Bioimpacts. 14(2):29955.
doi: 10.34172/bi.2023.29955
Original Article
Benchmarking different docking protocols for predicting the binding poses of ligands complexed with cyclooxygenase enzymes and screening chemical libraries
Sara Shamsian Investigation, Writing – original draft, 1, 2 
Babak Sokouti Investigation, Supervision, Writing – original draft, 3
Siavoush Dastmalchi Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing, 2, 3, 4, *
Author information:
1Student Research Committee, Tabriz University of Medical Sciences, Tabriz, 5165665931, Iran
2Department of Medicinal Chemistry, School of Pharmacy, Tabriz University of Medical Sciences, Tabriz, 5166414766, Iran
3Biotechnology Research Center, Tabriz University of Medical Sciences, Tabriz, 5165665813, Iran
4Faculty of Pharmacy, Near East University, POBOX:99138, Nicosia, North Cyprus, Mersin 10, Turkey
Abstract
Introduction:
Non-steroidal anti-inflammatory drugs (NSAIDs) constitute an important class of pharmaceuticals acting on cyclooxygenase COX-1 and COX-2 enzymes. Due to their numerous severe side effects, it is necessary to search for new selective, safe, and effective anti-inflammatory drugs. In silico design of novel therapeutics plays an important role in nowadays drug discovery pipelines. In most cases, the design strategies require the use of molecular docking calculations. The docking procedure may require case-specific condition for a successful result. Additionally, many different docking programs are available, which highlights the importance of identifying the most proper docking method and condition for a given problem.
Methods:
In the current work, the performances of five popular molecular docking programs, namely, GOLD, AutoDock, FlexX, Molegro Virtual Docker (MVD) and Glide to predict the binding mode of co- crystallized inhibitors in the structures of known complexes available for cyclooxygenases were evaluated. Furthermore, the best performers, Glide, AutoDock, GOLD and FlexX, were further evaluated in docking-based virtual screening of libraries consisted of active ligands and decoy molecules for cyclooxygenase enzymes and the obtained docking scores were assessed by receiver operating characteristics (ROC) analysis.
Results:
The results of docking experiments indicated that Glide program outperformed other docking programs by correctly predicting the binding poses (RMSD less than 2 Å) of all studied co-crystallized ligands of COX-1 and COX-2 enzymes (i.e., the performance was 100%). However, the performances of the other studied docking methods for correctly predicting the binding poses of the ligands were between 59% to 82%. Virtual screening results treated by ROC analysis revealed that all tested methods are useful tools for classification and enrichment of molecules targeting COX enzymes. The obtained AUCs range between 0.61-0.92 with enrichment factors of 8 – 40 folds.
Conclusion:
The obtained results support the importance of choosing appropriate docking method for predicting ligand-receptor binding modes, and provide specific information about docking calculations on COXs ligands.
Keywords: Non-steroidal anti-inflammatory drugs, Molecular docking, ROC analysis, Drug discovery
Copyright and License Information
© 2024 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) exert their therapeutic effects by inhibiting cyclooxygenase (COX) enzymes and are among the most commonly used medications due to their wide range of uses.1-3 At least two COX isoforms, namely COX-1 and COX-2, are known, which share 60% sequence similarity.4 There is also another form called COX-3, which is a splice variant of COX-1.5
COX-1 is the constitutive integral membrane isoform found in the endoplasmic reticulum of most cell types, whereas COX-2 is primarily the inducible isoform.6 COX-1 is responsible for the synthesis of prostaglandins such as PGE2 and PGI2, which play important cytoprotective effects on GI functions such as induction of bicarbonate secretion and reduction of gastric acid secretion. On the other hand, COX-2 is expressed in response to pro-inflammatory cytokines like TNF-α, IL-1β, and IL-6. Classical NSAIDs inhibit both enzymes COX-1 and COX-2 and their use leads to gastrointestinal side effects. Therefore, the development of selective COX-2 inhibitors can significantly reduce these side effects.7
Introducing a new drug to the market is a complex and time-consuming process.8 There are numerous stages for this process, such as target identification and validation, lead compound identification, lead optimization, preclinical drug development, clinical trials and finally post-market monitoring for drug safety. The application of computer-aided drug design (CADD) is an essential approach for developing new drugs.9 Structure-based drug design (SBDD) and ligand-based drug design (LBDD) are two major types of CADD used in modern drug discovery.10 If the 3D structure of the target is available through experimental or prediction methods, SBDD approaches can reveal the binding modes of the ligands to their target.11 The most common methods used in SBDD are molecular docking, structure-based virtual screening (SBVS) and molecular dynamics (MD) simulations.12
Molecular docking is an in silico method that samples conformations of small molecules in protein binding sites.13 Molecular docking is a fast and efficient technique that predicts the binding mode and binding affinity between a ligand and a target at the atomic level.14
During the past decades, several docking programs have been developed within both academia and industry, such as DOCK,15 AutoDock,16 AutoDock Vina,17 GOLD,18 Glide,19 FlexX,20 Surflex,21 Molegro Virtual Docker,22 ICM,23 Cdocker, LigandFit,24 MOE-Dock,25 LeDock,26 rDock,27 UCSF Dock,28 and many others.
It is evident that the systematic investigation of existing docking approaches would be helpful in selecting those algorithms and scoring functions that are optimal for a given molecular docking task. Moreover, many studies have recently compared the performance of numerous docking and screening techniques.29-31
For a docking program, two basic components are the sampling algorithm and the scoring function.32 The goal of a sampling algorithm is to generate putative ligand orientations/conformations (usually called poses) at the binding site of a protein. Three main types of ligand sampling algorithms exist: shape matching, systematic search and stochastic algorithms.33 The objectives of scoring function are to evaluate and rank the poses generated in docking simulations. As the most important component of molecular docking, scoring functions have three major applications: binding mode identification, binding affinity prediction and virtual database screening.12
A suitable docking program should be able to reproduce the experimental binding modes of ligands. The root mean square deviation (RMSD) between corresponding atoms of docked pose of the ligand and its experimental binding mode is one of the widely used criteria for testing the quality of a docking calculation. The RMSD value less than 2 Å suggests proper docking outcome.34
Virtual screening (VS) is a powerful technique for identifying hit molecules from large chemical libraries. VS is divided into two broad categories, namely ligand-based (LBVS) and structure-based (SBVS) virtual screening methods. LBVS can be performed by similarity search, ligand-based pharmacophores and quantitative structure-activity relationship (QSAR). On the other hand, SBVS can be done mainly through docking calculations.35
The receiver operating characteristics (ROC) curve with the calculation of the area under the curve (AUC) is a practical way of measuring the overall performance of diagnostic tests. In the case of testing docking algorithms, ROC curves can allow a direct comparison of different virtual screening workflows. In virtual drug screening, ROC curves are often used to visualize the efficiency of the used application to separate active ligands from inactive molecules. In general, the higher the AUC, the more effective the virtual screening workflow is in discriminating active compounds from inactive compounds. In this context, sensitivity (Se) is the percentage of truly active compounds being selected. On the other hand, specificity (Sp) is the percentage of truly inactive compounds that are discarded.36,37 Due to the importance of molecular docking in drug design and discovery, the current study aimed to evaluate a set of popular docking programs, namely GOLD, AutoDock, LeadIT (FlexX), Molegro Virtual Docker (MVD) and Glide, for their performance to correctly predict the poses of bound ligands to COX-1 and COX-2 enzymes determined experimentally. In addition to this, AutoDock, GOLD, Glide and FlexX were evaluated by comparing their effectiveness in selecting active compounds from a database of decoys in virtual screening.
Materials and Methods
Data set collection and protein preparation
The crystal structures of cyclooxygenase-ligand complexes available in Protein Data Bank at RCSB (https://www.rcsb.org/) were downloaded. Rofecoxib (RCX molecule in structure 5KIR) was considered as the reference ligand. All complexes were superimposed onto 5KIR structure using DeepView software (Version 4.1.0). Those complexes with ligands that did not occupy the same site as with rofecoxib or did not have a drug like structure were excluded from the study. Finally, 51 complexes containing COX-1 and COX-2 enzymes were selected. The list of PDB identifier codes and crystallography resolutions of the structures are given in Table 1. The downloaded protein structures were edited with DeepView software to remove redundant chains, ligands, waters, cofactors and ions. Next, a heme molecule was added to those structures that do not have heme. Eventually, a single-chain protein was used as an input to the docking programs.
Table 1.
List of druglike ligands containing COX-1 and COX-2 structures
|
PDB code
|
Resolution (Å)
|
PDB code
|
Resolution (Å)
|
PDB code
|
Resolution (Å)
|
| 1CQE |
3.10 |
3N8W |
2.75 |
4RRZ |
2.57 |
| 1CX2 |
3.00 |
3N8Y |
2.60 |
4RS0 |
2.81 |
| 1EQG |
2.61 |
3N8Z |
2.90 |
5F1A |
2.38 |
| 1EQH |
2.70 |
3NT1 |
1.73 |
5IKQ |
2.41 |
| 1HT5 |
2.75 |
3NTG |
2.19 |
5IKR |
2.34 |
| 1HT8 |
2.69 |
3PGH |
2.50 |
5IKT |
2.45 |
| 1PGE |
3.50 |
3Q7D |
2.40 |
5IKV |
2.51 |
| 1PGF |
4.50 |
3QMO |
3.00 |
5JVZ |
2.62 |
| 1PGG |
4.50 |
3RR3 |
2.84 |
5JW1 |
2.82 |
| 1PTH |
3.40 |
4COX |
2.90 |
5KIR |
2.70 |
| 1PXX |
2.90 |
4FM5 |
2.81 |
5U6X |
2.93 |
| 1Q4G |
2.00 |
4M11 |
2.45 |
5W58 |
2.27 |
| 2AYL |
2.00 |
4O1Z |
2.40 |
5WBE |
2.75 |
| 3KK6 |
2.75 |
4OTY |
2.35 |
6BL3 |
2.22 |
| 3LN0 |
2.20 |
4PH9 |
1.81 |
6BL4 |
2.22 |
| 3LN1 |
2.40 |
4RRW |
2.57 |
6COX |
2.80 |
| 3MQE |
2.80 |
4RRX |
2.78 |
6V3R |
2.66 |
HyperChem (Version 8.0.6, Hypercube Inc.) was used to generate and energy minimize ligand structures using MM+ empirical force field38 followed by semi-empirical quantum mechanics PM3,39 which is a reparametrization of AM1 method, available in HyperChem. Open Babel software (version 3.1.1) was used for file format conversion.
Docking calculations
Docking with GOLD
To do the docking calculations, first Hermes (Version 1.7.0, 2015) visualization interface to GOLD (Version 5.3.0, 2014 Cambridge Crystallographic Data Centre) software was used for receptor preparation. The center of the active (binding) site was defined by selecting one of the central atoms of the co-crystallized ligand and then the active site was identified as a 10 Å radius sphere from that point. All atoms of the receptor within the sphere were considered active in the calculations. The ligand and reference ligand (co-crystallized ligand) were specified. All four scoring functions available for GOLD including ChemPLP, GoldScore, ChemScore and ASP were tested in separate runs. For each structure, 10 independent runs were carried out where the early termination option was turned on, and poses were ranked by scoring functions. The early termination option instructs GOLD to terminate docking runs on a given ligand if the top 3 solutions are within 1.5 Å. Also, for each scoring function, a docking run was performed without applying early termination option and top 10 scoring poses were saved for analysis.
Docking with AutoDock
AutoDock 4.2 was used for molecular docking. The protein structure was imported into the AutoDockTools (Version 1.5.7, Molecular Graphics Laboratory, The Scripps Research Institute) workspace and polar hydrogen atoms and Kollman charges were computed for the protein. The protein was saved in PDBQT format. For ligand preparation, Gasteiger partial charges were added and rotatable bonds were defined. Finally, the prepared ligands were saved in PDBQT format. A grid box was built with a spacing of 0.375 Å and size of 84 × 84 × 84 grid points. The Lamarckian genetic algorithm was selected for the ligand conformational search. After the preparation of grid (.gpf) and docking (.dpf) parameter files, molecular docking was conducted using Cygwin as a Linux-like environment for windows. In the current study, the numbers of 10 and 50 GA runs were used. In the case of virtual screening experiments, AutoDock Vina algorithm was used by applying AutoDock 4 force field. The center of grid box (20 × 20 × 20 Å3) was defined based on coordinates of the active ligand provided for the target protein by the database (co-crystalized ligand extracted from the complex).
Docking with LeadIT platform (FlexX)
FlexX tool available in LeadIT platform (Version 2.1.8, 2014, BioSolveIT, GmbH) was employed for docking investigation. Reference ligand was extracted from the PDB file of protein-ligand complex. Then it was converted to MOL2 format using Open Babel software. Single-chain protein was loaded into LeadIT and heme molecule and Fe atom were selected as the receptor components. Binding site was defined by selecting amino acids within 10 Å radius of reference ligand. The default docking and scoring parameters remained unchanged.
Docking with MVD
Molecular docking by using Molegro Virtual Docker (MVD) software (Version 6.0, 2013, CLC Bio) was undertaken by introducing PDB files of protein and ligand into the workspace. Before initiation of docking operation, structure of protein and ligand were prepared. Using the detect cavity option, the possible binding pocket(s) on the protein were identified. Reference ligand was used for RMSD calculations. MVD default settings were used including MolDock score [GRID], a grid resolution of 0.30 Å for grid generation, 10 Å radius from the template as the binding site and MolDock SE as the search algorithm. The number of runs was set to 10. After docking, energy minimization and optimization of hydrogen bonds for each pose was done.
Docking with Glide
Before performing docking calculations with Glide (Version 8.8, Schrodinger, 2021), protein and ligand structures were prepared using protein preparation and LigPrep wizards, respectively. The receptor grid was centered on the active site residues of the receptor. Glide docking was carried out in standard-precision (sp) mode. The OPLS3e force field was used in simulations and the settings were left to their default values. For virtual screening calculations, Virtual Screening Workflow (VSW) of Schrodinger software suite was applied using default parameters and GlideScore scoring function.
Evaluation criteria
Root-mean-square-deviation (RMSD) between the matching heavy atoms of the predicted pose and those of the crystal structure was used to assess ligand placement accuracy of docking calculation.
Virtual screening
Virtual screening calculations were carried out using docking programs, GOLD, Glide, AutoDock Vina (using AutoDock 4 force field) and FlexX. The screening was based on two target proteins, namely COX-1 and COX-2. The Database of Useful Decoys-Enhanced (DUD-E) was used in the experiments.40 It includes, for each target, a PDB file and a set of active compounds and decoys with an average actives/decoys proportion of 2%.
Results and Discussion
NSAIDs are among the widely used pharmaceuticals prescribed as analgesic, antipyretic and anti-inflammatory agents.41 Worldwide sale of NSAIDs in 2019 was $US 15.58 billion and it is expected to reach $US 24.35 billion by 2027 with 5.8% annual growth rate. The increased prevalence of disorders such as rheumatoid arthritis, osteoarthritis, migraine, and other pain associated conditions is leading to increased consumption of NSAIDs.1 The intake of NSAIDs never been without side effects. Although the use of low doses of some NSAIDs may be regarded as safe, their standard doses are associated with the increased risk of cardiovascular and life-threatening gastrointestinal tract bleeding.42 Consequently, the need to develop new nonsteroidal anti-inflammatory drugs acting on COXs and even downstream targets such as prostaglandin E (PGE) receptors is being always felt necessary.43
The diversity of the expertise and cutting-edge technologies involved during a drug discovery and development project since the very beginning of the initial idea until the approval of a drug for a given condition makes it a very expensive and time-consuming process.44,45 Drug discovery and development projects usually start with the identification of lead compounds showing activity against an already identified suitable target followed by optimization of the lead compound mainly through medicinal chemistry activities. Then, after many years of research during the rest of the discovery stage, one or two promising drug candidates may enter clinical phases of the development stage. Eventually, if successful, the newly introduced pharmaceutical will be monitored during post marketing surveillance period. Many computational methods were developed particularly during last few decades to speed up drug design and discovery processes while reducing the cost and rate of failure. Nowadays, computational studies such as protein structure modeling, molecular mechanics calculations, molecular dynamics simulations, QSAR analyses, pharmacophore modeling, and molecular docking are extensively practiced at different steps of drug design and discovery pipeline including target identification and optimization, target structure prediction, virtual library generation and screening, lead identification and optimization and activity prediction.46-51 One of the widely used computational methods which is applied at different stages of drug discovery and development is molecular docking. Molecular docking is a computational method to predict binding conformation and orientation of a ligand (usually a small molecule) when interacts with its receptor (protein or DNA).52 It can be used to investigate mechanisms of drug binding to targets. Different molecular docking methods vary in terms of the size of ligand molecules (organic molecules, peptides, and proteins) they can handle,53 the algorithms they use for sampling the conformational space (search method) and the scoring function which is used to prioritize different poses sampled by the search method.54 Many docking algorithms from both academia and industry are available to scientific community and from time to time they are evaluated in terms of their performance.55-57 Different methods may perform differently when dealing with different problems. Therefore, for more reliable results, it would be appropriate to identify the best performing method for a given molecular docking task.
Reproducing experimentally known binding poses of COX inhibitors
In this study, we have evaluated the performances of five well respected docking software in predicting the experimentally known poses of 51 COX-1 and COX-2 co-crystallized ligands. Table 2 summarizes the docking results for the studied protein-ligand complexes. The percentages of top-ranked solutions close to the corresponding experimentally determined structures with a range of defined RMSD values are reported in the table. According to the results, collectively, Glide docking method performed the best, while FlexX program accomplished the poorest results. GOLD docking method using GoldScore scoring function was the least successful pose predictor among the studied docking conditions when the RMSD cutoff criterion was set to ≤ 2 Å. Glide method predicted the conformations successfully for 94% and 100% of ligands with RMSD cutoffs of ≤ 1 Å and ≤ 2 Å, respectively. Consequently, it can be considered the best performing docking algorithm when dealing with the prediction of binding poses of inhibitors of COX enzymes. The second-best performer was AutoDock (50 runs), which were able to predict the conformations of the ligands with 49% and 82% success rates for ≤ 1 Å and ≤ 2 Å RMSD cutoffs, respectively. One of the interesting aspects of the obtained data presented in Table 2 is the substantial improvement in the percentages of the correct predictions by all docking methods when the RMSD cutoff value was increased from ≤ 1 Å to ≤ 2 Å. The results indicated that by accepting RMSD cutoff value of ≤ 2 Å, as the criterion of correct prediction of ligands poses, on average more than 75% of the studied complexes were predicted correctly. At this level of accuracy, the minimum (59%) and maximum (100%) prediction efficiencies were achieved by GOLD (GoldScore) and Glide docking methods, respectively. Moreover, the results in Table 2 shows that, all used docking methods, except Glide, predicted enzyme-bound conformation of some ligands very incorrectly (i.e., RMSD values > 4, 5, and 6 Å). This indicates that there are ligands which most of the docking schemes were not able to predict their bound poses at the acceptable level of accuracy. It is obvious that Glide successfully predicted correct conformation for all ligand-enzyme complexes. And once again, the second-best performer, i.e., AutoDock (with 50 runs) was superior than the others with the least rates (excluding Glide) of failures. There are good reciprocal correlations between percentages of correct and incorrect pose predictions. The results presented in Table 2 are graphically illustrated in Fig. 1 for easy visual inspection.
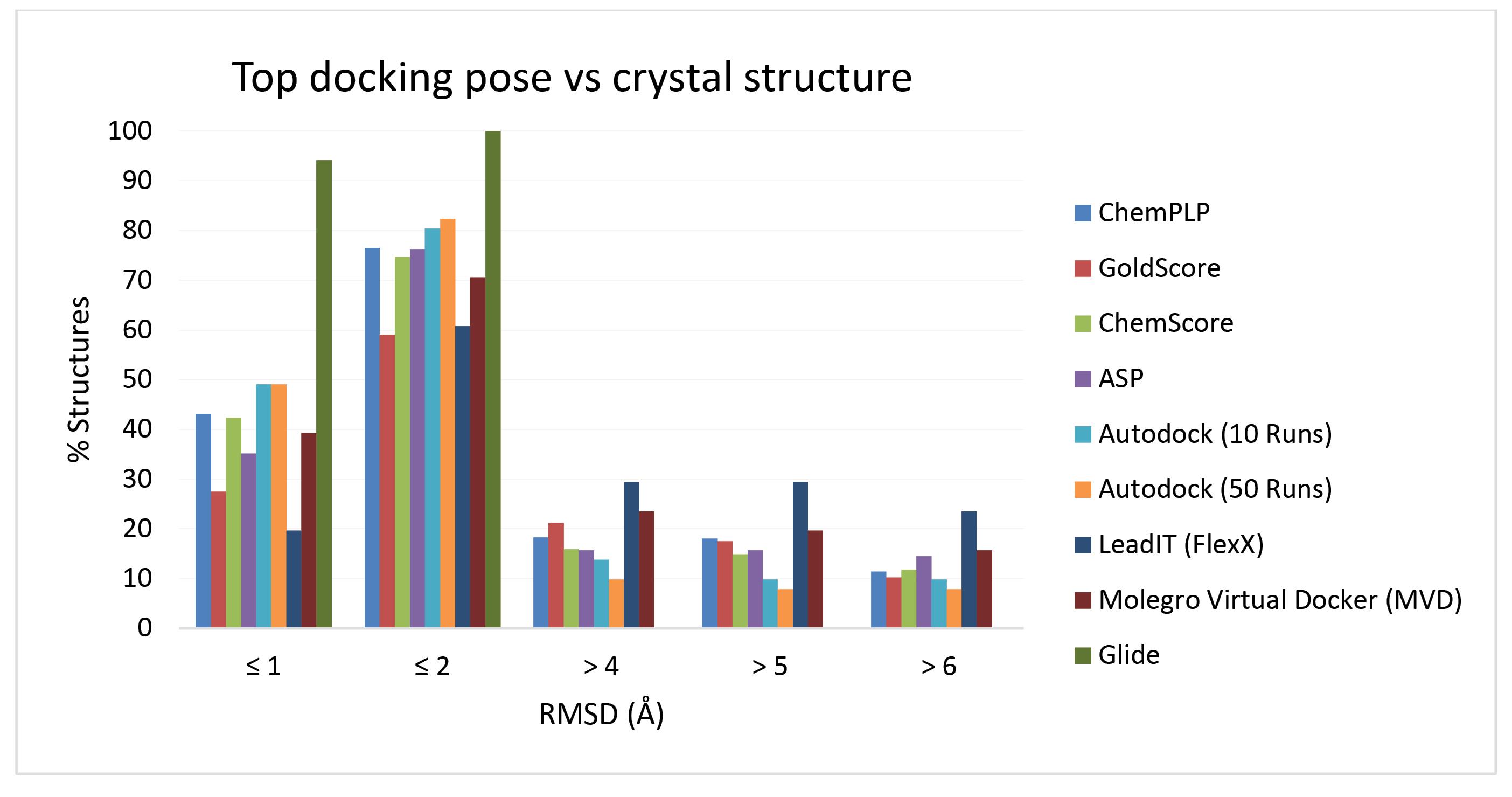
Fig. 1.
Distribution of RMSD between the top-ranked docking pose and the corresponding experimentally determined pose in crystal structures. x axis: RMSD cutoffs y axis: percentage of top-ranked docking poses within a given RMSD cutoff values of the crystallographic pose.
.
Distribution of RMSD between the top-ranked docking pose and the corresponding experimentally determined pose in crystal structures. x axis: RMSD cutoffs y axis: percentage of top-ranked docking poses within a given RMSD cutoff values of the crystallographic pose.
As shown in Table 2 and Fig. 1, according to the success rates observed based on RMSD ≤ 2 Å for the top ranked poses, the order of performance of docking programs on 51 COX-1/COX-2 enzyme-inhibitor complexes is Glide (100 %) ˃ AutoDock (50 Runs) (82.35 %) ˃ AutoDock (10 Runs) (80.39 %) ˃ GOLD (ChemPLP) (76.47 %) ≈ GOLD (ASP) (76.27 %) ˃ GOLD (ChemScore) (74.1 %) ˃ MVD (70.58 %) ˃ LeadIT (FlexX) (60.78 %) ˃ GOLD (GoldScore) (59.01 %). It is worth mentioning that increasing the number of runs from 10 to 50 in AutoDock calculations has led to only a marginal improvement in docking performance. Therefore, one may take into account the trade-off between small gain in docking accuracy and speed of calculations.
Table 2.
Performance of docking programs on 51 protein-ligand complexes containing COX-1 and COX-2 enzymes with co-crystalized ligands in terms of percentages of docking solutions deviating from the experimental conformation of the ligands at different RMSD cutoff values
|
Docking method
|
RMSD
|
|
% ≤ 1 Å
|
% ≤ 2 Å
|
% > 4 Å
|
% > 5 Å
|
% > 6 Å
|
| GOLD (ChemPLP)* |
43.13 |
76.47 |
18.03 |
18.03 |
11.37 |
| GOLD (GoldScore)* |
27.45 |
59.01 |
21.17 |
17.45 |
10.19 |
| GOLD (ChemScore)* |
42.35 |
74.7 |
15.88 |
14.9 |
11.76 |
| GOLD (ASP)* |
35.09 |
76.27 |
15.68 |
15.68 |
14.5 |
| AutoDock (10 Runs) |
49.01 |
80.39 |
13.72 |
9.8 |
9.8 |
| AutoDock (50 Runs) |
49.01 |
82.35 |
9.8 |
7.84 |
7.84 |
| LeadIT (FlexX) |
19.6 |
60.78 |
29.41 |
29.41 |
23.52 |
| MVD |
39.21 |
70.58 |
23.52 |
19.6 |
15.68 |
| Glide |
94.12 |
100 |
0 |
0 |
0 |
*Figures for GOLD algorithm using different scoring functions are the average of 10 separate runs with early termination option turned on.
In the case of GOLD docking algorithm, four different scoring functions were used to predict top 10 solutions for 51 enzyme-inhibitor complexes. Comparison of the results showed that at the accuracy level of RMSD ≤ 2 Å, ChemPLP scoring function successfully predicted the experimental poses for 42 out of 51 inhibitors (0.82%). The RMSD of the first-rank solutions (i.e., the highest scoring pose) compared to their corresponding experimental solutions for 6 inhibitors were greater than 2 Å, and in the mean time for these inhibitors the closest solutions were also greater than 2 Å. (In some cases, the first rank solution is also the closest solution to the reference pose.) For instance, the top scoring pose obtained by GOLD (ChemPLP) docking method for ligand FF8 deviated 3.99 Å from its enzyme bound position solved by X-ray crystallography (PDB structure 5W58). In this case, the top scoring solution is also the closest solution to the experimental pose. These 6 cases are called “general failures”. Likewise, for the remaining 3 inhibitors, GOLD was not successful in predicting good solutions for their enzyme bound poses. In fact, in each of latter 3 cases, GOLD was able to find a pose with RMSD ≤ 2 Å, but ChemPLP scoring function failed to rank that as the best solution, and instead a wrong solution with high RMSD relative to the reference pose (experimental data) was scored the best. This type of misprediction is termed “ranking failure”. This failure can be represented by the top scoring pose obtained by ChemPLP function in GOLD docking for ligand P6A in structure 5U6X, which showed a RMSD of 10.29 Å relative to its experimentally obtained pose. However, the closest pose among 10 generated solutions had a very small RMSD of 1.56 Å. The results of this analysis for ChemPLP and other scoring functions available in GOLD are shown in Fig. 2, where the RMSD of the closest observed solution is plotted against the RMSD of top ranked solution. Simply, in “General failures” no solutions with RMSD ≤ 2.0 Å were observed, whereas in “ranking failures” solutions that could have been considered successes were generated but not correctly ranked. As is clear from the plots in Fig. 2, ChemPLP scoring function outperforms other three functions available in GOLD docking algorithm, with highest success rate and less likelihood of failures.
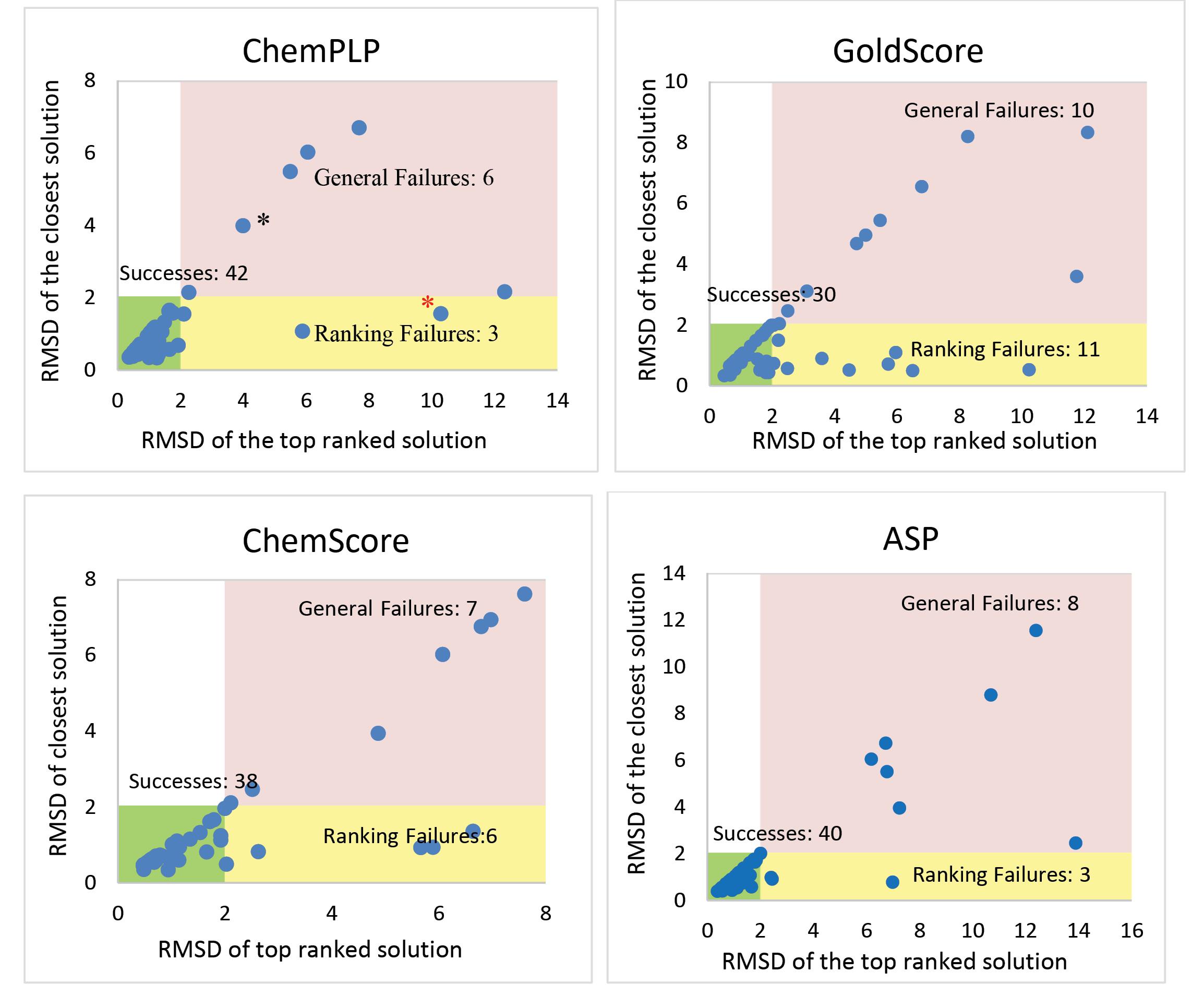
Fig. 2.
Distribution of RMSDs for top-ranked and closest solutions for top 10 docking poses generated with GOLD algorithm using ChemPLP, GoldScore, ChemScore and ASP scoring functions. Data points labeled with black and red asterisks are FF8 and P6A ligands in 5W58 and 5U6X PDB structures, respectively. They are examples of general and ranking failures for docking calculations using GOLD (ChemPLP) method.
.
Distribution of RMSDs for top-ranked and closest solutions for top 10 docking poses generated with GOLD algorithm using ChemPLP, GoldScore, ChemScore and ASP scoring functions. Data points labeled with black and red asterisks are FF8 and P6A ligands in 5W58 and 5U6X PDB structures, respectively. They are examples of general and ranking failures for docking calculations using GOLD (ChemPLP) method.
The results of docking calculations were inspected by dividing them into two groups based on COX-1 and COX-2 isoforms. The performance of studied docking methods on 18 COX1-inhibitor complexes and 32 COX2-inhibitor complexes are illustrated in Fig. 3 and reported in detail in Tables S1 and S2 of Supplementary file 1. As shown in Fig. 3, all studied docking methods (except Glide) predicted poses of COX1 inhibitors with marginally higher success rate than that of COX2 inhibitors.
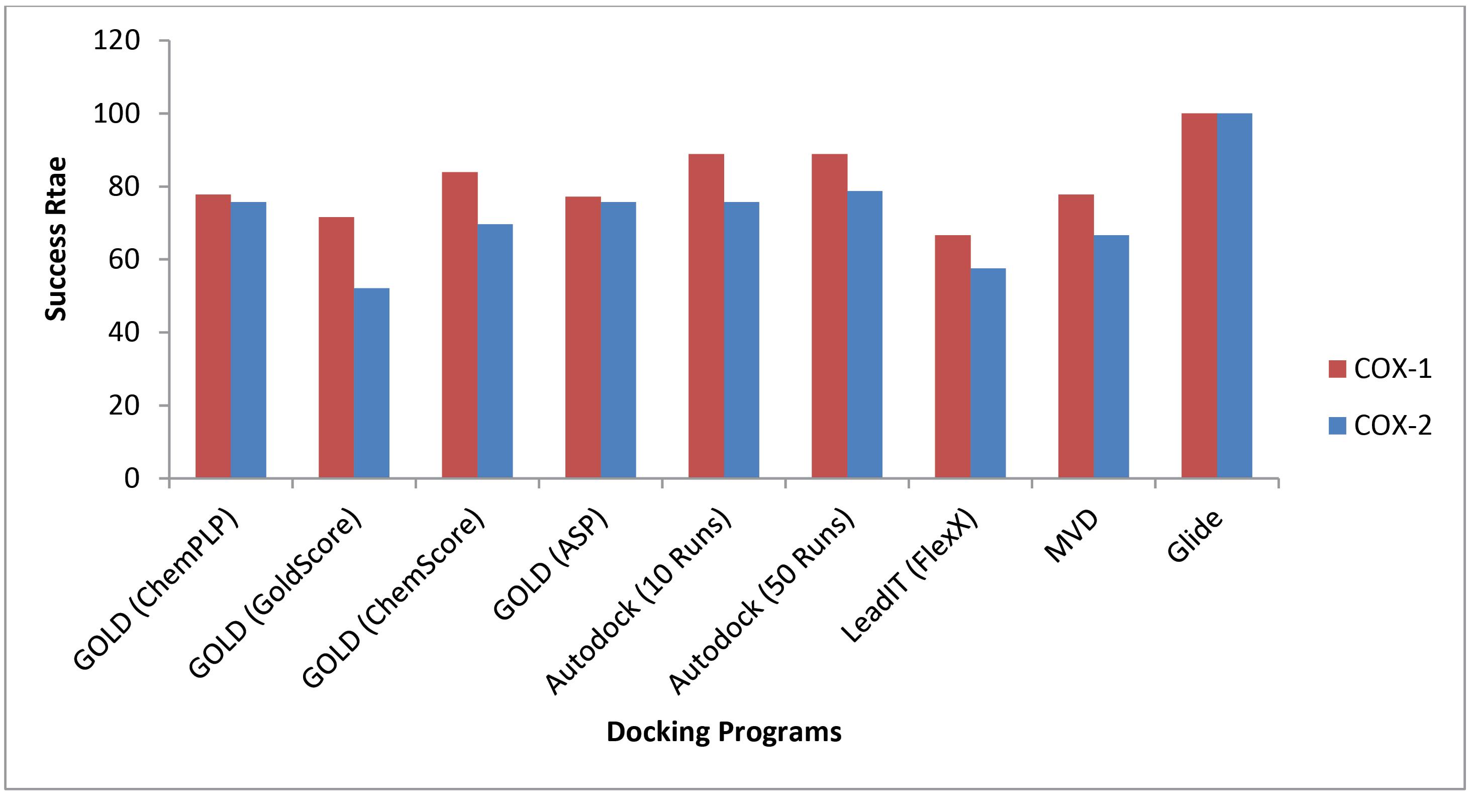
Fig. 3.
Comparison of the performances of different docking methods on 18 COX1-inhibitor complexes and 32 COX2-inhibitor complexes presented in terms of percentages of docking solutions at RMSD cutoff level of ≤ 2 Å.
.
Comparison of the performances of different docking methods on 18 COX1-inhibitor complexes and 32 COX2-inhibitor complexes presented in terms of percentages of docking solutions at RMSD cutoff level of ≤ 2 Å.
Screening of chemical structures for identifying COX inhibitors by docking methods
In order to further compare the usefulness of the docking methods in design and discovery of novel anti-inflammatory agents, some of the studied methods namely, GOLD (ChemPLP), Glide, AutoDock Vina (using AutoDock 4 force field) and FlexX were evaluated for their ability in identifying COX1/COX2 active ligands from decoy structures using docking-based virtual screening analyses. From different scoring functions available in GOLD program, ChemPLP was used for screening experiments due to its better performance in predicting experimentally solved COX1/COX2-inhibitor complexes. Table 3 shows the results of ROC analyses for the docking screening calculations. Commonly, the area under the curve (AUC) is used as the measure of the capability of a binary classifier to differentiate between classes in ROC analysis, where the higher AUC indicates a better performance for the model in correctly assigning the membership to classes. The AUC values in this study range between 0.6115 to 0.9275 obtained for ROC analyses of docking-based screening and classification calculations shown for GOLD on COX1 dataset and Glide on COX2 dataset, respectively. According to the AUC values found in this work, all studied docking methods performed better on COX2 dataset than the COX1 dataset, which means that, in general, the docking methods score active ligands higher than the corresponding decoys and they do this with better performance for COX2 ligands. The best discrimination of active ligands over decoys was obtained for Glide method on COX2 dataset with AUC value of 0.9275. However, AutoDock gave the best results for COX1 dataset (i.e., AUC value of 0.8978). Once again, AutoDock outperforms the others considering combined COX1 and COX2 datasets.
Table 3.
Statistics of ROC analyses on the results of docking screening calculations using different docking methods
|
|
Enzyme
|
Statistics
|
|
AUC
|
Std. error
|
95% CI
|
P
value
|
| Docking method |
Glide |
COX1 |
0.7041 |
0.01426 |
0.6762 to 0.7321 |
<0.0001 |
| COX2 |
0.9275
|
0.007629 |
0.9126 to 0.9425 |
<0.0001 |
| Combined |
0.8055 |
0.007556 |
0.7907 to 0.8203 |
<0.0001 |
| GOLD |
COX1 |
0.6115 |
0.01834 |
0.5755 to 0.6474 |
<0.0001 |
| COX2 |
0.8576 |
0.01032 |
0.8374 to 0.8778 |
<0.0001 |
| Combined |
0.7609 |
0.01058 |
0.7402 to 0.7817 |
<0.0001 |
| AutoDock |
COX1 |
0.8978
|
0.04347 |
0.8127 to 0.9830 |
<0.0001 |
| COX2 |
0.9050 |
0.008964 |
0.8875 to 0.9226 |
<0.0001 |
| Combined |
0.8926
|
0.009512 |
0.8740 to 0.9112 |
<0.0001 |
| FlexX |
COX1 |
0.6747 |
0.02004 |
0.6354 to 0.7139 |
<0.0001 |
| COX2 |
0.7698 |
0.01247 |
0.7454 to 0.7943 |
<0.0001 |
| Combined |
0.7379 |
0.01079 |
0.7168 to 0.7591 |
<0.0001 |
Fig. 4 shows the illustrative representation of the results for ROC analyses performed on docking scores obtained for docking screening (i.e., results for docking library of COX1 and COX2 active ligands and matching decoy structures into the binding site of COX1 and COX2 enzymes, respectively) by Glide software as the representative (See Fig. S1 for ROC curves and the corresponding statistical analyses for all used docking methods).
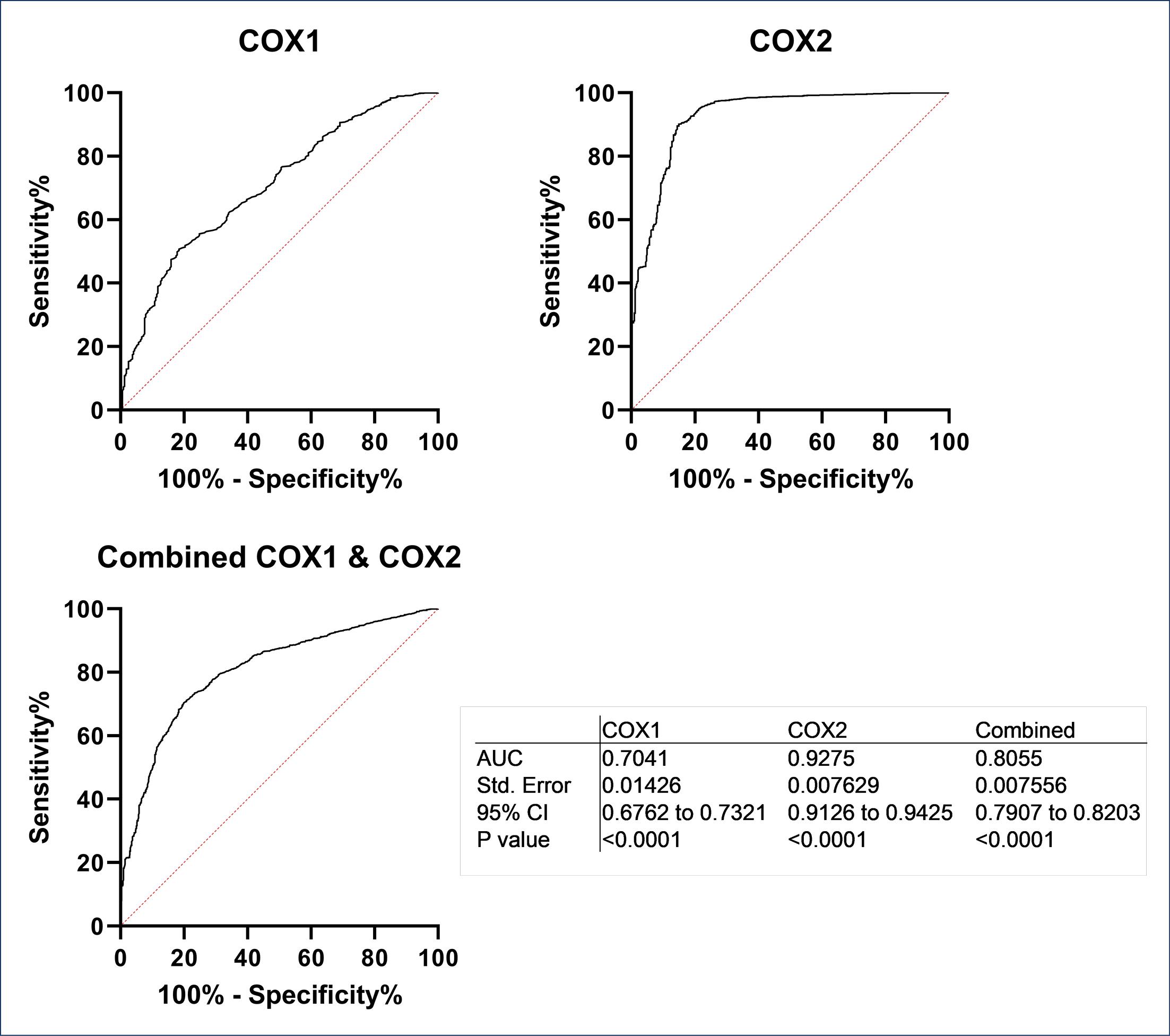
Fig. 4.
The results of ROC analyses on docking scores for COX1 and COX2 active ligands and decoys obtained by Glide docking method. The plots show AUC curves for COX1, COX2, and combined COX1 and COX2 date sets. The statistics for ROC analyses are given in bottom right hand panel.
.
The results of ROC analyses on docking scores for COX1 and COX2 active ligands and decoys obtained by Glide docking method. The plots show AUC curves for COX1, COX2, and combined COX1 and COX2 date sets. The statistics for ROC analyses are given in bottom right hand panel.
Close investigation of the results presented so far provides interesting information regarding the application of docking calculation on ligands targeting COX enzymes. Glide and AutoDock performed better in reproducing the binding pose of COX ligands with structurally known ligand-enzyme complexes (higher percentages of docking solutions within the given RMSD cutoffs shown in Table 2) and also these two methods have led to better AUC values in ROC analyses. The agreement between the two different docking performance assessment strategies used in this study is more pronounced when dealing with COX2 dataset. The correlations between AUC values and the percentages of docking solutions within the given RMSD cutoffs for COX2 dataset using Glide, GOLD, AutoDock and FlexX are very high as shown in Fig. 5. The correlations are even higher (R2 close to unity) for the combined data excluding AutoDock. However, for COX1 dataset no such correlation was observed.
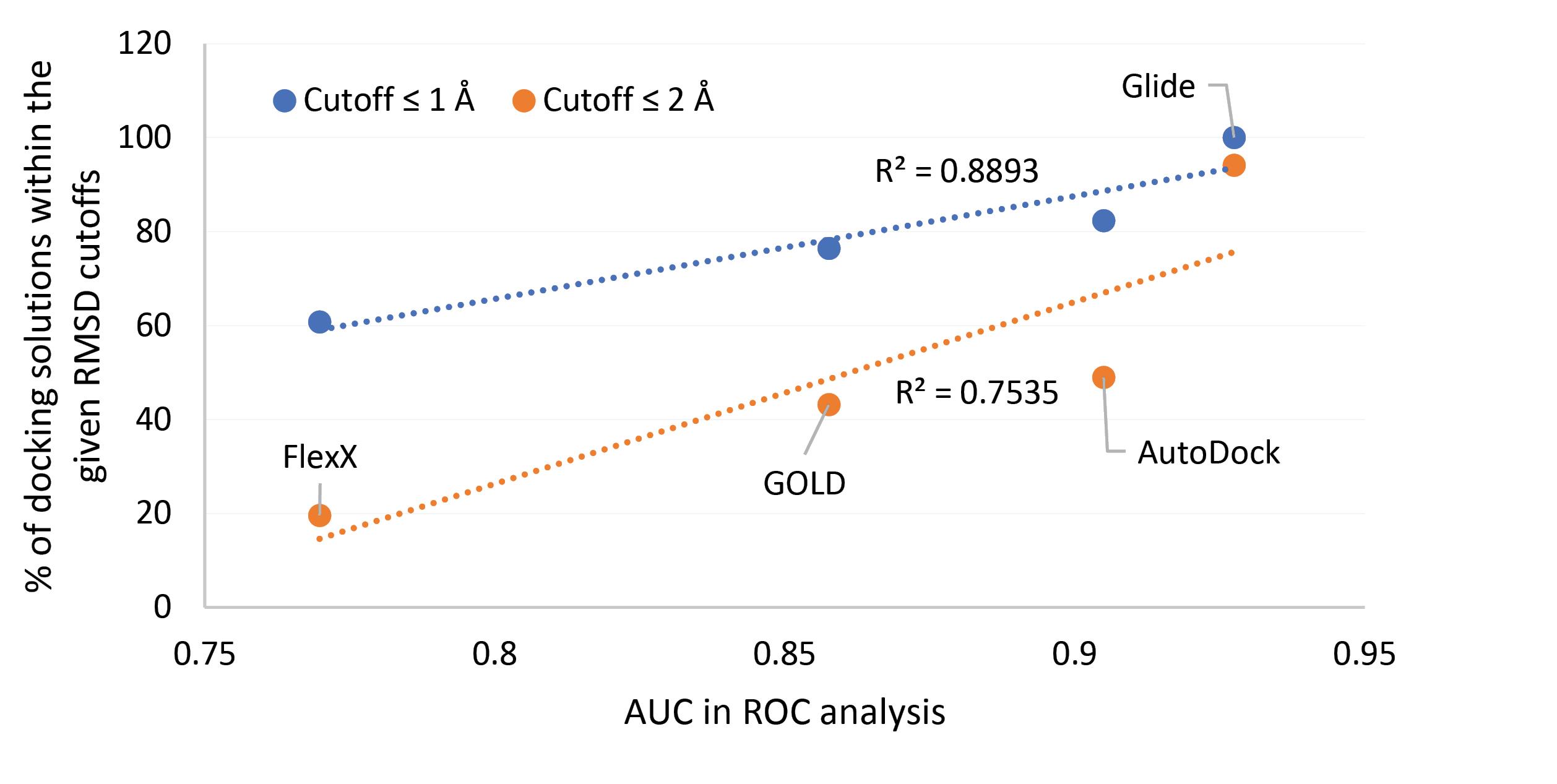
Fig. 5.
Correlations between AUC values in ROC analyses on docking screening results and the percentages of docking solutions within the given RMSD cutoffs from the experimentally known positions of structurally known ligand-COX complexes using Glide, GOLD, AutoDock and FlexX software.
.
Correlations between AUC values in ROC analyses on docking screening results and the percentages of docking solutions within the given RMSD cutoffs from the experimentally known positions of structurally known ligand-COX complexes using Glide, GOLD, AutoDock and FlexX software.
Another point which can be deduced from the results is that the studied software performed marginally better in reproducing the experimental poses of co-crystalized COX2 ligands compared to the co-crystalized COX1 ligands for the structurally solved complexes. In contrast, in large scale docking-based screening calculations the opposite was the case, and ranking COX2 dataset was more precise than that for COX1 dataset. This may be due to the relatively bigger size of COX2 active ligands/decoys dataset and may allude to the fact that one may use docking screening with more confidence for the identification of COX2 ligands. Enrichment factor is a reliable criterion to assess the performance of a classification method. In this regard the enrichment factors (EFs) were calculated for large-scale docking experiments on COX1 and COX2 active ligands/decoys datasets (Table S3). It is noteworthy to mention that EFs for 0.1% of top-ranking structures (ligands and decoys) for different docking methods (Glide, GOLD, AutoDock and FlexX) range between 8 and 40 determined for GOLD on COX1 and AutoDock on COX1 datasets respectively. This means that all studied docking protocols are able to efficiently enrich the active ligands among top-scoring structures. The estimated EFs (at the level of 0.1% top-ranking scores) for COX1 dataset are 40, 29, 19 and 8 for AutoDock, FlexX, Glide and GOLD, respectively. For COX2 dataset, EFs are 31, 23, 20 and 12 for AutoDock, GOLD, Glide and FlexX.
Conclusion
Based on the results obtained in this study, one may conclude that in the case of COX enzymes, which are the prime targets for NSAIDs and are involved in different conditions such as inflammation, arthritis, cardiovascular diseases and cancer. Glide docking method outperformed other methods by successfully predicting the binding poses for all structurally known ligand-COX complexes. For large scale docking studies for the identification of COX active ligands all studied docking methods showed certain degrees of success. For example, Glide performed the best based on the ROC analysis of the scores for docking active ligand/decoy structures on COX2 enzyme, while AutoDock was the best on COX1 dataset and combined COX1 and COX2 datasets. All tested methods were able to enrich active ligands at 0.1% of top-scoring structures, however, AutoDock exhibited the highest performance in this regard as well. In general, the results obtained in this study reinforce the notion that docking methods are valuable tools in predicting ligand-receptor binding modes, and provide specific information about docking in ligand-COX enzyme systems. However, they do not substitute experiments and all docking methods have their specific limitations which should be realized by users to better appreciate the strengths and weaknesses of each approach.
Research Highlights
What is the current knowledge?
√ Docking protocols are valuable means of predicting 3D structures of ligand-target complexes.
√ Docking protocols perform differently on different ligand-target systems.
What is new here?
√ Glide docking method performed the best on predicting binding poses of ligands on COX enzymes.
√ Glide and AutoDock showed better performances for ranking ligands (actives and decoys) targeting COX2 and COX1 enzymes, respectively.
Acknowledgements
Authors would like to thank Research Office and Biotechnology Research Center of Tabriz University of Medical Sciences for providing financial support and laboratory facilities.
Competing Interests
Authors declare no conflict of interests.
Ethical Statement
None to be declared.
Funding
This work was funded by Tabriz University of Medical Sciences (Grant number: 66498).
Supplementary files
Supplementary file 1 contains Tables S1-S3 and Figure S1.
(pdf)
References
- Brennan R, Wazaify M, Shawabkeh H, Boardley I, McVeigh J, Van Hout MC. A Scoping Review of Non-Medical and Extra-Medical Use of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs). Drug Safety 2021; 44:917-28. doi: 10.1007/s40264-021-01085-9 [Crossref] [ Google Scholar]
- Non-steroidal Anti-inflammatory Drugs Market Size USD 31.45 Billion by 2030. BioSpace; 2022 [cited 2023 August 07]; Available from: www.biospace.com/article/non-steroidal-anti-inflammatory-drugs-market-size-usd-31-45-billion-by-2030/.
- Non-steroidal anti-inflammatory drugs (NSAIDs) market size, share and industry analysis. Fortune Business Insights; [cited 2023 August 07]; Available from: www.fortunebusinessinsights.com/non-steroidal-anti-inflammatory-drugs-nsaids-market-102823.
- Oniga SD, Pacureanu L, Stoica CI, Palage MD, Crăciun A, Rusu LR. COX Inhibition Profile and Molecular Docking Studies of Some 2-(Trimethoxyphenyl)-Thiazoles. Molecules 2017; 22:1507. doi: 10.3390/molecules22091507 [Crossref] [ Google Scholar]
- Stachowicz K. Stachowicz KDeciphering the mechanisms of regulation of an excitatory synapse via cyclooxygenase-2A review. BiochemPharmacol 2021; 192:114729. doi: 10.1016/j.bcp.2021.114729 [Crossref] [ Google Scholar]
- Thiruchenthooran V, Sánchez-López E, Gliszczyńska A. Perspectives of the Application of Non-Steroidal Anti-Inflammatory Drugs in Cancer Therapy: Attempts to Overcome Their Unfavorable Side Effects. Cancers (Basel) 2023; 15. 10.3390/cancers15020475.
- Kaya Çavuşoğlu B, Sağlık BN, Acar Çevik U, Osmaniye D, Levent S, Özkay Y. Design, synthesis, biological evaluation, and docking studies of some novel chalcones as selective COX-2 inhibitors. Arch Pharm (Weinheim) 2021; 354:e2000273. doi: 10.1002/ardp.202000273 [Crossref] [ Google Scholar]
- Vemula D, Jayasurya P, Sushmitha V, Kumar YN, Bhandari V. CADD, AI and ML in drug discovery: A comprehensive review. Eur J Pharm Sci 2023; 181:106324. doi: 10.1016/j.ejps.2022.106324 [Crossref] [ Google Scholar]
- Dar KB, Bhat AH, Amin S, Hamid R, Anees S, Anjum S. Modern Computational Strategies for Designing Drugs to Curb Human Diseases: A Prospect. Curr Top Med Chem 2018; 18:2702-19. doi: 10.2174/1568026619666190119150741 [Crossref] [ Google Scholar]
- Rudrapal M, Chetia D. Virtual Screening, Molecular Docking and QSAR Studies in Drug Discovery and Development Programme. Journal of Drug Delivery and Therapeutics 2020; 10:225-33. doi: 10.22270/jddt.v10i4.4218 [Crossref] [ Google Scholar]
- Wang X, Song K, Li L, Chen L. Structure-Based Drug Design Strategies and Challenges. Curr Top Med Chem 2018; 18:998-1006. doi: 10.2174/1568026618666180813152921 [Crossref] [ Google Scholar]
- Batool M, Ahmad B, Choi S. A Structure-Based Drug Discovery Paradigm. Int J Mol Sci 2019; 20:2783. doi: 10.3390/ijms20112783 [Crossref] [ Google Scholar]
- Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH. A critical assessment of docking programs and scoring functions. J Med Chem 2006; 49:5912-31. doi: 10.1021/jm050362n [Crossref] [ Google Scholar]
- Xu S, Wang L, Pan X. An evaluation of combined strategies for improving the performance of molecular docking. J BioinformComput Biol 2021; 19:2150003. doi: 10.1142/s0219720021500037 [Crossref] [ Google Scholar]
- Ewing TJA, Kuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. Journal of Computational Chemistry 1997; 18:1175-89. doi: 10.1002/(SICI)1096-987X(19970715)18:9<1175::AIDJCC6>3.0.CO;2-O [Crossref] [ Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009; 30:2785-91. doi: 10.1002/jcc.21256 [Crossref] [ Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010; 31:455-61. doi: 10.1002/jcc.21334 [Crossref] [ Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997; 267:727-48. doi: 10.1006/jmbi.1996.0897 [Crossref] [ Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT. Glide: a new approach for rapid, accurate docking and scoring1Method and assessment of docking accuracy. J Med Chem 2004; 47:1739-49. doi: 10.1021/jm0306430 [Crossref] [ Google Scholar]
- Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol 1996; 261:470-89. doi: 10.1006/jmbi.1996.0477 [Crossref] [ Google Scholar]
- Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem 2003; 46:499-511. doi: 10.1021/jm020406h [Crossref] [ Google Scholar]
- Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem 2006; 49:3315-21. doi: 10.1021/jm051197e [Crossref] [ Google Scholar]
- Schapira M, Abagyan R, Totrov M. Nuclear hormone receptor targeted virtual screening. J Med Chem 2003; 46:3045-59. doi: 10.1021/jm0300173 [Crossref] [ Google Scholar]
- Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model 2003; 21:289-307. doi: 10.1016/s1093-3263(02)00164-x [Crossref] [ Google Scholar]
- Corbeil CR, Williams CI, Labute P. Variability in docking success rates due to dataset preparation. J Comput Aided Mol Des 2012; 26:775-86. doi: 10.1007/s10822-012-9570-1 [Crossref] [ Google Scholar]
- Zhao H, Caflisch A. Discovery of ZAP70 inhibitors by high-throughput docking into a conformation of its kinase domain generated by molecular dynamics. Bioorg Med Chem Lett 2013; 23:5721-6. doi: 10.1016/j.bmcl.2013.08.009 [Crossref] [ Google Scholar]
- Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, Garmendia-Doval AB, Juhos S, Schmidtke P. rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoSComput Biol 2014; 10:e1003571. doi: 10.1371/journal.pcbi.1003571 [Crossref] [ Google Scholar]
- Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT. DOCK 6: Impact of new features and current docking performance. J Comput Chem 2015; 36:1132-56. doi: 10.1002/jcc.23905 [Crossref] [ Google Scholar]
- Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases1Evaluation of different docking/scoring combinations. J Med Chem 2000; 43:4759-67. doi: 10.1021/jm001044l [Crossref] [ Google Scholar]
- Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem 1999; 42:5100-9. doi: 10.1021/jm990352k [Crossref] [ Google Scholar]
- Knegtel RM, Bayada DM, Engh RA, von der Saal W, van Geerestein VJ, Grootenhuis PD. Comparison of two implementations of the incremental construction algorithm in flexible docking of thrombin inhibitors. J Comput Aided Mol Des 1999; 13:167-83. doi: 10.1023/a:1008014604433 [Crossref] [ Google Scholar]
- Xu X, Huang M, Zou X. Docking-based inverse virtual screening: methods, applications, and challenges. Biophys Rep 2018; 4:1-16. doi: 10.1007/s41048-017-0045-8 [Crossref] [ Google Scholar]
- Huang SY, Zou X. Advances and challenges in protein-ligand docking. Int J Mol Sci 2010; 11:3016-34. doi: 10.3390/ijms11083016 [Crossref] [ Google Scholar]
- Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins 2003; 52:609-23. doi: 10.1002/prot.10465 [Crossref] [ Google Scholar]
- Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr Med Chem 2013; 20:2839-60. doi: 10.2174/09298673113209990001 [Crossref] [ Google Scholar]
- Lätti S, Niinivehmas S, Pentikäinen OT. Rocker: Open source, easy-to-use tool for AUC and enrichment calculations and ROC visualization. J Cheminform 2016; 8:45. doi: 10.1186/s13321-016-0158-y [Crossref] [ Google Scholar]
- Triballeau N, Acher F, Brabet I, Pin JP, Bertrand HO. Virtual screening workflow development guided by the "receiver operating characteristic" curve approachApplication to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem 2005; 48:2534-47. doi: 10.1021/jm049092j [Crossref] [ Google Scholar]
- Allinger NL. Allinger NLConformational analysis130MM2A hydrocarbon force field utilizing V1 and V2 torsional terms. Journal of the American Chemical Society 1977; 99:8127-34. doi: 10.1021/ja00467a001 [Crossref] [ Google Scholar]
- Stewart JJ. Optimization of parameters for semiempirical methods IV: extension of MNDO, AM1, and PM3 to more main group elements. J Mol Model 2004; 10:155-64. doi: 10.1007/s00894-004-0183-z [Crossref] [ Google Scholar]
- Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. Journal of Medicinal Chemistry 2012; 55:6582-94. doi: 10.1021/jm300687e [Crossref] [ Google Scholar]
- Mullan J, Weston KM, Bonney A, Burns P, Mullan J, Rudd R. Consumer knowledge about over-the-counter NSAIDs: they don't know what they don't know. Aust N Z J Public Health 2017; 41:210-4. doi: 10.1111/1753-6405.12589 [Crossref] [ Google Scholar]
- McCarberg B, Gibofsky A. Need to develop new nonsteroidal anti-inflammatory drug formulations. Clin Ther 2012; 34:1954-63. doi: 10.1016/j.clinthera.2012.08.005 [Crossref] [ Google Scholar]
- Mahesh G, Anil Kumar K, Reddanna P. Overview on the Discovery and Development of Anti-Inflammatory Drugs: Should the Focus Be on Synthesis or Degradation of PGE(2)?. J Inflamm Res 2021; 14:253-63. doi: 10.2147/jir.S278514 [Crossref] [ Google Scholar]
- Lionta E, Spyrou G, Vassilatis DK, Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem 2014; 14:1923-38. doi: 10.2174/1568026614666140929124445 [Crossref] [ Google Scholar]
- Krogsgaard-Larsen P, Madsen U. Introduction to drug design and discovery. In: Strømgaard K, P Krogsgaard-Larsen, U Madsen, editors. Textbook of Drug Design and Discovery. 5th ed: Taylor & Francis Group; 2017. p. 14.
- Wang G, Bai Y, Cui J, Zong Z, Gao Y, Zheng Z. Computer-Aided Drug Design Boosts RAS Inhibitor Discovery. Molecules 2022; 27. 10.3390/molecules27175710.
- Sun Y, Jiao Y, Shi C, Zhang Y. Deep learning-based molecular dynamics simulation for structure-based drug design against SARS-CoV-2. Comput Struct Biotechnol J 2022; 20:5014-27. doi: 10.1016/j.csbj.2022.09.002 [Crossref] [ Google Scholar]
- Govindaraj RG, Thangapandian S, Schauperl M, Denny RA, Diller DJ. Recent applications of computational methods to allosteric drug discovery. Front Mol Biosci 2022; 9:1070328. doi: 10.3389/fmolb.2022.1070328 [Crossref] [ Google Scholar]
- Bekker GJ, Kamiya N. Advancing the field of computational drug design using multicanonical molecular dynamics-based dynamic docking. Biophys Rev 2022; 14:1349-58. doi: 10.1007/s12551-022-01010-z [Crossref] [ Google Scholar]
- Cavasotto CN, Scardino V. Machine Learning Toxicity Prediction: Latest Advances by Toxicity End Point. ACS Omega 2022; 7:47536-46. doi: 10.1021/acsomega.2c05693 [Crossref] [ Google Scholar]
- Johnson TO, Akinsanmi AO, Ejembi SA, Adeyemi OE, Oche JR, Johnson GI. Modern drug discovery for inflammatory bowel disease: The role of computational methods. World J Gastroenterol 2023; 29:310-31. doi: 10.3748/wjg.v29.i2.310 [Crossref] [ Google Scholar]
- Zhu H, Zhang Y, Li W, Huang N. A Comprehensive Survey of Prospective Structure-Based Virtual Screening for Early Drug Discovery in the Past Fifteen Years. Int J Mol Sci 2022; 23. 10.3390/ijms232415961.
- Lee AC-L, Harris JL, Khanna KK, Hong J-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. International Journal of Molecular Sciences 2019; 20:2383. doi: 10.3390/ijms20102383 [Crossref] [ Google Scholar]
- Hamzeh-Mivehroud M, Sokouti B, Dastmalchi S. Molecular Docking at a Glance. In: Dastmalchi S, M Hamzeh-Mivehroud, B Sokouti, editors. Methods and Algorithms for Molecular Docking-Based Drug Design and Discovery. IGI Global; 2016. p. 1-38.
- Cross JB, Thompson DC, Rai BK, Baber JC, Fan KY, Hu Y. Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J Chem Inf Model 2009; 49:1455-74. doi: 10.1021/ci900056c [Crossref] [ Google Scholar]
- Ivanova L, Karelson M. The Impact of Software Used and the Type of Target Protein on Molecular Docking Accuracy. Molecules 2022; 27:9041. doi: 10.3390/molecules27249041 [Crossref] [ Google Scholar]
- Xu M, Shen C, Yang J, Wang Q, Huang N. Systematic Investigation of Docking Failures in Large-Scale Structure-Based Virtual Screening. ACS Omega 2022; 7:39417-28. doi: 10.1021/acsomega.2c05826 [Crossref] [ Google Scholar]