Bioimpacts. 2025;15:30095.
doi: 10.34172/bi.30095
Original Article
A procedure for DNA methylation assessment in osteoporosis-related gene promoters of umbilical cord blood: A study on the Prospective Epidemiological Research Studies in Iran (PERSIAN) birth cohort
Sadegh Baradaran Mahdavi Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing, 1, 2, 3 
Seyed Morteza Javadirad Data curation, Formal analysis, Investigation, Methodology, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing, 4, *
Mahsa Rafieian Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – original draft, 2, 3
Parnian Poursafa Investigation, Methodology, 2
Vajihe Azimian Zavareh Investigation, Methodology, Resources, 3
Seyede Shahrbanoo Daniali Methodology, 2
Motahar Heidari-Beni Methodology, 2
Masoomeh Goodarzi-Khoigani Methodology, 2
Babak Vahdatpour Supervision, 1
Hossein Mirhendi Methodology, Resources, Supervision, 3, 5
Roya Kelishadi Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Visualization, 2, *
Author information:
1Department of Physical Medicine and Rehabilitation, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran
2Child Growth and Development Research Center, Research Institute for Primordial Prevention of Non-Communicable Disease, Isfahan University of Medical Sciences, Isfahan, Iran
3Core Research Facilities (CRF), Isfahan University of Medical Sciences, Isfahan, Iran
4Department of Cell and Molecular Biology, Faculty of Biological Science and Technology, University of Isfahan, Isfahan, Iran
5Department of Medical Parasitology and Mycology, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran
Abstract
Introduction:
It is believed that DNA methylation can modify disease susceptibility in response to environmental factors as early as the perinatal period. In this study, we aimed to present a streamlined DNA methylation analysis procedure for osteoporosis-related gene promoters in the umbilical cord blood.
Methods:
The Prospective Epidemiological Research Studies in Iran (PERSIAN) birth cohort was established in 2016. In this study, a total of 300 umbilical cord blood samples were collected at the time of delivery. For all samples, DNA was extracted and converted using sodium bisulfite. Multiple primer sets were designed for Wnt1, Wnt10b, β-catenin, OPG, and RANKL gene promoters in the online MethPrimer platform. Next, bisulfite sequencing PCR (BSP), as the gold standard method for exploring methylated and unmethylated cytosines, was performed in a gradient-controlled setting. The PCR products were then purified and directly sequenced. Subsequently, the chromatograms were interpreted.
Results:
For Wnt10b, β-catenin, and OPG genes, the converted DNA could be successfully amplified. The frequency of acceptable chromatograms for analysis was 195 for Wnt10b (195/300, 0.65%), 198 for β-catenin (198/300, 0.66%), and 50 for OPG (50/50, 100%).
Conclusion:
BSP can be efficiently used to investigate the methylation of target gene promoters in umbilical cord blood DNA.
Keywords: NA methylation, Epigenomics, Promoter regions, Birth cohort, Prenatal exposure delayed effects, Non-communicable diseases
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This study was registered in and funded by the Iranian National Institute for Medical Research Development (NIMAD) (Grant No. 4001752, awarded to the first and last authors in the clinician-scientist program).
Introduction
DNA methylation is assumed to be a determinant of non-communicable diseases (NCDs).1 The NCDs are recognized as a group of heterogeneous diseases in terms of pathophysiology, fatality rate, and non-fatal outcomes. Evidence suggests that the burden of NCDs is increasing globally, making them a major public health concern.2,3 According to the Developmental Origin of Health and Disease (DOHaD) concept, the life course approach predicts that environmental exposure in early life may modify the risk of chronic diseases later in life.4 Although several genes have been identified as potential contributors to the development of NCDs, it has been suggested that the combined effects of these genetic factors cannot significantly increase the population-level risk.5 Accordingly, the development of NCDs is believed to result from a complex interplay between genetic, environmental, and behavioral factors.6
Growing evidence suggests that epigenetic mechanisms play a key role in mediating the effects of early environmental exposure on human development later in life. DNA methylation, a well-studied epigenetic modification, involves the addition of a methyl group to cytosine residues at the carbon-5 (C-5) position of cytosine, most frequently within CpG dinucleotides.7 The CpG-rich islands are mainly concentrated in the 5' promoter region of genes. Therefore, DNA methylation is generally associated with gene repression and may contribute to the modification of disease risk in response to environmental factors.8
Today, a variety of technologies are available for analyzing DNA methylation patterns, including methods for the assessment of global DNA methylation, in situ DNA methylation, and site-specific measurements. Bisulfite methods, followed by polymerase chain reaction (PCR) amplification of bisulfite-modified DNA, are routinely employed to determine the methylation status of CpG dinucleotides, each providing some information on the methylation status. As a gold standard method, bisulfite sequencing PCR (BSP) can be used in site-specific approaches to directly identify methyl cytosines.9
The crucial role of Wnt signaling pathways in cell differentiation and proliferation during embryonic development is well-documented. The Wnt protein family consists of a large group of glycoproteins with the capacity to activate signaling cascades. Due to the diverse biological functions of the Wnt pathway, mutations or disruptions in this pathway have been associated with a range of diseases in humans and animals, including congenital malformations, cancer, type II diabetes, cardiovascular diseases, and bone-related disorders.10,11 Human and animal studies have shown that the Wnt/β-catenin pathway plays an important role in bone homeostasis and development.12 It is known that activation of Wnt/β-catenin signaling increases osteoblastogenesis and bone mass. It also increases the transcription of osteoprotegerin (OPG), an antagonist of the receptor activator of nuclear factor-B ligand (RANKL). RANKL, which is primarily produced by osteocytes, is a key regulator of osteoclast differentiation and bone resorption. Several Wnt proteins, including Wnt1 and Wnt10b, are associated with skeletal development.13 Besides, mutation in the Wnt1 gene has been attributed to severe early-onset osteoporosis.14
Emerging evidence suggests differences in DNA methylation in the bone and whole blood tissues of osteoporotic patients compared to healthy individuals.15 Considering the contribution of epigenetic changes to the development of osteoporosis,16 our perspective was to assess early DNA methylation modifications in bone-forming signaling pathways implicated in the development of osteoporosis. Therefore, our primary objective in this study was to describe the application of BSP in investigating the methylation status of DNA obtained from umbilical cord blood. For this purpose, we evaluated the promoters of genes involved in the Wnt/β-catenin pathway.
Methods
Study design and ethical aspects
This study was conducted on the nationwide Prospective Epidemiological Research Studies in Iran (PERSIAN) birth cohort in Isfahan. In 2016, the PERSIAN birth cohort was established to evaluate the effects of sociodemographic factors, diet, lifestyle, and environmental exposure on various maternal and neonatal health outcomes and physical health, as well as genetic and epigenetic factors. Detailed information related to the study design, study sample, and data collection method has been published previously.17 In brief, the participants of this study included Iranian pregnant women who had lived in Isfahan or its satellite cities for at least one year, had no history of infertility, and planned to give birth in the hospitals of Isfahan.
The PERSIAN birth cohort study and the current sub-study of this birth cohort were conducted in accordance with the Declaration of Helsinki. The study objectives and protocol were explained to the participants, and they were assured that their information would be kept confidential. All the participants provided informed consent for themselves and their children, both for the initial recruitment and the subsequent follow-up stages of the study. The informed consent covered various aspects of the study, including the completion of questionnaires, the collection of biological samples, and the handling of personal information. For the current PERSIAN birth cohort sub-study, the Ethics Committee of Isfahan University of Medical Sciences approved the study protocol before starting the recruitment process and epigenetic investigation on 03/12/2019 (ethical number: IR.MUI.RESEARCH.REC.1398.507). Fig. 1 presents an overview of the consecutive steps of DNA methylation analysis performed in this study, which are described in detail below.
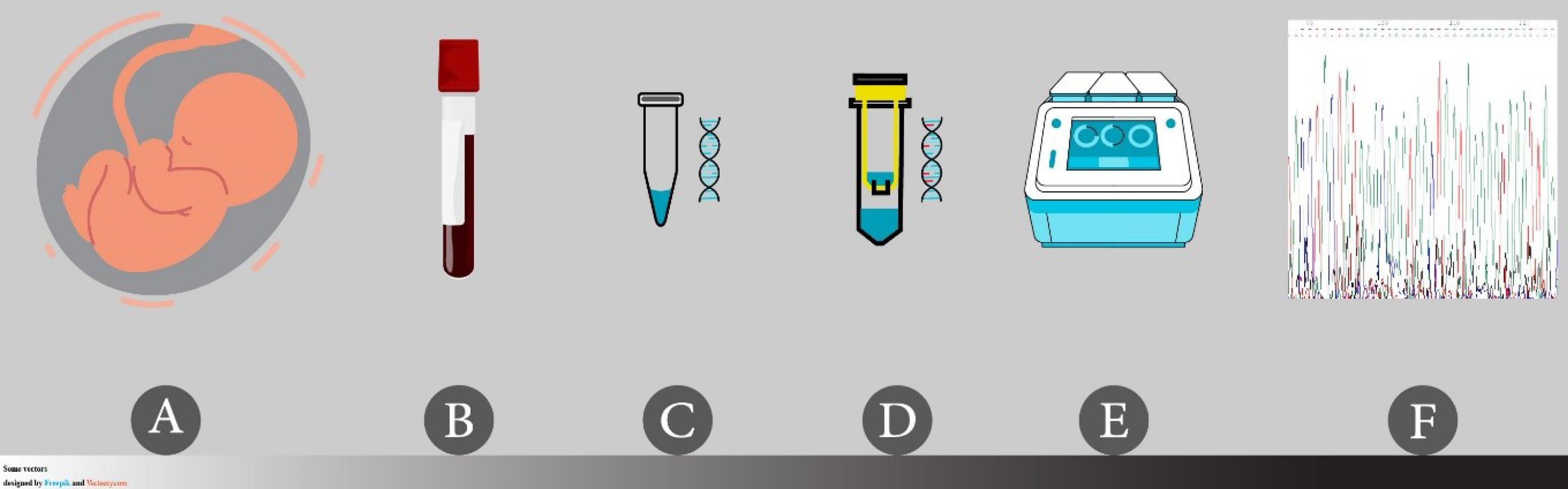
Fig. 1.
Steps of genomic DNA methylation analysis for gene promoters in the PERSIAN birth cohort study.(A) Umbilical cord blood collection at delivery, (B) processing and storage of blood samples, with and without anticoagulants, in a temperature-controlled environment (biobank), (C) DNA extraction from blood samples using a column-based nucleic acid extraction kit, (D) highly efficient sodium bisulfite conversion of DNA using a column-based bisulfite conversion kit, (E) amplification of templates using BSP method in a gradient-controlled setting specific for the designed primers, and (F) direct bisulfite sequencing using a DNA sequencer and software analysis.
.
Steps of genomic DNA methylation analysis for gene promoters in the PERSIAN birth cohort study.(A) Umbilical cord blood collection at delivery, (B) processing and storage of blood samples, with and without anticoagulants, in a temperature-controlled environment (biobank), (C) DNA extraction from blood samples using a column-based nucleic acid extraction kit, (D) highly efficient sodium bisulfite conversion of DNA using a column-based bisulfite conversion kit, (E) amplification of templates using BSP method in a gradient-controlled setting specific for the designed primers, and (F) direct bisulfite sequencing using a DNA sequencer and software analysis.
Umbilical cord blood collection and storage
Using proper operating room setups and trained staff, the entire umbilical cord blood was collected from 300 mothers at the time of delivery (Fig. 1A). Whole blood samples were collected in tubes, with or without anticoagulant EDTA, in volumes of 7 mL or 6 mL, respectively (Fig. 1B). Next, the tubes containing the anticoagulant were inverted three to four times, and the tubes without the anticoagulant were left at room temperature for 30 to 40 minutes. The tubes were then centrifuged for 10-15 minutes at 3000 g to separate aliquots of serum, plasma, and cells within 24 hours. For long-term sample storage, system-compatible freezer boxes, which are cold- and humidity-resistant, were used. Cryotubes labeled with barcodes were immediately stored at -80 °C in a freezer. Detailed records of each sample’s characteristics were stored in the biobank system at the School of Health, Isfahan University of Medical Sciences. Before use for DNA extraction, the remaining frozen blood, without the anticoagulant, was allowed to thaw at room temperature.
DNA extraction and purification
DNA extraction was carried out following the manufacturer’s protocol, using a column-based DNA extraction kit (ExgeneTM Blood SV Mini, Cat. No.: 105-101, Korea) (Fig. 1C). The chemicals and tools used for DNA extraction and the subsequent processes included absolute ethanol, pipets, pipet tips, microcentrifuge tubes (1.5 mL), collection tubes, racks, an incubator, a spinner, a vortex mixer, and a microcentrifuge. Proteinase K, buffer BL, buffer BW, buffer TW, and buffer AE were also provided by the kit; the manufacturer’s website has detailed information on the DNA extraction procedure.18 In this step, the entire process consisted of cell lysis, DNA binding to a matrix, and washing.
In brief, DNA extraction began by preparing 200 µL of blood, followed by lysis with 20 mg/mL of proteinase K. The samples were then washed with buffer BW and buffer TW, and DNA was eluted using 20 µL of buffer. This volume of buffer AE was added to ensure the recovery of higher DNA concentrations. The eluate was then stored in new 1.5-ml microcentrifuge tubes. Parafilm was used to create a physical barrier for the microtubes to reduce evaporation and prevent contamination of the solution. As an alternative, screw cap microcentrifuge tubes (1.5 mL) could be used for enhanced protection. In the following steps, DNA solutions were kept at -20 °C.19 Additionally, a NanoDrop 1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) was used to measure DNA concentrations. The absorbance ratios were read at 260 nm and 280 nm to assess the purity/quality of DNA.
Sodium bisulfite conversion of DNA
The complete conversion (deamination) of unmethylated cytosine to uracil is necessary for a successful bisulfite DNA analysis and requires the incubation of sample DNA in bisulfite salt at high temperatures and low pH levels.20 However, it may result in DNA fragmentation, amplification of short amplicons, and the introduction of experimental artifacts.21 On the other hand, less aggressive treatment methods can lead to an overestimation of methylation status due to the identification of non-converted cytosine.22 The Qiagen EpiTect Bisulfite Kit (48) (Cat. No.: 59104, Germany) was utilized for DNA conversion (Fig. 1D).23
The required equipment and reagents for bisulfite conversion included absolute ethanol, pipets, pipet tips, PCR tubes, a thermal cycler, microcentrifuge tubes (1.5 ml), racks, a vortex mixer, a microcentrifuge, and a heating block (optional). Also, the kit provided Bisulfite Mix, DNA Protect Buffer, RNase-free water, spin columns, buffer BL, buffer BW, buffer BD, buffer EB, and carrier RNA. By using this kit, small amounts of DNA could be converted (1 ng to 2 μg in a volume of up to 20 μL) during this process. The manufacturer claims that in accordance with the bisulfite treatment protocol, the conversion rate of cytosine is greater than 99%, which is more than adequate.21
Two main steps were used to convert unmethylated cytosine to uracil: 1) incubation of DNA with sodium bisulfite, and 2) desulfonation and cleanup of converted DNA. In step 1, bisulfite reactions were prepared and placed in the thermal cycler for bisulfite conversion. Bisulfite reaction components consisted of DNA solution, RNase-free water, Bisulfite Mix, and DNA Protect Buffer. The addition of DNA Protect Buffer from this kit to bisulfite reactions in PCR tubes helped prevent DNA fragmentation and ensured efficient DNA denaturation, which is necessary for complete conversion. When preparing the bisulfite reactions, the total volume of DNA solution combined with RNase-free water should be 20 µL. Accordingly, we added 15 µL of RNase-free water to 5 µL of DNA solution (with an average concentration of 100 ng/µL). It should be noted that depending on the concentration of starting DNA, alternative preparations may be used.
In step 2, the converted DNA was bound to spin columns and then washed to remove any impurities. Next, it was eluted from the column membrane of each sample by adding 20 µL of buffer EB at the end of the cleanup procedure. Step 1 could be completed within a single working day, while step 2 could be completed on the following day for up to 48 reactions, as provided by the kit. The purified converted DNA was kept at -20 °C. According to the kit instructions, the templates may be kept at -20 °C for at least three years, without any significant changes in the quality of DNA. After conversion, when evaluating DNA quality using a NanoDrop spectrophotometer, the template should not be considered double-stranded genomic DNA. Instead, it is comparable to RNA.24
Amplification of templates using the BSP method
The studied genes in this study were as follows: Wnt family member 1 (WNT1, Ensembl: ENSG00000125084), Wnt family member 10B (WNT10B, Ensembl: ENSG00000169884), Catenin beta 1 (CTNNB1 or β-catenin, Ensembl: ENSG00000168036), TNF receptor superfamily member 11b (TNFRSF11B or OPG, Ensembl: ENSG00000164761), and TNF superfamily member 11 (TNFSF11 or RANKL, Ensembl: ENSG00000120659). The gene sequences and detailed information about their promoter regions were obtained from the UCSC Genome Browser database (https://genome.ucsc.edu/index.html), and the CpG sites within the promoter region were detected.
The bisulfite sequencing primer sets were designed using the online MethPrimer platform (https://www.urogene.org/methprimer/)25 with the following parameters: primer size of 20 bp to 30 bp, product size of 100 bp to 300 bp, and primer melting temperature (Tm) of 50 °C to 60°C. The minimum number of CpGs in products was set at four, and the primers were selected around the predicted CpG sites. Generally, it is crucial to design a primer for BSP to achieve artifact-free downstream sequencing; this step is more difficult in BSP than in conventional PCR. The CpG dinucleotides are advised not to be included in the primer, and if necessary, they should only be used at the 5' end. It is also recommended that the amplified sequence be 150 bp to 500 bp in length21,26 due to DNA fragmentation mentioned earlier. Primer stocks were prepared and dissolved in water and then stored at a temperature of -20 °C.
The PCR step in this study required the following materials: PCR-grade water, converted template DNA, forward and reverse primer sets, Ampliqon Taq DNA Polymerase 2X Master Mix RED (Cat. No.: A180301, Denmark), and a thermal cycler (preferably with a gradient setting). Generally, adherence to the laboratory sterilization protocols is strongly advised when preparing PCR reactions to prevent undesirably contaminated converted DNA. To produce clear and distinct bands, the total PCR reaction volume was set at 12.5 µL, and a PCR annealing gradient was used to determine the ideal temperature for each set of primers (Fig. 1E).
A volume of PCR products (2-3 µL) was subjected to 1.5% agarose gel electrophoresis (voltage of 100 V for 20-30 minutes). The gel solution was mixed with 1 µL of YTA safe stain (Cat. No.: YT0001) to detect nucleic acids in agarose gel. No additional loading dye was added to the PCR product because the inert red dye of Master Mix RED sufficed.
Direct bisulfite sequencing
Once the PCR products appeared as strong bands of expected size on the electrophoresis gel, they would be purified. For the subsequent stages of PCR, such as sequencing or cloning, a quick and straightforward purification of nucleic acids was necessary to remove impurities, such as enzymes, salts, nucleotides, primer dimers, and small DNA fragments. Generally, it is much more effective to use direct sequencing.27 Direct purification of PCR products, extraction of DNA bands from agarose gel, and manual protocols available in molecular laboratories are some of the techniques used to remove unused products from template DNA. We recommend using a kit or procedure similar to ours for low-volume PCR products (about 10 μL out of a total of 12.5 μL), with amplicon sizes ranging from 150 bp to 500 bp. The Applied Biosystems ExoSAP-ITTM PCR Product Clean-up Reagent28 was used in this study, which allowed us to process at least 5 μL of the PCR product.29
Sequencing was performed using an ABI 3730 DNA Analyzer, and Chromas Version 2.5 was used to display and interpret the chromatogram data (.abl) (Fig. 1F). In this step, we used the same forward and reverse primers as those used in the PCR step for each gene at a concentration of 5 μM. The information of each chromatogram sample included the number of individual peaks corresponding to each nucleotide base. Except for the precise location of unmethylated cytosines, which are converted to uracil during bisulfite treatment and then to thymine in the subsequent PCR reaction, all nucleotide sequences were similar to those of the reference gene.
The degree of methylation could be calculated at CpG sites in a specific sample. At specific cytosine sites, there might be two independent peaks corresponding to cytosine or thymine. Unmethylated cytosine was converted to uracil during bisulfite treatment, as previously mentioned, while methylated cytosine did not change. The cytosine signals were then correlated with the methylation level at each CpG site. The level of methylation at each CpG site was measured based on the cytosine and thymine peak heights. Fundamental equations, including (C/(C + T)*100) or (G/(G + A)*100), were computed to determine the degree of methylation when forward or reverse primers were utilized for DNA sequencing, respectively. For the analysis of the methylation level for each gene in each sample, the part of the sequence data with the least amount of noise was selected. The number of cytosines that remained unchanged, as well as converted cytosines, was then counted after they were compared to the complementary reference sequence obtained from the National Center for Biotechnology Information (NCBI).
Results
We collected the umbilical cord blood samples of 300 mothers for the DNA methylation analysis. DNA was extracted from all the samples. An average DNA concentration of 100 ng/μL was measured based on the spectrophotometer analyses. The absorbance ratios were within the range of 1.8-2.2. There was no significant difference in the concentration of DNA extracted from blood samples with EDTA compared to those without an anticoagulant. Therefore, in the next steps, we used blood samples without the anticoagulant. All of the samples were then treated with sodium bisulfite.
Multiple sets of primers were designed for our candidate genes (five for each of Wnt1, Wnt10b, β-catenin, OPG, and RANKL genes), using the online MethPrimer software. A number of these primers were synthesized (five for Wnt1, two for Wnt10b, four for β-catenin, two for OPG, and two for RANKL genes) and tested (five for Wnt1, two for Wnt10b, two for β-catenin, one for OPG, and two for RANKL genes) for the PCR setup of each gene. Table 1 indicates the reaction elements, concentrations, and reaction circumstances in our research.
Table 1.
Modified PCR characteristics for bisulfite sequencing of DNA extracted from umbilical cord blood samples
|
Component |
Volume/Reaction
|
Final Concentration
|
| Taq DNA Polymerase 2x Master Mix RED |
6.25 μL |
1x |
| Primer Forward (10 μM) |
0.25-0.5 μL * |
0.2-0.4 μM |
| Primer Reverse (10 μM) |
0.25-0.5 μL * |
0.2-0.4 μM |
| H2O |
X μL |
- |
| Template DNA |
1-1.5 μL |
100-150 ng |
| Total Volume |
12.5 μL |
- |
|
Cycles
|
Time
|
Temperature
|
| 1 |
2-5 minutes |
95 °C |
| 40 |
20-30 seconds |
95 °C |
| 1 minute |
50-65 °C (determined with gradient setting for each primer set) |
| 30 seconds |
72 °C |
| 1 |
15 minutes |
72 °C |
* In the case of primer dimer formation, the volume may be set at 0.25 μL.
For Wnt10b, β-catenin, and OPG genes, we could successfully amplify the converted DNA. Fig. 2 presents the MethPrimer results for these genes. The MethPrimer outputs are provided in Supplementary files 1-3. Additionally, information on the primers used for amplification in this step is presented in Table 2.
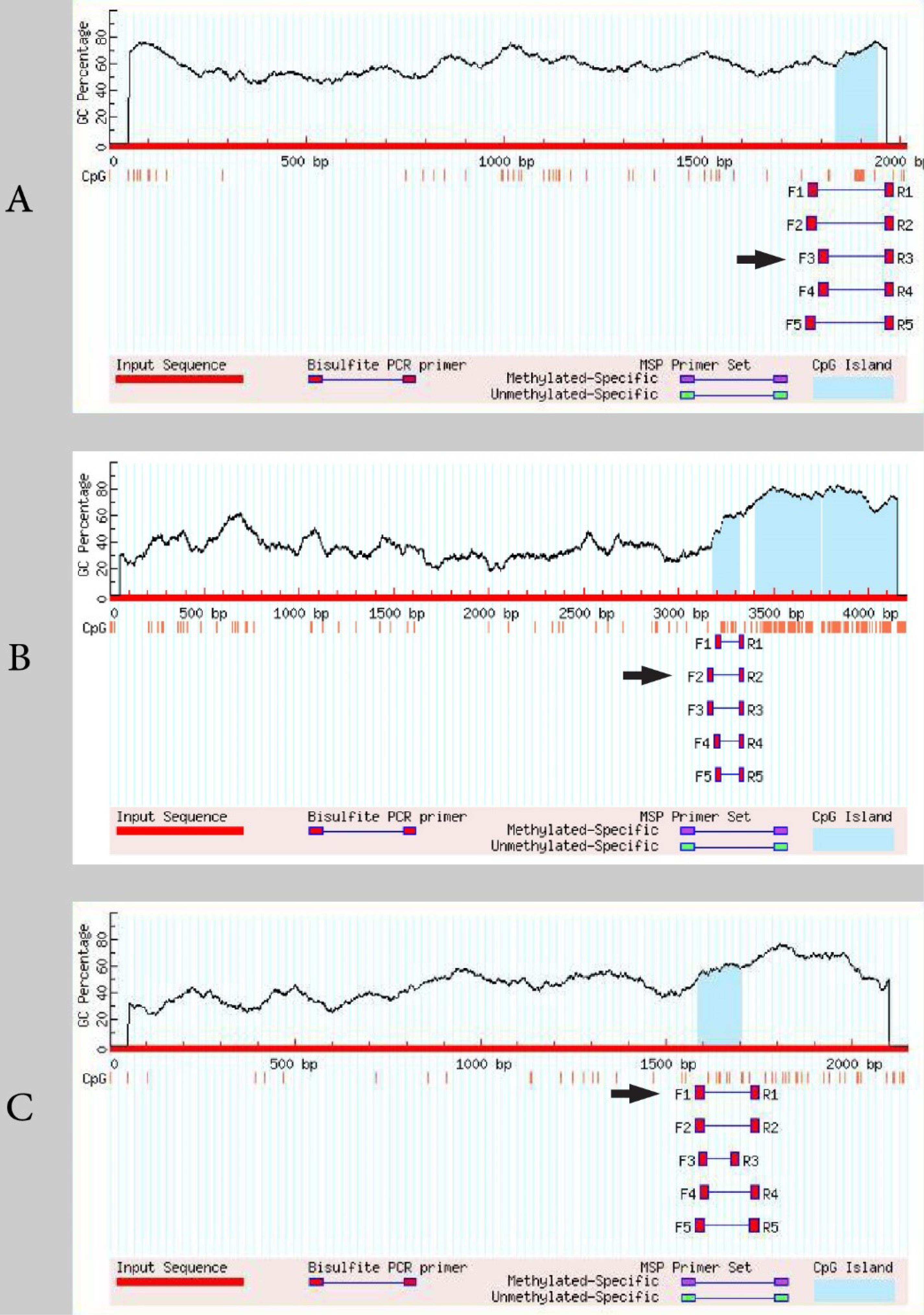
Fig. 2.
The MethPrimer results for Wnt10b (A), β-catenin (B), and OPG (C) genes. The arrow shows the primer replicating the sequence favorably (see Table 2).
.
The MethPrimer results for Wnt10b (A), β-catenin (B), and OPG (C) genes. The arrow shows the primer replicating the sequence favorably (see Table 2).
Table 2.
List of primers used for bisulfite sequencing PCR
Gene symbol
(Ensembl accession number) |
Forward and Reverse primers (5' > 3')1
|
Product size (bp)
|
Annealing temperatures tested (°C)
|
Best annealing temperature (°C)
|
Number of CpGs in PCR product2
|
Chromosome
|
UCSC Genome Browser start-stop positions
|
WNT10B
(ENSG00000169884) |
F: TAGTGGGGTTAAGTTTGAATTTAGG |
190 |
59, 60.5, 61.4, 62.4, 63.3, 64.5
62.5 |
62.5 |
15 |
12 |
48971768-48971957 |
| R: ACCTAAAACTTCCCCACCCTAC |
CTNNB1
(ENSG00000168036) |
F: GTTTTGGAGTTAATTTATTTTTATTAGTTA |
189 |
51.5, 52.6, 54 |
52.6 |
9 |
3 |
41198603-41198791 |
| R: TAAATCCCACCTCTCAACAAACTAT |
TNFRSF11B
(ENSG00000164761) |
F: TATAAAGAGGGGTTTTGTAATTTGAG |
173 |
57.3, 58.6, 60.1, 61.4, 62.4, 63.3
61.2 |
61.2 |
9 |
8 |
118952285-118952457 |
| R: TTATATCTCCTCCACCCTAAAAATC |
1: F: Forward primer, R: Reverse primer.
2: Obtained from MethPrimer results.
Meanwhile, the results of gel electrophoresis revealed that the PCR products for Wnt1 and RANKL genes were unfavorable and unreproducible using the synthesized primers (the primer information and gel results are not provided). Fig. 3 shows the gel electrophoresis results for Wnt10b, β-catenin, and OPG genes. Our experience shows that a specific primer set may only amplify the desired sequence at a specific annealing temperature, which is consistent with other research findings.24 It is important to note that if the starting template is limited, the reaction conditions can be improved by either increasing the extension time or performing the PCR reaction twice.21,30 Additionally, the volume of forward and reverse primers may be decreased in the presence of primer dimers. In our study, optimization of PCR conditions had minimal effects on the results, with no bands or smeared bands. Under this condition, a different set of primers might be used.
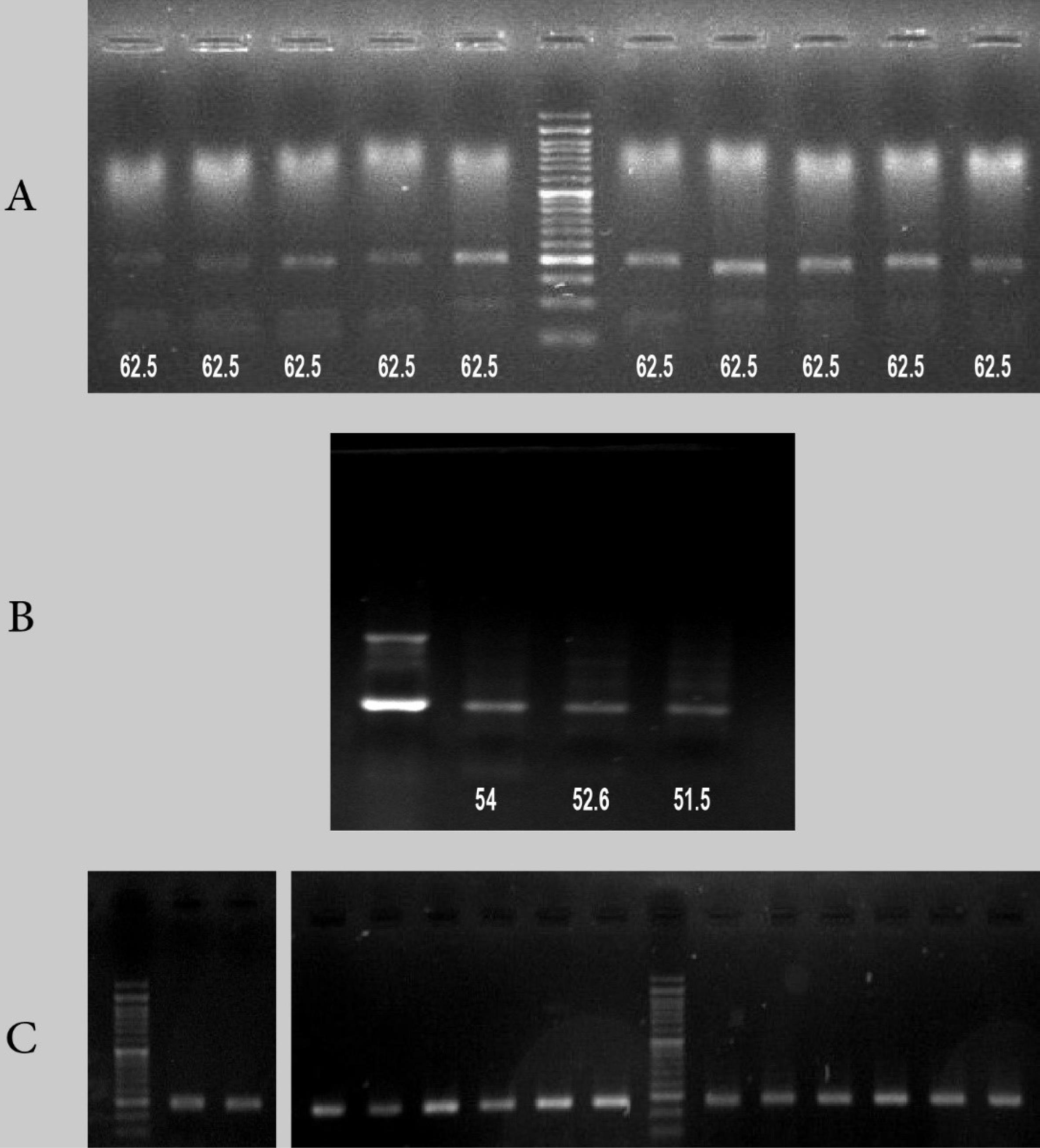
Fig. 3.
A) The gel electrophoresis results of Wnt10b gene in 10 different samples at a fixed annealing temperature. B) The gel electrophoresis results of β-catenin for one sample using an annealing gradient. C) Right: the gel electrophoresis results of OPG gene for two samples using an annealing gradient. Left: the gel electrophoresis results of OPG gene for two samples at 61.2 °C.
.
A) The gel electrophoresis results of Wnt10b gene in 10 different samples at a fixed annealing temperature. B) The gel electrophoresis results of β-catenin for one sample using an annealing gradient. C) Right: the gel electrophoresis results of OPG gene for two samples using an annealing gradient. Left: the gel electrophoresis results of OPG gene for two samples at 61.2 °C.
After purification of the PCR products, 300 templates were sequenced for Wnt10b, 300 templates for β-catenin, and 50 templates for OPG genes. Based on the DNA sequencing results, cleaner chromatograms were obtained using the reverse primer.The number (frequency) of acceptable chromatograms for analysis was 195 for Wnt10b (195/300, 0.65%), 198 for β-catenin (198/300, 0.66%), and 50 for OPG (50/50, 100%). For Wnt10b, β-catenin, and OPG, a total of 13, 5, and 3 CpG sites were considered within the gene promoters for the DNA methylation analysis, respectively. Fig. 4 shows the CpG sites in gene promoters as displayed in the chromatogram outputs.
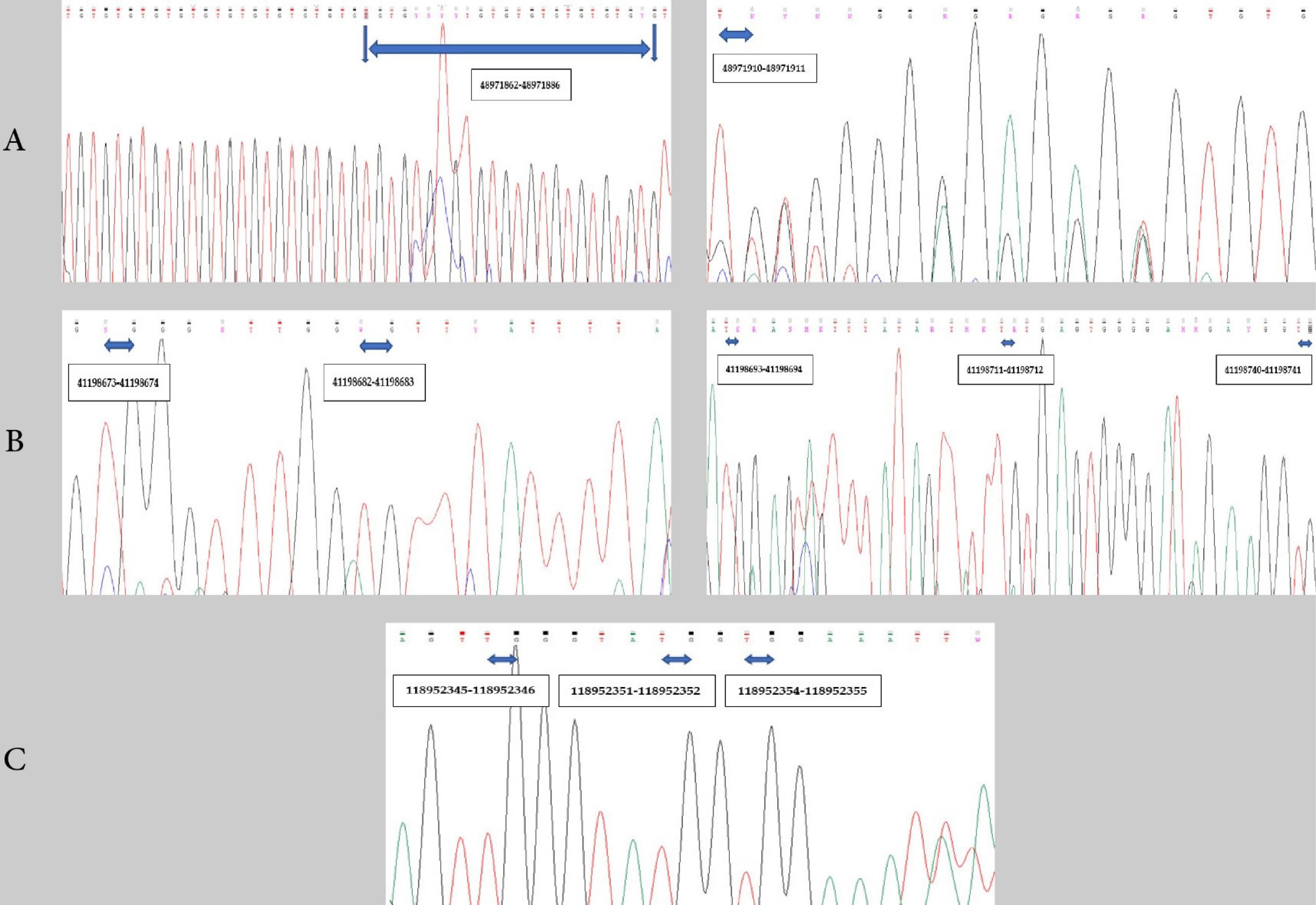
Fig. 4.
The Chromas view of a part of DNA sequence after direct bisulfite sequencing using the reverse primer, indicating 13 CpG sites for Wnt10b, five CpG sites for β-catenin, and three CpG sites for OPG. The numbers in black boxes represent the UCSC Genome Browser positions of CpG sites: A) Wnt10b, B) β-catenin, and C) OPG.
.
The Chromas view of a part of DNA sequence after direct bisulfite sequencing using the reverse primer, indicating 13 CpG sites for Wnt10b, five CpG sites for β-catenin, and three CpG sites for OPG. The numbers in black boxes represent the UCSC Genome Browser positions of CpG sites: A) Wnt10b, B) β-catenin, and C) OPG.
Discussion
In this study, we described a basic method for DNA methylation analysis of candidate gene promoters in umbilical cord blood samples. We initially conducted a DNA methylation analysis for some candidate genes involved in the pathogenesis of osteoporosis. Due to the determinant role of epigenetic modifications in the development of age-related diseases, recent research has focused on bone homeostasis and osteoporosis.
In an epigenome-wide association study by Cheishvili et al, the level of whole-genome methylation in blood samples from 22 women with osteoporosis was compared to that of 22 healthy women in a control group, using the Illumina Infinium Human Methylation 450K analysis. In this study, different CpG sites were found in osteoporotic patients, which were methylated in different ways compared to the healthy group. The authors were finally able to introduce five genes involved in bone biology and suggested that whole blood methylation analysis could be used to identify individuals at risk of developing osteoporosis at an earlier stage.31 Additionally, the role of DNA methylation in regulating the differentiation and function of bone cells has been reported in the literature. In osteoblasts, the methylation process regulates the expression of a large number of genes involved in cell function. These genes include alkaline phosphatase, sclerostin, osterix, distal-less homeobox 5, estrogen receptor 1, osteopontin, RANKL, OPG, and leptin.32
The contribution of canonical Wnt (β-catenin dependent) and RANKL/OPG/RANK pathways to bone homeostasis has been extensively described in humans.33 In the present study, using BSP, we were able to directly determine the DNA methylation pattern and degree at the target CpG sites within the promoters of Wnt10b, β-catenin, and OPG genes. Moreover, Wang et al. studied the DNA methylation status of RANKL and OPG promoters in bone tissues obtained from a group of osteoporotic fracture patients (OPF) and a group of non-osteoporotic fracture patients (non-OPF) using BSP. After running on agarose gel, the product bands were dissolved, purified, and then subjected to ligation and cloning using a vector cloning kit. In their research, the region of analysis contained 32 CpG sites in the RANKL promoter and nine CpG sites in the OPG promoter region. For RANKL, the degree of methylation was 96.77% and 9.69% in the non-OPF and OPF groups, respectively. In contrast, for OPG, the degree of methylation was 3.7% and 7.41% in the non-OPF and OPF groups, respectively.34 Similarly, Kalkan and Becer studied the whole blood samples of a group of obese and non-obese individuals to determine the association between the methylation status of RANKL gene and obesity, using the methylation-sensitive high resolution melting (MS-HRM) analysis, according to the EpiTect® HRMTM PCR Handbook. They found a significant difference between the obese and control groups concerning the methylation status of RANKL gene. Based on their findings, 20% and 80% of RANKL gene methylation were detected in obese and non-obese individuals, respectively.35
The procedure described in our study may be reproducible for other targets or genes of importance after designing the corresponding primers. In this regard, Parrish et al. previously used this method to investigate the methylation patterns of DNA isolated from brain tissues.29 Besides, this method might be applied to peripheral blood samples rather than umbilical cord blood samples. The degree of methylation measured by this method, along with the perinatal data, could be analyzed as a variable for each participant, using appropriate statistical models for the study sample.
In the PERSIAN birth cohort, it is planned to follow-up the mother-offspring pairs for at least 10 years. Ongoing research suggests that maternal lifestyle and environmental factors can impact epigenetic modifications at the time of delivery, potentially affecting long-term health outcomes. In a previous study on 115 cord blood samples using pyrosequencing, it was found that prenatal exposure to drinking water disinfection by-products is associated with decreased Alu methylation and decreased LINE-1 methylation.36 In another study, childhood body composition was linked to epigenetic modifications of multiple genes in the umbilical cord tissue.37 Moreover, there were associations between the methylation status of specific CpG sites within the endothelial nitric oxide synthase (eNOS) promoter region at birth and the child’s bone mineral content at nine years, suggesting the potential role of eNOS in bone metabolism.38 In a previous investigation on a number of mother-offspring pairs, methylation at a specific site in retinoid X receptor alpha (RXRA) gene was inversely associated with the maternal vitamin D index. Also, a higher site-specific methylation level in the RXRA promoter region was associated with a lower bone mineral content in children at the age of four years.39 However, the results of a prospective cohort study on Finnish children until 15 years of age revealed no significant difference in the umbilical cord DNA methylome of children who progressed to type 1 diabetes and the control group.40 Since there are various sources of heterogeneity between studies, such as the sample size or DNA methylation analysis method, further research is required to clarify the role of epigenetic changes in the development of NCDs.
Some PCR-based assays have been proposed to assess the effectiveness of bisulfite conversion, the degree of DNA degradation, and recovery due to DNA degradation.22,41 Additionally, the conversion efficacy of Qiagen EpiTect Bisulfite Kit was 98.7% in a previous study, evaluating the performance of nine commercially available bisulfite conversion kits, including the Qiagen EpiTect Bisulfite Kit and the Qiagen EpiTect Fast DNA Bisulfite Kit. Compared to kits with a rapid bisulfite reaction time, such as the Qiagen EpiTect Fast DNA Bisulfite Kit, kits with a longer bisulfite reaction time (e.g., Qiagen EpiTect Bisulfite Kit) have shown higher fragmentation rates.42
Direct sequencing, methylation-specific PCR, methylation-sensitive single-nucleotide primer extension (MS-SnuPE), and combined bisulfite restriction analysis (COBRA) are some bisulfite-based methods that have been used to study DNA methylation patterns.9 Nevertheless, bisulfite-based DNA methylation methods have shown higher quantitative accuracy and sensitivity compared to restriction enzyme methods. Therefore, in multiple studies, bisulfite sequencing has been introduced as the gold standard method due to its ability to accurately distinguish 5-methylcytosine from cytosine.20,30,43,44
After bisulfite conversion of DNA and PCR assay, it is necessary to purify the product by using commercial PCR purification kits or by isolating specific bands from electrophoresis gel. Subsequently, the final product may be sequenced directly. Alternatively, sub-cloning of the PCR product may be performed before clone sequencing. Compared to plasmid cloning, direct sequencing may save some time and cost for researchers. However, direct sequencing may be associated with the loss of sequence length due to primer attachment. It should be noted that previous research has reported failure rates for both methods.45 Therefore, the selection of the most efficient and cost-effective approach to investigate the methylation pattern depends on the study question and design, as well as the information provided by each method. In this study, we used a site-specific assessment method, facilitating a rapid and direct investigation of the methylation status. Moreover, Fraga and Estellar previously reviewed the methods and applications of DNA methylation analysis.9
In the current study, we described the challenges of BSP application in a prospective cohort to evaluate the epigenetic modifications at birth. This investigation could provide valuable information on the origins of chronic diseases as early as the perinatal period. Generally, epigenetic modifications are tissue-specific. Despite the maintenance of exposure memory by epigenetic marks, the temporality of modifications (i.e., Chrono dimension) related to aging or the development of human diseases may explain the complex relationships between the environment, genetics, and epigenetic marks in the life course.46
Limitations
According to the present findings, even after long-term storage at -80°C, cord blood samples can provide enough DNA for epigenetic analysis. No other fresh blood samples were examined using our extraction kit to investigate differences in DNA concentrations between fresh blood and stored cord blood samples. Also, we did not use a second extraction kit to validate the cord blood DNA concentrations, and further studies are needed to address these issues. Moreover, total blood samples, with and without EDTA anticoagulant, provided us with approximately equal DNA concentrations for the analysis of DNA methylation. However, we continued our study with blood samples without the anticoagulant and did not compare them in the conversion or PCR steps. Additionally, we could not perform neonatal bone marrow aspiration due to ethical issues, and cord blood samples were used to detect early-onset DNA methylation changes. Finally, our research found that some genes had high failure rates during sequencing, possibly due to DNA fragmentation after conversion, the used primers, or sequencing artifacts. Improvements could be made by using different primer pairs, optimizing PCR and sequencing conditions, and sub-cloning PCR products.
Conclusion
BSP, as a common method of DNA methylation analysis, can be streamlined for umbilical cord blood samples in the context of large birth cohort studies. This method can provide valuable information about potential epigenetic modifications involved in the pathogenesis of chronic diseases in response to environmental factors.
Research Highlights
What is the current knowledge?
-
A complex interaction between genetics and epigenetic marks (including DNA methylation) exists in response to environmental factors in developing chronic diseases.
-
Bisulfite sequencing genomic PCR (BSP), is a gold standard method to explore methylated and unmethylated cytosines in CpG islands.
What is new here?
-
We presented a streamlined DNA methylation analysis procedure for osteoporosis-related gene promoters in the umbilical cord blood.
-
Perinatal exposure to environmental factors may contribute to DNA methylation modifications at birth and possibly leads to long-term health outcomes.
Competing Interests
None to declare.
Ethical Statement
The PERSIAN birth cohort study and the current sub-study of the PERSIAN birth cohort have been conducted in accordance with the Declaration of Helsinki. For the current PERSIAN Birth Cohort sub-study, the ethics committee of Isfahan University of Medical Sciences has approved the protocol before starting the recruitment process and epigenetic investigation on 03/12/2019 (ethical number: IR.MUI.RESEARCH.REC.1398.507). The study objectives and protocol were explained to participants, and they were reassured that their information would be kept confidential. All of them signed the consent forms for the recruitment step and the follow-up stages for themselves and their children. The informed consent consisted of various points related to questionnaires, biological samples, and personal information.
Supplementary files
Supplementary file 1. Primer picking results for bisulfite sequencing PCR (MethPrimer-WNT10B).
(pdf)
Supplementary file 2. Primer picking results for bisulfite sequencing PCR (MethPrimer- CTNNB1).
(pdf)
Supplementary file 3. Primer picking results for bisulfite sequencing PCR (MethPrimer- TNFRSF11B).
(pdf)
Acknowledgements
We want to thank the large team at the Research Institute for Primordial Prevention of Non-communicable Disease, Isfahan University of Medical Sciences, Isfahan, Iran, and Core Research Facilities (CRF), Isfahan University of Medical Sciences, Isfahan, Iran, especially Dr. Shima Gharibi, Mrs. Maryam Karimian, Ms. Nasim Rafiei and Mr. Mohammad Reza Pourreza for their mutual contributions.
References
- Sharp GC, Relton CL. Epigenetics and noncommunicable diseases. Epigenomics 2017; 9:789-91. doi: 10.2217/epi-2017-0045 [Crossref] [ Google Scholar]
- Charalampous P, Gorasso V, Plass D, Pires SM, von der Lippe E, Mereke A. Burden of non-communicable disease studies in Europe: a systematic review of data sources and methodological choices. Eur J Public Health 2022; 32:289-96. doi: 10.1093/eurpub/ckab218 [Crossref] [ Google Scholar]
- Gong JB, Yu XW, Yi XR, Wang CH, Tuo XP. Epidemiology of chronic noncommunicable diseases and evaluation of life quality in elderly. Aging Med (Milton) 2018; 1:64-6. doi: 10.1002/agm2.12009 [Crossref] [ Google Scholar]
- Baird J, Jacob C, Barker M, Fall CH, Hanson M, Harvey NC. Developmental origins of health and disease: a lifecourse approach to the prevention of non-communicable diseases. Healthcare (Basel) 2017; 5:14. doi: 10.3390/healthcare5010014 [Crossref] [ Google Scholar]
- Morris AP, Voight BF, Teslovich TM, Ferreira T, Segrè AV, Steinthorsdottir V. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012; 44:981-90. doi: 10.1038/ng.2383 [Crossref] [ Google Scholar]
- Hunter DJ, Reddy KS. Noncommunicable diseases. N Engl J Med 2013; 369:1336-43. doi: 10.1056/NEJMra1109345 [Crossref] [ Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6-21. doi: 10.1101/gad.947102 [Crossref] [ Google Scholar]
- Yamada L, Chong S. Epigenetic studies in developmental origins of health and disease: pitfalls and key considerations for study design and interpretation. J Dev Orig Health Dis 2017; 8:30-43. doi: 10.1017/s2040174416000507 [Crossref] [ Google Scholar]
- Fraga MF, Esteller M. DNA methylation: a profile of methods and applications. Biotechniques 2002; 33:632, 4, 6-49. doi: 10.2144/02333rv01 [Crossref] [ Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17:9-26. doi: 10.1016/j.devcel.2009.06.016 [Crossref] [ Google Scholar]
- Luo J, Chen J, Deng ZL, Luo X, Song WX, Sharff KA. Wnt signaling and human diseases: what are the therapeutic implications?. Lab Invest 2007; 87:97-103. doi: 10.1038/labinvest.3700509 [Crossref] [ Google Scholar]
- Regard JB, Zhong Z, Williams BO, Yang Y. Wnt signaling in bone development and disease: making stronger bone with Wnts. Cold Spring HarbPerspect Biol 2012; 4:a007997. doi: 10.1101/cshperspect.a007997 [Crossref] [ Google Scholar]
- Manolagas SC. Wnt signaling and osteoporosis. Maturitas 2014; 78:233-7. doi: 10.1016/j.maturitas.2014.04.013 [Crossref] [ Google Scholar]
- Mäkitie RE, Haanpää M, Valta H, Pekkinen M, Laine CM, Lehesjoki AE. Skeletal characteristics of WNT1 osteoporosis in children and young adults. J Bone Miner Res 2016; 31:1734-42. doi: 10.1002/jbmr.2841 [Crossref] [ Google Scholar]
- Visconti VV, Cariati I, Fittipaldi S, Iundusi R, Gasbarra E, Tarantino U. DNA methylation signatures of bone metabolism in osteoporosis and osteoarthritis aging-related diseases: an updated review. Int J Mol Sci 2021; 22:4244. doi: 10.3390/ijms22084244 [Crossref] [ Google Scholar]
- Holroyd C, Harvey N, Dennison E, Cooper C. Epigenetic influences in the developmental origins of osteoporosis. Osteoporos Int 2012; 23:401-10. doi: 10.1007/s00198-011-1671-5 [Crossref] [ Google Scholar]
- Zare Sakhvidi MJ, Danaei N, Dadvand P, Mehrparvar AH, Heidari-Beni M, Nouripour S. The prospective epidemiological research studies in IrAN (PERSIAN) birth cohort protocol: rationale, design and methodology. Longitudinal and Life Course Studies 2021; 12:241-62. doi: 10.1332/175795920x16062247639874 [Crossref] [ Google Scholar]
- GeneAll Bldg. ExgeneTM Blood SV. Korea: GeneAll Biotechnology Co Ltd; 2022. Available from: https://geneall.com/en/sub/products/prod.asp?mode=view&idx=325&s_keyword=&s_cate=20&s_filter=&s_browser=&s_display=&s_align=0. Accessed December 13, 2022.
- Lee SB, Crouse CA, Kline MC. Optimizing storage and handling of DNA extracts. Forensic Sci Rev 2010; 22:131-44. [ Google Scholar]
- Li Y, Tollefsbol TO. DNA methylation detection: bisulfite genomic sequencing analysis. Methods Mol Biol 2011; 791:11-21. doi: 10.1007/978-1-61779-316-5_2 [Crossref] [ Google Scholar]
- Warnecke PM, Stirzaker C, Song J, Grunau C, Melki JR, Clark SJ. Identification and resolution of artifacts in bisulfite sequencing. Methods 2002; 27:101-7. doi: 10.1016/s1046-2023(02)00060-9 [Crossref] [ Google Scholar]
- Ehrich M, Zoll S, Sur S, van den Boom D. A new method for accurate assessment of DNA quality after bisulfite treatment. Nucleic Acids Res 2007; 35:e29. doi: 10.1093/nar/gkl1134 [Crossref] [ Google Scholar]
- QIAGEN. EpiTect Bisulfite Kits. For Complete Bisulfite Conversion and Cleanup of DNA for Methylation Analysis. QIAGEN; 2022. Available from: https://www.qiagen.com/us/products/discovery-and-translational-research/epigenetics/dna-methylation/bisulfite-conversion-assays/epitect-bisulfite-kits. Accessed December 13, 2022.
- Zymo Research. Learn More About Bisulfite Conversion. Zymo Research Corporation; 2022. Available from: https://www.zymoresearch.com/pages/bisulfite-beginner-guide. Accessed December 13, 2022.
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002; 18:1427-31. doi: 10.1093/bioinformatics/18.11.1427 [Crossref] [ Google Scholar]
- New England Biolabs. FAQ: What Are the General Recommendations for Designing Primers for Bisulfite-Treated/Deaminated DNA? USA: New England Biolabs; 2022. Available from: https://international.neb.com/support/international-ordering-support#Current%20Location. Accessed December 13, 2022.
- Ma H, Difazio S. An efficient method for purification of PCR products for sequencing. Biotechniques 2008; 44:921-3. doi: 10.2144/000112809 [Crossref] [ Google Scholar]
- Thermo Fisher Scientific. ExoSAP-ITTM PCR Product Cleanup Reagent. Germany: Thermo Fisher Scientific; 2022. Available from: https://www.thermofisher.com/order/catalog/product/78201.1.ML#:~:text=ExoSAP%2DIT%E2%84%A2%20PCR%20Product%20Cleanup%20Reagent%20is%20used%20for,nucleotide%20polymorphism%20(SNP)%20analysis. Accessed December 13, 2022.
- Parrish RR, Day JJ, Lubin FD. Direct bisulfite sequencing for examination of DNA methylation with gene and nucleotide resolution from brain tissues. CurrProtocNeurosci 2012; Chapter 7: Unit 7.24. 10.1002/0471142301.ns0724s60.
- Tusnády GE, Simon I, Váradi A, Arányi T. BiSearch: primer-design and search tool for PCR on bisulfite-treated genomes. Nucleic Acids Res 2005; 33:e9. doi: 10.1093/nar/gni012 [Crossref] [ Google Scholar]
- Cheishvili D, Parashar S, Mahmood N, Arakelian A, Kremer R, Goltzman D. Identification of an epigenetic signature of osteoporosis in blood DNA of postmenopausal women. J Bone Miner Res 2018; 33:1980-9. doi: 10.1002/jbmr.3527 [Crossref] [ Google Scholar]
- Vrtačnik P, Marc J, Ostanek B. Epigenetic mechanisms in bone. Clin Chem Lab Med 2014; 52:589-608. doi: 10.1515/cclm-2013-0770 [Crossref] [ Google Scholar]
- Gogakos AI, Cheung MS, Bassett JD, Williams GR. Bone signaling pathways and treatment of osteoporosis. Expert Rev Endocrinol Metab 2009; 4:639-50. doi: 10.1586/eem.09.38 [Crossref] [ Google Scholar]
- Wang P, Cao Y, Zhan D, Wang D, Wang B, Liu Y. Influence of DNA methylation on the expression of OPG/RANKL in primary osteoporosis. Int J Med Sci 2018; 15:1480-5. doi: 10.7150/ijms.27333 [Crossref] [ Google Scholar]
- Kalkan R, Becer E. RANK/RANKL/OPG pathway is an important for the epigenetic regulation of obesity. Mol Biol Rep 2019; 46:5425-32. doi: 10.1007/s11033-019-04997-z [Crossref] [ Google Scholar]
- Yang P, Zhou B, Cao WC, Wang YX, Huang Z, Li J. Prenatal exposure to drinking water disinfection by-products and DNA methylation in cord blood. Sci Total Environ 2017; 586:313-8. doi: 10.1016/j.scitotenv.2017.01.224 [Crossref] [ Google Scholar]
- Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes 2011; 60:1528-34. doi: 10.2337/db10-0979 [Crossref] [ Google Scholar]
- Harvey NC, Lillycrop KA, Garratt E, Sheppard A, McLean C, Burdge G. Evaluation of methylation status of the eNOS promoter at birth in relation to childhood bone mineral content. Calcif Tissue Int 2012; 90:120-7. doi: 10.1007/s00223-011-9554-5 [Crossref] [ Google Scholar]
- Harvey NC, Sheppard A, Godfrey KM, McLean C, Garratt E, Ntani G. Childhood bone mineral content is associated with methylation status of the RXRA promoter at birth. J Bone Miner Res 2014; 29:600-7. doi: 10.1002/jbmr.2056 [Crossref] [ Google Scholar]
- Laajala E, Kalim UU, Grönroos T, Rasool O, Halla-Aho V, Konki M. Umbilical cord blood DNA methylation in children who later develop type 1 diabetes. Diabetologia 2022; 65:1534-40. doi: 10.1007/s00125-022-05726-1 [Crossref] [ Google Scholar]
- Hong SR, Shin KJ. Bisulfite-converted DNA quantity evaluation: a multiplex quantitative real-time PCR system for evaluation of bisulfite conversion. Front Genet 2021; 12:618955. doi: 10.3389/fgene.2021.618955 [Crossref] [ Google Scholar]
- Holmes EE, Jung M, Meller S, Leisse A, Sailer V, Zech J. Performance evaluation of kits for bisulfite-conversion of DNA from tissues, cell lines, FFPE tissues, aspirates, lavages, effusions, plasma, serum, and urine. PLoS One 2014; 9:e93933. doi: 10.1371/journal.pone.0093933 [Crossref] [ Google Scholar]
- Al Harrasi I, Al-Yahyai R, Yaish MW. Detection of differential DNA methylation under stress conditions using bisulfite sequence analysis. Methods Mol Biol 2017; 1631:121-37. doi: 10.1007/978-1-4939-7136-7_7 [Crossref] [ Google Scholar]
- Zheng X, Wu Q, Wu H, Leung KS, Wong MH, Liu X. Evaluating the consistency of gene methylation in liver cancer using bisulfite sequencing data. Front Cell Dev Biol 2021; 9:671302. doi: 10.3389/fcell.2021.671302 [Crossref] [ Google Scholar]
- Linder JE, Plachco TE, Libster R, Miller EK. Sequencing human rhinoviruses: direct sequencing versus plasmid cloning. J Virol Methods 2015; 211:64-9. doi: 10.1016/j.jviromet.2014.09.020 [Crossref] [ Google Scholar]
- Oh ES, Petronis A. Origins of human disease: the chrono-epigenetic perspective. Nat Rev Genet 2021; 22:533-46. doi: 10.1038/s41576-021-00348-6 [Crossref] [ Google Scholar]