Bioimpacts. 12(6):501-513.
doi: 10.34172/bi.2022.23733
Original Research
Changing the daily injection of glatiramer acetate to a monthly long acting product through designing polyester-based polymeric microspheres
Fatima Molavi 1  , Mohammad Barzegar-Jalali 1 , Hamed Hamishehkar 2, *
, Mohammad Barzegar-Jalali 1 , Hamed Hamishehkar 2, *
Author information:
1Biotechnology Research Center, Student Research Committee, Department of pharmaceutics, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
2Drug Applied Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
Abstract
Introduction:
Glatiramer acetate (GA) is a newly emerged therapeutic peptide to reduce the frequency of relapses in multiple sclerosis (MS). Despite its good performance in controlling MS, it is not widely used due to daily or biweekly subcutaneous injections due to rapid degradation and body clearance. Therefore, implant design with sustained release leads to prolonged biological effects by gradually increasing drug exposure and protecting GA from rapid local degradation.
Methods:
Different emulsion methods, PLGA type, surfactant concentration, drug/polymer ratio, drying processes, stirring method, and other variables in preliminary studies modified the final formulation. The release kinetics were studied through mechanistic kinetic models such as zero-order, Weibull, Higuchi, etc. In this study, all challenges for easy scale-up, methodological detail, and a simple, feasible setup in mass production were discussed.
Results:
The optimized formulation was obtained by 1:6 drug/PLGA, 0.5% w/w polyvinyl alcohol, and 0.75% w/w NaCl in the external aqueous phase, 1:10 continuous phase to dispersed phase ratio, and without any surfactant in the primary emulsion. The final freeze-dried particles presented a narrow distributed size of 1-10 µm with 7.29% ± 0.51 drug loading and zero-order release behavior with appropriate regression correlation (R2 98.7), complete release, and only 7.1% initial burst release.
Conclusion:
Therefore, to achieve improvement in patient compliance through better and longer efficacy, designing the parenteral sustained release microspheres (MPSs) of this immune modulator is a promising approach that should be considered.
Keywords: Peptide and protein, Drug delivery, Polymeric microparticles, Multiple sclerosis, Controlled release, Poly(D,L-lactic-co-glycolic acid)
Copyright and License Information
© 2022 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Controlled release formulations have been considered to direct the protein and peptide drugs toward clinical application, i.e., improvement of the pharmacotherapy regime and elimination of the frequent administration.
1,2
The important biological impact of designing long acting polymeric particulate systems is increasing the biomaterials' drug exposure.
3-5
Glatiramer acetate (GA) was selected as a drug model for peptide formulation because of A) hydrophilic amphiphilic nature and being in class III in the BCS classification (high solubility and low permeability), and B) as a low potent peptide, a large amount of drug (80 mg, according to Mapi pharma studies) is needed to encapsulate for one month consistent effect. GA has currently been administered to reduce the frequency of relapses in multiple sclerosis (MS) patients
6
in 20 and 40 mg prefilled syringes for daily and biweekly usage, respectively. The fast bio-distribution and clearance cause multiple injections, which consequently restrict their clinical application.
There are different methods to prepare the microspheres (MPSs), which must control particle size, size distribution and improve release kinetics.
7
Several techniques including physicochemical processes (evaporation, phase separation methods and self-healing encapsulation) and mechanical methods (e.g., extrusion process, spray drying, microfluidic approach
8
and supercritical fluid methods
9
) are potentially useful for the MPS preparation.
10
In this study, emulsification evaporation methods were applied.
To focus on other studies, myriad studies have been developed on peptide-loaded MPS for polymeric MPSs (e.g., octreotide, lutein, goserelin, etc) and clinically tested in different methods. For example, a spray drying method was developed to make an injectable poly lactide-co-glycolide acid (PLGA) MPSs encapsulating leuprolide (LD) and was compared with the benchmark product LD MPSs.
11
In another study,
12
LD MPSs were prepared by water in oil in water (W/O/W) techniques and in vitro in vivo correlation was investigated. The in vivo release profiles presented similar behavior to in vitro release profiles in PBS,
13
although with low initial burst release and slightly faster in vivo release rates. The low burst release is attributed to the masking absorption-phase effect from the intramuscular (IM) site.
12,14
In the study of developing an octreotide MPSs through emulsification method
15
, derived silk fibroin
16
was applied and high DL (8–10 wt %) was achieved. In another case, goserelin MPSs were prepared with Poloxamer hydrogel in PLGA and compared with Zoladex® implant, high Encapsulation efficiency (EE) (94.16%), low burst release (less than 2%) and 9.36-fold enhanced relative bioavailability were showed in 49 days by providing alternatives to the Zoladex®.
17
Overall, it is not difficult to imagine that a better formulation can be developed. Despite the expiration date of patent products, till now, no generic product has been approved for the long-acting products (e.g., LD), probably due to the complexity of manufacturing processes and lack of comprehensive studies.
18
Recently, there has been tremendous interest and consequently, competitive activities in the development of injectable sustained release GA by pharmaceutical industries. This study aimed to optimize the preparation and evaluation of a one-month long-acting GA MPSs with appropriate peptide loading and ideal drug release (i.e., minimized burst release, zero-order drug release, and complete release) and reproducible industrial approaches by using a feasible manufacturing technique. On the other hand, the comprehensive evaluation of different variables that were investigated simultaneously in this study had not been previously studied in other studies. The results may lead a formulator to pick out the correct way to adjust the formulation parameters, thus quickly achieving the goal with the least possibility of wasting expensive polymers and peptides. The result of this study may even be used in the development of other injectable sustained release peptides and proteins that are estimated to be the future of medications.
Materials and Methods
Materials
GA was provided by Tofigh Daru Research & Engineering Company, Tehran, Iran. Evonic, Germany, provided PLGA, (Resomer 502H, Resomer 502 and 752H). Poly (vinyl alcohol) (PVA, molecular weight (MW), 75 KD), ortho-phthalaldehyde (OPA), and 3-Mercaptopropionic acid (3-MPA) were purchased by Sigma, ALDRICH, Germany. Dichloromethane (DCM or methylene chloride), Span 80®, paraffin, acetonitrile, and sodium hydroxide were purchased from Merck Chemicals Co., Darmstadt, Germany. Boric acid was obtained from E.R. SQUIBB & Sons, NY, USA. Other chemical reagents and used solvents were analytical grades.
Preparation of MPSs
Three techniques prepared MPSs: (a) W/O/W, (b) solid in oil in water (S/O/W), and c) Solid in oil in oil (S/O/O).
W/O/W double emulsion
The primary emulsion was prepared by dispersing the aqueous solution of the drug (~27 mg in 200 µL water) into the polymeric phase (PLGA in ~3 g DCM), drug/polymer ratio as mentioned in Table 1. This mixture was entirely treated with ultrasonic (UP 200H. Hielscher, Germany) (power 120 W, working power 20%, 5 cycles of 7 seconds under the use of icy water to avoid evaporating of DCM and destroying peptide) to form a homogeneous milky solution. After cooling the obtained emulsion, the double emulsion was produced by dispersing the primary emulsion to the external aqueous phase, containing 0.5% (w/w) PVA and 0.75% (w/w) NaCl (1:10 continuous phase to dispersed phase (CP/DP) by using a homogenizer at the speed of 21000 rpm for 5 minutes (Silent Crusher, Heidolph, Germany) to form a W/O/W emulsion. To prevent the double layer particles from changing to single layer, the stability of the primary W1/O emulsion was analyzed in the presence of hydrophilic emulsifier (PVA). For this purpose, the droplet size and appearance of the emulsion were investigated for one day before transferring the primary emulsion to the second emulsion. After removing organic solvent, the solidified particles were obtained by centrifuging at 900 g (Universal 320, Pole ideal Tajhiz CO., Iran).
Table 1.
The applied independent variables in the preparation of microspheres through W1/O/W2 emulsion method
|
Formulation
|
PLGA type
|
Drug/polymer ratio
|
Presence of PVA in W
1
/O emulsion
|
|
F1a
|
502H |
1:10 |
No |
|
F2a
|
502H |
1:10 |
yes |
| F3 |
502H |
1:10 |
No |
| F4 |
502H |
1:10 |
Yes |
| F5 |
502 |
1:10 |
No |
| F6 |
752H |
1:10 |
No |
| F7 |
502 |
1:6 |
No |
| F8 |
752H |
1:6 |
No |
| F9 |
502H |
1:6 |
No |
| F10 |
502H |
1:6 |
Yes |
aThe drying process of these two formulations were on the tray at room temperature. PLGA; Poly lactic-co-glycolic acid, PVA; Poly (vinyl alcohol).
Then after washing twice with water, the particles were dried in two different ways, freeze-drying and on a tray at room temperature. All the methods were done in triplicate. Table 1 shows the different applied variables in the production of MPSs via the W/O/W emulsion method.
S/O/W double emulsion
The initial suspension of the drug was prepared by dispersing the powder of GA (~30 mg) in the polymeric phase (PLGA in ~3 g DCM) under stirring. Then the final emulsion was prepared by dispersing the S/O phase in the external water phase (30 mL) followed by solvent extraction. The rest of the process is similar to the W/O/W method. All the procedures were done in triplicate.
S/O/O double emulsion
The first part was prepared by dispersing the powder of the drug into the homogeneous solution of the polymeric solution. In detail, 30 mg GA was added to the polymeric solution in ~3 mL pure acetonitrile or 95: 5 Acetonitrile: water and tried to uniformly dispersed by mixing it on a hot plate stirrer (MR 300IK, Heidolph, Germany) at 1400 rpm to form the drug suspension. Then the resulting suspension was gradually injected into the 30 mL oily solution (paraffin), containing 3% Span® 80 while being mixed via a A) rotor mixer at 2000 rpm (VIBRAX VXR basic, IKA®, Germany) and, B) hot plate stirrer at 1400 rpm (Table 2) for 2 hours at ambient temperature to remove the organic solvent. The solidified particles were obtained by centrifugation at 3500 g, after washing two times with n-hexane and water. Finally, the particles were reconstituted by 5 mL water. After that, it was dried by a freeze-drying process. All the methods were done in triplicate.
Table 2.
The applied independent variables in the preparation of microspheres through S/O/O emulsion method
|
Formulation
|
PLGA type
|
Drug/polymer ratio
|
Type of stirrer
|
| F11 |
502H |
1:5 |
Rotor type |
| F12 |
752H |
1:5 |
Rotor type |
| F13 |
502H |
1:6 |
Rotor type |
| F14 |
502 |
1:6 |
Rotor type |
| F15 |
752H |
1:6 |
Rotor type |
| F16 |
752H |
1:6 |
Magnetic type |
| F17 |
502 |
1:6 |
Magnetic type |
| F18 |
502H |
1:6 |
Magnetic type |
|
F19*
|
502H |
1:10 |
Magnetic type |
| F20 |
502H |
1:10 |
Magnetic type |
* In this formulation, the effect of solvent in drug-polymer suspension (95:5 Acetonitrile: water) on EE and particle size was evaluated. PLGA; Poly lactic-co-glycolic acid.
The effect of polymer type, drying process, surfactant in primary W1/O emulsion, and drug/polymer ratio and agitation rate in S/O/O preparation methods were performed to achieve the optimized small and stable MPSs.
Drying method
After centrifugation and washing, MPS formulations were dried by two different methods, freeze-drying process and on the tray at room temperature. Particles were lyophilized by following re-suspension of sediment in purified water. The sample had been frozen for 12 hours at -70°C, and freeze-dried (CHRIST LOC-1m, Alfa 1-4 LSC, Germany) for 24 hours at pressure 0.3 mbar and -20°C. In the tray method, re-suspension of sediment of formulations (F1 and F2) were filtered and situated in an incubator at 25°C for 6 hours. After evaluating the water content, the formed MPSs were used for further investigations.
Peptide content
To analyze the GA content, 3 mg of the formed GA-loaded MPSs (GA-MPS) were suspended in a 1 mL organic solvent (acetonitrile) to dissolve the polymer. After centrifugation, the precipitant was dissolved with either 0.1 M borate buffer or purified water to measure GA via spectroscopic and chromatographic methods, respectively.
UV spectrophotometer
To analyze the GA as a basis peptide with an ultraviolet (UV) spectrophotometer (Ultrospec 2000, Pharmacia Biotech, UK), the derivative product of amino acid is needed. For this purpose, a 0.1 M borate buffer with pH 9.3 and OPA solution as a bio-reagent are applied. To prepare the borate buffer, 1 M boric acid, 0.75 M Sodium chloride (NaCl), and 4 M Sodium hydroxide (NaOH) (for adjusting the pH to 9.3) were applied. Moreover, to obtain OPA working solution (1 mg/mL), 5.0 mg OPA was added to 5.0 mL 0.05 M NaOH solution, and then after mixing thoroughly, a 25 µL 3-MPA was added to the resulting solution. The solution was carefully mixed and kept at 2-8°C for up to two weeks. To prepare the stock solution for the calibration curve (1 mg/mL), 10 mg of GA was dissolved with a 10 mL of 0.1 M borate buffer. A calibration curve was performed in the range from 4 to 62 µg/mL. From each calibration solution, 1 mL was transferred into a 1.5-2 mL tube, and after that 60 µL of OPA solution was added into the solutions. The solution had carefully been mixed and kept at ambient temperature for 30 minutes. The absorption of the prepared solution was read at 339 nm by a UV spectrophotometer. Linearity was assessed based on statistical calculations, which were performed for six calibration curves. The method was checked concerning validation parameters.
HPLC method development
High-performance liquid chromatography (HPLC) system equipped with gel permeation chromatography (GPC) module was used to analyze average molecular weight. Chromatographic analysis was accomplished at 30°C using a PWXL TSKgel G3000PWXL 7.8 ID × 30.0 L 500-8000 chromatographic column. The mobile phase consisted of 500 mL sodium acetate 4 M, and adjusted to pH 6.0 with a glacial acetic acid solution. This buffer is applicable for 2 days. The aqueous phase was eluted with a 0.5 mL/min flow rate and read at 235 nm. From prepared solution, 20 µL was applied as injection volume. The fresh, filtered, and degassed mobile phase was applied. Assay of each sample was performed in triplicate. According to ICH guideline, the method was checked by concerning validation parameters i.e., specificity, suitability, linearity, precision, the limit of detection, and the limit of quantitation.
EE and drug loading (DL) measurement
Calculating the concentration of the final active pharmaceutical ingredient (API) in the particles, in addition to providing the exact dose, should be evaluated for two main reasons: first, the safety concerns of using polymers, and second, calculating the syringeability by evaluating the solid percentage. For this purpose, 3 mg of the prepared particles were dispersed in 1.0 mL acetonitrile. After mixing vigorously with vortex and ultra-sonication bath, the sample was centrifuged for 6 minutes at 4500 g. Then, the dried sediment was dissolved in a 1.0 mL of the borate buffer 0.1 M and 60 µL OPA working solution. Then after complete mixing, the solution was incubated for 30 minutes at room temperature. The absorption of the prepared solution was analyzed by a UV spectrophotometer at 339 nm. EE and DL percentages of the MPSs were calculated by the following equation:
Particle characterization
Apart from GA analysis (EE and DL percentages, and peptide integrity determination by analyzing MW), the various descriptive techniques were used to study the physicochemical properties of MPSs, such as particle size and size distribution, particle charge, particle morphology, drug and polymer interaction, crystallography, residual solvent, release behavior, flow properties and syringeability, and finally stability studies.
Size and charge of the microspheres
Particle size, size distribution, and surface charge are essential factors to monitor MPSs in vitro and in vivo performance. Particle size and zeta potential were analyzed using light scattering mechanism (LS) (SALD 2101, Shimadzu particle size, Japan) and zeta analyzer (Zetasizer Nano ZS, Malvern, UK) Phase Analysis Light Scattering (M3-PALS)-based techniques, respectively. The analysis was performed in triplicate, and the mean value was reported. The span value for the size distribution was reported according to the following equation:
Here, the numbers 10, 50, and 90 reveal the percentages of particles in diameters up to those numbers. The narrow size distribution has a small span value.
Morphology of particles
The morphology of the dried particles was studied by scanning electron microscope (SEM) (ZEISS, Sigma VP, Germany) after the palladium/gold coating of the particles on an aluminum stub.
Fourier transform infrared (FT-IR)
The FT-IR of blank MPS, GA-MPSs, and pure GA were evaluated to see the reactions in the carriers. In addition, the FT-IR of pure GA, GA extracted from PLGA MPSs associated with PLGA polymers were assessed to understand drug degradation during the W/O/W and S/O/O encapsulation processes. FT-IR spectra of the powders were conducted on a (Bruker, TENSOR 27, Germany) at room temperature (25 °C) at a resolution of 4 cm-1 that were used to record the spectra. The FT-IR spectra were recorded between 400 to 4000 cm-1.
X-Ray diffraction (XRD)
XRD diffractograms of the pure peptide, dried blank MPSs and dried extracted peptide from MPSs were obtained at ambient temperature using a XRD (MPD 3000, Ital structure, Italy), equipped with a Cu/Kα (λ = 0.154 nm) X-ray source (40kV and 30 mA) and the materials were scanned from 5° to 40° 2θ.
Release behavior study
A certain amount of drug (equivalent to 50 µg/mL according to the LC value of each formula) was taken to study release behavior. A known quantity of the drug (equivalent amount 50 µg/mL considering the LC value of each formula) was taken for release behavior study. A 50 µg/mL GA solution was considered as a control solution. 20 mL of PBS pH 7.4 containing 0.05% sodium azide (NaN3) was chosen as the release media.
19,20
Then, the MPS suspensions were incubated at 37°C with agitation at 200 rpm. Following centrifugation at 1500 g for 4 minutes, the supernatants (500 µL) were collected and after adding borate buffer (500 µL) and OPA (60 µL) and thoroughly mixing, the amount of released GA was determined by UV spectrophotography as described in UV spectrophotometer section. The release media was fully replaced by adding 500 µL to the residual of centrifuged samples at specific time intervals; minutes 5, 30, hours 3, 6, 24 and days 3, 6, 12, 20 up to the day 30. To study drug release profile of an extended-release dosage form, at least three time points are essential. Different sampling times may be required for drug approval and regulatory purposes.
21
The first time point indicates the initial burst release, which is usually within the first hour or two. The second time is selected to show the drug release behavior, and a final time is considered to show the final and complete release of the API from the final dosage forms.
22,23
Mechanistic kinetic models
In vitro drug release can provide insight into the behavior of a complex biological system at both the absorption and effect sites and its function in in vivo. In macromolecules, especially in sustained-release formulations, the efficiency studies and kinetic mathematical models based on statistical analysis
24
will be more helpful to predict in vivo behavior. For the best model among other kinetic models including zero order, first order, Higuchi, power law, Hixson-Crowell, square root of mass, Weibull, and reciprocal powered time, the experimental data were evaluated by adjusting the data on their equations.
25,26
The proposed model is expected to have a high R-squared (RSQ) and minimum error to predict drug behavior in the biological system.
Stability studies
The stability of a pharmaceutical product in a final container closure system (CCS) defines the durability of formulations over the physicochemical and microbiological changes in a defined storage condition. Therefore, the CCS should not have any physical or chemical interaction with the final product. To evaluate the stability of both incorporated GA in loaded MPSs and MPSs, the accelerated and long-term stability studies were performed for all dried and final products. Stability tests should include particle size and morphology, assay analysis or MW (content and integrity of peptide and protein), drug release profile, and moisture content. However, in this study, particle size, morphology and assay studies have been tripled to determine the shelf life of the prepared products. Long-term stability studies were conducted at 5°C ± 2°C/75% ± 5% relative humidity (RH) for 6 months using a locked and temperature-controlled fridge (Himalia R405, Iran) and accelerated stability was conducted at 25°C ± 2°C/45% ± 5% RH for 6 months which was kept on the bench under controlled temperature and humidity condition. According to ICH guidelines, the samples were withdrawn at predetermined intervals of 0, 30, 60, 120, and 180 days.
Syringeability study
A syringe study was performed to analyze the ease of passage of the reconstituted suspension through a hypodermic needle. Ease of injection, blockage, foaming tendencies, uniformity and the solid percentage of the reconstituted suspension was analyzed. Analyzing the solid percentage was conducted for all formulations by the following equation:
Acceptance criteria less than (NMT) 30% were considered. Two essential features are the length and size of the needle. Standard muscle products (IMs) typically require needles from 23G to 18G in thickness and 2.5 to 7.5 cm in length compared to smaller needles from 25G to 23G and 1.5 to 2 cm in thickness and length, respectively, which are most frequently used for standard subcutaneous products (SCs).
27
Diluent preparation
The design of isotonic diluent by evaluating viscosity, clogging and reconstitution ability of the final product is critical step that should be presented for the pharmaceutical regulation. Tonicity modifiers, preservatives, surfactants, and water for injection are the primary agents of diluent formulations.
7
Here, two diluent compositions were designed based on the existing commercial products such as LD, triptorelin, pasireotide, and octreotide. Table 3 shows the composition of two different prepared diluents.
Table 3.
Formulation of diluents for prepared microspheres
|
Diluent composition
|
1
|
2
|
|
Compound
|
Amount (w/v %)
|
Compound
|
Amount (w/v %)
|
| Tonicity modifier |
Mannitol |
9 |
Mannitol |
7.4 |
| Surfactant |
NaCMC |
1.4 |
NaCMC |
2.6 |
| Surfactant |
Poloxamer 188 |
0.4 |
Polysorbate 80 |
0.17 |
For both formulations, the solvent was water for injection. NaCMC; Sodium carboxymethylcellulose.
Statistics
The effect of variables on MPSs was assessed by two-way analysis of variance (ANOVA). A level of P < 0.05 was considered statistically significant. All analyses were carried out at 95% confidence.
Results
MPS preparation
The results of W/O/W and S/O/O emulsion methods have been compared and for better comparison, twenty formulations' results are shown in Table 4. Data show that the GA-MPSs can load up to 1:6 or even up to 1:5 in S/O/O method. The result was consistent with the other studies
28,29
On the other hand, the S/O/W method was not suitable for GA in which particles tend to aggregate and formed sticky agglomerates during the production process.
Table 4.
Characterization of prepared particles. Data were presented as mean ± standard deviation (n=3)
|
|
Particle size (µm)
|
Span
|
EE (%)
|
DL (%)
|
Appearance
|
| F1 |
23.4 ± 3.81 |
0.80 ± 0.05 |
23.4 ± 3.56 |
1.93 ± 0.29 |
A spherical, porous surface |
| F2 |
24.2 ± 6.20 |
0.98 ± 0.46 |
24.7 ± 1.70 |
2.03 ± 0.14 |
A spherical, porous surface |
| F3 |
10.0 ± 0.00 |
1.07 ± 0.07 |
52.2 ± 8.30 |
4.93 ± 0.68 |
A spherical and aggregate, slightly porous surface |
| F4 |
12.0 ± 2.54 |
2.67 ± 1.46 |
66.6 ± 6.32 |
5.47 ± 0.52 |
ND |
| F5 |
20.3 ± 0.49 |
1.29 ± 0.39 |
90.8 ± 1.07 |
7.45 ± 0.09 |
A spherical, porous surface |
| F6 |
6.65 ± 0.49 |
1.20 ± 0.56 |
36.5 ± 0.03 |
3.00 ± 0.00 |
A spherical and slightly aggregate, smooth surface |
| F7 |
21.9 ± 2.80 |
1.15 ± 0.01 |
50.8 ± 7.75 |
7.12 ± 1.09 |
A spherical, porous surface |
| F8 |
8.77 ± 0.49 |
1.70 ± 0.42 |
46.2 ± 9.19 |
6.48 ± 1.29 |
ND |
| F9* |
6.55 ± 0.35 |
1.91 ± 0.45 |
52.1 ± 3.67 |
7.29 ± 0.51 |
A spherical, slight porous surface |
| F10 |
10.7 ± 1.54 |
1.80 ± 0.37 |
50.1 ± 4.61 |
7.02 ± 0.65 |
ND |
| F11 |
27.0 ± 7.77 |
1.17 ± 0.69 |
30.5 ± 3.25 |
5.06 ± 0.54 |
Both spherical and Irregular, smooth surface |
| F12 |
48.9 ± 1.55 |
2.07 ± 0.11 |
30.5 ± 1.11 |
4.66 ± 0.39 |
Irregular and aggregate, smooth surface |
| F13 |
22.5 ± 0.14 |
0.85 ± 0.96 |
22.4 ± 2.54 |
3.13 ± 0.35 |
ND |
| F14 |
27.2 ± 7.63 |
0.77 ± 0.01 |
42.8 ± 1.09 |
6.00 ± 0.15 |
ND |
| F15 |
17.7 ± 5.28 |
1.50 ± 0.04 |
20.4 ± 1.96 |
2.86 ± 0.27 |
ND |
| F16 |
17.1 ± 4.38 |
1.41 ± 0.01 |
21.0 ± 0.69 |
2.95 ± 0.09 |
A spherical, smooth surface |
| F17 |
37.6 ± 1.53 |
0.88 ± 0.40 |
49.5 ± 4.06 |
6.93 ± 0.56 |
A spherical, smooth surface |
| F18 |
47.7 ± 3.18 |
1.18 ± 0.41 |
31.4 ± 1.46 |
4.39 ± 0.20 |
ND |
| F19 |
22.2 ± 2.99 |
1.18 ± 0.44 |
58.7 ± 4.54 |
4.81 ± 0.37 |
A fine spherical, smooth surface |
| F20 |
10.6 ± 0.94 |
1.25 ± 0.35 |
31.1 ± 0.87 |
2.55 ± 0.07 |
ND |
*Optimized formulation. ND; Not determined, EE; Encapsulation efficiency, DL; Drug loading.
Peptide content and characterization
HPLC method development
HPLC is widely used to study peptides and protein instabilities by MW analysis of macromolecules, identification of GA-induced degradation products, and assessing peptide integrity.
30
This method was validated by linearity (R2 0.9864), specificity (2 ± 0.2 % RSD), precision (2.17% RSD), and accuracy (0.18% Error). GA chromatograms extracted from MPSs produced by W/O/W and S/O/O methods and pure GA are presented in Fig. 1. The absence of further peaks, peak shape, and retention time (approximately at the 16th minute) indicate that the peptide was quite stable during the production process and chemical structure of GA has not changed.
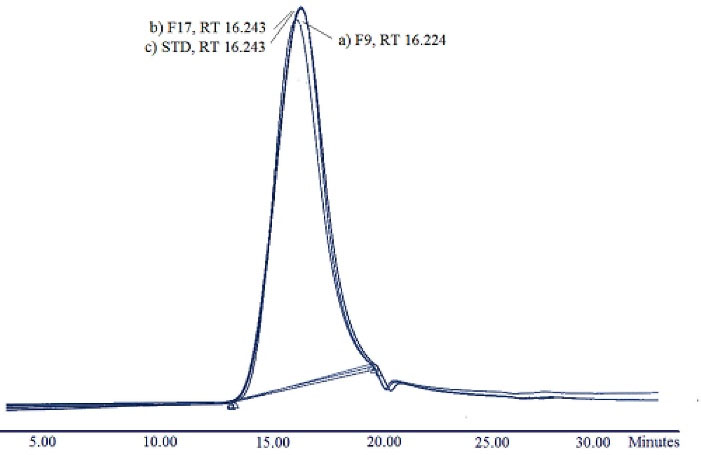
Fig. 1.
Glatiramer acetate (GA) chromatograms analyzed by Gel permeation chromatography; a) Extracted GA from Poly lactide-co-glycolide (PLGA) 502H Microspheres (F9), b) Extracted GA from PLGA 502 Microspheres (F17), and c) standard (unprocessed) GA.
.
Glatiramer acetate (GA) chromatograms analyzed by Gel permeation chromatography; a) Extracted GA from Poly lactide-co-glycolide (PLGA) 502H Microspheres (F9), b) Extracted GA from PLGA 502 Microspheres (F17), and c) standard (unprocessed) GA.
UV spectrophotography
The optical density of the obtained solution was assessed at 339 nm by UV spectrophotometer. Calibration curve ranging from 4 to 62 µg/mL was calculated by linearity acceptance (R2: 0.98) accuracy (7.7 %Error), LOD (0.083 µg/mL), and the difference between the results for duplicate samples at each concentration was less than 4%.
EE and loading percentage
The EE and loading results of particles prepared by W/O/W and S/O/O and optimized particles are shown in Table 4. GA is a water-soluble peptide that is supposed to have low EE due to the leakage into the external water phase.
31
The optimized MPS presented high levels of peptide DL 7.29% and EE 52% (F9).
MPS characterization
Morphology of particles
Particle size and particle morphology have effect on loading, product injectability, release kinetics, pharmacokinetics,
7
and even in nano scale particles targeting ability, phagocytic and cellular uptake.
32
F9 was the optimized formulation (Fig. 2G-H) with a spherical and relatively smooth surface. The average particle size of MPSs prepared by W/O/W was 10-20 µm with a narrow size distribution, suitable for IM and SC injections (Fig. 2A-H). In contrast, aggregation and broad particle size distribution (20-50 µm) was observed in MPSs, prepared by S/O/O methods (Fig. 2I-L). Almost all prepared MPS, produced by W/O/W, have visible porosity on MPS surfaces due to the aqueous phase in the W/O/W emulsion. However, it was observed that MS with polymer that has a high MW has a smooth surface (Fig. 2D). Use of rotor type mixer (2000 rpm) instead of magnetic stirrer (1400 rpm) leads to a non-significant decrease in the size of particles (formulation 11-15) which is a remarkable result in the industry. The use of water, associated with the organic solvent in the F19 formulation, reduces the particle size and size distribution, significantly.

Fig. 2.
SEM results of microspheres (A-H and I-L belong to particles prepared by W/O/W and S/O/O emulsion methods, respectively), A) Blank PLGA 502H Microspheres, B) F1, C) F5, D) F6, E) F7, F) F7 in 1 µm scale, G) F9, H) F9 in 1 µm scale, I) Blank PLGA 502 Microspheres, J) F16, K) F17, L) F12.
.
SEM results of microspheres (A-H and I-L belong to particles prepared by W/O/W and S/O/O emulsion methods, respectively), A) Blank PLGA 502H Microspheres, B) F1, C) F5, D) F6, E) F7, F) F7 in 1 µm scale, G) F9, H) F9 in 1 µm scale, I) Blank PLGA 502 Microspheres, J) F16, K) F17, L) F12.
The SEM results show the particle size in dried form, relating to the shelf life of the particles, on the contrary, LS shows the hydrodynamic diameter including the core and molecules adsorbed or attached on the surface. The LS method is more appropriate because it is used medically. However, stability data shows that particle growth on dried particles did not occur for 6 months.
Surface charge of particles
The zeta potential of final and optimized formulation was measured by Nano ZS, and the value of -23.8 mV was reported for optimized MPSs. In SC administered microparticulate drug delivery systems, the charge of MPSs is important in enhancing the rapid and uniform suspension following reconstitution of freeze-dried powder with lack of agglomeration and consequently proper syringe ability. Both positively and negatively charged particles at the zeta potential of 20 to 30 mV will prevent particle aggregation by creating an effective repulsion between the particles and apparently provides stable particle dispersion,
33
and is formed with slight shaking the homogeneous suspension. GA seems to have a large net positive charge.
34,35
Therefore, a zero-order prolonged release observed for optimized formulation may be contributed to the opposite charge of the carrier and drug. Although the small size of MPSs is desired to guarantee appropriate syringe ability of formulation during SC injection via thin needles,
36
the prolonged release of drugs through these MPSs will be threatened. Therefore, the opposite charge of carrier and drug will be crucial to provide a sustained drug release via small size MPSs.
Fourier transform infrared
The FT-IR spectra of blank MPS, GA-MPSs, pure GA, and GA extracted from PLGA MPSs (F9 and F17) were assessed and presented in Fig. 3. The spectra of loaded MPSs show three separate peaks at wavenumbers 1656, 1542, and 3298 cm -1, indicating amide I, amide II, and carboxylic acid regions, respectively. The peak related to amide II was broadened (Fig. 3 spectrum e), and the phenol peak shifted slightly to the higher wavelengths (e.g., blue change, 3298 to 3308 cm -1) with a slight expansion. In comparison, these peaks were not seen in blank MPSs (Fig. 3 spectrum d).
Fig. 3 spectra b-c show the extracted GA spectra that present drug stability during the W/O/W and S/O/O processes, respectively. As seen in these spectra and standard GA (Fig. 3 spectrum a), the peaks of phenol and amides (I and II) were at around 3298 cm -1 and 1546 cm -1 and 1656 cm -1, respectively. Since there is no change in the position and shape of the peaks, it is assumed that neither the peptide is hydrolyzed nor the decomposition occurs through the microencapsulation process.
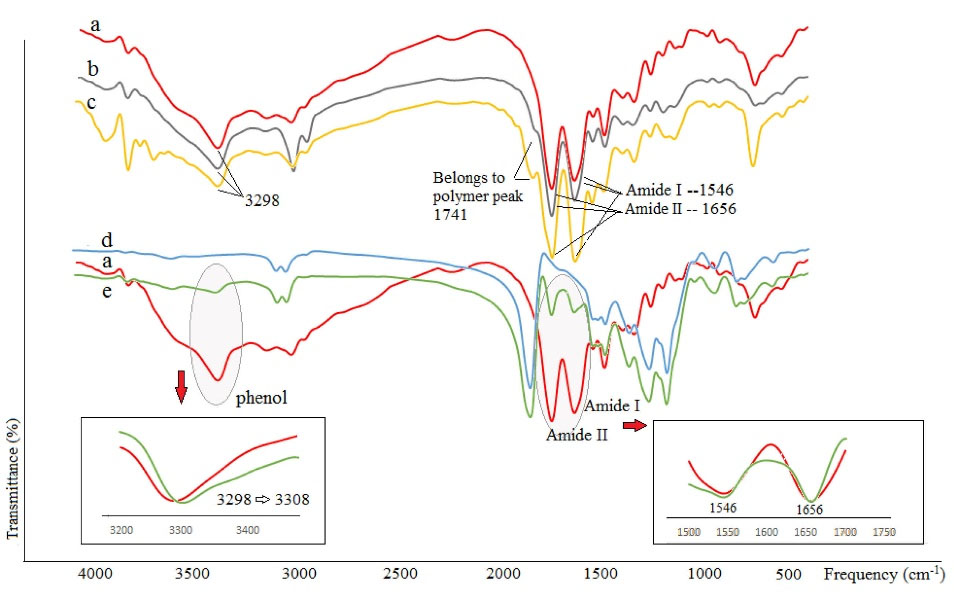
Fig. 3.
FT-IR spectra of a) standard Glatiramer acetate (GA) (unprocessed), b) extracted GA from PLGA 502H MPSs produced by W/O/W process, c) extracted GA from PLGA 502 MPSs produced by S/O/O process, d) Blank MPSs, e) GA-loaded MPSs (F9). MPSs; Microspheres.
.
FT-IR spectra of a) standard Glatiramer acetate (GA) (unprocessed), b) extracted GA from PLGA 502H MPSs produced by W/O/W process, c) extracted GA from PLGA 502 MPSs produced by S/O/O process, d) Blank MPSs, e) GA-loaded MPSs (F9). MPSs; Microspheres.
X-Ray diffraction
Fig. 4 presents the results of the X-ray diffractions of pure GA (as a reference), extracted peptide (GA) from MPSs prepared by W/O/W and S/O/O emulsification methods, and two GA-MPSs formulations (F9 and F16). As can be seen, the diffraction patterns of the extracted GA and GA powders reveal the amorphous form of peptides with the same pattern.
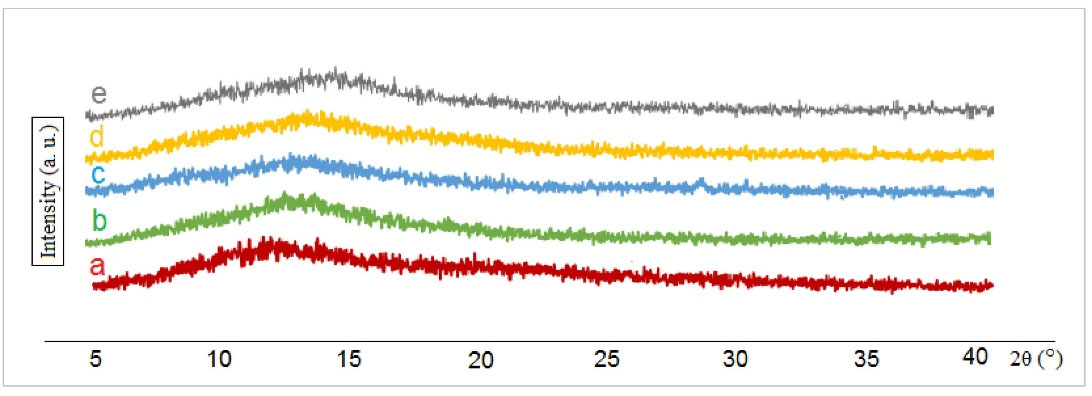
Fig. 4.
Wide-angle XRD with scattering angle 5° < 2θ < 40°. Profiles of a) pure GA, b) Extracted GA from microspheres (MPSs) F9, c) Extracted GA from MPSs F16, d) GA loaded-MPSs (F9) and, e) GA loaded -MPSs (F16).
.
Wide-angle XRD with scattering angle 5° < 2θ < 40°. Profiles of a) pure GA, b) Extracted GA from microspheres (MPSs) F9, c) Extracted GA from MPSs F16, d) GA loaded-MPSs (F9) and, e) GA loaded -MPSs (F16).
Release profile study
The in vitro analysis is essential to assure batch reproducibility for reliable biological activity. The result of peptide release from the prepared particles and the optimized formulation (F9) were presented in Fig. 5A-C. Moreover, Fig. 5D shows the SEM results of MPSs following in vitro release over one month (Initial time and after the first day and 30 days). Repeated release experiments of F9 showed (a) less burst release (~7.1 %), (b) less lag time (slowly started), (c) zero-order release behavior (k=0.001), and (d) complete release within one month (~94%) with good reproducibility.
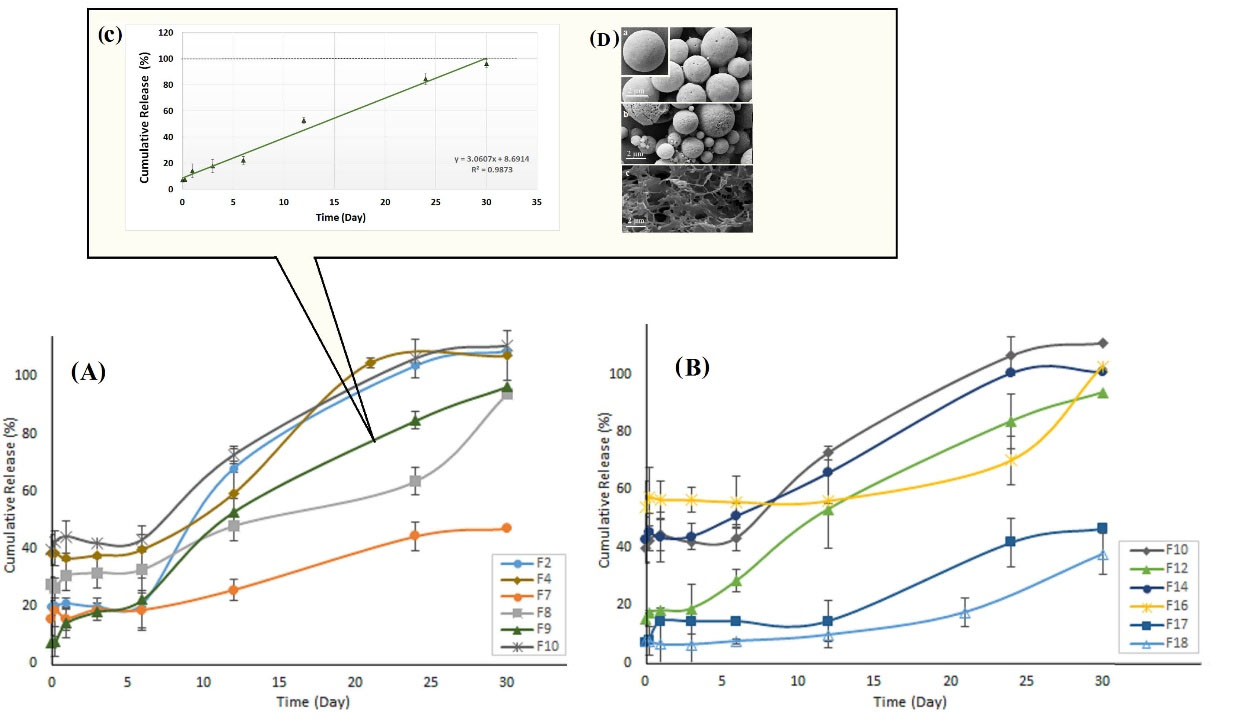
Fig. 5.
The release behavior of Glatiramer acetate microspheres produced by A) W/O/W process, B) S/O/O process, C) The zero-order release profile of optimized formulation (F9) with ~ 7% initial burst release. D) SEM results of microspheres following in vitro release over one-month: a) initial time and after b) first day, c) 30 days. W/O/W; Water in oil in water, S/O/O; Solid in oil in oil.
.
The release behavior of Glatiramer acetate microspheres produced by A) W/O/W process, B) S/O/O process, C) The zero-order release profile of optimized formulation (F9) with ~ 7% initial burst release. D) SEM results of microspheres following in vitro release over one-month: a) initial time and after b) first day, c) 30 days. W/O/W; Water in oil in water, S/O/O; Solid in oil in oil.
Mechanistic kinetic models
Kinetic models try to predict the in vivo behavior of GA-MPSs through in vitro profile release at the injection site. Zero-order model was the best fitting model for F9 formulation with RSQ 0.987 and an error of 0.76, which was obtained from equation F= K0t+b, where the K0 is the zero-order release constant, although it was almost nonlinear in the first 20-30% of the release profile that followed Higuchi release model (RSQ of 0.969). It is usually observed when diffusion-erosion mechanisms occur.
37
The Fick’s first diffusion law and the Noyes-Whitney dissolution law represent a predictable pharmacological response at the sites of effect.
Stability studies
The MPSs are particularly interesting because of their high stability. A comparison of the particle stability results prepared by W/O/W and S/O/O and the optimized formulation was presented in Fig. 6 and supplementary data. Stability data showed that the optimized formulation (F9) was completely stable during the real-time stability tests within 6 months, while the peptide content was slightly reduced under the accelerated stability test. However, if the product is prepared in sterile conditions, the stability of the particles will be more improved and controlled. To analyze the effect of the drying process (F1 vs. F3), when particles on the tray (F1) were dried instead of freeze dryer (F3), the particle size increased, indicating that the freeze-drying process prevents particle aggregation (P value = 0.026). Under the similar condition, the use of PVA in both primary and secondary aqueous media (F4 vs. F3 and F10 vs. F9), the particle size was increased, and drug loading was significantly decreased over 6 months, which may be due to PVA-induced aggregation. The type of mixing in S/O/O methods (rotor mixer or magnetic stirrer) has no significant effect (P value = 0.978) on stability test. SEM results also confirmed this observation. However, the particles produced by the magnetic stirrer are significantly smaller and tend to be denser on stability test. According to the results, large-scale adjustment (i.e., the modifying shaft of the blender) will be essential.
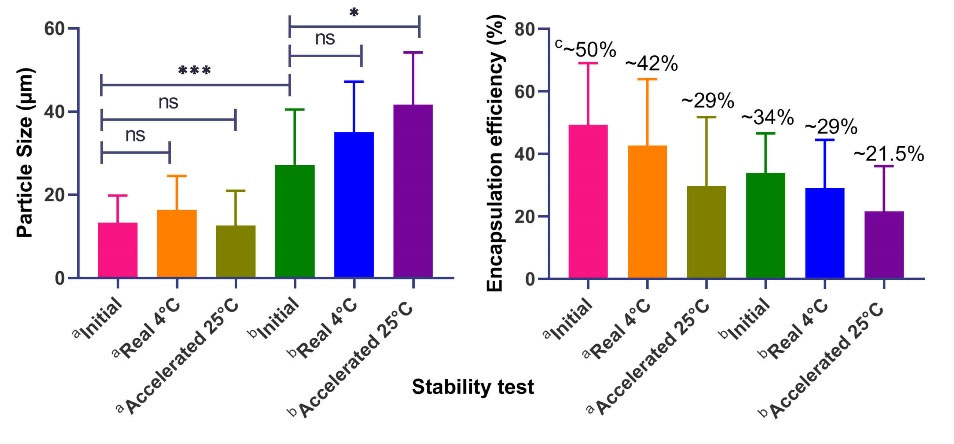
Fig. 6.
The effect of stability conditions on A) Particle size and B) Encapsulation efficiency (%). a,b Stability results of particles prepared by W/O/W (F1-F10), and S/O/O methods (F11-F20), respectively; c Mean value of encapsulation efficiency; ns Non-significant (P value ≥ 0.05); * Significant (P value: 0.01 to 0.05); ** Very significant (P value: 0.001 to 0.01); *** Extremely significant (P value: 0.0001 to 0.001). W/O/W; Water in oil in water, S/O/O; Solid in oil in oil.
.
The effect of stability conditions on A) Particle size and B) Encapsulation efficiency (%). a,b Stability results of particles prepared by W/O/W (F1-F10), and S/O/O methods (F11-F20), respectively; c Mean value of encapsulation efficiency; ns Non-significant (P value ≥ 0.05); * Significant (P value: 0.01 to 0.05); ** Very significant (P value: 0.001 to 0.01); *** Extremely significant (P value: 0.0001 to 0.001). W/O/W; Water in oil in water, S/O/O; Solid in oil in oil.
Syringeability study
The chosen diluents should be able to dissolve the freeze-dried products easily and be compatible with the final product, and should not affect the stability of the reconstituted product. The solid percentage for the optimized formulation (F9) in the 4 mL diluent was 27.43%, tonicity (Osmolality) of diluent and suspension were 247 mOsm/kg and 280-310 mOsm/kg, respectively. Besides, with the slight shaking, the homogeneous suspension was formed and easily discharged through the needles. The pH of the formulation after dissolving in the diluent was 6.8 ± 0.2.
Discussion
In developing innovative methods to fabricate polymeric particles, slight changes in manufacturing process or formulation show noteworthy differences in physicochemical properties of particles such as loading percentage and release behavior.
It is generally more difficult to encapsulate the hydrophilic drugs, and in most cases, the amount is unacceptable.
38
Regardless of the type of polymer, formulations with a lower polymer content following the IIA FDA database are preferred. In the W/O/W methods, the use of salt
7
and surfactant could overcome this obstacle. Besides, as in the S/O/O emulsion technique, the external phase is mainly paraffin leakage to the external phase is also limited. Moreover, in the S/O/O technique, deletion of using a homogenizer, which is a threatening factor for peptide and protein stability during MPS fabrication, was very helpful in increasing the encapsulation rate.
39
The method of S/O/W resulted in sticky agglomerates. This strange result was observed several times. It might be attributed to the reaction of the positively charged GA (which instantly dissolved in external aqueous phase because of instable initial S/O dispersion) with negatively charged PLGA. In the W/O/W formulations based on 502H and 502 PLGA (F4 versus F10 and F5 versus F7), as the ratio increases from 1:6 to 1:10, the amount of loading increases due to the low amount of drug. In addition, the percentage of loading was significantly reduced (P-Value 0.017) when the MPSs were dried on the tray (F1 and F2) compared to those which were dried by the freeze dryer (F3 and F4) indicating that the freeze dryer prevents the drug loss during the process. The use of PVA as a non-ionic surfactant in both primary (2%) and secondary (0.5%) aqueous media affected the amount of loading, specifically when a low amount of peptide (1:10) was used (F2 vs. F1 and F4 vs. F3). Turning now to the experimental evidence, the presence of PVA in primary aqueous media in the higher ratio (1:10) has a more positive effect on loading than when not applied (25 vs. 23 and 67 vs. 52) (P value=0.044). The surfactant used in the primary W/O emulsion should have a low HLB. In contrast, using the surfactant in the secondary emulsion should have a high HLB to ease the secondary emulsification. PVA, as a hydrophilic polymeric surfactant, has a high HLB near 18. Hence, the use of PVA in the primary emulsion has not had much effect, and on the contrary, it has been demonstrated that only nano/MPSs, which were formed by using PVA in secondary emulsion, have a spherical appearance without aggregation. To demonstrate the negative effect of the PVA application in the primary aqueous phase, both F4 and F10 showed a higher initial burst release than other W/O/W formulations. In addition, the use of surfactants in low proportions only in the secondary phase prevents problematic release kinetics while combining alkaline and nuclear drugs with acidic polymers.
7
In several studies, W/O/W techniques have been used to prepare biomaterials MPSs; still, low loading amount is a drawback. The researchers have found different ways to overcome this problem; (a) using a free base instead of the salt form of the drug.
40
(b) Conformational rigidification or hydrophobization using polyelectrolytes.
41
(c) By adding salts such as NaCl, NaBr to the external aqueous phase. (d) Adjusting the pH of the aqueous phase upper than pKa of the peptide.
42
(f) Applying amphiphilic polymer, i.e., PLA grafted to the hydrophilic backbone.
43
(g) By blocking acylation through PEGylation on the different amino groups.
44
(h) The addition of lysine or arginine as positively charged amino acids to the primary aqueous phase. (i) Using surfactant in preparation of both emulsion layers in the W/O/W method and finally, (j) Designing a feasible S/O/O method.
39
Particles made by PLGA 502 through S/O/O methods (F14 and F17) have higher loading than other formulations based on PLGA 502H and 752H. This result could be attributed to the stability of the particles during the preparation process. On the other hand, the peptide in the MPSs prepared by the uncapped polymers was placed on the particle’ surface and was thoroughly rinsed during the washing step. In formulation F19, due to the use of a small amount of water with acetonitrile, the loading rate increased, possibly being bound by soluble peptide and acidic end group of polymer (502H) in the presence of water.
In the FT-IR results, the widening peak of the amide II range and observations in the phenol region are signs of hydrogen bonding between nitrogen (N) of GA in the amide II region and the carboxylic group PLGA through the fabrication process. These results are consistent with the findings of other studies, such as insulin.
45
As there is no change in the position and shape of the peaks, it is assumed to be complete extraction of the peptide, and neither hydrolysis nor degradation peptide appears through the microencapsulation process.
The proof of amorphousness of the particles through XRD results, in addition to increasing the solubility and improving the release behavior, due to the absence of any sign of crystallinity,
46
indicates the uniformity of the content within the particles.
47
Besides, direct interaction among peptide molecules is relatively weak in formulations that prove the minimum loss of in vivo activity. The chemical structure of the used polymer has a significant effect on the release kinetics, especially in particles prepared by the S/O/O method. In the formulations F16, F17, and F18, the other parameters are similar regardless of the effect of loading. In formulation F16 (PLGA 752H (MW 4-15 kDa), DL ~3%) high burst release (~50%) has seen in contrast to formulation F17 (DL ~7%) and F18 (DL ~4.5%) with PLGA 502 and 502H (average Mw 12 kDa), which had relatively low burst release, ~20% and ~10%, respectively. This event may be related to high viscosity with 752H polymer; same concentration but higher viscosity. On the other hand, due to in the S/O/O technique, the medium is oily, and PLGA 752H is more hydrophobic than other polymers; it loses the tendency to encapsulate better, then the drug settles on the formed particles instead of inside. In addition, end group differences in F17 and F18 (502H and 502, acid vs. ester) may influence the binding of the peptide to the PLGA matrix. Although in this case, when polymer degradation begins, the chain scission will rapidly increases the number of free acidic groups, and no major difference is expected in the binding of the basic peptides to the acidic lactate or glycolide groups.
A simplified model based on pharmacokinetic simulation optimizes the release kinetics from sustained-release formulations. In designing the Prednisone MPSs, the researchers found that the release mechanism of low MW PLGA (502, 502H) is mostly follows power law and involves solubilization, diffusion, and erosion.
25,48
The experimental data in this study, as shown in Table 5, confirms previous findings and contributes additional evidence that suggests preparation method also has a significant effect on release mechanism regarding the results of F16 and F18. Most of the particles that prepared by S/O/O method have a long lag time (Fig. 5), possibly attributed to the Fickian diffusion mechanism. In this study, the highest correlation was reported for PLGA 502H MPS prepared by W/O/W method (F9).
Table 5.
Evaluation of drug release of different GA-Microspheres through zero-order kinetics mechanistic model
|
Formulation
|
F7
|
F8
|
F9
|
F16
|
F17
|
F18
|
| Polymer used |
502 |
752H |
502H |
752H |
502 |
502H |
| Slope |
1.090 |
1.986 |
3.060 |
1.238 |
1.26 |
0.863 |
| RSQ |
0.9671 |
0.9474 |
0.9873 |
0.7465 |
0.9230 |
0.7656 |
Comparisons are provided for both microspheres made by W/O/W (F7-F9), and S/O/O (F16-F18) methods, formulation parameters are similar other than the polymer effect.
In both W/O/W and S/O/O methods, the type of polymer had no significant effect on particle stability that would be entirely controlled by adjusting the condition. In general, apart from production processes and formulation parameters, CCS, storage and shipment condition, and even condition of environment between preparing reconstitute, and usage are the factors that affect the final stability of the pharmaceutical product.
27
Both 23G and 21G needles can be used as packaging materials. Local reactions such as Eligard® and Zoladex® are common in both SC and IM injections of PLGA formulations. However, topical treatment and the use of ice at the injection site are ways to reduce the pain caused by large needles.
27
In contrast, another blind study showed that the experienced pain caused by Zoladex® (Goserelin, 16G needle) and Prostap® (leuprorelin, 23G needle) was not significantly different.
49
Research Highlights
What is the current knowledge?
√ GA has currently been administered to reduce the frequency of relapses in MS patients for daily and biweekly usage.
√ The fast degradation and clearance cause multiple injections, which consequently restrict the clinical application.
√ Sustained release products increase patient compliance and effectiveness of pharma-cotherapy.
What is new here?
√ A novel one-month injection formulation of GA-MPSs was prepared and characterized.
√ The optimized formulation had high encapsulation percentage and ideal release behavior.
√ The feasible and reproducible approach can be a means to produce abundant sustained release products.
√ GA stability assessment was carried out using a validated HPLC-GPC method.
√ Microspheres were stable during a 6-month accelerated stability study.
√ GA-MPSs will improve the efficiency of GA through increasing the drug exposure that leads to patient compliance.
Conclusion
This study was performed to design stable peptide MPSs with high loading percentage and zero-order release behavior with a proper dosage form. GA-MPSs were prepared using modified W/O/W and S/O/O double emulsion methods. Repeated release experiments of the optimal F9 formulation indicated acceptable and reproducible behavior for laboratory and even for scale-up batches in the future. Several advantages of this formulation for other products include high loading percentage, ideal release profile, content uniformity, stability, and high syringe ability. XRD results confirmed the amorphous nature of GA-MPSs, which is suitable for protein solubility and assuring biological activity.
This formulation is intended for local administration. The released GA will directly compete with numerous myelin antigens to bind to the major histocompatibility complex (MHC) molecules on the antigen-presenting cell surface. Therefore, the released GA will maintain the same function as the free GA through continuous drug exposure by protecting GA from rapid local degradation. The produced formulation is exciting because it paves the way for industrial-scale preparation of stabilized MPSs suspensions. In further research, as a comprehensive formulation, this data can be a means to produce abundant sustained-release products in the market that have not been produced so far.
Acknowledgments
The authors would like to thank the Tabriz University of Medical Sciences for providing a grant (Grant No: 60004, 2018) to support this work (Ph.D. thesis, No. 141).
Ethical statement
There is none to be stated.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Authors’ contribution
HH and FM designed the idea. The experiments, data collection, statistical analyses were conducted by FM. Data interpretation were performed by MBJ, HH and FM. The project supervised by HH and MBJ. The manuscript was drafted by FM and revised by HH and MBJ.
Supplementary Materials
Supplementary file 1 contains Table S1.
(pdf)
References
- Zirak N, Bolandparvaz Jahromi A, Salahinejad E. Vancomycin release kinetics from Mg–Ca silicate porous microspheres developed for controlled drug delivery. Ceram Int 2020; 46:508-12. doi: 10.1016/j.ceramint.2019.08.290 [Crossref] [ Google Scholar]
- Huang F, Sun L, Zhao C, Qiu Z, Zhao Y, Jin W. Protein microcapsules integrated hierarchical scaffolds for local treatment of acute myocardial infarction model. Appl Mater Today 2021; 22:100901. doi: 10.1016/j.apmt.2020.100901 [Crossref] [ Google Scholar]
- Jirwankar P, Shah D, Shao J. Protection of Protein Drugs by Self-Emulsified Nanoemulsion Against Proteolysis. J Pharm Sci 2020; 109:2615-21. doi: 10.1016/j.xphs.2020.05.005 [Crossref] [ Google Scholar]
- Sung YK, Kim SW. Recent advances in polymeric drug delivery systems. Biomaterials Research 2020; 24:12. doi: 10.1186/s40824-020-00190-7 [Crossref] [ Google Scholar]
- Fathi M, Barar J. Perspective highlights on biodegradable polymeric nanosystems for targeted therapy of solid tumors. Bioimpacts 2017; 7:49-57. doi: 10.15171/bi.2017.07 [Crossref] [ Google Scholar]
- Lalive PH, Neuhaus O, Benkhoucha M, Burger D, Hohlfeld R, Zamvil SS. Glatiramer acetate in the treatment of multiple sclerosis: emerging concepts regarding its mechanism of action. CNS Drugs 2011; 25:401-14. doi: 10.2165/11588120-000000000-00000 [Crossref] [ Google Scholar]
- Molavi F, Barzegar-Jalali M, Hamishehkar H. Polyester based polymeric nano and microparticles for pharmaceutical purposes: A review on formulation approaches. J Control Release 2020; 320:265-82. doi: 10.1016/j.jconrel.2020.01.028 [Crossref] [ Google Scholar]
- Silva ACQ, Vilela C, Santos HA, Silvestre AJD, Freire CSR. Recent trends on the development of systems for cancer diagnosis and treatment by microfluidic technology. Appl. Mater. Today 2020; 18(#):100450. doi: 10.1016/j.apmt.2019.100450 [Crossref] [ Google Scholar]
- Della Porta G, Campardelli R, Cricchio V, Oliva F, Maffulli N, Reverchon E. Injectable PLGA/Hydroxyapatite/Chitosan Microcapsules Produced by Supercritical Emulsion Extraction Technology: An In Vitro Study on Teriparatide/Gentamicin Controlled Release. J Pharm Sci 2016; 105:2164-72. doi: 10.1016/j.xphs.2016.05.002 [Crossref] [ Google Scholar]
- Martins KF, Messias AD, Leite FL, Duek EAR. Preparation and characterization of paclitaxel-loaded PLDLA microspheres. Mat Res 2014; 17:650-6. doi: 10.1590/S1516-14392014005000028 [Crossref] [ Google Scholar]
- Shi N-Q, Zhou J, Walker J, Li L, Hong JKY, Olsen KF. Microencapsulation of luteinizing hormone-releasing hormone agonist in poly (lactic-co-glycolic acid) microspheres by spray-drying. J Control Release 2020; 321(#):756-72. doi: 10.1016/j.jconrel.2020.01.023 [Crossref] [ Google Scholar]
- Andhariya JV, Jog R, Shen J, Choi S, Wang Y, Zou Y. Development of Level A in vitro-in vivo correlations for peptide loaded PLGA microspheres. J Control Release 2019; 308:1-13. doi: 10.1016/j.jconrel.2019.07.013 [Crossref] [ Google Scholar]
-
J-M. Cardot1, E. Beyssac, M.Alric. In Vitro–In Vivo Correlation: Importance of Dissolution in IVIVC. Dissolution Technol 2007; 15-9. 10.14227/DT140107P15.
- Rhee Y-S, Sohn M, Woo BH, Thanoo BC, DeLuca PP, Mansour HM. Sustained-release delivery of octreotide from biodegradable polymeric microspheres. AAPS PharmSciTech 2011; 12:1293-301. doi: 10.1208/s12249-011-9693-z [Crossref] [ Google Scholar]
- Gong H, Wang J, Zhang J, Wu J, Zheng Z, Xie X. Control of octreotide release from silk fibroin microspheres. Mater Sci Eng C Mater Biol Appl 2019; 102:820-8. doi: 10.1016/j.msec.2019.05.004 [Crossref] [ Google Scholar]
- Zhang H, Liu Y, Chen C, Cui W, Zhang C, Ye F. Responsive drug-delivery microcarriers based on the silk fibroin inverse opal scaffolds for controllable drug release. Appl Mater Today 2020; 19:100540. doi: 10.1016/j.apmt.2019.100540 [Crossref] [ Google Scholar]
- Qi P, Bu R, Zhang H, Yin J, Chen J, Zhang A. Goserelin Acetate Loaded Poloxamer Hydrogel in PLGA Microspheres: Core-Shell Di-Depot Intramuscular Sustained Release Delivery System. Mol Pharm 2019; 16:3502-13. doi: 10.1021/acs.molpharmaceut.9b00344 [Crossref] [ Google Scholar]
- Zhou J, Hirota K, Ackermann R, Walker J, Wang Y, Choi S. Reverse Engineering the 1-Month Lupron Depot®. AAPS J 2018; 20:105. doi: 10.1208/s12248-018-0253-2 [Crossref] [ Google Scholar]
-
Marques. MRC, Loebenberg. R, Almukainzi. M. Simulated Biological Fluids with Possible Application in Dissolution Testing. Dissolution Technol. 2011; 15-28. 10.14227/DT180311P15.
- Shen J, Burgess DJ. Accelerated in vitro release testing of implantable PLGA microsphere/PVA hydrogel composite coatings. Int J Pharm 2012; 422:341-8. doi: 10.1016/j.ijpharm.2011.10.020 [Crossref] [ Google Scholar]
- Clark. BC, Dickinson. PA, Pyrah. IT. In Vitro=In Vivo Release from Injectable Dispersed Systems. In: Burgess DJ, editor. Injectable Dispersed Systems Formulation, Processing, and Performance. Taylor and francis group; 2005. p. 34.
- Convention USP. USP 42, NF 37: The United States Pharmacopeia, the National Formulary: United States Pharmacopeial Convention; 2018.
-
Shah. VP, DeMuth. J, Hunt. DG. Performance Test for Parenteral Dosage Forms. Dissolution Technol. 2015; 16-21. 10.14227/DT220415P16.
- Maderuelo C, Zarzuelo A, Lanao JM. Optimization of Release Kinetics from Sustained-Release Formulations using Model-Independent Pharmacokinetic Simulation. J Pharm Sci 2011; 100:3260-7. doi: 10.1002/jps.22565 [Crossref] [ Google Scholar]
- Barzegar-Jalali M, Adibkia K, Valizadeh H, Shadbad MR, Nokhodchi A, Omidi Y. Kinetic analysis of drug release from nanoparticles. J Pharm Pharm Sci 2008; 11:167-77. doi: 10.18433/j3d59t [Crossref] [ Google Scholar]
- Molavi F, Hamishehkar H, Nokhodchi A. Impact of Tablet Shape on Drug Dissolution Rate Through Immediate Released Tablets. Adv Pharm Bull 2020; 10:656-61. doi: 10.34172/apb.2020.079 [Crossref] [ Google Scholar]
- Schwendeman SP, Shah RB, Bailey BA, Schwendeman AS. Injectable controlled release depots for large molecules. J Control Release 2014; 190:240-53. doi: 10.1016/j.jconrel.2014.05.057 [Crossref] [ Google Scholar]
- Yang Z, Liu L, Su L, Wu X, Wang Y, Liu L. Design of a zero-order sustained release PLGA microspheres for palonosetron hydrochloride with high encapsulation efficiency. Int J Pharm 2020; 575:119006. doi: 10.1016/j.ijpharm.2019.119006 [Crossref] [ Google Scholar]
- Pathak S, Gupta B, Poudel BK, Tran TH, Regmi S, Pham TT. Preparation of High-Payload, Prolonged-Release Biodegradable Poly(lactic-co-glycolic acid)-Based Tacrolimus Microspheres Using the Single-Jet Electrospray Method. Chem Pharm Bull (Tokyo) 2016; 64:171-8. doi: 10.1248/cpb.c15-00799 [Crossref] [ Google Scholar]
- Vaishya RD, Mandal A, Patel S, Mitra AK. Extended release microparticle-in-gel formulation of octreotide: Effect of polymer type on acylation of peptide during in vitro release. Int J Pharm 2015; 496:676-88. doi: 10.1016/j.ijpharm.2015.11.002 [Crossref] [ Google Scholar]
- Ramazani F, Chen W, van Nostrum CF, Storm G, Kiessling F, Lammers T. Strategies for encapsulation of small hydrophilic and amphiphilic drugs in PLGA microspheres: State-of-the-art and challenges. Int J Pharm 2016; 499:358-67. doi: 10.1016/j.ijpharm.2016.01.020 [Crossref] [ Google Scholar]
- Barar J. Bioimpacts of nanoparticle size: why it matters?. Bioimpacts 2015; 5:113-5. doi: 10.15171/bi.2015.23 [Crossref] [ Google Scholar]
- Lengyel M, Kállai-Szabó N, Antal V, Laki AJ, Antal I. Microparticles, Microspheres, and Microcapsules for Advanced Drug Delivery. Sci Pharm 2019; 87:20. doi: 10.3390/scipharm87030020 [Crossref] [ Google Scholar]
- Song JY, Larson NR, Thati S, Torres-Vazquez I, Martinez-Rivera N, Subelzu NJ. Glatiramer acetate persists at the injection site and draining lymph nodes via electrostatically-induced aggregation. J Control Release 2019; 293:36-47. doi: 10.1016/j.jconrel.2018.11.007 [Crossref] [ Google Scholar]
- Jalilian B, Einarsson HB, Vorup-Jensen T. Glatiramer acetate in treatment of multiple sclerosis: a toolbox of random co-polymers for targeting inflammatory mechanisms of both the innate and adaptive immune system?. Int J Mol Sci 2012; 13:14579-605. doi: 10.3390/ijms131114579 [Crossref] [ Google Scholar]
- Schwendeman SP, Shah RB, Bailey BA, Schwendeman AS. Injectable controlled release depots for large molecules. J Control Release 2014; 190:240-53. doi: 10.1016/j.jconrel.2014.05.057 [Crossref] [ Google Scholar]
- Papadopoulou V, Kosmidis K, Vlachou M, Macheras P. On the use of the Weibull function for the discernment of drug release mechanisms. Int J Pharm 2006; 309:44-50. doi: 10.1016/j.ijpharm.2005.10.044 [Crossref] [ Google Scholar]
- Leelakanok N, Geary S, Salem A. Fabrication and Use of Poly(d,l-lactide-co-glycolide)-Based Formulations Designed for Modified Release of 5-Fluorouracil. J Pharm Sci 2018; 107:513-28. doi: 10.1016/j.xphs.2017.10.012 [Crossref] [ Google Scholar]
- Emami J, Hamishehkar H, Najafabadi AR, Gilani K, Minaiyan M, Mahdavi H. A novel approach to prepare insulin-loaded poly(lactic-co-glycolic acid) microcapsules and the protein stability study. J Pharm Sci 2009; 98:1712-31. doi: 10.1002/jps.21544 [Crossref] [ Google Scholar]
- Guo W, Quan P, Fang L, Cun D, Yang M. Sustained release donepezil loaded PLGA microspheres for injection: Preparation, in vitro and in vivo study. Asian J Pharm Sci 2015; 10:405-14. doi: 10.1016/j.ajps.2015.06.001 [Crossref] [ Google Scholar]
- Devrim B, Bozkır A. Design and Evaluation of Hydrophobic Ion-Pairing Complexation of Lysozyme with Sodium Dodecyl Sulfate for Improved Encapsulation of Hydrophilic Peptides/Proteins by Lipid-Polymer Hybrid Nanoparticles. J NanomedNanotechnol 2015; 6:259. doi: 10.4172/2157-7439.1000259 [Crossref] [ Google Scholar]
- Ramazani F, Chen W, Van Nostrum CF, Storm G, Kiessling F, Lammers T. Formulation and characterization of microspheres loaded with imatinib for sustained delivery. Int J Pharm 2015; 482:123-30. doi: 10.1016/j.ijpharm.2015.01.043 [Crossref] [ Google Scholar]
- Zhou YX, Li SL, Fu HL, Cheng SX, Zhang XZ, Zhuo RX. Fabrication and in vitro drug release study of microsphere drug delivery systems based on amphiphilic poly-alpha,beta-[N-(2-hydroxyethyl)-L-aspartamide]-g-poly(L-lactide) graft copolymers. Colloids Surf B 2008; 61:164-9. doi: 10.1016/j.colsurfb.2007.08.001 [Crossref] [ Google Scholar]
- Zhang Y, Sophocleous AM, Schwendeman SP. Inhibition of peptide acylation in PLGA microspheres with water-soluble divalent cationic salts. Pharm Res 2009; 26:1986-94. doi: 10.1007/s11095-009-9914-2 [Crossref] [ Google Scholar]
- Baghban Taraghdari Z, Imani R, Mohabatpour F. A Review on Bioengineering Approaches to Insulin Delivery: A Pharmaceutical and Engineering Perspective. MacromolBiosci 2019; 19:e1800458. doi: 10.1002/mabi.201800458 [Crossref] [ Google Scholar]
- Liu J, Grohganz H, Rades T. Influence of polymer addition on the amorphization, dissolution and physical stability of co-amorphous systems. Int J Pharm 2020; 588:119768. doi: 10.1016/j.ijpharm.2020.119768 [Crossref] [ Google Scholar]
- Štukelj J, Svanbäck S, Agopov M, Löbmann K, Strachan CJ, Rades T. Direct Measurement of Amorphous Solubility. Anal Chem 2019; 91:7411-7. doi: 10.1021/acs.analchem.9b01378 [Crossref] [ Google Scholar]
- Giovagnoli S, Blasi P, Ricci M, Schoubben A, Perioli L, Rossi C. Physicochemical characterization and release mechanism of a novel prednisone biodegradable microsphere formulation. J Pharm Sci 2008; 97:303-17. doi: 10.1002/jps.21073 [Crossref] [ Google Scholar]
- Montgomery BS, Borwell JP, Higgins DM. Does needle size matter? Patient experience of luteinising hormone-releasing hormone analogue injection. Prostate Cancer Prostatic Dis 2005; 8:66-8. doi: 10.1038/sj.pcan.4500778 [Crossref] [ Google Scholar]