Bioimpacts. 13(5):373-382.
doi: 10.34172/bi.2023.24180
Original Article
Platinum(IV) compounds as potential drugs: a quantitative structure-activity relationship study
Jurica Novak Conceptualization, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, 1, 2, * 
Alena R. Zykova Conceptualization, Writing – original draft, 3
Vladimir A. Potemkin Conceptualization, Funding acquisition, Methodology, Writing – review & editing, †
Vladimir V. Sharutin Conceptualization, 3
Olga K. Sharutina Conceptualization, Writing – review & editing, 3
Author information:
1Department of Biotechnology, University of Rijeka, Rijeka, Croatia
2Center for Artificial Intelligence and Cyber security, University of Rijeka, Rijeka, Croatia
3Faculty of Chemistry, Department of Theoretical and Applied Chemistry, South Ural State University, Chelyabinsk, Russia
†Deceased September 1, 2021
Abstract
Introduction:
Machine learning methods, coupled with a tremendous increase in computer power in recent years, are promising tools in modern drug design and drug repurposing.
Methods:
Machine learning predictive models, publicly available at chemosophia.com, were used to predict the bioactivity of recently synthesized platinum(IV) complexes against different kinds of diseases and medical conditions. Two novel QSAR models based on the BiS algorithm are developed and validated, capable to predict activities against the SARS-CoV virus and its RNA dependent RNA polymerase.
Results:
The internal predictive power of the QSAR models was tested by 10-fold cross-validation, giving cross-R2 from 0.863 to 0.903. 38 different activities, ranging from antioxidant, antibacterial, and antiviral activities, to potential anti-inflammatory, anti-arrhythmic and anti-malarial activity were predicted for a series of eighteen platinum(IV) complexes.
Conclusion:
Complexes 1, 3 and 13 have high generalized optimality criteria and are predicted as potential SARS-CoV RNA dependent RNA polymerase inhibitors.
Keywords: SARS-CoV, Drug repurposing, Platinum(IV) complexes, Generalized optimality criterion, RNA dependent RNA polymerase inhibitor
Copyright and License Information
© 2023 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Platinum(IV) complexes are promising compounds that demonstrate anti-inflammatory, antimicrobial (L. monocytogenes, S. aureus, E. coli, S. typhi, B. abortus, M. luteus), and antifungal (A. fumigatus, C. albicans) properties.1,2 They also demonstrated high antitumor activity against certain tumor cell lines, such as lung cancer, ovarian carcinomas, colon carcinomas,3-5 and bladder cancer.6 From a synthetic point of view and with respect to the method of administration of drugs into the body, platinum(IV) complexes have a number of advantages over platinum(II) complexes. First, the kinetic inertness of platinum(IV) complexes reduces the possibility of adverse reactions in vivo.7 This is because they are more stable in acidic environments forming as a result of the activity of bacteria (the production of folic acid for DNA synthesis), and are more active against these bacterial cultures.2 Secondly, these compounds are able to penetrate cells intact and undergo reduction inside the cell. The reduction of platinum(IV) to platinum(II) by biological agents is necessary for their antitumor activity.8,9 Finally, platinum(IV) complexes do not lose potency against cancer cells under hypoxic conditions, therefore, they are potential candidates for the treatment of avascular tumors.10 Recently, hexachloroplatinate(IV) complexes with pyridinium and benzimidazole groups were tested and were shown to inhibit two carcinoma cell lines (A549 and CNE-2).11
The potential of platinum compounds in combating viral diseases is currently being investigated by several groups. Castro et al reviewed platinum compounds showing promising antiviral properties.12 They showed that different ligands in the coordination sphere of platinum may have a significant impact on the efficacy and pharmacokinetic profiles of new potential antiviral drugs. One of the platinum-based antiviral agents, TriplatinNC, protects cells from enterovirus 71 and human metapneumovirus infection.13 It blocks viral entry by shielding cells from virus attack, opening new directions for the development of metalloshielding antiviral drugs. Another approach in the development of antiviral metal preparations for the treatment of COVID-19 is considering metallodrug interactions with biomolecules related to viral replication.14 Thus, the antiviral activity of platinum coordination compounds and their use as antiviral agents is of special interest, especially during the COVID-19 pandemic.
Globally, as of 8 February 2022, there have been over 396 million confirmed cases of COVID-19, including more than 5.7 million deaths, reported to WHO.15 COVID-19 is a contagious disease caused by a new member of the Coronaviridae family, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Recently, the 3D structure of SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) has been resolved by cryo-electron microscopy.16 The pivotal role of RdRp in the replication of a virus’s genome makes it a perfect target for drug design. Several studies have used computational chemistry techniques trying to repurpose existing drugs and identify potential RdRp inhibitors.17-20 Based on a molecular docking study, Elfiky identified ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir as potential anti-COVID-19 drugs.17 Byléhn et al were interested in inhibition mechanisms19 and showed that remdesivir follows a similar interaction pattern as natural base adenine, and proposed a delayed termination mechanism.
A variety of statistical methods, such as linear, polynomial, and nonlinear regressions, partial least squares, or principal component analysis, are used for QSAR model design. Recently, with advances in computer power, machine learning entered and revolutionized the field of computationally aided drug design.21-23 Following the recommendations of Tropsha et al24–26 for QSAR model design and validation, like selecting only accurate, precise and consistent experimental data for the dataset, and rigorous validation of predictive models, it is possible to design models with high predictive power.
For a drug to show therapeutic effects, it has to interact with some molecule of the targeted organism, e.g., a peptide, an enzyme, or DNA. With a proper description of those interactions, it is possible to design a decision rule for classifying molecules as active or inactive. On www.chemosophia.com,27 an on-line platform for cheminformatics, bioinformatics, and drug design, two approaches for the prognosis of bioactivities are adopted, based on 3D QSAR molecular interior and exterior models. Since all calculations were performed using the ChemoSophia platform, we briefly revise both techniques in the Methods section. Recently, a successful QSAR model using a machine learning model was designed and validated.28 It was applied in a drug-repurposing study, and among more than 6000 medical compounds from the DrugBank database potential SARS-CoV 3CLpro inhibitors were shortlisted. Motivated by the success of this study, we develop two models capable of predicting the biological activity of unknown compounds against SARS viruses. A series of platinum complexes are tested for potential biological activity, with the aim of finding new medical application for them.
Materials and Methods
A variety of QSAR models were used to predict the bioactivity of platinum complexes against a series of 38 medical conditions. 36 of them are available on chemosophia.com, while we developed and validated two novel models against SARS-CoV (all mechanisms) and SARS-CoV RNA dependent RNA polymerase as a part of the present study. Since all these models are based on the algorithms used for the reconstruction of a molecular field of the model receptor, a short overview of those algorithm is presented.
The reconstruction of the molecular field of the model receptor is a cornerstone of a Biological Substrate Search (BiS) algorithm.29 The molecular field is represented by Coulomb and van der Waals potentials on the molecular surface. Pseudo-atoms of the model receptor are calculated based on the complementarity principle between a ligand and the receptor. The orientation of each subsequent molecule in the training dataset is optimized in the obtained field, with the minimal overall probability (P) with
being the function to be minimized. M is the total number of pseudo-atoms of the receptor, and pm equals
The contributions to the Em are calculated using the MERA force field. After each molecule, the complementary receptor field is updated. For technical details see references.29-31 Once the receptor’s molecular field has been reconstructed and the desirability function defined, it is possible to place a new molecule into the receptor, reorient it, calculate the interactions, and classify it as being active (P > 0.5) or inactive (P ≤ 0.5). The BiS algorithm was tested using different small molecule datasets and for various kinds of bioactivity and proved to be a high-quality classification scheme, with cross-validation quality usually above 0.9.30,32-36 The algorithm calculates the entire spectrum of interaction characteristics, including interaction energies (Ej), forces (Fj), and force constants (kj, elastic component)27,37:
(3)
where
and
are Coulomb and Van der Waals energies of the interaction of each m-th atom of the molecule with the j-th pseudo-atom of the receptor. Uj is the elastic energy of interaction of the molecule with the j-th pseudo-atom
∆rj is a deviation of the j-th pseudo-atom of the receptor from the average position when interacting with the molecule of the dataset. In addition, when equating the force constants to zero,
the algorithm can simulate an unlimitedly expandable receptor. This property is useful since it imitates receptor pockets, which are characterized by a large variation in size.
The use of a self-consistent field in BiS identifies the optimal arrangement of molecules in the complementary receptor until a constant energy value and values of the forces of intermolecular interactions equal to zero are achieved. In the general case, in the algorithm, the energy includes Coulomb, van der Waals interactions, and the elastic energy of intermolecular interactions—the latter depends on the force constants that determine the flexibility and the extensibility of the pseudo-receptor.
CoMIn is a 3D QSAR interior based method, with potentials (φ) at the junctions of the 3D lattice as descriptors.30,37 Besides classical potentials, such as Coulomb and van der Waals, the potential of hydrogen bonds or their combination weighted by factor wi, quantum descriptors (equation 6)
and its derivative (equation 7)
(7)
can also be used. wij is i-th weight factor of atom j (atomic charge, volume, distribution of electron density, the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), the difference of distribution of LUMO and HOMO, multiplied by the corresponding energy of the orbitals, and the products of these weight factors), rjm is the distance of atom j from the lattice junction m, and αj and βj are explained in Potemkin et al study.29 Those potentials are the descriptors used to design QSAR models. The electron density version of CoMIn algorithm is named ConGO.
The first step in the algorithm includes the superposition of the molecules from the training set and the computation of the generalized field described by potentials at the lattice junctions. The results include the Coulomb and van der Waals contributions (equations 3 and 4) and the potential of hydrogen bonds or their combination weighted by specific factors (equations 6 and 7) at the junctions of the 3D lattice in the space between the ligand molecule and the atoms of the pseudo-receptor. Those potentials are the descriptors used in the second step where the QSAR model is designed, correlating biological activity and the descriptors obtained by BiS or CoMIn algorithms. This is done by regression analysis (multiple linear, polynomial, non-linear, or transcendent) or by the partial least square technique. The final step is the estimation of the quality of the model, its validation, and the evaluation of its predictive power. Additionally, by analyzing the potentials it is possible to identify the pharmacophoric and anti-pharmacophoric fragments of the molecule. The whole procedure using the example of 5-HT1A agonists is described in detail in Potemkin et al study.37 Molecular orbitals were calculated using B3LYP functional.38 For light atoms 6-31+G(d,p) basis set was used, while for platinum, the LANL2DZ basis set with effective core potential was used.39 The visualization of molecular orbitals was performed in Gabedit.40
To design QSAR models capable of predicting bioactivity against the SARS-CoV virus and the inhibition potential against SARS-CoV RdRp, the pIC50 values characterizing the compounds’ bioactivity against the specific targets are were collected from the ChEMBL database.41,42 Data curation was performed according to the suggestions of Fourches et al,43 so salts and mixtures were removed. For duplicate inputs, more optimistic values were removed. After this process, the pIC50 values were within the 3.00 and 7.30 range for SARS-CoV RdRp and within 3.55 and 6.64 for SARS-CoV test sets. The SARS-CoV training set had 236 compounds (Table S1), while SARS-CoV RdRp had 154 (Table S2). 3D structures were generated by GP Global software available at chemosophia.com, and optimized using the MultiGen approach30,35,44 for the global minimization of energy without changing the geometries of stereo centers. To validate the performance of the methods, 10-fold cross-validation was applied. The final result of the QSAR model for SARS-CoV is a geometrical mean of neural network and multiple regression models desirability functions. Technical details about QSAR model design and validation are summarized in Table 1, while the basic steps are presented in Fig. 1.
Table 1.
Parameters and validation of QSAR models for a prognosis of anti-SARS-CoV bioactivity
|
QSAR model |
SARS-CoV
|
SARS-CoVRdRp
|
| Method |
1) Neural network (3 layers)
2) Multiple regression |
Neural network |
| Algorithm |
1) CoMIn
2) BiS |
CoMIn |
| Potential |
1) HOMO distribution
2) Coulomb, van der Waals and elastic energy |
The LUMO distribution derivative |
| Cross-R2 |
1) 0.903
2) 0.881 |
0.863 |

Fig. 1.
Workflow from data collection to the successful QSAR model design.
.
Workflow from data collection to the successful QSAR model design.
Since all compound geometries used to generate models were optimized by the MultiGen approach30,35,44 and MM3 force field,45 the structures of platinum complexes were minimized using the same approach, with water as a solvent. Although it is expected that the difference in the structures of the complexes in crystal and in water solution are substantial, as an initial geometry estimate, geometries obtained by X-ray diffraction experiments were used. ChemoSophia,27 an online application for chemoinformatics, bioinformatics, and computational toxicology research available at chemosophia.com, was used to predict 38 types of bioactivities (Table S3) and ADMET properties for minimum energy complexes.
The partial desirability functions for metabolism on isoforms 3A4 and 2D6 of cytochrome P450, and cytotoxicity, are determined from the desirability function:
where x is 3A4 (probability of metabolism on 3A4 isoform) or 2D6 (probability of metabolism on 2D6 isoform) or cyt (probability of cytotoxicity); a and b are parameters determined from the system of two equations. The first equation, when
(
is the mean value of x) the dx = 0.5. The second equation, when the value x = x + ∆ (∆ is the confidence interval of x at a confidence level of 0.95) the dx = 0.95.
Since logP should be in the range from 0 to 5, according to Lipinski’s rules, i.e., it has a lower and an upper bound, a two-sided desirability function is used:
(9)
where a, b and c are parameters determined from the system of three equations. The first equation, when logP = 0 (lower bound) the dlogP = 0.05. The second equation, when the value logP = 5 (upper bound) the dlogP = 0.05. The third equation, when the value logP= 2.5 (mean value) the dlogP = 0.95.
The generalized optimality criterion (d) is calculated as the geometric mean of partial desirability functions
(10)
Results and Discussion
Thirty-six out of the 38 QSAR models used in this study are publicly available at chemosophia.com. Those models are based on BiS, CiS and CoMIn algorithms used to calculate specific potentials (see equations 3–7) that serve as features in the subsequent step where regression or machine learning models were used to build the prediction model. For example, in reference,30 the authors go step by step through the algorithm and validation process with detailed explanations for four different models, including anti-tumor DNA-antimetabolites, factor Xa inhibitors, inhibitors of 5-HT1A and α1-AR receptors. Following the same approach, recently Novak and Potemkin published a model predicting bioactivity against the SARS-CoV main protease (3CLpro), with a cross-R2 value of 0.91.28
The investigated platinum(IV) complexes 1-18 were synthesized and characterized earlier (Table 2).46–50
Table 2.
Platinum complexes whose bioactivity were studied with references to their synthesis and characterization
| No. |
Formula
|
Reference
|
|
1
|
[(C2H5)4N]2[PtCl6] |
46
|
|
2
|
[(C2H5)2NH2]2[PtCl6] |
47
|
|
3
|
[(CH3)3NH]2[PtCl6] |
48
|
|
4
|
[Ph4P]2[PtCl6]×CH3CN |
48
|
|
5
|
[Ph3PCH=CHCH3]2[PtCl6] |
50
|
|
6
|
[Ph3PCH2OCH3]2[PtCl6] |
50
|
|
7
|
[Ph3PC2H5]2[PtBr6] |
48
|
|
8
|
[Ph3P(cyclo-C3H5)]2[PtBr6] |
48
|
|
9
|
[Ph3PCH2Ph]2[PtBr6] |
48
|
|
10
|
[Ph3PCH3]2[PtBr6] |
49
|
|
11
|
[Ph3PCH=CH2]2[PtBr6] |
49
|
|
12
|
[Ph3PCH2CH=CH2]2[PtBr6] |
49
|
|
13
|
[(C2H5)4N][PtCl5(DESO-S)] |
48
|
|
14
|
[Ph3PCH2CH=CHCH2PPh3][PtCl5(DMSO-S)] |
48
|
|
15
|
[Ph3PC2H5][PtCl5(DESO-S)] |
48
|
|
16
|
[Ph3PCH2OCH3][PtCl3(DMSO-S)] |
48
|
|
17
|
cis-[PtCl2(DESO-S)(PPh3)] |
48
|
|
18
|
[Ph3PCH2Ph][PtBr5(DMSO-S)] |
48
|
According to the X-ray analysis, the crystals in complexes 1-12 are ionic pairs consisting of two tetraorganylammonium or organyltriphenylphosphonium tetrahedral cations and one octahedral anion [PtCl6]2– or [PtBr6]2–. The phosphorus and nitrogen atoms in the cations have a slightly distorted tetrahedral coordination. The C-N-C and С-Р-С valence angles approach the ideal tetrahedral value, being between 109.0(16)° and 115.1(3)° for 1-3, and 106.8(2)° and 113.7(3)° for 4-12. The N–С bonds differ slightly from each other and are from 1.441(2) Å to 1.494(3) Å (1–3). The P–C bond lengths are from 1.785(6) Å to 1.842(8) Å for 4-12 and are slightly lower than the sum of the covalent radii of phosphorus and carbon atoms (1.88 Å 44). The center of the symmetry is located on the Pt(IV) atom of the hexachloroplatinate or hexabromoplatinate anions. Therefore, the asymmetric part of the reported structures contains half of the anion. A small distortion of the octahedral geometry was found for the anions: cis-angles Cl-Pt-Cl and Br-Pt-Br in the asymmetric part vary from 88.09(2)° to 91.47(3)°. Trans-angles Сl-Pt-Cl and Br-Pt-Br are almost equal to the theoretical value 180.0°. The cations of complexes (13-16, 18) have similar bond lengths and bond angles as (1-12). Anions have a slightly distorted octahedral configuration.
In water solutions, organic cations surround the Pt(IV) anion. In complex 1, equatorial Pt-Cl bond lengths are 2.277 Å, and axial 2.282 Å, with just minor deviations from a perfect octahedron. The organic part of the complex, the tetraethylammonium cations, have direct van der Waals contacts with four chlorine anions. The substitution of bulky tetraethylammonium cations by trimethylammonium cations (complex 3) is reflected in Pt-Cl bond lengths, which are longer compared to 1.
By replacing a chlorine anion with a diethyl sulfoxide, complex 13 is obtained. Compound 13, with desirability equal to 0.54, is a complex with octahedral geometry in water solution. Diethyl sulfoxide is in a coordination sphere of Pt(IV), together with five chlorine anions. Pt-Cl bond length, where chlorine is in the axial position (or trans relative to Sulphur), is 2.346 Å and it is the longest of all Pt-Cl bonds. Pt-S distance is slightly longer (2.350 Å), and the angle S-Pt-Cl is 175.7°. The tetraethylammonium cation is positioned closer to the diethyl sulfoxide, having close contact with one of the equatorial chlorine anions.
After geometry optimization for all 18 complexes, 38 kinds of bioactivities were predicted, together with ADMET properties. The results of ADMET property predictions are presented in Table 3. Most of the complexes have high scores for being metabolized on at least one of two cytochrome P450 isoforms (1 = a high probability of metabolism, 0 = low probability of metabolism). This fact by itself does not mean that a complex cannot be used as a drug. It can, if it remains after metabolism sufficiently to exploit the therapeutic effect. Generalized optimality criterion (d) is a criterion based on particular desirability functions, with the most desirable molecule having a score of 1. Only four complexes (1-3, 13) satisfy Lipinski’s recommendation for lipophilicity, usually expressed as a partition coefficient in 1-octanol or a water system, with logP values in the range between 0.9 (2) and 4.2 (13). Chemosophia’s toxicity classification model identified all Pt complexes as cytotoxic. Toxicity resulting from cisplatin administration, one of the most widely used and successful chemotherapeutic agents, includes kidney damage, hearing loss, severe nausea and vomiting, peripheral nerve damage, and bone marrow suppression.51 As the lethal dose is significantly higher than their therapeutic concentrations, those compounds might have practical therapeutic potential. Therefore, additional research is needed. The general desirability of a complex as a potential drug (d) is calculated as the geometrical mean of particular (property based) desirabilities. According to the desirability rule, the top three platinum complexes with the best ADMET properties are 1, 3, and 13. Compounds with organyltriphenylphosphonium moiety have undesirable ADMET properties.
Table 3.
Predicted ADMET properties of Pt(IV) complexes and their desirability as potential drugs
|
|
3A4
|
2D6
|
cyt
|
logP
|
d
3A4
|
d
2D6
|
d
logP
|
d
cyt
|
d
|
|
1
|
0.7756 |
0.2238 |
1.0 |
1.4500 |
0.9774 |
0.9942 |
0.9002 |
0.1268 |
0.5771 |
|
2
|
0.9077 |
0.9026 |
1.0 |
0.9100 |
0.6260 |
0.0000 |
0.7666 |
0.1268 |
0.0330 |
|
3
|
0.0000 |
0.4593 |
1.0 |
1.1500 |
1.0000 |
0.9237 |
0.8454 |
0.1268 |
0.5610 |
|
4
|
0.9540 |
0.6527 |
1.0 |
6.8400 |
0.2586 |
0.5077 |
0.0000 |
0.1268 |
0.0000 |
|
5
|
0.9713 |
0.4839 |
1.0 |
7.7367 |
0.1341 |
0.9011 |
0.0000 |
0.1268 |
0.0000 |
|
6
|
0.9795 |
0.6722 |
1.0 |
6.6233 |
0.0886 |
0.4310 |
0.0000 |
0.1268 |
0.0000 |
|
7
|
0.9891 |
0.1287 |
1.0 |
8.1337 |
0.0488 |
0.9980 |
0.0000 |
0.1268 |
0.0000 |
|
8
|
0.9893 |
0.0006 |
1.0 |
7.9337 |
0.0482 |
0.9995 |
0.0000 |
0.1268 |
0.0000 |
|
9
|
0.9803 |
0.6156 |
1.0 |
8.4203 |
0.0848 |
0.6382 |
0.0000 |
0.1268 |
0.0000 |
|
10
|
0.8906 |
0.8469 |
1.0 |
7.8337 |
0.7281 |
0.0029 |
0.0000 |
0.1268 |
0.0000 |
|
11
|
0.9826 |
0.8268 |
1.0 |
8.1337 |
0.0740 |
0.0093 |
0.0000 |
0.1268 |
0.0000 |
|
12
|
0.9231 |
0.6885 |
1.0 |
8.3337 |
0.5135 |
0.3649 |
0.0000 |
0.1268 |
0.0000 |
|
13
|
0.0297 |
0.3044 |
1.0 |
4.2413 |
1.0000 |
0.9859 |
0.6914 |
0.1268 |
0.5422 |
|
14
|
0.9392 |
0.6887 |
1.0 |
7.4500 |
0.3818 |
0.3638 |
0.0000 |
0.1268 |
0.0000 |
|
15
|
0.9902 |
0.6244 |
1.0 |
7.6667 |
0.0454 |
0.6094 |
0.0000 |
0.1268 |
0.0000 |
|
16
|
0.9555 |
0.5986 |
1.0 |
5.0150 |
0.2466 |
0.6895 |
0.0430 |
0.1268 |
0.1745 |
|
17
|
0.7633 |
0.0000 |
1.0 |
8.8900 |
0.9829 |
0.9995 |
0.0000 |
0.1268 |
0.0000 |
|
18
|
0.5802 |
0.5934 |
1.0 |
7.5250 |
0.9997 |
0.7039 |
0.0000 |
0.1268 |
0.0000 |
3A4, 2D6 – isoforms of cytochrome P450, cyt – cytotoxicity, logP – lipophilicity, d –desirability function.
Complexes 1, 3, and 13 (Fig. 2) show potential activity against a variety of conditions (Table 4) and have the highest generalized optimality criterion. Since all three of them are toxic, it is not of special importance to suggest them against conditions that are not serious or for which less toxic drugs already exist.
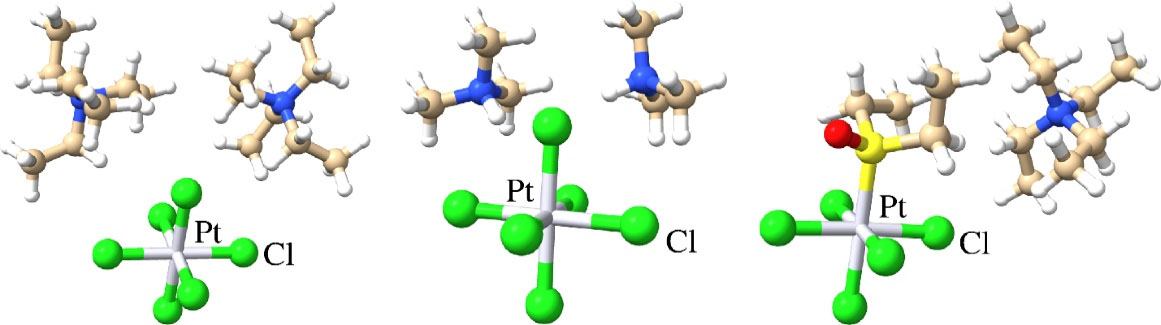
Fig. 2.
Structures of Pt(IV) complexes with favorable drug desirability. 1 (left), 3 (middle) and 13 (right).
.
Structures of Pt(IV) complexes with favorable drug desirability. 1 (left), 3 (middle) and 13 (right).
Table 4.
Biological activity of platinum complexes with high desirability function
|
Target/condition
|
1
|
3
|
13
|
| α1 Adrenergic receptor inhibitors |
Yes |
No |
No |
| Conduction anesthesia |
Yes |
Yes |
No |
| DNA gyrase inhibition |
No |
Yes |
No |
| Antioxidant activity |
No |
No |
Yes |
| Anti-arrhythmic agents |
Yes |
No |
No |
| SARS-CoV RdRp inhibitors |
Yes |
No |
Yes |
| Benzodiazepine receptor inhibitors |
Yes |
No |
No |
| Human factor Xa inhibitors |
No |
No |
No |
| HIV1 |
No |
Yes |
Yes |
| LOX5 inhibitors |
No |
Yes |
No |
| Influenza A |
Yes |
No |
Yes |
| Influenza B |
No |
Yes |
No |
There are several hundred proteins and double stranded DNA fragments complexed with coordinative compounds of platinum in the RSCB PDB database (www.rscb.org). Zhao et al52 recently designed, synthesized and measured bioactivity against cancer cell lines for three biotinylated platinum(IV) complexes. Their molecular docking studies confirmed that non-covalent interactions are responsible for the effective binding of the biotin moieties of all the studied platinum(IV) complexes for streptavidin, while platinum did not affect their binding abilities. Platinum introduces additional stabilization to the complex, compared to the protein-ligand complex without platinum. Sankarganesh et al53 performed spectroscopic titrations, viscometric measurements, and molecular docking analysis to show that gold and platinum complexes intercalate to circulating tumor DNA. Here, we focus on anti-SARS activity, where Pt(IV) complexes may act as RdRp inhibitors. Compounds 1 and 13 exhibit potential bioactivity against SARS-CoV RdRp and are further analyzed. Bioactivity predictions for all Pt(IV) complexes are collected in Table S4 in Supplementary file 1.
The pharmacophoric and antipharmacophoric fragments are obtained by analysis of the generalized field.37 Fig. 3 shows pharmacophoric (red) and antipharmacophoric (blue) fragments of complexes 1 and 13 in the modelled complementary field of SARS-CoV RdRp. It can be seen that metallic center (platinum) and organic ligand exhibit mostly antipharmacophoric properties. Just like in the example of sodium chloride, where combining reactive metallic sodium and poisonous gaseous chlorine forms table salt (sodium chloride) with completely different properties from original compounds, here one can also observe ‘the whole is bigger than the sum of its parts’ effect. Although separately platinum and the organic ligand do not show bioactivity, when they interact, they are predicted to have significant bioactivity against a variety of conditions. We believe that the emergence of bioactivity is a consequence of the interaction of valence shell electrons from the platinum with chlorine and that this interaction is modulated further by ligand and/or diethyl sulfoxide. Fig. 3 shows that equatorial chlorine anions, closest to the ethylene moiety of compound 1, are pharmacophoric, together with axial one, pointing away from ligands. The analysis of HOMO, presented in Fig. 4, shows that there are 3p orbitals centered on those specific chlorine anions and the
orbital of platinum. The energy difference between HOMO and HOMO-1 orbitals is only 0.027 eV, while the HOMO–LUMO gap is 4.01 eV.
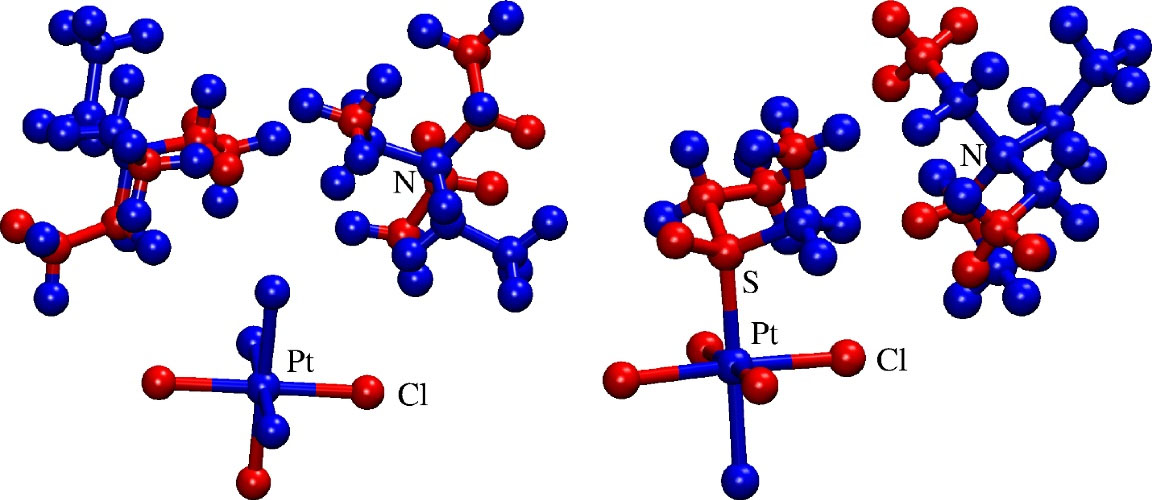
Fig. 3.
The distribution of pharmacophore (red) and anti-pharmacophore (blue) parts of compounds 1 (left) and 13 (right), identified by the generalized field of SARS-CoV RNA dependent RNA polymerase.
.
The distribution of pharmacophore (red) and anti-pharmacophore (blue) parts of compounds 1 (left) and 13 (right), identified by the generalized field of SARS-CoV RNA dependent RNA polymerase.

Fig. 4.
Frontier molecular orbitals of complex 1, calculated on B3LYP/LanL2DZ level of theory.
.
Frontier molecular orbitals of complex 1, calculated on B3LYP/LanL2DZ level of theory.
The scientific community and medical practice offer proof of the importance of platinum compounds, such as cisplatin, carboplatin, oxaliplatin, nedaplatin, and lobaplatin.54 Several platinum-based agents are well documented, and much is known about the mechanism of action in anticancer treatments.55 Some insights are also obtained about their interaction with the target.52,53 Considering the good quality of the training sets and the robustness and successful practical demonstrations of the applied methods,28,30 our results suggest that platinum complexes could be used against a wider range of medical conditions, including viruses. Nevertheless, although QSAR modeling is a valuable tool to screen large compound libraries and identify drug candidates, we hope that our study will attract interest and motivate experimental groups to conduct research in complex in vitro and in vivo experimental systems.
Conclusion
Cisplatin [cis-diamminedichloroplatinum(II)] was the first platinum complex registered as a chemotherapeutic agent against various types of cancers. Further investigations resulted in a new class of antineoplastic drugs—carboplatin and oxaliplatin. A computationally aided drug designed coupled with artificial intelligence might be a game changer, lowering the costs and speeding up the process of drug development. Present day models are capable of predicting bioactivities, cytotoxicity, and ADMET properties, opening doors toward the design of selective drugs with minimal, or ideally without, side effects. Platinum(IV) complexes might have an important role in this process.
In the first part of the paper, two new QSAR models predicting activity against SARS-CoV and its RdRp are developed. 10-fold cross-validation indicates their robustness and high predictive ability. Those models are based on the re-creation of the complementary receptor’s molecular field by BiS or CoMIn algorithms and are being implemented at chemosophia.com. To reach the second goal, the biological activity was predicted for a series of recently synthesized and characterized platinum(IV) complexes. Altogether, 38 different activities, ranging from antioxidant, antibacterial, and antiviral activities, to potential anti-inflammatory, anti-arrhythmic, and anti-malarial agents were predicted. Geometries of platinum complexes obtained by X-ray structure analysis were optimized, and the most stable structures in water solution were obtained. In the second stage, pharmacokinetic and cytotoxicity properties were predicted, and drug desirability properties were calculated. Three platinum complexes with a desirability factor above 0.5, although being predicted as toxic, are identified as hit molecules and their potential application against 12 medical conditions is shortlisted. Compounds 1 and 13 have high scores as potential inhibitors of SARS-CoV RdRp. Although a closer analysis of pharmacophoric parts of the complex reveals that both platinum(IV) and a majority of ligand atoms are antipharmacophoric in nature, we believe that biological activity arises dominantly from the interaction of electrons from platinum(IV) and chlorine anions, but interaction with a ligand cannot be ruled out.
Research Highlights
What is the current knowledge?
√ Cisplatin [cis-diamminedichloroplatinum(II)] was the first platinum complex registered as a chemotherapeutic agent against various types of cancers.
√ Present day models are capable of predicting bioactivities, cytotoxicity and ADMET properties, opening the door toward the design of selective drugs with minimal, or ideally without, side effects.
What is new here?
√ The cytotoxicity, ADMET properties and bioactivities against 38 different kinds of diseases and medical conditions have been predicted for a series of platinum(IV) complexes.
√ Two QSAR models for predicting activity against SARS-CoV (cross-R2 = 0.903 and 0.881) and SARS-CoV RNA dependent RNA polymerase (cross-R2 = 0.863) have been designed and validated.
√ Pt(IV) complexes with organyltriphenylphosphonium moiety have undesirable ADMET properties.
√ Compounds 1, 3 and 13 have a favorable desirability function and have predicted activity against various medical conditions, including inhibition potential against SARS-CoV RNA dependent RNA polymerase.
√ The analysis of (anti)pharmacophoric fragments reveal that majority of the atoms are antipharmacophoric, indicating that bioactivity is a property that emerges as a result of interaction between platinum(IV), halogen anions and organic moieties.
Funding sources
This work was supported by the RFBR, DST, CNPq and SAMRCA under Grant 20-53-80002.
Competing interests
The authors declare no conflict of interests.
Supplementary files
Supplementary file 1 contains Tabales S1- S4.
(pdf)
References
- Watanabe T, Takano M, Ogasawara A, Mikami T, Kobayashi T, Watabe M, et al. Anti-Candida Activity of a New Platinum Derivative. Antimicrob Agents Chemother 2000; 44: 2853–4. 10.1128/AAC.44.10.2853-2854.2000.
- Öğütçü H, Yetim NK, Özkan EH, Eren O, Kaya G, Sarı N, et al. Nanospheres caped Pt(II) and Pt (IV): synthesis and evaluation as antimicrobial and Antifungal Agent. Pol J Chem Technol 2017; 19: 74–80. 10.1515/pjct-2017-0011.
- Gramatica P, Papa E, Luini M, Monti E, Gariboldi MB, Ravera M. Antiproliferative Pt(IV) complexes: synthesis, biological activity, and quantitative structure–activity relationship modeling. J Biol Inorg Chem 2010; 15:1157-69. doi: 10.1007/s00775-010-0676-4 [Crossref] [ Google Scholar]
- Varbanov HP, Jakupec MA, Roller A, Jensen F, Galanski M, Keppler BK. Theoretical Investigations and Density Functional Theory Based Quantitative Structure–Activity Relationships Model for Novel Cytotoxic Platinum(IV) Complexes. J Med Chem 2013; 56:330-44. doi: 10.1021/jm3016427 [Crossref] [ Google Scholar]
- Hizal S, Hejl M, Jakupec MA, Galanski M, Keppler BK. Synthesis, characterization, lipophilicity and cytotoxic properties of novel bis(carboxylato)oxalatobis(1-propylamine)platinum(IV) complexes. InorganicaChim Acta 2019; 491:76-83. doi: 10.1016/j.ica.2019.03.036 [Crossref] [ Google Scholar]
- Łakomska I, Wojtczak A, Sitkowski J, Kozerski L, Szłyk E. Platinum(IV) complexes with purine analogsStudies of molecular structure and antiproliferative activity in vitro. Polyhedron 2008; 27:2765-70. doi: 10.1016/j.poly.2008.05.032 [Crossref] [ Google Scholar]
- Galanski M, Jakupec M, Keppler B. Update of the Preclinical Situation of Anticancer Platinum Complexes: Novel Design Strategies and Innovative Analytical Approaches. Curr Med Chem 2005; 12:2075-94. doi: 10.2174/0929867054637626 [Crossref] [ Google Scholar]
- Hall MD, Hambley TW. Platinum(IV) antitumour compounds: their bioinorganic chemistry. Coord Chem Rev 2002; 232:49-67. doi: 10.1016/S0010-8545(02)00026-7 [Crossref] [ Google Scholar]
- Weaver EL, Bose RN. Platinum(II) catalysis and radical intervention in reductions of platinum(IV) antitumor drugs by ascorbic acid. J InorgBiochem 2003; 95:231-9. doi: 10.1016/S0162-0134(03)00136-3 [Crossref] [ Google Scholar]
- Hall MD, Mellor HR, Callaghan R, Hambley TW. Basis for Design and Development of Platinum(IV) Anticancer Complexes. J Med Chem 2007; 50:3403-11. doi: 10.1021/jm070280u [Crossref] [ Google Scholar]
- Zhao J, Chen F, Han Y, Chen H, Luo Z, Tian H. Hydrogen-bonded organic–inorganic hybrid based on hexachloroplatinate and nitrogen heterocyclic cations: Their synthesis, characterization, crystal structures, and antitumor activities in vitro. Molecules 2018; 23:1397. doi: 10.3390/molecules23061397 [Crossref] [ Google Scholar]
- de Castro F, de Luca E, Benedetti M, Fanizzi FP. Platinum compounds as potential antiviral agents. Coord Chem Rev 2022; 451:214276. doi: 10.1016/j.ccr.2021.214276 [Crossref] [ Google Scholar]
- Bailly B, Gorle AK, Dirr L, Malde AK, Farrell NP, Berners-Price SJ. Platinum complexes act as shielding agents against virus infection. Chem Comm 2021; 57:4666-9. doi: 10.1039/d1cc01593a [Crossref] [ Google Scholar]
- de Paiva REF, Marçal Neto A, Santos IA, Jardim ACG, Corbi PP, Bergamini FRG. What is holding back the development of antiviral metallodrugs? A literature overview and implications for SARS-CoV-2 therapeutics and future viral outbreaks. Dalton Trans 2020; 49:16004-16033. doi: 10.1039/d0dt02478c [Crossref] [ Google Scholar]
- WHO Coronavirus (COVID-19) Dashboard [accessed on 2021 Nov 22] https://covid19.who.int/.
- Hillen HS, Kokic G, Farnung L, Dienemann C, Tegunov D, Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020; 584:154-6. doi: 10.1038/s41586-020-2368-8 [Crossref] [ Google Scholar]
- Elfiky AA. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci 2020; 253:117592. doi: 10.1016/j.lfs.2020.11759 [Crossref] [ Google Scholar]
- Indu P, Rameshkumar MR, Arunagirinathan N, Al-Dhabi NA, Valan Arasu M, Ignacimuthu S. Raltegravir, Indinavir, Tipranavir, Dolutegravir, and Etravirine against main protease and RNA-dependent RNA polymerase of SARS-CoV-2: A molecular docking and drug repurposing approach. J Infect Public Health 2020; 13:1856-61. doi: 10.1016/j.jiph.2020.10.015 [Crossref] [ Google Scholar]
- Byléhn F, Menéndez CA, Perez-Lemus GR, Alvarado W, de Pablo JJ. Modeling the Binding Mechanism of Remdesivir, Favilavir, and Ribavirin to SARS-CoV-2 RNA-Dependent RNA Polymerase. ACS Cent Sci 2021; 7:164-74. doi: 10.1021/acscentsci.0c01242 [Crossref] [ Google Scholar]
- Cannalire R, Cerchia C, Beccari AR, Di Leva FS, Summa V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J Med Chem 2022; 65:2716-2746. doi: 10.1021/acs.jmedchem.0c01140 [Crossref] [ Google Scholar]
- Lo YC, Rensi SE, Torng W, Altman RB. Machine learning in chemoinformatics and drug discovery. Drug Discov Today 2018; 23:1538-46. doi: 10.1016/j.drudis.2018.05.010 [Crossref] [ Google Scholar]
- Ekins S, Puhl AC, Zorn KM, Lane TR, Russo DP, Klein JJ. Exploiting machine learning for end-to-end drug discovery and development. Nat Mater 2019; 18:435-41. doi: 10.1038/s41563-019-0338-z [Crossref] [ Google Scholar]
- Mak KK, Pichika MR. Artificial intelligence in drug development: present status and future prospects. Drug Discov Today 2019; 24:773-80. doi: 10.1016/j.drudis.2018.11.014 [Crossref] [ Google Scholar]
- Golbraikh A, Tropsha A. Beware of q2!. J Mol Graph Model 2002; 20:269-76. doi: 10.1016/S1093-3263(01)00123-1 [Crossref] [ Google Scholar]
- Tropsha A, Gramatica P, Gombar VK. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb Sci 2003; 22:69-77. doi: 10.1002/qsar.200390007 [Crossref] [ Google Scholar]
- Tropsha A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol Inform 2010; 29:476-88. doi: 10.1002/minf.201000061 [Crossref] [ Google Scholar]
- Potemkin V, Grishina M. Grid-Based Technologies for In Silico Screening and Drug Design. Curr Med Chem 2018; 25:3526-37. doi: 10.2174/0929867325666180309112454 [Crossref] [ Google Scholar]
- Novak J, Potemkin VA. A new glimpse on the active site of SARS-CoV-2 3CLpro, coupled with drug repurposing study. Mol Divers 2022; 26:2631-45. doi: 10.1007/s11030-021-10355-8 [Crossref] [ Google Scholar]
- Potemkin VA, Pogrebnoy AA, Grishina MA. Technique for energy decomposition in the study of “receptor-ligand” complexes. J Chem Inf Model 2009; 49:1389-406. doi: 10.1021/ci800405n [Crossref] [ Google Scholar]
- Potemkin VA, Grishina MA. A new paradigm for pattern recognition of drugs. J Comput Aided Mol Des 2008; 22:489-505. doi: 10.1007/s10822-008-9203-x [Crossref] [ Google Scholar]
- Potemkin V, Grishina M. Principles for 3D/4D QSAR classification of drugs. Drug Discov Today 2008; 13:952-9. doi: 10.1016/j.drudis.2008.07.006 [Crossref] [ Google Scholar]
- Grishina MA, Pogrebnoi AA, Potemkin VA, Zrakova TY. Theoretical study of the substrate specificity of cytochrome P-450 isoforms. Pharm Chem J 2005; 39:509-13. doi: 10.1007/s11094-006-0011-0 [Crossref] [ Google Scholar]
- Potemkin VA, Grishina MA, Fedorova OV, Rusinov GL, Ovchinnikova IG, Ishmetova RI. Theoretical investigation of the antituberculous activity of membranotropic podands. Pharm Chem J 2003; 37:468-72. doi: 10.1023/B:PHAC.0000008246.07413.d9 [Crossref] [ Google Scholar]
- Mikuchina K, Potemkin V, Grishina M, Laufer S. 3D QSAR analysis and pharmacophore modelling of p38 MAP kinase inhibitors using BiS algorithm. Arch Pharm Pharm Med Chem 2002; 335:74. [ Google Scholar]
- Potemkin VA, Arslambekov RM, Bartashevich EV, Grishina MA, Belik AV, Perspicace S. Multiconformational method for analyzing the biological activity of molecular structures. J Struct Chem 2002; 43:1126-30. [ Google Scholar]
- Potemkin VA, Grishina MA, Belik AV, Chupakhin ON. Quantitative relationship between structure and antibacterial activity of quinolone derivatives. Pharm Chem J 2002; 36:22-5. doi: 10.1023/A:1015744707357 [Crossref] [ Google Scholar]
- Potemkin AV, Grishina MA, Potemkin VA. Grid-based Continual Analysis of Molecular Interior for Drug Discovery, QSAR and QSPR. Curr Drug Discov Technol 2017; 14:1-25. doi: 10.2174/1570163814666170207144018 [Crossref] [ Google Scholar]
- Becke AD. Becke ADDensity-functional thermochemistryIIIThe role of exact exchange. J Chem Phys 1993; 98:5648. doi: 10.1063/1.464913 [Crossref] [ Google Scholar]
- Hay PJ, Wadt WR. Hay PJ, Wadt WRAb initio effective core potentials for molecular calculationsPotentials for K to Au including the outermost core orbitals. J Chem Phys 1985; 82:299-310. doi: 10.1063/1.448975 [Crossref] [ Google Scholar]
- Allouche AR. Gabedit-A graphical user interface for computational chemistry softwares. J Comput Chem 2011; 32:174-82. doi: 10.1002/jcc.21600 [Crossref] [ Google Scholar]
- Davies M, Nowotka M, Papadatos G, Dedman N, Gaulton A, Atkinson F. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res 2015; 43:W612-20. doi: 10.1093/nar/gkv352 [Crossref] [ Google Scholar]
- Gaulton A, Hersey A, Nowotka ML, Patricia Bento A, Chambers J, Mendez D. The ChEMBL database in 2017. Nucleic Acids Res 2017; 45(D1):D945-54. doi: 10.1093/nar/gkw1074 [Crossref] [ Google Scholar]
- Fourches D, Muratov E, Tropsha A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J Chem Inf Model 2010; 50:1189-204. doi: 10.1021/ci100176x [Crossref] [ Google Scholar]
- Bartashevich EV, Potemkin VA, Grishina MA, Belik AV. A method for multiconformational modeling of the three-dimensional shape of a molecule. J Struct Chem 2002; 43:1033-9. [ Google Scholar]
- Potemkin VA, Bartashevich EV, Belik AV. A New Approach to Predicting the Thermodynamic Parameters of Substances from Molecular Characteristics. Russ J Phys Chem 1996; 70(3):411-6. [ Google Scholar]
- Tkacheva A. Synthesis and Structure of the Platinum Complex [(C2H5)4N]2[PtCl6]. Bull South Ural State Univ Ser Chem 2017; 9:74-6. doi: 10.14529/chem170412 [Crossref] [ Google Scholar]
- Tkacheva AR, Sharutin VV, Sharutina OK, Shlepotina NM, Kolesnikov ОL, Shishkova YS. Tetravalent Platinum Complexes: Synthesis, Structure, and Antimicrobial Activity. Russ J Gen Chem 2020; 90:655-9. doi: 10.1134/S1070363220040155 [Crossref] [ Google Scholar]
- Zykova A. Synthesis and Structure of Aryl Phosphorus Compounds. Bull South Ural State Univ Ser Chem 2020; 12:5-50. doi: 10.14529/chem200401 [Crossref] [ Google Scholar]
- Zykova AR, Sharutin V V. Zykova AR, Sharutin V V, Sharutina OKNew Organyltriphenylphosphonium Hexabromoplatinates [Ph3PR]2[PtBr6], R = CH3, CH=CH2, CH2CH=CH2. Russ J Inorg Chem 2021; 66(1):56-60. doi: 10.1134/S0036023621010149 [Crossref] [ Google Scholar]
- Tkacheva AR, Sharutin VV, Sharutina OK, Slepukhin PA. Synthesis and Structure of Hexachloroplatinate Complexes. Russ J Gen Chem 2019; 89(9):1816-21. doi: 10.1134/S1070363219090147 [Crossref] [ Google Scholar]
- Johnstone TC, Pil PM, Lippard SJ. Cisplatin and Related Drugs. In: Reference Module in Biomedical Sciences. Elsevier; 2015. p. 1–14. 10.1016/B978-0-12-801238-3.98740-3.
- Zhao J, Hua W, Xu G, Gou S. Biotinylated platinum(IV) complexes designed to target cancer cells. J InorgBiochem 2017; 176:175-80. doi: 10.1016/j.jinorgbio.2017.08.017 [Crossref] [ Google Scholar]
- Sankarganesh M, Dhaveethu Raja J, Sakthikumar K, Solomon RV, Rajesh J, Athimoolam S. New bio-sensitive and biologically active single crystal of pyrimidine scaffold ligand and its gold and platinum complexes: DFT, antimicrobial, antioxidant, DNA interaction, molecular docking with DNA/BSA and anticancer studies. Bioorg Chem 2018; 81:144-56. doi: 10.1016/j.bioorg.2018.08.006 [Crossref] [ Google Scholar]
- Zhou J, Kang Y, Chen L, Wang H, Liu J, Zeng S, et al. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front Pharmacol 2020; 20; 11. 10.3389/fphar.2020.00343.
- Hato SV, Khong A, de Vries IJM, Lesterhuis WJ. Molecular Pathways: The Immunogenic Effects of Platinum-Based Chemotherapeutics. Clin Cancer Res 2014; 20(11):2831-7. doi: 10.1158/1078-0432.CCR-13-3141 [Crossref] [ Google Scholar]