Bioimpacts. 12(4):371-391.
doi: 10.34172/bi.2022.23871
Review
Advance trends in targeting homology-directed repair for accurate gene editing: An inclusive review of small molecules and modified CRISPR-Cas9 systems
Forough Shams 1  , Hadi Bayat 2, 3, Omid Mohammadian 4, 3, Somayeh Mahboudi 5, Hassan Vahidnezhad 6, 7, Mohsen Soosanabadi 8, Azam Rahimpour 4, 3, *
, Hadi Bayat 2, 3, Omid Mohammadian 4, 3, Somayeh Mahboudi 5, Hassan Vahidnezhad 6, 7, Mohsen Soosanabadi 8, Azam Rahimpour 4, 3, *
Author information:
1Department of Medical Biotechnology, School of Advanced Technologies in Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran
2Department of Molecular Genetics, Faculty of Biological Sciences, Tarbiat Modares University, Tehran, Iran
3Department of Tissue Engineering and Applied Cell Sciences, School of Advanced Technologies in Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran
4Medical Nano-Technology and Tissue Engineering Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran
5Department of Food Science and Technology, School of Agriculture, Shiraz University, Shiraz, Iran
6Department of Dermatology and Cutaneous Biology, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA, USA
7Jefferson Institute of Molecular Medicine, Thomas Jefferson University, Philadelphia, PA, USA
8Department of Medical Genetics, Semnan University of Medical Sciences, Semnan, Iran
Abstract
Introduction:
Clustered regularly interspaced short palindromic repeat and its associated protein (CRISPR-Cas)-based technologies generate targeted modifications in host genome by inducing site-specific double-strand breaks (DSBs) that can serve as a substrate for homology-directed repair (HDR) in both in vitro and in vivo models. HDR pathway could enhance incorporation of exogenous DNA templates into the CRISPR-Cas9-mediated DSB site. Owing to low rate of HDR pathway, the efficiency of accurate genome editing is diminished. Enhancing the efficiency of HDR can provide fast, easy, and accurate technologies based on CRISPR-Cas9 technologies.
Methods:
The current study presents an overview of attempts conducted on the precise genome editing strategies based on small molecules and modified CRISPR-Cas9 systems.
Results:
In order to increase HDR rate in targeted cells, several logical strategies have been introduced such as generating CRISPR effector chimeric proteins, anti-CRISPR proteins, modified Cas9 with donor template, and using validated synthetic or natural small molecules for either inhibiting non-homologous end joining (NHEJ), stimulating HDR, or synchronizing cell cycle. Recently, high-throughput screening methods have been applied for identification of small molecules which along with the CRISPR system can regulate precise genome editing through HDR.
Conclusion:
The stimulation of HDR components or inhibiting NHEJ can increase the accuracy of CRISPR-Cas-mediated engineering systems. Generating chimeric programmable endonucleases provide this opportunity to direct DNA template close proximity of CRISPR-Cas-mediated DSB. Small molecules and their derivatives can also proficiently block or activate certain DNA repair pathways and bring up novel perspectives for increasing HDR efficiency, especially in human cells. Further, high throughput screening of small molecule libraries could result in more discoveries of promising chemicals that improve HDR efficiency and CRISPR-Cas9 systems.
Keywords: Modified CRISPR-Cas9, Small molecule, Genome editing, HDR
Copyright and License Information
© 2022 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
The progress of programmable nucleases has considerably augmented the field of precision genome editing and provided a novel promising avenue for gene therapy approaches. Until now, four major classes of programmable nucleases have been established based on introducing a site-specific double-strand break (DSB); including i) meganucleases or homing endonucleases which are achieved from microbial mobile genetic compounds, ii) zinc finger (ZF) nucleases which are inspired by eukaryotic transcription factors, iii) transcription activator-like effector (TALE) nucleases which are originated from Xanthomonas bacteria, and iv) RNA-guided DNA endonucleases which are human codon-optimized form of archaeal and most bacterial adaptive immune systems, CRISPR-Cas.
1,2
CRISPR-Cas systems are categorized into two basic classes and six types. Because of the diversity and practicability of CRISPR-Cas9, which is categorized in class II-type 2, most attractions have been concentrated on this system and considered as an impressive genome editing technology in eukaryotic cells. In meganucleases, ZFN and TALEN, specific DNA binding is developed by protein-DNA interactions, while Cas9 is recruited to target DNA sequences by a single-guide RNA (sgRNA) molecule.
3,4
These four categories of genome editing tools have the common state of generating a site-specific DSB in the targeted genome (Fig. 1). Naturally occurred DSBs are repaired by two distinct pathways, error-prone nonhomologous end-joining (NHEJ) or precise homology-directed repair (HDR).
5,6
Since NHEJ naturally is an error-prone repair pathway, by inhibiting the expression or function of essential components that are involved in this pathway, we could ameliorate the nuclease-mediated HDR efficiency.
7,8
In this regard, various small molecules have been recognized for modulating the efficiency of genome editing by either suppressing NHEJ or elevating the activity of HDR pathway. Using small molecules would develop a simple method that has several advantages for enhancing precision genome engineering. This review was focused on the mechanisms and effects of small molecules in DSB repair to progress the HDR pathway for improving the efficiency of precision genome editing in both in vivo and in vitro settings. Furthermore, we aimed to highlight the recent progress in enhancing HDR efficiency through using overlapping sequences and also applying novel Cas9 chimeric variants.
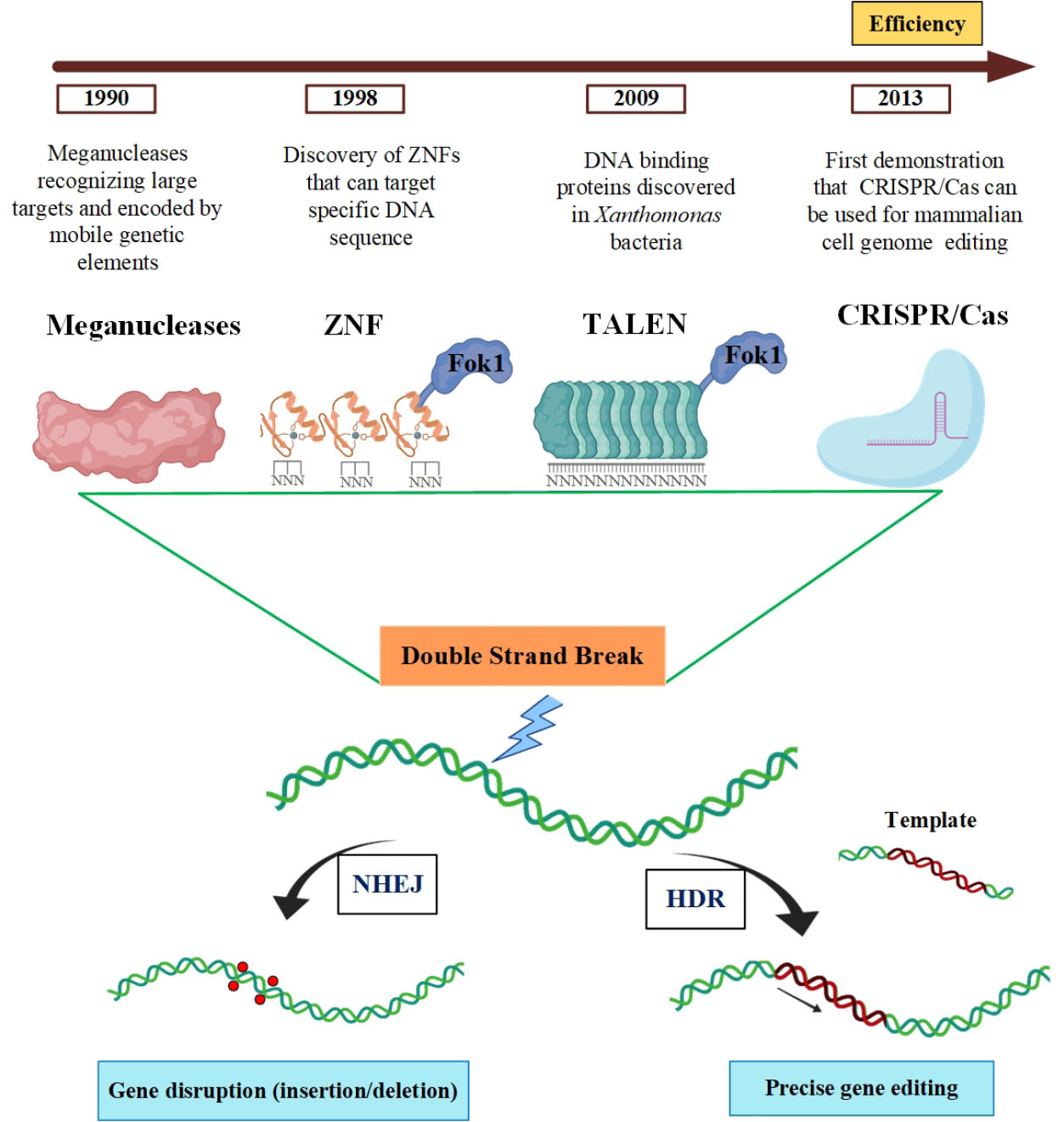
Fig. 1.
The mechanisms and gene editing platforms for repairing DSB.
DSBs which are induced by Meganucleases, ZFNs, TALENs, and CRISPR-Cas9 at specific sites could be repaired by NHEJ or HDR pathway. Figure was created with BiorRender (https://biorender.com).
.
The mechanisms and gene editing platforms for repairing DSB.
DSBs which are induced by Meganucleases, ZFNs, TALENs, and CRISPR-Cas9 at specific sites could be repaired by NHEJ or HDR pathway. Figure was created with BiorRender (https://biorender.com).
DNA repair pathways
By introducing a DSB, several factors such as BRCA1 (breast cancer type 1 susceptibility protein), 53BP1 (p53-binding protein1), and receptor-associated protein 80 (RAP80) are recruited to the damaged site and constitute ionizing radiation induced foci (IRIF). It is elucidated that a complex network of molecular interactions activating BRCA1 or 53BP1 derives DSB to HDR or NHEJ repair pathway, respectively. These two antagonizing factors permanently are acting contrary to each other at the DSB site. In order to identify the effect of small molecules to increase HDR pathway for more efficient and precise gene editing, an overview of the DNA repair pathways and their key factors is presented.
9
NHEJ pathway
Following the induction of DSB, the first reaction usually occurs through the NHEJ pathway. In mammalian cells, almost three quarter of DSBs are repaired via NHEJ and its defect results in various developmental disorders and enhances the rate of DSB-related chromosomal mutagenesis.
10
NHEJ is a broad term and commonly classified into two types:
-
Canonical NHEJ (c-NHEJ): it generally acts in end-joining and for a long time was characterized by its association with Ku, DNA ligase IV, and dependent factors.
10
-
Non-canonical NHEJ: several homology-independent repairs triggered by the c-NHEJ dysfunction which need DNA Ligase I/III and is known as “alternative NHEJ” (alt-NHEJ/A-NHEJ), “microhomology-mediated end joining” (MMEJ), or “backup NHEJ”.
10,11
According to distinct DNA ends, NHEJ uses different strategies and is initiated by phosphorylation of 53BP1 at DSBs through protein kinase ataxia-telangiectasia mutated (ATM). 53BP1 is a chromatin-binding protein and an important part of DSB signaling repair in mammalian cells that during G1 promotes NHEJ. The basic effector proteins for 53BP1 are DNA polymerase zeta processivity subunit (REV7), PAX transactivation domain-interacting protein, and RAP1- interacting factor 1 (RIF1) (Fig. 2A).
12
RIF1 is acting together with REV7 for recruiting a large complex to DSB.
13
This large complex is called shieldin which consists of four components including REV7, SHLD1 (induces NHEJ while decreasing the HR by constraining DNA end resection), SHLD2 (an effector of REV7), and SHLD3.
14,15
Furthermore, the core NHEJ factor recognizes broken ends and keeps them next to each other so that the other processing factors can be activated.
16
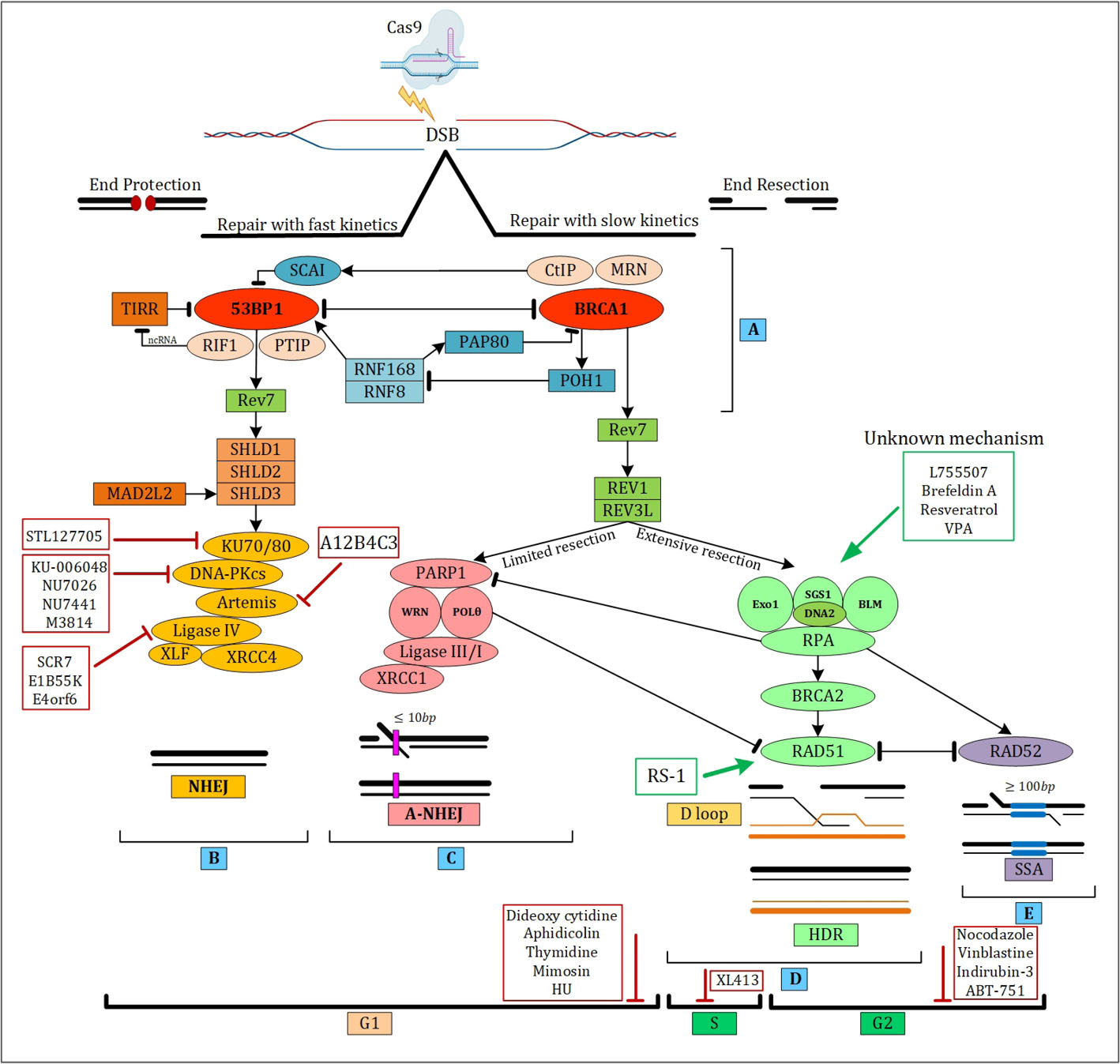
Fig. 2.
The interaction of repair pathways at the DNA break site in mammals and comparison of c-NHEJ, a-NHEJ, HDR, and SSA mechanisms.
(A) c-NHEJ key factor (53BP1) and HDR key factor (BRCA1) are motivated by complex interactions. (B) For DSB repair the first pathway choice in mammalian cells is NHEJ which is occurred in all cell cycle phases. When in DSB the terminuses are preserved from the incision and then ligated, NHEJ is promoted. (C) The microhomologous sequences which are adjacent to DSB are annealed in alt-NHEJ pathway. (D) HDR repair pathway employs a repair template such as sister chromatid to reliably amend the DSB. (E) SSA is a DSB repair pathway for fixing DSB by annealing lengthy homologous sequences at flanking sites. Figure was created with BiorRender (https://biorender.com).
.
The interaction of repair pathways at the DNA break site in mammals and comparison of c-NHEJ, a-NHEJ, HDR, and SSA mechanisms.
(A) c-NHEJ key factor (53BP1) and HDR key factor (BRCA1) are motivated by complex interactions. (B) For DSB repair the first pathway choice in mammalian cells is NHEJ which is occurred in all cell cycle phases. When in DSB the terminuses are preserved from the incision and then ligated, NHEJ is promoted. (C) The microhomologous sequences which are adjacent to DSB are annealed in alt-NHEJ pathway. (D) HDR repair pathway employs a repair template such as sister chromatid to reliably amend the DSB. (E) SSA is a DSB repair pathway for fixing DSB by annealing lengthy homologous sequences at flanking sites. Figure was created with BiorRender (https://biorender.com).
In c-NHEJ, predominantly Ku 80 interacts and promotes the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and creates a permanent complex that remains bound to the DSB ends.
16-18
DNA-PK can modulate the activity of different enzymes through autophosphorylation or phosphorylation and lead to DNA end processing by Artemis which cuts DNA overhangs for making blunt ends.
19
Then, DNA polymerase µ and λ can add missing nucleotides at the DSB ends.
16,20,21
The next step is ligation of blunt end that is accomplished by Ligase IV (Lig IV). Ligase IV usually makes a complex with X-ray repair cross-complementing 4 (XRCC4) and XRCC4-like factor (XLF) and induces related downstream pathways (Fig. 2B).
22
Alt-NHEJ pathway is generally active during the S and G2 phases of the cell cycle and repairs DNA DSBs through microhomology (MH)-mediated end joining (MMEJ) (Fig. 2C).
23
With regard to the annealing of the flanking MHs, MMEJ is classified into three distinct steps: pre-annealing, annealing, and post-annealing.
24
The initial step starts from end resection to subject MHs flanking DSBs through joining of poly [ADP-ribose] polymerase 1 (PARP1) to DSB ends for facilitating the resection factors [BRCA1/CtIP and MRN complex].
25,26
Intermediate annealing is reclaimed by the XPF/ERCC1 structure-specific nuclease complex, which is similar to XRCC4 in c-NHEJ. Then, this complex cuts the non-homologous ends and generates 3’- hydroxyl overhang that is desirable for developing by DNA polymerase. Finally, the DNA ligation is catalyzed by Ligase II and III.
27
Homology-directed repair (HDR) pathway
HDR, as a constant repair mechanism, comes into action in the S- or G2-phase and needs homologous DNA sequences.BRCA1 by suppressing 53BP1, is a pivotal initiating factor for HDR. Therefore, various inner and outer factors are involved in this case such as tudor-interacting repair regulator (TIRR) which is able to mask the H4K20me2 binding surface (a specific target for 53BP1).
28
It is reported that overexpression of TIRR leads to 53BP1 reduction at the DSB site through competing with RIF1 to bind 53BP1.
28,29
Furthermore, the special complex of MRN along with BRCA1 could identify dsDNA and create a 15–20 bp nick from the 5′-ends of the DSB.
30
MRE11 is an Mn2+ dependent endonuclease that nicks the DNA upstream from the break site and involves in DNA DSB repair homologous recombination for maintenance of telomere (Fig. 2A, D).
31,32
SCAI is a related protein to induce separation of 53BP1-RIF1. Dramatically the relation of SCAI-RIF1 leads to some of the BRCA1 activity. Hence, a great number of HR-factors such as BRCA1, MRN, CtIP, and so on are inhibited by knocking out of SCAI.
29,33
A deubiquitinating enzyme (DUB), POH1, increases the removal of 53BP1 from the damaged locus by preparing interaction between RAP80 and the BRCA1 BRCT domain.
34
This enzyme is a part of proteasome that can neutralize RNF8/RNF168 (E3 ubiquitin ligases)-dependent ubiquitination activity.
29,34,35
As above mentioned, the non-canonical NHEJ pathway is extremely complicated and needs numerous biochemical factors. Interestingly, HDR and A-NHEJ have a similar initial steps. However, it is not elucidated how these two repair pathways are segregated later.
The initiation of HDR is continued by a two-step end processing. First, it starts with the MRN complex and the CtIP nucleases that bind to the DNA DSB and second, in order to create longer 3′-ends, the second stage for deep resection is occurred by EXO1 and SGS1-DNA2 nucleases.
36,37
The impermanent single-stranded DNA (ssDNA) overhang needs to be concealed by replication protein A (RPA) to be protected from exonuclease activities. Then, Rad51 replaces heterotrimeric RPA and for simplifying a quest for a homologous donor, generates a nucleoprotein presynaptic filament.
38,39
The 3′ DNA filament will be coated with many proteins, and then attacks homologous duplex DNA to develop the D-loop structure as an exchange intermediate. The D-loop structures are used for the synthesis of identical DNA sequences.
34,40,41
The second end joins the D-loop and starts the development of a double Holliday Junction (HJ) structure.
42
Indeed, 53BP1 and BRCA1- mediated mechanisms are complex systems that many factors and several antagonized points are involved in their inviolability. In this line, a plethora of targets exists for manipulating the efficacy of the HDR pathway.
Single-strand annealing (SSA) is an important subtype of HDR because of its specific mechanism. This pathway is observed when DSBs occur between two repeat sequences (Fig. 2E).
43
SSA in terms of homological loci is similar to MMEJ. Although the applied mechanism is the same for both of them, the involving proteins in SSA are similar to HDR.
40,41
Importance of small molecules in DNA repair pathways
Certain DNA repair pathways could be successfully activated or blocked by small-molecule compounds.
44
During the past decade, multiple studies have shown that small molecules are a straightforward strategy for increasing precision genome engineering.
7
Several advantages are mentioned for small molecules such as their high penetrant effects that lead to a rapid and controlled response. In addition, easy titration of small molecules provides optimal concentrations of inhibitors for delivering to the cell with extremely successful consequences.
45
The pharmacological approach to obtain a functional small-molecule usually contains: i) screening a library of chemical compounds to recognize lead scaffolds; ii) examining substitution places of the small molecule in terms of medicinal chemistry because replacements may result in modifications in specificity or sensitivity; and iii) deriving additional formatives for optimizing the efficacy of the small molecule. Generally, pharmacological procedures have been successful for recognizing various classes of potent inhibitor proteins such as proteases, nuclear hormone receptors, kinases, channels, and G protein-coupled receptors. Several efforts have been established to chemically scale up the efficiency of HDR pathway.
46
The nuclear domain knock-in screening that is described by multiple studies develops an idea that indicting simple means of quickly appraising small molecules that can elevate the efficiency of HDR is mediated by CRISPR-Cas9 technology.
7,45
For example, Yu et al applied a high-throughput chemical screening test based on the reporter system for exploiting HDR efficiency. It was reported that using the candidate small molecules could increased the efficiency of HDR for large fragments and point mutations 3- and 9-fold, respectively. L755507 and Brefeldin A were two small molecules whose positive effects were elucidated in this high-throughput chemical screening test. Moreover, many small molecules that inhibit HDR and could elevate indel mutations mediated by NHEJ were also reported.
7
Therefore, research to screen other small molecule libraries has been going on and introduced a strategy that simplifies precise CRISPR-Cas gene editing for clinical applications and biomedical experiments.
Inhibition of key NHEJ factors
Recently numerous small molecules have been validated to enhance the efficiency of HDR, which is mediated by CRISPR-Cas9 in various cells.
46
Several studies have shown that DSBs, introduced by CRISPR-Cas9 system, are mostly repaired by NHEJ. Hence, it seems reasonable, by inhibiting the key enzymes of NHEJ, the HDR efficiency would be increased. A comprehensive list of small molecules with NHEJ inhibitory effects is summarized in Table 1.
Table 1.
Small molecules involved in inhibition of NHEJ and stimulation of HDR
|
Targeted protein
|
Small molecule
|
Cell type
|
Locus/Gene
|
Dose of substance
|
Study method
|
Observed effects
|
Ref.
|
|
Inhibiting NHEJ
|
| DNA-PK |
Nu7026 |
iPSC |
CALD1
KATNA1
SLITRK1
|
20 µM |
In vitro
|
1.5
2.6
2.5-fold increase in HDR
|
46
|
|
|
Nu7026 |
HEK293 |
HPRT |
20 µM |
In vitro
|
3.0-fold increase in HDR |
46
|
|
|
Nu7026 |
K562 |
HPRT |
20 µM |
In vitro
|
4.0-fold increase in HDR |
46
|
|
|
Nu7026 |
CD4+ T
|
HPRT |
20 µM |
In vitro
|
3.0-fold increase in HDR |
46
|
|
|
Nu7026 |
CD34+ progenitor cells
|
HPRT |
20 µM |
In vitro
|
1.7-fold increase in HDR |
46
|
|
|
Nu7026 |
HEK293 |
GFP |
30 µM |
In vitro
|
2.5 -fold increase in HDR |
47
|
|
|
Nu7026 |
HepG2-1.1merHBV |
HBV genotype D |
7.5 µM |
In vitro
|
Increased A-NHEJ |
48
|
|
|
NU7026 |
K562 |
GFP |
3 µM |
In vitro
|
Modest increase in HDR (1.1-fold) |
49
|
|
|
NU7441 |
K562 |
GFP |
3 µM |
In vitro
|
2.4-fold increase in HDR (8.6% to 21.5%) |
49
|
|
|
NU7441 |
HSPC |
PTPRC |
3 µM |
In vitro
|
2-fold increase in HDR (12 to 24%) |
49
|
|
|
NU7441 |
iPSCs |
CTNNB1 |
2 µM |
In vivo
|
Modest increase in HDR 1.2(16% vs. 13% in control) |
50
|
|
|
NU7441 |
HEK293/ TRL |
GFP |
2 µM |
In vitro
|
2-fold increase in HDR |
51
|
|
|
NU7441 |
MEFs |
TP53 |
2 µM |
In vitro
|
10- fold increase in HDR |
51
|
|
|
KU-0060648 |
HEK293/ TRL |
GFP |
250 nM |
In vitro
|
2.1-fold increase in HDR |
51
|
|
|
M3814 |
409B2 hiPSC1
|
FRMD7 |
2 µM |
In vitro
|
Increased in HDR (18% to 81% ) |
52
|
| Ku complex |
STL127705 |
SF-767 cells |
|
2.5 µM |
In vitro
|
Not tested |
53
|
| Ku complex |
STL127705 |
PrEC cells |
|
2.5 µM |
In vitro
|
Not tested |
53
|
|
ATR2
|
VE-822 |
hiPSC |
OCT4
ALBUMIN
|
1 µM |
In vitro
|
4-fold increase in HDR by CRISPR-Cas9 and CRISPR-Cpf1- mediated targeting
3.5-fold increase in HDR by CRISPR-Cpf1 5-fold increase by CRISPR-Cas-mediated targeting
|
54
|
| ATM |
Trichostatin A |
iPSCs |
CALD1
KATNA1
SLITRK1
|
0.01 µM |
In vitro
|
1.5
2.2
1.8-fold increase in HDR
|
46
|
| CRISPY mix |
Trichostatin A
Nu7026
|
iPSCs |
CALD1
KATNA1
SLITRK1
|
0.01 µM
20µM
|
In vitro
|
1.8
2.5
3.1-fold increase in HDR
|
46
|
| Ligation |
SCR-7 |
HCT-116 cells |
AAVS1 |
10 µM |
In vitro
In vivo
|
1.7-fold increase in β-catenin gene (14.6% vs. 8.4% in control %) |
55
|
|
|
SCR-7 |
Mouse embryos |
lgkc |
1000 µM |
In vitro
|
4.5- fold increase in HDR (22.7% vs. 5% in control) |
8
|
|
|
SCR-7 |
Mouse embryos |
Kell |
1000 µM |
In vitro
|
2.2-fold increase in HDR (59.3 % vs. 26.6% in control) |
8
|
|
|
SCR-7 |
CHO cells |
COSMC FUT8 |
0.1–20 µM + 10 mM LiCl |
In vitro
|
None |
56
|
|
|
SCR7 |
iPSCs |
CALD1
KATNA1
SLITRK1
|
1 µM |
In vitro
|
None |
46
|
|
|
SCR-7 |
Mouse ESCs3
|
Actb |
1 µM |
In vitro
|
Promoted Tild-CRISPR-mediated knock-in |
57
|
|
|
SCR-7 |
HEK293T cells |
MALAT1 |
1-10 µM |
In vitro
|
Modest fold increase in HDR (13.6% vs. 12.5 in control %) |
58
|
|
|
SCR-7 |
Murine zygotes |
Tex15 |
50 µM |
In vivo
|
9.7-fold increase in HDR (56.2% vs. 5.8% in control) |
59
|
|
|
SCR-7 |
HEK293A |
LMNA |
1μM |
In vitro
|
Modest increase in HDR (11.7% vs. 9.9% in control) |
45
|
|
|
SCR-7 |
Porcine fetal fibroblasts |
GFP |
200,50 µM |
In vitro
|
2-fold increase in HDR (11.2% vs. 5.6% in control) |
60
|
|
|
SCR-7 |
Porcine fetal fibroblasts |
ROSA26 |
100 µM |
In vitro
|
1.9-fold increase in HDR with neomycin selection (49.7% vs.26.2% in control) |
60
|
|
|
SCR-7 |
Zebrafish embryos |
Ybx1S82A
|
20µM |
In vivo
|
3.6 -fold increase in HDR (55% vs. 15% in control ) |
61
|
|
|
SCR-7 |
COS-7 cells |
PAH |
15 µM |
In vitro
|
2.5-fold increase in HDR (22.1% vs. 8.8% in control ) |
62
|
|
|
SCR-7 |
HEK293T cells |
GFP |
1 µM |
In vitro
|
1.8- fold increase in HDR |
51
|
| CRISPY mix |
SCR-7
KU-0060648
|
HEK293T cells |
GFP |
1 µM
250 µM
|
In vitro
|
2.9- fold increase in HDR |
51
|
| CRISPY mix |
SCR-7
NU7441
|
HEK293T cells |
GFP |
1 µM
2 µM
|
In vitro
|
3- fold increase in HDR |
51
|
|
Stimulating HDR
|
| RAD51 |
RS-1 |
K562 |
GFP |
3 µM |
In vitro
|
2.2-fold increase in HDR (17.6% vs. 8.6% in control) |
49
|
|
|
RS-1 |
Zebrafish embryos |
eBFP2 |
30 µM |
In vivo
|
1.5-fold increase in HDR by Cas9-mediated |
63
|
|
|
RS-1 |
U2OS |
LMNA |
10 µM |
In vitro
|
Modest increase in HDR (2.5% vs. 1.9 in control %) |
45
|
|
|
RS-1 |
HEK293 A cells |
LMNA |
10 µM |
In vitro
|
6-fold increase in HDR
(21% vs. 3.5 in control %)
|
45
|
|
|
RS-1 |
HEK293 A cells |
PML |
10 µM |
In vitro
|
4- fold increase in HDR(40% vs. 10% in control %) |
45
|
|
|
RS-1 |
Zebrafish embryos |
Ybx1S82A
|
20 µM |
In vivo
|
1.6- fold increase in HDR (24% vs. 15% in control %) |
61
|
|
|
RS-1 |
iPSCs |
CALD1
KATNA1
SLITRK1
|
1 µM |
In vitro
|
None |
46
|
|
|
RS-1 |
iPSCs |
CTNNB1
PRDM14
|
10 µM |
In vivo
|
None |
50
|
|
|
RS-1 |
Bovine zygotes |
XbaI |
7.5 µM |
In vitro
|
2.1-fold increase in HDR (53% vs. 25% in control %) |
64
|
1 Human iPS cells; 2 ATM and Rad3-related; 3Embryonic stem cells.
DNA-PK
The first component of NHEJ for recognizing and binding to DSBs is DNA-PK. This holoenzyme is a compound of a 460 kD catalytic subunit, DNA-PKcs, Ku70, and Ku80 subunits (regulatory heterodimer). It has been revealed that recruiting libraries of these compounds could develop several molecules for inhibiting DNA-PK activity. Since NHEJ repair relies on DNA-PK activity, detrimental genetic mutations or using small molecules with inhibitory effects on DNA-PK could result in an increase in HDR occurrence. Targeting the ATP binding site of DNA-PK by small molecules is the most successful procedure for inhibiting this pivotal factor in NHEJ. For the phosphate transfer reaction by irreversible alkylation, lysine 802 in DNA-PKcs active site could be targeted by Wortmannin which is a small molecule with an inhibitory effect on PI3 kinases. Wortmannin naturally originated from a furanosteroid metabolite from Penicillium funiculosum. Although the experimental efficacy of wortmannin has been proven, multiple obstacles such as poor solubility, lack of specificity, and in vivo toxicity restrict its clinical application.
65
LY294002 is a competitive inhibitor of DNA-PK and PI3 kinase. It is reversibly able to interact with the kinase domain. This small molecule is originated from the plant flavonoid as a morpholine derivative. LY294002 leads to a delay in DSB repair, which might attribute largely to inhibition of DNA-PK. Because of fast-metabolic clearance and in vivo toxicity, it is impossible to clinically evaluate the effects of LY294002. However, LY294002 has been demonstrated to be a productive and leading compound which by acquiring biochemical alterations, a series of recombinant compounds with more appropriate features would be established. A modified form of LY294002 is NU7026 which is more selective for DNA-PK compared to other PI3 kinases such as ATM and ATR that had an IC50 of 13µM for PI3K, 0.23µM for DNA-PK, and >100µM for ATR or ATM. Nonetheless, for obtaining potent and selective DNA-PK inhibitors, the substitution of 2-morpholin-4-yl and alteration of thiopyran-4-ones or pyran-4-ones is proposed.
66
NU7441 is another agent that originates from LY294002 with remarkably improved potency compared to NU7026. It strongly inhibits DNA-PK with an IC50 of 0.3 µM in cell lines. Tavecchio et al reported that in the presence of ionizing radiation (IR) and NU7026, the induced DSBs continued for a long time and the activeness of HR enhanced moderately.
67
Development of NU7441 continues and leads to the identification of KU-0060648, which has greater solubility against DNA-PK and is a binary inhibitor of PI-3K and DNA-PK in vitro. Munck et al showed that the inhibitory effect of KU-0060648 on DNA-PK in MCF7 is approximately 8-fold higher than in SW620 cells.
68
Other non-toxic compounds based on the LY294002 structure, such as IC86621, IC87102, and IC87361 have expanded the application of compounds derived from this small molecul.
66
NU7026, NU7441, and KU-0060648 provide high efficiency for improving the HDR rate in genome editing compared to other DNA-PK inhibitors. Previously it was demonstrated that the efficiency of knock-in is augmented by NU7026 in hiPSCs. Likewise, this small molecule is a crucial complex in the NHEJ pathway. Riesenberg et al reported that NU7026 was the only small molecule that clearly increased the efficiency of targeted gene fragment insertion in HEK293 (by 3-fold), K562 cells (by 4-fold), CD4+ T cell (by 3-fold), and CD34+ progenitor cells (by 1.7-fold).
46
Besides, Robert et al described that treating HEK293 TLR with NU7441 and KU-0060648 leads to, respectively, a 3- and 4-fold increase in HDR efficiency, and an approximately 2-fold reduction in the NHEJ repair. They also demonstrated that oligonucleotide-mediated HDR, as a repair template, at the endogenous site could be stimulated by both NU7441 and KU-0060648. Therefore the results of using additional DNA-PK inhibitors lead to compatibility with the Cas9 editing system.
51
Moreover, combining proteins or siRNA along with small molecules could be more effective. They showed that Adenovirus 4 (Ad4), E1B55K and E4orf6 proteins with KU-0060648 or NU7441 could induce HDR approximately up to 5-fold in HEK293T cells.
51
Aksoy et al reported the initial application of NU7441 as a powerful HDR enhancer with an increase in HDR efficiency up to 13.4-fold in zebrafish embryos genome-edited by CRISPR-Cas9 system.
63
Recently, M3814 was introduced as a new selective pharmacological inhibitor of DNA-PK which has not been applied in genome editing before. M3814 is a highly potent molecule that showed acceptable activity in preclinical models. This molecule was also introduced as a practical therapeutic strategy in cancer treatment.
69
For instance, in combination with cisplatin and etoposide, M3814 has represented promising activity in lung cancer in vivo models.
70
On the other hand, Sun et al showed that the repair of radiation-induced DSBs could fruitfully be shut off by M3814. Furthermore, this small molecule can efficiently increase activation and phosphorylation of p53.
71
Additionally, M3814 was used in phase Ib/II clinical trials for the treatment of several cancers such as rectal cancer (NCT03770689) and small cell lung cancer (NCT03116971). Riesenberg et al used M3814 in genome editing for the first time. It was used to transiently inhibit NHEJ and increase HDR from 18% to 81% in K562 cells while demonstrating moderate toxicity.
52
In line with these premises, M3814 could be applied in gene therapy, where high HDR performances may be required to achieve therapeutic goals. VE-822, a specific inhibitor of ATR, that was recently used by Ma et al enhances the efficiency of HDR efficiency in combination with a plasmid donor and a ssODN donor by 5.9-fold and 3-fold, respectively. Furthermore, combining AZD-7762, a specific inhibitor of checkpoint kinase CHEK1 and the ATR inhibitor could remarkably boost the specificity of CRISPR-Cas9 genome editing.
54
Ku70/80 complex
The heterodimeric Ku complex is the most logical choice for inhibition of the entire NHEJ process. This protein shows a great binding affinity for dsDNA termini and has a ring-like shape which upon binding DNA, cannot tolerate any substantial structural variation. This ring-like structure leads to the interaction of Ku protein with DNA that is crucial for the activation of kinase and in this regard, it is also an appropriate target for intervention.
65
Although in the initiation of the NHEJ pathway, Ku has a key role, limited studies have addressed this protein and only one inhibitor of Ku protein was obtained (STL127705).
44
For the first time, Weterings et al in the low micro-molar range, confirmed the compound with Ku-inhibitory activity. However, the ability of this molecule to block NHEJ is not well documented.
53
Recently, Gavande et al represented novel small molecule inhibitors that bind to the Ku–DNA protein to block the protein–DNA interaction. These specific Ku–DNA binding inhibitors (Ku-DBi’s) block Ku-DNA interaction, the activity of DNA-PK kinase, and in vitro NHEJ by directly binding to Ku protein. Moreover, Ku-DBi’s increase the cellular activity of radiomimetic agents and IR.
72
DNA-end processing enzymes
The DNA-end processing step in NHEJ is an attractive issue for studying and various nucleic acid enzymes involved in this step are recognized. However, no remarkable effort has been made to validate these enzymes as potential targets. Because of the hardness in purifying the proteins and the requirements for complex assay, no inhibiting agent has been identified for Artemis as DNA-PK dependent endonuclease. Polynucleotide kinase/phosphatase (PNKP) can bind to DNA 5'-end and dephosphorylate DNA 3'-end in the NHEJ pathway. Furthermore, PNKP is essential for both single- and DSB repairs. Therefore identifying small molecule inhibitors for this enzyme seems very useful to regulate NHEJ.
65
Five compounds with remarkable inhibitory effect on PNKP phosphatase activity was recognized by Freschauf et al.
73
A12B4C3 is one of these compounds with IC50 value of 0.06 Mmol/L, showing the highest noncompetitive inhibitory effect on phosphatase activity of PNKP by obstructing its secondary structure.
73
There is no more information about the effect of this inhibitor on NHEJ pathway. Therefore, identifying and introducing additional small molecules for inhibition of DNA-end processing enzyme is required.
Ligation process enzymes
The ligation process of NHEJ particularly the DNA ligase enzyme is an attractive target for regulating NHEJ. L189 was the first recognized compound by Chen et al for inhibiting the DNA ligase.
74
Although this molecule had a poor specificity, it represented a promising inhibitory effect on Ligase I, III, and IV.
74
One of the L189 derivatives, SCR7, was synthesized as a more specific and putative inhibitor of NHEJ. This compound blocks end-joining through intervening with the connection of Ligase IV to DNA, consequently, the accumulation of DSBs within the cells and increasing cytotoxicity were accrued.
75
Most investigations showed a considerable dose-dependent decline of NHEJ in various models, both in vitro and in vivo. Some groups demonstrated that a modest increasing efficiency of HDR-mediated gene insertion could be achieved by combining SCR7 with enhancing genome engineering techniques such as synchronization or optimized Cas9 delivery.
76,77
The SCR7 effects in CRISPR-Cas9 experiments and subsequently the increased HDR rate was demonstrated in various cells such as HEK293T (by 2-fold),
51
A549 (by 3-fold), MelJuSo (by 19-fold),
8
and HEK293 (by 5-fold).
77
However, complete inefficiencies of Scr7 or slightly increasing the HDR rate were reported in cells such as human embryonic stem cells,
78
iPSCs,
50
Porcine fetal fibroblasts,
79
rabbit embryos,
80
HEK293A,
45
and CHO cells.
56
Srivastava et al demonstrated that the reason for these inconsistency effects of SCR7 is related to different expressions of LigIV in various cell cultures, and the cells with high expression level of LigIV are more sensitive to SCR7.
40,75
Contrary to the results for SCR7 reported by Raghavan et al, others discovered several differences in the original structure of SCR7.
75,81
Greco et al performed a broad investigation on structural determination of SCR7 and confirmed the structure of SCR7-R and its closely related derivative, SCR7-G, that is generated by the synthesis protocol described by Raghavan (Fig. 3).
81
They found that commercially available SCR7 (SCR7-X) structurally is similar to SCR7-G. Indeed, both SCR7-G and SCR7-R have weak inhibitory effects on LigIV while providing stronger activity with regard to LigI and LigIII/XRCC1. Hence, they suggested that the effect of increasing HDR by SCR7, particularly at a low concentration (about <200 uM) is occurred by other mechanisms.
81
Despite these differences, it is confirmed that at the defined concentration of SCR7-X/SCR7-G (1 µM), the efficiency of CRISPR-Cas9 gene editing could be enhanced.
75,81
By focusing on embryos, it was observed that SCR7 increases HDR efficiency in the mouse embryos,
8,82
but not in the rabbit embryos.
80
Likewise, Singh et al reported the increased rate of HDR (by 10-fold) in murine zygotes
82
and Maruyama et al also reported correcting homozygous editing in addition to increased HDR efficiency.
8
In order to improve the efficiency of SCR7, combining this small molecule with other NHEJ inhibitors is a logical suggestion. Chu et al showed that using Scr7 simultaneously with XRCC6 and LIG4 knock-out models resulted in 5-fold increase in HDR efficiency,
77
while others have not observed such increasing effects.
83
Moreover, it was determined that a combination of SCR7 with KU-0060648 or NU7441 in HEK293 cell (CRISPY mix) could stimulate HDR efficiency by 3-fold.
51
In general, inhibiting the key factors of NHEJ causes the unrepaired DSB accumulation in the cell, as a result, apoptosis and cell death would be increased. In this line, this approach could increase cytotoxicity and should be applied with precaution.

Fig. 3.
Different structures of the Ligase IV inhibitor, SCR7.
(A) L189 as a human DNA ligase inhibitor. (B) Less stable parental SCR7 reported by Srivastava et al can get autocyclized into a more stable form (SCR7-R). (C) Having the same molecular weight, number of protons, molecular mass, and melting point. Pending dehydrogenation, SCR7-R gets converted into SCR7-G (D), the compound sold as SCR7 by XcessBio (SCR7-X), possessing a different molecular weight, number of protons, molecular mass, and melting point.
.
Different structures of the Ligase IV inhibitor, SCR7.
(A) L189 as a human DNA ligase inhibitor. (B) Less stable parental SCR7 reported by Srivastava et al can get autocyclized into a more stable form (SCR7-R). (C) Having the same molecular weight, number of protons, molecular mass, and melting point. Pending dehydrogenation, SCR7-R gets converted into SCR7-G (D), the compound sold as SCR7 by XcessBio (SCR7-X), possessing a different molecular weight, number of protons, molecular mass, and melting point.
Supporting HDR key factors
HDR is a substantial pathway for certifying the precise repair of nicks and DSBs. Therefore, another way for changing DNA repair from NHEJ to HDR is instigation of HDR key components.
84
A comprehensive list of small molecules, which are involved in stimulation of HDR, is represented in Table 1.
Rad51, a factor for exchanging DNA strands, is the central and target point for HDR, and by the accumulation of other proteins on ssDNA, it forms presynaptic filaments.
85
Indeed, it has an extensive role in the formation of a RAD51–ssDNA filament by replacing RPA, and interactingwith ssDNA. In order to enhance RAD51 binding to DNA, Jayathilaka et al developed a high-throughput microplate-based assay for evaluation of the filament formation of hRAD51 on ssDNA.
86
By applying this approach, a library with 10,000 compounds was screened that resulted in the recognition of a small molecule called RAD51-stimulatory compound 1 (RS-1).
87
In different biochemical conditions, RAD51 binding could be enhanced by RS-1. After the ultrastructural assay of developed filaments on ssDNA, it was determined that RS-1 could increase protein–DNA complex lengths and the pitch of helical filament turns. Moreover, the experiment of RS-1 by salt titration demonstrated increasing filament stability. According to experiments, RS-1 depending on the condition can stimulate the activity of RAD51-mediated homologous strand (D-loop) at least 5- to 11-fold.
86
Actually, the organization of active presynaptic filaments is boosted by RS-1 that can increase the homologous recombination activity of RAD51 by about 2-fold.
86
Therefore, Jayathilaka et al showed that RS-1 could motivate the performance of homologous recombination repair; thereby, it is functional in both medical and research settings.
86
In research, RS-1 in HEK293A could increase the HDR rate up to 6-fold and 4-fold by using Cas9 and Cas9 nickase, respectively.
88
Song et al reported impressive results in both in vivo and in vitro. They indicated that RS-1 enhanced the HDR rate by 6-fold for the ROSA26-like locus (RLL) in vitro and by 2.4-fold for the CFTR gene in rabbit embryos.
80
Combinatorial treatment using RS-1 and NU7441 in zebrafish embryos
63
or applying both SCR7 and RS-1 with/without L755507 in HEK293 cells did not show a significant increase in HDR efficiency compared to RS1 alone.
45
Similar results were also observed when L755507, SCR7, and RS-1 were used together in stem cells.
46
However, in one report it was revealed that the combination of SCR7 and RS1 increased HDR efficiency up to 74% in zebrafish embryos.
61
Unfortunately, contradictory outcomes have been achieved for both small drugs, SCR7 and RS-1, in various cell types. In this regard, the highest efficiency of RS1 was observed in bovine zygotes.
64
In addition to RS-1, MLN4924 is another small molecule for stimulating HDR. It is a Nedd8 activating enzyme (NAE) inhibitor that can block the neddylation of CtIP and result in a rise in the extent of DNA end cutting at strand breaks. MLN4924 through inhibition of neddylation artificially increases the CtIP expression level which in turn promotes HDR efficiency.
46
Although the effect of some small molecules was unknown or was observed in different stages of DNA repair (multiple effects), they could provide increasing HDR efficiency. A comprehensive list of small molecules with multiple or unknown effects is summarized in Table 2.
Table 2.
Small molecules with multiple or unknown effects
|
Targeted point
|
Small molecule
|
Cell type
|
Locus/ Gene
|
Dose of substance
|
Study method
|
Observed effects
|
Ref.
|
| β3-adrenergic receptor agonist |
L755507 |
K562 |
GFP |
3 µM |
In vitro
|
1.5-fold increase in HDR |
49
|
|
|
L755507 |
HeLa |
ACTA2 |
5 µM |
In vitro
|
1.5-fold increase in HDR (5.1% vs. 3.5% in control) |
7
|
|
|
L755507 |
hiPSCs |
SOD1 |
5 µM |
In vitro
|
8.9-fold increase in HDR (3.13% vs. 0.35% in control) |
7
|
|
|
L755507 |
Mouse ESCs |
NANOG |
5 µM |
In vitro
|
1.9-fold fold increase in HDR (33.3% vs. 17.7% in control) |
7
|
|
|
L755507 |
HUVEC |
ACTA2 |
5 µM |
In vitro
|
2-fold increase in HDR (1.8% vs. 0.9% in control) |
7
|
|
|
L755507 |
K562 |
ACTA2 |
5 µM |
In vitro
|
1.6-fold increase in HDR (1.3% vs. 0.8% in control) |
7
|
|
|
L755507 |
Fibroblast CRL-2097 |
ACTA2 |
5 µM |
In vitro
|
1.7-fold increase in HDR (6.0% vs. 3.5% in control) |
7
|
|
|
L755507 |
Human ES cell-derived (neural stem cells) |
ACTA2 |
5 µM |
In vitro
|
1.3-fold increase in HDR (1.0% vs. 0.8% in control) |
7
|
|
|
L755507 |
iPSCs |
CTNNB1
PRDM14
|
5 µM |
In vivo
|
None |
50
|
|
|
L755507 |
Porcine fetal fibroblasts |
GFP |
40μM |
In vitro
|
1.9- fold increase in HDR (10.9% vs. 5.6% in control) |
60
|
|
|
L755507 |
Porcine fetal fibroblasts |
ROSA26 |
40μM |
In vitro
|
2- fold increase in HDR with neomycin selection (51.6% vs. 26.2% in control) |
60
|
|
|
L755507 |
Porcine fetal fibroblasts |
GFP |
20μM |
In vitro
|
1.6 fold increase in HDR (9% vs. 5.6% in control) |
60
|
|
|
L755507 |
HEK293A |
LMNA |
5 µM |
In vitro
|
Modest increase in HDR
(10.1% vs. 9.9% in control)
|
45
|
|
|
L755507 +RS-1 |
HEK293A |
LMNA |
5 µM + 10μM |
In vitro
|
1.7 fold increase in HDR (17.1% vs. 9.9% in control) |
45
|
|
|
L755507 +RS-1+SCR-7 |
HEK293A |
LMNA |
5 µM + 10μM+1μM |
In vitro
|
1.4 fold increase in HDR ( 14% vs. 9.9% in control) |
45
|
Inhibition of intracellular
transport from ER to Golgi
|
Brefeldin A |
Mouse ES cells |
Nanog |
0.1 µM |
In vitro
|
2-fold increase in HDR (27.2% vs. 17.7% in control) |
7
|
|
|
Brefeldin A |
iPSCs |
CTNNB1 |
0.1 µM |
In vivo
|
1.3-fold increase in HDR (17% vs. 13% in control) |
50
|
|
|
Brefeldin A |
iPSCs |
PRDM14 |
0.1 µM |
In vivo
|
Modest increase in HDR
(8.5% vs. 7% in control)
|
50
|
| Broad range of biological activities |
Resveratrol |
Porcine fetal fibroblasts |
GFP |
50μM |
In vitro
|
2.7-fold increase in HDR (15% vs. 5.6% in control) |
60
|
|
|
Resveratrol |
Porcine fetal fibroblasts |
GFP |
200μM |
In vitro
|
2.3-fold increase in HDR (13% vs. 5.6% in control) |
60
|
|
|
Resveratrol |
Porcine fetal fibroblasts |
ROSA26 |
50μM |
In vitro
|
1.8-fold increase in HDR with neomycin selection (46%
vs. 26.2% in control)
|
60
|
|
|
Resveratrol |
iPSCs |
CALD1
KATNA1
SLITRK1
|
1 µM |
In vitro
|
None |
46
|
Synchronizing cell-cycle
HDR and NHEJ are dominant repair pathways at different phases of cell-cycle. Synchronization of cells is reversible and provides to be widely used while it does not affect the pluripotency of stem cells. One approach for increasing HDR efficiency in CRISPR-Cas9 genome-edited cells is using small molecules for cell-cycle synchronization. In this line, various small molecules have been introduced for arresting cell cycle at different phases. For example,Lovastatin hampers cell cycle at early G1 and partly at G2/M phase; L-mimosine, Hydroxyurea (HU), Aphidicolin, Dideoxycytidine (ddC), and Thymidine could apprehend cells at G1-S frontier before starting the DNA replication; and antineoplastic microtubule polymerization inhibitor (Nocodazole), Indirubin-3-monoxime, Vinblastine, and ABT block cell cycle at G2/M phase.Since HDR is restricted to the S and G2 phases, synchronizing the cell cycle in these two stages might be beneficial for increasing the HDR rate. Some small molecules arrest cells at the G1-S border before the onset of DNA replication by inhibiting the effective enzyme of A-NHEJ pathways. Some molecules arrest cells at the G2/M phase, thereby providing modulation of HDR-mediated Cas9 genome-editing through cell cycle synchronization.
Arrest at the G1-S border
Aphidicolin is a reversible chemical inhibitor that inhibits the DNA polymerase-α, δ, and blocks cell cycle in S phase.
89
Several reports have estimated that Aphidicolin increases HDR rate in HCT116 cells, embryonic stem cells (ESCs), and primary neonatal fibroblasts by 3-fold, 1.6-fold, and 1.3-fold, respectively.
90,91
Furthermore, the frequency of HDR was enhanced with thymidine treatment in H9 human embryonic stem cells (hES) and human primary neonatal fibroblasts (neoFB).
91
It is determined that HU arrests cell cycle in S-phase by inhibiting ribonucleotide reductase enzyme. Tsakraklides et al reported that cells, by using HU before transformation process in five yeast strains, were synchronized in S-phase and the efficiency of gene targeting was increased.
92
2′,3′-dideoxycytidine is another promising small molecule that slows down the replication fork movement and results in S-phase extension. Brachman et al reported that this small molecule develops the S phase up to 70% and increases HDR rate by 3-fold in DLD-1 cells.
93
Recently, XL413, a new small molecule targeting cell division cycle 7(CDC7) plays an important role during initiation of DNA replication, arrests primary T cells in S phase, and enhances the HDR efficiency up to 3.5-fold.
94
According to these observations, designing experiments for identifying the most optimal small molecules is necessary because different small molecules that intervene in cell synchronization have varied effects on various cells.
Arrest at G2/M
G2/M phase is another crucial hot spot in cell cycle for being regulated by small molecules to increase HDR efficiency. Lin et al reported that Nocodazole is able to arrest cell cycle at specific phases and increase HDR rate with CRISPR-Cas9 by 1.4-6-fold in HEK293T cells.
91
By applying Nocodazole or ABT-751, Yang et al could successfully enhance HDR-mediated knock-ins by 3- to 6-fold.
78
The treatment with Nocodazole reverses the synchronization of cell cycle in different cell models such as iPSCs and human pluripotent stem cells (hPSCs),
50,78,91
albeit not observed in ESCs and primary neonatal fibroblasts.
91
Combinatorial treatment with CCND1, a cell cycle regulatory subunit in G1/S transition, and Nocodazole increased the HDR efficiency up to 30% in iPSCs.
50
Vinblastine is another useful small molecule for cell cycle synchronization in the G2/M phases through binding to tubulins and blocking microtubule dynamics.
95
It is elucidated that Vinblastine increases HDR rate by 6- to 10-fold in HeLa, U-2OS, and HT-1080 cell lines. Likewise, Indirubins by inhibition of several cyclin-dependent kinases, leads to cell-cycle prevention in G1/S or G2/M in different cell models.
96
Rahman et al reported that by utilizing meganuclease I-SceI and ZFNs together with indirubin-3′-monoxime, the HDR rate increased by 2- to 5-fold in Hela, U-2OS, and HT-1080 cell lines. Incredibly, indirubin-3′-monoxime results in an enhancement in HDR by 10-fold in primary umbilical cord–derived mesenchymal stromal cells (UC-MSCs). Finally, it was concluded that indirubin-3′-monoxime is a promising small molecule for enhancing adeno-associated virus/ZFN-mediated gene editing especially in UC-MSCs for clinical approaches.
97
The effective strategy of modified CRISPR-Cas Systems for boosting HDR activity
HDR pathway leads to the precise insertion of the donor template at the DSB site in the presence of a homologous donor strand. Induction of the HDR pathway for repairing DSBs which is introduced by CRISPR-Cas9 is a suitable approach for increasing the accuracy of knocking in approaches. This procedure provides plenty of technical capabilities for researchers and industries such as developing insertions or deletions models, inserting sequences for epitope tags, generating SNPs, and inserting whole genes into target site to produce modified organisms. Although the HDR pathway along with CRISPR-Cas9 is an easy way to introduce a site-specific rectification, HDR activity is very low that has a negative and direct effect on precision genome engineering functions.
98,99
Enhancing the efficiency of HDR provides fast, easy, and accurate technologies based on CRISPR-Cas9.
44,100
In this regard, engineering strategies are required for inducing the HDR pathway for accurate gene modification.
HDR-fusion Cas9
According to previous findings, each of NHEJ and HDR pathways is dominated in different cell-cycle phases separately.
40,101
In this line, precisely activating Cas9 in S and G2 phases results in an increased HDR rate. The main studied fusion motifs include CtIP, Rad52, DN1S, mSA, and Geminin. These engineered variants usually enhance knock-in occurrence in both in vivo and in vitro (Fig. 4). It is reported that by fusing a N-terminal fragment of CtIP, which is a key protein in HDR initiation, to Cas9 protein, the CtIP was forced to DSB site that increasing the efficiency of HDR by 2-folds. Moreover, when comparing the patterns of indels introduced by wild-type Cas9 and engineered Cas9, it was speculated that existence of different patterns could originate from a different balance of DSB repair pathways.
102
According to this observation, recently Tran et al designed an experiment to establish Cas9 fusion with other factors which are involved in HDR pathway. They reported that similar to CtIP, fusing Cas9 to Mre11 and Rad52 but not Rad51C, could scale up the efficiency of HDR by 2-fold. Moreover, it was indicated that fusing CtIP to gRNA through MS2 binding loops resembling Cas9-CtIP variant is able to extend the ratio of HDR/NHEJ approximately by 6-folds (Fig. 4A). Remarkably, when simultaneously using Cas9-CtIP with MS2-CtIP, the ratio of HDR/NHEJ boosted up to 14.9-fold.
103
However, there are several limitations in using these chimeric proteins broadly. For example, Mre11 and CtIP have complex post-translational regulation and the prediction of protein interactions is confusing. This phenomenon could not be profitable because it originates from endogenous cellular components.
104
53BP1 plays a key role in the initiation of NHEJ. In this line, by inhibiting this protein the efficiency of HDR would be increased (Fig. 4B). It is indicated that fusing Cas9 to a truncated piece of p53 named DN1S, a dominant-negative mutant of 53BP1, could significantly reduce NHEJ and increase the efficiency of HDR up to 86% especially at DSB sites introduced by CRISPR-Cas9 system in different human cell lines.
105
A challenging limitation for using chimeric Cas9 proteins for increasing HDR efficiency is their cellular toxicity. It is indicated that Cas9-DNS1, in reality, increases cellular toxicity by about 10%. Finally, it should be considered that these chimeric proteins would not be effective for increasing HDR ratio because none of these engineered proteins are involved in regulating long range 5’ to 3’ end resection.
106
To address these limitations, Hackley et al fused Cas9 to human Exo1 and reported that this chimeric protein compared to wild-type, Cas9 is able to increase the rate of HDR by 2.5-fold and reduce cellular toxicity about 4-fold (Fig. 4C).
107
Furthermore, miCas9 is the recently developed small-size HDR-fusion variant that increases HDR capacity. Fusion motif, Brex27 of the engineered variant interacts with RAD51 and enhances the chance of RAD51 presence at the target locus. MiCas9 increases the knock-in rates of large size genes which are mediated by dsDNA, consistently lessens insertion occurrences and off-target deletion, and keeps or enhances single-stranded oligodeoxynucleotide (ssODN)-mediated precise gene editing rates (Fig. 4D).
108
Anti-CRISPR proteins, which are naturally derived from bacteriophages, are promising inhibitory proteins for controlling the activity of CRISPR-Cas systems (Fig. 4E). Recently, Matsumoto et al observed that during phase S and phase G2 the fused human Cdt1 is degraded where HDR is dominant. When AcrIIA4, the natural anti-CRISPR protein, was fused to the N-terminus region of Cdt1, engineered CRISPR-Cas system was activated at the intended phases, S/G2, and DNA repair was boosted through HDR.
109

Fig. 4.
Accumulation of fusion Cas9 to improve the efficiency of the HDR pathway.
Generally, knock-in events are less frequent than template-free indels. (A) Accumulating CtIP via MS2 and association with special sgRNA loops of Cas9 increased knock-in ratio significantly. (B) Fusing Cas9 to DN1S, a truncated piece of p53 reduces NHEJ and increases the efficiency of HDR. (C) Fusing Cas9 to human Exo1 by inhibiting NHEJ increases the rate of HDR. (D) To improve the HDR capacity of Cas9, the miCas9 was created by adding thirty-six amino acids to spCas9. Adding Brex27 to miCas9 results in enrichment of edited region. (E) Activating specific Cas9 system by Anti-CRISPR AcrIIA4 by binding to Cas9-sgRNA acts as an inhibitor for SpyCas9 in G1 phase. In S/G2/M phases, degradation of Cdt1 (S phase degradation domain) leads to AcrIIA4 degradation and consequently, activates the SpyCas9-sgRNA complex. Figure was created with BiorRender (https://biorender.com).
.
Accumulation of fusion Cas9 to improve the efficiency of the HDR pathway.
Generally, knock-in events are less frequent than template-free indels. (A) Accumulating CtIP via MS2 and association with special sgRNA loops of Cas9 increased knock-in ratio significantly. (B) Fusing Cas9 to DN1S, a truncated piece of p53 reduces NHEJ and increases the efficiency of HDR. (C) Fusing Cas9 to human Exo1 by inhibiting NHEJ increases the rate of HDR. (D) To improve the HDR capacity of Cas9, the miCas9 was created by adding thirty-six amino acids to spCas9. Adding Brex27 to miCas9 results in enrichment of edited region. (E) Activating specific Cas9 system by Anti-CRISPR AcrIIA4 by binding to Cas9-sgRNA acts as an inhibitor for SpyCas9 in G1 phase. In S/G2/M phases, degradation of Cdt1 (S phase degradation domain) leads to AcrIIA4 degradation and consequently, activates the SpyCas9-sgRNA complex. Figure was created with BiorRender (https://biorender.com).
Modified sgRNA, Cas9 and/or donor template
For simplifying CRISPR approaches, enhancing the efficiency of HDR seems laborious. Another helpful application is employing the assembly of ssDNA template, sgRNA, and Cas9 along with donor DNA at the targeted site to a homologous sequence. These strategies are implemented to increase the ratio of HDR through bringing donor template close to the introduced DSB site by CRISPR-Cas9 system. Several reports have corroborated the simplicity of increasing the HDR efficiency by advancing the assembly of these components individually or in a complex.
In agrobacteria, Vir proteins are able to transfer T-DNA construct into plant cells. VirD2 relaxase is a component of Vir proteins that covalently binds to the 5’-site of single-stranded T-DNA and by using its nuclear localization signal, directs attached T-DNA into nucleus and integrates it in the plant genome. When VirD2 was fused to Cas9, this chimeric protein was able to recruit repair template in close proximity to the introduced DSB by Cas9 and increased HDR rate up to 6-fold more than repair with Cas9 alone (Fig. 5A). This finding opens up novel promising opportunities for precise plant genome editing.
110
Several attempts have been taken to recruit donor oligonucleotides to introduce DSB by Cas9. It is indicated that donor template could be fused into gRNA and by using this strategy HDR efficiency was increased three times.
56
Several studies also used the advantage of biotin-streptavidin interaction for directing donor template close proximity to Cas9- mediated DSB.
111,112
Moreover, the high-affinity biotin-streptavidin interaction was also used for increasing HDR rate at DSB induced by CRISPR-Cas9 ribonucleoproteins (RNPs). RNPs have shorter half-life compared to their plasmid counterparts, so by using these components the off-target effects would be reduced significantly. Recently, it is elucidated that by combining two cells homologous recombination CRISPR genome editing with a modified biotin–streptavidin approach, in a site-specific manner, the knock-in efficiency of any large fragment could increase up to 95% compared to standard methods in mouse embryos. Although the function of sgRNA may be interrupted by linking an aptamer, engineered Cas9 could be recruited to the biotinylated DNA template. Ma et al fused avidin to Cas9 in mouse embryos for enhancing the efficiency of accurate knock-in from 0 to 15-22%. On the other hand, it was indicated that when wild-type Cas9 is used all mutations contained indels, generated through NHEJ (Fig. 5B).
111
Furthermore, Gu et al reported the use of mSA fused to Cas9 for localizing donor DNAs to target sites. Fusing mSA, because of its monomer structure, to Cas9 demonstrates a biotin-binding domain to recruit repair templates without accumulating the nuclease (Fig. 5C). By incorporating reporter genes at 20 different loci in various cell types, this design was evaluated.
113
Several studies were developed that consistent with other observations have revealed the crucial role of donor localization to increase HDR pathway.
114-116
Another strategy for directing donor template to close proximity of Cas9-mediated DSB is to bind donor template to Cas9 through covalent linkage. In this line, covalent tethering was reported between RNP complex form of CRISPR-Cas9 and ssODN via a fusion of porcine circovirus 2 (PCV) target sequence with HUH endonuclease. The need to alter donor ssDNA or the sgRNA is resolved via HUH-tagged recombinant protein (Fig. 5D). It is estimated that this strategy augmented HDR efficiency up to 30-fold.
116
In addition, Savic et al reported that donor template can physically link to Cas9 enzyme through a SNAP-tag (Fig. 5E). They indicated that O6-benzylguanine (BG)-labeled DNA oligos are able to covalently bind to Cas9-SNAP chimeric proteins and the efficiency of HDR increased up to 24-fold.
115
There are several limitations for these introduced methods, such as i) there are technical and commercial difficulties for obtaining terminally modified long ssOND; and ii) the expression level of Cas9 or its delivery efficiency might be changed by fusing a functional domain or linker and also maybe a susceptible chimeric protein to protease cleavage.
In order to overcome these limitations, recently a novel innovative strategy known as genetic code–expansion technology was developed. This technology is established by using chemically modified Cas9 mutant including an azide-containing noncanonical amino acid which is able to recruit modified or unmodified repair template to the DSB site (Fig. 5F). Such modifications enable conjugation of alkyne-azide cycloaddition by dibenzylcyclooctyne (DBCO)-DNA adaptor or dibenzylcyclooctyne modified donor ssODN, which in turn facilitates recruitment of the repair template to the cleavage complex for the HDR pathway. A universal platform is developed by these Cas9 conjugates for using unmodified ssODNs which are commercially available at high purity and a low cost to the RNP complex by base pairing. This strategy increased the HDR efficiency up to 10-fold in both mouse zygotes and human cell culture.
117
As mentioned about the advantage of biotin-streptavidin interaction, this approach was only successful for knocking-in large fragments into a limited number of targeted loci. Two-cell stage has an exceptional long G2 phase in embryos. In addition, the chromatin state in this extended phase is open and provides a golden opportunity for editing enzymes and repair template to easily reach targeted sites. Recently, S1mplex, a modular RNA aptamer-streptavidin strategy (Fig. 6), was developed for delivering RNP form of purified CRISPR-Cas9 system together with an engineered sgRNA containing a streptavidin-binding aptamer into target cell for in vitro or ex vivo genome editing. It is reported that this system increased precise editing up to 18-fold and augmented pool cells including multiplexed precise edits by 42-fold.
112
Furthermore, new genetic information can help to increase HDR efficiency by utilizing timed delivery of CRISPR-RNP complexes along with various drugs which are arresting the cell cycle.
118
In addition, recently inhibiting main factors in NHEJ and stimulating proteins participating in HDR by using small molecules are highly regarded.
40

Fig. 5.
Variation methods for tethering the ssODN donor template to modified Cas9 for enhancing gene editing efficiency
. Modified Cas9 along with sgRNA were joined to ssODN donor template to enhance the rate of precise repair via the targeted HDR pathway (A-F). Figure was created with BiorRender (https://biorender.com).
.
Variation methods for tethering the ssODN donor template to modified Cas9 for enhancing gene editing efficiency
. Modified Cas9 along with sgRNA were joined to ssODN donor template to enhance the rate of precise repair via the targeted HDR pathway (A-F). Figure was created with BiorRender (https://biorender.com).
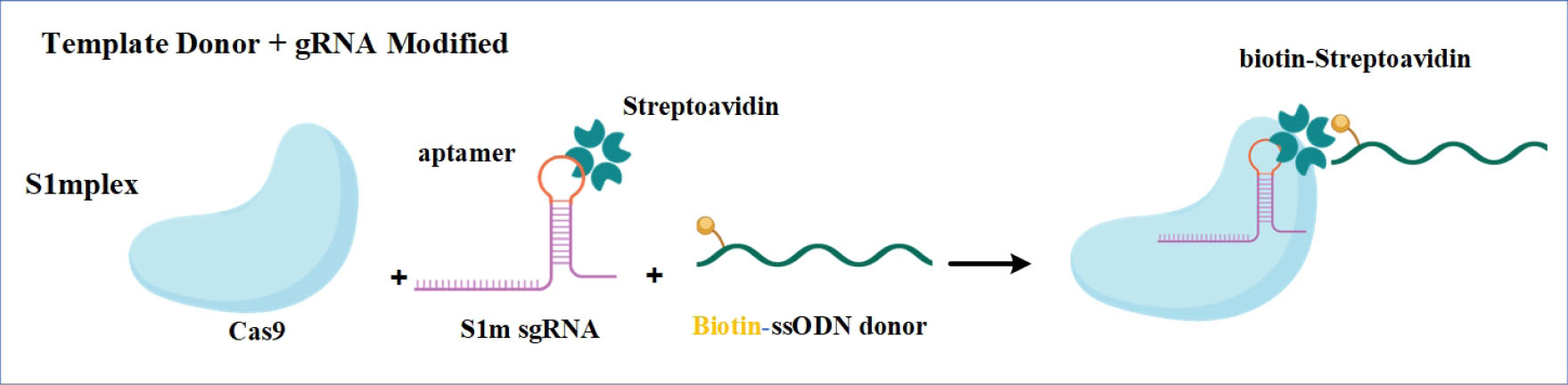
Fig. 6.
Engineered sgRNA which is harboring a streptavidin aptamer that contains streptavidin-binding aptamer was joined to a biotinylated ssODNA donor providing an efficient method of recruiting biotinylated DNA.
Figure was created with BioRender (https://biorender.com).
.
Engineered sgRNA which is harboring a streptavidin aptamer that contains streptavidin-binding aptamer was joined to a biotinylated ssODNA donor providing an efficient method of recruiting biotinylated DNA.
Figure was created with BioRender (https://biorender.com).
Small molecules impact on improving CRISPR-Cas-mediated gene or transcript editing
Over the past decade, several studies have been done on the effect of small molecules on improving the genome editing systems. Pruett-Miller et al used small molecules to enhance the rate of gene targeting by ZFN and reduced its toxicity. Indeed, they could increase transiently expression level of the modified ZFNs, by adding a manipulated destabilizing FKBP12 domain to the N-terminus, fusing a ubiquitin moiety to the N-terminus, linking a modified destabilizing FKBP12 domain to the N-terminus, adding MG132 proteasome inhibitor and Shield1 synthetic ligand.
119
Intuition of the CRISPR-cas9 system has revolutionized genomic editing approaches. Using small molecules for regulating Cas9 activity to improve the efficiency of this system have been recently investigated in both direct and indirect approaches. In the direct approach, Davis et al generated a Cas9 endonuclease with impaired cleavage activity by incorporating a 4- hydroxytamoxifen (4-HT)- responsive intein sequence at specific positions. In the presence of 4-HT, this small molecule binds to intein, enforces conformational changes, provokes protein splicing, and restores the DNA cleavage activity of inactive Cas9. This approach showed that conditionally activated Cas9 corrects target genomic sites with higher specificity (up to 25-fold) than wild-type Cas9 in human cells.
120
The indirect approach has relied on the regulation of Cas9 activity by using small molecules that target endogenous cellular processes.
91
By analyzing 4000 small molecules with known function, Brefeldin A, a small molecule inhibiting protein transportation between endoplasmic reticulum to Golgi apparatus and L755507, an β3-adrenergic receptor agonist, increased targeted reporter gene integration by 2-fold and 3-fold, respectively.
7
It is indicated that SCR7 is able to enhance the proficiency of the genome editing which is performed by CRISPR-Cas9 technology.
77
Maruyama et al demonstrated that by using CRISPR-Cas9 technology, SCR7 is able to increase the HDR efficiency approximately to 19-fold in mammalian cells and mouse embryos. Indeed, this increased efficiency has a positive dual function on the HDR pathway and CRISPR-Cas9 system.
8
Moreover, the small molecules involved in the stimulation of the HDR pathway, such as RS-1, are able to increase the CRISPR-Cas9 efficiency. Pinder et al showed that RS-1 increases the performance of both Cas9 and HDR up to 3-fold.
45
Moreover, the combination of small molecules known as "CRISPY mix", such as NU7026, MLN4924, inhibiting neddylation of CtIP, Trichostatin A, and NSC15520, in collaboration with Cas9 nickase, enhanced the efficiency of knock-in experiments.
46
Besides all successful achievements in increasing HDR efficiency through stimulation of HDR or inhibition of NHEJ, it should be considered that these strategies are harmful to genome integrity.
121
On one hand, inhibition of NHEJ would increase premature aging and the incidence of cancers. On the other hand, stimulation of RAD51 would lead to an augmentation in spontaneous recombination especially across widespread repetitive sequences which in turn, could result in loss of key genetic information.
122,123
In this line, discovering small molecules with minimal off-target effects on global genome stability sounds extremely crucial. Recently, Zhang et al established a novel and easy-to-score screening system by doing a mechanistic study and analyzing 722 natural small molecules which are commonly used in traditional Chinese medicine. It is elucidated that farrerol, isolated from Rhododendron dauricum, with anti-inflammatory and anti-bacterial properties, scaled up homologous recombination without any effect on NHEJ. Furthermore, this natural small molecule enhanced CRISPR-Cas9-mediated genome editing in different in vitro and in vivo models.
124
Precise control over exposure time and expression level of CRISPR-Cas9 system during gene editing approaches is extremely important for enhancing its application. To address this serious challenge, Wu et al generated a chimeric endonuclease by fusing small molecule-assisted shut-off (SMASh) to Cas9 protein. It was well-demonstrated that in the presence of hepatitis C virus (HCV) protease inhibitor asunaprevir (ASV), a clinically approved small molecule for HCV, this chimeric endonuclease degraded but upon ASV removal, this phenomenon reversed. Generating chimeric endonuclease such as Cas9-SMASH fusion could increase the accuracy and versatility of genome editing approach based on CRISPR-Cas9 technology.
125
Although CRISPR-Cas9 technology has presented high efficiency in various in vitro and in vivo models, knock-in efficiency by this technology has not been successful in hESC. Cas12a (also known as Cpf1) is a CRISPR effector which is categorized in class II- type V. CRISPR-Cas12a has shown promising potential for expanding genome editing toolbox. One special feature of this system is introducing a staggered DSB in targeted regions which seems to have increased the HDR rate.
126
A high-throughput chemical screening for identifying the candidate small molecules elucidated that AZD-7762 and VE-822 are able to improve CRISPR-Cas12a -mediated precise genome engineering.
54
Furthermore, the design of a new paradigm for stimulation of the structure of Cas protein and/or small molecules interaction with Cas-DNA seems a logical approach for the enhancement of the efficiency of the CRISPR-Cas mediated gene editing. In this line, Li et al provided a new paradigm for modulating the efficiency of CRISPR-Cas12a-mediated gene editing by small molecules. According to their results, a rational small molecule, quinazoline-2,4(1H,3H)-dione, is able to work like a molecular glue between Acidaminococcus Cas12a (AsCas12a) and crRNA and stabilize endonuclease-crRNA complex which in turn, would improve the efficiency of gene editing which is mediated by AsCas12a mammalian cells.
127
Manipulation of genetic information flow at the RNA level provides the opportunity to change the expression level of genes without long-term modification to the host genome. Cas13 proteins are CRISPR effectors with RNA targeting activity and are classified into class II- type VI. CRISPR-Cas13 has shown promising results in RNA targeting strategies and RNA-based diagnostic tests.
128
The large size (~100-130 kDa) and bacterial origin restrain the applications of CRISPR-Cas13 technology in both research and therapeutic developments. Recently, a novel CRISPR-Cas-inspired RNA targeting system (CIRTS) was generated to overcome these limitations. CIRTS is an engineered RNP that is able to recruit protein cargo site-selectively to target the transcript through using Watson-Crick-Franklin base-pair interactions with a gRNA.
129
In order to temporally control CIRTS dynamics, a small molecule-inducible RNA-targeting platform was established based on this effector by coupling heterodimerization domains of the abscisic acid small molecule system with CIRTS. This system provides an opportunity to easily target any desired transcript under a small molecule inducible-CIRTS platform.
130
CRISPR-Cas applications in in vivo preclinical and clinical models
Currently, most of clinical applications which are mediated by CRISPR-Cas technology for monogenic disorders are ex vivo approaches. During last years, several in vivo gene editing studies have been represented that target both HDR and NHEJ pathways. Since the efficiency of in vivo editing is lower compared to in vitro, a screening approach for detecting the modified cells through accurate editing boosts the feasibility of HDR approach in clinicalmodels.
131
Although CRISPR-Cas9 has been used in ex vivo clinical trials related to human immunodeficiency virus-1 (HIV) infection, cancers, b-thalassemia (ClinicalTrials.gov: NCT03655678), and sickle cell disease(ClinicalTrials.gov: NCT03745287), mentioned strategies such as synchronization, modified CRISPR-Cas Systems, and small molecules in the clinical models could be promising in future. Some of the recent applications based on HDR gene editing in preclinical and clinical models are presented in Table 3. Hereon, we emphasize several HDR-based accurate in vivo gene editing experiments utilizing CRISPR-Cas that can be potentially applied to human in future.
Table 3.
List of the recent therapeutic gene editing studies based on HDR in preclinical models
|
Human disease
|
Organ
|
Editing strategy
|
Delivery system
|
Gene editing tool
|
Ref.
|
| Hemophilia A and B |
Mouse liver |
HDR-dependent gene insertion |
Systemic injection of AAV8 |
ZFN |
132
|
| Hemophilia A and B |
Mouse liver |
HDR-based corrective gene editing |
Systemic injection of AAV8 |
ZFN |
133
|
| Hemophilia B |
Mouse liver |
HDR-based point mutation correction |
Injection of AAV8 |
CRISPR-Cas9 |
134
|
| Hemophilia B |
Mouse liver |
HDR-mediated insert into mFIXinto murine ROSA26 safe harbor |
Intravenous injection of AAV8 |
CRISPR-Cas9 |
135
|
| Hemophilia B |
Mouse liver |
HDR-based point mutation correction in F9 locus
|
Intravenous injection of adenovirus |
CRISPR-Cas9 |
136
|
| Hunter's syndrome |
Mouse liver |
HDR-mediated integration into albumin locus |
Systemic injection of AAV8 |
ZFN |
137
|
HTI
|
Mouse liver |
HDR-based point mutation correction |
Intravenous injection of AAV2/8 and LNP |
CRISPR-Cas9 |
138
|
HTI
|
Rat liver |
HDR-based point mutation correction |
Intravenous injection of adenovirus |
CRISPR-Cas9 |
139
|
| AATD |
Mouse liver |
HDR-based point mutation correction |
Intravenous and intraperitoneal injection of AAV |
CRISPR-Cas9 |
140
|
| DMD |
Mouse muscle |
HDR-mediated dystrophin gene correction |
Intramuscular and retro-orbital injection of dual AAV6 |
CRISPR-Cas9 |
141
|
| DMD |
Mouse muscle |
HDR-based point mutation correction |
Intramuscular injection of Gold nanoparticle Cas9 RNP and donor DNA |
CRISPR-Cas9 |
142
|
| Retinitis pigmentosa |
Mouse Eye |
HDR-based point mutation correction |
Subretinal and electroporation of RecA-MS2 Plasmid and ssDNA donor |
CRISPR-Cas9/ RecA |
143
|
| OTCdeficiency |
Mouse liver |
HDR-based point mutation correction |
Intravenous injection of AAV8 |
CRISPR-Cas9 |
144
|
| OTC deficiency |
Mouse liver |
HDR- mediated insert codon optimized human OTC into intron 4 of mouse OTC locus
|
Intravenous injection of AAV8 |
CRISPR-Cas9 |
145
|
| MPS type I |
Multiorgan |
HDR- mediated insert Idua into murine ROSA26 safe harbor |
Intravenous injection of cationic liposomes |
CRISPR-Cas9 |
146
|
AAV, adeno-associated virus; mFIX, factor IX; HTI, hereditary tyrosinemia; LNP, lipid nanoparticle; AATD, alpha-1 antitrypsin deficiency; DMD, Duchenne muscular dystrophy; OTC, ornithine transcarbamylase; MPS, mucopolysaccharidosis.
Review Highlights
What is the current knowledge?
√ HDR is attractive for its high fidelity; the choice of repair pathway is biased in a biological context.
√ Mammalian cells preferentially employ NHEJ over and HDR is restricted.
What is new here?
√ Facilitating the development of a modified CRISPR-Cas9 system to achieve more precise gene editing.
√ Suggesting the remarkable advantage of small molecules for establishing drug inducible genome editing platforms.
√ Drug inducible genome editing platform would help to control editing approaches dose- and exposure time-dependently.
Conclusion
The stimulation of HDR pathway can increase the accuracy of CRISPR-Cas-mediated engineering systems. Generating chimeric programmable endonucleases provides this opportunity to direct DNA template close proximity of CRISPR-Cas-mediated DSB. However, it should be considered that by fusing a functional domain or linker, the expression level of Cas9 or its delivery efficiency might be changed and also the generated fusion protein may be prone to protease cleavage. Small molecules and their derivatives are able to proficiently block or activate certain DNA repair pathways. Most small molecules increase precise genome editing through HDR. The results of most studies suggest that for increasing HDR-driven knock-in events, blockade of NHEJ is more efficient than improving HDR. Furthermore, the targeting upstream protein in NHEJ provides a greater potential for increasing the HDR-mediated gene insertion. Nonetheless, small molecules have unequal and even diverse influences on the efficiency of precise genome editing in various cell models. Hence, it is necessary to survey the efficacy of this approach in certain cells, different DNA loci, and in vivo studies. In summary, inhibitors of DNA-PK and ligase complex enhance the efficiency of precise genome editing efficiency in various cell types. There is serious consideration about genome integrity by inhibiting NHEJ or stimulating HDR components. In this line, discovering safe natural products such as farrerol, which is able to increase HDR with no effect on NHEJ, brings up a novel perspective for increasing the efficiency of genome editing approach. Ultimately, high throughput screening of small molecule libraries could result in more discoveries of promising chemicals that improve HDR efficiency and CRISPR-Cas9 systems. Another remarkable advantage of small molecules is their usefulness for establishing drug-inducible genome editing platforms. This finding would help researchers temporally control the dose and exposure time of genome or transcriptome editing tools.
Acknowledgments
The authors wish to express their gratitude for financial support from Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Funding sources
Shahid Beheshti University of Medical Sciences, Tehran, Iran (grant No. 11451).
Ethical statement
Not applicable.
Competing interests
Authors declare they do not have any conflict of interests.
Authors’ contribution
FSh: conceptualization and draft preparation, writing and reviewing. HD: study consultation, writing and reviewing. OM: conceptualization and draft preparation. SM, HV, MS: study consultation and validation. AR: supervision, writing and reviewing, and project administration.
References
- Karagyaur MN, Rubtsov YP, Vasiliev PA, Tkachuk VA. Practical Recommendations for Improving Efficiency and Accuracy of the CRISPR/Cas9 Genome Editing System. Biochem (Mosc) 2018; 83:629-42. doi: 10.1134/S0006297918060020 [Crossref] [ Google Scholar]
- Bayat H, Omidi M, Rajabibazl M, Sabri S, Rahimpour A. The CRISPR Growth Spurt: from Bench to Clinic on Versatile Small RNAs. J Microbiol Biotechnol 2017; 27:207-18. doi: 10.4014/jmb.1607.07005 [Crossref] [ Google Scholar]
- Golchin A, Shams F, Karami F. Advancing Mesenchymal Stem Cell Therapy with CRISPR/Cas9 for Clinical Trial Studies. Adv Exp Med Biol 2020; 1247:89-100. doi: 10.1007/5584_2019_459 [Crossref] [ Google Scholar]
- Bayat H, Naderi F, Khan AH, Memarnejadian A, Rahimpour A. The Impact of CRISPR-Cas System on Antiviral Therapy. Adv Pharm Bull 2018; 8:591-7. doi: 10.15171/apb.2018.067 [Crossref] [ Google Scholar]
- Salmaninejad A, Jafari Abarghan Y, Bozorg Qomi S, Bayat H, Yousefi M, Azhdari S. Common therapeutic advances for Duchenne muscular dystrophy (DMD). Int J Neurosci 2021; 131:370-89. doi: 10.1080/00207454.2020.1740218 [Crossref] [ Google Scholar]
- Janik E, Niemcewicz M, Ceremuga M, Krzowski L, Saluk-Bijak J, Bijak M. Various Aspects of a Gene Editing System—CRISPR–Cas9. Int J Mol Sci 2020; 21:9604. doi: 10.3390/ijms21249604 [Crossref] [ Google Scholar]
- Yu C, Liu Y, Ma T, Liu K, Xu S, Zhang Y. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015; 16:142-7. doi: 10.1016/j.stem.2015.01.003 [Crossref] [ Google Scholar]
- Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol 2015; 33:538-42. doi: 10.1038/nbt.3190 [Crossref] [ Google Scholar]
- Kakarougkas A, Jeggo PA. DNA DSB repair pathway choice: an orchestrated handover mechanism. Br J Radiol 2014; 87:20130685. doi: 10.1259/bjr.20130685 [Crossref] [ Google Scholar]
- Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol 2013; 5:a012757. doi: 10.1101/cshperspect.a012757 [Crossref] [ Google Scholar]
- Bae S, Kweon J, Kim HS, Kim JS. Microhomology-based choice of Cas9 nuclease target sites. Nat Methods 2014; 11:705-6. doi: 10.1038/nmeth.3015 [Crossref] [ Google Scholar]
- Callen E, Faryabi RB, Luckey M, Hao B, Daniel JA, Yang W. The DNA damage- and transcription-associated protein paxip1 controls thymocyte development and emigration. Immunity 2012; 37:971-85. doi: 10.1016/j.immuni.2012.10.007 [Crossref] [ Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 2013; 49:858-71. doi: 10.1016/j.molcel.2013.01.002 [Crossref] [ Google Scholar]
- Findlay S, Heath J, Luo VM, Malina A, Morin T, Coulombe Y. SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J 2018; 37:e100158. doi: 10.15252/embj.2018100158 [Crossref] [ Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 2018; 560:122-7. doi: 10.1038/s41586-018-0362-1 [Crossref] [ Google Scholar]
- Waters CA, Strande NT, Wyatt DW, Pryor JM, Ramsden DA. Nonhomologous end joining: a good solution for bad ends. DNA Repair (Amst) 2014; 17:39-51. doi: 10.1016/j.dnarep.2014.02.008 [Crossref] [ Google Scholar]
- Pawelczak KS, Turchi JJ. A mechanism for DNA-PK activation requiring unique contributions from each strand of a DNA terminus and implications for microhomology-mediated nonhomologous DNA end joining. Nucleic Acids Res 2008; 36:4022-31. doi: 10.1093/nar/gkn344 [Crossref] [ Google Scholar]
- Radhakrishnan SK, Lees-Miller SP. DNA requirements for interaction of the C-terminal region of Ku80 with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). DNA Repair (Amst) 2017; 57:17-28. doi: 10.1016/j.dnarep.2017.06.001 [Crossref] [ Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 2017; 18:495-506. doi: 10.1038/nrm.2017.48 [Crossref] [ Google Scholar]
- Daley JM, Laan RL, Suresh A, Wilson TE. DNA joint dependence of pol X family polymerase action in nonhomologous end joining. J Biol Chem 2005; 280:29030-7. doi: 10.1074/jbc.M505277200 [Crossref] [ Google Scholar]
- Paull TT. Saving the ends for last: the role of pol mu in DNA end joining. Mol Cell 2005; 19:294-6. doi: 10.1016/j.molcel.2005.07.008 [Crossref] [ Google Scholar]
- Andres SN, Vergnes A, Ristic D, Wyman C, Modesti M, Junop M. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res 2012; 40:1868-78. doi: 10.1093/nar/gks022 [Crossref] [ Google Scholar]
- McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet 2008; 24:529-38. doi: 10.1016/j.tig.2008.08.007 [Crossref] [ Google Scholar]
- Sinha S, Villarreal D, Shim EY, Lee SE. Risky business: Microhomology-mediated end joining. Mutat Res 2016; 788:17-24. doi: 10.1016/j.mrfmmm.2015.12.005 [Crossref] [ Google Scholar]
- Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A 2013; 110:7720-5. doi: 10.1073/pnas.1213431110 [Crossref] [ Google Scholar]
- Rahal EA, Henricksen LA, Li Y, Williams RS, Tainer JA, Dixon K. ATM regulates Mre11-dependent DNA end-degradation and microhomology-mediated end joining. Cell Cycle 2010; 9:2866-77. doi: 10.4161/cc.9.14.12363 [Crossref] [ Google Scholar]
- Ahmad A, Robinson AR, Duensing A, van Drunen E, Beverloo HB, Weisberg DB. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol Cell Biol 2008; 28:5082-92. doi: 10.1128/mcb.00293-08 [Crossref] [ Google Scholar]
- Drane P, Brault ME, Cui G, Meghani K, Chaubey S, Detappe A. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 2017; 543:211-6. doi: 10.1038/nature21358 [Crossref] [ Google Scholar]
- Anuchina AA, Lavrov AV, Smirnikhina SA. TIRR: a potential front runner in HDR race-hypotheses and perspectives. Mol Biol Rep 2020; 47:2371-9. doi: 10.1007/s11033-020-05285-x [Crossref] [ Google Scholar]
- Symington LS. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol 2014; 6:a016436. doi: 10.1101/cshperspect.a016436 [Crossref] [ Google Scholar]
- Paull TT. Mechanisms of ATM Activation. Annu Rev Biochem 2015; 84:711-38. doi: 10.1146/annurev-biochem-060614-034335 [Crossref] [ Google Scholar]
- Kijas AW, Lim YC, Bolderson E, Cerosaletti K, Gatei M, Jakob B. ATM-dependent phosphorylation of MRE11 controls extent of resection during homology directed repair by signalling through Exonuclease 1. Nucleic Acids Res 2015; 43:8352-67. doi: 10.1093/nar/gkv754 [Crossref] [ Google Scholar]
- Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 2011; 25:350-62. doi: 10.1101/gad.2003811 [Crossref] [ Google Scholar]
- Kakarougkas A, Ismail A, Katsuki Y, Freire R, Shibata A, Jeggo PA. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res 2013; 41:10298-311. doi: 10.1093/nar/gkt802 [Crossref] [ Google Scholar]
- Butler LR, Densham RM, Jia J, Garvin AJ, Stone HR, Shah V. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J 2012; 31:3918-34. doi: 10.1038/emboj.2012.232 [Crossref] [ Google Scholar]
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet 2011; 45:247-71. doi: 10.1146/annurev-genet-110410-132435 [Crossref] [ Google Scholar]
- Kim KP, Mirkin EV. So similar yet so different: The two ends of a double strand break. Mutat Res 2018; 809:70-80. doi: 10.1016/j.mrfmmm.2017.06.007 [Crossref] [ Google Scholar]
- Bell JC, Kowalczykowski SC. Mechanics and Single-Molecule Interrogation of DNA Recombination. Annu Rev Biochem 2016; 85:193-226. doi: 10.1146/annurev-biochem-060614-034352 [Crossref] [ Google Scholar]
- Pellegrini L, David SY, Lo T, Anand S, Lee M, Blundell TL. Insights into DNA recombination from the structure of a RAD51–BRCA2 complex. Nature 2002; 420:287-93. doi: 10.1038/nature01230 [Crossref] [ Google Scholar]
- Smirnikhina SA, Anuchina AA, Lavrov AV. Ways of improving precise knock-in by genome-editing technologies. Hum Genet 2019; 138:1-19. doi: 10.1007/s00439-018-1953-5 [Crossref] [ Google Scholar]
- Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 2016; 26:52-64. doi: 10.1016/j.tcb.2015.07.009 [Crossref] [ Google Scholar]
- Liu M, Rehman S, Tang X, Gu K, Fan Q, Chen D. Methodologies for Improving HDR Efficiency. Front Genet 2018; 9:691. doi: 10.3389/fgene.2018.00691 [Crossref] [ Google Scholar]
- Sung P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem 1997; 272:28194-7. doi: 10.1074/jbc.272.45.28194 [Crossref] [ Google Scholar]
- Pawelczak KS, Gavande NS, VanderVere-Carozza PS, Turchi JJ. Modulating DNA Repair Pathways to Improve Precision Genome Engineering. ACS Chem Biol 2018; 13:389-96. doi: 10.1021/acschembio.7b00777 [Crossref] [ Google Scholar]
- Pinder J, Salsman J, Dellaire G. Nuclear domain 'knock-in' screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res 2015; 43:9379-92. doi: 10.1093/nar/gkv993 [Crossref] [ Google Scholar]
- Riesenberg S, Maricic T. Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Nat Commun 2018; 9:2164. doi: 10.1038/s41467-018-04609-7 [Crossref] [ Google Scholar]
- Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016; 540:144-9. doi: 10.1038/nature20565 [Crossref] [ Google Scholar]
- Kostyushev D, Kostyusheva A, Brezgin S, Zarifyan D, Utkina A, Goptar I. Suppressing the NHEJ pathway by DNA-PKcs inhibitor NU7026 prevents degradation of HBV cccDNA cleaved by CRISPR/Cas9. Sci Rep 2019; 9:1847. doi: 10.1038/s41598-019-38526-6 [Crossref] [ Google Scholar]
- Jayavaradhan R, Pillis DM, Malik P. A Versatile Tool for the Quantification of CRISPR/Cas9-Induced Genome Editing Events in Human Hematopoietic Cell Lines and Hematopoietic Stem/Progenitor Cells. J Mol Biol 2019; 431:102-10. doi: 10.1016/j.jmb.2018.05.005 [Crossref] [ Google Scholar]
- Zhang JP, Li XL, Li GH, Chen W, Arakaki C, Botimer GD. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol 2017; 18:35. doi: 10.1186/s13059-017-1164-8 [Crossref] [ Google Scholar]
- Robert F, Barbeau M, Ethier S, Dostie J, Pelletier J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med 2015; 7:93. doi: 10.1186/s13073-015-0215-6 [Crossref] [ Google Scholar]
- Riesenberg S, Chintalapati M, Macak D, Kanis P, Maricic T, Paabo S. Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res 2019; 47:e116. doi: 10.1093/nar/gkz669 [Crossref] [ Google Scholar]
- Weterings E, Gallegos AC, Dominick LN, Cooke LS, Bartels TN, Vagner J. A novel small molecule inhibitor of the DNA repair protein Ku70/80. DNA Repair (Amst) 2016; 43:98-106. doi: 10.1016/j.dnarep.2016.03.014 [Crossref] [ Google Scholar]
- Ma X, Chen X, Jin Y, Ge W, Wang W, Kong L. Small molecules promote CRISPR-Cpf1-mediated genome editing in human pluripotent stem cells. Nat Commun 2018; 9:1303. doi: 10.1038/s41467-018-03760-5 [Crossref] [ Google Scholar]
- Hu Z, Shi Z, Guo X, Jiang B, Wang G, Luo D. Ligase IV inhibitor SCR7 enhances gene editing directed by CRISPR-Cas9 and ssODN in human cancer cells. Cell Biosci 2018; 8:12. doi: 10.1186/s13578-018-0200-z [Crossref] [ Google Scholar]
- Lee JS, Grav LM, Pedersen LE, Lee GM, Kildegaard HF. Accelerated homology-directed targeted integration of transgenes in Chinese hamster ovary cells via CRISPR/Cas9 and fluorescent enrichment. Biotechnol Bioeng 2016; 113:2518-23. doi: 10.1002/bit.26002 [Crossref] [ Google Scholar]
- Yao X, Zhang M, Wang X, Ying W, Hu X, Dai P. Tild-CRISPR Allows for Efficient and Precise Gene Knockin in Mouse and Human Cells. Dev Cell 2018; 45:526-36 e5. doi: 10.1016/j.devcel.2018.04.021 [Crossref] [ Google Scholar]
- Gutschner T, Haemmerle M, Genovese G, Draetta GF, Chin L. Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Cell Rep 2016; 14:1555-66. doi: 10.1016/j.celrep.2016.01.019 [Crossref] [ Google Scholar]
- Singh P, Schimenti JC, Bolcun-Filas E. A mouse geneticist's practical guide to CRISPR applications. Genetics 2015; 199:1-15. doi: 10.1534/genetics.114.169771 [Crossref] [ Google Scholar]
- Li G, Zhang X, Zhong C, Mo J, Quan R, Yang J. Small molecules enhance CRISPR/Cas9-mediated homology-directed genome editing in primary cells. Sci Rep 2017; 7:8943. doi: 10.1038/s41598-017-09306-x [Crossref] [ Google Scholar]
- Zhang Y, Zhang Z, Ge W. An efficient platform for generating somatic point mutations with germline transmission in the zebrafish by CRISPR/Cas9-mediated gene editing. J Biol Chem 2018; 293:6611-22. doi: 10.1074/jbc.RA117.001080 [Crossref] [ Google Scholar]
- Pan Y, Shen N, Jung-Klawitter S, Betzen C, Hoffmann GF, Hoheisel JD. CRISPR RNA-guided FokI nucleases repair a PAH variant in a phenylketonuria model. Sci Rep 2016; 6:35794. doi: 10.1038/srep35794 [Crossref] [ Google Scholar]
- Aksoy YA, Nguyen DT, Chow S, Chung RS, Guillemin GJ, Cole NJ. Chemical reprogramming enhances homology-directed genome editing in zebrafish embryos. Commun Biol 2019; 2:198. doi: 10.1038/s42003-019-0444-0 [Crossref] [ Google Scholar]
- Lamas‐Toranzo I, Martínez‐Moro A, Millán‐Blanca G, Sánchez J, Lonergan P, Bermejo‐Álvarez P. RS‐1 enhances CRISPR‐mediated targeted knock‐in in bovine embryost. Mol Reprod Dev 2020; 87:542-549. doi: 10.1002/mrd.23341 [Crossref] [ Google Scholar]
- Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS. DNA repair targeted therapy: The past or future of cancer treatment?. Pharmacol Ther 2016; 160:65-83. doi: 10.1016/j.pharmthera.2016.02.003 [Crossref] [ Google Scholar]
- Davidson D, Amrein L, Panasci L, Aloyz R. Small Molecules, Inhibitors of DNA-PK, Targeting DNA Repair, and Beyond. Front Pharmacol 2013; 4:5. doi: 10.3389/fphar.2013.00005 [Crossref] [ Google Scholar]
-
Tavecchio M, Munck JM, Cano C, Newell DR, Curtin NJ. Further characterisation of the cellular activity of the DNA-PK inhibitor, NU7441, reveals potential cross-talk with homologous recombination. Cancer Chemother Pharmacol 2012; 69: 155-64. 10.1007/s00280-011-1662-4
- Munck JM, Batey MA, Zhao Y, Jenkins H, Richardson CJ, Cano C. Chemosensitization of cancer cells by KU-0060648, a dual inhibitor of DNA-PK and PI-3K. Mol Cancer Ther 2012; 11:1789-98. doi: 10.1158/1535-7163.MCT-11-0535 [Crossref] [ Google Scholar]
- Zenke FT, Zimmermann A, Sirrenberg C, Dahmen H, Kirkin V, Pehl U. Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol Cancer Ther 2020; 19:1091-101. doi: 10.1158/1535-7163.MCT-19-0734 [Crossref] [ Google Scholar]
- Sirrenberg C, Zimmermann A, Grombacher T, Vassilev LT, Damstrup L, Zenke FT. A novel selective DNA-PK inhibitor, M3814, as a potential combination partner of Etoposide and Cisplatin in the therapy of lung cancer. AACR 2017; 77:4183. doi: 10.1158/1538-7445.AM2017-4183 [Crossref] [ Google Scholar]
- Sun Q, Guo Y, Liu X, Czauderna F, Carr MI, Zenke FT. Therapeutic Implications of p53 Status on Cancer Cell Fate Following Exposure to Ionizing Radiation and the DNA-PK Inhibitor M3814. Mol Cancer Res 2019; 17:2457-68. doi: 10.1158/1541-7786.MCR-19-0362 [Crossref] [ Google Scholar]
- Gavande NS, VanderVere-Carozza PS, Pawelczak KS, Mendoza-Munoz P, Vernon TL, Hanakahi LA. Discovery and development of novel DNA-PK inhibitors by targeting the unique Ku-DNA interaction. Nucleic Acids Res 2020; 48:11536-50. doi: 10.1093/nar/gkaa934 [Crossref] [ Google Scholar]
- Freschauf GK, Karimi-Busheri F, Ulaczyk-Lesanko A, Mereniuk TR, Ahrens A, Koshy JM. Identification of a small molecule inhibitor of the human DNA repair enzyme polynucleotide kinase/phosphatase. Cancer Res 2009; 69:7739-46. doi: 10.1158/0008-5472.CAN-09-1805 [Crossref] [ Google Scholar]
- Chen X, Zhong S, Zhu X, Dziegielewska B, Ellenberger T, Wilson GM. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res 2008; 68:3169-77. doi: 10.1158/0008-5472.CAN-07-6636 [Crossref] [ Google Scholar]
- Srivastava M, Nambiar M, Sharma S, Karki SS, Goldsmith G, Hegde M. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012; 151:1474-87. doi: 10.1016/j.cell.2012.11.054 [Crossref] [ Google Scholar]
- Ma Y, Chen W, Zhang X, Yu L, Dong W, Pan S. Increasing the efficiency of CRISPR/Cas9-mediated precise genome editing in rats by inhibiting NHEJ and using Cas9 protein. RNA Biol 2016; 13:605-12. doi: 10.1080/15476286.2016.1185591 [Crossref] [ Google Scholar]
- Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 2015; 33:543-8. doi: 10.1038/nbt.3198 [Crossref] [ Google Scholar]
- Yang D, Scavuzzo MA, Chmielowiec J, Sharp R, Bajic A, Borowiak M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci Rep 2016; 6:21264. doi: 10.1038/srep21264 [Crossref] [ Google Scholar]
- Gerlach M, Kraft T, Brenner B, Petersen B, Niemann H, Montag J. Efficient Knock-in of a Point Mutation in Porcine Fibroblasts Using the CRISPR/Cas9-GMNN Fusion Gene. Genes (Basel) 2018; 9:269. doi: 10.3390/genes9060296 [Crossref] [ Google Scholar]
- Song J, Yang D, Xu J, Zhu T, Chen YE, Zhang J. RS-1 enhances CRISPR/Cas9-and TALEN-mediated knock-in efficiency. Nat Commun 2016; 7:10548. doi: 10.1038/ncomms10548 [Crossref] [ Google Scholar]
- Greco GE, Matsumoto Y, Brooks RC, Lu Z, Lieber MR, Tomkinson AE. SCR7 is neither a selective nor a potent inhibitor of human DNA ligase IV. DNA Repair 2016; 43:18-23. [ Google Scholar]
- Singh AM, Adjan Steffey VV, Yeshi T, Allison DW. Gene Editing in Human Pluripotent Stem Cells: Choosing the Correct Path. J Stem Cell Regen Biol 2015; 1.
- Shy BR, MacDougall MS, Clarke R, Merrill BJ. Co-incident insertion enables high efficiency genome engineering in mouse embryonic stem cells. Nucleic Acids Res 2016; 44:7997-8010. doi: 10.1093/nar/gkw685 [Crossref] [ Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 2008; 77:229-57. doi: 10.1146/annurev.biochem.77.061306.125255 [Crossref] [ Google Scholar]
- Mason JM, Dusad K, Wright WD, Grubb J, Budke B, Heyer WD. RAD54 family translocases counter genotoxic effects of RAD51 in human tumor cells. Nucleic Acids Res 2015; 43:3180-96. doi: 10.1093/nar/gkv175 [Crossref] [ Google Scholar]
- Jayathilaka K, Sheridan SD, Bold TD, Bochenska K, Logan HL, Weichselbaum RR. A chemical compound that stimulates the human homologous recombination protein RAD51. Proc Natl Acad Sci U S A 2008; 105:15848-53. doi: 10.1073/pnas.0808046105 [Crossref] [ Google Scholar]
- Budke B, Lv W, Kozikowski AP, Connell PP. Recent developments using small molecules to target RAD51: how to best modulate RAD51 for anticancer therapy?. ChemMedChem 2016; 11:2468-73. doi: 10.1002/cmdc.201600426 [Crossref] [ Google Scholar]
- Pinder J, Salsman J, Dellaire G. Nuclear domain ‘knock-in’screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res 2015; 43:9379-92. doi: 10.1093/nar/gkv993 [Crossref] [ Google Scholar]
- Pérez-Benavente B, Farràs R. Cell synchronization techniques to study the action of CDK inhibitors. Methods Mol Biol 2016; 1336:85-93. doi: 10.1007/978-1-4939-2926-9_8 [Crossref] [ Google Scholar]
- Rivera-Torres N, Strouse B, Bialk P, Niamat RA, Kmiec EB. The position of DNA cleavage by TALENs and cell synchronization influences the frequency of gene editing directed by single-stranded oligonucleotides. PLoS One 2014; 9:e96483. doi: 10.1371/journal.pone.0096483 [Crossref] [ Google Scholar]
- Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014; 3:e04766. doi: 10.7554/eLife.04766 [Crossref] [ Google Scholar]
- Tsakraklides V, Brevnova E, Stephanopoulos G, Shaw AJ. Improved gene targeting through cell cycle synchronization. PLoS One 2015; 10:e0133434. doi: 10.1371/journal.pone.0133434 [Crossref] [ Google Scholar]
- Brachman EE, Kmiec EB. Gene repair in mammalian cells is stimulated by the elongation of S phase and transient stalling of replication forks. DNA Repair (Amst) 2005; 4:445-57. doi: 10.1016/j.dnarep.2004.11.007 [Crossref] [ Google Scholar]
- Wienert B, Nguyen DN, Guenther A, Feng SJ, Locke MN, Wyman SK. Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Nat Commun 2020; 11:2109. doi: 10.1038/s41467-020-15845-1 [Crossref] [ Google Scholar]
- Fernandez-Garcia B, Casado P, Prado MA, Ugarte-Gil LJ, Artime N, Cabal-Hierro L. Proteomic analysis of annexin A2 phosphorylation induced by microtubule interfering agents and kinesin spindle protein inhibitors. J Proteome Res 2010; 9:4649-60. doi: 10.1021/pr100377v [Crossref] [ Google Scholar]
- Eisenbrand G, Hippe F, Jakobs S, Muehlbeyer S. Molecular mechanisms of indirubin and its derivatives: novel anticancer molecules with their origin in traditional Chinese phytomedicine. J Cancer Res Clin Oncol 2004; 130:627-35. doi: 10.1007/s00432-004-0579-2 [Crossref] [ Google Scholar]
- Rahman SH, Bobis-Wozowicz S, Chatterjee D, Gellhaus K, Pars K, Heilbronn R. The nontoxic cell cycle modulator indirubin augments transduction of adeno-associated viral vectors and zinc-finger nuclease-mediated gene targeting. Hum Gene Ther 2013; 24:67-77. doi: 10.1089/hum.2012.168 [Crossref] [ Google Scholar]
- Moehle EA, Rock JM, Lee Y-L, Jouvenot Y, DeKelver RC, Gregory PD. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc Natl Acad Sci U S A 2007; 104:3055-60. doi: 10.1073/pnas.0611478104 [Crossref] [ Google Scholar]
- Ebrahimi V, Hashemi A. Challenges of in vitro genome editing with CRISPR/Cas9 and possible solutions: A review. Gene 2020; 753:144813. doi: 10.1016/j.gene.2020.144813 [Crossref] [ Google Scholar]
- Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol 2015; 208:44-53. doi: 10.1016/j.jbiotec.2015.04.024 [Crossref] [ Google Scholar]
- Yang H, Ren S, Yu S, Pan H, Li T, Ge S. Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks. Int J Mol Sci 2020; 21:6461. doi: 10.3390/ijms21186461 [Crossref] [ Google Scholar]
- Charpentier M, Khedher AHY, Menoret S, Brion A, Lamribet K, Dardillac E. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat Commun 2018; 9:1133. doi: 10.1038/s41467-018-03475-7 [Crossref] [ Google Scholar]
- Tran NT, Bashir S, Li X, Rossius J, Chu VT, Rajewsky K. Enhancement of Precise Gene Editing by the Association of Cas9 With Homologous Recombination Factors. Front Genet 2019; 10:365. doi: 10.3389/fgene.2019.00365 [Crossref] [ Google Scholar]
- Tomimatsu N, Mukherjee B, Harris JL, Boffo FL, Hardebeck MC, Potts PR. DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair. J Biol Chem 2017; 292:10779-90. doi: 10.1074/jbc.M116.772475 [Crossref] [ Google Scholar]
- Jayavaradhan R, Pillis DM, Goodman M, Zhang F, Zhang Y, Andreassen PR. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat Commun 2019; 10:2866. doi: 10.1038/s41467-019-10735-7 [Crossref] [ Google Scholar]
- Canny MD, Moatti N, Wan LCK, Fradet-Turcotte A, Krasner D, Mateos-Gomez PA. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat Biotechnol 2018; 36:95-102. doi: 10.1038/nbt.4021 [Crossref] [ Google Scholar]
- Hackley CR. A Novel Set of Cas9 Fusion Proteins to stimulate Homologous Recombination: Cas9-HRs. CRISPR J 2020; 4:253-263. doi: 10.1101/2020.05.17.100677 [Crossref] [ Google Scholar]
- Ma L, Ruan J, Song J, Wen L, Yang D, Zhao J. MiCas9 increases large size gene knock-in rates and reduces undesirable on-target and off-target indel edits. Nat Commun 2020; 11:6082. doi: 10.1038/s41467-020-19842-2 [Crossref] [ Google Scholar]
- Matsumoto D, Tamamura H, Nomura W. A cell cycle-dependent CRISPR-Cas9 activation system based on an anti-CRISPR protein shows improved genome editing accuracy. Commun Biol 2020; 3:1-10. doi: 10.1038/s42003-020-01340-2 [Crossref] [ Google Scholar]
- Ali Z, Shami A, Sedeek K, Kamel R, Alhabsi A, Tehseen M. Fusion of the Cas9 endonuclease and the VirD2 relaxase facilitates homology-directed repair for precise genome engineering in rice. Commun Biol 2020; 3:44. doi: 10.1038/s42003-020-0768-9 [Crossref] [ Google Scholar]
- Ma M, Zhuang F, Hu X, Wang B, Wen XZ, Ji JF. Efficient generation of mice carrying homozygous double-floxp alleles using the Cas9-Avidin/Biotin-donor DNA system. Cell Res 2017; 27:578-81. doi: 10.1038/cr.2017.29 [Crossref] [ Google Scholar]
- Carlson-Stevermer J, Abdeen AA, Kohlenberg L, Goedland M, Molugu K, Lou M. Assembly of CRISPR ribonucleoproteins with biotinylated oligonucleotides via an RNA aptamer for precise gene editing. Nat Commun 2017; 8:1711. doi: 10.1038/s41467-017-01875-9 [Crossref] [ Google Scholar]
- Gu B, Posfai E, Rossant J. Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat Biotechnol 2018; 36:632-7. doi: 10.1038/nbt.4166 [Crossref] [ Google Scholar]
- Lee K, Mackley VA, Rao A, Chong AT, Dewitt MA, Corn JE. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. Elife 2017; 6:e25312. doi: 10.7554/eLife.25312 [Crossref] [ Google Scholar]
- Savic N, Ringnalda FC, Lindsay H, Berk C, Bargsten K, Li Y. Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Elife 2018; 7:e33761. doi: 10.7554/eLife.33761 [Crossref] [ Google Scholar]
- Aird EJ, Lovendahl KN, St Martin A, Harris RS, Gordon WR. Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun Biol 2018; 1:54. doi: 10.1038/s42003-018-0054-2 [Crossref] [ Google Scholar]
- Ling X, Xie B, Gao X, Chang L, Zheng W, Chen H. Improving the efficiency of precise genome editing with site-specific Cas9-oligonucleotide conjugates. Sci Adv 2020; 6:eaaz0051. doi: 10.1126/sciadv.aaz0051 [Crossref] [ Google Scholar]
- Nunez JK, Harrington LB, Doudna JA. Chemical and Biophysical Modulation of Cas9 for Tunable Genome Engineering. ACS Chem Biol 2016; 11:681-8. doi: 10.1021/acschembio.5b01019 [Crossref] [ Google Scholar]
- Pruett-Miller SM, Reading DW, Porter SN, Porteus MH. Attenuation of zinc finger nuclease toxicity by small-molecule regulation of protein levels. PLoS Genet 2009; 5:e1000376. doi: 10.1371/journal.pgen.1000376 [Crossref] [ Google Scholar]
- Davis KM, Pattanayak V, Thompson DB, Zuris JA, Liu DR. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat Chem Biol 2015; 11:316-8. doi: 10.1038/nchembio.1793 [Crossref] [ Google Scholar]
- Vartak SV, Raghavan SC. Inhibition of nonhomologous end joining to increase the specificity of CRISPR/Cas9 genome editing. FEBS J 2015; 282:4289-94. doi: 10.1111/febs.13416 [Crossref] [ Google Scholar]
- Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell 2005; 120:497-512. doi: 10.1016/j.cell.2005.01.028 [Crossref] [ Google Scholar]
- Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst) 2008; 7:686-93. doi: 10.1016/j.dnarep.2007.12.008 [Crossref] [ Google Scholar]
- Zhang W, Chen Y, Yang J, Zhang J, Yu J, Wang M. A high-throughput small molecule screen identifies farrerol as a potentiator of CRISPR/Cas9-mediated genome editing. Elife 2020; 9:e56008. doi: 10.7554/eLife.56008 [Crossref] [ Google Scholar]
- Wu Y, Yang L, Chang T, Kandeel F, Yee JK. A Small Molecule-Controlled Cas9 Repressible System. Mol Ther Nucleic Acids 2020; 19:922-32. doi: 10.1016/j.omtn.2019.12.026 [Crossref] [ Google Scholar]
- Bayat H, Modarressi MH, Rahimpour A. The Conspicuity of CRISPR-Cpf1 System as a Significant Breakthrough in Genome Editing. Curr Microbiol 2018; 75:107-15. doi: 10.1007/s00284-017-1406-8 [Crossref] [ Google Scholar]
- Li W, Chan C, Zeng C, Turk R, Behlke MA, Cheng X. Rational Design of Small Molecules to Enhance Genome Editing Efficiency by Selectively Targeting Distinct Functional States of CRISPR-Cas12a. Bioconjug Chem 2020; 31:542-6. doi: 10.1021/acs.bioconjchem.0c00062 [Crossref] [ Google Scholar]
- Xu D, Cai Y, Tang L, Han X, Gao F, Cao H. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci Rep 2020; 10:1794. doi: 10.1038/s41598-020-58104-5 [Crossref] [ Google Scholar]
- Rauch S, He E, Srienc M, Zhou H, Zhang Z, Dickinson BC. Programmable RNA-Guided RNA Effector Proteins Built from Human Parts. Cell 2019; 178:122-34 e12. doi: 10.1016/j.cell.2019.05.049 [Crossref] [ Google Scholar]
- Rauch S, Jones KA, Dickinson BC. Small Molecule-Inducible RNA-Targeting Systems for Temporal Control of RNA Regulation. ACS Cent Sci 2020; 6:1987-96. doi: 10.1021/acscentsci.0c00537 [Crossref] [ Google Scholar]
- Dasgupta I, Flotte TR, Keeler AM. CRISPR/Cas-Dependent and Nuclease-Free In Vivo Therapeutic Gene Editing. Hum Gene Ther 2021; 32:275-93. doi: 10.1089/hum.2021.013 [Crossref] [ Google Scholar]
- Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 2011; 475:217-21. doi: 10.1038/nature10177 [Crossref] [ Google Scholar]
- Anguela XM, Sharma R, Doyon Y, Miller JC, Li H, Haurigot V. Robust ZFN-mediated genome editing in adult hemophilic mice. Blood 2013; 122:3283-7. doi: 10.1182/blood-2013-04-497354 [Crossref] [ Google Scholar]
- Wang L, Yang Y, Breton CA, White J, Zhang J, Che Y. CRISPR/Cas9-mediated in vivo gene targeting corrects hemostasis in newborn and adult factor IX–knockout mice. Blood 2019; 133:2745-52. doi: 10.1182/blood.2019000790 [Crossref] [ Google Scholar]
- Stephens CJ, Lauron EJ, Kashentseva E, Lu ZH, Yokoyama WM, Curiel DT. Long-term correction of hemophilia B using adenoviral delivery of CRISPR/Cas9. J Control Release 2019; 298:128-41. doi: 10.1016/j.jconrel.2019.02.009 [Crossref] [ Google Scholar]
- Guan Y, Ma Y, Li Q, Sun Z, Ma L, Wu L. CRISPR/Cas9‐mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol Med 2016; 8:477-88. doi: 10.15252/emmm.201506039 [Crossref] [ Google Scholar]
- Laoharawee K, DeKelver RC, Podetz-Pedersen KM, Rohde M, Sproul S, Nguyen H-O. Dose-dependent prevention of metabolic and neurologic disease in murine MPS II by ZFN-mediated in vivo genome editing. Mol Ther 2018; 26:1127-36. doi: 10.1016/j.ymthe.2018.03.002 [Crossref] [ Google Scholar]
- Yin H, Song C-Q, Dorkin JR, Zhu LJ, Li Y, Wu Q. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol 2016; 34:328-33. doi: 10.1038/nbt.3471 [Crossref] [ Google Scholar]
- Shao Y, Wang L, Guo N, Wang S, Yang L, Li Y. Cas9-nickase–mediated genome editing corrects hereditary tyrosinemia in rats. J Biol Chem 2018; 293:6883-92. doi: 10.1074/jbc.RA117.000347 [Crossref] [ Google Scholar]
- Bjursell M, Porritt MJ, Ericson E, Taheri-Ghahfarokhi A, Clausen M, Magnusson L. Therapeutic genome editing with CRISPR/Cas9 in a humanized mouse model ameliorates α1-antitrypsin deficiency phenotype. EBioMedicine 2018; 29:104-11. doi: 10.1016/j.ebiom [Crossref] [ Google Scholar]
- Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun 2017; 8:14454. doi: 10.1038/ncomms14454 [Crossref] [ Google Scholar]
- Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng 2017; 1:889-901. doi: 10.1038/s41551-017-0137-2 [Crossref] [ Google Scholar]
- Cai Y, Cheng T, Yao Y, Li X, Ma Y, Li L. In vivo genome editing rescues photoreceptor degeneration via a Cas9/RecA-mediated homology-directed repair pathway. Sci Adv 2019; 5:eaav3335. doi: 10.1126/sciadv.aav3335 [Crossref] [ Google Scholar]
- Yang Y, Wang L, Bell P, McMenamin D, He Z, White J. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol 2016; 34:334-8. doi: 10.1038/nbt.3469 [Crossref] [ Google Scholar]
- Wang L, Yang Y, Breton C, Bell P, Li M, Zhang J. A mutation-independent CRISPR-Cas9–mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci Adv 2020; 6:eaax5701. doi: 10.1126/sciadv.aax5701 [Crossref] [ Google Scholar]
- Schuh RS, Poletto É, Pasqualim G, Tavares AMV, Meyer FS, Gonzalez EA. In vivo genome editing of mucopolysaccharidosis I mice using the CRISPR/Cas9 system. J Control Release 2018; 288:23-33. doi: 10.1016/j.jconrel.2018.08.031 [Crossref] [ Google Scholar]