Bioimpacts. 14(3):28876.
doi: 10.34172/bi.2023.28876
Original Article
Unravelling benzazepines and aminopyrimidine as multi-target therapeutic repurposing drugs for EGFR V774M mutation in neuroglioma patients
Jitender Singh Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing, 
Krishan L Khanduja Writing – review & editing,
Pramod K Avti Conceptualization, Project administration, Supervision, Validation, Writing – review & editing, , *
Author information:
Department of Biophysics, Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh, India – 160012
Abstract
Introduction:
Neuroglioma, a classification encompassing tumors arising from glial cells, exhibits variable aggressiveness and depends on tumor grade and stage. Unraveling the EGFR gene alterations, including amplifications (unaltered), deletions, and missense mutations (altered), is emerging in glioma. However, the precise understanding of emerging EGFR mutations and their role in neuroglioma remains limited. This study aims to identify specific EGFR mutations prevalent in neuroglioma patients and investigate their potential as therapeutic targets using FDA-approved drugs for repurposing approach.
Methods:
Neuroglioma patient’s data were analyzed to identify the various mutations and survival rates. High throughput virtual screening (HTVS) of FDA-approved (1615) drugs using molecular docking and simulation was executed to determine the potential hits.
Results:
Neuroglioma patient samples (n=4251) analysis reveals 19% EGFR alterations with most missense mutations at V774M in exon 19. The Kaplan-Meier plots show that the overall survival rate was higher in the unaltered group than in the altered group. Docking studies resulted the best hits based on each target's higher docking score, minimum free energy (MMGBSA), minimum kd, ki, and IC50 values. MD simulations and their trajectories show that compounds ZINC000011679756 target unaltered EGFR and ZINC000003978005 targets altered EGFR, whereas ZINC000012503187 (Conivaptan, Benzazepine) and ZINC000068153186 (Dabrafenib, aminopyrimidine) target both the EGFRs. The shortlisted compounds demonstrate favorable residual interactions with their respective targets, forming highly stable complexes. Moreover, these shortlisted compounds have drug- like properties as assessed by ADMET profiling.
Conclusion:
Therefore, compounds (ZINC000012503187 and ZINC000068153186) can effectively target both the unaltered/altered EGFRs as multi-target therapeutic repurposing drugs towards neuroglioma.
Keywords: Allele frequencies, EGFR mutations, Gibbs free energy, Glioma, Missense-mutation, Multi-target
Copyright and License Information
© 2024 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
A brain tumor is an intracranial neoplasm that develops in the brain or central spinal canal.1 Glioblastoma (GBM) is the most frequent malignant of the brain and other CNS tumors accounting for 47.7% of all cases, and 3.21 cases occur for every 100,000 people.2 The World Health Organization (WHO) differentiates glioma into two types based on their histology and prognosis: low-grade and high-grade gliomas.3 Low-grade gliomas include grade I (pylociticastrocytomas) and grade II (diffuse astrocytomas) tumors that are slow-growing and have a favorable prognosis.4 Grade III (anaplastic astrocytomas) and grade IV GBM are the most common and aggressive CNS cancers, with 99% of cases being deemed incurable.4 Despite significant improvements in treating some cancers, treating GBM has been challenging for over a decade.5 The incidence of glioma in adults is 3/100,000 per year, making up 52% of all primary brain tumors.6 Despite aggressive treatment, the prognosis for GBM patients is abysmal, with a median overall survival rate of just 10% for 12 months and five years.5 Poor patient outcomes result from intrinsic or acquired tumor resistance to radiation or chemotherapy.7 Receptor tyrosine kinases (RTKs) are strongly implicated in developing resistance to radiation and chemotherapy in GBM.8 Numerous RTK inhibitors are tested with conventional medicines, but none has demonstrated a therapeutic advantage with increased toxicity.9 The EGFR gene is known to overexpress in various cancers.10,11 It is observed that EGFR is overexpressed in most primary GMBs and some secondary ones and indicates more aggressive GBM characteristics.12 EGFR gene over-expression and amplification are present in about 40% of primary GBMs.10 In addition, overexpression of EGFR is associated with (63–75%) of the gene expression changes in both wild-type/non-mutant and mutant EGFR1 types.13
In GBM mutations in the EGFR (epidermal growth factor receptor) are frequent and cause disease progression and aggressiveness.8 and might have a causative role in cancer development and therapeutic resistance.14 GBM gene amplification and overexpression are standard in low-grade gliomas, suggesting that aberrant EGFR signaling plays a causative role in the pathogenesis of GBM. EGFRvIII type is the most frequent EGFR mutant found in neuroglioma.10 This mutant is formed by erasing 2–7 exons of the EGFR gene, resulting in the deletion of 267 amino acid residues from the receptor's extracellular domain. EGFRvIII does not bind to ligands but instead signals inherently and is an essential process in gliomagenesis.15 The EGFRvIII mutant is more tumorigenic than its wild-type receptor.13 Several tumor biology aspects, including the patient's survival rate, cell proliferation, metastatic spread, motility rate, and treatment susceptibility, may be influenced by increased EGFRvIII expression.13
Tyrosine kinase inhibitors (TKIs), antibodies, and vaccinations are utilized to block the EGFR signaling network, making it a popular target for medicinal therapies.16 Recent studies suggest that in Erlotinib-treated patients, co-expression of PTEN and EGFRvIII are not associated with increased survival.16 Erlotinib is an EGFR inhibitor with a low response rate and patient survival rate.16 So, there is an urgent need to identify the new mutation of EGFR having high-frequency rates in neuroglioma and target them with specific and effective drugs. In the drug development process, in silico computational approaches, in recent times with significant technological advancements, have become invincible tools for discovering and repurposing purposes with the shortest times and greater probability of achieving the new or repurposed drugs.17 Molecular docking and molecular dynamics simulation are advanced computational techniques that enable high-throughput virtual screening (HTVS) of large molecular libraries.18 These approaches assess the binding affinity between ligands and target receptors and generate scores to evaluate the results. In addition, they provide valuable structural insights into how ligands interact with target receptors, offering a view on the blocking mechanism. This information is crucial for identifying and confirming effective ligands with potential therapeutic applications. Overall, these methods surpass conventional techniques in their ability to generate structural hypotheses and aid in identifying promising ligands. The primary aim of this study was to identify a specific mutation in EGFR frequently observed in patients with neuroglioma. Further, the mutated form and wild-type EGFR was investigated as potential targets for assessing molecular drug interaction changes. A repurposing approach was also employed to screen FDA-approved drugs in order to identify potential therapeutic agents for further evaluation.
Materials and Methods
Analysis of clinical data
Genomic data retrieval and analysis of EGFR
This study accessed complete genomic information for the EGFR gene in neurogliomas from the cBioPortal cancer database.19 The cBioPortal is a user-friendly tool that allows for interactive interpretation of multiscale data, providing information on genomic and proteomic alterations, transcriptomics, and microarray data profiling. The main objective of the cBioPortal database is to facilitate direct access to genomic profile datasets and enable analysis of disease expression. We searched the cBioPortal database using the keywords, "CNS/Brain" and focused specifically on neuroglioma in order to filter the data and select relevant studies.
Cancer types and mutational analysis
In this study, complete genomic information for the EGFR gene in various forms of GBM was obtained from the cBioPortal database. The database allows users to search for specific genes in different cancer types. To evaluate the EGFR data, we searched using the "CNS/Brain" keywords, which generated results from 17 other datasets. The oncoprint visualization in the cBioPortal provided a concise overview of the queried genes, displaying them in a color-coded format to represent different types of mutations and genetic alterations. This visualization summarized the genomic and proteomic alterations in the selected genes. Plot diagrams were also used to illustrate the different mutation types of the queried genes, using color codes.
Survival analysis
The study utilized the Kaplan-Meier estimator plots to evaluate the overall and relapse-free survival rates of two distinct groups based on genomic alterations in the EGFR gene. Genomic alteration data for the EGFR gene was collected for the study participants. The unaltered EGFR group consisted of individuals without any identified alterations in the EGFR gene, while the altered EGFR group included individuals with various genomic alterations. Using the collected data, Kaplan-Meier estimator plots were generated to visualize the survival probabilities over time for each group. The Kaplan-Meier curves were analyzed to compare the overall and relapse-free survival rates between the unaltered and altered EGFR groups. This analysis provided insights into the impact of other genomic alterations in the EGFR gene on patient survival outcomes.
Expression pattern analysis of EGFR in different brain regions
The GTEx model was employed to examine tissue-specific gene expression and regulation of EGFR in various brain regions. Violin plots were utilized to depict the localization of EGFR expression across different brain regions. Additionally, the BEST program was used to generate a spatiotemporal expression heatmap, allowing the evaluation of EGFR gene expression patterns in other brain regions at various ages.
Structural analysis
Ligand preparation
We utilized the ZINC Database to retrieve the FDA-approved drug library's three-dimensional (3D) structures for docking studies. We downloaded a total of 1615 FDA-approved drugs in single sdf file format from the Zinc Database and included all of them for screening purposes. The open babel and PyRx programs were employed to optimize and minimize the ligands (FDA Drug molecules). The selection of the ZINC database in this methodology was based on several factors specific to the study. These factors included: a) well-documented chemical properties of the compounds in the database; b) structural diversity exhibited by these compounds; and c) availability of the compounds for screening purposes. Collectively, these factors ensured a robust and diverse set of compounds for subsequent screening and analysis.
Assessment of the structure and accurate binding site prediction
In this study, the unaltered three-dimensional (3D) structure of the EGFR protein was retrieved from the RCSB-PDB using X-ray crystallography data. The Protein Data Bank ID 3POZ was used for this purpose. To create the altered form of EGFR (V774M), we employed the Discovery Studio Visualizer software. Within the software's amino acid residues section, we replaced the Valine (V) amino acid residue with Methionine (M). The resulting modified structure was then saved in PDB format. Additionally, we utilized the Swiss Model homology method to generate the most accurate 3D model of the altered EGFR. the primary sequence of the altered EGFR was submitted to the Swiss Model database. A search was conducted within the database to find the template with the highest similarity to the altered EGFR sequence. This identified template was then utilized to generate the 3D model of the altered EGFR. To facilitate comparative analysis between the unaltered and altered EGFR structures, we aligned the modelled structure of the altered EGFR with the 3D structure of the unaltered EGFR. This alignment was performed using the Discovery Studio Visualizer, which enabled us to identify any conformational changes resulting from the missense mutation. Furthermore, Using Discovery Studio Visualizer and PyRx program, the 3D structures were prepared by removing hydrogen atoms (H atoms), heteroatoms, and non-essential water molecules from the target receptor 3D structures. The meta-pocket program was used to estimate the reliable ligand binding sites in EGFR.20 Further, ConSurf Server was used to identify the conserved residues in both the EGFR receptors (altered and unaltered) (3POZ id).21
Structure validation of mutant EGFR
To validate the modelled structure of the mutant EGFR receptor, this study utilized the structural analysis and verification server. The server was employed to assess the structural geometric features of the mutant EGFR receptor, including residue interaction compatibility and backbone conformation.22 The structural analysis involved examining the compatibility of interactions between individual residues and verifying the overall conformation of the protein backbone. The Ramachandran plot statistics, determined using the PROCHECK program, were employed to evaluate the quality of the modelled structures.22 The psi [Ψ] and phi [ϕ] angles of the protein backbone were analyzed to ensure that they fell within acceptable ranges, indicating a favorable stereochemical quality of the modelled structures. The ERRAT program was utilized to investigate and improve the non-bonded atomic interactions within the modelled structures. This analysis helped identify and resolve any unfavourable atomic interactions that may have occurred during the modelling process. Furthermore, the WHAT IF program was employed to address atomic collisions, also known as bumps, within the modelled structures. The program achieved this by adjusting the side chain torsion angles (chi1, chi2, and chi3) to eliminate any steric clashes between atoms.22
Ligand screening and docking
The high throughput virtual screening and molecular docking process were performed using PyRx software with a UFF force field.23 To create the accurate grid map and find the best docking interaction confirmations, a grid map with coordinates of x:15,9005, y:4252, and z:8,7171 was set for both EGFR receptors. With the molecular docking method, the eight poses retrieved from the docking runs for each ligand. Lamarck's genetic algorithm and empirical free energy were selected in the PyRx program. An empirical scoring function was used in the docking process to determine the binding affinity of a specific protein-ligand complex. Docking complexes with the best docking score and various binding interactions were further evaluated using the discovery studio visualizer (2.1.0).
Molecular dynamic simulation (MD) and trajectory analysis
The molecular dynamics study of the target receptor-ligand complexes was simulated using Schrodinger Maestro Version 20.1 (Desmond module). The OPLS-2005 force field was applied to optimize the protein structures for a simulation system. For MD simulation studies, the TIP3P three-site water solvation model, with orthorhombic shape boundary condition [10 Å × 10 Å × 10 Å distance] and the buffer methods (physiological concentration of monovalent ions) was used. The system builder module used TIP3P to achieve exceptional computational efficiency while considering the actual shape of the water molecules. The system was configured for simulation using a predetermined water model in an orthorhombic box with periodic boundary constraints determining the form and size of the box. An orthorhombic border box was used to cover the target receptor-ligand complexes, and further sodium-calcium ions were added to neutralize them. The salt concentration (0.15 M), positive salt ion (Na+), and negative salt ion (Cl-) were set to proceed further in the ion parameter settings. In the molecular dynamic simulation step, 100 ns time was selected with 0.0 elapsed time and 10000 frames. The temperature was set to 310 K with a pressure of 1.01325 bar, and the NPT ensemble class was chosen. The Martyna-Tobias-Klein Barostat model and nose-hoover chain thermostat model were used, each with a 1.0 and 2.0 ps relaxation time, respectively.
Furthermore, a 0 to 10000 (ps) step interval was used in the randomized velocities. The simulation process was carried out at 100 ns. Dynamic cross-correlation matrix (DCCM) and principal component analysis (PCA) were studied to understand the dynamic behavior of residue motions in the conformations. Principal component analysis was used to calculate the critical dynamics, which reflect the fundamental protein movements required for enzymatic activity and residue displacement in the docked complex concerning time.24
The DCCM, on the other hand, was built to find correlated motions of residues. The information between the i and j atoms of a time-correlated protein is shown in the matrix (Cij). While negative values signify atomic displacement in the opposite direction, positive values denote motions that are parallel to or correlated with one another. Using the Maestro wizard visualizer, a radius of gyration algorithm was performed to assess the receptor compactness and overall stability.
ADMET properties and therapeutic application of drug molecules
ADMET profiling is a crucial step in drug discovery and development, allowing for the evaluation of pharmacokinetic and toxicological properties of potential drug candidates. The SwissADME database is a widely utilized tool for conducting ADMET profiling.25 In this study, the ADMET profiling of selected FDA approved compounds was performed using the SwissADME database. Initially, the chemical SMILE structures of the compounds were retrieved from PubChem databases. Subsequently, these structures were input into the SwissADME database, which generated comprehensive data on various pharmacokinetic and physiochemical properties of the compounds. The SwissADME database provides valuable insights into the pharmacokinetic quality of therapeutic candidates, encompassing parameters such as solubility, permeability, metabolic stability, and toxicity. Toxicity classification was performed using PROTOX method.26 This classification system allowed for a standardized assessment of their safety profiles. Furthermore, the study explored the therapeutic applications of the FDA-shortlisted drugs from the PubChem Database.
Free energy, kd, ki, and IC50 calculation
An MMGBSA method determined the minimum free energy of the docked complex of altered and unaltered EGFR.27 The MMGBSA was calculated using the following formula:
(1)
ΔGsol represents the difference in solvation energy between the receptor-drug complex and the individual solvation energies of the receptor and drug. ΔEMM indicates the difference in energy between the receptor-drug complex and the combined energies of the unliganded receptor and drug. ΔG (SA) refers to the difference in surface area energy between the complex, the drug molecule, and the unliganded receptor.
The experimental dissociation constant (kd), inhibition constant (ki), and inhibitory concentration (IC50) were examined using data from the PubChem database. The dissociation constant kd reflects the affinity between the receptor and ligand when they are in equilibrium. A low kd indicates a high affinity, meaning the receptor readily binds to the ligand, while a high kd suggests a lower affinity and less binding.
The in silico kd dissociation constant is calculated by the following equation:
Where R is the gas constant (R=1.985 × 10−3 kcal mol−1 K−1), T is the temperature in Kelvin.
Similarly, the following formula was used to determine the inhibition constant (ki) from the binding energy (∆G):
Where (R=1.985 × 10−3 kcal mol−1 K−1) is the universal gas constant and Temperature (T=298.15 K).
Results
Analysis of clinical data
Genomic data retrieval and analysis of EGFR
The current study observed 17 different study data sets with 4251 clinical samples retrieved from the cBioPortal cancer genomic dataset. This information also included the types of neurogliomas, number of genomic profile samples, gene mutation numbers, structural variant genes, and patients’ survival rates. Additionally, the search term “EGFR” was used to filter the target gene data and examined at different data scale analyses, including oncoprint, neurogliomas types, mutation analysis, and patient's overall and disease-free survival rates.
Cancer types and mutational analysis
There were 19% genetic alterations occurring on EGFR genes in neuroglioma patients, including missense mutations, amplifications, and deep deletions (Fig. S1). The analysis of EGFR gene mutational data also revealed 432 missense, 5 truncating, 130 SV fusion, and 16 splice variant mutations (Fig. S2). We also identified that different types of neurogliomas have varying frequencies of gene alterations (Fig. 1). Genomic data were obtained from two groups: one with altered EGFR and the other with unaltered EGFR, comprising different samples and patient studies (Fig. S3). Further, the altered EGFR patient group exhibited a significantly higher frequency of alterations than the unaltered EGFR group (Fig. 2A-B). The mutation V774M on exon 19, with the highest allele frequency (0.97%), was selected for additional in silico computational studies as it represents the most significant risk for neuroglioma patients.
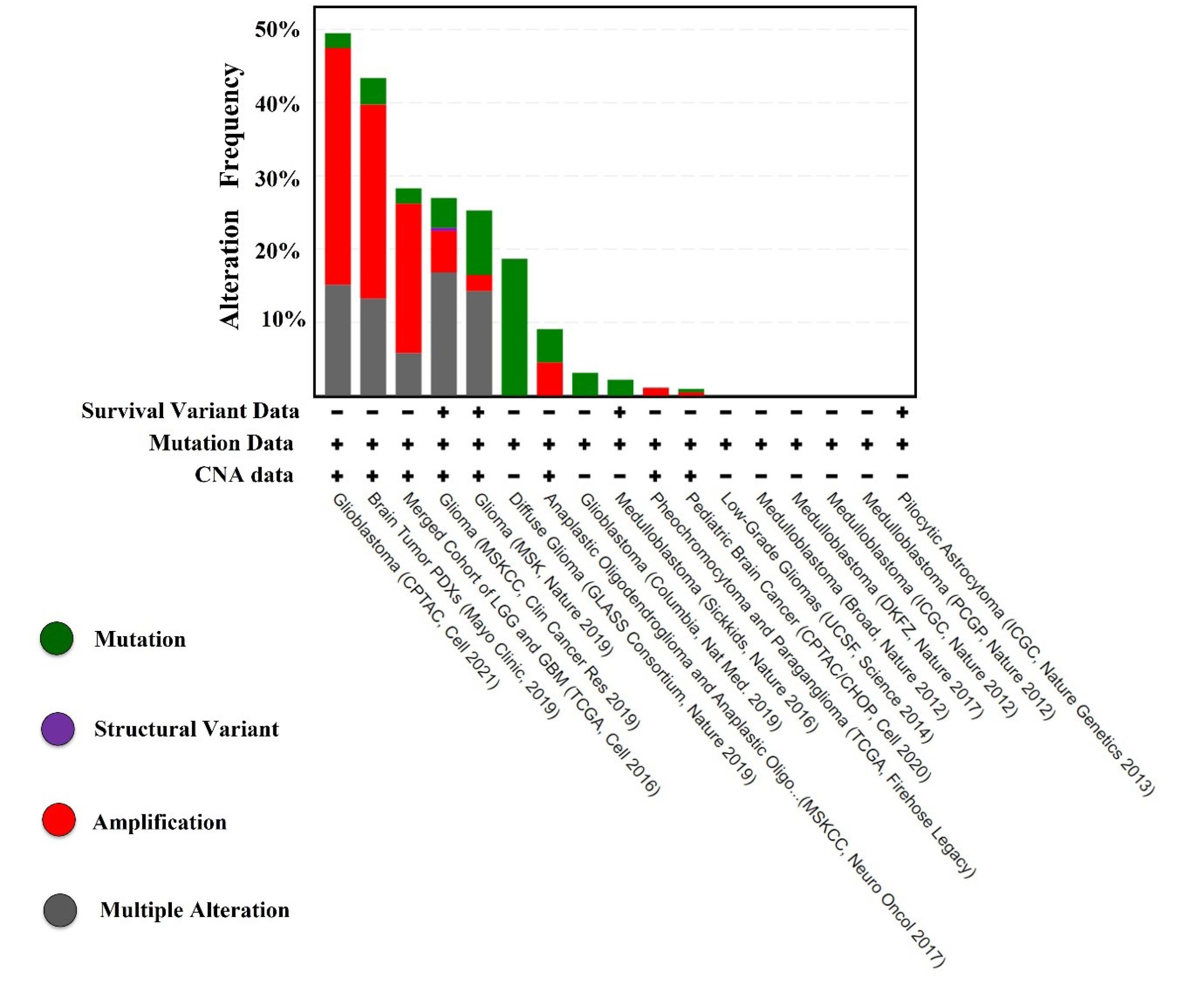
Fig. 1.
Histogram showing the EGFR alteration frequency in various neuroglioma types.
.
Histogram showing the EGFR alteration frequency in various neuroglioma types.
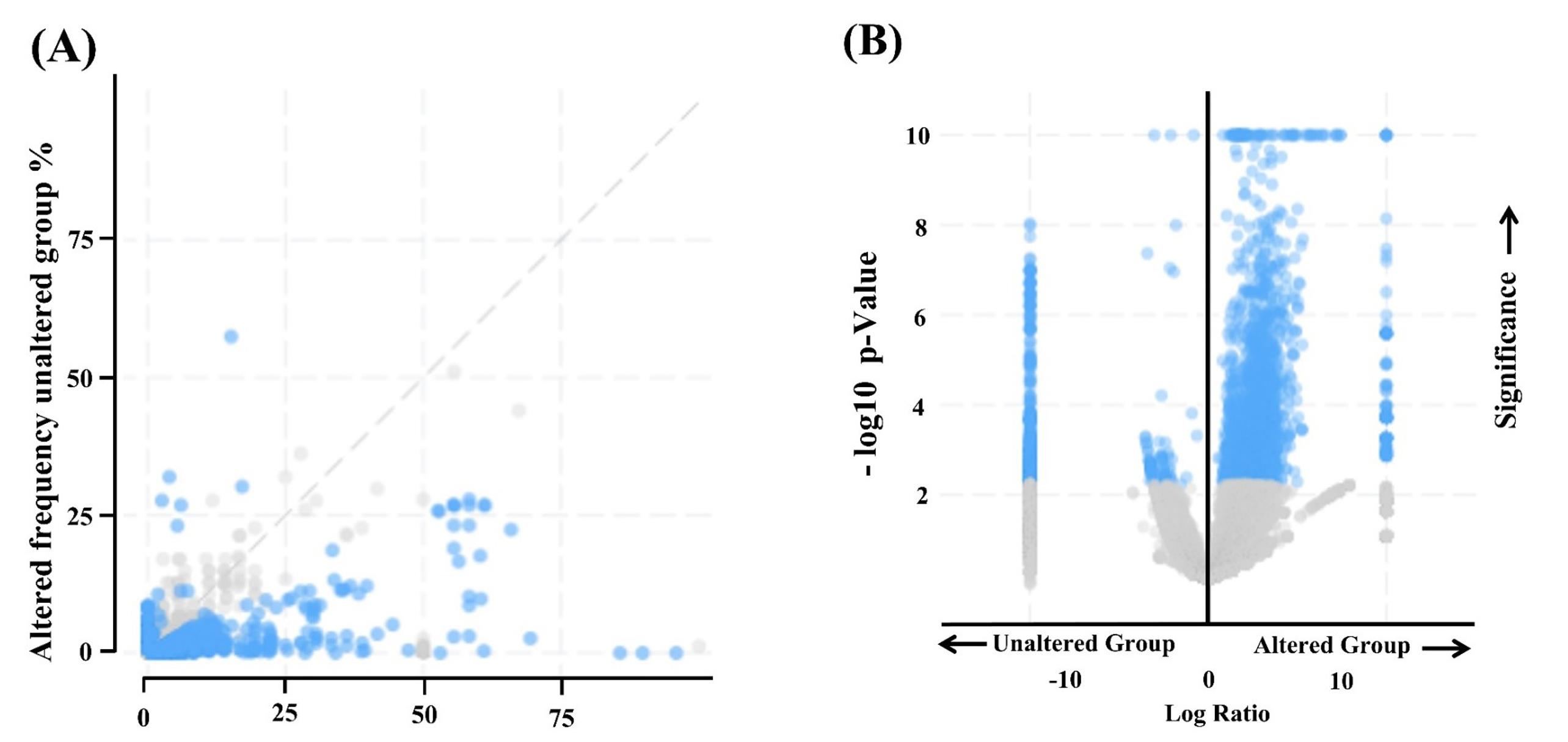
Fig. 2.
(A) Kaplan–Meier plots show overall survival and progression-free. (B) survival analysis of altered and unaltered EGFR.
.
(A) Kaplan–Meier plots show overall survival and progression-free. (B) survival analysis of altered and unaltered EGFR.
Survival analysis
Kaplan-Meier plots for overall survival and relapse-free survival were produced to compare the survival rates of the EGFR-altered and unaltered groups. According to the analysis, the altered EGFR group's overall survival rate was less than the unaltered group's, with a log-rank P value of 0.00 (Fig. 3A). In the altered EGFR group, the life span of patients is only 120 months compared to the unaltered group, which is 340 months (Fig. 3A). According to the results of relapse-free analyses, the unaltered group has a better survival rate than the altered EGFR group, which suggests that the altered group had a considerably shorter lifespan. The relapse-free group's log-rank p-value was p-value = 0.00 (Fig. 3B). The survival months of patients with altered EGFR is 120 months compared to the unaltered EGFR patients group, which is 250 months (Fig. 3B).
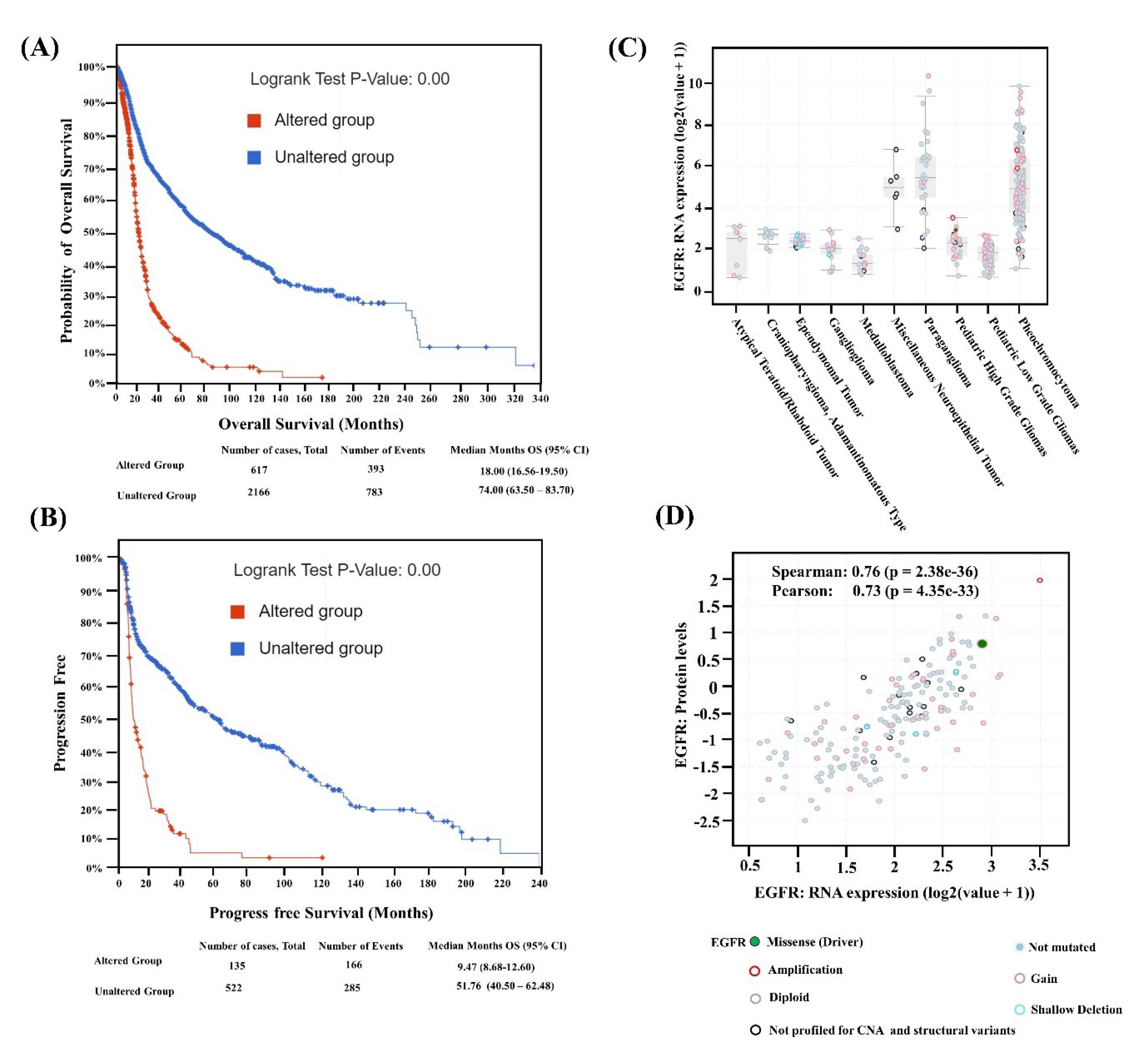
Fig. 3.
(A) Alteration frequency of unaltered and altered patient's groups, (B) log p value of the altered and unaltered patient group. (C) EGFR mRNA expression in various cancer types of neuroglioma with mutation types, (D) correlation of EGFR protein level expression vs. RNA expression in different mutation types.
.
(A) Alteration frequency of unaltered and altered patient's groups, (B) log p value of the altered and unaltered patient group. (C) EGFR mRNA expression in various cancer types of neuroglioma with mutation types, (D) correlation of EGFR protein level expression vs. RNA expression in different mutation types.
Expression analysis of EGFR gene in brain
The differential expression of EGFR mRNA vs. neuroglioma cancer types shows higher gain, amplification, and non-mutated EGFR in pheochromocytoma and paraganglioma glioma types (Fig. 3C). Similarly, the missense mutation expression is only present in pediatric high-grade gliomas compared to the other glioma cancer types (Fig. 3C). In pediatrics with low-grade glioma types, only gain and non-mutated alteration types are expressed in higher numbers (Fig. 3C). Moreover, shallow deletion shows expression in gangliogliomas and ependymoma tumors. Furthermore, the findings show that missense mutation and amplification are highly expressed at the mRNA and protein levels. The gain and non-mutated expression are significantly related when comparing the expression of different alteration types at the protein and mRNA levels (Fig. 3D). The spatiotemporal expression heat map results demonstrate that the striatum, amygdala, hippocampus, occipital cortex, cerebellum, and thalamus all have significant EGFR expression (Fig. S4A). The EGFR expression pattern represented a violin plot, with a substantial change in the brain caudate (basal ganglia) and brain nucleus accumbens than in another part of the brain (Fig. S4B).
Structural analysis
Assessment of the structure and accurate binding site prediction
In the current study, the PDB ids of EGFR receptors were screened from the RCSB database, using the filters such as x-ray crystallography and species name - homosapiens, selected from 2010-2021 and resolution from 2.5 to 1.0. Å (Table S1). A total of 21 PDB ids were shortlisted from such search criteria. After careful analysis, 5 PDB IDs are shortlisted based on the non-mutation and single chain of the protein. Multiple sequencing alignment results suggest that 5 PDB ids, when aligned sequentially, found 4 IDs with 100% identity (Fig. S5). Finally, the high resolution (1.50 Å) of the 3D X-crystallography structure of the target EGFR with PDB ID - 3POZ was chosen over all the other shortlisted ids (Table S1). As determined from the findings of the mutational analysis, the position of the amino acid (V774M) that was altered in the 3POZ structure reflects the mutated EGFR. The sequence and structural identity of the 3D design created with the Discovery Studio Visualizer was 99.69% identical to that of the 3POZ (Fig. S6B). When superimposed, the 3D structures of the unaltered and altered EGFR revealed a slight conformational change at the reside number (V774M) (Fig. S6B). Meta-pocket methods predicted the ligand-binding site of both the unaltered and altered EGFR (Table S2). According to the conservancy results, the top three regions are identified: variable, average, and highly conserved. Most of the residues were identified as highly conserved, which were also found in the catalytic and binding regions of the EGFR PDB id: 3POZ (Table S2).
Structure validation
Before proceeding further with the virtual screening process, the modeled 3D structure of altered EGFR is validated, and the results reveal the overall quality factor of 97.4 for the modeled structure (Fig. S7A). The overall quality factor of the 3D modeled structure was 97.40 (Fig. S7A). In the 3D/1D profile, 80% of the amino acid residues received a score of 0.2 (Fig. S7B). Most residues fall in the Ramachandran plot's favorable region, with an average Z score of 0.273 (Fig. S7C &D).
Ligand screening and docking
Based on the virtual screening generated shortlist, each receptor's top ten best-hit compounds were selected based on their minimum docking score and the presence of at least one or more hydrogen bonds (Table 1). In addition, two compounds currently used in the treatment of neuroglioma (Erlotinib and Gefitinib) were also included in the docking analysis. The results indicate that the shortlisted compounds exhibited a lower minimum free energy (-11.3 to -12.4 kcal/mol) compared to Erlotinib and Gefitinib (-7.8 to -8.7 kcal/mol) for both unaltered and altered EGFR (Table 1) suggesting that these compounds have a tremendous potential for binding and interacting with the receptors as compared to the currently used drugs. Furthermore, all compounds showed stable ligand contacts with the active site residues and minimal Gibbs free energy (ΔG). Based on the minimum free energy and hydrogen bonding interaction criteria, each receptor's top four lead compounds (altered and unaltered EGFR) were further shortlisted (Fig. S8 A-B). Figs. 4A-4L visually illustrate the 2D interactions between the top shortlisted docked structures, including the currently used drugs (Erlotinib and Gefitinib), towards unaltered and altered EGFR receptors. These figures provide insights into the molecular interactions responsible for the compounds' binding and potential therapeutic activity. The 2D interactions highlight specific receptor residues involved in crucial interactions with the compounds, such as hydrogen bonding, electrostatic interactions, and hydrophobic interactions. Analysis of the 2D interactions reveals that the currently used drugs, Erlotinib and Gefitinib, exhibit a lower number of hydrogen bonding interactions (0-4) with both unaltered and altered EGFR receptors compared to the shortlisted compounds (4-8). The 3D docked complexes and their interactions diagram of the shortlisted top hits for respective unaltered and altered EGFR are presented in Fig. 5A-H. The docking score of top lead candidate FDA drug molecules in the case of unaltered EGFR includes ZINC000012503187 (∆G = -12.0kcal/mol), ZINC000006716957 (∆G = -11.6kcal/mol), ZINC000011679756 (∆G = -11.3kcal/mol), and ZINC000068153186 (∆G = -11.3kcal/mol), Erlotinib (∆G = -7.8 kcal/mol), and Gefitinib (∆G = -8.7kcal/mol) (Fig. S8A). For altered EGFR, the top lead compounds having minimum free energy include ZINC000006716957 (∆G = -12.4 kcal/mol), ZINC000012503187 (∆G = -12.0 kcal/mol), ZINC000003978005 (∆G = -11.7 kcal/mol) and ZINC000068153186 (∆G= -11.6 kcal/mol), Erlotinib ((∆G = -7.7 kcal/mol), and Gefitinib (∆G = -8.1 kcal/mol) (Fig. S8B). These results confirmed that 4 compounds for each receptor out of 1615 FDA drug molecules could strongly target both the forms of EGFR receptors (unaltered and altered). These results also suggest that three shortlisted compounds are common for unaltered and altered EGFR, such as ZINC000012503187, ZINC000068153186, and ZINC000006716957.
Table 1.
Docking score of top 10 shortlisted FDA-approved and standard drugs
|
Sr. No.
|
ZINC id
|
Docking score (∆G) kcal/mol.
|
No of H bonds
|
|
Unaltered EGFR
|
| 1 |
ZINC000164760756 |
-12.3 |
5 |
| 2 |
ZINC000012503187 |
-12.0 |
4 |
| 3 |
ZINC000004099008 |
-11.6 |
7 |
| 4 |
ZINC000006716957 |
-11.6 |
8 |
| 5 |
ZINC000003978005 |
-11.5 |
4 |
| 6 |
ZINC000052955754 |
-11.5 |
5 |
| 7 |
ZINC000003932831 |
-11.3 |
4 |
| 8 |
ZINC000011679756 |
-11.3 |
4 |
| 9 |
ZINC000068153186 |
-11.3 |
7 |
| 10 |
ZINC000242548690 |
-11.2 |
7 |
| 11 |
Erlotinib |
-7.8 |
4 |
| 12 |
Gefitinib |
-8.7 |
3 |
|
Altered EGFR
|
| 1 |
ZINC000006716957 |
-12.4 |
3 |
| 2 |
ZINC000012503187 |
-12 |
3 |
| 3 |
ZINC000253632968 |
-12 |
4 |
| 4 |
ZINC000164760874 |
-11.8 |
6 |
| 5 |
ZINC000003978005 |
-11.7 |
4 |
| 6 |
ZINC000068153186 |
-11.6 |
3 |
| 7 |
ZINC000164760756 |
-11.6 |
4 |
| 8 |
ZINC000052955754 |
-11.5 |
4 |
| 9 |
ZINC000011616882 |
-11.3 |
2 |
| 10 |
ZINC000001550477 |
-11.1 |
4 |
| 11 |
Erlotinib |
-7.7 |
0 |
| 12 |
Gefitinib |
-8.1 |
4 |
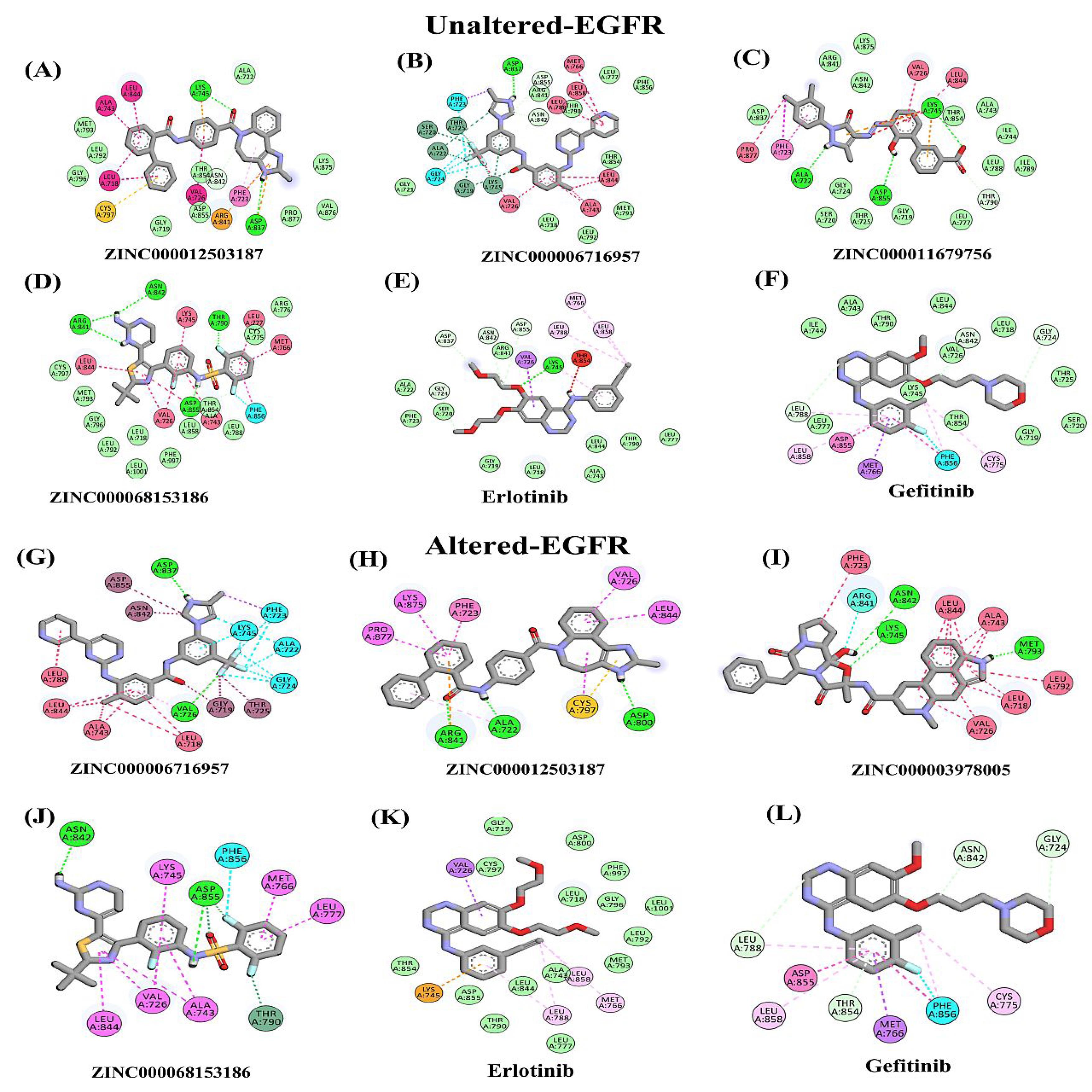
Fig. 4.
2D Interaction analysis of docked complexes with bonds (A-F) unaltered EGFR showing interactions with compounds (A) ZINC000012503187, (B) ZINC000006716957, (C) ZINC000011679756, (D) ZINC000068153186, (E) Erlotinib and (F) Gefitinib, (G-L) mutant/altered EGFR showing residual interactions with shortlisted compounds (G) ZINC000006716957, (H) ZINC000012503187, (I) ZINC000003978005, (J) ZINC000068153186, (K) Erlotinib and (L) Gefitinib.
.
2D Interaction analysis of docked complexes with bonds (A-F) unaltered EGFR showing interactions with compounds (A) ZINC000012503187, (B) ZINC000006716957, (C) ZINC000011679756, (D) ZINC000068153186, (E) Erlotinib and (F) Gefitinib, (G-L) mutant/altered EGFR showing residual interactions with shortlisted compounds (G) ZINC000006716957, (H) ZINC000012503187, (I) ZINC000003978005, (J) ZINC000068153186, (K) Erlotinib and (L) Gefitinib.
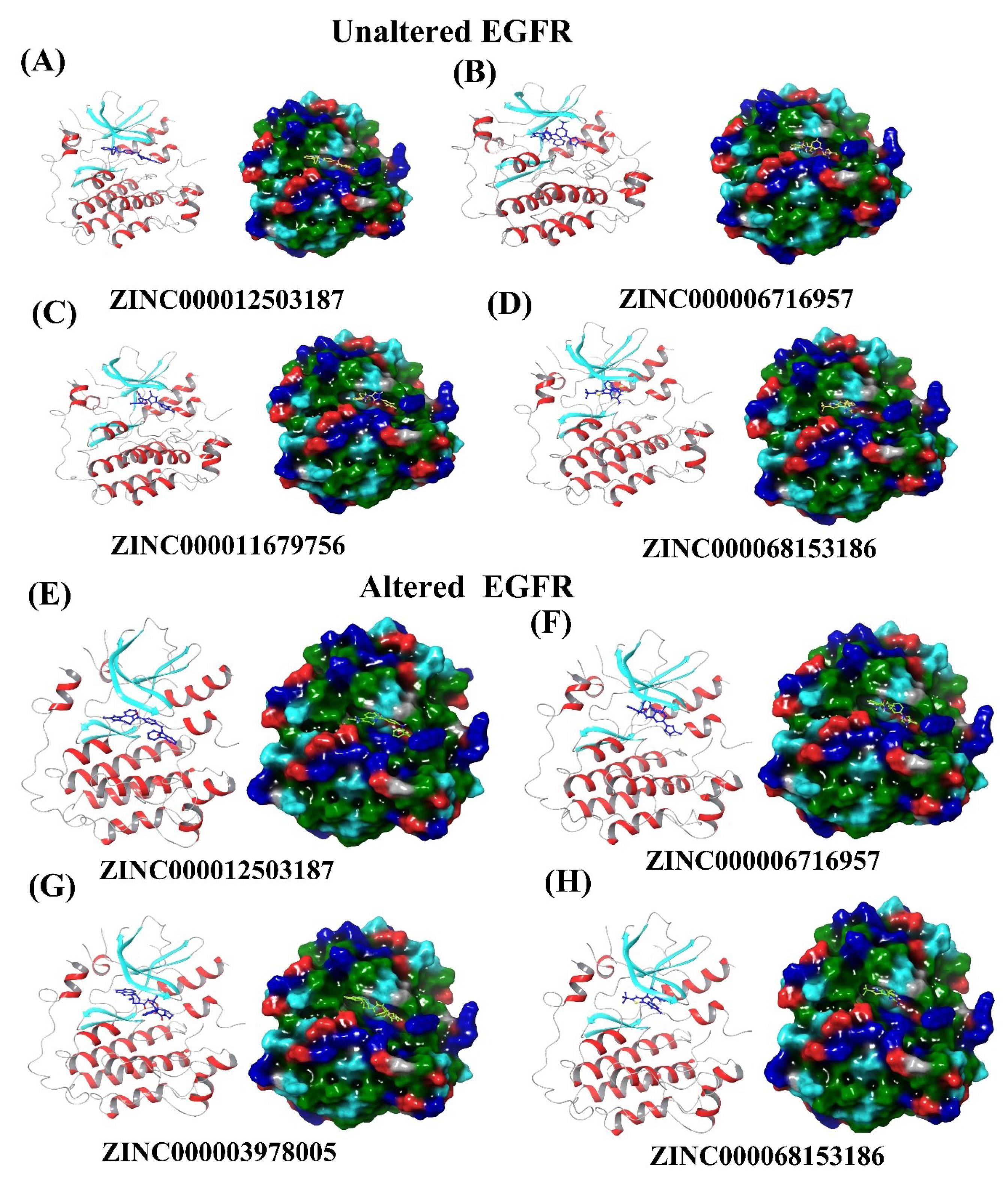
Fig. 5.
3D and surface view of docked complexes with shortlisted compounds a-d) docked complexes of unaltered EGFR (A) ZINC000012503187 (B) ZINC000006716957 (C) ZINC000011679756 (D) ZINC000068153186 (E-H) mutant/altered EGFR showing residual interactions with shortlisted compounds (E) ZINC000006716957 (F) ZINC000012503187 (G) ZINC000003978005 (H) ZINC000068153186.
.
3D and surface view of docked complexes with shortlisted compounds a-d) docked complexes of unaltered EGFR (A) ZINC000012503187 (B) ZINC000006716957 (C) ZINC000011679756 (D) ZINC000068153186 (E-H) mutant/altered EGFR showing residual interactions with shortlisted compounds (E) ZINC000006716957 (F) ZINC000012503187 (G) ZINC000003978005 (H) ZINC000068153186.
Molecular dynamic simulation
Molecular dynamics simulation is used to understand better the physical motion of the atoms and molecules in the top hits system. The considerable average RMSD of unaltered EGFR complex with compounds (ZINC Database ids ZINC000012503187 = 2.3Å, ZINC000006716957 = 3.5Å ZINC000011679756 = 2.2Å, and ZINC000068153186 = 2.1Å) show promising results (Fig. 6A & Table 2).Similarly, the RMSF values of unaltered EGFR complexes were ZINC000012503187 = 3.5Å, ZINC000006716957 = 3.1Å ZINC000011679756 = 3.4Å, and ZINC000068153186 = 3.3Å (Fig. 6B & Table 2). The altered/mutant EGFR complex has the best average RMSD values (include ZINC000006716957 = 3.6Å, ZINC000012503187 = 2.5Å, and ZINC000003978005 = 2.0Å and ZINC000068153186 =2.2Å) (Fig. 6C & Table 2). The average RMSF value of altered EGFR complexes was ZINC000006716957 = 3.0Å, ZINC000012503187 = 3.9Å, ZINC000003978005 = 3.6Å and ZINC000068153186 = 3.5 Å (Fig. 6D & Table 2). Results also confirm the most common amino acid residues (Phe723, Val726, Lys745, Asp837, Asn842, and Asp855) for both the unaltered and altered EGR interacting with these top compounds having 4-7 H- bond interactions within the active site (Fig. S9A-F & Table 2). These results indicate the drugs ZINC000012503187, ZINC000011679756, and ZINC000068153186 for unaltered and ZINC000003978005, ZINC000012503187, and ZINC000068153186 for altered EGFR formed strong stabilize complexes as compared to the ZINC000006716957 drug complexes. Moreover, two drugs, ZINC000068153186 and ZINC000012503187, intensely target both the EGFR-altered and unaltered forms.
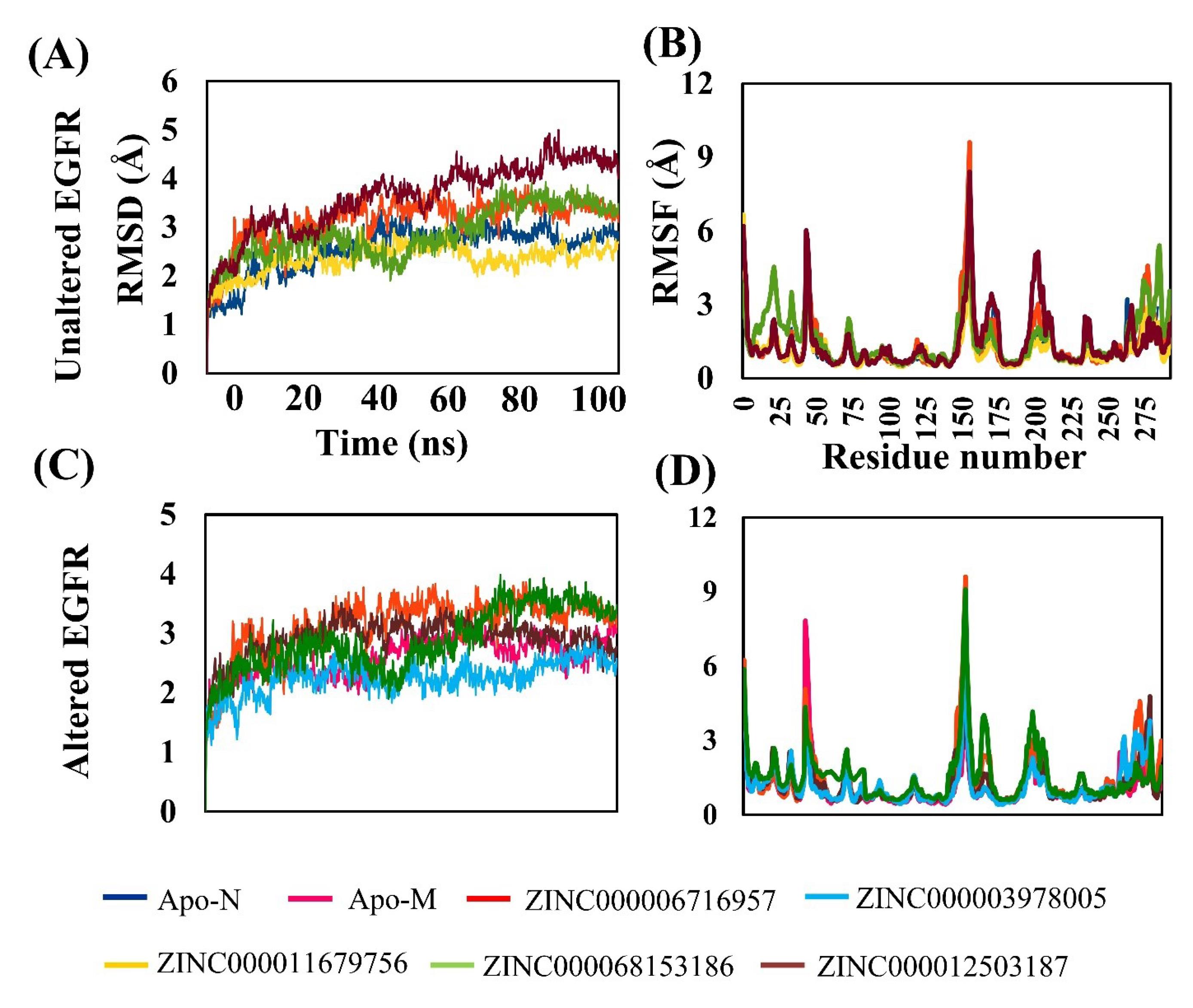
Fig. 6.
RMSD and RMSF of unaltered and altered EGFR of top selected FDA drug compound comparison with APO forms (A) RMSD values of unaltered EGFR with ZINC000012503187, ZINC000006716957, ZINC000011679756, and ZINC000068153186. (B) RMSF values of unaltered EGFR withZINC000012503187, ZINC000006716957, ZINC000011679756, and ZINC000068153186. (C) RMSD values of altered EGFR withZINC000006716957, ZINC000012503187, ZINC000003978005, and ZINC000068153186.
.
RMSD and RMSF of unaltered and altered EGFR of top selected FDA drug compound comparison with APO forms (A) RMSD values of unaltered EGFR with ZINC000012503187, ZINC000006716957, ZINC000011679756, and ZINC000068153186. (B) RMSF values of unaltered EGFR withZINC000012503187, ZINC000006716957, ZINC000011679756, and ZINC000068153186. (C) RMSD values of altered EGFR withZINC000006716957, ZINC000012503187, ZINC000003978005, and ZINC000068153186.
Table 2.
Average RMSD and RMSF of top hits with altered and unaltered EGFR
|
Receptor
|
Compound name
|
Average RMSD (Å)
|
Average RMSF (Å)
|
Interacting residues
|
| Unaltered EGFR |
ZINC000012503187 |
2.3 |
3.5 |
Leu718, Phe723, Val726, Ala743, Lys745, Cys797, Asp837, Arg841, Leu844 |
| ZINC000006716957 |
3.5 |
3.1 |
Gly719, Ser720, Phe723, Val726, Ala743, Lys745, Met766, Asp837, Leu844 |
| ZINC000011679756 |
2.2 |
3.4 |
Ala722, Phe723, Val726, Lys745, Thr854, Asp855, Pro877 |
| ZINC000068153186 |
2.1 |
3.3 |
Val726, Ala743, Lys745, Met766, Thr790, Arg841, Asn842, Leu844 |
| Altered EGFR |
ZINC000003978005 |
2.0 |
3.6 |
Leu718, Phe723, Val726, Ala743, Lys745, Leu792, Met793, Arg841, Asn842, Leu844 |
| ZINC000012503187 |
2.5 |
3.9 |
Ala722, Phe723, Val726, Cys797, Asp800, Arg841, Leu844, Lys875, Pro877 |
| ZINC000006716957 |
3.6 |
3.0 |
Gly719, Ala722, Phe723, Gly724, Thr725, Val726, Ala743, Lys745, Asp837, Asn842, Leu844, Asp855 |
| ZINC000068153186 |
2.2 |
3.5 |
Val726, Ala743, Lys745, Met766, Leu777, Thr790, Asn842, Leu844, Asp855, Phe856 |
Moreover,the radius of gyration was also evaluated for both forms of EGFR. The secondary structures' degree of compactness within a protein's 3D structure is determined by the radius of gyration as a result of the molecule's center of mass, a high radius of gyration implies the loose packing of proteins. In contrast, a low radius of gyration suggests tight and robust protein packaging. After the 100 ns simulation, both the apo structure and receptor-ligand complex structures show a high degree of compactness in the complex form. The radius of gyration for all top-best complexes in the unaltered and altered EGFR lies between 19 and 20.5 (Figs. 7A and B).
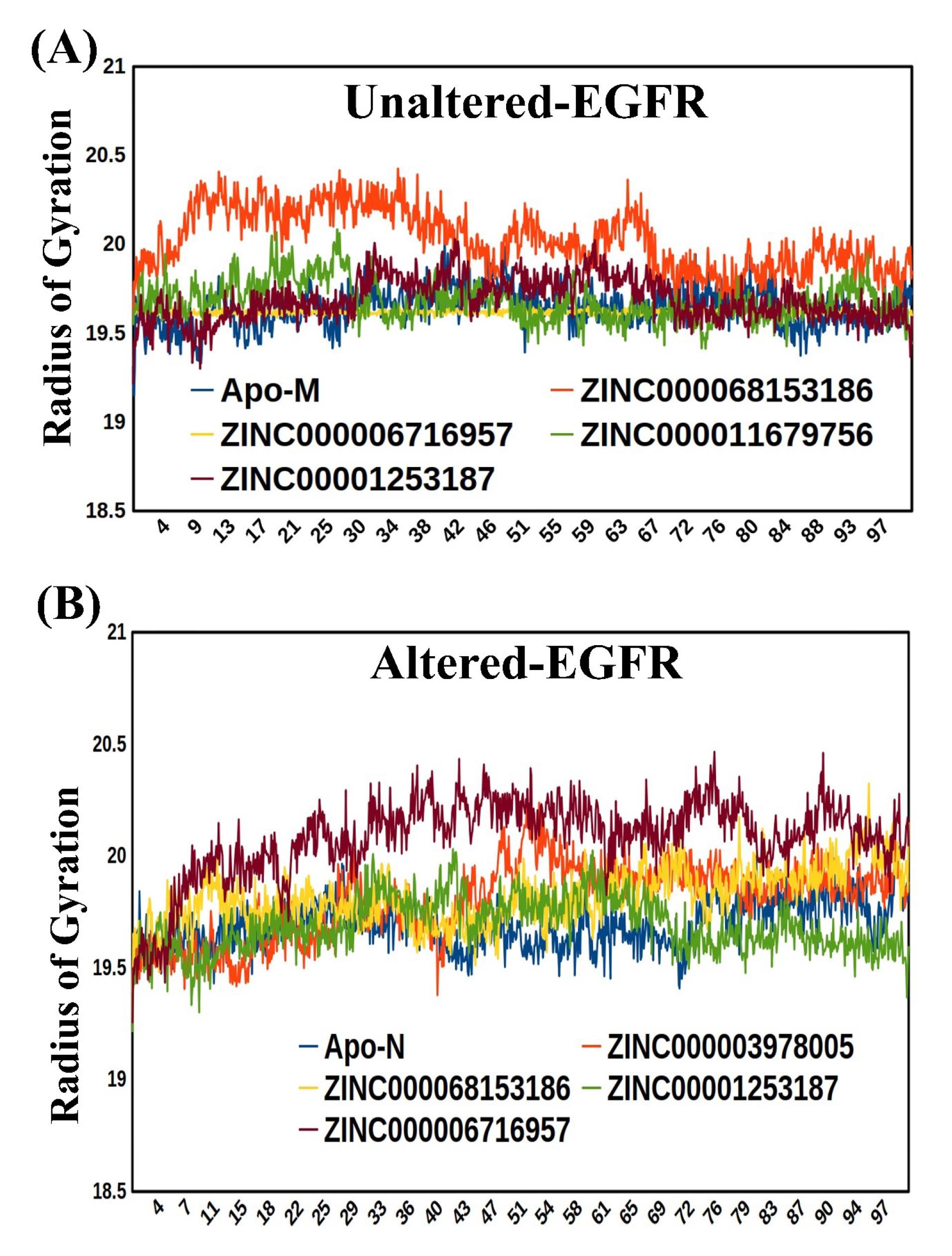
Fig. 7.
(A) The Radius of gyration of the Mutant/altered form of EGFR with Apo-mutant (B) Radius of gyration of the unaltered form of EGFR with Apo-unaltered.
.
(A) The Radius of gyration of the Mutant/altered form of EGFR with Apo-mutant (B) Radius of gyration of the unaltered form of EGFR with Apo-unaltered.
Principal component investigations
In both the unaltered and altered EGFR receptor forms, the atoms' most significant displacements or motions are determined using the principal component analysis method. The initial few eigenvectors represent the consolidated primary movements, but their frequency diminishes, resulting in progressively confined and localized oscillations. The PCA results for unaltered EGFR complexes with ZINC000006716957, ZINC000011679756, ZINC000068153186, and ZINC000012503187 show that the first three PCs account for 55.3%, 53.5.6%, 50.0 %, and 59.2% of the motion of deviation (Fig. S10A-D).For the altered EGFR, the compounds (ZINC000003978005, ZINC000006716957, ZINC000012503187, and ZINC000068153186) in the first 3 PCs account for 57.6%, 59.3%, 50.8%, and 45.7% residues motion observed complexes (Fig. S11A-D). Based on our findings, the amplitude of PC1 was found to be more significant in the altered EGFR than in the unaltered one. The graph displayed each protein conformation as a dot, representing the variation in the distribution of protein conformation. The continuous color display, ranging from blue to white to red, highlighted the various changes observed in the conformation of residues within the protein. This indicates that the altered EGFR exhibited more pronounced conformational variations than the unaltered one.
Dynamical cross-correlation matrix analysis
The different ligand-binding site residuals' functional motion/displacement of atoms as a time scale are examined using the dynamical cross-correlation matrix (DCCM). Bio3D algorithm assesses receptor families' sequence, conformational heterogeneity, and structural changes. Additionally, several utility functions are included to allow the R environment's statistical and graphical ability to interact with biological sequence and structure data. The DCCM graphs exhibit positive and negative correlation effects of the ligand-binding-related amino acid residues of unaltered and altered EGFR complexes and have different related motion patterns (Fig. 8A-H). The DCCM graph visualized in this study utilized dark cyan, white, and pink colors to represent specific patterns. Pink color represented anti-correlated pairs, indicating opposite motions of amino acid residues. Cyan color represented full cross-correlation pairs, indicating coordinated motions of residues. The analysis also showed that all the shortlisted compounds in the unaltered and altered EGFR docked complexes exhibited favorable residual correlated motions, indicating strong interaction and stability between the drugs and receptor residues (Fig. 8A-H).
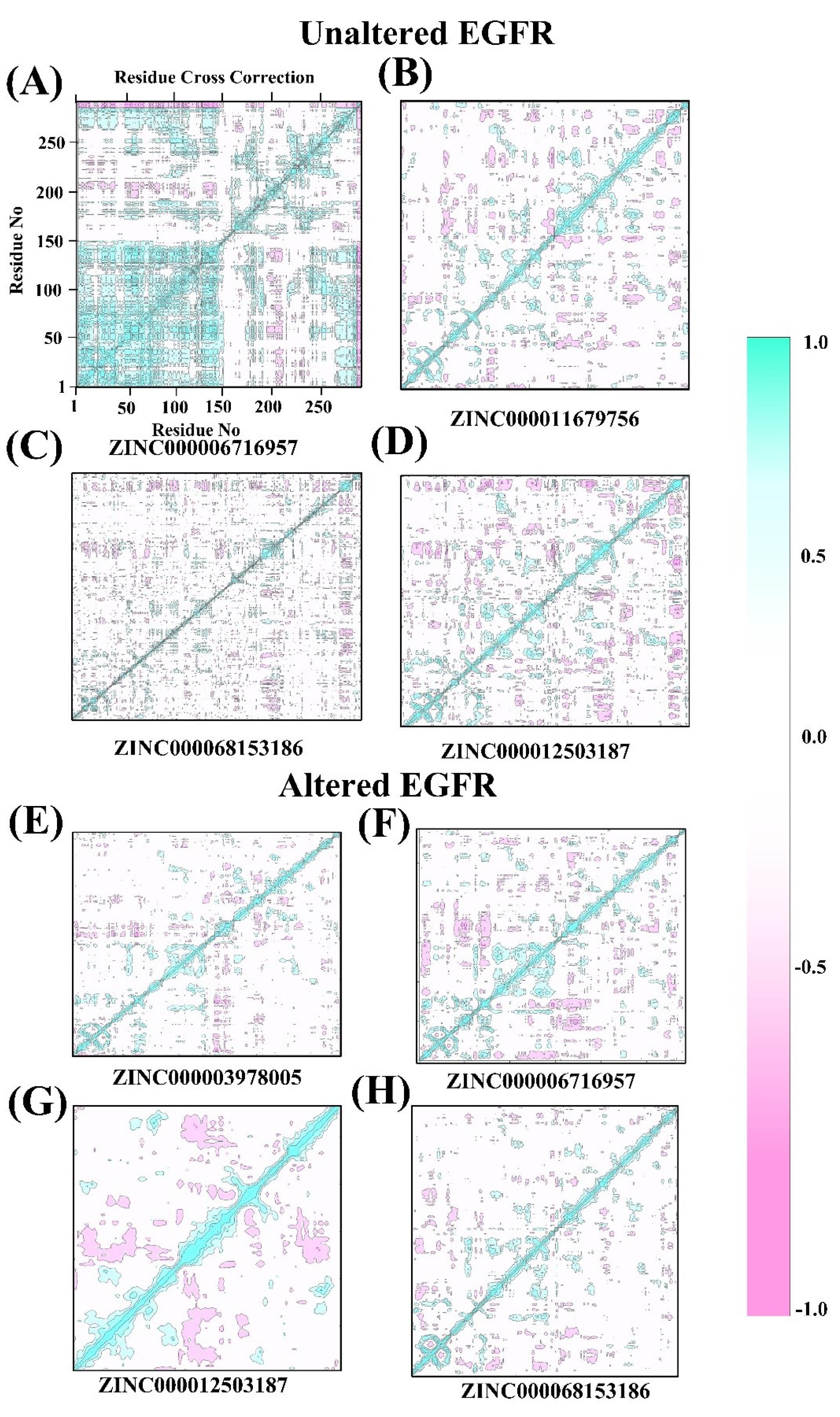
Fig. 8.
Dynamical cross-correlation matrix (DCCM) analysis of unaltered and mutant/altered EGFR, (A-D) dynamic correlation of unaltered EGFR with FDA drugs: ZINC000006716957, ZINC000011679756, ZINC000068153186, and ZINC000012503187. (E-H) dynamic correlation of mutant EGFR with FDA drugs: ZINC000003978005, ZINC000006716957, ZINC000012503187, and ZINC000068153186.
.
Dynamical cross-correlation matrix (DCCM) analysis of unaltered and mutant/altered EGFR, (A-D) dynamic correlation of unaltered EGFR with FDA drugs: ZINC000006716957, ZINC000011679756, ZINC000068153186, and ZINC000012503187. (E-H) dynamic correlation of mutant EGFR with FDA drugs: ZINC000003978005, ZINC000006716957, ZINC000012503187, and ZINC000068153186.
ADMET properties and therapeutic application of drug molecules
All the FDA-shortlisted drugs in this study demonstrated favorable pharmacokinetic characteristics, as they fully complied with the Lipinski rule of five (ROL5). They further showed non-mutagenic and non-carcinogenic properties and exhibited blood-brain barrier (BBB) permeability. Moreover, they were classified as Class III with minimal toxicity (Table S3). In addition to their potential for neuroglioma treatment, these drug molecules have also been utilized for various other medical conditions, including hepatitis C, hypervolemia or euvolemia, hypernatremia, acute lymphoblastic leukemia, migraine, histaminic cephalalgia, acute urinary retention, and promoting platelet synthesis in the bone marrow (Table S4).
Free energy, kd, ki, and IC50 calculation
Based on the results of the free energy calculation using MMGBSA, we observed that all of the shortlisted compounds exhibited a minimum free energy (ΔG) ranging from -37.8 to -63.25 kcal/mol (Table 3). These findings indicate that all these compounds possess a significantly considerable minimum free energy in both unaltered and unaltered EGFR receptors. The number of interacting amino acid residues involved in the drug binding mechanism of the top shortlisted drug molecules for each receptor altered EGFR and unaltered (Table 3). The dissociation and inhibition constants (kd & ki) result show minimum values between (0.0041 to 0.02818µM) and (0.00036 to 0.0049µM) (Table 4). Similarly, the IC50 of all shortlisted top best compounds was identified in the range of 0.00033 (minimum) to 0.69µM (maximum) (Table 4). Similarly, in silico kd and ki data shows a strong binding affinity of these drug molecules towards altered and unaltered EGFR (Table 4).
Table 3.
Free energy calculation (MMGBSA) of top best-docked hits and number of interacting residues involved in the drug binding mechanism
|
|
Compound name
|
Docking score
|
MMGBSA
(∆G kcal/mol)
|
Interacting residues
|
| Unaltered EGFR |
ZINC000012503187 |
-12.0 |
-58.8 |
Leu718, Phe723, Val726, Ala743, Lys745, Cys797, Asp837, Arg841, Leu844 |
| ZINC000006716957 |
-11.6 |
-39.09 |
Gly719, Ser720, Phe723, Val726, Ala743, Lys745, Met766, Asp837, Leu844, Asp855 |
| ZINC000011679756 |
-12.3 |
-52.53 |
Ala722, Phe723, Val726, Lys745, Asp837, Arg841, Thr854, Asp855, Pro877 |
| ZINC000068153186 |
-11.3 |
-51.66 |
Val726, Ala743, Lys745, Met766, Thr790, Arg841, Asn842, Leu844 |
| Altered EGFR |
ZINC000003978005 |
-11.7 |
-57.33 |
Leu718, Phe723, Val726, Ala743, Lys745, Leu792, Met793, Arg841, Asn842, Leu844 |
| ZINC000012503187 |
-12 |
-48.89 |
Ala722, Phe723, Val726, Cys797, Asp800, Arg841, Leu844, Lys875, Pro877 |
| ZINC000006716957 |
-12.4 |
-37.91 |
Gly719, Ala722, Phe723, Gly724, Thr725, Val726, Ala743, Lys745, Asp837, Asn842, Leu844, Asp855 |
| ZINC000068153186 |
-11.6 |
-63.25 |
Val726, Ala743, Lys745, Met766, Leu777, Thr790, Asn842, Leu844, Asp855, Phe856 |
Table 4.
The experimental and in silico, dissociation constant (kd), inhibition constant (ki), and inhibitory concentration (IC50) of top best hits
|
|
Compound name
|
Experimental
|
In silico
|
|
ki (µM)
|
Kd (µM)
|
IC50 (µM)
|
Bioassay AID
|
ki (µM)
|
Kd (µM)
|
| Unaltered EGFR |
ZINC000012503187 |
0.00036 |
- |
0.003 |
483982, 483984 |
0.00156 |
20 |
| ZINC000006716957 |
0.0049 |
0.0041 |
0.00033 |
428367, 1386089, 624983 |
0.00307 |
19.6 |
| ZINC000011679756 |
8.48 |
- |
0.69 |
1217113, 1282019 |
0.00510 |
20.8 |
| ZINC000068153186 |
8.48 |
0.013 |
0.0004 |
1424941, 1298014 |
0.00510 |
19.1 |
| Altered EGFR |
ZINC000003978005 |
0.000377 |
0.02818 |
0.000659 |
625190, 625190, 166825 |
0.00260 |
19.8 |
| ZINC000012503187 |
0.00036 |
- |
0.003 |
483982, 483984 |
0.00156 |
20 |
| ZINC000006716957 |
0.0049 |
0.0041 |
0.00033 |
428367, 1386089, 624983 |
0.000796 |
21.1 |
| ZINC000068153186 |
8.48 |
0.013 |
0.69 |
1217113, 1282019 |
0.00307 |
19.1 |
Discussion
The most prevalent form of cancer of the CNS is an astrocytoma, a glioma originating from astrocytes.28 Various mutations, such as deletions, amplifications, missense mutations, and single nucleotide polymorphisms (SNPs), have been found in the EGFR gene in gliomas, particularly in GBMs.29 This study employed computational approaches to investigate genomic alterations in EGFR gene and their impact on survival rates in neuroglioma patients. It also utilized molecular docking and drug screening techniques to identify potential lead drug molecules from the FDA-approved library for improved and targeted neuroglioma treatment with repurposing approach. Clinical analysis of 17 neuroglioma data sets for a total of 4251 patient samples suggests that 19% of genomic alterations were observed with the highest missense mutation counts (432), followed by amplification and mutational counts as compared to the other data sets (Figs. S1 & S2; Figs. 1 & 2). The missense mutation (V774M) in exon 19 with the highest allele frequency (0.97%), which reflects the highest risk for neuroglioma patients, was chosen for further analyses. This also indicates that mutated EGFR contributes significantly to these neuroglioma patients. Identifying specific mutations, such as the V774M missense mutation, which has the highest allele frequency and represents a high-risk factor for neuroglioma patients, provides crucial insights for understanding the underlying molecular mechanisms of the disease. These findings contribute to our knowledge of the genetic landscape of neuroglioma and can potentially aid in the development of targeted therapeutic strategies. In about 40% of tumors, neuroglioma exhibits EGFR gene mutations, amplification, and overexpression, a poignant characteristic of this disease.10 Scientific evidence suggests that abnormal/mutant EGFR signaling is involved in cancer development and is also responsible for therapeutic resistance, showing variable survival rates.30,31 According to the survival analysis, patients with alterations in the EGFR gene had lower overall survival and progression-free rates than patients with unaltered EGFR (Fig. 3). In the overall survival rate, the life span of the altered EGFR patient group was 160 months lower as compared to the unaltered EGFR. These findings also emphasize the need for targeted therapies that specifically address EGFR alterations, as they can potentially improve patient outcomes and survival rates.
The progression-free survival rate of the altered EGFR group was found to be 50% lower compared to the unaltered EGFR group. These results suggest that altered EGFR expression in neuroglioma patients is associated with a higher risk and potentially shorter lifespan compared to patients with unaltered EGFR expression. These results also indicate that mutant/altered EGFR is responsible for reducing overall patient survival and poor prognosis. Therefore, finding these particular high-frequency EGFR mutations and amplifications in neuroglioma patients will enable us to target the disease and evaluate its condition. So, it is necessary to assess the differential expression of such mutation types at the mRNA and protein levels to check their roles at the molecular level (Fig. 3).
Further, EGFR mRNA expression analysis suggests its expression is high in pheochromocytoma and paraganglioma cancer types with gain, amplification, and non-mutated mutation types compared to other neuroglioma cancer types (Fig. 3A). Similarly, the missense mutation in EGFR shows high expression in only pediatric high-grade gliomas compared to different neuroglioma subtypes (Fig. 3A). The outcomes of these results shed light on the prognostic value of EGFR alterations, identify high-frequency mutations and amplifications, and highlight the importance of assessing EGFR expression at the molecular level. The findings also contribute to a deeper understanding of neuroglioma pathogenesis and have the potential to shape future treatment approaches, ultimately improving patient outcomes in this challenging disease.
Furthermore, both the mRNA and protein levels of the EGFR missense mutation and amplification are highly expressed, indicating the development of the disease and poor clinical outcomes (Fig. 3B). The higher expression of missense mutations also suggests that such mutations have an essential role in disease progression and development.32 Evidence also indicates that novel missense mutations account for 13.6% (18/132) identified in the EGFR gene in GBM patients and 12.5% (1/8) in GBM cell lines.33 From our analysis, it is also evident that 72% of missense mutations occur in the neuroglioma patients of the EGFR receptor compared to the other mutation types. Identifying novel missense mutations in the EGFR gene in neuroglioma patients is a significant finding with important implications. It expands our knowledge of the genetic landscape of neuroglioma and highlights the diverse mutational profiles within the EGFR receptor. Understanding the specific mutations in neuroglioma patients, particularly the prevalence of missense mutations, provides valuable insights into the underlying genetic mechanisms driving the disease. Our findings also provide valuable insight for further research and the potential development of future treatment approaches targeting these specific mutations. By recognizing the importance of such novel mutations, we can potentially improve diagnostic accuracy and prognostic assessments and ultimately develop more effective and targeted therapies for neuroglioma patients.
Furthermore, it is crucial to understand the expression pattern of EGFR in different brain regions, which is more susceptible to missense mutations in the case of neuroglioma. One of the contributing factors for the missense mutations of EGFR in the brain to form the neuroglioma is its high expression in the striatum, amygdala, hippocampus, cortex, cerebellum, and thalamus (Fig. S4).Hence, the missense mutation observed at V774M, which accounts for a significantly higher percentage of all the mutations observed in 4251 patient samples, is chosen as the putative target. The mutation (especially missense) occurs in the intracellular EGFR kinase domain, that may result in increased ATP binding and account for chemotherapy drug resistance.16 Also, it is already known that altered EGFR signaling plays a role in glioma's high resistance to chemotherapy and radiation.12,34
Hence, it is crucial to identify that the new drug molecules can directly bind with the altered and unaltered EGFR, regulating expression in neuroglioma patients and increase patient survival. The mutation (V774M-EGFR) in exon 19 with the highest allele frequency was selected in the current investigation for additional computational in silico analyses. The 3POZ PDB id chosen for EGFR has the highest resolution (1.50 Å), is a non-mutant, and has a high sequence identity as compared to other PDB ids for EGFR (Figure S5, Table S1). Therefore, the mutant (V774M) structure of EGFR was modeled to obtain a 3D structure. The 3D structures of the altered and unaltered EGFR superimposed show a slight conformational change at the reside number (V774M) (Figure S6), and both structures are highly compact (Fig. S6). The mutant 3D EGFR model shows best-validated scores suggesting the overall good quality of the 3D structural model that can be used for further studies (Fig. S7). These results provide a foundation for exploring the potential targeting of altered and unaltered EGFR receptors in neuroglioma treatment and pave the way for further studies to develop novel therapeutic interventions based on the identified structural characteristics.
The analysis of amino acid residues involved in the interaction with drug molecules reveals that the altered and unaltered EGFR receptors share common residues in their binding sites (Table S2), indicating overlapping regions where the drugs can interact and potentially exert their effects. In drug discovery, assessing the structural conservation of the receptor, mainly the active site, is crucial.35 Our study shows that conservation analysis indicates that the residues forming the active site exhibit a high degree of conservation. These conserved residues are predominantly located in the catalytic site of the altered and unaltered EGFR receptors (Table S2). Understanding the conservative areas of these receptors, including the active site, can aid in selecting effective drug candidates for regulating the receptor's mechanism involved in the pathophysiology. This information enhances the understanding of the receptor's structure-function relationship and aids in designing and developing effective drugs for therapeutic interventions.
The docking studies demonstrate that the top shortlisted compounds, selected from 1614 FDA-screened compounds, exhibit strong interactions with the active sites of both unaltered and altered EGFR receptors, accompanied by significant hydrogen bonding interactions (Fig. S8 and Table 1). These findings also indicate that six specific amino acid residues (Phe723, Val726, Lys745, Asp837, Asn842, and Asp855) engage in interactions with the shortlisted drug molecules (Figs. 4-5 and Fig. S9). In contrast, the currently used drugs for neuroglioma treatment, Erlotinib, and Gefitinib, employed in EGFR target therapy, display lower docking scores and fewer hydrogen bonding interactions when compared to the shortlisted drugs for both unaltered and altered EGFR cases (Fig. 4 and Fig. S8). Hydrogen bonding is essential for stabilizing ligand-receptor complexes and influencing the binding affinity and effectiveness of the compounds as potential therapeutic agents. This finding indicates that the identified compounds have a higher potential for forming stable hydrogen bonding interactions with the receptor, which may contribute to their improved efficacy in neuroglioma treatment. The disparity in the number of hydrogen bond interactions emphasizes their importance in drug-receptor binding. It also highlights the potential advantages of these compounds over the currently used drugs. These findings further indicate that these compounds have the potential to be more effective in neuroglioma treatment compared to Erlotinib and Gefitinib.
The study also revealed that the V774M missense mutation, despite being present in the active site, did not play a role in interacting with the FDA-screened drug molecules. This observation suggests that the top common shortlisted compounds can effectively interact with the active site region of both altered and unaltered EGFR receptors. Bonding interactions between the receptors and drugs were identified as critical mechanisms in the interaction process, facilitating the formation of an efficient number of bonds. These interactions are crucial as they contribute to stabilize the ligand-target receptor interaction.36 In a recent study, isatin analogs were screened against five EGFR receptors (PDB ID 2J5F, 1M17, 2ITW, 2ITX, and 2ITY) with common interacting residues, though with differential interaction affinities.37 This study only presents the interaction ability of isatin analogs in the expressed EGFR receptors. However, our study not only evaluated the active site sequence homology and conservative regions of the altered and unaltered EFGR mutation having missense mutations, but also observed common drugs interactions with both receptors showing a multitargeting approach.
Further, the molecular dynamic simulation analysis suggests the overall stability of the docked complex structure. Molecular dynamic simulation studies (RMSD and RMSF) indicate that each target's top shortlisted compounds for respective EGFR targets showed stabilized ligand-target complexes. These findings suggest that drugs ZINC000012503187, ZINC000011679756, and ZINC000068153186 strongly stabilize the unaltered EGFR and drugs ZINC000003978005, ZINC000012503187, and ZINC000068153186 stabilize the altered EGFR as compared to the ZINC000006716957 drug. The drug ZINC000006716957 shows less complex stability towards altered and unaltered EGFR. However, only two common compounds ZINC000012503187 and ZINC000068153186 effectively stabilized both the altered and unaltered EGFR complexes with considerable RMSD and RMSF values (Fig. 6 and Table 2). These findings suggest that the selected drugs can promote stable interactions with their respective target receptors, leading to the overall stability of the ligand-target complexes. Such stability is crucial for the drugs' potential efficacy in regulating receptor activity and exhibiting therapeutic effects in neuroglioma treatment. The Radius of gyration suggests how these stable docked complex structures can be tightly packed into a three-dimensional (3D) structure.37 Proteins have the highest Radius of gyration among the protein sizes taken into account, indicating a less tightly packed structure than a low radius of gyration (ROG).38 As determined by the Radius of gyration studies, all the top shortlisted FDA-approved drug compounds, upon binding with the active site, show the tightly packed structures of both EGFR complexes (unaltered and altered) (Fig. 7).This suggests that these drugs effectively occupy and interact with the active sites of the receptors, leading to a compact arrangement of the complexes.
Once the stabilized and compact structures are obtained, further understanding of the motion of the amino acid residues and their displacement in the equilibrium state is necessary to deduce the overall binding interaction of the complex. Principal component analysis (PCA) tells the overall motion of atoms and their displacement in an equilibrium state, reflected as the clusters of tightly packed complexes in the 3D structural complex.39 Results reveal that the molecular interactions of the ligands with the receptors show thermodynamically distinct movements of atoms with a significant energy barrier within the altered and unaltered EGFR-docked complexes. However, three compounds, each of unaltered and altered EGFR complex, having two compounds in common, show a gradual conformational shift in the clusters from the initial blue cluster to the white cluster to the red color cluster (Figs. S10 and S11). With visual pattern recognition, the DCCM is a three-dimensional graphic representation of time-correlated data for all amino acid residues that bind ligands.40 According to our findings, all of the shortlisted drugs, including the two drugs that were tested on both receptors (the altered EGFR and the unaltered EGFR), showed excellent residual correlated motions and displacement of binding residues, indicating a strong interaction ability of such drugs towards the residues in the active sites (Fig. 8). These findings emphasize the favorable interaction potential between the selected drugs and the binding residues, enhancing our comprehension of the dynamic behavior exhibited by the drug-receptor complexes. Moreover, these outcomes offer valuable insights into the motion, clustering, and dynamic interaction patterns observed within drug-receptor complexes.It is also observed that the shortlisted drugs have been found to have BBB permeability, which suggests that they can cross the blood-brain barrier and potentially be used to treat neurological conditions such as neuroglioma (Table S3). The shortlisted drugs have also been utilized to treat a diverse range of illnesses, including hepatitis C, hypervolemia or euvolemia, hypernatremia, acute lymphoblastic leukemia, migraine, histaminic cephalalgia, acute urinary retention, and to promote platelet synthesis in the bone marrow (Table S4). These results suggest that the shortlisted drugs exhibit potential efficacy in neuroglioma treatment, can cross the blood-brain barrier, and have demonstrated effectiveness in treating a wide range of other illnesses. These findings contribute to their potential value as a possible therapeutic option for neuroglioma.
Additionally, free energy calculations (MMGBSA) present the strength and stability of the binding of docked drug molecules with their respective receptors. The MMGBSA results show that the top shortlisted compounds exhibit the least free energy, demonstrating the high binding affinity of these drugs for both receptors except a single compound (ZINC000006716957) (Table 3). Moreover, free energy calculations suggest that the most conserved active site residues interact with the shortlisted drug compounds and have a high binding affinity towards altered and unaltered EGFR (Table 4). These outcomes demonstrate the selected drug molecules robust binding potential and ability to interact effectively with the target receptors.
The binding affinity of drug molecules can be experimentally validated using the dissociation constant (kd), inhibition constant (ki), and inhibitory concentrations. In our study, the experimental results demonstrated that the FDA-approved drug compounds shortlisted for investigation exhibited the lowest kd and ki values (as shown in Table 4). Moreover, computational calculations of kd and ki also indicated a strong affinity between these shortlisted drug molecules and their respective altered and unaltered EGFR receptors (Table 4). Further, the inhibitory concentration (IC50) of the top shortlisted compounds was found to be low, indicating that even at minimal concentrations, these compounds effectively inhibited the expression of altered and unaltered EGFR, consequently impeding the proliferation of neuroglioma cells (Table 4). The results suggest that the selected drug molecules successfully bind to altered and unaltered EGFR, contributing to stable formation of the drug-receptor complex. The minimum free energy values obtained from the drug binding mechanism support this observation. These findings also suggest that the identified FDA-approved drugs in this study possess a high affinity for their target receptors, namely altered and unaltered EGFR. As a result, these drugs show the potential to effectively regulate the expression of these receptors in neuroglioma patients, showcasing a promising multi-target-based therapeutic drug repurposing approach.
Conclusion
The EGFR overexpression and their mutations are amongst the most distinguishing characteristics of GBM, but the cell-biological complexity of this target has long outpaced therapeutic advancements. According to the new understanding, the analysis of the neuroglioma patient data reveals that the EGFR (altered expression) is mutated in various ways, with a higher percentage of missense mutations occurring at V774M in exon 19 of the EGFR. The altered EGFR expression groups have lower overall survival and progression-free survival rates than the unaltered EGFR groups of neuroglioma patients. The top shortlisted FDA-approved drugs (3 for each receptor (2-common)) are vital and efficiently bind to the active site residues of unaltered and altered EGFR receptors. These compounds exhibit favorable docking scores, indicating a strong binding affinity, compared to the currently used drugs, Erlotinib and Gefitinib. The efficient binding of the top shortlisted drugs to the active site residues suggests their potential effectiveness in targeting the EGFR receptors. By interacting with specific amino acid residues within the receptor's active site, these compounds have the potential to modulate the receptor's function and inhibit signaling. Molecular dynamic simulation studies (RMSD and RMSF) suggest that the top 3 compounds for each target exhibit stable ligand-target complex structures. The Radius of gyration analysis indicates that the shortlisted molecules have the most tightly packed complex structures of altered and unaltered EGFR. PCA and DCCM results indicate thermodynamically different motions of atoms and good residual correlated motions that indicate a strong affinity for the ligands and residues in the active site of the altered and unaltered EGFR. Minimum free binding energy (∆G, MMGBSA) values suggest the strong binding affinity of these drugs for both receptors. The minimum kd and ki values indicate the greater binding affinity of common drug molecules and a smaller amount of drug needed to inhibit the respective EGFR receptors' activity.
Furthermore, the low IC50 values of shortlisted FDA-drug molecules indicate that these 3 shortlisted compounds for each receptor (2-common) are effective at low concentrations suggesting their high potential use. These results support useful and informative signs for identifying accurate therapeutic agents targeting the unaltered and altered EGFR receptors. Therefore, this study identifies 3 each efficient drug candidate molecules (ZINC000011679756, ZINC000012503187, and ZINC000068153186 (Conivaptan-Benzazepine and Dabrafenib-aminopyrimidine) for unaltered EGFR) and ZINC000003978005, ZINC000012503187, and ZINC000068153186 for altered EGFR). Furthermore, compounds ZINC000012503187 and ZINC000068153186 (Conivaptan-Benzazepine and Dabrafenib-aminopyrimidine) are the common drug candidates that show high potential towards their development as therapeutic repurposing drugs for both the altered and unaltered EGFR receptor for neuroglioma patients.
Research Highlights
What is the current knowledge?
√ The EGFR (unaltered) and its mutated (altered) forms are emerging targets due to their specific altered expressions in neuroglioma patients.
What is new here?
√ Neuroglioma patient samples genomic and proteomic analyses (of 3835 patients/4251 samples, CBioPortal) reveal 19% genetic alterations (missense mutations (V774M), exon 19) in EGFR with high allele frequency.
√ The Kaplan–Meier survival plots indicate unaltered group's overall relapse-free survival rate was higher than the altered group.
√ Both the unaltered and altered EGFR were screened for FDA-approved (1615 tested, ZINC database) drugs to choose a lead compound.
√ The lead compound has the lowest Gibbs free energy, showing minimum kd, ki, and IC50 values for each target.
√ Molecular dynamic simulation studies suggest ZINC00001253187 can effectively target both the unaltered/altered EGFR and a therapeutic drug of choice for glioma.
Data Availability Statement
Data will be made available on reasonable request.
Competing Interests
All the authors declare no conflict of interest.
Ethical Statement
Not applicable.
Funding
None.
Supplementary files
Supplementary file 1 contains Figs. S1-S11 and Tables S1-S4.
(pdf)
References
- Burel-Vandenbos F, Benchetrit M, Miquel C, Fontaine D, Auvergne R, Lebrun-Frenay C. EGFR immunolabeling pattern may discriminate low-grade gliomas from gliosis. J Neurooncol 2011; 102:171-8. doi: 10.1007/s11060-010-0308-4 [Crossref] [ Google Scholar]
- Cahill DP, Sloan AE, Nahed BV, Aldape KD, Louis DN, Ryken TC. The role of neuropathology in the management of patients with diffuse low grade glioma: A systematic review and evidence-based clinical practice guideline. J Neurooncol 2015; 125:531-49. doi: 10.1007/s11060-015-1909-8 [Crossref] [ Google Scholar]
- Lundy P, Domino J, Ryken T, Fouke S, McCracken DJ, Ormond DR. The role of imaging for the management of newly diagnosed glioblastoma in adults: a systematic review and evidence-based clinical practice guideline update. J Neurooncol 2020; 150:95-120. doi: 10.1007/s11060-020-03597-3 [Crossref] [ Google Scholar]
- Claus EB, Walsh KM, Wiencke JK, Molinaro AM, Wiemels JL, Schildkraut JM. Survival and low-grade glioma: the emergence of genetic information. Neurosurg Focus 2015; 38:E6. doi: 10.3171/2014.10.FOCUS12367 [Crossref] [ Google Scholar]
- Hanif F, Muzaffar K, Perveen K, Malhi SM, Simjee Sh U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac J Cancer Prev 2017; 18:3-9. doi: 10.22034/APJCP.2017.18.1.3 [Crossref] [ Google Scholar]
- Zhang Y, Liu G, Lang M, Zhang J, Geng J. Patients treatment with neuroglioma by teniposide and semustine and its influence on Twist and E-cadherin expression. Saudi Pharm J 2016; 24:299-304. doi: 10.1016/j.jsps.2016.04.001 [Crossref] [ Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017; 168:707-23. doi: 10.1016/j.cell.2017.01.017 [Crossref] [ Google Scholar]
- Alexandru O, Horescu C, Sevastre AS, Cioc CE, Baloi C, Oprita A. Receptor tyrosine kinase targeting in glioblastoma: performance, limitations and future approaches. Contemp Oncol (Pozn) 2020; 24:55-66. doi: 10.5114/wo.2020.94726 [Crossref] [ Google Scholar]
- Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B. Combination therapy in combating cancer. Oncotarget 2017; 8:38022-43. doi: 10.18632/oncotarget.16723 [Crossref] [ Google Scholar]
- Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010; 12:675-84. doi: 10.1593/neo.10688 [Crossref] [ Google Scholar]
- Singh J, Sangwan N, Chauhan A, Avti PK. Integrative network and computational simulation of clinical and genomic data for the identification of mutated EGFR in breast cancer patients for therapeutic targeting using purine analogues. Molecular Simulation 2022; 48:1548-60. doi: 10.1080/08927022.2022.2107638 [Crossref] [ Google Scholar]
- Westphal M, Maire CL, Lamszus K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017; 31:723-35. doi: 10.1007/s40263-017-0456-6 [Crossref] [ Google Scholar]
- Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J 2013; 280:5350-70. doi: 10.1111/febs.12393 [Crossref] [ Google Scholar]
- Sabbah DA, Hajjo R, Sweidan K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr Top Med Chem 2020; 20:815-34. doi: 10.2174/1568026620666200303123102 [Crossref] [ Google Scholar]
- An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene 2018; 37:1561-75. doi: 10.1038/s41388-017-0045-7 [Crossref] [ Google Scholar]
- Yamaoka T, Kusumoto S, Ando K, Ohba M, Ohmori T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int J Mol Sci 2018; 19. 10.3390/ijms19113491.
- Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 2011; 7:146-57. doi: 10.2174/157340911795677602 [Crossref] [ Google Scholar]
- Singh J, Sangwan N, Chauhan A, Sarma P, Prakash A, Medhi B. Screening and identification of phytochemical drug molecules against mutant BRCA1 receptor of breast cancer using computational approaches. Mol Cell Biochem 2022; 477:885-96. doi: 10.1007/s11010-021-04338-4 [Crossref] [ Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1. doi: 10.1126/scisignal.2004088 [Crossref] [ Google Scholar]
- Huang B. MetaPocket: a meta approach to improve protein ligand binding site prediction. OMICS 2009; 13:325-30. doi: 10.1089/omi.2009.0045 [Crossref] [ Google Scholar]
- Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 2016; 44:W344-50. doi: 10.1093/nar/gkw408 [Crossref] [ Google Scholar]
- Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 1993; 2:1511-9. doi: 10.1002/pro.5560020916 [Crossref] [ Google Scholar]
- Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol 2015; 1263:243-50. doi: 10.1007/978-1-4939-2269-7_19 [Crossref] [ Google Scholar]
- Grant BJ, Skjaerven L, Yao XQ. The Bio3D packages for structural bioinformatics. Protein Sci 2021; 30:20-30. doi: 10.1002/pro.3923 [Crossref] [ Google Scholar]
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017; 7:42717. doi: 10.1038/srep42717 [Crossref] [ Google Scholar]
- Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res 2018; 46:W257-W63. doi: 10.1093/nar/gky318 [Crossref] [ Google Scholar]
- Wang Z, Pan H, Sun H, Kang Y, Liu H, Cao D. fastDRH: a webserver to predict and analyze protein-ligand complexes based on molecular docking and MM/PB(GB)SA computation. Brief Bioinform 2022; 23:bbac201. doi: 10.1093/bib/bbac201 [Crossref] [ Google Scholar]
- Krouwer HG, Davis RL, McDermott MW, Hoshino T, Prados MD. Gangliogliomas: a clinicopathological study of 25 cases and review of the literature. J Neurooncol 1993; 17:139-54. doi: 10.1007/BF01050216 [Crossref] [ Google Scholar]
- Cimino PJ, Bredemeyer A, Abel HJ, Duncavage EJ. A wide spectrum of EGFR mutations in glioblastoma is detected by a single clinical oncology targeted next-generation sequencing panel. Exp Mol Pathol 2015. 98: 568-73. 10.1016/j.yexmp.2015.04.006.
- Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol 2018; 12:3-20. doi: 10.1002/1878-0261.12155 [Crossref] [ Google Scholar]
- Azuaje F, Tiemann K, Niclou SP. Therapeutic control and resistance of the EGFR-driven signaling network in glioblastoma. Cell Commun Signal 2015; 13:23. doi: 10.1186/s12964-015-0098-6 [Crossref] [ Google Scholar]
- Stefl S, Nishi H, Petukh M, Panchenko AR, Alexov E. Molecular mechanisms of disease-causing missense mutations. J Mol Biol 2013; 425:3919-36. doi: 10.1016/j.jmb.2013.07.014 [Crossref] [ Google Scholar]
- Lee JC, Vivanco I, Beroukhim R, Huang JH, Feng WL, DeBiasi RM. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med 2006; 3:e485. doi: 10.1371/journal.pmed.0030485 [Crossref] [ Google Scholar]
- Amelia T, Kartasasmita RE, Ohwada T, Tjahjono DH. Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors. Molecules 2022; 27:819. doi: 10.3390/molecules27030819 [Crossref] [ Google Scholar]
- Panjkovich A, Daura X. Assessing the structural conservation of protein pockets to study functional and allosteric sites: implications for drug discovery. BMC Struct Biol 2010; 10:9. doi: 10.1186/1472-6807-10-9 [Crossref] [ Google Scholar]
- Suvarna BS. Drug-receptor interactions. Kathmandu Univ Med J (KUMJ) 2011; 9:203-7. doi: 10.3126/kumj.v9i3.6306 [Crossref] [ Google Scholar]
- Cheke RS, Patil VM, Firke SD, Ambhore JP, Ansari IA, Patel HM. Therapeutic Outcomes of Isatin and Its Derivatives against Multiple Diseases: Recent Developments in Drug Discovery. Pharmaceuticals (Basel) 2022; 15:272. doi: 10.3390/ph15030272 [Crossref] [ Google Scholar]
- Lobanov M, Bogatyreva NS, Galzitskaia OV. [Radius of gyration is indicator of compactness of protein structure]. Mol Biol (Mosk) 2008; 42:701-6. [ Google Scholar]
- David CC, Jacobs DJ. Principal component analysis: a method for determining the essential dynamics of proteins. Methods Mol Biol 2014; 1084:193-226. doi: 10.1007/978-1-62703-658-0_11 [Crossref] [ Google Scholar]
- Dash R, Ali MC, Dash N, Azad MAK, Hosen SMZ, Hannan MA. Structural and Dynamic Characterizations Highlight the Deleterious Role of SULT1A1 R213H Polymorphism in Substrate Binding. Int J Mol Sci 2019; 20:6256. doi: 10.3390/ijms20246256 [Crossref] [ Google Scholar]