Bioimpacts. 14(1):27774.
doi: 10.34172/bi.2023.27774
Original Article
Synthesis and initial in vitro evaluation of olmutinib derivatives as prospective imaging probe for non-small cell lung cancer
Muammar Fawwaz Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing, 1, 2, * 
Kenji Mishiro Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing, 3, *
Arwansyah Arwansyah Formal analysis, Investigation, Methodology, Software, 4
Ryuichi Nishii Conceptualization, Methodology, Resources, 5
Kazuma Ogawa Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing, 2, 3, *
Author information:
1Laboratory of Pharmaceutical Chemistry, Faculty of Pharmacy, Universitas Muslim Indonesia, Urip Sumoharjo KM. 5, Makassar 90-231, Indonesia
2Graduate School of Medical Sciences, Kanazawa University, Kakuma-machi, Kanazawa, Ishikawa 920-1192, Japan
3Institute for Frontier Science Initiative, Kanazawa University, Kakuma-machi, Kanazawa, Ishikawa 920-1192, Japan
4Department of Chemistry Education, Faculty of Teacher Training and Education, Universitas Tadulako, Palu, Indonesia
5Biomedical Imaging Sciences, Department of Integrated Health Sciences, Graduate School of Medicine, Nagoya University, Higashi-ku, Nagoya 461-8673, Japan
Abstract
Introduction:
Imaging a non-small cell lung cancer (NSCLC) using radiolabeled tyrosine kinase inhibitors (TKIs) has attracted attention due to their unique interaction with the target epidermal growth factor receptor (EGFR). Olmutinib (OTB) is one of the third-generation EGFR TKIs, which selectively inhibit EGFR L858R/T790M mutation. In this study, we aim to estimate the interaction of the iodinated OTB (I-OTB)-receptor complex by molecular docking. Furthermore, we will synthesize the I-OTB and evaluate its activity toward EGFR L858R/T790M by in vitro cytotoxicity assay.
Methods:
A molecular docking simulation was carried out using an AutoDock Vina program package to estimate the interaction of the ligand-receptor complex. The I-OTB, N-{3-iodo-5-[(2-{[4-(4-methylpiperazin-1-yl)phenyl]aminothieno{3,2-d}pyrimidin-4-yl)oxy]phenyl} acrylamide, was synthesized by introducing an iodine atom in the phenyl group in the 3-aryloxyanilide structure. The half inhibitory concentration (IC50) was determined by employing a 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H tetrazolium monosodium salt (WST-8) assay to evaluate the activity of I-OTB.
Results:
The docking study exhibited that I-OTB could take an interaction similar to that of the parent compound. We successfully synthesized I-OTB and confirmed its structure by instrumental analysis. The binding energy of OTB and I-OTB in complex with EGFR T790M are -8.7 and -7.9 kcal/mol, respectively. The cytotoxicity assay showed that I-OTB also has an affinity towards the EGFR L858R/T790M mutation with the IC50 10.49 ± 5.64 𝜇M compared to the EGFR wild type with the IC50 over than 10 𝜇M.
Conclusion:
The cytotoxicity effect of I-OTB was comparable to that of OTB. This result indicates that the iodine substituent in OTB did not alter the parent compound selectivity toward double mutations EGFR L858R/T790M. Therefore, I-OTB is prominent for radioiodination, and [123/124I] I-OTB may be a promising candidate for EGFR L858R/T790M mutation imaging.
Keywords: Cytotoxicity, L858R/T790M, Molecular docking, Tyrosine kinase inhibitor
Copyright and License Information
© 2024 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Various imaging radiopharmaceuticals with positron emission tomography (PET) or single-photon emission computed tomography (SPECT) are essential in precision medicine.1,2 These techniques are noninvasive and provide quantitative details of biological functions with high sensitivity. Furthermore, in response to limitations found in an invasive biopsy limitation and in response to any bias resulting from tumor heterogeneity,3,4 molecular imaging employing PET or SPECT can stratify patients based on their susceptibilities to various diseases and may be classified into the genetic, molecular, or cellular profiles.5
The imaging probes labeled with radioiodine are of great attraction because of the availability of several radioiodine isotopes, such as 123I with a half-life (t1/2) of 13.2 hours for SPECT imaging, 131I with t1/2 of 8.0 days for radionuclide therapy, and 124I with t1/2 of 4.2 days for PET imaging, also 125I for various preclinical types of research and auger therapy.6-10
Radioiodination of tyrosine kinase inhibitors (TKIs) for imaging non-small cell lung cancer (NSCLC) has attracted a lot of scientists’ attention due to their unique interaction with the target epidermal growth factor receptor (EGFR).11-14 So far, various TKI imaging probes have been developed to assess the EGFR mutation status in NSCLC. However, the radiotracers employed in the imaging of double mutations of EGFR T790M/L858R, such as [11C]osimertinib, [11C]rociletinib, [125I]ICO1686, [77Br]BrCO1686, [125I]I-osimertinib, and [77Br]Br-osimertinib have some limitations, such as low accumulation in the targeted tumor, which results in insufficient tumor-to-blood ratios or tumor-to-nontarget tissue ratios.11-13,15
Olmutinib (OlitaTM, OTB, Fig. 1) is one of the orally active third-generation TKIs and selectively inhibits the EGFR with L858R and T790M mutations, sparing a wild type of EGFR.16,17 In 2016, it was approved to treat some NSCLC patients suffering from double mutations EGFR L858R/T790M in South Korea.16,18 The chemical structure of OTB contains three crucial functional groups, namely, the pyrimidine group, N‐methyl piperazine ring, and acrylamide group (Fig. 2).19,20 The pyrimidine is essential for binding to an ATP binding site in the EGFR.21 Considering the anticipated effect on the affinity to the EGFR and synthetic accessibility, we aimed to estimate the interaction of the iodinated OTB (I-OTB)-receptor complex by molecular docking. Furthermore, we will synthesize the novel I-OTB and evaluate its activity toward EGFR L858R/T790M by in vitro cytotoxicity assay.
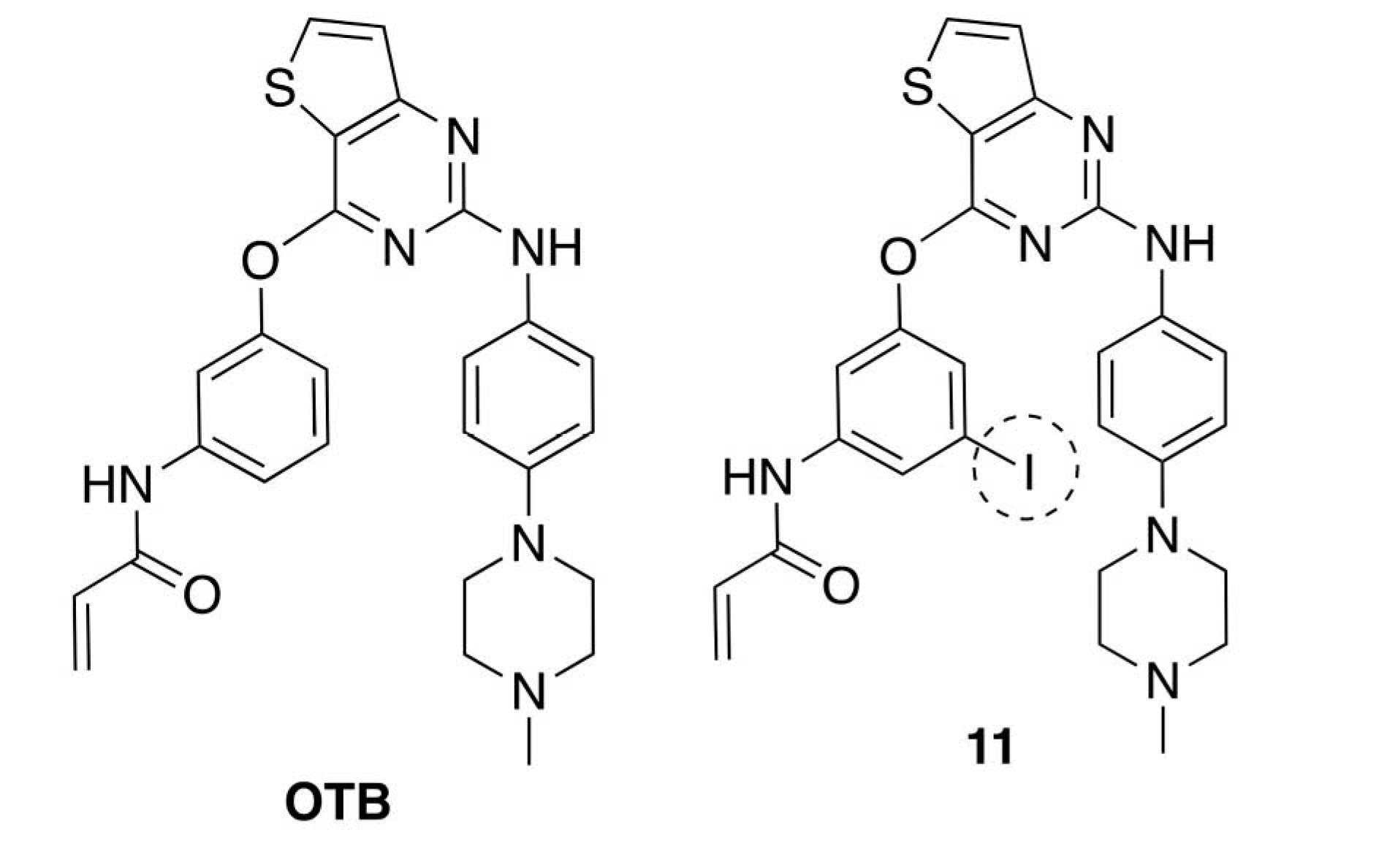
Fig. 1.
Structural formula of OTB and 11.
.
Structural formula of OTB and 11.
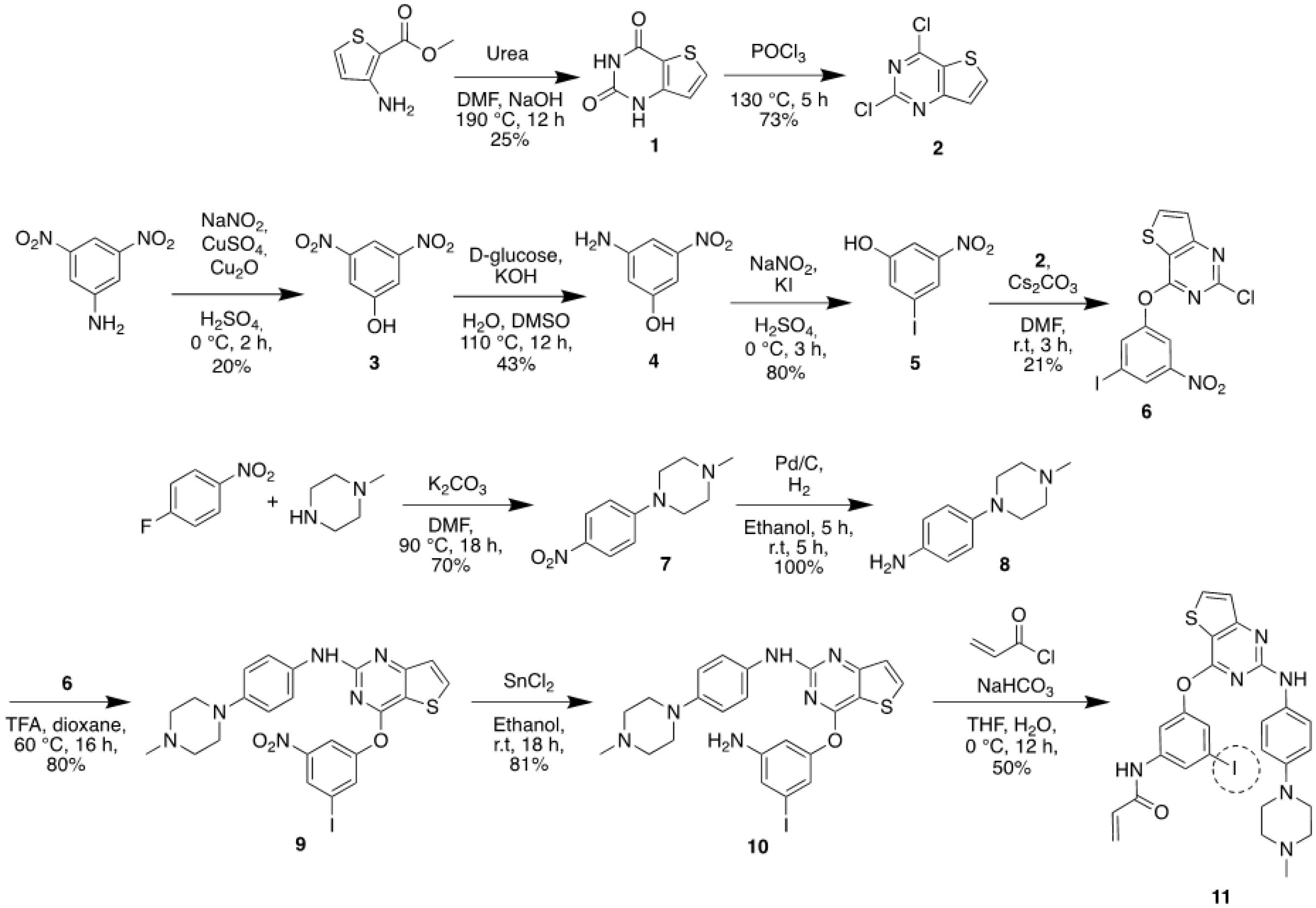
Fig. 2.
Synthetic scheme of iodinated compound 11.
.
Synthetic scheme of iodinated compound 11.
Here, a novel non-radioactive iodinated N-{3-iodo-5-[(2-{[4-(4-methylpiperazin-1-yl)phenyl]amino}thieno{3,2-d}pyrimidin-4-yl)oxy]phenyl}(I-OTB, 11) (Fig. 2) was synthesized. Moreover, EGFR subtype specificity of I-OTB was evaluated by employing three kinds of human NSCLC cell lines, namely H1975 (double mutations EGFR L858R/T790M), H3255 (active mutant EGFR L858R), and H441 (wild-type). This preliminary study is for developing a radioiodinated probe for companion diagnosis to select patients with the third generation of EGFR-TKIs.
Materials and Methods
Chemicals
Trypsin-ethylenediaminetetraacetic acid (EDTA) 0.25%, phosphate-buffered saline (PBS), penicillin-streptomycin (PS), Dulbecco's Modified Eagle Medium (DMEM)/Ham’s F-12 and RPMI-1640 medium were acquired from Nacalai Tesque, Inc. (Kyoto, Japan). Fetal bovine serum (FBS) was obtained from Biowest (Nuaillé, France). Solvents and reagents were purchased from Nacalai Tesque, Inc. (Kyoto, Japan), Merck (Darmstadt, Germany), Fujifilm Wako (Osaka, Japan), Kanto Chemical, Co., Inc. (Tokyo, Japan), and Tokyo Chemical Industry, Co., Ltd. (Tokyo, Japan).
Instrumentations
Auto Gamma System ARC-7010B (Hitachi, Ltd., Tokyo, Japan) recorded radioactivity. Thin-layer chromatography (TLC) on silica plates 60 F254 (Merck, Darmstadt, Germany) was used to monitor the reactions. High-performance liquid chromatography (HPLC) of SPD-20A system (Shimadzu Corp., Kyoto, Japan), using a Cosmosil® 5SL-II (20 ID × 250 mm) column (Nacalai Tesque, Inc., Kyoto, Japan). Nuclear magnetic resonance (NMR) spectroscopy was conveyed on JNM-ECS 400 and JNM-ECA 600 (JEOL Ltd., Tokyo, Japan). Direct analysis in real-time mass spectrometry (DART-MS) and electrospray ionization mass spectrometry (ESI-MS) were conducted on JMS-T100TD (JEOL Ltd., Tokyo, Japan). The optical density in 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H tetrazolium monosodium salt (WST-8) assay was conducted on Infinite® F200 Pro microplate reader (TECAN, Männedorf, Switzerland).
Molecular docking
In this current study, molecular docking was performed using the AutoDock Vina package to estimate the interaction of the ligand-receptor complex.22 The ligand molecule of OTB was retrieved from the PubChem database (CID: 54758501). The ligand was downloaded and saved to sdf extension. Meanwhile, a structure of 11 was generated by substituting a hydrogen atom in an aromatic ring of OTB with an iodine atom using MarvinSketch program packages. The structure was converted to 3D and saved to a protein data bank (PDB) file. Then, we applied Open Babel 2.4.1 program packages to convert sdf and pdb files to pdbqt formats.23 The chemical structures of these ligands are shown in Fig. 1. In receptor preparation, the three-dimensional structure of EGFR was obtained from the RCSB database (PDB ID: 3ika) (Fig. 3). The polar hydrogen and Kollman's united atom charges were computed to the receptor. Afterward, the receptor was saved in pdbqt format. All the docking results were processed and visualized in Open-Source PyMOL v 2.3 software.24
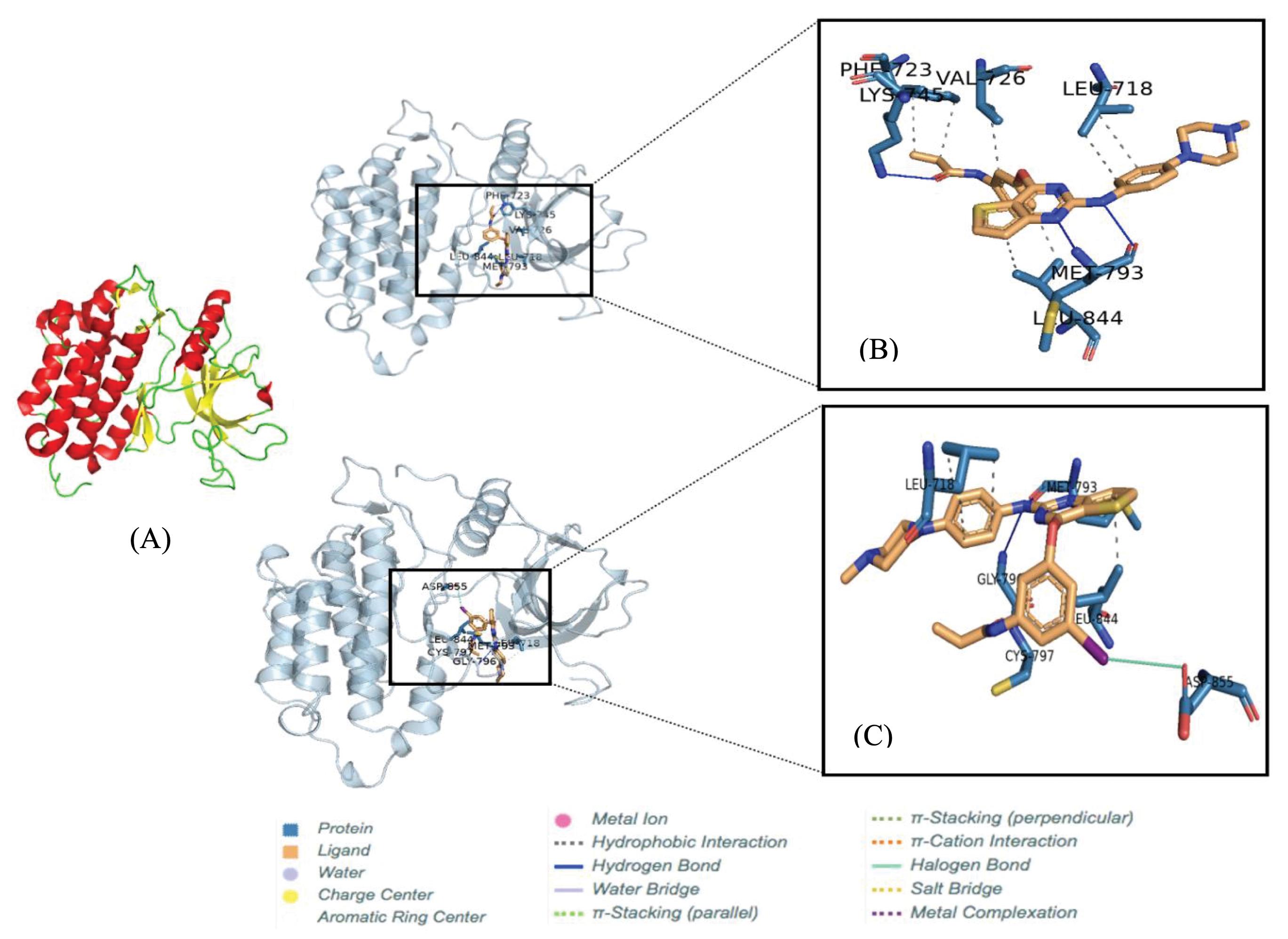
Fig. 3.
The tertiary structure of EGFR T790M. The alpha-helix and beta-sheet structures are presented by red and yellow colors in cartoon models, respectively (A). The binding poses between ligand and receptor (B) OTB (C) 11. The conformation pose of each complex was visualized by PLIP program combined with Pymol v 2.3 program packages.
.
The tertiary structure of EGFR T790M. The alpha-helix and beta-sheet structures are presented by red and yellow colors in cartoon models, respectively (A). The binding poses between ligand and receptor (B) OTB (C) 11. The conformation pose of each complex was visualized by PLIP program combined with Pymol v 2.3 program packages.
To perform molecular docking, the grid box parameter was required to limit the position and conformation of the ligand around the receptor site. The grid box was set on 24 × 22 × 22 with a grid spacing of 1.00 Å. Meanwhile, the grid box center was assigned at the coordinates -3.764 × 21.197 × 30.443. The exhaustiveness was computed at 100. Other parameters were computed as the default of AutoDock Vina. The Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm was utilized as a search parameter. All parameters were set using AutoDock Tools 1.5.6 created by Morris and co-workers.25 The docking parameters were computed according to similar protocols to our previous works.26-28
Probe synthesis
Fig. 2 shows the synthesis procedures of 11,12,29-31
and Supplementary file 1 (Figures S1-S12) supplies the 1H NMR spectra of all intermediate compounds and the final product. Additionally, the MS spectra and HPLC chromatogram confirmed the final product.
Synthesis of thieno(3,2-d)pyrimidine-2,4(1H,3H)-dione (1)
To a stirred mixture of methyl 3-aminothiophene-2-carboxylate (C6H7NO2S) (200 mg, 1.27 mmol, 1 eq.) and N,N-dimethylformamide (DMF) (500 µL), urea (450 mg, 7.62 mmol, 6 eq.) was added, and the mixture was continued stirring at 190 °C for 12 hours in a sealed tube. The reaction mixture was cooled to room temperature after the reaction was completed. After that, to the reaction mixture, 1 M aqueous sodium hydroxide (NaOH) solution was added, and insoluble material was removed by suction filtration. The filtrate was neutralized using 1 M aqueous hydrochloric acid (HCl) solution. The reaction mixture was extracted with ethyl acetate. The organic phase was separated, dried using sodium sulfate (Na2SO4), filtered, and concentrated by a rotary evaporator to afford 1 (50 mg, 25%) as a grey solid. 1H NMR (400 MHz, (CD3)2SO): δ 6.91 (1H, d, J = 5.2 Hz), 8.04 (1H, d, J = 4.8 Hz), 11.23 (1H, s), 11.57 (1H, s). MS (DART–) calculated for C6H4N2O2S [M–H]–: m/z = 167.0, found 167.6.
Synthesis of 2,4-dichlorothieno(3,2-d)pyrimidine (2)
The mixture of 1 (30 mg, 0.17 mmol, 1 eq.) and phosphorus oxychloride (POCl3) (200 µL) was stirred at 130 °C for 5 hours. The reaction mixture was cooled to room temperature after the reaction was completed. After that, to the reaction mixture, distilled water was added. The mixture was extracted with ethyl acetate, and the organic phase was separated, dried with Na2SO4, filtered, and concentrated by a rotary evaporator to afford 2 (22 mg, 73%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.56 (1H, d, J = 5.2 Hz), 8.13 (1H, d, J = 6.0 Hz). MS (DART–) calculated for C6H2Cl2N2S [M–H]–: m/z = 202.9, found 202.9.
Synthesis of 3,5-dinitrophenol (3)
To a stirred mixture of 3,5-dinitroaniline (C6H5N3O4) (91 mg, 0.5 mmol, 1.0 eq.) and sulfuric acid (H2SO4) (80 µL, 1.5 mmol, 3 eq.), sodium nitrite (NaNO2) in water 3 mL (38 mg, 0.55 mmol, 1.1 eq.) was added at 0 °C. TLC monitored the reaction to ensure the formation of the diazonium salt. Then saturated aqueous copper (II) sulfate (CuSO4) (50 mL) was added to the mixture, followed by the addition of copper (I) oxide (Cu2O) (72 mg, 0.5 mmol, 1 eq.). The mixture was stirred at room temperature for 2 hours. After the reaction was completed, the reaction mixture was neutralized with saturated aqueous sodium bicarbonate (NaHCO3). The mixture was extracted with ethyl acetate, and the organic phase was separated, dried with Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel (eluent: hexane/ethyl acetate = 7/3) to afford 3 (18 mg, 20%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 6.69 (1H, s), 8.02 (2H, d, J = 2.0 Hz), 8.63 (1H, t, J = 2.0 Hz). MS (DART–) calculated for C6H4N2O5 [M–H]–: m/z = 183.0, found 183.7.
Synthesis of 3-amino-5-nitrophenol (4)
To a stirred solution of 3 (5 mg, 0.05 mmol, 1.0 eq.) in H2O/DMSO (1/1) (400 µL), potassium hydroxide (KOH) (6 mg, 0.21 mmol, 4 eq.) and d-glucose (10 mg, 0.1 mmol, 2 eq.) were added at room temperature. The mixture was stirred at 110 °C for 12 hours. The mixture was cooled to room temperature and extracted with ethyl acetate. The organic phase was separated, dried with Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel (eluent: chloroform) to afford 4 (2 mg, 43%) as a pale yellow solid. 1H NMR (400 MHz, (CD3)2SO): δ 7.67 (1H, t, J = 2.4 Hz), 7.85 (2H, s), 8.06 (1H, t, J = 2.4 Hz), 8.42 (2H, td, J = 7.6, 1.6 Hz). MS (DART–) calculated for C6H6N2O3 [M–H]–: m/z = 153.0, found 153.4.
Synthesis of 3-iodo-5-nitrophenol (5)
To a stirred mixture of 4 (30 mg, 0.19 mmol, 1.0 eq.) and H2SO4 (30 µL), NaNO2 in water 1 mL (20 mg, 0.29 mmol, 1.5 eq.) was added at 0 °C for 3 hours. The reaction was monitored by TLC to ensure the formation of the diazonium salt, and then potassium iodide (KI) (80.51 mg, 0.48 mmol, 2.5 eq.) was added to the mixture at room temperature. After 3 hours, The reaction mixture was neutralized using saturated aqueous NaHCO3. The mixture was extracted with ethyl acetate, and the organic phase was separated, dried with Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified with column chromatography on silica gel (eluent: chloroform) to afford 5 (41 mg, 80%)as a pale yellow solid. 1H NMR (400 MHz, (CDCl3): δ 5.54 (1H, s), 7.54 (1H, dd, J = 2.4, 1.2 Hz), 7.66 (1H, t, J = 2.4 Hz), 8.14 (1H, t, J = 2.0 Hz). MS (DART–) calculated for C6H4INO3 [M–H]–: m/z = 263.9, found 263.9.
Synthesis of 2-chloro-4-(3-iodo-5-nitrophenoxy)thieno(3,2-d)pyrimidine (6)
To a stirred mixture of 2 (24 mg, 0.11 mmol, 1 eq.) and DMF (1 mL), 5 (29 mg, 0.11 mmol, 1 eq.), and cesium carbonate (Cs2CO3) (71 mg, 0.22 mmol, 2 eq.) were added at room temperature. The mixture was stirred at room temperature for 3 hours, and the reaction was monitored by TLC. The reaction mixture was quenched with water and then extracted with ethyl acetate. The organic phase was separated, dried with Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel (eluent: chloroform) to afford 6 (10 mg, 21%) as a pale yellow solid. 1H NMR (400 MHz, (CDCl3): δ 7.55 (1H, d, J = 5.6 Hz), 8.00 (1H, s), 8.08 (1H, d, J = 4.0 Hz), 8.17 (1H, t, J = 1.6 Hz), 8.54 (1H, s). MS (DART+) calculated for C6H5INO3 [M+H]+: m/z = 433.9, found 433.7.
Synthesis of 1-methyl-4-(4-nitrophenyl)piperazine (7)
To a stirred mixture of 1-methylpiperazine (C5H12N2) (0.61 mL, 5.45 mmol) in DMF (6.25 mL), 4-fluoronitrobenzene (C6H4FNO2) (0.75 ml, 7.07 mmol) and potassium carbonate (K2CO3) (1.13 g, 8.18 mmol) were added, and the mixture was stirred at 90 °C for 18 hours. The mixture was cooled to room temperature and extracted with ethyl acetate. The organic phase was separated, dried with Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel (eluent: chloroform) to afford 7 (1.1 g, 70%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 2.36 (3H, s), 2.56 (4H, t, J = 5.6 Hz), 3.44 (4H, t, J = 5.6 Hz), 6.82 (2H, dt, J = 9.6, 2.4 Hz), 8.12 (2H, dt, J = 9.6, 2.4 Hz). MS (DART+) calculated for C11H15N3O2 [M+H]+: m/z = 222.1, found 222.1.
Synthesis of 4-(4-methylpiperazin-1-yl)aniline (8)
To a stirred mixture of 7 (100 mg, 0.45 mmol, 1.0 eq.) and ethanol (4.50 mL), palladium on carbon (Pd/C 10%) (24 mg) was added under a nitrogen atmosphere. The nitrogen was exchanged with hydrogen, and the mixture was stirred under the hydrogen atmosphere (1 atm) for 5 hours. The mixture was filtrated through a pad of Celite® to remove the catalyst, and a rotary evaporator concentrated the filtrate to afford 8 (98 mg, 100%) as a purple solid. 1H NMR (400 MHz, (CDCl3): δ 2.34 (3H, s), 2.58 (4H, t, J = 4.8 Hz), 3.07 (4H, t, J = 4.8 Hz), 6.65 (2H, dt, J = 9.2, 2.4 Hz), 6.81 (2H, dt, J = 8.4, 1.6 Hz). MS (DART+) calculated for C11H17N3 [M+H]+: m/z = 192.1, found 192.0.
Synthesis of 4-(3-nitro-5-iodophenoxy)-N-[4-(4-methylpiperazin-1-yl)phenyl]thieno(3,2-d)pyrimidin-2-amine (9)
To a stirred mixture of 6 (10 mg, 0.02 mmol) and 2.0 M trifluoroacetic acid (TFA)/dioxane (200 µL), 8(4.4 mg, 0.02 mmol) was added, and the mixture was stirred at 60 °C for 16 hours. The reaction mixture was neutralized with saturated aqueous NaHCO3 and extracted with ethyl acetate. The organic phase was separated, dried with Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel (eluent: chloroform) to afford 9 (9.3 mg, 80%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 2.38 (3H, s), 2.63 (4H, t, J = 5.0 Hz), 3.18 (4H, t, J = 5.0 Hz), 6.80–6.85 (3H, m), 7.28 (1H, d, J = 5.6 Hz), 7.33 (1H, d, J = 8.8 Hz), 7.87 (1H, d, J = 5.6 Hz), 8.01 (1H, t, J = 1.8 Hz), 8.16 (1H, t, J = 2.0 Hz), 8.50 (1H, t, J = 1.8 Hz). MS (DART+) calculated for C23H21IN6O3S [M+H]+: m/z = 589.0, found 588.8.
Synthesis of 4-(3-amino-5-iodophenoxy)-N-[4-(4-methylpiperazin-1-yl)phenyl]thieno(3,2-d)pyrimidin-2-amine (10)
To a stirred mixture of 9 (13 mg, 0.02 mmol, 1 eq.) and ethanol (2.0 mL), SnCl2 (39.7 mg) was added under a nitrogen atmosphere at room temperature. After stirring for 18 hours, the reaction mixture was neutralized with saturated aqueous NaHCO3 and extracted with ethyl acetate. The organic phase was separated, dried with Na2SO4, filtered, and concentrated by a rotary evaporator to afford 10 (10 mg, 81%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 2.36 (3H, s), 2.59 (4H, t, J = 4.4 Hz), 3.16 (4H, t, J = 4.8), 6.55 (1H, t, J = 1.6 Hz), 6.88 (1H, s), 6.89 (1H, d, J = 9.2 Hz), 6.99 (1H, td, J = 1.6, 2.0 Hz), 7.04 (1H, t, J = 1.6 Hz), 7.25 (1H, d, 5.6 Hz), 7.42 (1H, d, J = 8.8 Hz), 7.81 (1H, t, J = 4.8 Hz). MS (ESI+) calculated for C23H23IN6OS [M+H]+: m/z = 559.1, found 559.2.
Synthesis of N-{3-iodo-5-[(2-{[4-(4-methylpiperazin-1-yl)phenyl]amino}thieno{3,2-d}pyrimidin-4-yl)oxy]phenyl} (11)
To a stirred mixture of 10 (5 mg, 0.01 mmol, 1.0 eq.), N,N-Diisopropylethylamine (DIPEA) (20 µL), and dichloromethane (DCM) (300 µL), acryloyl chloride (0.72 µL, 0.01 mmol, 1.0 eq.) was added at −30 °C and the mixture was stirred for 2 hours. The crude product was purified by HPLC with mobile phase system chloroform/methanol = 9/1, using a Cosmosil® 5SL-II (20 ID × 250 mm) column, flow rate 9.5 mL/min to afford 11 (1.1 mg, 10%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 2.42 (3H, s), 2.65 (4H, br, s), 3.19 (4H, br, s), 5.80–6.70 (3H, m), 6.80–6.85 (3H, m)a, 7.29 (1H, d, J = 4.8 Hz), 7.33 (2H, d, J = 8.0 Hz), 7.56 or 7.78 (1H, s)b, 7.88 (1H, d, J = 5.2 Hz), 8.01 (1H, t, J = 1.6 Hz), 8.16 (1H, t, J = 2.0 Hz), 8.50 (1H, t, J = 2.0 Hz). MS (ESI+) calculated for C26H25IN6O2S [M+H]+: m/z = 613.1, found 613.2. aThree protons derived from an impurity are also observed in this area. bOne of these two singlet signals is derived from an impurity.
Cytotoxicity assays
The half inhibitory concentration (IC50) of 11 and OTB toward NSCLC cell lines (H1975, H3255, and H441) was determined by WST-8 assay. These cell lines were kindly provided by Dr. Juri G. Gelovani, formerly of the Department of Experimental Diagnostic Imaging at the University of Texas, MD Anderson Cancer Center, Houston, TX., USA. The cells were seeded in a medium enriched with 10% FBS and 100 IU/mL PS in 96-well culture plates. The cells were allowed to reach a density of 2.5 × 103 cells/well (H1975 and H441) and 1 × 104 cells/well (H3255) by incubated for 24 h, 5% CO2, at 37 °C. After incubation, concentrations (0.01–100 µM) of 11 or OTB treated the cells for 48 h, and the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) assessed cell viability following the manufacturer’s protocol.
Statistical analysis
The data were statistically analyzed by unpaired Student’s t-test using GraphPad Prism 8.4.3 software (GraphPad Software, San Diego, CA, USA). All values are presented as mean ± standard deviation (SD), and P < 0.05 was considered statistically significant.
Results
Molecular docking
A molecular docking simulation was carried out to explore the binding mode of compound 11 to the active site of EGFR T790M (PDB code: 3ika). The binding energy of OTB and 11 in complex with EGFR T790M are –8.7 and –7.9 kcal/mol, respectively. These results indicate that the energy required for OTB and 11 to bind to the target receptor is similar. Thus, the modification of 11 does not affect its affinity for the T790M. Fig. 3 visualizes the binding pose of ligands at the site of the receptor. Moreover, Tables 1 and 2 summarize the detailed information on the ligand-receptor interactions by hydrogen and hydrophobic bond, respectively.
Table 1.
Hydrogen bonds of ligands in complex with the receptor
|
Model
|
Residue
|
AA
|
Distance H-A (Å)
|
Distance D-A (Å)
|
Donor angle
|
Donor atom
|
Acceptor atom
|
| OTB |
745 |
LYS |
1.90 |
2.82 |
147.40 |
470 [N] |
6025 [O2] |
|
|
793 |
MET |
2.21 |
3.06 |
139.93 |
6001 [N] |
904 [O2] |
|
|
793 |
MET |
2.24 |
3.21 |
160.17 |
900 [N] |
5993 [N2] |
|
11
|
793A |
MET |
1.83 |
2.80 |
171.38 |
6001 [N] |
904 [O2] |
|
|
793A |
MET |
2.25 |
3.21 |
155.29 |
900 [N] |
5993 [N2] |
|
|
796A |
GLY |
3.36 |
3.97 |
120.13 |
928 [N] |
6001 [Npl] |
|
|
797A |
CYS |
2.67 |
3.34 |
123.70 |
933 [N] |
6027 [O2] |
Table 2.
Hydrophobic interactions of ligands in complex with receptor
|
Model
|
Residue
|
AA
|
Distance
|
Ligand atom
|
Protein atom
|
| OTB |
718A |
LEU |
3.95 |
6007 |
228 |
|
|
718A |
LEU |
3.82 |
6008 |
230 |
|
|
723A |
PHE |
3.75 |
6027 |
266 |
|
|
723A |
PHE |
3.80 |
6028 |
262 |
|
|
726A |
VAL |
3.75 |
6018 |
288 |
|
|
844A |
LEU |
3.60 |
6022 |
1432 |
|
|
844A |
LEU |
3.38 |
5999 |
1431 |
|
11
|
718A |
LEU |
3.73 |
6007 |
228 |
|
|
718A |
LEU |
3.75 |
6008 |
230 |
|
|
718A |
LEU |
3.56 |
6005 |
231 |
|
|
844A |
LEU |
3.24 |
5999 |
1431 |
Probe synthesis
According to the reported crystal structure, the essential substituents playing a vital role in the binding of OTB to EGFR L858R/T790M are the pyrimidine group, N‐methyl piperazine ring, and acrylamide group.19,20 We hypothesized that compound 11, having an iodine atom at C(5) of the phenyl group in the 3-aryloxyanilide structure of OTB, could retain the original affinity of OTB toward EGFR L858R/T790M. A derivative of OTB, an iodinated compound of 11, was prepared by conducting several multistep reactions starting from a commercially-available material based on the reported procedure.12,29-31
Cytotoxicity assays
The IC50 of 11 was determined by employing a WST-8 assay. Although the cytotoxicity of 11 against H1975 was a little less than OTB, as shown in Table 3, the selectivity of 11 toward dual mutations EGFR cell line (H1975) was better than the parent compound, OTB.
Table 3.
The IC50 of 11, OTB, and gefitinib toward NSCLC cell lines by WST-8 assay
|
Cells lines
|
Mutation status
|
IC50 (μM)
|
|
11
|
OTB
|
Gefitinib*
|
| H1975 |
L858R/T790M |
10.49 ± 5.64 |
5.67 ± 2.19 |
> 10 |
| H3255 |
L858R |
14.55 ± 1.62 |
1.23 ± 1.23 |
0.02 ± 0.02 |
| H441 |
Wild type |
> 10 |
> 10 |
> 10 |
Data represent the mean ± SD of three separate experiments.
*Data from reference Fawwaz et al.12
Discussion
Several radiolabeled EGFR TKIs have been developed to serve as imaging agents to create therapeutic effects in the TKIs therapy for NSCLC. Some have been developed to monitor tumors using an EGFR L858R or EGFR del19.32,33 However, most agents did not show satisfactory results when stratifying the NSCLC patients with double mutations of EGFR L858R/T790M.34 Therefore, various scientists have continued their studies to develop a genuinely-effective imaging agent employed to diagnose and stratify the NSCLC patients with EGFR L858R/T790M regarding their therapeutic response to TKIs.
To overcome the TKI resistance caused by the T790M mutation, the third-generation TKIs were designed to possess a hydrophobic part that binds to a hydrophobic pocket modified by the mutation.35 Additionally, the acrylamide group was employed to form a covalent bond with the thiol of cysteine 797, resulting in irreversible inhibition of the EGFR.36 Many studies have shown that OTB selectively inhibits EGFR with mutations, especially the double mutations of L858R/T790M. In addition, OTB has fewer adverse effects, such as gastrointestinal toxicity, compared to first-generation and second-generation TKIs.18 Owing to the high selectivity of OTB, we hypothesized that a radioiodinated variant of OTB could overcome the limitation observed in the previously developed imaging agents for EGFR double mutations. Because the iodinated variant is required to retain the high selectivity of OTB, the iodine atom should be introduced into a structure such as a phenyl group in the 3-aryloxyanilide structure, which does not essentially participate in binding to the EGFR.7,37
The docking study showed that 11 has an interaction similar to that of the parent compound OTB with the EGFR T790M mutation. The estimated binding energy of 11 toward the EGFR was comparable to that of OTB. As seen in many EGFR kinase inhibitors,35 the pyrimidine ring is the most important group. Thus, we choose the best binding pose depending on the lowest binding energy between the ligand and the receptor by considering the position of the pyrimidine. In addition, the interaction between the ligand and the receptor by hydrogen and hydrophobic binding is also considered.
Both compounds exhibited hydrogen bonds with Met-793 in the hinge region (Table 1 and Fig. 3). The hydrophobic interaction of the thienopyrimidine group with a hydrophobic pocket composed of Ala-743, Met-790, and Leu-844 was also essential for binding the ligand and the EGFR T790M (Table 2). This result supported an experimental finding that a molecule associating with the Met-790 in an ATP-binding pocket of the T790M EGFR is critical for overcoming T790M EGFR drug resistance to first- and second-generation EGFR TKIs.35 Additionally, the diaminophenyl group showed hydrophobic interaction with Leu-718. In the found conformation, the acrylamide group was located at a position appropriate for the Michael addition by thiol in Cys-797. Unexpectedly, the introduced iodine showed a halogen bond interaction with Asp-855; hence, substituting iodine at this position could increase affinity 11 toward EGFR.
The synthesis of 11 was conducted by employing multistep reactions starting from a commercially-available material. In the final product analysis using NMR, several peaks arise in the chemical shift corresponding to protons in the chemical structure 11. The characteristic peak 11 was similar to OTB's, wherein protons bound to the aromatic and alkyl groups in both compounds appear in the downfield and upfield areas, respectively. The peak in the piperazine group appeared in the same chemical shift at 2.65 – 3.19 ppm.
To support the molecular docking research, it was necessary to study the cytotoxic activity in vitro. In this study, three different kinds of human NSCLC cell lines such as H1975 (EGFR L858R/T790M mutation), H3255 (EGFR L858R active mutant), and H441 (wild type EGFR) were employed. Although the IC50 of 11 toward double mutations EGFR was less than OTB (Table 3), 11 showed selectivity toward EGFR L858R/T790M compared to OTB. The decrease in activity 11 against H1975 was not necessarily caused by iodination but could be caused by impurities that contaminated the product. After HPLC purification, the data from the HPLC chromatogram, 1H NMR spectrum, and ESI-MS spectrum indicated the presence of 11 as a major compound. However, 1H NMR spectrum indicated contamination with almost an equimolar amount of acrylamide-related impurity (Fig. S11). To confirm the precise activity of 11, further purification or improvement of the synthetic procedure is required.
The cytotoxicity of 11 toward cancer cells with double mutations of EGFR was higher than that of gefitinib. Moreover, the IC50 of gefitinib toward H3255 as EGFR L858R was lower than 11. Since the cytotoxicity of all compounds toward H441 was not observed, we could prove that 11 and gefitinib had a high selectivity towards the EGFR L858R/T790M mutations and EGFR L858R, respectively. These data indicated that the iodine substituent in OTB increased selectivity toward H1975 and decreased activity against H3255. These characteristics served as a positive value for 11 and proved beneficial to 11 since they could improve the contrast in the imaging application and facilitate the stratification of the mutation status.
The biodistribution profile of OTB in the human body exhibited its rapid absorption and distribution into the organs. Moreover, OTB showed a fast plasma clearance and was excreted via the kidney in intact form.38 These characteristics are suitable for a radiotracer. Considering its high selectivity toward the L858R/T790M double mutations of EGFR compared to the wild-type EGFR and its fast plasma clearance, we expected that 11 would highly accumulatein the targeted tumor with EGFR L858R/T790M mutations. Therefore, we are strongly encouraged to conduct research using a radioiodinated variant of 11 since it is expected to show similar in vivo characteristics to OTB.
Conclusion
The OTB analog, 11, was successfully synthesized by multistep reactions. The molecular docking and in vitro cytotoxicity assays indicated that 11 has higher cytotoxicity and affinity towards the EGFR L858R/T790M mutation than wild-type EGFR. Therefore, we prove that introducing iodine into OTB does not alter the parent compound’s selectivity toward double mutations EGFR L858R/T790M. Therefore, [123/124I]I-OTB, the radioiodinated variant of 11, could be a promising candidate for EGFR L858R/T790M mutation imaging.
Research Highlights
What is the current knowledge?
√ OTB is the TKIs’ third generation, selectively inhibiting the activated double mutation EGFR L858R/T790M in lung cancer.
What is new here?
√ The OTB derivative was successfully synthesized and showed high selectivity toward EGFR L858R/T790M mutation compared to EGFR L858R and wild-type.
Acknowledgments
This work was supported by Terumo Life Science Foundation and MEXT KAKENHI Grant-in-Aid for Scientific Research (16H05397). The authors greatly acknowledge the Ministry of Research, Technology and Higher Education, Indonesia, along with the Institute of Research and Resource Development (LP2S), Universitas Muslim Indonesia, Makassar, Indonesia, for supporting the finalization of this study.
Competing Interests
The authors declare no conflicts of interest.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.
Ethical Statement
The authors declare no ethical issues to be considered.
Supplementary files
Supplementary file 1 contains Figs. S1-S12.
(pdf)
References
- Manning HC. World Molecular Imaging Congress 2015: precision medicine visualized. Mol Imaging Biol 2015; 17:295-6. doi: 10.1007/s11307-015-0855-3 [Crossref] [ Google Scholar]
- Waaijer SJH, Kok IC, Eisses B, Schröder CP, Jalving M, Brouwers AH. Molecular Imaging in Cancer Drug Development. J Nucl Med 2018; 59:726-32. doi: 10.2967/jnumed.116.188045 [Crossref] [ Google Scholar]
- Peled N, Roisman LC, Miron B, Pfeffer R, Lanman RB, Ilouze M. Subclonal Therapy by Two EGFR TKIs Guided by Sequential Plasma Cell-free DNA in EGFR-Mutated Lung Cancer. J Thorac Oncol 2017; 12:e81-e4. doi: 10.1016/j.jtho.2017.02.023 [Crossref] [ Google Scholar]
- Tan DS, Yom SS, Tsao MS, Pass HI, Kelly K, Peled N. The International Association for the Study of Lung Cancer Consensus Statement on Optimizing Management of EGFR Mutation-Positive Non-Small Cell Lung Cancer: Status in 2016. J Thorac Oncol 2016; 11:946-63. doi: 10.1016/j.jtho.2016.05.008 [Crossref] [ Google Scholar]
- Sun X, Xiao Z, Chen G, Han Z, Liu Y, Zhang C. A PET imaging approach for determining EGFR mutation status for improved lung cancer patient management. Sci Transl Med 2018; 10:1-13. doi: 10.1126/scitranslmed.aan8840 [Crossref] [ Google Scholar]
- Cavina L, van der Born D, Klaren PHM, Feiters MC, Boerman OC, Rutjes F. Design of Radioiodinated Pharmaceuticals: Structural Features Affecting Metabolic Stability towards in Vivo Deiodination. European J Org Chem 2017; 2017:3387-414. doi: 10.1002/ejoc.201601638 [Crossref] [ Google Scholar]
- Dubost E, McErlain H, Babin V, Sutherland A, Cailly T. Recent Advances in Synthetic Methods for Radioiodination. J Org Chem 2020; 85:8300-10. doi: 10.1021/acs.joc.0c00644 [Crossref] [ Google Scholar]
- Liu N, Shi Y, Guo J, Li H, Wang Q, Song M. Radioiodinated tyrosine based carbon dots with efficient renal clearance for single photon emission computed tomography of tumor. Nano Research 2019; 12:3037-43. doi: 10.1007/s12274-019-2549-7 [Crossref] [ Google Scholar]
- Patel N, Duffy BA, Badar A, Lythgoe MF, Årstad E. Bimodal Imaging of Inflammation with SPECT/CT and MRI Using Iodine-125 Labeled VCAM-1 Targeting Microparticle Conjugates. Bioconjug Chem 2015; 26:1542-9. doi: 10.1021/acs.bioconjchem.5b00380 [Crossref] [ Google Scholar]
- Ogawa K, Takeda T, Mishiro K, Toyoshima A, Shiba K, Yoshimura T. Radiotheranostics Coupled between an At-211-Labeled RGD Peptide and the Corresponding Radioiodine-Labeled RGD Peptide. ACS Omega 2019; 4:4584-91. doi: 10.1021/acsomega.8b03679 [Crossref] [ Google Scholar]
- Fawwaz M, Mishiro K, Nishii R, Makino A, Kiyono Y, Shiba K. A Radiobrominated Tyrosine Kinase Inhibitor for EGFR with L858R/T790M Mutations in Lung Carcinoma. Pharmaceuticals (Basel) 2021; 14:1-14. doi: 10.3390/ph14030256 [Crossref] [ Google Scholar]
- Fawwaz M, Mishiro K, Nishii R, Sawazaki I, Shiba K, Kinuya S. Synthesis and Fundamental Evaluation of Radioiodinated Rociletinib (CO-1686) as a Probe to Lung Cancer with L858R/T790M Mutations of Epidermal Growth Factor Receptor (EGFR). Molecules (Basel, Switzerland) 2020; 25:2914. doi: 10.3390/molecules25122914 [Crossref] [ Google Scholar]
- Mishiro K, Nishii R, Sawazaki I, Sofuku T, Fuchigami T, Sudo H. Development of Radiohalogenated Osimertinib Derivatives as Imaging Probes for Companion Diagnostics of Osimertinib. J Med Chem 2022; 65:1835-1847. doi: 10.1021/acs.jmedchem.1c01211 [Crossref] [ Google Scholar]
- Tang L, Peng C, Tang B, Li Z, Wang X, Li J. Radioiodinated Small-Molecule Tyrosine Kinase Inhibitor for HER2-Selective SPECT Imaging. J Nucl Med 2018; 59:1386-91. doi: 10.2967/jnumed.117.205088 [Crossref] [ Google Scholar]
- Ballard P, Yates JWT, Yang Z, Kim D-W, Yang JC-H, Cantarini M. Preclinical Comparison of Osimertinib with Other EGFR-TKIs in EGFR-Mutant NSCLC Brain Metastases Models, and Early Evidence of Clinical Brain Metastases Activity. Clin Cancer Res 2016; 22:5130-40. doi: 10.1158/1078-0432.Ccr-16-0399 [Crossref] [ Google Scholar]
- Kim ES. Olmutinib: First Global Approval. Drugs 2016; 76:1153-7. doi: 10.1007/s40265-016-0606-z [Crossref] [ Google Scholar]
- Lee K-O, Cha MY, Kim M, Song JY, Lee J-H, Kim YH. Abstract LB-100: Discovery of HM61713 as an orally available and mutant EGFR selective inhibitor. Cancer Res 2014; 74:LB-100. doi: 10.1158/1538-7445.Am2014-lb-100 [Crossref] [ Google Scholar]
- Kim DW, Lee DH, Han JY, Lee J, Cho BC, Kang JH. Safety, tolerability, and anti-tumor activity of olmutinib in non-small cell lung cancer with T790M mutation: A single arm, open label, phase 1/2 trial. Lung Cancer 2019; 135:66-72. doi: 10.1016/j.lungcan.2019.07.007 [Crossref] [ Google Scholar]
- Attwa MW, Kadi AA, Abdelhameed AS. Detection and characterization of olmutinib reactive metabolites by LC–MS/MS: Elucidation of bioactivation pathways. J Sep Sci 2020; 43:708-18. doi: 10.1002/jssc.201900818 [Crossref] [ Google Scholar]
- Limban C, Nuţă DC, Chiriţă C, Negreș S, Arsene AL, Goumenou M. The use of structural alerts to avoid the toxicity of pharmaceuticals. Toxicol Rep 2018; 5:943-53. doi: 10.1016/j.toxrep.2018.08.017 [Crossref] [ Google Scholar]
- Ricordel C, Friboulet L, Facchinetti F, Soria JC. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann Oncol 2018; 29:i28-i37. doi: 10.1093/annonc/mdx705 [Crossref] [ Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010; 31:455-61. doi: 10.1002/jcc.21334 [Crossref] [ Google Scholar]
- O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform 2011; 3:33. doi: 10.1186/1758-2946-3-33 [Crossref] [ Google Scholar]
- Delano DL. The PyMOL Molecular Graphics System, Version 2.3. 2020. https://pymol.org/.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009; 30:2785-91. doi: 10.1002/jcc.21256 [Crossref] [ Google Scholar]
- Arwansyah A, Arif AR, Ramli I, Kurniawan I, Sukarti S, Nur Alam M. Molecular modelling on SARS-CoV-2 papain-like protease: an integrated study with homology modelling, molecular docking, and molecular dynamics simulations. SAR QSAR Environ Res 2021; 32:699-718. doi: 10.1080/1062936X.2021.1960601 [Crossref] [ Google Scholar]
- Arwansyah A, Arif AR, Syahputra G, Sukarti S, Kurniawan I. Theoretical studies of Thiazolyl-Pyrazoline derivatives as promising drugs against malaria by QSAR modelling combined with molecular docking and molecular dynamics simulation. Molecular Simulation 2021; 47:988-1001. doi: 10.1080/08927022.2021.1935926 [Crossref] [ Google Scholar]
- Sumaryada T, Arwansyah, Roslia AW, Ambarsari L, Kartono A, editors. Molecular docking simulation of mangostin derivatives and curcuminoid on maltase- glucoamylase target for searching anti-diabetes drug candidates. 2016 1st International Conference on Biomedical Engineering (IBIOMED); 2016 5-6 Oct.
- Kristianslund R, Vik A, Hansen TV. A convenient synthesis of phenols. Synthetic Communications 2018; 48:2809-14. doi: 10.1080/00397911.2018.1496263 [Crossref] [ Google Scholar]
- Kumar M, Sharma U, Sharma S, Kumar V, Singh B, Kumar N. Catalyst-free water mediated reduction of nitroarenes using glucose as a hydrogen source. RSC Adv 2013; 3:4894-8. doi: 10.1039/C3RA40771C [Crossref] [ Google Scholar]
- Taniguchi T, Imoto M, Takeda M, Nakai T, Mihara M, Iwai T. Hydrolysis of Diazonium Salts Using a Two-Phase System (CPME and Water). Heteroatom Chemistry 2015; 26:411-6. doi: 10.1002/hc.21275 [Crossref] [ Google Scholar]
- Gelovani JG. Molecular imaging of epidermal growth factor receptor expression-activity at the kinase level in tumors with positron emission tomography. Cancer Metastasis Rev 2008; 27:645-53. doi: 10.1007/s10555-008-9156-5 [Crossref] [ Google Scholar]
- Yeh HH, Ogawa K, Balatoni J, Mukhapadhyay U, Pal A, Gonzalez-Lepera C. Molecular imaging of active mutant L858R EGF receptor (EGFR) kinase-expressing nonsmall cell lung carcinomas using PET/CT. Proc Natl Acad Sci U S A 2011; 108:1603-8. doi: 10.1073/pnas.1010744108 [Crossref] [ Google Scholar]
- Fawwaz M, Pratama M, Aminuddin AH, Baits M. Radiolabeled EGFR Tyrosine Kinase for the Detection of Dual Mutations EGFR L858R/T790M in NSCLC. Biointerface Res Appl Chem 2023; 13:1-11. doi: 10.33263/BRIAC135.500 [Crossref] [ Google Scholar]
- Yan X-E, Zhu S-J, Liang L, Zhao P, Choi HG, Yun C-H. Structural basis of mutant-selectivity and drug-resistance related to CO-1686. Oncotarget 2017; 8:53508-17. doi: 10.18632/oncotarget.18588 [Crossref] [ Google Scholar]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005; 2:57-61. doi: 10.1371/journal.pmed.0020017 [Crossref] [ Google Scholar]
- Ogawa K, Shiba K, Akhter N, Yoshimoto M, Washiyama K, Kinuya S. Evaluation of radioiodinated vesamicol analogs for sigma receptor imaging in tumor and radionuclide receptor therapy. Cancer Sci 2009; 100:2188-92. doi: 10.1111/j.1349-7006.2009.01279.x [Crossref] [ Google Scholar]
- Noh YS, Yoon S, Kim SR, Lee K-T, Jang I-J. A safety, pharmacokinetic, pharmacogenomic and population pharmacokinetic analysis of the third-generation EGFR TKI, olmutinib (HM61713), after single oral administration in healthy volunteers. Basic & Clinical Pharmacology & Toxicology 2019; 125:370-81. doi: 10.1111/bcpt.13262 [Crossref] [ Google Scholar]