Bioimpacts. 14(1):27513.
doi: 10.34172/bi.2023.27513
Original Article
Design and computational analysis of an effective multi-epitope vaccine candidate using subunit B of cholera toxin as a build-in adjuvant against urinary tract infections
Maryam Rezaei Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Software, Visualization, Writing – original draft, Writing – review & editing, 
Mehri Habibi Methodology, Software, Validation,
Parasoo Ehsani Validation,
Mohammad Reza Asadi Karam Conceptualization, Resources, Supervision, Visualization, , *
Saeid Bouzari Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing – review & editing, , *
Author information:
Molecular Biology Department, Pasteur institute of Iran, Tehran, Iran
Abstract
Introduction:
Urinary tract infection (UTI) is one of the most common infections, usually caused by uropathogenic Escherichia coli (UPEC). However, antibiotics are a usual treatment for UTIs; because of increasing antibiotic-resistant strains, vaccination can be beneficial in controlling UTIs. Using immunoinformatics techniques is an effective and rapid way for vaccine development.
Methods:
Three conserved protective antigens (FdeC, Hma, and UpaB) were selected to develop a novel multi-epitope vaccine consisting of subunit B of cholera toxin (CTB) as a mucosal build-in adjuvant to enhance the immune responses. Epitopes-predicted B and T cells and suitable linkers were used to separate them and effectively increase the vaccine's immunogenicity. The vaccine protein's primary, secondary, and tertiary structures were evaluated, and the best 3D model was selected. Since CTB is the TLR2 ligand, molecular docking was made between the vaccine protein and TLR2. Molecular dynamic (MD) simulation was employed to evaluate the stability of the vaccine protein-TLR2 complex. The vaccine construct was subjected to in silico cloning.
Results:
The designed vaccine protein has multiple properties in the analysis. The HADDOCK outcomes show an excellent interaction between vaccine protein and TLR2. The MD results confirm the stability of the vaccine protein- TLR2 complex during the simulation. In silico cloning verified the expression efficiency of our vaccine protein.
Conclusion:
The results of this study suggest that our designed vaccine protein could be a promising vaccine candidate against UTI, but further in vitro and in vivo studies are needed.
Keywords: Urinary tract infection, Multi-epitope vaccine, Molecular dynamic simulation
Copyright and License Information
© 2024 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Urinary tract infection (UTI) is the most common infection in adult women and the second most common infection in humans after respiratory tract infection.1,2 The results of different studies have shown that around 50% of women and 12% of men experience UTI, at least once in their life.3 Although UTIs are often treated empirically with antibiotics, the definitive antibiotic therapy in antibiotic resistance era may not be able to eliminate causative agents of UTI.4,5 Increasing rate of antibiotic-resistant lead to more mortality and high healthcare cost.6 These reasons highlight the need for alternative strategy to UTI treatment.7
Precaution approaches such as UTI vaccination can be helpful in solutions against threats.2
Much effort has been made to develop the UPEC vaccine, but no ideal vaccine is currently available for global use. One of the main reasons for their failure is the short-term protection that may be due to using specific virulence factors or whole cells in their preparation.8 Furthermore, live attenuated, killed organisms or subunit vaccines consist of a single protein, have large amounts of antigenic components that cause non-specific or allergic responses. The multi-epitope vaccine can be a good option for solving the problem.9 The vaccine design based on informatics tools and computational methods facilitates the identification of immunodominant B-cell and T-cell epitopes that can specifically stimulate both cellular and humoral immune responses.9-11
Epitope prediction and vaccine design based on immunoinformatics tools are more precise, reliable, affordable, and less time-consuming than conventional vaccines.11-13
Choosing the appropriate antigen is a critical step in the vaccine design process. Antigens involved in iron acquisition systems, adhesions, and highly conserved antigens are among the most promising vaccine candidate against UPEC.14 Therefore, our selection was made based on these antigens and considering the inhibition of colonization in the entire urinary tract.
The built-in adjuvant is a suitable alternative to the traditional adjuvant incorporated into the designed vaccine to overcome the problem of low immunogenicity of the vaccine.13,15 The subunit B of cholera toxin (CTB) was used as a built-in mucosal adjuvant in our vaccine design. CTB is a potent mucosal adjuvant that stimulates mucosal antibody responses; in addition, the coupling of CTB and antigens is known to increase immunogenicity against them by inducing B and T cell responses.16
The first stage of the innate immune response is detecting pathogen-associated molecular patterns (PAMPs) by pattern-recognition receptors (PRRs). Toll-like receptors (TLRs) are the most studied subset of PRRs. TLRs are expressed on innate immune cells, particularly antigen-presenting cells (APCs) such as dendritic cells (DCs).12,15,16 The TLR family is classified into two subfamilies: 1- cell surface TLRs, and 2- intracellular TLRs, each of which binds to different ligands.17
TLR2 is an essential receptor in the innate immune that initiates immune responses. Heat-labile enterotoxins (B-subunit) of Escherichia coli and V. cholera are among the PAMPs that bind to TLR2.16,18,19 Therefore, in our study, molecular docking was performed between CTB and TLR2.
This study aims to design an efficient multi-epitope subunit vaccine against UTI by considering: (I) Population coverage, (II) Evaluating various physicochemical properties and structural conformations, and (III) Evaluating the stability of the vaccine protein-receptor complex by conducting molecular dynamics (MD).
Materials and Methods
Antigen and adjuvant selection
According to previously published literature and considering the features required to be known as protective antigens, FdeC (factor adherence E. coli), Hma (Putative outer membrane receptor for iron compound or colicin), and UpaB (Adhesion outer membrane autotransporter barrel) were selected for our vaccine design. Our study used the cholera toxin B subunit (CTB) as a built-in adjuvant.
Protein sequences retrieval
The protein sequences of FdeC (Accession: AAN78896.1), Hma (Accession: AAN80941.1), and Upa B (Accession: AAN78907.1) of E. coli subsp. Cft073 and CtB (Accession: AET14210.1) of Vibrio cholerae serotype O1 (strain ATCC 39315 / El Tor Inaba N16961) were retrieved from Universal Protein Resource (Uniprot) database (http://www.uniprot.org/) in FASTA format.
Epitopes prediction
The different servers that have been used to predict B cell and T cell epitopes are mentioned below:
B cell epitopes prediction
B lymphocytes are responsible for secreting antibodies that are necessary for long-term protection.12 IEDB, ABCpred, BepiPred v2.0 and IgPred were used to predict B-cell linear epitopes of FdeC, Hma, and UpaB. The Immune Epitope Database (IEDB) is an online server that represents the data of immune epitopes on the antibody, T cell, and MHC binding in all species related to infectious, allergic, and autoimmune diseases and transplantation. IEDB tools also are used for epitope prediction and analysis.20 In the IEDB server, parameters such as hydrophilicity, flexibility, accessibility, turns, exposed surface, polarity, and the antigenic propensity of polypeptide chains were studied with the default window and the threshold value.21 ABCpred has used a systemic method based on a neural network to predict linear B cell epitopes.22 This server set the amino acid length at 20 mer and the scoring threshold at 0.51.
Cytotoxic T cell epitope predictions
Intracellular pathogens such as bacteria are recognized by cytotoxic T-cell receptor (TCR) of (CTL) in complex with MHC I molecules. MHC I molecules found on the surfaces of infected cells bind to the nine amino acid long peptides generated from proteins in the cells' cytosol and display these intracellular proteins to cytotoxic T cells. There are different allelic variants of MHCs, and each binds specific peptides.23 In our study, T cell epitope prediction was carried out using IEDB, NetMHC I, Epijen, and rankpep servers. HLA class I alleles A01:01, A02:01, A03:01, A11:01, and A24 in the human population, and H2Dd, H2Ld, and H2Kd in mice were chosen. Since our goal was to design a vaccine for humans, but our in vivo study would be done on mice, we tried to select epitopes that cover both the MHC of the human and mouse population. To predict MHC binding and T Cell epitope processing on the IEDB server, the peptide length was set at 9 mer.
Epitopes with percentile rank <0.5 in MHC binding and MHC IC50 < 500 nM in MHC I processing were chosen. The Net MHCI server predicts the binding of peptides to 81 different Human MHC alleles, including HLA-A, -B, -C, and –E, and 41 animals (Monkey, Cattle, Pig, and Mouse) based on artificial neural networks.23
On Net MHC I, the threshold for solid binders was set at 0.5%. Epijen server identifies T cell epitopes from protein sequence by considering proteasome cleavage, transporter-associated antigen processing (TAP) transport, MHC binding, and epitope selection. Passing through these steps concludes no more than 5% of the whole input protein sequence but will include 85% of the true epitopes.24 On Epijen, 5% was selected for an output cut-off. The rankpep server employ position specific scoring matrices method to predict peptide binding affinity to MHCI and II.25 On the rankpep server, the peptide length was set at 9 mer, and the other items were investigated by windows defaults.
Helper T lymphocyte epitope prediction
Helper T cells (HTLs) play a crucial role in stimulating both humoral and cellular immunity. Peptides derived from extracellular proteins are recognized by MHC class II on the surface of professional APCs. Many peptide-MHC complexes are presented to HTLs, but the T-Cell receptor only recognizes the HTL epitopes. HTL activates B cells to produce antibodies and also helps the CTL to eradicate infected cells.26-28
To predict T helper epitopes, MHCII alleles DRB1 01:01, DRB1 03:01, DRB1 04:01, DRB1 07:01, DRB1 11:01, DRB1 13:02, DRB1 15:01 and H21-Ad, H21-Ed were selected in human and mouse populations respectively. The length of the peptide was set at 15 mer. Prediction by IEDB tools was done on the basis of IC50<500 nM. NetMHCII and rankpep were also employed to predict MHCII binding epitopes by windows default. The NetMHCII server indicates the binding of peptides to MHC class II alleles with artificial neuron networks.28
Evaluation of HLA allele frequencies
The distribution and expression of HLA alleles may vary between regions and societies, and these discrepancies need to be considered in vaccine development.27 The Allele Frequency Net Database (AFND) contains information such as HLA alleles frequencies around the world.29 Therefore, the Allele frequencies server (http://www.allelefrequencies.net/) was used to evaluate the frequency of all types of HLA class I and II in almost all geographical regions of Iran.
Evaluation of epitopes antigenicity
Antigenicity evaluation is a fundamental step in vaccine development. The VaxiJen server evaluated the antigenicity of the selected epitopes. The VaxiJen server predicts the antigenicity of the query sequence based on physicochemical properties and is 70-89% accurate depending on the organism.30,31
Vaccine construction
To design a multi-epitope vaccine that can stimulate both humoral and cellular responses, selected B cell and T cell epitopes are linked together by GPGPG linkers. Furthermore, CTB was added to our vaccine construct by EAAAK linker as a built-in adjuvant to increase immune responses. Twelve constructions were designed based on epitope orders and CTB at the N-terminus or C-terminus of vaccine constructs.
Physicochemical characterization of vaccine construct
Various physicochemical properties of the vaccine construct, such as: 1- The number of amino acids, 2-Molecular weight, 3- Theoretical PI, 4- Extinction coefficient, 5- Estimated half-life, 6-instability index, 7- Aliphatic index, and 8- Grand average of hydropathicity, were evaluated by the ExPASy ProtParam tool. The physicochemical parameters of the given protein are calculated by comparing the pK values of different amino acids of the query sequence.
The aliphatic index of the protein shows the volume occupied by the aliphatic side chain (alanine, valine, isoleucine, and leucine). GRAVY is computed by the hydropathicity of all the amino acids divided by the number of amino acids in the protein sequence.32
Evaluation of tertiary structure
The I-TASSER server uses the threading technique to predict structure templates of query amino acid sequence from Protein Data Bank.33
Tertiary structure refinement and validation
The YASARA energy minimization server was used to refine the 3D model. In the next step, the quality of the best model was confirmed by the ProSA-web and the Ramachandran plot obtained from Swiss. Prot. In addition, the MolProbity web service calculated the crash score to carry out an all-atom contact analysis.
The ProSAweb is a tool to assess the overall quality of the generated 3D model of the protein based on the statistical analysis of the atomic coordinates of the protein model compared to all available protein structures using NMR and X-ray methods. The z-scores, outside the database average for native proteins, show errors in protein structure.34,35 Ramachandran plot classifies the amino acid residues in the favored, allowed, and outlier regions based on analysis of the phi-psi torsion angles of the protein backbone.36
mRNA structure prediction
The mfold program predicted the structure of the mRNA.
Evaluation of antigenicity, allergenicity, and solubility of protein vaccine
VaxiJen, AlgPred and AllerTOP servers, with a default parameter, were used to assess the antigenicity and avoid the allergic reaction of the designed vaccine. The solubility of the vaccine protein was analyzed at www.biotech.ou.edu/ protein sol server.
The AlgPred server uses multiple algorithms such as BLAST, IgE epitope, MAST, and SVMc to evaluate the allergenicity of the query sequence with high accuracy (85% precision at -0.4 threshold).12,36
The Protein-Sol server predicts protein solubility using a sequence-based method. This method, 35 sequence-based properties are calculated based on the differences between soluble and insoluble proteins from various databases. The solubility of the query sequence is analyzed and determined as the scaled solubility value (Query Sol).32,37
Evaluation of secondary structure
The secondary structure of the vaccine protein was analyzed with the PSIPRED workbench and the GOR4 server to determine the percentage of the helix, strand, and coil. The three-dimensional (3D) modeling was performed with the I-TASSER server. The 3D modeled structures were visualized by the programs PyMol and the UCSF Chimera.
PSIPRED uses the neural network method to investigate the secondary structure of the protein from amino acid sequences.38 The GOR4 server predicts the secondary structure based on information theory and using the amino acid sequence database with known secondary structure.39
Defining the binding site and molecular docking
The tertiary structure of TLR2, as a receptor of CTB, was obtained from Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB:2Z7X). A molecular docking study was carried out by
Haddock server to confirm the binding affinity of the designed vaccine protein withTLR2.The PRODIGY server was employed to predict the binding free energy (ΔG) and dissociation constant (Kd).
The software Accelrys Discovery Studio evaluated the hydrophobic region of vaccine protein.
Visualization and analysis of docking results were performed using PyMol software and LigPlot+ v.1.4.5.
Molecular dynamic simulation studies
To check the stability of vaccine protein and TLR2, GROMACS 2020.3 was employed.
Docked complex of the designed vaccine and TLR2 was submitted to MD simulations using the all-atom optimized potentials for liquid simulations (OPLS-AA) force field.40
The solvation of the system was analyzed with the SPC water model in a cubic box with a distance of 1Å between the molecule and the box edges. The system was neutralized with NA++ and Cl-. Energy minimization of the system was executed by the steepest descent minimization integrator and 5000 minimization steps. The maximum minimization force was less than 100 kJ.mol-1 nm-1. The system was equilibrated using NVT (constant number of particles, volume, and temperature) and NPT (endless number of particles, pressure, and temperature) algorithms.41 Finally, the MD simulation was carried out for 100 ns with 2 femtoseconds (fs) time step until the system reached the steady state. Output trajectories based on root mean square deviation (RMSD), fluctuation (RMSF), and gyration radius (Rg) were analyzed. The graphs were visualized by Xmgrace software.
Codon optimization and in silico cloning
The codon optimization based on codon usage in the expression host is necessary to achieve a high expression level. In our study, the DNA sequence of the vaccine protein was optimized using the Java Codon adaptation tool (JCAT). The codon adaptation index (CAI) and the GC content were calculated. The CAI ranges from 0 to 1; the higher CAI shows better expression in the expression host. Furthermore, the overall GC content between 30% to 70% is necessary for high-level expression in the host.42
The NcoI and XhoI restriction sites at the N- and C-termini of the final optimized vaccine sequence were added, and this sequence was cloned into the pET 28a(+) vector using the vector NTI and Snap-gene.
Immune simulation
In silico immunostimulation was performed using the C-ImmSim server to assess the immune response profile relative to the designed vaccine protein. C-ImmSim server is an online, dynamic immune simulation server that uses the Celada-Seiden model to estimate the adaptive immune humoral and cellular response against a given antigen.43 In our study, four injections through 28 days with 63 stimulation steps (1-time step equal to 8 hours) were considered to analyze immune responses. The production of antibodies, cytokines, and interferon after injection of the vaccine protein was calculated.
Results
Sequences retrieval
The protein sequences of FdeC, Hma, and UpaB of E. coli subsp. Cft073 and CtB of Vibrio cholerae serotype O1 (strain ATCC 39315/El Tor Inaba N16961) were driven from the Uniprot database with the following accession number: AAN78896.1, AAN80941.1, AAN78907.1, and AET14210.1 respectively. The FdeC protein consists of three regions (A, B, and C); region B (residues 597 to 1008) was reported as the antigenic part of FdeC.44 UpaB consists of the signal sequence (residues 1 to 37), an α-domain (residues 38 to 500), and a β-domain (residues 501-776). According to previous studies, the α-domain (residues 38 to 500) is a functional domain45, so the epitopes in our study were selected from FdeCB and αUpaB.
Hma and CtB protein sequences, without signal peptide, were used.
Epitopes prediction
B cell epitopes prediction
Linear B cell epitopes in FdeC, Hma, and UpaB were selected by comparing different prediction algorithms. A threshold of 0.4 was chosen in VaxiJen. The selected epitopes are summarized in Table 1.
Table 1.
B cell linear epitopes with their vaxijen scores
|
Protein
|
Start
|
End
|
Peptide sequence
|
Length
|
VaxiJen
|
| FdeC |
190 |
210 |
LSSNQSKVSVAEDHVKAGEST |
21 |
1.2 |
| Hma |
283 |
302 |
ANEVKTTQPVPSPMIQALTVH |
21 |
0.78 |
| UpaB |
230 |
270 |
GAGSATQNLNLSVADSTIYSDVLALSESENSASTTTNVNMN |
41 |
1.2 |
T cell epitopes prediction
The use of different epitopes prediction servers resulted in many epitopes. They were filtered by considering some essential properties such as covering most HLA I (based on binding and processing properties) and HLA II alleles in humans and mice, which have been predicted by the most employed servers and have been known as an antigen in VaxiJen. T cell epitopes are given in Table 2.
Table 2.
T cell epitopes, related HLA alleles, and vaxijen scores
|
Protein
|
Start
|
End
|
Peptide sequence
|
MHC
|
Length
|
VaxiJen
|
| FdeC |
342 |
374 |
TGSERPSAGNWTEKGNGVYVSTLTLGSAAGQLS |
Ad,Ed,DRB10401,
DRB11101,DRB10101
DRB10701 |
33 |
0.8692 |
| Hma |
647 |
680 |
NEDQYRINLALNNLTDKRYRTAHETIPAAGFNAA |
A2402, H2Kd, H-2-IAd
DRB1-0101,DRB1-0401
DRB1-1302 |
34 |
0.9561 |
| UpaB |
411 |
461 |
TTEGGLYQYDLTQGADGNFYFVKNTHKASNASSVIQAMAAAPANVANLQAD |
H-2-Dd, A0101, A0201
H2IAd, H2IEd
DRB1-0101,DRB1-1302 DRB1-1501,DRB1-1101 DRB50101,DRB10701 DRB10401 |
51 |
0.7442 |
Evaluation of HLA allele frequencies
The frequency of MHC alleles was evaluated in http://www.allelefrequencies.net and is shown in Fig. S1. Our goal was a selection of reported alleles from different geographical regions.
Selection and joining epitopes
Twelve different constructs were designed based on epitope orders and placement of CTB at the N-terminus or C-terminus of the vaccine construct. The primary sequence of the selected construct is shown in Fig. 1A.
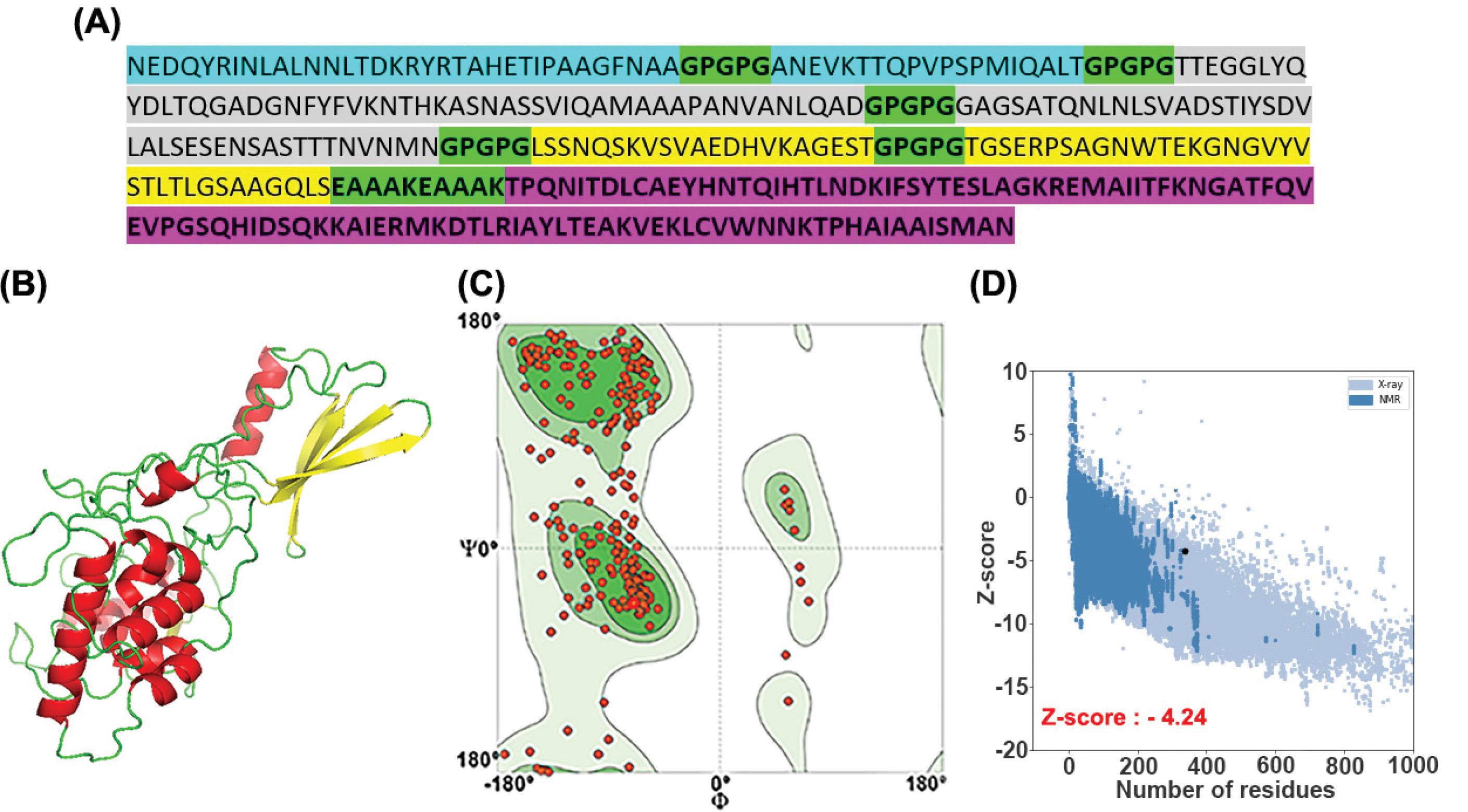
Fig. 1.
(A) The sequence of vaccine-protein candidates based on epitope orders and CTB placement. The epitopes of Hma, UpaB, and FdeC antigens are shown in blue, grey, and yellow, respectively. All the linkers are colored in green, and CTB (build-in adjuvant) is shown in pink. (B) Representation of the 3D structure of the vaccine protein with PyMol. The respective elements، such as the loops, are given in green; the helices are in red, while the beta-sheets are colored in yellow. (C) Ramachandran plot of the vaccine protein: 85.07% of the residues are in the favored region, 3.08%of the residues are in the allowed, and only 1.49%residuesare in the outlier region. (D) ProSA plot of the vaccine protein. The Z-score is shown in a large black dot. The Z-score represents the overall quality of the model.
.
(A) The sequence of vaccine-protein candidates based on epitope orders and CTB placement. The epitopes of Hma, UpaB, and FdeC antigens are shown in blue, grey, and yellow, respectively. All the linkers are colored in green, and CTB (build-in adjuvant) is shown in pink. (B) Representation of the 3D structure of the vaccine protein with PyMol. The respective elements، such as the loops, are given in green; the helices are in red, while the beta-sheets are colored in yellow. (C) Ramachandran plot of the vaccine protein: 85.07% of the residues are in the favored region, 3.08%of the residues are in the allowed, and only 1.49%residuesare in the outlier region. (D) ProSA plot of the vaccine protein. The Z-score is shown in a large black dot. The Z-score represents the overall quality of the model.
Physicochemical characterization of the vaccine construct
The amino acid number, molecular weight, Grand Average of Hydropathicity (GRAVY) values, and aliphatic index of the vaccine protein were 337 a.a, 35108 Da, -0.437, and 69.64, respectively. The theoretical PI of constructs 7,8,10, and 11 was calculated to be 5.68, and the PI of the others was 5.67.The half-life of all the designed constructs was not the same:
The half-life of the first six constructs, construct nine and 12, was estimated to be 7.2 hours in mammalian reticulocytes, more than 20 hours in yeast, and more than 10 hours in E. coli. The half-life of constructs 7 and 11 were defined as 5.5 hours in mammalian reticulocytes, 3 min in yeast, and 2 minutes in E. coli. The estimated half-life of constructs 8 and 10 was 1.4 hours in mammalian reticulocytes, 3 minutes in yeast, and more than 10 hours in E. coli.
Tertiary structure prediction
The final 3D model of the vaccine protein was selected based on the C-Score, the template modeling (TM) –score, and the root-mean-square deviation (RMSD) estimated by the I-Tasser server for each uploaded template. The final predicted model's c-score, TM, and RMSD were 0.00, 0.71±0.11 and 6.4±3.9 Å, respectively. The standard range of the C-score is between -5 to 2. The 3D model of the vaccine protein is shown in Fig. 1B.
Tertiary structure refinement and validation
The refined 3D model of the vaccine protein is shown in Fig. 2. The Ramachandran plot verified that 85.07% of residues of the developed model were in the favored region, 3.08% were allowed, and only 1.49% were in the outlier area. The analysis of the refined model on the ProSA-web server resulted in a Z-score value of -4.24. The clash score of all atoms was calculated to be 1.23, which was within the normal range.The plots were obtained from swiss. Pro and ProSA-web are given in Figs. 1C and 1D.
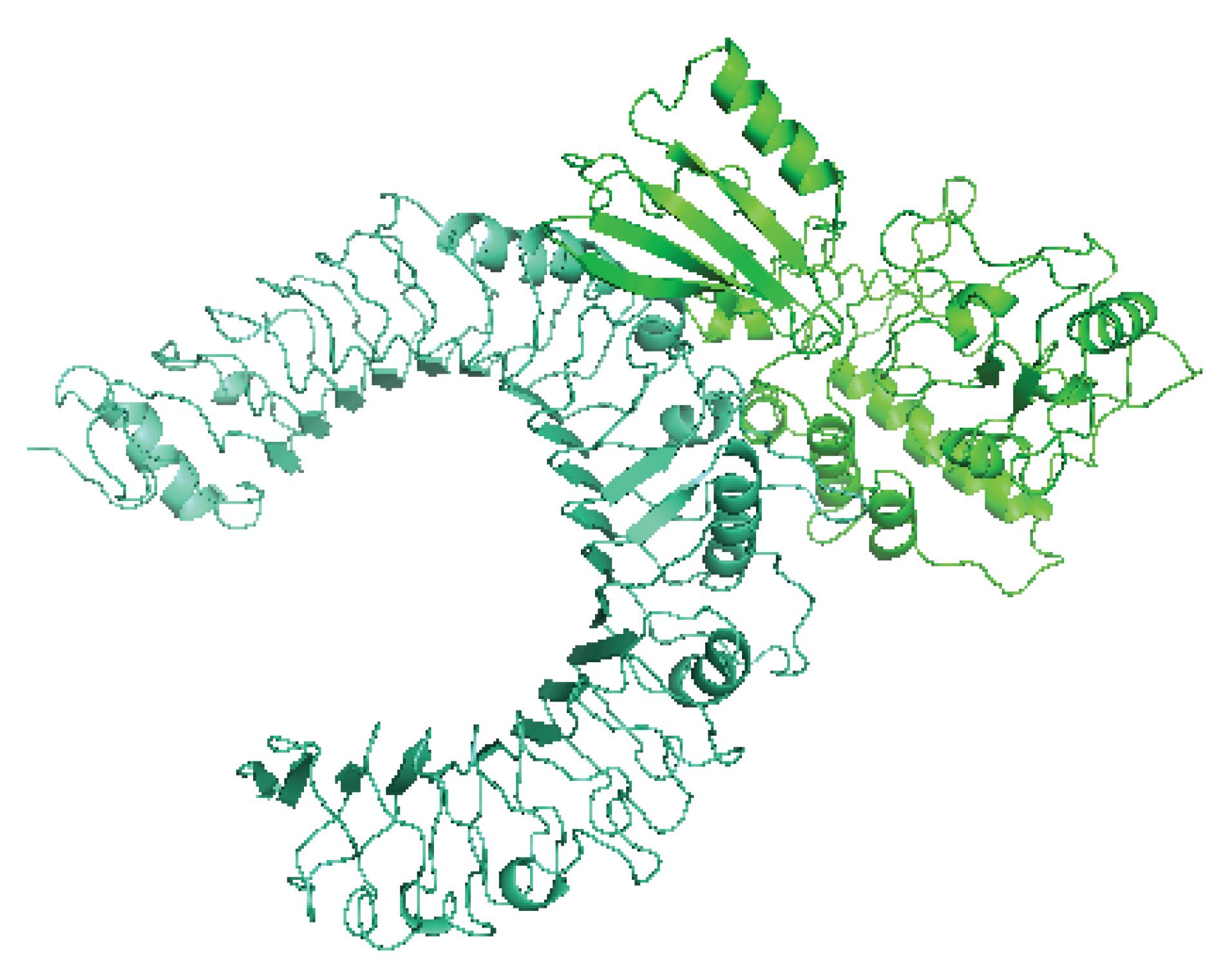
Fig. 2.
Adocked complex of vaccine protein and TLR2. Vaccine protein is shown in green color, and TLR2 is shown in cyan color. The 3D structure of the docked complex of vaccine protein and TLR2 is visualized with PyMol.
.
Adocked complex of vaccine protein and TLR2. Vaccine protein is shown in green color, and TLR2 is shown in cyan color. The 3D structure of the docked complex of vaccine protein and TLR2 is visualized with PyMol.
mRNA structure prediction
The best-predicted mRNA structure (ΔG=-252.17 kcal/mol) is shown in Fig. S2 (Supplementary file 1). No hairpin or pseudoknot was seen at the site of the first nucleotides at the 5' end. Furthermore, Table S1 (Supplementary file 1) shows free energy details of the 5' end of chimeric gene mRNA structure.
Evaluation of antigenicity, allergenicity, and solubility of the vaccine protein
The antigenicity and allergenicity assessment found that the designed vaccine protein is a protective antigen (=0.9195) and non-allergen for humans. The solubility analysis of the vaccine sequence showed that the vaccine protein has (100%) solubility when overexpressed in E. coli. The solubility score of the designed vaccine was 0.612, the Protein-sol server predicted the solubility of proteins as the scaled solubility value (Query Sol), and the average solubility for the population experimental dataset (PopAvrSol) was 0.45. Consequently, any solubility value greater than 0.45 is assumed to be more soluble than the intermediate soluble E. coli protein from the experimental solubility dataset.46
Secondary structure prediction
According to the PSIPRED and GOR4, the designed vaccine protein contains 32.05% α-helix, 0.00% β- sheet, 14.84% strand, and 53.12% coils. The prediction of the secondary structure is represented in Fig. 3.

Fig. 3.
Predict the secondary structure of the designed vaccine.
.
Predict the secondary structure of the designed vaccine.
Molecular docking of the vaccine with TLR2
In molecular docking studies and subsequent MD assay, TLR2 was considered the receptor. Hydrophobic protein fragments were evaluated to define the binding region of the vaccine protein. The hydrophobic region belonging to CTB may interact with TLR2. In previous studies of TLR2, residues 266-355 belong to the ligand-binding and dimerization region.13
The top 32 docked complexes were investigated for intramolecular interactions with the LigPlot+ software. The complex with the highest number of hydrogen bonds and hydrophobic contacts and the lowest interaction energy, was selected for further inspection in the MD simulation step. The selected docked complex is shown in Fig. 2. The docking results of the vaccine protein and TLR2 by the HADDOCK server are summarized in Table 3. The LigPlot analysis of the TLR2-vaccine protein is outlined in Table 4. The two-dimensional diagram of the vaccine-receptor docked complex is shown in Fig. S3 (Supplementary file 1).
Table 3.
Docking result of the vaccine protein and TLR2 by HADDOCK server
|
Score
|
ΔG interaction* (kcal mol-1) |
Kd(M)
|
ΔE electrostatic (kcal mol-1) |
ΔE desolvation (kcal mol-1) |
ΔE VdW (kcal mol-1) |
| 70.3 ± 8.4 |
-15.7 |
2.9E-12 |
70.3 ± 8.4 |
-6.8 ± 4.3 |
-87.6 ± 7.3 |
*ΔG interaction was computed by Prodigy webserver.
Table 4.
Interacting amino acids of Vaccine protein-TLR2 by LigPlot+ software
|
Complex
|
Interacting amino acids of ligand
|
Interacting amino acids of receptor
|
| Vaccine protein- TLR2 |
Gln250, Ile251, His252, Lys268, Ala331, Ala332, Ile333, Ser334, Ala336, Asn337 |
Val292, Ala297, Ser298, Asp299, Asn300, Asp301, Arg302, Val303, Leu324, Tyr326, Ser329, Tyr332, Ser333, Glu336, Leu354 |
Molecular dynamic simulation
The process equilibrated the simulation system's energy components, temperature, pressure, density, and volume in the equilibration steps to ensure the stability of the simulation system throughout the MD simulation. The dynamic process was carried out at a temperature of ~ 300 K and a pressure of ~1 bar. The MD of TLR2 and the vaccine protein complex's output represent this complex's high stability during 100 ns of simulation (Fig. 4). The RMSD (root means square deviation) plot analysis of complex stability indicates that this complex reached a steady state around 60 ns of simulation (Fig. 4A). The Rg (radius of gyration) plot, an indicator of the system's stable and relaxed folding structure, figured out that the TLR2-vaccine protein complex has reached the relaxed and fold design in the last 20ns of simulation (Fig. 4B). The RMSF plot was applied to investigate the variations of each amino acid of TLR2 and vaccine protein during simulation. Data showed that residues 235-337 of the vaccine protein were in interaction with the 266-355 region of TLR2, which belongs to the binding region of the TLR2, have low and identical fluctuation in 75, 95, and 99 ns of simulation, which suggests that small changes for TLR2 and vaccine protein happened during the simulation (Fig. 4C, 4D). The merged tertiary structure of the TLR2-vaccine protein complex and LigPlot+ analysis was represented in Fig. S4 and Table S2 (Supplementary file 1). The tertiary structure of the TLR2-vaccine protein is shown in Fig. 5, indicating that the vaccine protein still interacts effectively with TLR2 during the simulation time (Fig. 5A). The LigPlot+ software suggested Gln250, Ile251, and His252 in vaccine protein and Arg302, Asp301, Val292, and Leu324 in TLR2 as the pivotal interacting amino acids, which are the same before and after stimulation (Fig. 5B). The binding affinity (ΔG) and dissociation constant (Kd) predicted value of the vaccine-TLR2 complex after MD simulation was -11.2 kcal mol-1 and 5.6E-09 M, respectively.
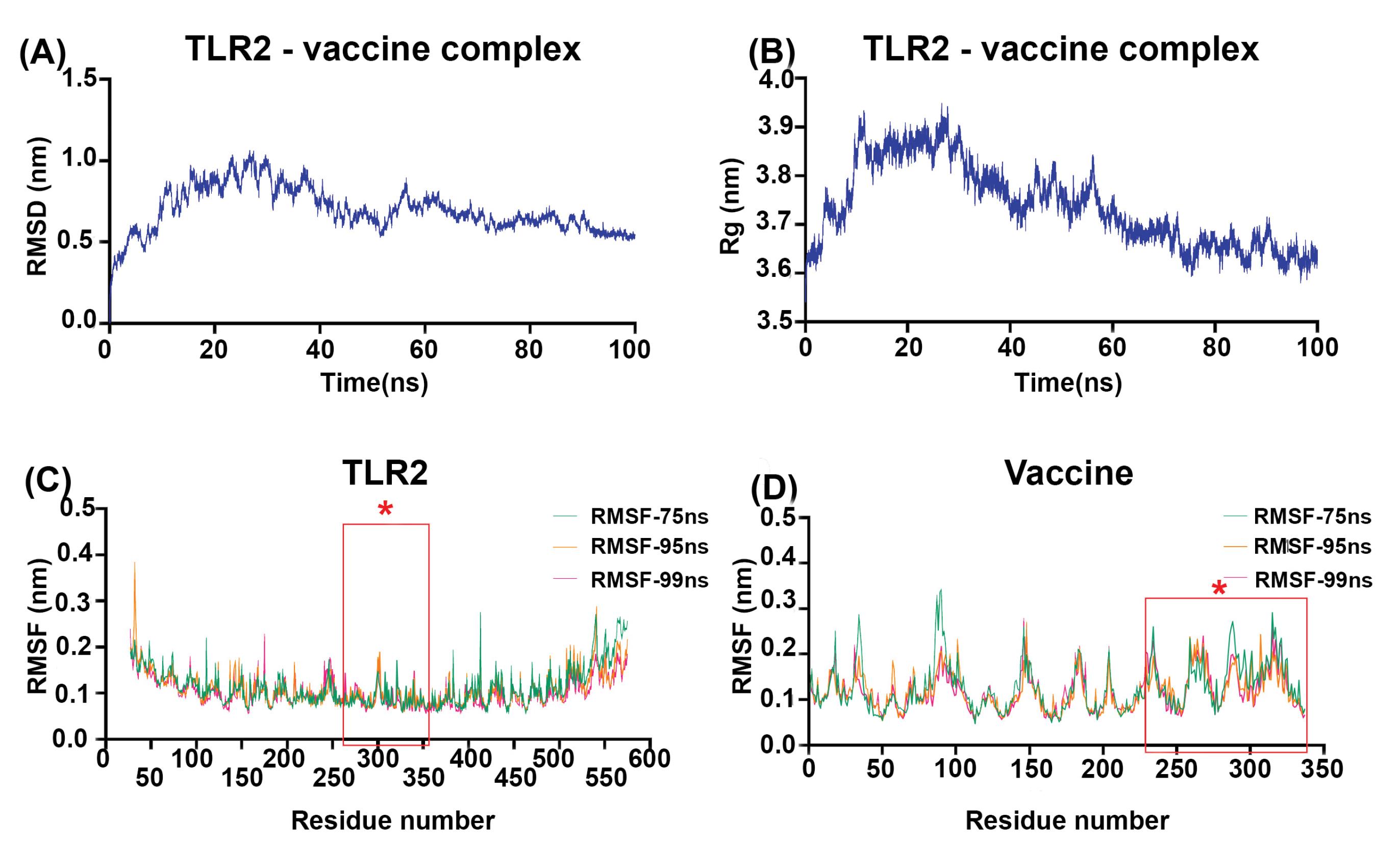
Fig. 4.
Molecular dynamic simulation analysis of the vaccine protein-TLR2 complex over 100 ns. (A) RMSD plot showing the steady state of the complex from 60 ns. (B) The radius of gyration (Rg) plot indicates the compactness of the vaccine-TLR2 complex in the last 20ns of the MD simulation. (C) RMSF graph of TLR2 shows less than 0.25 nm of fluctuation for most residues, particularly residues 266-355found in the binding region.The asterisks on the plot are amino acids of the binding site.(D) The RMSF graph of the vaccine protein displays a fluctuation of less than 0.25 nm for all residues, especially residues 235-337. The asterisks on the plot are amino acids of the binding site.
.
Molecular dynamic simulation analysis of the vaccine protein-TLR2 complex over 100 ns. (A) RMSD plot showing the steady state of the complex from 60 ns. (B) The radius of gyration (Rg) plot indicates the compactness of the vaccine-TLR2 complex in the last 20ns of the MD simulation. (C) RMSF graph of TLR2 shows less than 0.25 nm of fluctuation for most residues, particularly residues 266-355found in the binding region.The asterisks on the plot are amino acids of the binding site.(D) The RMSF graph of the vaccine protein displays a fluctuation of less than 0.25 nm for all residues, especially residues 235-337. The asterisks on the plot are amino acids of the binding site.
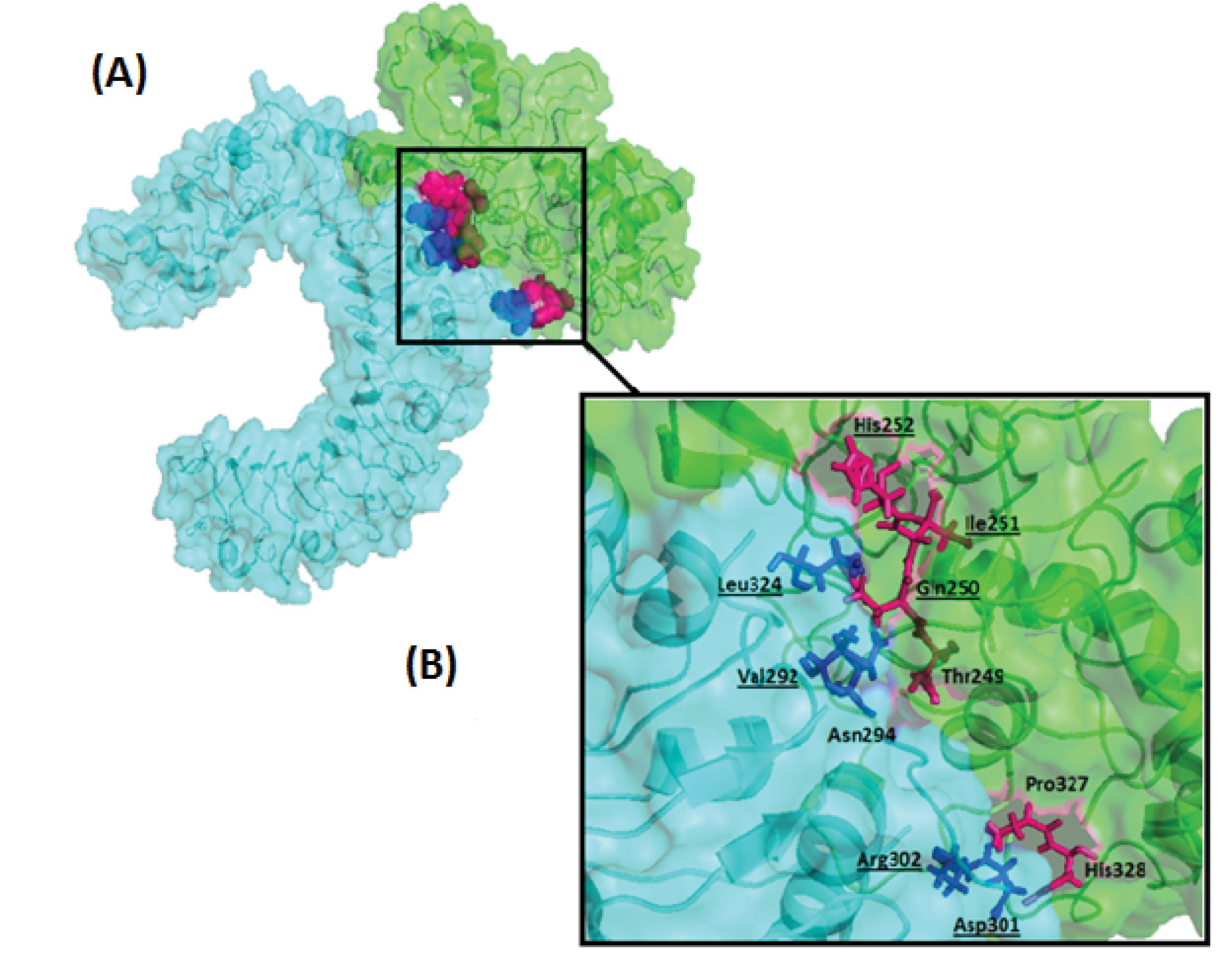
Fig. 5.
(A) The surface–cartoon representation of the vaccine protein-TLR2 complex after Molecular dynamic simulation. (B) close -up view of the stick representation of interacting amino acids: The interacting amino acids of the ligand are shown in pink. Interacting amino acids of receptors are shown in blue. The underlined amino acids are common in the vaccine-TLR2 complex before and after the MD simulation. The figure is generated with PyMol.
.
(A) The surface–cartoon representation of the vaccine protein-TLR2 complex after Molecular dynamic simulation. (B) close -up view of the stick representation of interacting amino acids: The interacting amino acids of the ligand are shown in pink. Interacting amino acids of receptors are shown in blue. The underlined amino acids are common in the vaccine-TLR2 complex before and after the MD simulation. The figure is generated with PyMol.
Also, during simulation time, RMSD Clustering analysis was performed to discover conformation changes in the TLR2-vaccine protein complex. This analysis generates a 10001-matrix structure with an energy of 65.7899, clustered into ninety-six numbers with RMSD values ranging from 0.0635253 to 1.09729 nm with an average of 0.335514. RMSD value result indicated that all clusters had no significant conformational changes during simulation. This finding is compatible with the tertiary structure alignment of protein structure in every 10 ns, which represented no structural changes of TLR2-vaccine complex during simulation (Fig. S5, Supplementary file 1).
Codon optimization and in silico cloning
The CAI and GC content of the optimized gene sequence was 1 and 52, respectively, which shows a high probability of expression in the host. An ideal CAI value for a good expression in the host is 0.8-1.
The vaccine sequence was cloned into the vector pET28a. The desired cloned sequence contains 1025 base pairs (Fig. 6).
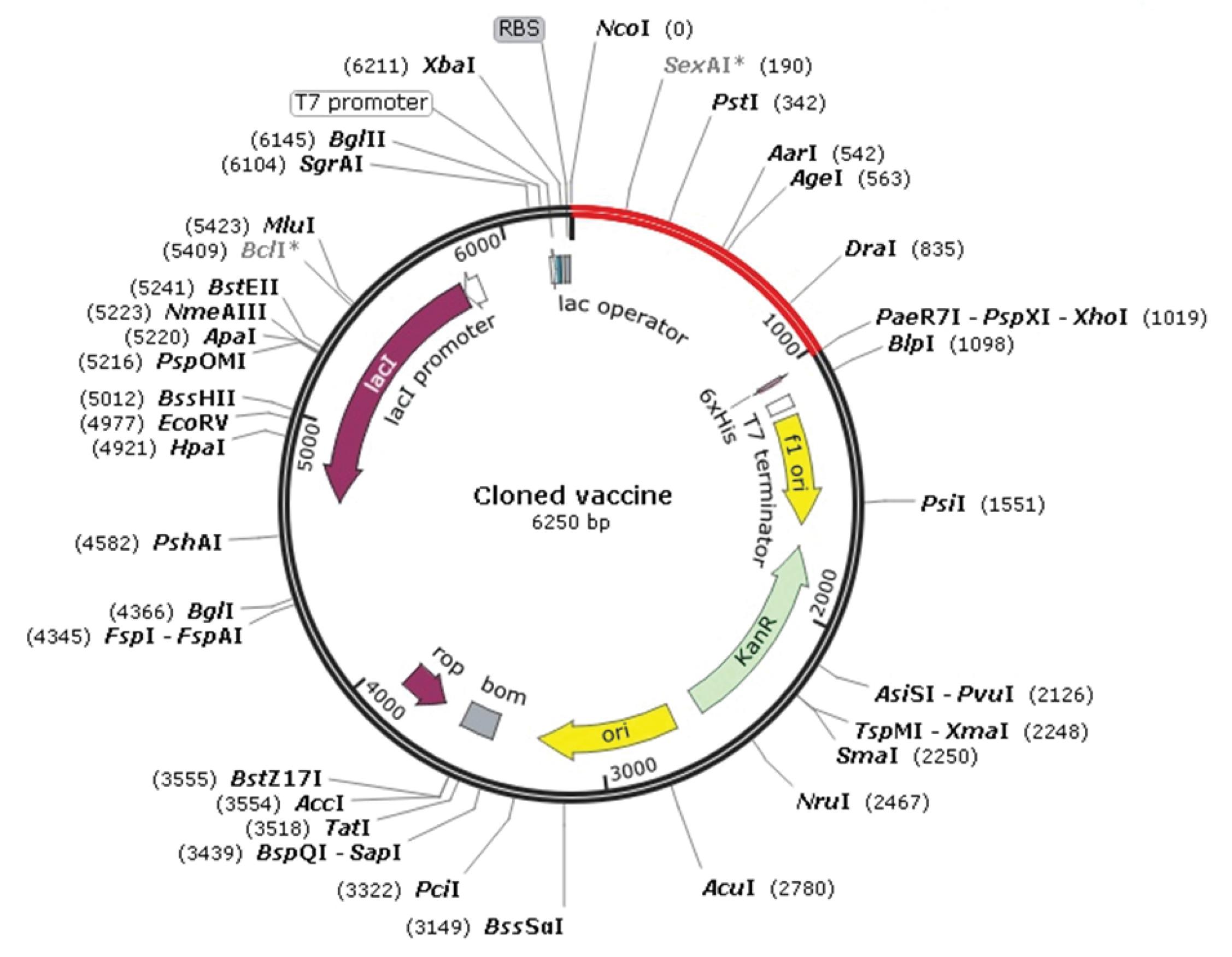
Fig. 6.
In silico cloning of optimized codons encoding the vaccine protein in the vector pET28 a (+) to confirm the expression of the vaccine protein in the E. coli as a host. Red colored indicating the optimized sequence (1025 bp). Fig. 6 is generated using snap-gene.
.
In silico cloning of optimized codons encoding the vaccine protein in the vector pET28 a (+) to confirm the expression of the vaccine protein in the E. coli as a host. Red colored indicating the optimized sequence (1025 bp). Fig. 6 is generated using snap-gene.
In silico immune simulation
Although the innate immune response against UTI is well-studied, less is known about the role of the adaptive immune response in this field.47 Prior experimental studies reported increased sensitivity to UTIs in B- and T-lymphocyte-depleted mice compared to the normal mice.48,49 Also, the transfer of T lymphocytes from infected mice to naïve mice can prevent UPEC colonization.50,51 Therefore, B and T lymphocytes are essential in immunity against UTI. Our study's in silico immune simulation (Fig. 7) shows our injections increased rate of B and T cells. Another important immune cell that plays an essential role in initiating adaptive immunity is the DC. The ImmSim server results also reported an increase in the DC population during the administration of our vaccine (Fig. 7A-D). As previously reported, a protective UTI vaccine must have the ability to stimulate humoral immunity.47 Our study's elevated IgM and IgG levels confirm that our vaccine can effectively induce humoral immunity (Fig. 7E). In addition, it also tested various cytokines levels. Previous studies have shown that interferon-gamma (IFN-γ) is essential for protecting against UTI52,53; herein (Fig. 7F), the IFN-γ concentration is reported to be high. Consequently, these results approve that the proposed vaccine protein could elicit an effective immune response to protect against UTIs.
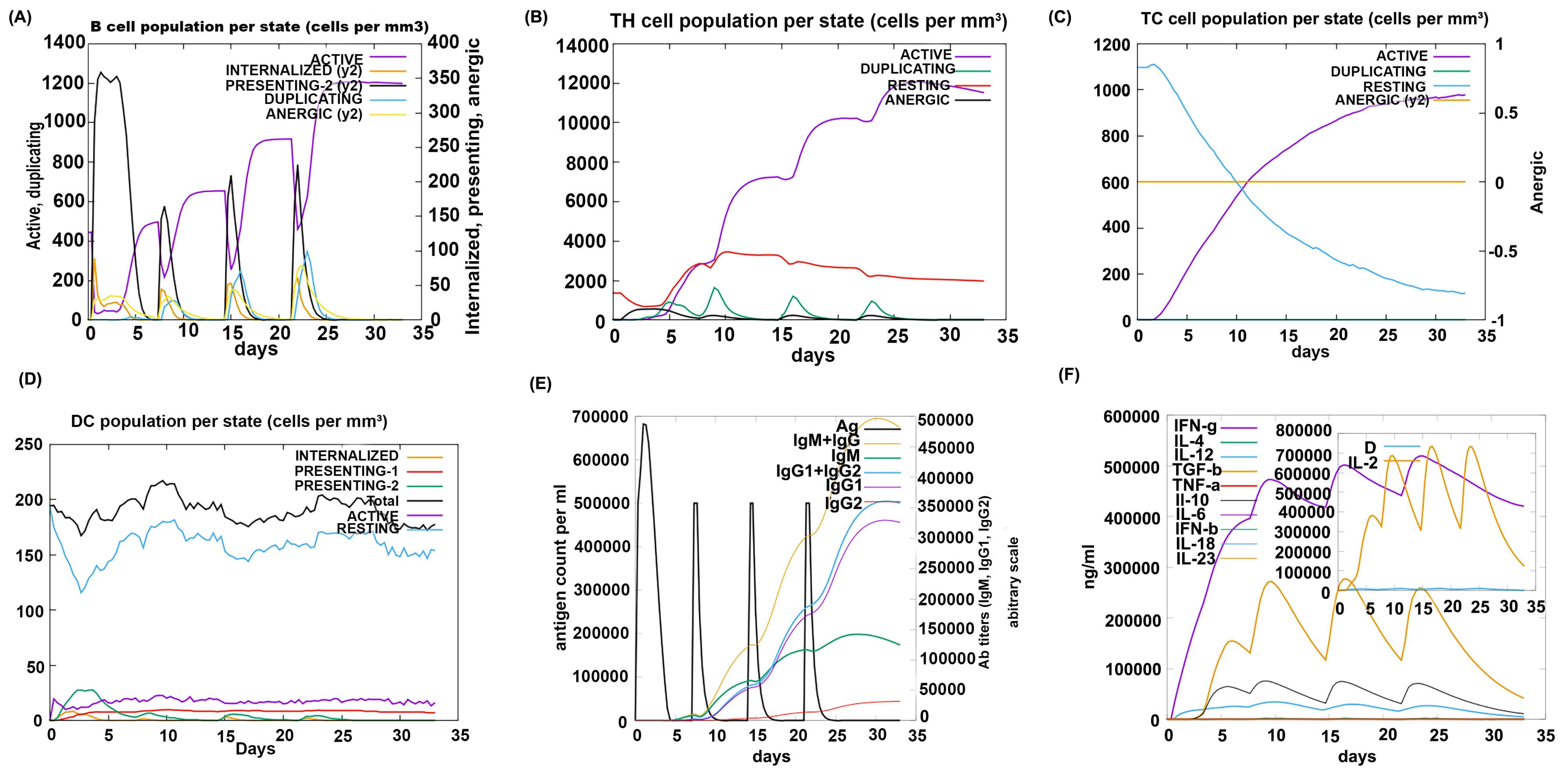
Fig. 7.
C-ImmSim presentation of an in silico immune simulation with the designed vaccine protein. (A, B, C, D) the graphs show the population of B cells, T helper cells, T cytotoxic cells, and Dendritic cells, respectively. (E) Antibodies production after injection of vaccine protein is shown (the black vertical lines are antigen). (F) Graph displaying cytokines levels after administration of vaccine protein.
.
C-ImmSim presentation of an in silico immune simulation with the designed vaccine protein. (A, B, C, D) the graphs show the population of B cells, T helper cells, T cytotoxic cells, and Dendritic cells, respectively. (E) Antibodies production after injection of vaccine protein is shown (the black vertical lines are antigen). (F) Graph displaying cytokines levels after administration of vaccine protein.
Discussion
Vaccination is a prophylactic method that is better than therapeutic approaches and is also known as the most effective method with a vast impact on public health.9,54 In recent years, cutting-edge bioinformatics methods with various tools in vaccinology-related software, statistical methods and databases facilitate the analysis of immunological data, the prediction of B cell and T cell epitopes, and the selection of efficient vaccine candidates, which all conclude to improve vaccine design,55,56
Among several candidate antigens introduced in previous studies,14 FdeC, Hma, and UpaB were selected for further investigation in our research. FdeC is an adhesion involved in colonizing the bladder and kidney,44,57 Hma is an outer membrane receptor that binds to hemin with high affinity. It escalates kidney colonization58 and UpaB is an autotransporter adhesion. The results of previous studies show that its removal reduces the colonization of UPEC in the mouse bladder.45 Since the bacteria that cause UTI can infect the bladder (lower UTI) or the kidneys (upper UTI),57 these antigens can be an excellent choice to prevent colonization in both sites.
Subunit vaccines cause a weaker immune response than other types of vaccines.59 Using a vaccine platform that contains vaccine antigen(s) and adjuvant properties may help overcome this deficiency. We use a mucosal adjuvant in our vaccine construct.60 CTB is one of the mucosal adjuvants known as the gold standard for its power of adjuvanticity.13 On the other hand, the first step in the UPEC pathogenesis process is to break the mucosal barriers,61 therefore, boosting mucosal immune responses can prevent UPEC colonization in the early stages.
EAAAK linkers were used to join the adjuvant and epitopes, and GPGPG linkers were employed to bind BCL and TCL epitopes. The above-mentioned rigid linkers were chosen to achieve an effective separation between adjuvant and epitopes and individual epitopes to increase the stability of our vaccine protein.62
In multi-epitopes vaccines, their order and position are also critical in addition to characteristics of the protein vaccine components (epitopes and adjuvant). Each segment's location can affect the other's role and function and ultimately affect the designed vaccine's physicochemical features and effectiveness.63 Our vaccine was constructed using three BCL epitopes, three TCL epitopes, an adjuvant, and appropriate linkers. Twelve constructs were made from the abovementioned segments, which differ in order and position.
Although there were more options for their arrangement, analysis of all possible constructs was impossible. Furthermore, our primary goal was to design a multi-epitope vaccine with a stable structure with suitable physicochemical properties and the ability to stimulate immune responses. According to our results, the final selected construct will lead us to this goal.
The physicochemical properties of the 12 constructs were biologically considerable. According to the theoretical PI, the protein is acidic, while the aliphatic index identifies it as thermostable, and the GRAVY score points out that the construct is hydrophilic. Furthermore, the instability index signifies it is a stable protein.
The CTB is the non-toxic subunit of cholera toxin that is classified as a PAMPs or TLR ligand. The antigen binding to CTB increases the possibility of receptor-mediated uptake and, subsequently, antigen presentation by the APCs.64 To result in a stable immune response, the interaction of the vaccine protein with specific immune cell receptors is necessary. TLRs have a vital role in the innate immune responses.27 A wide range of TLR2 ligands have been reported, such as lipids, lipoproteins, and proteins.17 Heat-labile enterotoxin B (LTB) is a protein that binds to TLR2. Since CTB has a high degree of sequence and structural similarity to LTB, TLR2 was considered a receptor for CTB in our study. According to previous research, the hydrophobic patches in CTB and highly frustrated regions in TLR2, which corresponded to interact with TLR2 ligands, were used for the docking process.13,18 Considering these data, the best docked model was selected.
MD simulation studies evaluate the dynamic behavior of proteins and interactions between molecules.31 The interaction between CTB as a build-in adjuvant of a multi-epitope vaccine and TLR2 as a receptor was studied by Nezafat et al.16 The analysis of interacting amino acids revealed that Gln250, Ile251and His252 of CTB and Val292, Asn294, Asp301, Leu302 and Leu324 of TLR2 before and after the MD simulation were common in our study. These residues were also reported as the interacting amino acids in the Nezafat et al study. The repetition of the same binding pattern in our research and Nezafat and colleagues’ analysis shows that the above amino acids play critical roles in the effective interaction between CTB and TLR2.
The PRODIGY server consequences confirmed that even after MD simulation, vaccine proteins interact with TLR2 with a high binding affinity (-11.2 kcal mol-1).
Overall, the MD simulation findings confirm the stability of the protein-ligand complex. In addition, these results highlighted the excellent interaction between the vaccine protein and TLR2.
Codon optimization of the designed vaccine was carried out to ensure an effective expression in E. coli host; the calculated GC content (52%) revealed the possibility of an efficient expression in E. coli. Furthermore, the vaccine protein was cloned in the expression vector pET-28a (+) by in silico cloning so that the expression of the vaccine in the bacterial system is possible.
Conclusion
This study designed a novel multi-epitope vaccine against urinary tract infection using immunoinformatics analysis to provide cellular and humoral immunity. Antigen selection was made based on conservation and protection to achieve this goal. The best linear B cell and T Cell epitopes were selected by considering HLA allele frequencies in the Iranian population. To increase the immune responses and reduce the concentration of the minimum antigen required to activate immune cells, CTB was used as a mucosal build-in adjuvant.15 To improve the stability and expression yield of the vaccine protein, linkers were used to separate the different segments.62 Since the TLR2 is an essential receptor in initiating immune responses that interact with various PAMPs such as CTB, therefore, molecular docking and dynamics study was performed using TLR2. Based on this study, our multi-epitope vaccine could be a promising vaccine candidate against urinary tract infections. Although In vitro and in vivo studies are required to confirm its prophylactic effects.
Research Highlights
What is the current knowledge?
√ UTI is the most common infection in adult women and the second most common infection in humans.
√ Although UTIs are often treated empirically with antibiotics, definitive antibiotic therapy may not be able to eliminate causative agents of UTIs.
√ The increasing rate of antibiotic-resistant lead to more mortality and high healthcare cost.
√ Much effort has been made to develop the UPEC vaccine, but no ideal vaccine is currently available for global use.
What is new here?
√ A novel multi-epitope vaccine against urinary tract infection was designed using immunoinformatics analysis to provide cellular and humoral immunity.
√ To increase the immune responses and reduce the concentration of the minimum antigen, CTB was used as a mucosal build-in adjuvant.
√ Population coverage, various physicochemical properties, and structural conformations were considered in our design.
√ Binding affinity and the stability of the build-in adjuvant and TLR2 were investigated.
√ Based on the results, the designed multi-epitope vaccine could be a promising vaccine candidate against urinary tract infections.
Acknowledgement
The authors wish to express their deep gratitude to all who provided support during the course.
Competing Interests
The authors declare no conflict of interest.
Ethical Statement
None to be declared.
Funding
This study was funded by Pasteur Institute of Iran as Ph.D. thesis (Grant Number: B-9635) and Iran National Science Foundation (INSF) (Grant number:98020317).
Supplementary files
Supplementary file 1 contains Tables S1-S2 and Figs. S1-S5.
(pdf)
References
- Price TK, Hilt EE, Dune TJ, Mueller ER, Wolfe AJ, Brubaker L. Urine trouble: should we think differently about UTI?. Int Urogyneco J 2018; 29:205-10. doi: 10.1007/s00192-017-3528-8 [Crossref] [ Google Scholar]
- Mobley HLT, Alteri CJ. Development of a Vaccine against Escherichia coli Urinary Tract Infections. Pathogens (Basel, Switzerland) 2015; 5:1. doi: 10.3390/pathogens5010001 [Crossref] [ Google Scholar]
- Rezaei-Tavirani M, Ghafourian S, Sayehmiri F, Pakzad R, Safiri S, Pakzad I. Prevalence of Cotrimoxazole Resistance Uropathogenic Bacteria in Iran: A Systematic Review and Meta-Analysis. Arch Clin Infect Dis 2018; 13:e63256. doi: 10.5812/archcid.63256 [Crossref] [ Google Scholar]
- Farajnia S, Alikhani MY, Ghotaslou R, Naghili B, Nakhlband A. Causative agents and antimicrobial susceptibilities of urinary tract infections in the northwest of Iran. Int J Infect Dis 2009; 13:140-4. doi: 10.1016/j.ijid.2008.04.014 [Crossref] [ Google Scholar]
- Asadi Karam MR, Habibi M, Bouzari S. Use of flagellin and cholera toxin as adjuvants in intranasal vaccination of mice to enhance protective immune responses against uropathogenic Escherichia coli antigens. Biologicals 2016; 44:378-86. doi: 10.1016/j.biologicals.2016.06.006 [Crossref] [ Google Scholar]
- Bryce A, Hay AD, Lane IF, Thornton HV, Wootton M, Costelloe C. Global prevalence of antibiotic resistance in paediatric urinary tract infections caused by Escherichia coli and association with routine use of antibiotics in primary care: systematic review and meta-analysis. BMJ 2016; 352:i939. doi: 10.1136/bmj.i939 [Crossref] [ Google Scholar]
- Durant L, Metais A, Soulama-Mouze C, Genevard J-M, Nassif X, Escaich S. Identification of Candidates for a Subunit Vaccine against Extraintestinal Pathogenic Escherichia coli. Infect Immun 2007; 75:1916-25. doi: 10.1128/iai.01269-06 [Crossref] [ Google Scholar]
- Hagan EC, Mobley HLT. Uropathogenic <em>Escherichia coli</em> Outer Membrane Antigens Expressed during Urinary Tract Infection. Infect Immun 2007; 75:3941. doi: 10.1128/IAI.00337-07 [Crossref] [ Google Scholar]
- Abbas G, Zafar I, Ahmad S, Azam SS. Immunoinformatics design of a novel multi-epitope peptide vaccine to combat multi-drug resistant infections caused by Vibrio vulnificus. Eur J Pharm Sci 2020; 142:105160. doi: 10.1016/j.ejps.2019.105160 [Crossref] [ Google Scholar]
- Jabbar B, Rafique S, Salo-Ahen OMH, Ali A, Munir M, Idrees M. Antigenic Peptide Prediction From E6 and E7 Oncoproteins of HPV Types 16 and 18 for Therapeutic Vaccine Design Using Immunoinformatics and MD Simulation Analysis. Front Immunol 2018; 9:3000. doi: 10.3389/fimmu.2018.03000 [Crossref] [ Google Scholar]
- Verma S, Sugadev R, Kumar A, Chandna S, Ganju L, Bansal A. Multi-epitope DnaK peptide vaccine against S.Typhi: An in silico approach. Vaccine 2018; 36:4012-22. doi: 10.1016/j.vaccine.2018.05.106 [Crossref] [ Google Scholar]
- Khan M, Khan S, Ali A, Akbar H, Sayaf AM, Khan A. Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci Rep 2019; 9:13321. doi: 10.1038/s41598-019-49354-z [Crossref] [ Google Scholar]
- Nezafat N, Eslami M, Negahdaripour M, Rahbar MR, Ghasemi Y. Designing an efficient multi-epitope oral vaccine against Helicobacter pylori using immunoinformatics and structural vaccinology approaches. Mol Biosyst 2017; 13:699-713. doi: 10.1039/c6mb00772d [Crossref] [ Google Scholar]
- Nesta B, Pizza M. Vaccines Against Escherichia coli. In: Frankel G, EZ Ron, editors. Escherichia coli, a Versatile Pathogen. Cham: Springer International Publishing; 2018. p. 213-42. 10.1007/82_2018_111.
- Lei Y, Zhao F, Shao J, Li Y, Li S, Chang H. Application of built-in adjuvants for epitope-based vaccines. PeerJ 2019; 6:e6185. doi: 10.7717/peerj.6185 [Crossref] [ Google Scholar]
- Nezafat N, Karimi Z, Eslami M, Mohkam M, Zandian S, Ghasemi Y. Designing an efficient multi-epitope peptide vaccine against Vibrio cholerae via combined immunoinformatics and protein interaction based approaches. Comput Biol Chem 2016; 62:82-95. doi: 10.1016/j.compbiolchem.2016.04.006 [Crossref] [ Google Scholar]
- Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol 2014; 5:461. doi: 10.3389/fimmu.2014.00461 [Crossref] [ Google Scholar]
- Kang JY, Nan X, Jin MS, Youn S-J, Ryu YH, Mah S. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 2009; 31:873-84. doi: 10.1016/j.immuni.2009.09.018 [Crossref] [ Google Scholar]
- Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik S-G. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007; 130:1071-82. doi: 10.1016./j.cell.2007.09.008 [Crossref] [ Google Scholar]
- Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res 2019; 47:D339-D43. doi: 10.1093/nar/gky1006 [Crossref] [ Google Scholar]
- Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 2019; 47:D339-d43. doi: 10.1093/nar/gky1006 [Crossref] [ Google Scholar]
- Wang X, Sun Q, Ye Z, Hua Y, Shao N, Du Y. Computational approach for predicting the conserved B-cell epitopes of hemagglutinin H7 subtype influenza virus. Exp Ther Med 2016; 12:2439-46. doi: 10.3892/etm.2016.3636 [Crossref] [ Google Scholar]
- Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-30: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res 2008; 36:W509-W12. doi: 10.1093/nar/gkn202 [Crossref] [ Google Scholar]
- Doytchinova IA, Guan P, Flower DR. EpiJen: a server for multistep T cell epitope prediction. BMC bioinformatics 2006; 7:1-11. doi: 10.1186/1471-2105-7-131 [Crossref] [ Google Scholar]
- Reche PA, Glutting J-P, Zhang H, Reinherz EL. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics 2004; 56:405-19. doi: 10.1007/s00251-004-0709-7 [Crossref] [ Google Scholar]
- Khan A, Khan S, Saleem S, Nizam-Uddin N, Mohammad A, Khan T. Immunogenomics guided design of immunomodulatory multi-epitope subunit vaccine against the SARS-CoV-2 new variants, and its validation through in silico cloning and immune simulation. Comput Biol Med 2021; 133:104420. doi: 10.1016/j.compbiomed.2021.104420 [Crossref] [ Google Scholar]
- Kar T, Narsaria U, Basak S, Deb D, Castiglione F, Mueller DM. A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep 2020; 10:1-24. doi: 10.1038./s41598-020-67749-1 [Crossref] [ Google Scholar]
- Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018; 154:394-406. doi: 10.1111/imm.12889 [Crossref] [ Google Scholar]
- Gonzalez-Galarza Faviel F, McCabe A, Santos Eduardo J Md, Jones J, Takeshita L, Ortega-Rivera Nestor D. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res 2020; 48:D783-D8. doi: 10.1093/nar/gkz1029 [Crossref] [ Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 2007; 8:1-7. doi: 10.1186/1471-2105-7-131 [Crossref] [ Google Scholar]
- Negahdaripour M, Nezafat N, Eslami M, Ghoshoon MB, Shoolian E, Najafipour S, et al. Structural vaccinology considerations for in silico designing of a multi-epitope vaccine. Infect Genet Evol 2018. 58: 96-109. 10.1016/j.meegid.2017.12.008.
- Hasan M, Ghosh PP, Azim KF, Mukta S, Abir RA, Nahar J. Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microb Pathog 2019; 130:19-37. doi: 10.1016/j.micpath.2019.02.023 [Crossref] [ Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC bioinformatics 2008; 9:1-8. doi: 10.1186/1471-2105-9-40 [Crossref] [ Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 2007; 35:W407-W10. doi: 10.1093/nar/gkm290 [Crossref] [ Google Scholar]
- Ikram A, Zaheer T, Awan FM, Obaid A, Naz A, Hanif R. Exploring NS3/4A, NS5A and NS5B proteins to design conserved subunit multi-epitope vaccine against HCV utilizing immunoinformatics approaches. Sci Rep 2018; 8:1-14. doi: 10.1038./s41598-018-34254-5 [Crossref] [ Google Scholar]
- Negahdaripour M, Eslami M, Nezafat N, Hajighahramani N, Ghoshoon MB, Shoolian E. A novel HPV prophylactic peptide vaccine, designed by immunoinformatics and structural vaccinology approaches. Infect Genet Evol 2017; 54:402-16. doi: 10.1016/j.meegid.2017.08.002 [Crossref] [ Google Scholar]
- Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J. Protein–Sol: a web tool for predicting protein solubility from sequence. Bioinformatics 2017; 33:3098-100. doi: 10.1093/bioinformatics/btx345 [Crossref] [ Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics 2000; 16:404-5. doi: 10.1093/bioinformatics/16.4.404 [Crossref] [ Google Scholar]
- Kouza M, Faraggi E, Kolinski A, Kloczkowski A. The GOR method of protein secondary structure prediction and its application as a protein aggregation prediction tool. Prediction of protein secondary structure: Springer; 2017. p. 7-24. 10.1007/978-1-4939-6406-2_2.
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015; 1-2:19-25. doi: 10.1016/j.softx.2015.06.001 [Crossref] [ Google Scholar]
- Mafakher L, Rismani E, Rahimi H, Enayatkhani M, Azadmanesh K, Teimoori-Toolabi L. Computational design of antagonist peptides based on the structure of secreted frizzled-related protein-1 (SFRP1) aiming to inhibit Wnt signaling pathway. J Biomol Struct Dyn 2020; 1-20. 10.1080/07391102.2020.1835718.
- Atapour A, Negahdaripour M, Ghasemi Y, Razmjuee D, Savardashtaki A, Mousavi SM. In silico designing a candidate vaccine against breast cancer. Int J Pept Res Ther 2020; 26:369-80. doi: 10.1007/s10989-019-09843-1 [Crossref] [ Google Scholar]
- Fathollahi M, Fathollahi A, Motamedi H, Moradi J, Alvandi A, Abiri R. In silico vaccine design and epitope mapping of New Delhi metallo-beta-lactamase (NDM): an immunoinformatics approach. BMC Bioinformatics 2021; 22:1-24. doi: 10.1186/s12859-021-04378-z [Crossref] [ Google Scholar]
- Nesta B, Spraggon G, Alteri C, Gomes Moriel D, Rosini R, Veggi D. FdeC, a Novel Broadly Conserved Escherichia coli Adhesin Eliciting Protection against Urinary Tract Infections. mBio 2012; 3:e00010-12. doi: 10.1128/mBio.00010-12 [Crossref] [ Google Scholar]
- Paxman JJ, Lo AW, Sullivan MJ, Panjikar S, Kuiper M, Whitten AE. Unique structural features of a bacterial autotransporter adhesin suggest mechanisms for interaction with host macromolecules. Nat Commun 2019; 10:1967. doi: 10.1038/s41467-019-09814-6 [Crossref] [ Google Scholar]
- Niwa T, Ying B-W, Saito K, Jin W, Takada S, Ueda T. Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. PNAS 2009; 106:4201-6. doi: 10.1073/pnas.0811922106 [Crossref] [ Google Scholar]
- Lacerda Mariano L, Ingersoll MA. The immune response to infection in the bladder. Nat Rev Urol 2020; 17:439-58. doi: 10.1038/s41585-020-0350-8 [Crossref] [ Google Scholar]
- Sivick KE, Mobley HL. Waging war against uropathogenic Escherichia coli: winning back the urinary tract. Infect Immun 2010; 78:568-85. doi: 10.1128/IAI.01000-09 [Crossref] [ Google Scholar]
- Weichhart T, Haidinger M, Hörl WH, Säemann MD. Current concepts of molecular defence mechanisms operative during urinary tract infection. Eur J Clin Invest 2008; 38:29-38. doi: 10.1111/j.1365-2362.2008.02006.x [Crossref] [ Google Scholar]
- Song J, Abraham SN. Innate and adaptive immune responses in the urinary tract. Eur J Clin Invest 2008; 38:21-8. doi: 10.1111/j.1365-2362.2008.02005.x [Crossref] [ Google Scholar]
- Thumbikat P, Waltenbaugh C, Schaeffer AJ, Klumpp DJ. Antigen-Specific Responses Accelerate Bacterial Clearance in the Bladder. J Immunol 2006; 176:3080-6. doi: 10.4049/jimmunol.176.5.3080 [Crossref] [ Google Scholar]
- Asadi Karam MR, Oloomi M, Mahdavi M, Habibi M, Bouzari S. Vaccination with recombinant FimH fused with flagellin enhances cellular and humoral immunity against urinary tract infection in mice. Vaccine 2013; 31:1210-6. doi: 10.1016/j.vaccine.2012.12.059 [Crossref] [ Google Scholar]
- Habibi M, Asadi Karam MR, Bouzari S. Evaluation of the effect of MPL and delivery route on immunogenicity and protectivity of different formulations of FimH and MrpH from uropathogenic Escherichia coli and Proteus mirabilis in a UTI mouse model. Int Immunopharmacol 2015; 28:70-8. doi: 10.1016/j.intimp.2015.05.027 [Crossref] [ Google Scholar]
- Rana A, Akhter Y. A multi-subunit based, thermodynamically stable model vaccine using combined immunoinformatics and protein structure based approach. Immunobiology 2016; 221:544-57. doi: 10.1016/j.imbio.2015.12.004 [Crossref] [ Google Scholar]
- Bahrami AA, Payandeh Z, Khalili S, Zakeri A, Bandehpour M. Immunoinformatics: In Silico Approaches and Computational Design of a Multi-epitope, Immunogenic Protein. Int Rev Immunol 2019; 38:307-22. doi: 10.1080/08830185.2019.1657426 [Crossref] [ Google Scholar]
- Salemi A, Pourseif MM, Omidi Y. Next-generation vaccines and the impacts of state-of-the-art in-silico technologies. Biologicals 2021; 69:83-5. doi: 10.1016/j.biologicals.2020.10.002 [Crossref] [ Google Scholar]
- Terlizzi ME, Gribaudo G, Maffei ME. UroPathogenic Escherichia coli (UPEC) Infections: Virulence Factors, Bladder Responses, Antibiotic, and Non-antibiotic Antimicrobial Strategies. Front Microbiol 2017; 8:1566. doi: 10.3389/fmicb.2017.01566 [Crossref] [ Google Scholar]
- Hagan EC, Mobley HL. Haem acquisition is facilitated by a novel receptor Hma and required by uropathogenic Escherichia coli for kidney infection. Mol Microbiol 2009; 71:79-91. doi: 10.1111/j.1365-2958.2008.06509.x [Crossref] [ Google Scholar]
- Patronov A, Doytchinova I. T-cell epitope vaccine design by immunoinformatics. Open biol 2013; 3:120139. doi: 10.1098/rsob.120139 [Crossref] [ Google Scholar]
- Pourseif MM, Masoudi-Sobhanzadeh Y, Azari E, Parvizpour S, Barar J, Ansari R. Self-amplifying mRNA vaccines: Mode of action, design, development and optimization. Drug Discov Today 2022; 27:103341. doi: 10.1016/j.drudis.2022.103341 [Crossref] [ Google Scholar]
- Bien J, Sokolova O, Bozko P. Role of uropathogenic Escherichia coli virulence factors in development of urinary tract infection and kidney damage. Int J Nephrol 2012; 2012. 10.1155/2012/681473.
- Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev 2013; 65:1357-69. doi: 10.1016/j.addr.2012.09.039 [Crossref] [ Google Scholar]
- Yu K, Liu C, Kim B-G, Lee D-Y. Synthetic fusion protein design and applications. Biotechnol Adv 2015; 33:155-64. doi: 10.1016/j.biotechadv.2014.11.005 [Crossref] [ Google Scholar]
- Yang J, Dai LX, Pan X, Wang H, Li B, Zhu J, et al. Protection against Helicobacter pylori infection in BALB/c mice by oral administration of multi-epitope vaccine of CTB-UreI-UreB. Pathog Dis 2015; 73. 10.1093/femspd/ftv026.