 were grown on a coverslip for 24 hours. The next day, the cells were treated for 2 hours with SHK 2.5, 5, and 10 μM. The treatments were removed from the samples washed twice and fixed with 2% Paraformaldehyde with Triton X. Then, the nuclei were stained with DAPI and the cellular membrane Wheat Germ Agglutinin, Alexa Fluor® 488 Conjugate according to the manufacturer's instruction. Samples were mounted with PloLong Diamond Antifade Mountant and images were taken with a Leica TCS SP8 confocal microscope (Leica Microsystems GmbH, Wetzlar, Germany).
β-Galactosidase (13-Gal) staining
To detect senescent cells, samples were stained for β-galactosidase. The cells were grown in 6 well plates 2.0 × 105 cells/well. The next day, the cells were treated with the full media and doxorubicin 100 nM (as the negative and positive controls, respectively), SHK 2.5 μM, 5 μM, and 10 μM for 4 hours After 4 hours treatment, the samples were washed with PBS and fixed with 2% paraformaldehyde at room temperature for 5 minutes. Then, they were washed and incubated with the fresh senescence-associated 3-Gal (SA-,3-Gal) stain solution 1 mg/mL at 37°C for 12 hours. After staining, cells were washed, air-dried, and images were taken using an inverted wide-field microscope.
JC1 staining
The cells were cultured in 6-well plates 5.0 × 105cells/ mL in a CO2 incubator overnight at 37°C. The next day, the cells were treated with full media (negative control), camptothecin 20 μM (positive control), SHK 0.3, 1.5, 2.5, and 5 μM. For the flow cytometry analysis, 5 hours after treatments, the samples were washed twice and 100 μL/mL JC-1 staining solution was added to each well. Samples were incubated in a CO2 incubator at 37ºC for 15 minutes. Samples were washed, harvested, resuspended in PBS, and analyzed promptly. In healthy cells with functional mitochondria, the red JC-1 molecules can be accumulated and detected in the FL2 channel. However, the apoptotic cells or the unhealthy ones with damaged mitochondria contain mainly green JC-1 monomers and are detectable in the FL-1 channel. For the fluorescence microscopy analysis, 5 hours after treatment, samples were washed twice and 100 μL/mL JC-1 staining solution was added to each sample, and samples were incubated in a CO2 incubator at 37ºC for 20 minutes. The cells were imaged directly in the culture medium since phenol red does not interfere with fluorescence staining. The accumulated JC-1 molecules in the healthy cells were detected using the Texas Red filter of the fluorescence microscope, while the JC1 monomers in the unhealthy/apoptotic cells were detected using the FITC filter of the fluorescence microscope.
Reactive oxygen species (ROS) formation evaluation
For ROS formation analysis, 2’,7’–dichlorofluorescein diacetate (DCFDA) staining was performed. TC1 cells were grown in 6-well plates to reach 70% confluency; then treated with SHK (2.5, and 5.0 µM) and H2O2 (100 mM) as the positive control, and incubated for 3 h before staining with DCFDA.
Before experiments, 20 mM DCFDA stock was prepared in absolute ethanol and kept at 4ºC until use. For flow cytometry, the cells were trypsinized and re-suspended at the density of 2×106 cells/mL. Then, 1 μL of 20 mM DCFDA was added to each sample (1 mL/sample). The samples were incubated at 37ºC in dark for 15 minutes, then treated with PBS as a control. The samples were washed (×2), then re-suspended in PBS, and analyzed through a flow cytometer equipped with a 488 nm argon-ion laser and a 525±10 nm bandpass emission filter. For fluorescent microscopy, the cells were washed twice with PBS. Next, 1 μL of 20 mM DCFH-DA was added to each sample (1 mL/sample). The samples were incubated at 37ºC in dark for 15 minutes. The cells were washed (×2) with PBS and images were captured through a fluorescence microscope (FITC filter).
Acridine orange staining
The cells were cultured in 6 well plates with a density of 5.0 ×105cells/mL in a CO2 incubator overnight at 37ºC for 24 hours. On the next day, the cells were received the following treatments, full media as the negative control, rapamycin 500 nm as the autophagy inducer (positive control), SHK with different concentrations (0.3 μM, 1.5 μM, 2.5 μM, and 5 μM) for 5 hours. Flow cytometry was performed to assess the generation of acidic vesicular organelles (AVOs). The cells were stained with acridine orange solution (1 μg/mL) for 15 minutes. They were then detached from the plate with trypsin-EDTA and resuspended in a phenol red-free growth medium (1.0×104 cells/mL). The samples were immediately read through a flow cytometer (BD Canto II) at excitation 488 nm, and red fluorescence >600 nm versus dot plot of green fluorescence at 530 nm. For imaging of the autophagic vacuoles, 5 hours post-treatment, samples were washed twice and then incubated with acridine orange solution (1 μg/mL) in serum-free medium at 37 ºC for 15 minutes. The acridine orange was removed, samples were washed (×2), and the cell images were acquired employing an inverted fluorescence microscope. For the analyses, the cytoplasm and nucleus of the cells were stained in bright green (detected with the FITC filter), whereas the acidic autophagic vacuoles were stained in bright red (detected with the Texas Red filter).
Flow cytometry analysis of apoptosis necrosis
To identify any transition from the apoptotic condition to the necrotic stage, a flow cytometry assay was applied utilizing phospholipid-binding annexin V protein that can show high affinity to phosphatidylserine in the presence of Ca2+, which is the major sign of early apoptosis.
50
To this end, the cultured cells in a six-well plate were treated with different concentrations of SHK alone or SHK NPs. About 12 hours post-treatment, the cells were delicately trypsinized and centrifuged at 200 × g for 5 minutes. Afterward, the cells (around 1.0 × 106 cells) were washed with 5.0 mL ice-cold PBS by centrifugation at 200 × g for 5 minutes. Then, they were resuspended in 500 μL of binding buffer (1x), which was supplemented with 5 μL of the apoptosis detection reagent (annexin V-EnzoGold) and 5 μL of the necrosis detection reagent, and incubated at room temperature for 15 minutes. To detect apoptosis and necrosis, the flow cytometry analyses were carried out using the FL2 and FL3 channels, respectively. The positive control cells (i.e., using UV irradiation for 15 minutes) and the untreated control cells were used for the comparison.
In vivo biodistribution analysis
To investigate the targeting efficacy of the 78Fc-armed, SHK-loaded NPs, we used a model developed previously.
27
PEGylated PLGA NPs loaded with IGC or free ICG (0.5 nmoles ICG in 1x DPBS) were injected (100 μL/injection) intravenously in female athymic nu/nu mice (strain 088 Nude -/-, 18g, 4-weeks, Charles River Laboratories) (n=4/group). Infrared detection images were taken before injection and immediately after injection, 50 minutes, 2 hours, 6 hours, 12 hours, 24 hours, 48 hours, and 72 hours after injection. About 72 hours after injection, no signal was detected, mice were sacrificed and organs (brain, heart, lungs, thymus, kidneys, spleen, liver, oviduct, ovaries) were harvested and an infrared image of the organs was taken to detect an infrared signal. Images were taken using the Li-COR Pearl Impulse instrumentation provided by the University of Pennsylvania Small Animal Imaging Facility (SAIF) Optical/Bioluminescence Core, supported by NIH grant CA016520.
Targeting efficacy in the mouse model
Human TEM1 highly- expressing MS1 and MS1 cell lines, both expressing Luciferase at the same level were provided by Dr. Chunsheng Li. Tumor vascular model subcutaneous grafts expressing hTEM1 were established by the injection of 2.0 × 107 MS1-hTEM1 or MS1 in each flank of nude female mice. Before tumors were implanted, the female athymic nu/nu mice (strain 088 Nude-/-, 18 g, 4-6 weeks, Charles River Laboratories) were acclimatized at the SAIF vivarium for 1 week, after which the mice were supplied with food and water ad libitum. Cellular growth was monitored by luminescence detection on a daily basis. Two weeks after the injection of cells, hemangiomas were established in both flanks, and treatments were injected intravenously into the retro-orbital complexes. The treatments were as follows: saline-control group; 5mg/kg free SHK; PLGA PEGylated empty NPs, PLGA PEGylated SHK-loaded NPs content of SHK/injection 5 mg/kg; 78Fc-armed PLGA PEGylated SHK-loaded NPs, content of SHK/injection 5 mg/kg; 100 μL/injection; n=7 mice/group. Treatments were injected intravenously on days 1, 3, 5, 7, and 9 after the hemangiomas were established. Small animal imaging detecting cellular luminesce and SHK fluorescence were being taken every other day using Perkin Elmer IVIS Spectrum instrumentation provided by the University of Pennsylvania SAIF Optical/Bioluminescence Core, supported by NIH grant CA016520. On day 14th, the animals were sacrificed and hemangiomas were collected. Mouse weight was monitored daily for any indication of weight loss and/or behavioral change.
Therapeutic efficacy in the mouse model
The subcutaneous tumors with the overexpression of endogenous murine TEM1 were developed through the injection of 4.0×104 TC1 cells into the right flank of female nude mice (4-6 weeks old). Before the implantation of tumors, female athymic nu/nu mice (strain 088 Nude-/-, 18 g, Charles River Laboratories) were acclimatized at the SAIF vivarium for 1 week. Mice were carefully supplied with food and water ad libitum. The injected TC1 cells expressed firefly luciferase, enabling BLI imaging. On day 7, after cell inoculation TC1 tumors were established. The animals were intravenously injected via the retro-orbital complexes with various treatments, including saline-control group; 5 mg/kg free SHK; PLGA PEGylated empty NPs; PLGA PEGylated SHK-loaded NPs content of SHK/injection 5 mg/kg; 78Fc-armed PLGA PEGylated SHK-loaded NPs, the content of SHK/injection 5 mg/kg; 100 μL/injection; n=7 mice/group. The treatments were administered on days 1, 3, 5, 7, and 9 after tumors were established, while, to detect luminescence and SHK fluorescence, images were taken every other day. Animal weight was monitored daily. On the day 14th, the animals were sacrificed and tumors were collected.
Therapeutic efficacy in the metastatic mouse model
The metastatic tumors in the lung were developed by intravenous injection of TC1 cells (1.0×105 luciferase-expressing TC1 cells) via the tail vein. These mice grow BLI visible lung tumors within 7 days after the cell inoculation, which otherwise succumbed in 3-4 weeks if no treatment is provided. Before tumors were implanted, female athymic nu/nu mice (strain 088 Nude-/-, 18 g, 4-6 weeks old, Charles River Laboratories) were acclimatized at the SAIF vivarium for 1 week. The animals were supplied with food and water ad libitum. On day 3 after TC1 cell injection, treatments were injected intravenously into the retro-orbital complexes, as follows: saline-control group; 5mg/kg free SHK; PLGA PEGylated empty NPs; PLGA PEGylated SHK-loaded NPs content of SHK/injection 5 mg/kg; 78Fc-armed PLGA PEGylated SHK-loaded NPs, the content of SHK/injection 5mg/kg; 100 μL/injection; n=7 mice/group. The treatments were administered on days 3, 5, 7, 9, and 11, while to detect luminescence and SHK fluorescence images were taken every other day. Animal weights were monitored on a daily basis. On day 16, the animals were sacrificed and the lungs were collected.
Therapeutic efficacy in the immunocompetent mice model
The subcutaneous tumors with the overexpression of endogenous murine TEM1 were established by the injection of 1.0×105 TC1 cells into the right flank of female 8-week-old C57BL/6 mice. Mice were acclimatized at the SAIF vivarium for 1 week before tumors were implanted. Seven days after cell inoculation tumors were established. Treatments were injected intravenously into the retro-orbital complexes, as follows: saline-control group; 5mg/kg free SHK; PLGA PEGylated empty NPs; PLGA PEGylated SHK-loaded NPs content of SHK/injection 5mg/kg; 78Fc-armed PLGA PEGylated SHK-loaded NPs, content of SHK/injection 5 mg/kg; 100 μL/injection; n=7 mice/group. The treatments were administered on days 1, 3, 5, 7, and 9 after tumors were established, while, to detect luminescence and SHK fluorescence, images were taken every other day. Animal weight was monitored daily. On day 14th, animals were sacrificed and tumors and spleens were collected. The experiment was repeated three times. For the TC1 re-challenge, the animals treated with 78Fc-armed NPs and were tumor-free on the last day of the experiment were collected and monitored. After a month, about 1.0×105 TC1 cells were injected into the right flank of these mice, and the tumor growth was monitored by luciferase detection every other day.
Therapeutic efficacy in the re-challenged animals
To further investigate if the immune system was indeed active after the therapy, instead of sacrificing the tumor-free mice, we collected and observe them for tumor growth. After 3 weeks, we attempted to re-challenge them by injecting TC1 cells subcutaneously. The tumor growth was monitored by luciferase detection, while no further treatment was given to the animals.
TUNEL assay
To detect the apoptotic dead cells in situ, the TUNEL assay was employed using the Cell/Death Detection Kit (Roche Applied Science; catalog no. 11684809910) according to the manufacturer’s guidelines. Briefly, frozen sections with about 6 μm thickness from the OCT-embedded tissue were allowed to air-dry under the hood for 30 minutes. The samples were then fixed with 2% paraformaldehyde in PBS at room temperature for 30 minutes. Each sample was treated with 100 μL of 3% H2O2-methanol solution at room temperature for 20 minutes and washed (×3) with PBS. To block the nonspecific binding sites, each sample was further treated with a 10% normal goat serum blocking solution at room temperature for 1 hour. The samples were analyzed through a Nikon Ti fluorescence microscope and a Leica TCS SP8 confocal microscope.
Immunofluorescence assay
Tumor samples were sliced using a cryostat, and tissue slides were frozen. Frozen tissue slides were allowed to air-dry for 30 minutes. The tissue slides were fixed in 4% paraformaldehydein 1x warm PBS (pH 7.4) at room temperature for 15 minutes. Slides were washed (×3) with 1x PBS. Samples were incubated in 3% H2O2 in methanol at room temperature for 20 minutes, followed by washing (×3) in PBS. Normal goat serum (10%) was used for blocking (1 hour incubation at room temperature). Blocking solution was aspirated and a mixture of primary antibodies (Abs) was added (150 μL final volume of anti-CD31 dilution 1:200 (suggested 1:300), anti-ICAM 1:500 dilution, and anti-VCAM 1:700 dilution, in 5% Goat serum). Samples were incubated with primary Abs at 4°C overnight. The following day, the slides were washed (×3) with PBS. Then, a mixture of the secondary Abs was added (5 μg/mL final concentration in 5% goat serum) and samples were incubated at room temperature in dark for 1 h. Slides were washed (×3) in PBS. Finally, the stained tissue was mounted with PloLong Diamond Antifade Mountant with DAPI.
IFN-γ enzyme-linked immunosorbent spot assay (ELISpot)
To carry out the ELISpot analysis, 96-well MAIP plates were coated with a 2.5 μg/mL solution of rat anti-mouse IFN-γ overnight. Bulk splenocytes were collected and plated at 1.0 × 106 cells/well in triplicate. The samples were then incubated with 2 μg/mL peptides at 37°C for 20 hours. Afterward, the samples were washed with PBS and 0.05% Tween 20. The samples were then exposed to anti-mouse biotin-conjugated anti–IFN-γ antibody and incubated at 4°C overnight. The following day, streptavidin-alkaline phosphatase conjugate was introduced for 30 minutes. The samples were developed through the addition of nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate. Finally, the spots were analyzed employing an automated ELISpot reader (Autoimmun Diagnostika GmbH, Strassberg, Germany).
Study approval
Before conducting any in vivo studies, all animal studies protocols were approved by the IACUC and University Laboratory Animal Resources at the University of Pennsylvania. Mice were treated following the Animal Care and Use Committee guidelines of the University of Pennsylvania.
Results
Development and characterization of shikonin nanoparticles
SHK-loaded PLGA NPs were prepared, PEGylated, and functionalized with anti-TEM1 78Fc scFv (Fig. S1), as described previously.
28
One-step PEGylation and 78Fc conjugation resulted in the production of NPs with a high drug content and a conjugation efficiency of ~90% (Table S1). The unmodified and 78Fc-armed functionalized PLGA NPs displayed smooth spherical morphology with an average size of around 130 nm and zeta potential of approximately −35 mV (Fig. S1). After the PEGylation and conjugation of 78Fc, DC%, DE%, DL%, and YE% of SHK-loaded NPs were somewhat reduced (Table S1). Among various kinetic models used to fit the SHK release profile, the SHK release profile was found to obey Wagner's log-probability (Table S2). The pattern of drug release exhibited that 20% of the drug molecules could be released within the first 2 hours. Nevertheless, about 80% of the drug was found to be released after 48 h. The release of SHK from the NPs appeared to be somewhat pH-dependent with the highest release at pH 7.4 (Table S2).
Internalization and cytotoxicity of SHK NPs
The cellular uptakes of the free SHK, SHK-loaded PEGylated PLGA NPs, and 78Fc-armed SHK-loaded PEGylated PLGA NPs in the mammalian cells (MS1HuTem1, MS1, and TC1 cell lines) are shown in Fig. 1. Confirming our previous findings,
28
the engineered NPs enhanced the entry of SHK into the cells, while the functionalization of NPs with 78Fc could further enhance the internalization of NPs in the TEM1-expressing cells such as MS1HuTem1 or TC1 cells, but not in the TEM1-negative MS1 cells (Fig. 1A, B and C).
Based on the cytotoxicity analysis, the SHK nanoformulation was found to display higher cytotoxicity in the MS1, MS1HuTem1, and TC1 cell lines (Fig. 1D, E, and F, respectively). The empty NPs did not induce any cytotoxic effects, indicating the safety of nanoformulation itself. As shown in Fig. 1, the TEM1-targeting NPs showed significantly higher cytotoxicity in the TEM1-overexpressing cells, implying the high capacity of the 78Fc-armed SHK-loaded PEGylated PLGA NPs in the active targeting of the TEM1-expressing cells in vitro. On the other hand, while the free SHK showed a higher impact on the T lymphocytes, the SHK nanoformulation resulted in significantly lower toxicity (Fig. 1G), indicating that the cytotoxic effect of the nanoformulation is largely dependent on the active targeting mechanism and internalization of NPs into the target cells.
Inhibition of cell migration and invasion
As the most relevant approach for the establishment of de novo strategies for the effective diagnosis and therapy of cancer, we looked at the SHK effects on the migration and invasiveness of the tumor cells (Fig. 2). To mimic the migration of cancer cells in vivo, we studied the effects of SHK on the cell movement through the extracellular matrices and found SHK to induce substantial inhibitory effects on the invasive migration as a fundamental function underlying the cellular processes, including metastasis and invasion of cancer cells (Fig. 2). SHK at a concentration of 5 μM (i.e., IC50) was able to effectively inhibit both the cell migration and invasion (Fig. 2B and C, respectively).
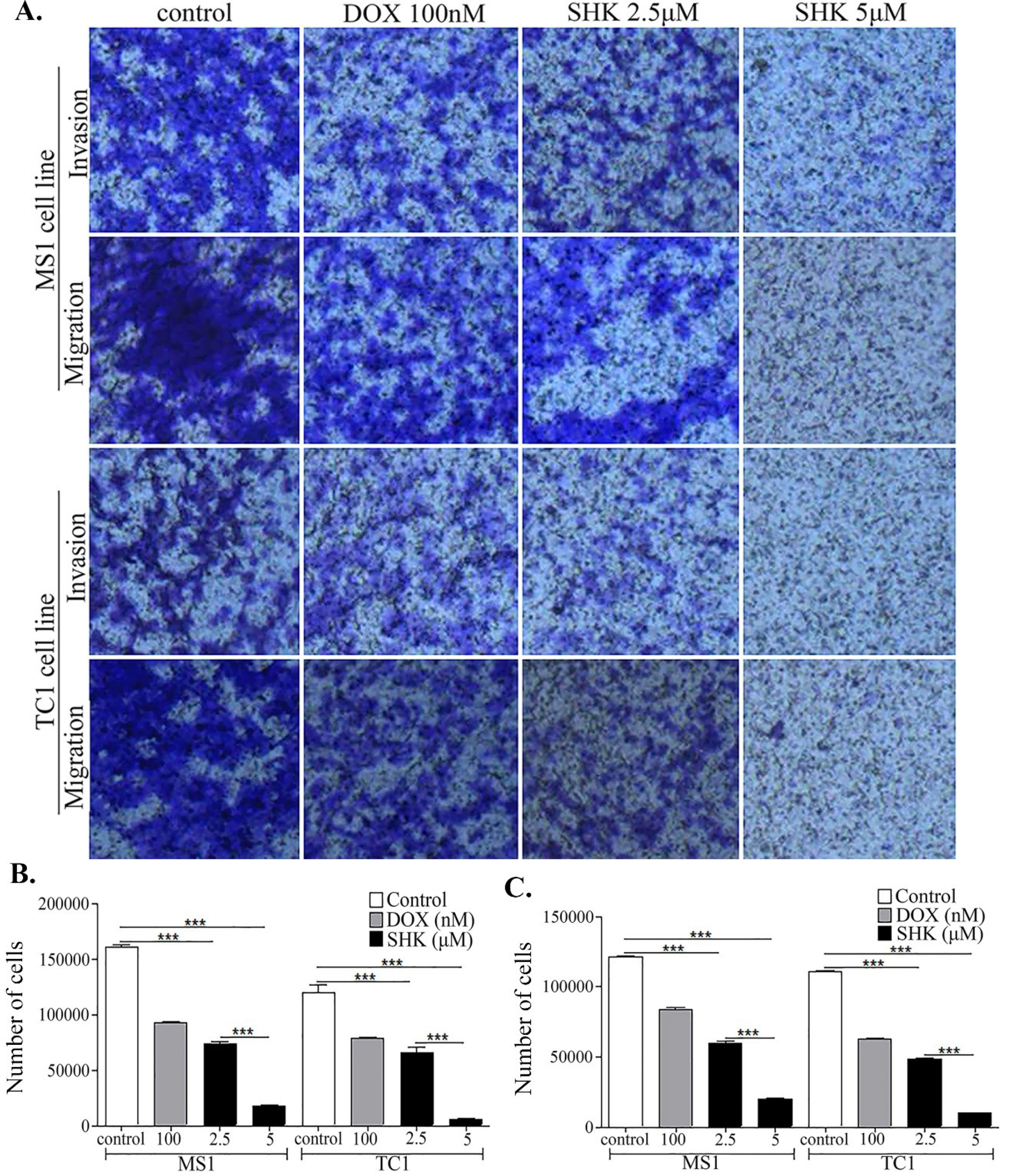
Fig. 2.
The effects of shikonin (SHK) on the tumor cells migration and invasion in different cell lines. A) The inhibitory effects of SHK on the migration and invasion abilities of the MS1 and TC1 cells cultivated in the Matrigel-coated transwells. B) The quantified inhibitory effects of SHK on the migration of cells. C) The quantified inhibitory effects on the invasion of cells.
.
The effects of shikonin (SHK) on the tumor cells migration and invasion in different cell lines. A) The inhibitory effects of SHK on the migration and invasion abilities of the MS1 and TC1 cells cultivated in the Matrigel-coated transwells. B) The quantified inhibitory effects of SHK on the migration of cells. C) The quantified inhibitory effects on the invasion of cells.
SHK effects on the formation of colony and heterochromatin foci and senescence
The colony formation analysis, for studying the anchorage-independent growth as a hallmark of carcinogenesis, revealed that SHK was able to inhibit the formation of colonies significantly as compared to DOX used as the positive control (Fig. 3A and B).
The cytotoxicity of SHK was further investigated to find the possible mechanisms(s) behind such cytotoxicity. The fluorescence microscopy analysis resulted in somewhat morphological changes in the nucleus of the cells upon treatment with SHK (Fig. 3C). Clear DNA-stained dense foci were observed, which were readily distinguishable from the chromatin in the normal cells resembling the senescence-associated heterochromatin foci (SAHF) in the SHK-treated cells (Fig. 3C), indicating of possible induction of the senescence and dilapidation by SHK. The senescence analysis through β-galactosidase staining revealed that only a few SHK-treated cells displayed increased β-galactosidase metabolism compared to the positive senescence inducer DOX-treated control cells (Fig. 3D). In the treated MS1 cells with 100 nM DOX, the whole cellular population showed increased β-galactosidase metabolism (Fig. 3E). These results indicate that SHK is not able to induce senescence.
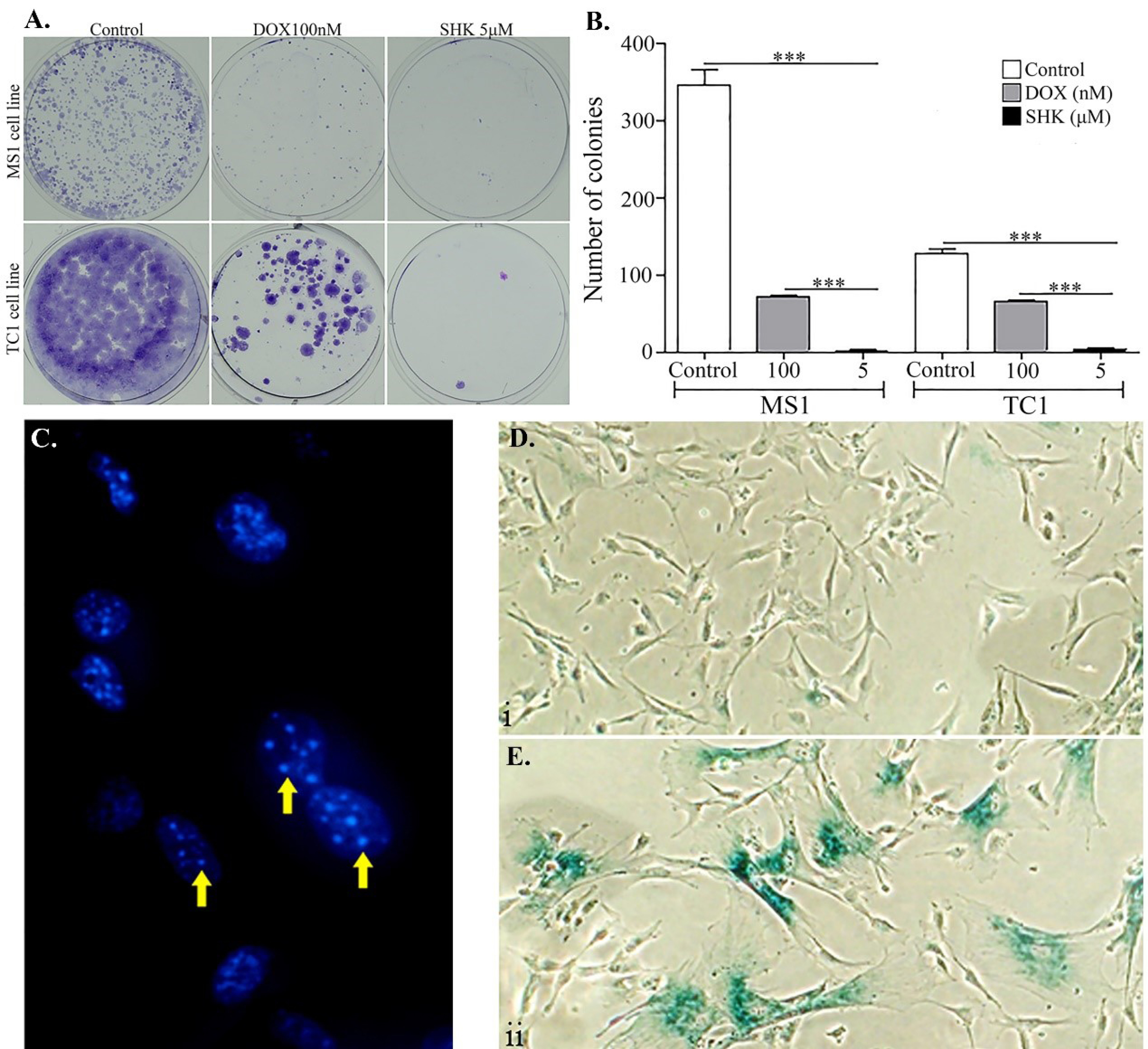
Fig. 3.
The SHK biological impacts. A) The formation colony in the treated MS1 and TC1cells 8 days after the treatment. Eight days after the treatment, the cells were fixed and stained with Crystal violet. Threshold 50 cells and higher were considered a single colony. B) The quantification of the colony formation in the MS1 and TC1 cells. C) The fluorescence microscopy image of DAPI staining of MS1 cells treated with SHK 2.5 μM. The DNA foci formation after SHK treatment is pointed with yellow arrows. D) β-galactosidase staining of MS1 cells treated with 2.5 μM SHK. E) β-galactosidase staining of MS1 cells treated with 100 nM DOX. Data are means ± SD of four independent experiments. *P < 0.05.
.
The SHK biological impacts. A) The formation colony in the treated MS1 and TC1cells 8 days after the treatment. Eight days after the treatment, the cells were fixed and stained with Crystal violet. Threshold 50 cells and higher were considered a single colony. B) The quantification of the colony formation in the MS1 and TC1 cells. C) The fluorescence microscopy image of DAPI staining of MS1 cells treated with SHK 2.5 μM. The DNA foci formation after SHK treatment is pointed with yellow arrows. D) β-galactosidase staining of MS1 cells treated with 2.5 μM SHK. E) β-galactosidase staining of MS1 cells treated with 100 nM DOX. Data are means ± SD of four independent experiments. *P < 0.05.
Induction of mitochondrial depolarization and autophagosomes by SHK
SHK, at a concentration above 2.5 μM, was found to induce some important morphological changes, including mitochondrial depolarization in the treated TC1 cells (Fig. 4A). The effect was found to be concentration-dependent (Fig. 4B). A strong signal of SHK was detected in the cytoplasm with an area of condensed chromatin in the nuclei of cells treated with SHK (Fig. 4C).
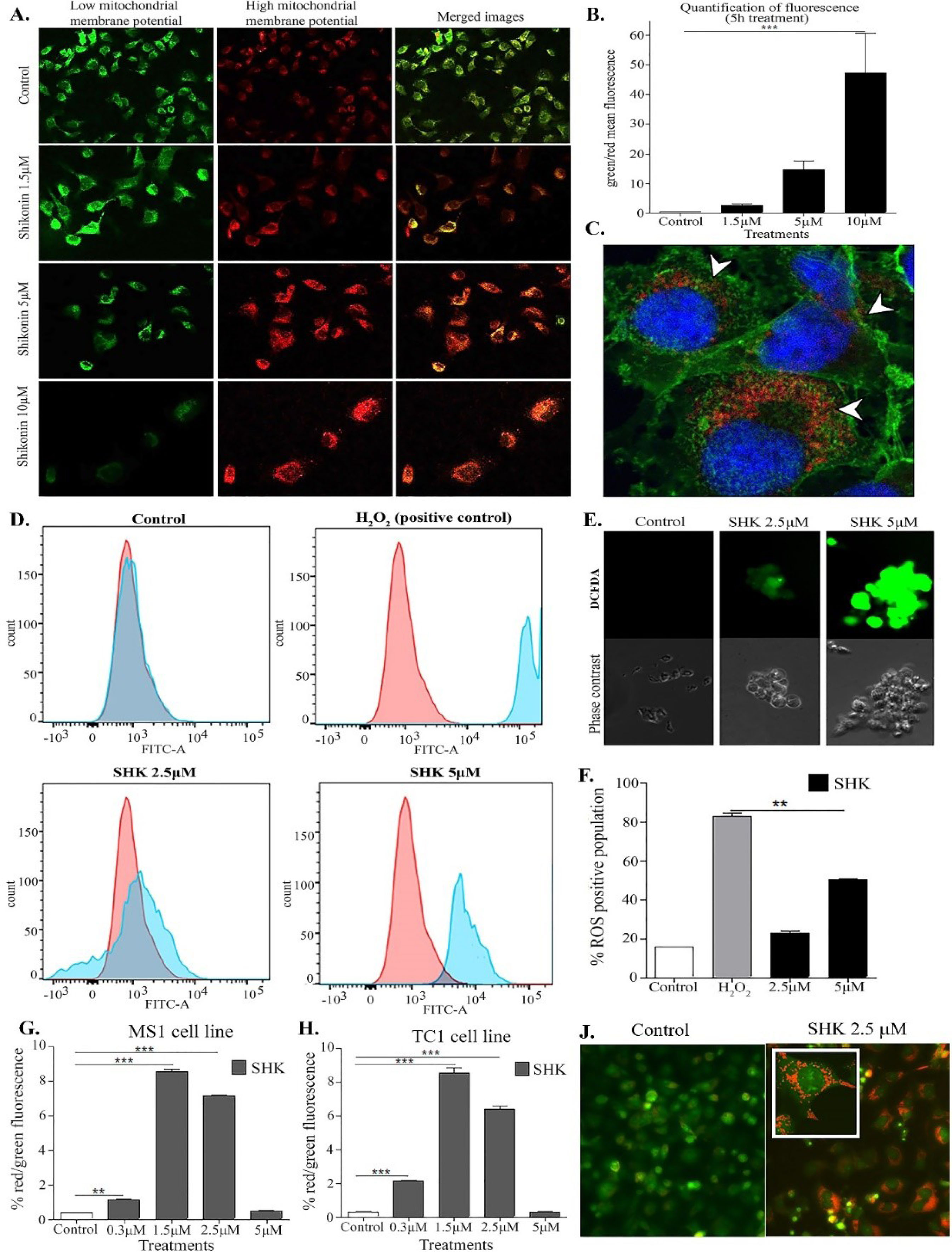
Fig. 4.
Induction of mitochondrial depolarization, reactive oxygen species (ROSs), and the acidic vacuolar organelles (AVO) in the cells treated with shikonin (SHK). A) Fluorescence microscopy images of the TC1 cells treated with SHK (1.5 µM, 5 µM, and 10 µM) for 5 h and stained with JC1. The depolarized and normal mitochondria are shown as green and red, respectively. B) Quantification of green/red fluorescence ratio of SHK-treated TC1 cells. C) Confocal microscopy image of the TC1 cells treated SHK (5 µM for 3 h). The cells were fixed and stained with Wheat germ agglutinin (membrane, green) and DAPI (nucleus, blue), and SHK (red). SHK localized in the cytoplasm. D) Flow cytometry results after 3 h treatment. E) Light and fluorescent microscopy images of DCFDA stained cells after 3h treatment. F) Quantification of ROS-positive cells treated with SHK. G and H) Flow cytometry analysis of fluorescence ratio in the TC1 cells and the MS1 cells, respectively. The cells were analyzed after 5h treatment with various concentrations of SHK. J) Fluorescence microscopy images of the untreated (left) and the SHK (2.5 mM)-treated (right) cells. *P < 0.05.
.
Induction of mitochondrial depolarization, reactive oxygen species (ROSs), and the acidic vacuolar organelles (AVO) in the cells treated with shikonin (SHK). A) Fluorescence microscopy images of the TC1 cells treated with SHK (1.5 µM, 5 µM, and 10 µM) for 5 h and stained with JC1. The depolarized and normal mitochondria are shown as green and red, respectively. B) Quantification of green/red fluorescence ratio of SHK-treated TC1 cells. C) Confocal microscopy image of the TC1 cells treated SHK (5 µM for 3 h). The cells were fixed and stained with Wheat germ agglutinin (membrane, green) and DAPI (nucleus, blue), and SHK (red). SHK localized in the cytoplasm. D) Flow cytometry results after 3 h treatment. E) Light and fluorescent microscopy images of DCFDA stained cells after 3h treatment. F) Quantification of ROS-positive cells treated with SHK. G and H) Flow cytometry analysis of fluorescence ratio in the TC1 cells and the MS1 cells, respectively. The cells were analyzed after 5h treatment with various concentrations of SHK. J) Fluorescence microscopy images of the untreated (left) and the SHK (2.5 mM)-treated (right) cells. *P < 0.05.
Upon analysis by using fluorogenic dye, DCFDA was used to measure the hydroxyl/peroxyl and other reactive oxygen species (ROS) activities within the cell, in which the formation of ROSs was observed in the SHK-treated cells as compared to the untreated negative and H2O2-treated positive cells (Fig. 4D). The SHK-induced mitochondrial depolarization appeared to elicit the formation of ROSs in a dose-dependent manner (Fig. 4E and 4F).
Based on the acridine orange staining analysis, the AVOs were formed in the cells treated with SHK, in large part because of the induction of autophagosomes elicited by ROS in the SHK-treated MS1 and TC1 cells (Fig. 4G and 4H, respectively). The autophagosomes were formed in the cells treated with lower concentrations of SHK (2.5 µM) but not in the cells treated with the toxic concentrations of SHK (Fig. 4J), indicating that the lower concentrations of SHK could function as an autophagy inducer. The breakdown of the mitochondrial transmembrane potential might be attributed to the mitochondrial permeability transition pores opening, which might result in the release of cytochrome c into the cytosol. Such a phenomenon, in turn, can prompt the activation of other downstream events in the apoptotic cascade. The induction of mitochondrial depolarization by SHK was further validated with the JC1 staining, which revealed that SHK could cause a shift in the mitochondrial membrane potential in a dose-dependent manner, resulting in enhanced mitochondrial depolarization (Fig. 5).
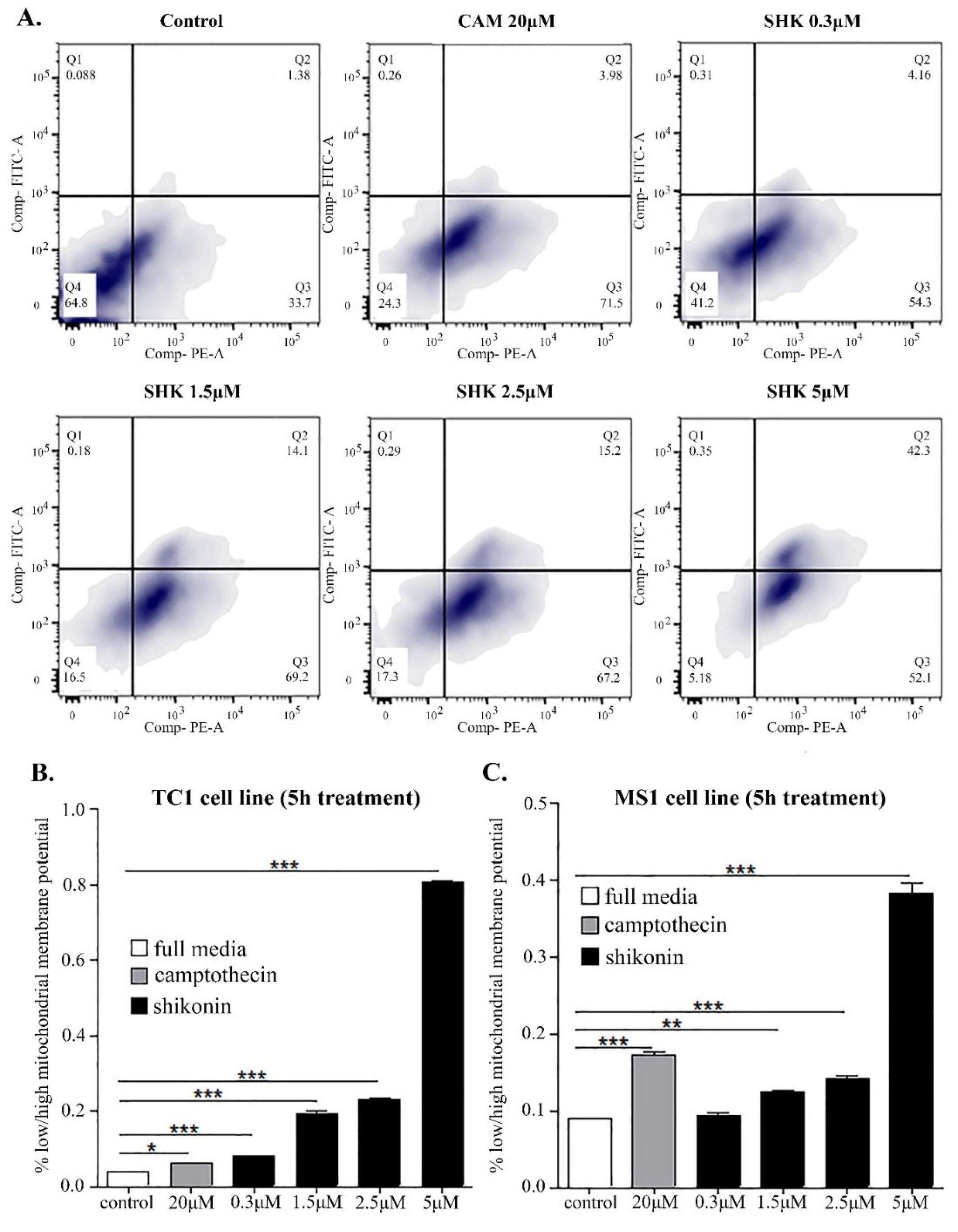
Fig. 5.
Flow cytometry analysis of JC1 staining for mitochondrial depolarization detection TC1 and MS1 cells. A) Mitochondrial depolarization is increased when SHK concentration is increased. In the SHK 5µM treated cells, ~50% of the cell population showed depolarized mitochondria. B) TC1 cells fluorescence ratio. C) MS1 cells fluorescence ratio. *P < 0.05.
.
Flow cytometry analysis of JC1 staining for mitochondrial depolarization detection TC1 and MS1 cells. A) Mitochondrial depolarization is increased when SHK concentration is increased. In the SHK 5µM treated cells, ~50% of the cell population showed depolarized mitochondria. B) TC1 cells fluorescence ratio. C) MS1 cells fluorescence ratio. *P < 0.05.
Apoptosis and necrosis effects of NPs in vitro
The apoptosis and necrosis analyses, performed using the annexin-V FITC/PI in MS1 and TC1 cell lines after treatment with SHK 5μM or staurosporine (apoptosis inducer) 1 μM at 37°C for 2 hours, three populations of cells observed in both cell lines (Fig. 6A). These cells included (i) the viable cells with no apoptotic or necrotic signs (i.e., annexin V-cyanine 3 and NDR negative); (ii) the apoptotic cells (i.e., annexin V-cyanine 3 positive and NDR negative); and (iii) the cells underwent the late-stage apoptosis and early necrosis (i.e, annexin V-cyanine 3 and NDR positive). The ratio of the apoptotic to the necrotic cells in the SHK-treated MS1 and TC1 cells was 1:1, while the ratio of the apoptotic to the necrotic cells treated with staurosporine was 2:1 (Fig. 6B and C). These findings suggest that SHK can induce both cell death mechanisms.
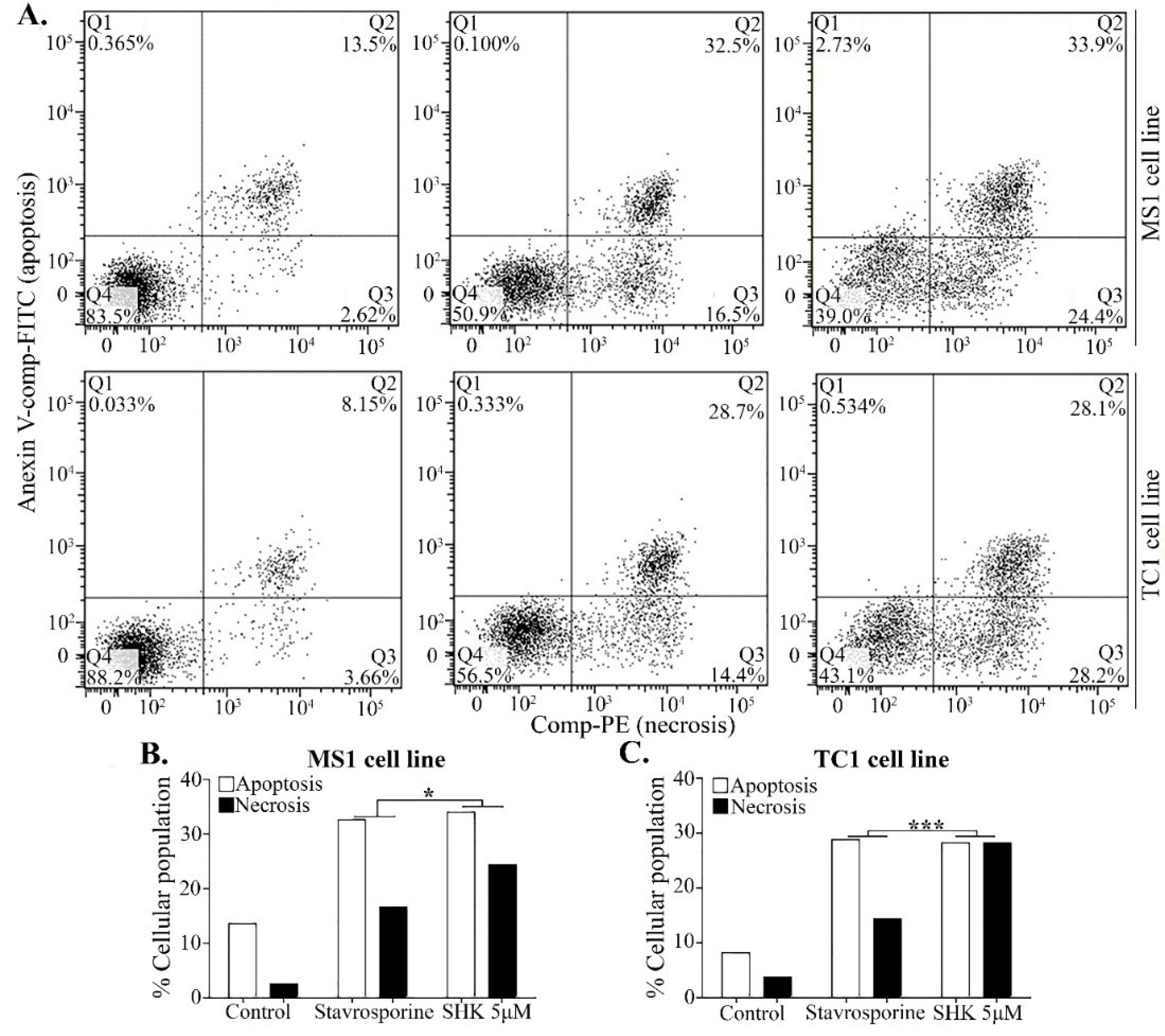
Fig. 6.
Apoptosis/necrosis assay by the annexin-V FITC/PI in MS1 and TC1 cell lines after SHK 5μM or staurosporine (apoptosis inducer) 1μM 2h treatment. A) Detection of Apoptosis/necrosis in MS1 and TC1 cell lines by flow cytometry. B) Evaluation of apoptotic/necrotic ratio of % cellular death in MS1 cell line. C) Evaluation of apoptotic/necrotic ratio of % cellular death in TC1 cell line. *P < 0.05.
.
Apoptosis/necrosis assay by the annexin-V FITC/PI in MS1 and TC1 cell lines after SHK 5μM or staurosporine (apoptosis inducer) 1μM 2h treatment. A) Detection of Apoptosis/necrosis in MS1 and TC1 cell lines by flow cytometry. B) Evaluation of apoptotic/necrotic ratio of % cellular death in MS1 cell line. C) Evaluation of apoptotic/necrotic ratio of % cellular death in TC1 cell line. *P < 0.05.
In vivo biodistribution and targeting efficacy in a mouse model
The biodistribution analysis of the engineered NPs loaded with indocyanine green (ICG) injected intravenously via the retro-orbital route revealed no signal of ICG in the mice after 24 hours. Once the organs were harvested, there was no high detection of ICG in any of the organs observed, suggesting that the NPs did not maintain by the liver after 72 hours. However, there was somewhat retention of the ICG in the liver in the control group (free ICG treated) (data not shown). The active targeting efficacy of the 78Fc-armed SHK-loaded NPs was confirmed using a mice model
27
subcutaneously injected with the MS1 and the MS1HuTem1 cells respectively at the right and left sides (Fig. 7A). The luciferase was detected on the first day of the detection and the last day of the experiment (Fig. 7A). On the first day of the experiment, the two sides appeared to have the same number of cells; however, after treatment with the 78Fc-armed NPs, the MS1HuTem1 cells were significantly eliminated (Fig. 7B), indicating the high in vivo targeting efficacy of the 78Fc-armed SHK-loaded NPs, as well as the efficacy of SHK as a cytotoxic agent. The luminescence quantification confirmed the results (Fig. 7B). The MS1 and MS1HuTem1 cells were developed to have the same growth rate in vivo and to express the same levels of luciferase (Fig. 7C and 7D). Mice weight was monitored daily, and neither any fluctuation of animal weight nor any behavioral change was observed, implying that this nano-therapy did not impose severe toxicity. As shown in Fig. 7E, the SHK high fluorescent signal was detected in the MS1HuTem1 hemangioma even 5 days after treatment, while we couldn’t detect any luciferase signal (live cells) in that area the same day. Furthermore, we noticed that when SHK was diluted in the serum, it could emit at 620 nm, indicating SHK as a compound easy to detect by in vivo imaging (Fig. 7F).
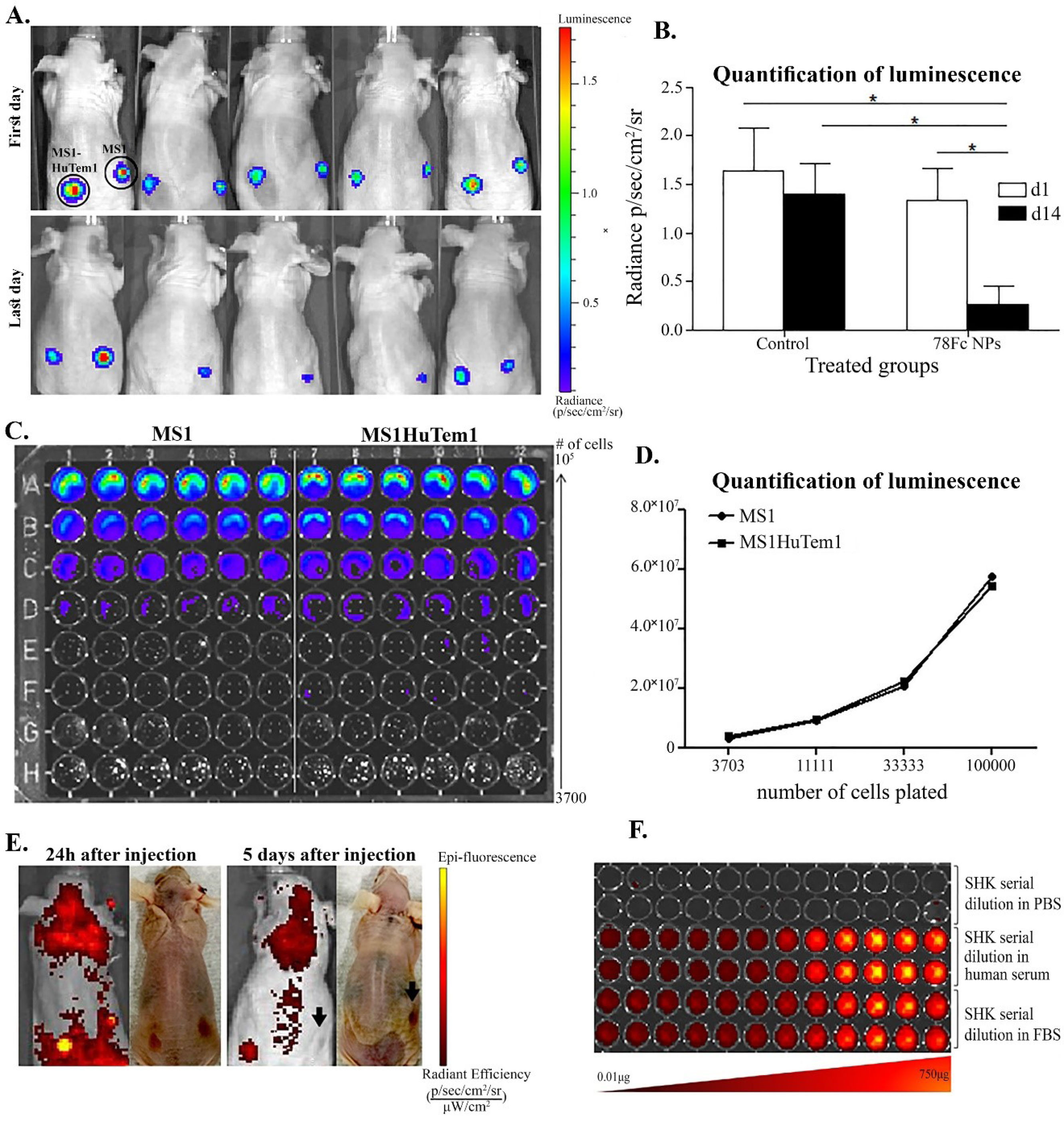
Fig. 7.
In vivo targeting efficacy of anti-TEM1 78Fc-armed SHK-loaded PLGA NPs. Mice were bearing MS1 cells subcutaneous at their right side and MS1HuTem1 cells at their left side. A) Representative image of luminescence detection of the MS1 and MS1-huTem1 cell lines. B) Quantification of cellular luciferase expression. C) Luminescence detection on the first (top) and the last day (bottom) of the experiment. D) Quantification of the MS1HuTem1/MS1 luminescence detection ratio of control and anti-TEM1 Fc armed SHK-loaded PLGA NPs treated groups. E) Detection of SHK fluorescence 24h after the first injection (left) and on the last day of the experiment (right). SHK high fluorescent signal is detected in the MS1-huTem1 hemangioma, even 5 days after treatments, while the MS1-huTem1 hemangioma disappeared. F) Representative image of detection of fluorescence of SHK serial dilutions in PBS (top) human serum (middle) and FBS (bottom). SHK: Shikonin, PLGA: poly(lactic-co-glycolic acid, NPs: nanoparticles. *P < 0.05.
.
In vivo targeting efficacy of anti-TEM1 78Fc-armed SHK-loaded PLGA NPs. Mice were bearing MS1 cells subcutaneous at their right side and MS1HuTem1 cells at their left side. A) Representative image of luminescence detection of the MS1 and MS1-huTem1 cell lines. B) Quantification of cellular luciferase expression. C) Luminescence detection on the first (top) and the last day (bottom) of the experiment. D) Quantification of the MS1HuTem1/MS1 luminescence detection ratio of control and anti-TEM1 Fc armed SHK-loaded PLGA NPs treated groups. E) Detection of SHK fluorescence 24h after the first injection (left) and on the last day of the experiment (right). SHK high fluorescent signal is detected in the MS1-huTem1 hemangioma, even 5 days after treatments, while the MS1-huTem1 hemangioma disappeared. F) Representative image of detection of fluorescence of SHK serial dilutions in PBS (top) human serum (middle) and FBS (bottom). SHK: Shikonin, PLGA: poly(lactic-co-glycolic acid, NPs: nanoparticles. *P < 0.05.
Therapeutic efficacy of NPs in the mouse and metastatic mouse models
The 78Fc-armed SHK-loaded NPs therapeutic efficacy analysis in the TC1 subcutaneously injected nu/nu mice model
51
showed the luciferase expression in the 78Fc-armed SHK-loaded NPs treated group in comparison with the PBS-treated as the control on the first and the last day of the experiment (Fig. 8A and B). The quantity of the luciferase expression in the treated group with the 78Fc-armed SHK-loaded PLGA NPs was significantly lower than that of the PBS treated group (Fig. 8C). The tumor weight and size in the treated group with the 78Fc-armed SHK-loaded PLGA NPs were significantly reduced as compared to the control group (Fig. 8D and E).
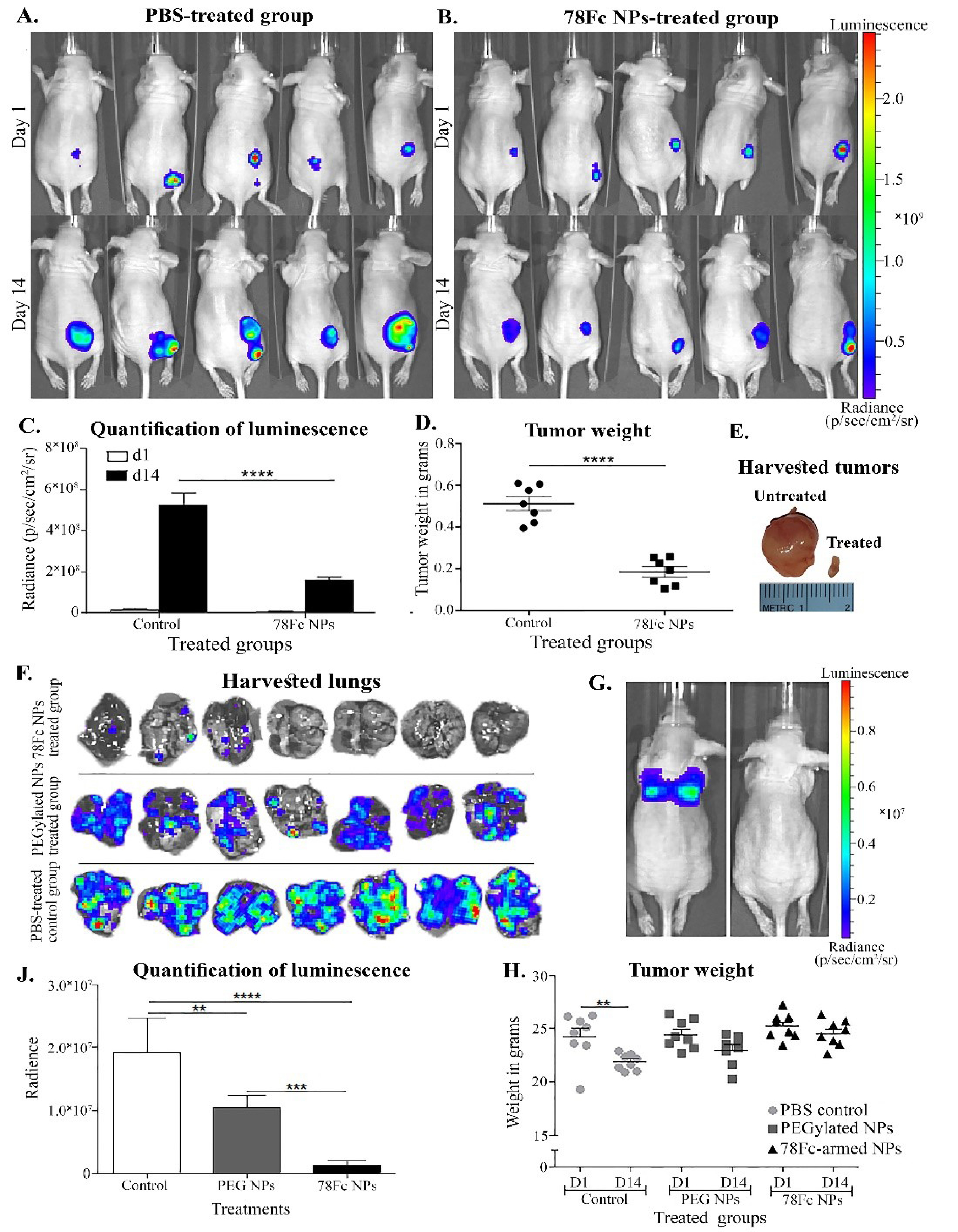
Fig. 8.
TC1 subcutaneous tumor model (panels A to E) and intravenous (panels F to J) tumor metastasis models tested in nu/nu mice. A) PBS-treated, control group at first (top) and last (bottom) day respectively. B) PEGylated SHK-loaded anti-TEM1 78Fc conjugated PLGA NPs treated group at first (top) and last (bottom) day respectively. C) Quantification of detected luminescence on day 1 and 14, in both treated groups. D) The weight of the harvested tumors. E) Image of TC1 harvested tumors left side PBS-treated control group, middle PLGA PEGylated SHK-loaded NPs-treated group, right side PLGA PEGylated 78Fc-armed SHK-loaded NPs treated group. F) Harvested lungs on the last day of the experiment. G) Luminescence detection image on day 14 of a mouse treated with PBS (left) and a mouse treated with PLGA PEGylated 78Fc-armed SHK-loaded NPS. H) Quantification of luminescence detected in the lungs and statistical analysis using t-test (comparison between groups). J) Weight monitoring on the first and the last day of the experiment. SHK: shikonin PLGA: poly(lactic-co-glycolic acid), NPs: nanoparticles.
.
TC1 subcutaneous tumor model (panels A to E) and intravenous (panels F to J) tumor metastasis models tested in nu/nu mice. A) PBS-treated, control group at first (top) and last (bottom) day respectively. B) PEGylated SHK-loaded anti-TEM1 78Fc conjugated PLGA NPs treated group at first (top) and last (bottom) day respectively. C) Quantification of detected luminescence on day 1 and 14, in both treated groups. D) The weight of the harvested tumors. E) Image of TC1 harvested tumors left side PBS-treated control group, middle PLGA PEGylated SHK-loaded NPs-treated group, right side PLGA PEGylated 78Fc-armed SHK-loaded NPs treated group. F) Harvested lungs on the last day of the experiment. G) Luminescence detection image on day 14 of a mouse treated with PBS (left) and a mouse treated with PLGA PEGylated 78Fc-armed SHK-loaded NPS. H) Quantification of luminescence detected in the lungs and statistical analysis using t-test (comparison between groups). J) Weight monitoring on the first and the last day of the experiment. SHK: shikonin PLGA: poly(lactic-co-glycolic acid), NPs: nanoparticles.
The 78Fc-armed SHK-loaded NPs' therapeutic efficacy in the TC1 lung metastasis model analysis revealed that the engineered NPs impose great efficacy in the treated animals (Fig. 8F and 8G). During the last day of the experiment, the tumor nodules at the lungs of the animals treated with the 78Fc-armed SHK-loaded PLGA NPs were significantly decreased or even diminished as compared to the control group (Fig. 8F and 8G). The quantity of luminescence revalidated such findings (Fig. 8J). The weight of the animals treated with the 78Fc-armed SHK-loaded PLGA NPs remained stable throughout the experiment as compared to the control group that showed weight loss and some behavioral changes (Fig. 8H).
Therapeutic efficacy of NPs in the immunocompetent mice
The SHK immunomodulatory capacity
37
was investigated in the C57BL6 mice subcutaneously injected with the TC1 cells with results showing a lowered/diminished luciferase expression in the treated animals with the 78Fc-armed SHK-loaded PLGA NPs on day 14 (Fig. 9A and 9B). The quantity of luminescence validated the results (Fig. 9C and D). The animals treated with the engineered NPs showed good weight stability as compared to the PBS-treated group (Fig. 9E and 9F). On the last day of the experiment, approximately 72% of the mice population treated with the 78Fc-armed SHK-loaded PLGA NPs were tumor-free, while the rest 28% of the mice population had extremely small tumors as compared to the control group or the PEGylated NPs group (Fig. 9G and 9H), indicating the therapeutic effects of the targeted nanoformulation in the immunocompetent C57BL6 mice.
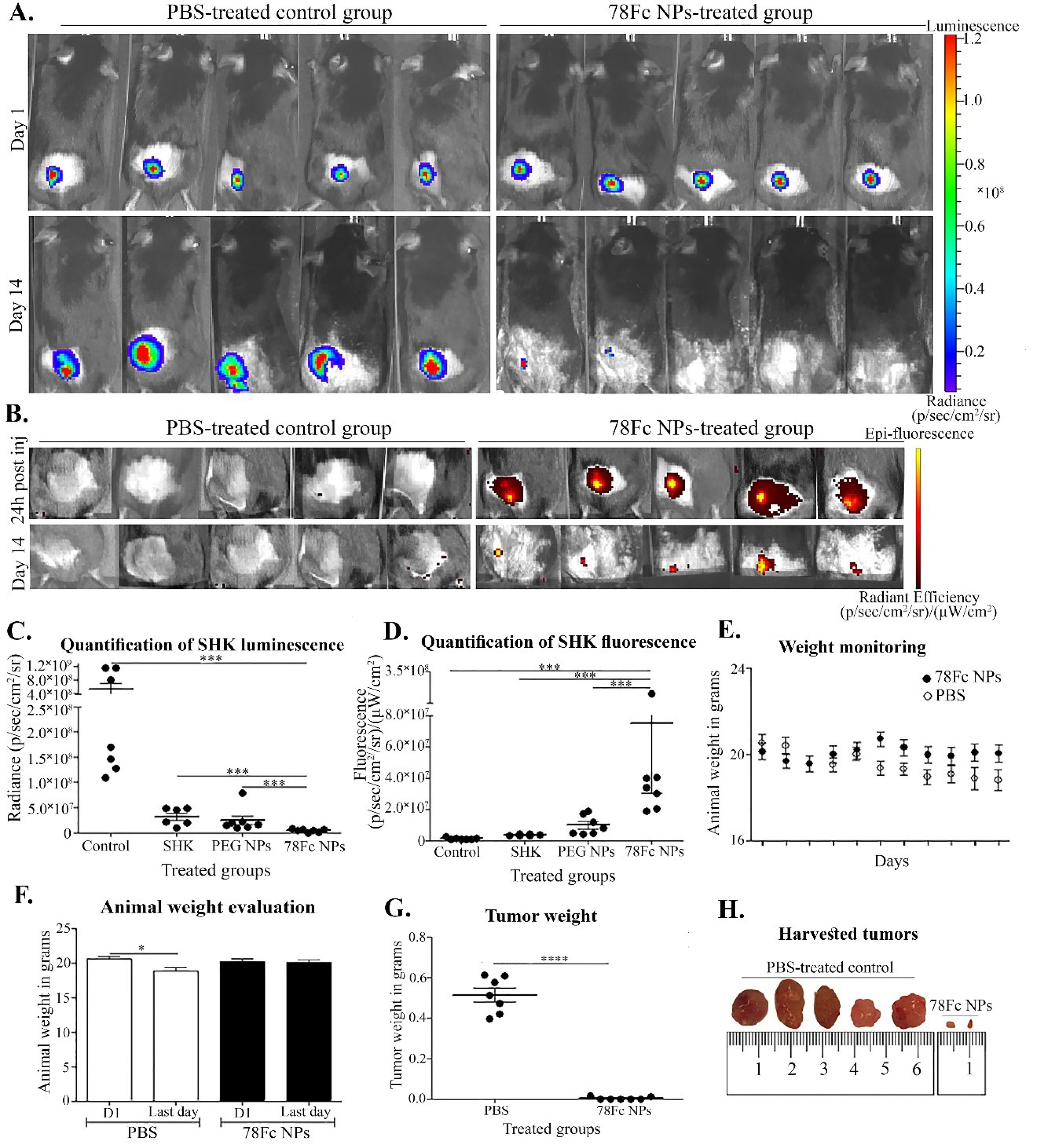
Fig. 9.
Small animal imaging and analysis of the treated animals. A) Luminescence detection before starting treatments and the last day of the experiment. The PBS-treated and 78Fc treated groups are shown in the left and right, respectively. B) Fluorescence detection 24h after the 1st injection and on the last day of the experiment (right) in comparison with the PBS-treated group (left). C) Quantification of luminescence detected. D) Quantification of SHK fluorescence detected. E) Animal weight monitoring throughout the experiment. F) Analysis of animal weight change between the first and last days. G) Analysis of tumor weight of PBS and 78Fc NPs treated groups. H) The isolated tumors from the PBS-treated group and the 78Fc NPs treated group. *P < 0.05.
.
Small animal imaging and analysis of the treated animals. A) Luminescence detection before starting treatments and the last day of the experiment. The PBS-treated and 78Fc treated groups are shown in the left and right, respectively. B) Fluorescence detection 24h after the 1st injection and on the last day of the experiment (right) in comparison with the PBS-treated group (left). C) Quantification of luminescence detected. D) Quantification of SHK fluorescence detected. E) Animal weight monitoring throughout the experiment. F) Analysis of animal weight change between the first and last days. G) Analysis of tumor weight of PBS and 78Fc NPs treated groups. H) The isolated tumors from the PBS-treated group and the 78Fc NPs treated group. *P < 0.05.
Therapeutic efficacy of NPs in re-challenged animals
A few days after the TC1 cells re-challenge, the cellular growth started decreasing and by day 14 the TC1 cells were eliminated (Fig. 10A). The analysis of the tumor site where TC1 cells were injected on the last day of the experiment validated the results (Fig. 10B), which were further validated by the quantity of luminescence (Fig. 10C). These results indicate that the immune system of the C57BL6 mice remained to be functional after the treatment with the 78Fc-armed SHK-loaded PLGA NPs, enabling animals to recognize and eliminate the tumor cells.
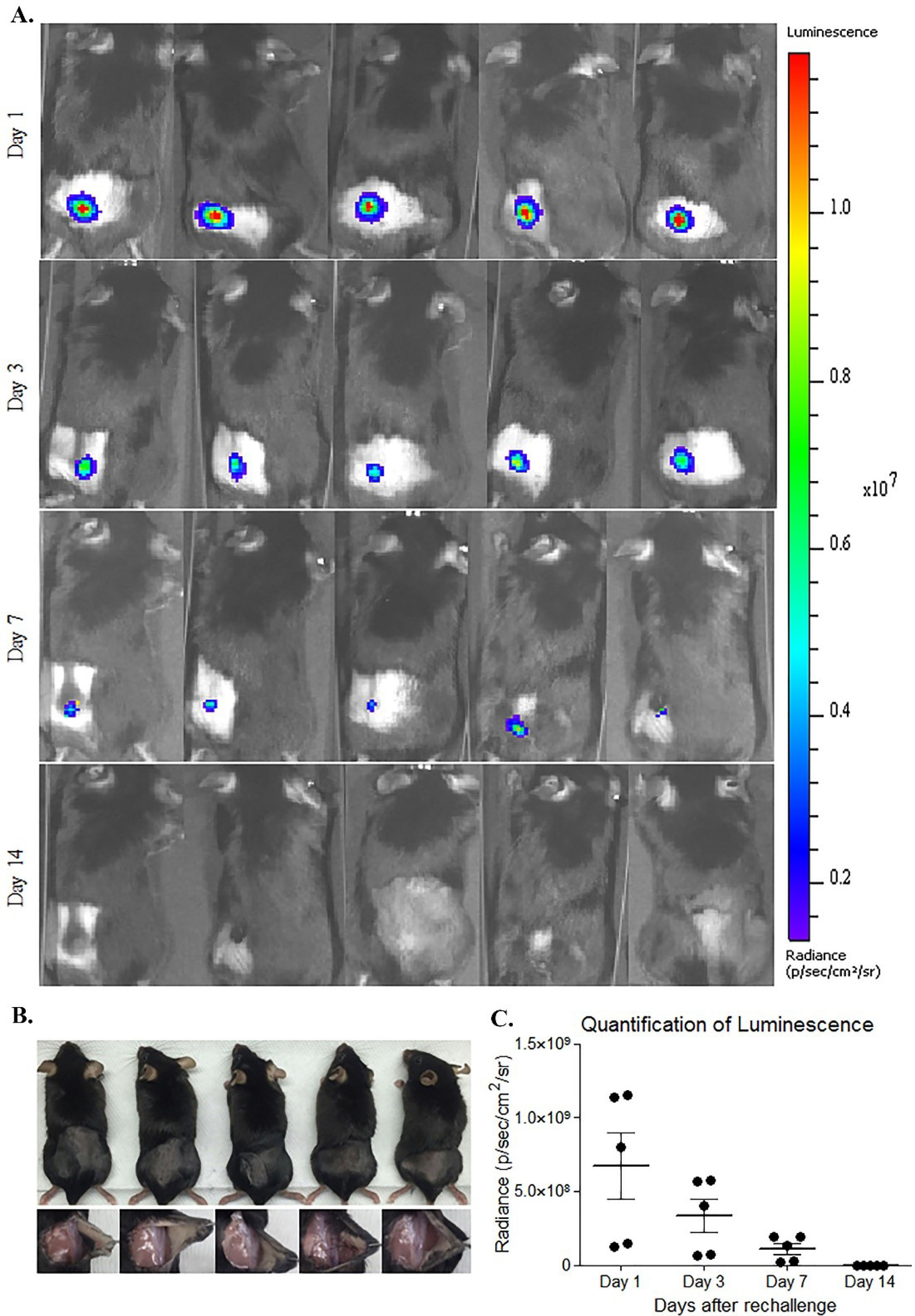
Fig. 10.
Small animal imaging of C57BL6 mice. A) The re-challenged animals with TC1 cells expressing luciferase. B) Photograph of the site where TC1 cells were injected on the last day of the experiment. C) Quantification of luminescence throughout the experiment.
.
Small animal imaging of C57BL6 mice. A) The re-challenged animals with TC1 cells expressing luciferase. B) Photograph of the site where TC1 cells were injected on the last day of the experiment. C) Quantification of luminescence throughout the experiment.
Induction of apoptosis and necrosis in vivo
The results of the TUNEL assay showed that the slides of frozen tissue samples treated with the 78Fc-armed SHK-loaded PEGylated PLGA NPs were stained positive for TUNEL reagent as compared to the PBS treated group as the control. This finding confirms the induction of apoptosis in vitro by the DNA fragmentation resultant from apoptotic signaling cascades (Fig. 11).
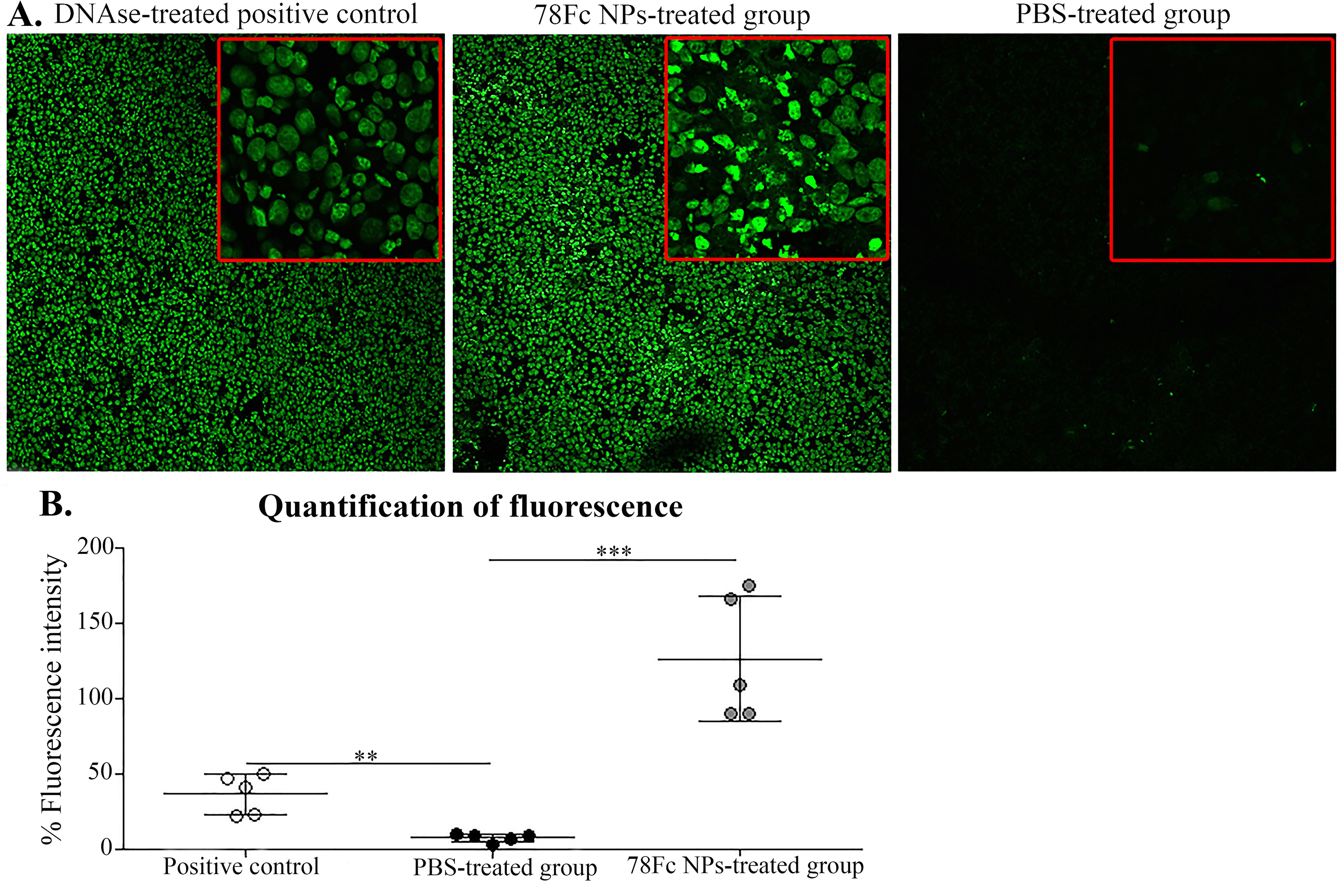
Fig. 11.
TUNEL assay on frozen tissue samples. A) The DNAse treated positive control (left), the 78Fc treated group sample (middle), the PBS-treated group (right). Images were taken using confocal microscope magnification 10x, images insert magnification is 100x. B) Tissue detected fluorescence intensity quantified using Fiji software.
.
TUNEL assay on frozen tissue samples. A) The DNAse treated positive control (left), the 78Fc treated group sample (middle), the PBS-treated group (right). Images were taken using confocal microscope magnification 10x, images insert magnification is 100x. B) Tissue detected fluorescence intensity quantified using Fiji software.
Further, based on the immunofluorescence and TUNEL analyses, the induction of apoptosis in vivo in the slides of frozen tissue samples treated with 78Fc-armed SHK-loaded NPs was detected as compared to the PBS treated group as the negative control. It was found that SHK treated cells could exhibit apoptotic: necrotic cell death at approximately 1:1 ratio (Fig. 12).
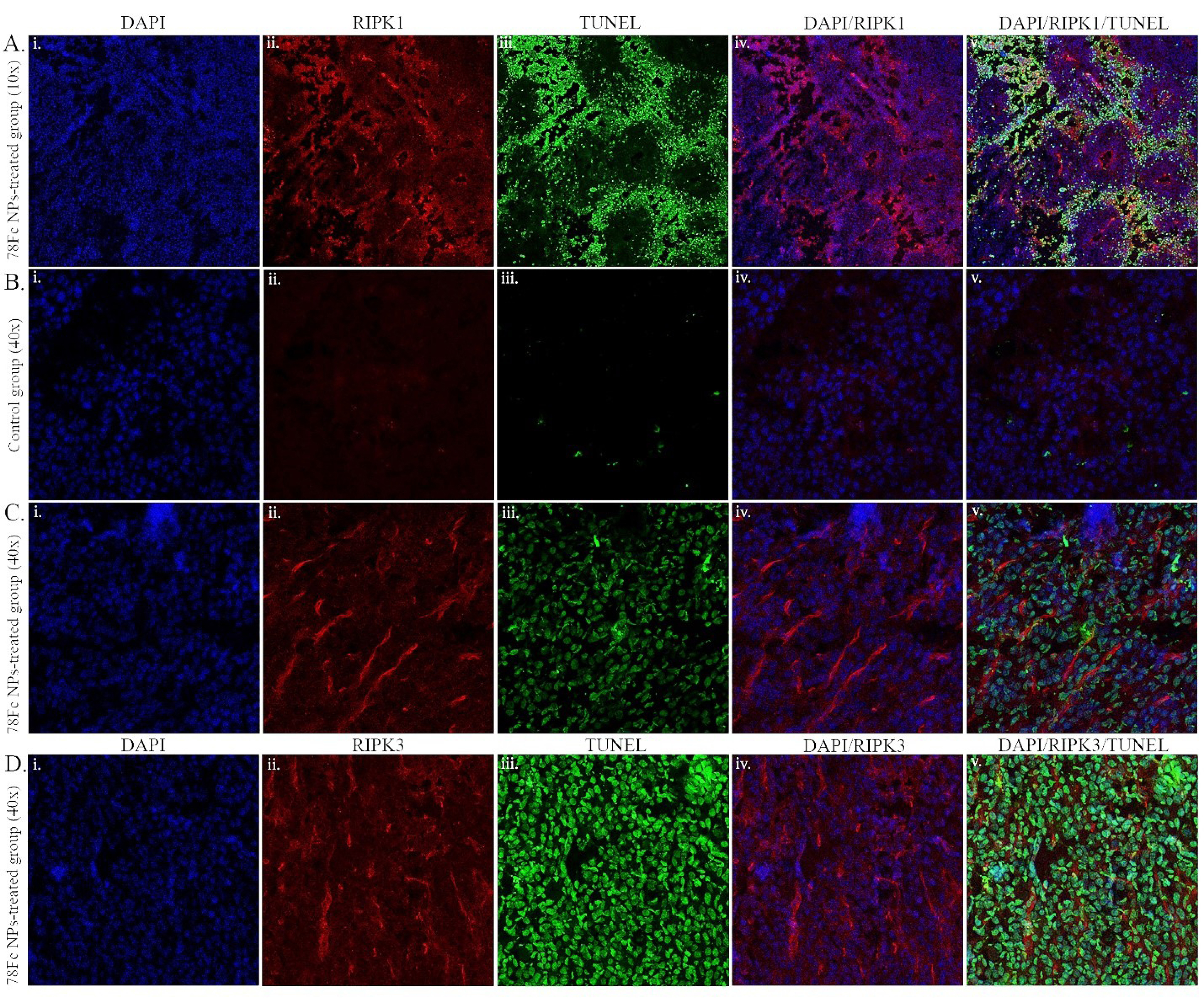
Fig. 12.
RIPK1/3 and TUNEL staining on frozen in vivo tissue samples. A) 78Fc-armed SHK-loaded NPs treated samples (10x magnification). (B) PBS-treated control group samples (40x magnification). C) 78Fc-armed SHK-loaded NPs treated samples (40x magnification). D) 78Fc-armed SHK-loaded NPs treated samples (40x magnification). (i) DAPI nuclear staining (blue), (ii) RIPK1/3 staining (red), (iii) TUNEL staining (green), (iv) merged DAPI (blue) and RIPK1/3 (red) channels, (v) merged DAPI (blue), RIPK1/3 (red), and TUNEL (green) images.TUNEL (green) images merged.
.
RIPK1/3 and TUNEL staining on frozen in vivo tissue samples. A) 78Fc-armed SHK-loaded NPs treated samples (10x magnification). (B) PBS-treated control group samples (40x magnification). C) 78Fc-armed SHK-loaded NPs treated samples (40x magnification). D) 78Fc-armed SHK-loaded NPs treated samples (40x magnification). (i) DAPI nuclear staining (blue), (ii) RIPK1/3 staining (red), (iii) TUNEL staining (green), (iv) merged DAPI (blue) and RIPK1/3 (red) channels, (v) merged DAPI (blue), RIPK1/3 (red), and TUNEL (green) images.TUNEL (green) images merged.
Further, the IFN-γ enzyme-linked immunosorbent spot assay analysis was conducted to study whether the murine immune system was able to recognize and eliminate the TC1 cells. The ELISpot analysis, which was performed on the splenocytes of the 78Fc-armed SHK-loaded NPs treated tumor-free mice, showed a weak response to the E6/E7 peptides which was not statistically significant as compared to the positive control.
Discussion
Despite the constant advances in the field of oncology, cancer remains to be one of the leading causes of death worldwide. After a conventional chemotherapy course, cancer patients are prone to relapse and/or develop drug resistance. Although chemotherapy modalities have been accepted as an effective treatment strategy for most types of cancers, such an intervention is often linked with some undesired side effects. The major reason for the occurrence of adverse reactions is the cytotoxic nature of most of the anticancer chemotherapeutic agents and their nonspecific distribution to healthy tissues/organs. Many studies have attempted to develop targeted DDS and achieve specific/selective delivery of chemotherapeutics into the TME. Given that the tumor neovasculature exhibits a fenestrated endothelial feature, in combination with the poor lymphatic drainage at the TME, nanomedicines can accumulate passively within the TME through the EPR effect. The nanoscale delivery system can further be decorated with a targeting moiety to serve as a nanocarrier with active targeting potential.
52
Different nanosized DDSs have been employed for the delivery of cytotoxic agents. Unlike non-biodegradable polymer-/lipid-based DDSs that are considered as toxic entities,
53-56
most of the biodegradable polymers like PLGA happen to be a much safer drug carrier for the delivery of cytotoxic chemotherapies.
57-60
In the present study, we developed an advanced nanoformulation platform as reported previously
28
for the targeted delivery of SHK – a known natural secondary metabolite with cytostatic, cytotoxic, and immunomodulatory impacts in a concentration-dependent manner.
61,62
The SHK was shown to activate both receptors and the mitochondrial-mediated apoptosis effectively
37
and circumvent the cancer drug resistance via the initiation of necroptotic cell death.
63
To limit the SHK cytotoxicity solely within the TME, we formulated the SHK-loaded PEGylated PLGA NPs armed with a TEM1-specific antibody fragment, 78Fc scFv (Fig. S1). Based on the composition (polymer and surfactant type and concentration) and formulation method,
28
PLGA NPs were formulated with good loading efficiency (Table S1) and a size range of 100-140 nm with a smooth surface (Fig. S1). The release of SHK from PLGA NPs in both PBS and the human serum was investigated at different pHs to disclose the release profile within the vesicular machinery of the target cells (e.g., endosomal compartments) and the TME, where the pH is acidic. The release rate was found to be slower at the lower pH, indicating a sustained-release profile in an acidic condition (Table S2). Such behavior can lead to the accumulation of drug molecules at the TME, where the pH is somewhat acidic. It has been reported that NPs of this size can actively accumulate at the tumor site in approximately 2 hours,
64,65
during which period the systemic release of SHK is speculated to be around 20%. However, the systemic release of a small amount of SHK might be in favor of the cancer therapy goals via stimulating the immune system.
39
Once the NPs reached the TME through size-dependent passive targeting mechanism, they can attach to the TEM1-expressing cells through the targeting agent and actively accumulate within the TME where the remaining ~80% of SHK load can be released. Given that the cytotoxic effects of SHK are non-specific, the main purpose of the current study was to deliver SHK actively specifically to the target cancerous cells and investigate its impacts on these cells. The PLGA NPs were PEGylated to become stealth and long-circulation particles, and conjugated with anti-TEM1 78Fc scFv recruiting the EDC/NHS chemistry for the active targeting purpose (Fig. S1), which could actively target the TEM1-positive, MS1HuTem1 endothelial cells (Fig. 1). Our findings showed that the 78Fc-armed SHK-loaded PEGylated PLGA NPs could considerably bind to the TEM1-overexpressing cells and induce markedly high toxicity in the target MS1HuTem1 cells (Fig. 1). Our previous results on the effects of SHK led us to seek the mechanisms underlying the toxic impacts of SHK.
28
Thus, we first looked at the cytotoxicity of SHK alone in the treated cells. We witnessed the inhibitory effects of SHK, at a concentration of 5μM (i.e., IC50), on the migration and invasion abilities of MS1 and TC1 cells cultivated in the Matrigel-coated transwells (Fig. 2). Besides, the malignant transformation analyses revealed that SHK could significantly inhibit the formation of colonies of the MS1 and TC1 cells comparable to that of DOX (Fig. 3). It can be assumed that the targeted delivery of SHK could impose its inhibitory impacts solely on the target cancer cells.
28
SHK was also found to elicit some morphological changes in the cellular nuclei (Fig. 4), perhaps through chromatin remodeling. Even if the SHK-induced heterochromatin foci resembled the SAHF, SHK did not induce profound senescence in the treated cells (Fig. 4). The morphological analysis showed that SHK at a concentration around IC50 can induce substantial cytotoxicity possibly through induction of ROSs (Fig. 4). Based on our findings, high amounts of SHK were able to accumulate in the cytoplasm and induce ROSs, which led us to assume that the mitochondria could be a possible biological target of SHK. The JC-1 staining experiments for studying the effect of SHK on the mitochondria showed a concentration impact of SHK resulting in depolarization of mitochondria (Figs. 4 and 5). It should be noted that SHK was able to induce ROS in a dose-dependent manner (Fig. 4). We also witnessed that the induction of ROSs by SHK induced somewhat mitochondrial degradation by autophagy and further mitochondrial depolarization and degradation.
68
Moreover, in recent years, a growing piece of evidence discusses the oxidative stress function(s) as the converging point of autophagy, in which ROS is counted as one of the major intracellular signal transducers that might sustain the autophagy.
69
Thus, we investigated the effect of SHK treatment in the formation of autophagosomes and found that it could induce the formation of autophagic vacuoles only in low (non-toxic) concentrations (Fig. 4). Further, SHK was able to induce both necrosis and apoptosis at a ratio 1:1 in the treated cellular population (Fig. 6). We ponder that the mitochondrial depolarization by SHK can result in apoptosis, which is deemed to be one of the SHK mechanisms of action (Figs. 5 and 6). This finding is in agreement with other researchers' findings.
66,67
Our results are in clear consensus with previous reports reporting on the potential of SHK in the induction of both necrosis and apoptosis (necroptosis) in the cancer cells.
70,71
To investigate the targeting efficacy of the 78Fc-armed SHK-loaded PEGylated PLGA NPs in vivo, we used an animal model developed previously.
27
The biodistribution study showed that PLGA NPs were cleared from the mice after 72h. At the targeting efficacy mouse model employed, after treatment with the 78Fc-armed SHK-loaded PEGylated PLGA NPs, the MS1HuTem1 cells were eliminated (Fig. 7). These results demonstrate the high in vivo targeting efficacy of the 78Fc-armed SHK-loaded NPs, as well as the therapeutic efficacy of SHK as a cytotoxic agent. Furthermore, we noticed that a high fluorescent signal of SHK was detected in the MS1-huTem1 hemangioma, even 5 days after the treatment (Fig. 7), while we could not detect any luciferase signal (live cells) in the same area on the last day. Such observations suggest that the 78Fc-armed SHK-loaded PEGylated PLGA NPs not only bind to their target actively but also retain within the tumor site for a few days. At the therapeutic efficacy mouse model employed in the current study, the tumors treated with the 78Fc-armed SHK-loaded PEGylated PLGA NPs were meaningfully reduced as compared to the PBS-treated control group (Fig. 7). In the metastatic model, we studied the 78Fc-armed SHK-loaded PEGylated PLGA NPs that imposed significant therapeutic efficacy. The tumor nodules at the lungs of the 78Fc-armed SHK-loaded PEGylated PLGA NPs treated groups were significantly decreased or even eliminated (Fig. 8). The therapeutic efficacy mouse model was further studied using immunocompetent C57BL6 mice. In this model, we noticed that the 78Fc-armed SHK-loaded PEGylated PLGA NPs treatment had an even better therapeutic outcome compared to the nu/nu strain (Fig. 9). Among other hypotheses to explain these results, we assumed that the better therapeutic outcome could be attributed to the synergy of the immune system with the engineered targeted nanoformulation. The results of the tumor re-challenge study indicated that the targeted nanoformulation of SHK could induce an immune response towards the TC1 cells (Fig. 10). However, additional investigations need to be accomplished for a better understanding of such modulation/stimulation of the immune system by SHK before its translation into clinical applications.
Previous studies have emphasized that various nanoscaled formulations loaded with anticancer agents can become the game-changer in the field of cancer therapy soon.
72-78
Given that the SHK was found to cause necroptosis in vitro, we decided to investigate the mechanism of death induced by SHK in vivo. Thus, frozen tissue samples from tumors treated with the 78Fc-armed SHK-loaded PEGylated PLGA NPs were stained with TUNEL reagent, anti-RIPK1, and anti-RIPK3 Abs. The slides from the 78Fc-armed SHK-loaded PEGylated PLGA NPs treated groups were stained positive for both TUNEL and the RIP kinases (Figs. 11 and 12). These data indicate that SHK nano delivery can induce both apoptosis and necroptosis in vivo.
Conclusion
SHK can bind to the mitochondria, which can cause mitochondrial depolarization leading to apoptosis. Further, SHK can induce the production of intracellular ROS and the appearance of autophagosomes in low concentrations. Moreover, SHK shows a great capability to induce apoptosis and necroptosis in the cell lines/models examined in the current study. Furthermore, SHK was able to inhibit cell migration and invasion in vitro. To confine the biological impacts of SHK on cancerous cells, we developed the 78Fc-armed SHK-loaded PEGylated PLGA NPs which showed extremely high targeting potential and therapeutic efficacy in all the in vivo models employed in the current study. Finally, the results of the tumor re-challenge experiment suggest that the targeted nanoformulation of SHK imposes immunostimulatory effects in vivo. Upon our findings, the engineered biodegradable PEGylated and anti-TEM1 78Fc scFv armed SHK-loaded PLGA NPs are proposed as a novel multifunctional nanomedicine for the targeted therapy of solid tumors with overexpression of TEM1.
Acknowledgments
The authors are thankful to Dr. Andrea Facciabene, Dr. Elizabeth Browning, and Xiaohui Peng (University of Pennsylvania, Philadelphia, PA, USA) for their technical support.
Funding sources
The in vivo imaging was performed at the University of Pennsylvania SAIF Optical/Bioluminescence Core, supported by NIH grant CA016520. This work was supported by NIH R01CA156695. SPORE in Ovarian Cancer Pilot P50 CA083638-14, and the Honorable Tina Brozman Foundation.
Ethical statement
All animal studies were approved by the IACUC and University Laboratory Animal Resources at the University of Pennsylvania. The authors declare that the raw data required to reproduce these reported results and the processed data required to reproduce the reported findings are available upon request.
Competing interests
YO and JB act as the Editor-in-Chief of the journal, and LEK is a member of the editorial board of the journal. The peer-review process of this work was carried out in a double-blind manner as part of the journal editorial policy.
Authors’ contribution
GC, CL, and YO designed the study. EIM conducted all the experiments. JB, YG, RS, and LEK assisted in data interpretation and analyses. EIM drafted the manuscript. EIM, JB, and YO revised the manuscript. GC, RS, and YO supervised the study.
Supplementary Materials
Supplementary file 1 contains Figure S1 and Tables S1-S2.
(pdf)
Research Highlights
What is the current knowledge?
simple
-
√ SHK shows unique pharmacological effects such as anti-inflammatory, antitumor, antioxidant, antithrombotic, antimicrobial, and wound healing effects.
-
√ SHK can induce significant cytotoxicity in vitro and in vivo.
What is new here?
simple
-
√ PEGylated PLGA NPs were functionalized anti-TEM1 antibody fragment (78Fc) and loaded with SHK.
-
√ The nanoformulation induced marked therapeutic effects in the MS1-xenograft mice.
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90. doi: 10.3322/caac.20107 [Crossref] [ Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74. doi: 10.1016/j.cell.2011.02.013 [Crossref] [ Google Scholar]
- Guddati AK. Ovarian cancer stem cells: elusive targets for chemotherapy. Med Oncol 2012; 29:3400-8. doi: 10.1007/s12032-012-0252-6 [Crossref] [ Google Scholar]
- Rocconi RP, Sullivan P, Long B, Blaize M, Brown J, Arbuckle J. Treatment of Chemotherapy-Induced Anemia in Ovarian Cancer Patients: Does the Use of Erythropoiesis-Stimulating Agents Worsen Survival?. Int J Gynecol Cancer 2012; 22. doi: 10.1097/IGC.0b013e31825104f4 [Crossref]
- Bookman MA. First-line Chemotherapy in Epithelial Ovarian Cancer. Clin Obstet Gynecol 2012; 55:96-113. doi: 10.1097/GRF.0b013e31824b45da [Crossref] [ Google Scholar]
- Meier W, du Bois A, Reuss A, Kuhn W, Olbricht S, Gropp M. Topotecan versus treosulfan, an alkylating agent, in patients with epithelial ovarian cancer and relapse within 12 months following 1st-line platinum/paclitaxel chemotherapy A prospectively randomized phase III trial by the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group (AGO-OVAR). Gynecol Oncol 2009; 114:199-205. doi: 10.1016/j.ygyno.2009.04.026 [Crossref] [ Google Scholar]
- Ferrero JM, Weber B, Geay JF, Lepille D, Orfeuvre H, Combe M. Second-line chemotherapy with pegylated liposomal doxorubicin and carboplatin is highly effective in patients with advanced ovarian cancer in late relapse: a GINECO phase II trial. Ann Oncol 2007; 18:263-8. doi: 10.1093/annonc/mdl376 [Crossref] [ Google Scholar]
- Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Controlled Release 2010; 148:135-46. doi: 10.1016/j.jconrel.2010.08.027 [Crossref] [ Google Scholar]
- Kafil V, Omidi Y. Cytotoxic impacts of linear and branched polyethylenimine nanostructures in A431 cells. Bioimpacts 2011; 1:23-30. doi: 10.5681/bi.2011.004 [Crossref] [ Google Scholar]
- Omidi Y. Smart multifunctional theranostics: simultaneous diagnosis and therapy of cancer. Bioimpacts 2011; 1:145-7. doi: 10.5681/bi.2011.019 [Crossref] [ Google Scholar]
- Ranjbar-Navazi Z, Eskandani M, Johari-Ahar M, Nemati A, Akbari H, Davaran S. Doxorubicin-conjugated D-glucosamine- and folate- bi-functionalised InP/ZnS quantum dots for cancer cells imaging and therapy. J Drug Target 2018; 26:267-77. doi: 10.1080/1061186X.2017.1365876 [Crossref] [ Google Scholar]
- Vandghanooni S, Eskandani M, Barar J, Omidi Y. AS1411 aptamer-decorated cisplatin-loaded poly(lactic-co-glycolic acid) nanoparticles for targeted therapy of miR-21-inhibited ovarian cancer cells. Nanomedicine (Lond) 2018; 13:2729-58. doi: 10.2217/nnm-2018-0205 [Crossref] [ Google Scholar]
- Vandghanooni S, Eskandani M, Barar J, Omidi Y. Antisense LNA-loaded nanoparticles of star-shaped glucose-core PCL-PEG copolymer for enhanced inhibition of oncomiR-214 and nucleolin-mediated therapy of cisplatin-resistant ovarian cancer cells. Int J Pharm 2020; 573:118729. doi: 10.1016/j.ijpharm.2019.118729 [Crossref] [ Google Scholar]
- Fathi M, Barar J, Erfan-Niya H, Omidi Y. Methotrexate-conjugated chitosan-grafted pH- and thermo-responsive magnetic nanoparticles for targeted therapy of ovarian cancer. Int J Biol Macromol 2020; 154:1175-84. doi: 10.1016/j.ijbiomac.2019.10.272 [Crossref] [ Google Scholar]
- Mooguee M, Omidi Y, Davaran S. Synthesis and in vitro release of adriamycin from star-shaped poly(lactide-co-glycolide) nano- and microparticles. J Pharm Sci 2010; 99:3389-97. doi: 10.1002/jps.22106 [Crossref] [ Google Scholar]
- Thamake SI, Raut SL, Gryczynski Z, Ranjan AP, Vishwanatha JK. Alendronate coated poly-lactic-co-glycolic acid (PLGA) nanoparticles for active targeting of metastatic breast cancer. Biomaterials 2012; 33:7164-73. doi: 10.1016/j.biomaterials.2012.06.026 [Crossref] [ Google Scholar]
- Davaran S, Omidi Y, Mohammad Reza R, Anzabi M, Shayanfar A, Ghyasvand S. Preparation and in vitro evaluation of linear and star-branched PLGA nanoparticles for insulin delivery. Journal of Bioactive and Compatible Polymers 2008; 23:115-31. doi: 10.1177/0883911507088276 [Crossref] [ Google Scholar]
- Omidi Y, Barar J, Heidari HR, Ahmadian S, Yazdi HA, Akhtar S. Microarray analysis of the toxicogenomics and the genotoxic potential of a cationic lipid-based gene delivery nanosystem in human alveolar epithelial A549 cells. Toxicol Mech Methods 2008; 18:369-78. doi: 10.1080/15376510801891286 [Crossref] [ Google Scholar]
- Omidi Y, Barar J. Induction of human alveolar epithelial cell growth factor receptors by dendrimeric nanostructures. Int J Toxicol 2009; 28:113-22. doi: 10.1177/1091581809335177 [Crossref] [ Google Scholar]
- Rettig WJ, Garin-Chesa P, Healey JH, Su SL, Jaffe EA, Old LJ. Identification of endosialin, a cell surface glycoprotein of vascular endothelial cells in human cancer. Proc Natl Acad Sci U S A 1992; 89:10832-6. doi: 10.1073/pnas.89.22.10832 [Crossref] [ Google Scholar]
- Christian S, Winkler R, Helfrich I, Boos AM, Besemfelder E, Schadendorf D. Endosialin (Tem1) Is a marker of tumor-associated myofibroblasts and tumor vessel-associated mural cells. Am J Pathol 2008; 172:486-94. doi: 10.2353/ajpath.2008.070623 [Crossref] [ Google Scholar]
- Croix BS, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E. Genes expressed in human tumor endothelium. Science 2000; 289:1197. doi: 10.1126/science.289.5482.1197 [Crossref] [ Google Scholar]
- Marty C, Langer-Machova Z, Sigrist S, Schott H, Schwendener RA, Ballmer-Hofer K. Isolation and characterization of a scFv antibody specific for tumor endothelial marker 1 (TEM1), a new reagent for targeted tumor therapy. Cancer Lett 2006; 235:298-308. doi: 10.1016/j.canlet.2005.04.029 [Crossref] [ Google Scholar]
- Teicher BA. Newer vascular targets: Endosialin (Review). Int J Oncol 2007; 30:305-12. doi: 10.3892/ijo.30.2.305 [Crossref] [ Google Scholar]
- Rouleau C, Curiel M, Weber W, Smale R, Kurtzberg L, Mascarello J. Endosialin protein expression and therapeutic target potential in human solid tumors: sarcoma versus carcinoma. Clin Cancer Res 2008; 14:7223. doi: 10.1158/1078-0432.CCR-08-0499 [Crossref] [ Google Scholar]
- Hinnen P, Eskens FALM. Vascular disrupting agents in clinical development. Br J Cancer 2007; 96:1159-65. doi: 10.1038/sj.bjc.6603694 [Crossref] [ Google Scholar]
- Li C, Wang J, Hu J, Feng Y, Hasegawa K, Peng X. Development, optimization, and validation of novel anti-TEM1/CD248 affinity agent for optical imaging in cancer. Oncotarget 2014; 5:6994-7012. doi: 10.18632/oncotarget.2188 [Crossref] [ Google Scholar]
- Matthaiou EI, Barar J, Sandaltzopoulos R, Li C, Coukos G, Omidi Y. Shikonin-loaded antibody-armed nanoparticles for targeted therapy of ovarian cancer. Int J Nanomedicine 2014; 9:1855-70. doi: 10.2147/IJN.S51880 [Crossref] [ Google Scholar]
- MacFadyen JR, Haworth O, Roberston D, Hardie D, Webster MT, Morris HR. Endosialin (TEM1,CD248) is a marker of stromal fibroblasts and is not selectively expressed on tumour endothelium. FEBS Lett 2005; 579:2569-75. doi: 10.1016/j.febslet.2005.03.071 [Crossref] [ Google Scholar]
- Davies GC, Giles H, Mansel RE, Mason MD, Jiang WG. Levels of expression of endothelial markers specific to tumour-associated endothelial cells and their correlation with prognosis in patients with breast cancer. Clin Exp Metastasis 2004; 21:31-7. doi: 10.1023/b:clin.0000017168.83616.d0 [Crossref] [ Google Scholar]
- Rmali KA, Puntis MCA, Jiang WG. Prognostic values of tumor endothelial markers in patients with colorectal cancer. World J Gastroenterol 2005; 11:1283-6. doi: 10.3748/wjg.v11.i9.1283 [Crossref] [ Google Scholar]
- Becker R, Lenter MC, Vollkommer T, Boos AM, Pfaff D, Augustin HG. Tumor stroma marker endosialin (Tem1) is a binding partner of metastasis-related protein Mac-2 BP/90K. FASEB J 2008; 22:3059-67. doi: 10.1096/fj.07-101386 [Crossref] [ Google Scholar]
- MacFadyen J, Savage K, Wienke D, Isacke CM. Endosialin is expressed on stromal fibroblasts and CNS pericytes in mouse embryos and is downregulated during development. Gene Expression Patterns 2007; 7:363-9. doi: 10.1016/j.modgep.2006.07.006 [Crossref] [ Google Scholar]
- Bagley RG, Rouleau C, St St. Martin T, Boutin P, Weber W, Ruzek M, et al Human endothelial precursor cells express tumor endothelial marker 1/endosialin/CD248. Mol Cancer Ther 2008; 7:2536. doi: 10.1158/1535-7163.MCT-08-0050 [Crossref] [ Google Scholar]
- Tomkowicz B, Rybinski K, Foley B, Ebel W, Kline B, Routhier E. Interaction of endosialin/TEM1 with extracellular matrix proteins mediates cell adhesion and migration. Proc Natl Acad Sci U S A 2007; 104:17965-70. doi: 10.1073/pnas.0705647104 [Crossref] [ Google Scholar]
- Nanda A, Karim B, Peng Z, Liu G, Qiu W, Gan C. Tumor endothelial marker 1 (Tem1) functions in the growth and progression of abdominal tumors. Proc Natl Acad Sci U S A 2006; 103:3351-6. doi: 10.1073/pnas.0511306103 [Crossref] [ Google Scholar]
- Chen C-H, Lin M-L, Ong P-L, Yang J-T. Novel multiple apoptotic mechanism of shikonin in human glioma cells. Ann Surg Oncol 2012; 19:3097-106. doi: 10.1245/s10434-012-2324-4 [Crossref] [ Google Scholar]
- Wiench B, Eichhorn T, Paulsen M, Efferth T. Shikonin directly targets mitochondria and causes mitochondrial dysfunction in cancer cells. Evid Based Complement Alternat Med 2012; 2012:726025. doi: 10.1155/2012/726025 [Crossref] [ Google Scholar]
- Long S, GuangZhi Y, BaoJie G, Wei X, YanYong H, YingLi W. Shikonin derivatives protect immune organs from damage and promote immune responses in vivo in tumour-bearing mice. Phytother Res 2012; 26:26-33. doi: 10.1002/ptr.3503 [Crossref] [ Google Scholar]
- Andujar I, Recio MC, Giner RM, Ríos JL. Traditional Chinese Medicine Remedy to Jury: The Pharmacological Basis for the Use of Shikonin as an Anticancer Therapy. Curr Med Chem 2013; 20:2892-8. doi: 10.2174/09298673113209990008 [Crossref] [ Google Scholar]
- Chen H-M, Wang P-H, Chen S-S, Wen C-C, Chen Y-H, Yang W-C. Shikonin induces immunogenic cell death in tumor cells and enhances dendritic cell-based cancer vaccine. Cancer Immunol Immunother 2012; 61:1989-2002. doi: 10.1007/s00262-012-1258-9 [Crossref] [ Google Scholar]
- Papageorgiou VA, Andreana N, Couladouros EA, Hepworth D, Nicolaou KC. The chemistry and biology of alkannin, shikonin, and related naphthazarin natural products. Angew Chem Int Ed Engl 1999; 38:270-301. doi: 10.1002/(SICI)1521-3773(19990201)38:3<270::AID-ANIE270>3.0.CO;2-0 [Crossref] [ Google Scholar]
- Papageorgiou VP, Assimopoulou AN, Ballis AC. Alkannins and shikonins: a new class of wound healing agents. Curr Med Chem 2008; 15:3248-67. doi: 10.2174/092986708786848532 [Crossref] [ Google Scholar]
- Li W, Liu J, Zhao Y. PKM2 inhibitor shikonin suppresses TPA-induced mitochondrial malfunction and proliferation of skin epidermal JB6 cells. Mol Carcinog 2014; 53:403-12. doi: 10.1002/mc.21988 [Crossref] [ Google Scholar]
- Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011; 30:4297-306. doi: 10.1038/onc.2011.137 [Crossref] [ Google Scholar]
- Wu Z, Wu L, Li L, Tashiro S-i, Onodera S, Ikejima T. p53-Mediated cell cycle arrest and apoptosis induced by shikonin via a caspase-9-dependent mechanism in human malignant melanoma A375-S2 cells. J Pharmacol Sci 2004; 94:166-76. doi: 10.1254/jphs.94.166 [Crossref] [ Google Scholar]
- Nam KN, Son M-S, Park J-H, Lee EH. Shikonins attenuate microglial inflammatory responses by inhibition of ERK, Akt, and NF-κB: neuroprotective implications. Neuropharmacology 2008; 55:819-25. doi: 10.1016/j.neuropharm.2008.06.065 [Crossref] [ Google Scholar]
- Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther 2007; 6:1641. doi: 10.1158/1535-7163.MCT-06-0511 [Crossref] [ Google Scholar]
- Barzegar-Jalali M, Adibkia K, Valizadeh H, Shadbad MR, Nokhodchi A, Omidi Y. Kinetic analysis of drug release from nanoparticles. J Pharm Pharm Sci 2008; 11:167-77. doi: 10.18433/j3d59t [Crossref] [ Google Scholar]
- Demchenko AP. Beyond annexin V: fluorescence response of cellular membranes to apoptosis. Cytotechnology 2013; 65:157-72. doi: 10.1007/s10616-012-9481-y [Crossref] [ Google Scholar]
- Facciponte JG, Ugel S, De Sanctis F, Li C, Wang L, Nair G. Tumor endothelial marker 1-specific DNA vaccination targets tumor vasculature. J Clin Invest 2014; 124:1497-511. doi: 10.1172/JCI67382 [Crossref] [ Google Scholar]
- Chowdhury MR, Schumann C, Bhakta-Guha D, Guha G. Cancer nanotheranostics: Strategies, promises and impediments. Biomed Pharmacother 2016; 84:291-304. doi: 10.1016/j.biopha.2016.09.035 [Crossref] [ Google Scholar]
- Roy I, Vij N. Nano-delivery in Airway Diseases: Challenges and Therapeutic Applications. Nanomedicine 2010; 6:237-44. doi: 10.1016/j.nano.2009.07.001 [Crossref] [ Google Scholar]
- Siafaka PI, Üstündağ Okur N, Karavas E, Bikiaris DN. Surface modified multifunctional and stimuli responsive nanoparticles for drug targeting: current status and uses. Int J Mol Sci 2016; 17:1440. doi: 10.3390/ijms17091440 [Crossref] [ Google Scholar]
- Thomas TJ, Tajmir-Riahi HA, Thomas T. Polyamine–DNA interactions and development of gene delivery vehicles. Amino Acids 2016; 48:2423-31. doi: 10.1007/s00726-016-2246-8 [Crossref] [ Google Scholar]
- Thaxton CS, Rink JS, Naha PC, Cormode DP. Lipoproteins and lipoprotein mimetics for imaging and drug delivery. Adv Drug Deliv Rev 2016; 106:116-31. doi: 10.1016/j.addr.2016.04.020 [Crossref] [ Google Scholar]
- Mokhtarzadeh A, Alibakhshi A, Yaghoobi H, Hashemi M, Hejazi M, Ramezani M. Recent advances on biocompatible and biodegradable nanoparticles as gene carriers. Expert Opin Biol Ther 2016; 16:771-85. doi: 10.1517/14712598.2016.1169269 [Crossref] [ Google Scholar]
- Khan I, Gothwal A, Sharma AK, Kesharwani P, Gupta L, Iyer AK. PLGA Nanoparticles and Their Versatile Role in Anticancer Drug Delivery. Crit Rev Ther Drug Carrier Syst 2016; 33:159-93. doi: 10.1615/CritRevTherDrugCarrierSyst.2016015273 [Crossref] [ Google Scholar]
- Li Z, Loh XJ. Recent advances of using polyhydroxyalkanoate-based nanovehicles as therapeutic delivery carriers. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2017; 9. doi: 10.1002/wnan.1429 [Crossref]
- Lakkireddy HR, Bazile D. Building the design, translation and development principles of polymeric nanomedicines using the case of clinically advanced poly(lactide(glycolide))-poly(ethylene glycol) nanotechnology as a model: An industrial viewpoint. Adv Drug Deliv Rev 2016; 107:289-332. doi: 10.1016/j.addr.2016.08.012 [Crossref] [ Google Scholar]
- Zare K, Khosrowshahli M, Nazemiyeh H, Movafeghi A, Azar AM, Omidi Y. Callus culture of Echium italicum L towards production of a shikonin derivative. Natural Product Research 2011; 25:1480-7. doi: 10.1080/14786410902804857 [Crossref] [ Google Scholar]
- Zare K, Nazemiyeh H, Movafeghi A, Khosrowshahli M, Motallebi-Azar A, Dadpour M. Bioprocess engineering of Echium italicum L: induction of shikonin and alkannin derivatives by two-liquid-phase suspension cultures. Plant Cell, Tissue and Organ Culture (PCTOC) 2010; 100:157-64. doi: 10.1007/s11240-009-9631-x [Crossref] [ Google Scholar]
- Hu X, Xuan Y. Bypassing cancer drug resistance by activating multiple death pathways--a proposal from the study of circumventing cancer drug resistance by induction of necroptosis. Cancer Lett 2008; 259:127-37. doi: 10.1016/j.canlet.2007.11.007 [Crossref] [ Google Scholar]
- Snehalatha M, Venugopal K, Saha RN, Babbar AK, Sharma RK. Etoposide Loaded PLGA and PCL Nanoparticles II: Biodistribution and Pharmacokinetics after Radiolabeling with Tc-99m. Drug Delivery 2008; 15:277-87. doi: 10.1080/10717540802006500 [Crossref] [ Google Scholar]
- Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Livaniou E, Evangelatos G. Effect of copolymer composition on the physicochemical characteristics, in vitro stability, and biodistribution of PLGA–mPEG nanoparticles. Int J Pharm 2003; 259:115-27. doi: 10.1016/S0378-5173(03)00224-2 [Crossref] [ Google Scholar]
- Liang W, Cui J, Zhang K, Xi H, Cai A, Li J. Shikonin induces ROS-based mitochondria-mediated apoptosis in colon cancer. Oncotarget 2017; 8:109094-106. doi: 10.18632/oncotarget.22618 [Crossref] [ Google Scholar]
- Shilnikova K, Piao MJ, Kang KA, Ryu YS, Park JE, Hyun YJ. Shikonin induces mitochondria-mediated apoptosis and attenuates epithelial-mesenchymal transition in cisplatin-resistant human ovarian cancer cells. Oncol Lett 2018; 15:5417-24. doi: 10.3892/ol.2018.8065 [Crossref] [ Google Scholar]
- Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012; 8:1462-76. doi: 10.4161/auto.21211 [Crossref] [ Google Scholar]
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 2015; 22:377-88. doi: 10.1038/cdd.2014.150 [Crossref] [ Google Scholar]
- Xuan Y, Hu X. Naturally-occurring shikonin analogues--a class of necroptotic inducers that circumvent cancer drug resistance. Cancer Lett 2009; 274:233-42. doi: 10.1016/j.canlet.2008.09.029 [Crossref] [ Google Scholar]
- Han W, Xie J, Li L, Liu Z, Hu X. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis 2009; 14:674-86. doi: 10.1007/s10495-009-0334-x [Crossref] [ Google Scholar]
- Siminzar P, Omidi Y, Golchin A, Aghanejad A, Barar J. Targeted delivery of doxorubicin by magnetic mesoporous silica nanoparticles armed with mucin-1 aptamer. J Drug Target 2020; 28:92-101. doi: 10.1080/1061186X.2019.1616745 [Crossref] [ Google Scholar]
- Fathi M, Abdolahinia ED, Barar J, Omidi Y. Smart stimuli-responsive biopolymeric nanomedicines for targeted therapy of solid tumors. Nanomedicine (Lond) 2020; 15:2171-200. doi: 10.2217/nnm-2020-0146 [Crossref] [ Google Scholar]
- Dolatkhah M, Hashemzadeh N, Barar J, Adibkia K, Aghanejad A, Barzegar-Jalali M. Graphene-based multifunctional nanosystems for simultaneous detection and treatment of breast cancer. Colloids Surf B Biointerfaces 2020; 193:111104. doi: 10.1016/j.colsurfb.2020.111104 [Crossref] [ Google Scholar]
- Akbarzadeh Khiavi M, Safary A, Barar J, Farzi-Khajeh H, Barzegari A, Mousavi R. PEGylated gold nanoparticles-ribonuclease induced oxidative stress and apoptosis in colorectal cancer cells. Bioimpacts 2020; 10:27-36. doi: 10.15171/bi.2020.04 [Crossref] [ Google Scholar]
- Akbarzadeh Khiavi M, Safary A, Barar J, Ajoolabady A, Somi MH, Omidi Y. Multifunctional nanomedicines for targeting epidermal growth factor receptor in colorectal cancer. Cell Mol Life Sci 2020; 77:997-1019. doi: 10.1007/s00018-019-03305-z [Crossref] [ Google Scholar]
- Vandghanooni S, Eskandani M, Barar J, Omidi Y. Aptamedicine: a new treatment modality in personalized cancer therapy. Bioimpacts 2019; 9:67-70. doi: 10.15171/bi.2019.09 [Crossref] [ Google Scholar]
- Abdolahinia ED, Nadri S, Rahbarghazi R, Barar J, Aghanejad A, Omidi Y. Enhanced penetration and cytotoxicity of metformin and collagenase conjugated gold nanoparticles in breast cancer spheroids. Life Sci 2019; 231:116545. doi: 10.1016/j.lfs.2019.116545 [Crossref] [ Google Scholar]