Bioimpacts. 2025;15:30333.
doi: 10.34172/bi.30333
Mini Review
Role of Toll-like receptors in exosome biogenesis and angiogenesis capacity
Parisa Hassanpour Writing – original draft, 1, 2 
Fatemeh Sadeghsoltani Writing – original draft, 3
Mir‑Meghdad Safari Software, 4
Sanya Haiaty Writing – original draft, 5
Reza Rahbarghazi Conceptualization, Data curation, Project administration, Validation, Writing – review & editing, 2, 6, *
Ali Mota Writing – original draft, 1
Mohamad Rahmati Conceptualization, Data curation, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing, 2, 1, *
Author information:
1Department of Clinical Biochemistry and Laboratory Medicine, School of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran
2Stem Cell Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
3Student Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran
4Virtual School of Medical Education and Management, Shahid Beheshti University of Medical Sciences, Tehran, Iran
5Infectious and Tropical Diseases Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
6Department of Applied Cell Sciences, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences, Tabriz, Iran
*These authors contributed equally to this work.
Abstract
Adaptive inflammation consists of multiple cellular changes and molecular reactions to protect host cells against several pathological conditions. Along with the activation of varied immune cells, the production and secretion of cytokines arrays can regulate the progression of inflammatory response in a paracrine manner. Among different molecular cascades, Toll-like receptors (TLRs) are activated in response to several pathological conditions and damage signals. It has been indicated that extracellular vesicles, especially exosomes (Exos) are key bioshuttles with specific cargoes and are involved in cell-to-cell communication. The role of Exos in the initiation, progression, and cession of inflammation has been previously addressed in terms of cytokine transmission. Whether and how the activation of TLRs can alter the Exo biogenesis and angiogenesis potential in immune cells and endothelial cells (ECs) remains to be elucidated. Here, the cross-talk between the TLRs, Exo biogenesis, and angiogenesis has been highlighted.
Keywords: Toll-like receptors, Exosomes, Exocytosis, Angiogenesis, Endothelial cells, Immune cells
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This study was supported by a grant (No: 66514) from Tabriz University of Medical Sciences. The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Introduction
Among different signaling cascades, Toll-like receptors) TLRs (and relevant downstream effectors participate in several cell bioactivities. These receptors belong to the pattern recognition receptors family (PRRs).1 In response to exogenous pathogens and damage signals, the activation of PRRs triggers a series of molecular pathways leading to the production of pro-inflammatory cytokines.2 Of 13 mammalian types of TLRs, murine and human cells can express TLR1-10, 12, and 13.3 Notably, the function of all TLRs was defined previously except the TLR10 (Fig. 1). Based on subcellular location, TLRs are classified into two groups. The cell plasma membrane-bound TLRs such as TLR 1, 2, 4, 5, 6, and 11 have the potential to bind microbial compounds, while endosomal TLRs such as TLR 3, 7, 8, and 9 can attach to intracellular pathogens.4 From a structural viewpoint, TLRs possess three distinct domains. The N-terminal extracellular domain is composed of a hydrophobic leucine-rich repeat, namely LRR, with the potential to recognize several pathogens. The middle part possesses a single helical motif that passes through a plasma membrane, and finally, the conserved cytoplasmic domain is juxtaposed to the Toll/interleukin-1 receptor (TIR) complex.5
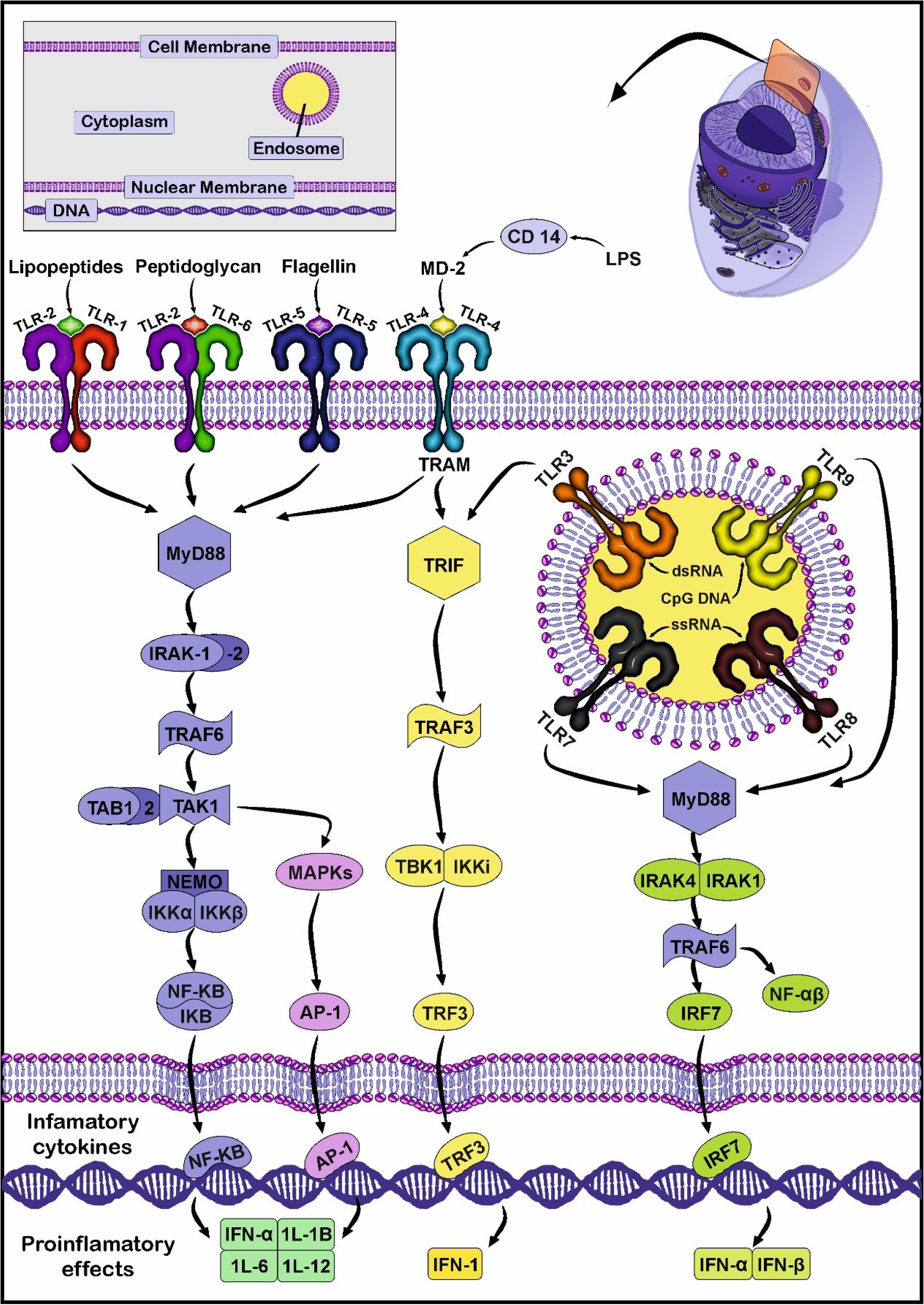
Fig. 1.
TLR signaling pathway. The homodimers of TLR5, TLR4, and heterodimers TLR2-TLR1 or TLR2-TLR6 recognize DAMPs and PAMPs on the surface of the plasma membrane. Inside the cells, TLR3, TLR7-TLR8, and TLR9 are located on the membrane of the endosomes and identify various intracellular genetic components. Following the close interaction of ligands and TLRs, downstream molecular cascades are initiated in MyD88-dependent or MyD88-independent (TIRAP axis) manners. In the MyD88-dependent signaling pathway, the recruitment of effectors such as IRAK4 and IRAK1/2 leads to the engagement and activation of TRAF6 and TAK1 complex, respectively. In the MyD88-dependent pathway, factors belonging to the IRAKs family such as IRAK4, and IRAK1/2 are recruited and phosphorylated. In the next step, IRAK1 can interact with TRAF6 which is followed by the addition of TAK1. TAKI can stimulate MAPK, and facilitate the translocation of NF-kB into the nucleus, leading to the production of several pro-inflammatory cytokines. In an alternative pathway, TLR3 and 4 can exclusively bind to TRIF without the participation of MyD88, resulting in the entry of TRF3 into the nucleus and INF expression. Created by CorelDRAW Graphics Suite Ver. 250.0.0.230.
.
TLR signaling pathway. The homodimers of TLR5, TLR4, and heterodimers TLR2-TLR1 or TLR2-TLR6 recognize DAMPs and PAMPs on the surface of the plasma membrane. Inside the cells, TLR3, TLR7-TLR8, and TLR9 are located on the membrane of the endosomes and identify various intracellular genetic components. Following the close interaction of ligands and TLRs, downstream molecular cascades are initiated in MyD88-dependent or MyD88-independent (TIRAP axis) manners. In the MyD88-dependent signaling pathway, the recruitment of effectors such as IRAK4 and IRAK1/2 leads to the engagement and activation of TRAF6 and TAK1 complex, respectively. In the MyD88-dependent pathway, factors belonging to the IRAKs family such as IRAK4, and IRAK1/2 are recruited and phosphorylated. In the next step, IRAK1 can interact with TRAF6 which is followed by the addition of TAK1. TAKI can stimulate MAPK, and facilitate the translocation of NF-kB into the nucleus, leading to the production of several pro-inflammatory cytokines. In an alternative pathway, TLR3 and 4 can exclusively bind to TRIF without the participation of MyD88, resulting in the entry of TRF3 into the nucleus and INF expression. Created by CorelDRAW Graphics Suite Ver. 250.0.0.230.
TLR signaling is stimulated after direct interaction of TLRs with TIR-domain containing adaptor protein, namely myeloid differential factor 88 (MyD88) (Fig. 1).6 It is believed that the attachment of specific ligands such as pathogen-associated molecular patterns (PAMP) such as RNA viruses, several bacteria species, and damage-associated molecular patterns (DAMPS) like miRNAs, heat shock proteins (HSPs), and high mobility group box 1 (HMGB1) can promote TLR signaling pathway.7 Bacterial lipopolysaccharides (LPS) can stimulate the TLR co-receptor CD14 and thus downstream effectors.8 Results have shown that the release of endogenous molecules after tissue injury can be easily recognized by different TLR types. Among them, HSP60 can activate TLRs at the early stages of tissue injury and inflammation.9 Based on the recruitment of distinct adaptor proteins, the TLR signaling pathway is classified into myeloid differentiation factor 88 (MyD88)-dependent and MyD88-independent axes. The latter pathway is promoted by TIR-domain-containing adaptor-inducing interferon-β (TRIF).10 Previous molecular investigations have shown that only TLR 3 can bind exclusively to TRIF without the activation of MyD88. By contrast, TLR4 can engage both MyD88 and TRIF effectors while other TLR types are dependent on MyD88 activity.11 Upon activation of the TLR signaling pathway, the transcription of certain factors such as nuclear factor-κB (NF-κB) mitogen-activated protein kinase (MAPK), and pro-inflammatory cytokines are stimulated (Fig. 1). Of note, TLRs in hetero or homo-dimer structures can bind to the target molecules.12 The close interaction of PAMPs or DAMPs with TLRs activates the TLR/MyD88 axis. In the next step, MyD88 recruits IL-1R-related kinases (IRAKs) family such as IRAK4 and 1. Both IRAK4 and 1 are phosphorylated, followed by the separation of IRAK1 from MyD88 and close interaction with TNF receptor-associated factor 6 (TRAF6).13,14 Thereafter, transforming growth factor-β-activated kinase 1 (TAKI) and subunits TAK1-binding protein (TAB1, 2, and 3) attach to TRAF6.15 The activated TAK1 can promote two signaling cascades, such as phosphorylation of IκB kinase (IKK) and activation of mitogen-activated protein kinase (MAPK).16 Under normal circumstances, IκB can inhibit nuclear factor NF-κB. After the activation of the IKK complex, IκB is phosphorylated and subsequently degraded. In the latter step, NF-κB translocates into the nucleus to up-regulate the expression of pro-inflammatory cytokines.17 In an alternative pathway, the activation of the MAPK signaling pathway triggers activator protein 1 (AP1). This factor in collaboration with NF-κB increases the synthesis of IL-1, 6, and TNF-α.18 The activation of the TRIF pathway by TLR3 and 4 stimulates another signaling pathway16,19 It should be noted that TRIF and TRAM (TRIF-related adaptor molecule) can be activated via the MyD88-independent pathway. TRAM acts as an adapter protein and connects TLR to TRIF, leading to TAK1, TRAF6, and NF-ƙB activation.18 It was suggested that the activation of TRAF6 leads to the promotion of TBK1 and IKK-ε to phosphorylate interferon regulatory factor 3 (IRF3). The procedure is followed by the translocation of phosphorylated IRF3 into the nucleus and the expression of type 1 interferon.18,20
Exosome biogenesis
Exosomes (Exos) are nano-sized extracellular vesicles with the potential to carry biological cargo such as lipids, proteins, short-length DNAs, and various RNA types from the host cells to acceptor cells. Based on different studies, Exos with a mean diameter size of 30-150 nm, are originated from the endosomal system.21-23 The phenomenon of Exo biogenesis is a multi-step process in which several intraluminal vesicles (ILVs) are generated via the invagination of plasma membrane inside late endosomes and multivesicular bodies (MVBs) (Fig. 2).24-26 In the next steps, the fusion of MVBs with the plasma membrane leads to the release of ILVs into the extracellular environment, hereafter known as Exos.27 Two distinct pathways have been proposed for MVBs in the latter steps. First, MVBs can fuse with lysosomes and are subjected to proteolytic degradation.28 Besides, MVBs can be oriented to the cell membrane where direct physical contact and fusion lead to the release of ILVs into the extracellular matrix (ECM). Evidence revealed that various factors, such as the endosomal sorting complex required for transport (ESCRT), lipids (ceramides), and tetraspanins participate in ILV formation within MVBs.29 Cargo sorting is orchestrated via ESCRT-dependent or ESCRT-independent pathways.26 ESCRT is a cytoplasmic protein that generates coated subdomains on the surface of endosomes. These features facilitate membrane budding and the formation of ILVs. From the molecular structure, the ESCRT complex is composed of four certain subsets, including ESCRT-0, -I, -II, and -III, that are in close relation with specific proteins ALIX, TSG101, and VPS4.30 These factors can help in the sequestration of certain cargo into the lumen of ILVs.30
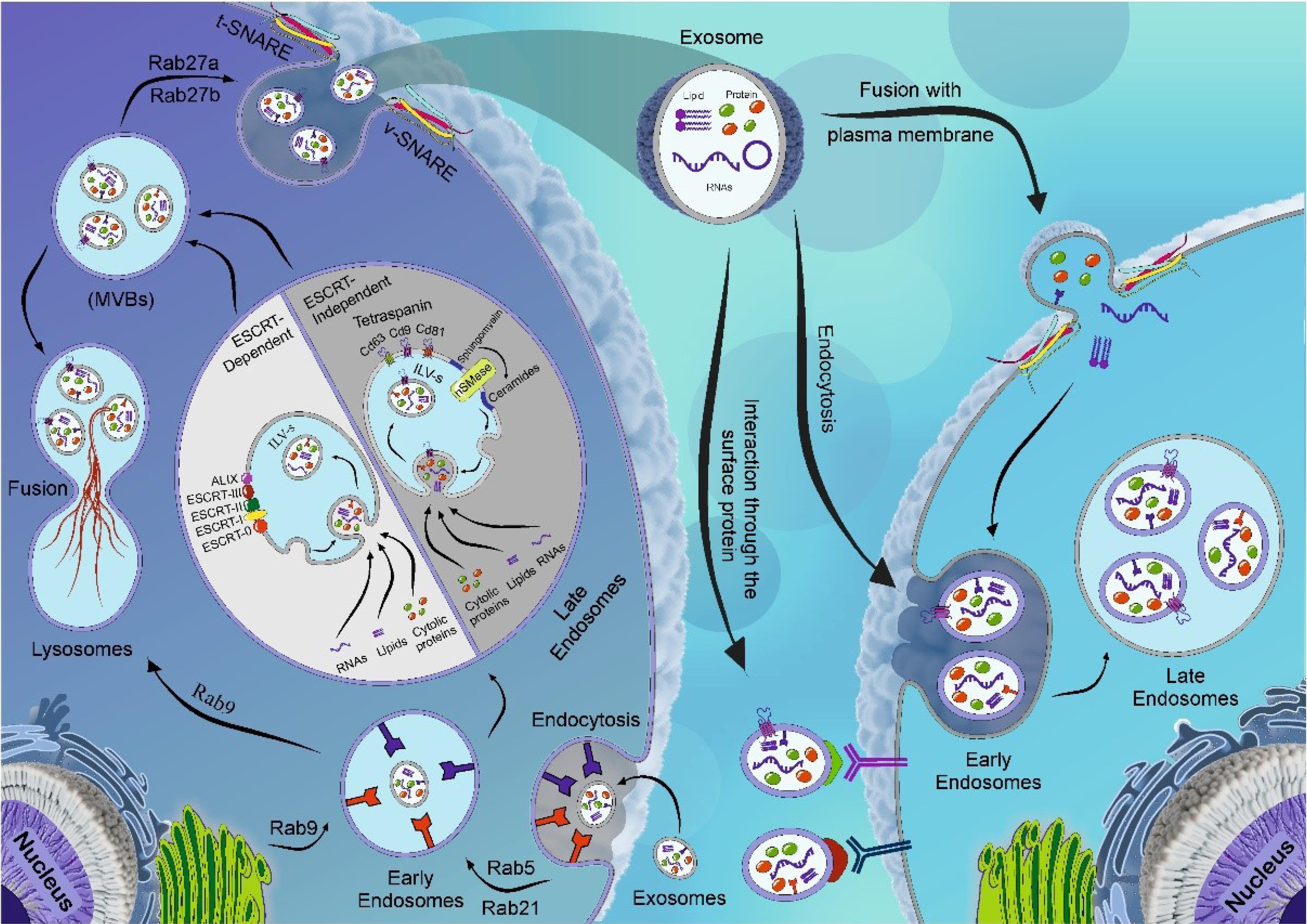
Fig. 2.
Schematic illustration of Exo biogenesis and abscission. Different effectors participate in the biogenesis of Exos. Following the internalization of cargo into the target cell, the early endosomes are generated. Early endosomes can mature into late endosomes after packing several signaling molecules such as proteins, lipids, and nucleic acids. This activity is performed via the invagination of endosomal membranes, resulting in the formation of numerous intraluminal vesicles (ILVs). In the next step, later endosomes mature into MVBs. The process of cargo sorting into the ILVs is done via the participation of ESCRT-dependent or ESCRT-independent pathways. MVBs can fuse with the lysosomes and undergo enzymatic digestion or maintain physical contact with the plasma membrane to release ILVs into the ECM, where hereafter known as Exos. Effectors such as RABs, v-SNARE, and t-SNARE are required for the fusion of MVBs with the plasma membrane. The released Exos can be guided to close or remote cells through direct fusion, receptor-mediated fusion, and endocytosis. Created using CorelDRAW Graphics Suite Ver. 250.0.0.230.
.
Schematic illustration of Exo biogenesis and abscission. Different effectors participate in the biogenesis of Exos. Following the internalization of cargo into the target cell, the early endosomes are generated. Early endosomes can mature into late endosomes after packing several signaling molecules such as proteins, lipids, and nucleic acids. This activity is performed via the invagination of endosomal membranes, resulting in the formation of numerous intraluminal vesicles (ILVs). In the next step, later endosomes mature into MVBs. The process of cargo sorting into the ILVs is done via the participation of ESCRT-dependent or ESCRT-independent pathways. MVBs can fuse with the lysosomes and undergo enzymatic digestion or maintain physical contact with the plasma membrane to release ILVs into the ECM, where hereafter known as Exos. Effectors such as RABs, v-SNARE, and t-SNARE are required for the fusion of MVBs with the plasma membrane. The released Exos can be guided to close or remote cells through direct fusion, receptor-mediated fusion, and endocytosis. Created using CorelDRAW Graphics Suite Ver. 250.0.0.230.
Generally, the ESCRT machinery has the main role in attaching, sorting, and clustering ubiquitinylated proteins into the MVBs.31 To initiate these procedures, ESCRT-0 recognizes and sequestrates ubiquitinated proteins into the endosomal membrane. In the next steps, the activation of ESCRT-I and -II subsets predisposes membrane deformation and invagination of ILVs.31,32 The close interaction of Alix with this complex promotes the recruitment of ESCRT-III. The addition of ESCRT-III accelerates the release of de novo ILVs into the endosomal lumen. The collaboration of Alix with exosomal proteins like the ESCRT complex can play a key role in budding and cutting the endosomal membrane through interaction with syndecan.33 Along with these changes, energy supply via accessory proteins such as Vps4 causes the recycling of ESCRT machinery.34 In addition to the ESCRT-dependent pathway, components of the ESCRT-independent pathway such as ceramides can also help the ILV generation. For example, the enzymatic activity of sphingomyelinase leads to the production of ceramide from sphingomyelin, and accumulated ceramide levels contribute to the formation of ILVs within MVBs.35,36 Other components of ESCRT-independent pathways are tetraspanins, which are involved in ILV formation. Tetraspanins consist of a large number of protein complexes in the plasma membrane of Exos, lysosomes, and endosomes.37,38 It has been thought that Exos harbor large contents of tetraspanins with several transmembrane domains.39 Tetraspanins are in close association with each other and different signaling proteins.40 Several studies have applied tetraspanins as integral biomarkers for Exo characterization and immunophenotyping. Besides, these factors are involved in migration, signaling, fusion, intercellular adhesion, and other functions.29,41 Tetraspanins mainly CD9, CD37, CD53, CD63, and CD81 can foster the generation of ILVs using various mechanisms. To be specific, CD81 facilitates Rac GTPase function while CD9 promotes the fusion process with the plasma membrane and has an important role in the release of exosomal β-catenin.42 Notably, CD63 is abundantly present in the exosomal membrane and actively participates in the biogenesis of Exos.43 The existence of distinct tetraspanin membranes such as CD55 and CD59 protects Exos from the complement system.44,45 Recent studies state that the ESCRT-independent pathway can also modify exosomal cargo.46 The secretion of Exos from the host cells is associated with the activity of several factors like molecular switch Rab GTPase, membrane fusion complexes such as soluble NSF attachment protein receptors (SNAREs), cytoskeleton proteins (actin, and myosin, and motor proteins kinesin, and dynein).47,48 As mentioned earlier, the Rab is a subset of the RAS family with GTPase activity. This protein is located on the cytosolic side of the plasma membrane and plays a crucial role in regulating molecular pathways and Exo trafficking.49 The stimulation of both RAB and SNARE complexes expedites the MVB docking and fusion with the plasma membrane, leading to Exo secretion (Fig. 2).50 For instance, Rab5 regulates the release of Exos by forming and fusion early endosomes. Rab27 regulates the movement and attachment of secretory vesicles with the plasma membrane via physical contact with specific factors.51 Likewise, microtubules and molecular motors are important in transporting MVBs towards the plasma membrane.52 These factors are required to transport, dock, and assimilate MVBs with the plasma membrane.53 A zipper-like structure is formed by pairing SNAREs (v-SNARE and t-SNARE) can promote the abscission phenomenon and the release of Exos ECM.25
TLRs and exocytosis
As a common belief, the induction of the TLR signaling pathway causes the translocation of NF-κB into the nucleus with the involvement of several factors and enzymes. It is thought that NF-κB activation can be done through proteasome-mediated proteolysis of ubiquitin (Fig. 3).54,55 Ubiquitin has a critical role in pro-inflammatory responses after cell exposure to microbial products and an array of cytokines. Under physiological conditions, NF-κB is inhibited by IκB within the cytosol. Soon after the attachment of stimulatory ligands to TLRs, IRAK1, and 2 are phosphorylated to stimulate TRAF6.56 In the following steps, the binding of TRAF6 to ubiquitin lysine residue per se promotes the kinase activity of TAK1. TAk1 and I kappa B kinase (IKK) simultaneously bind to non-canonical protein ubiquitination k-63 via phosphorylation and ubiquitination of the IKKg subunit (NEMO). These features lead to proteasomal degradation of IKB.57-59
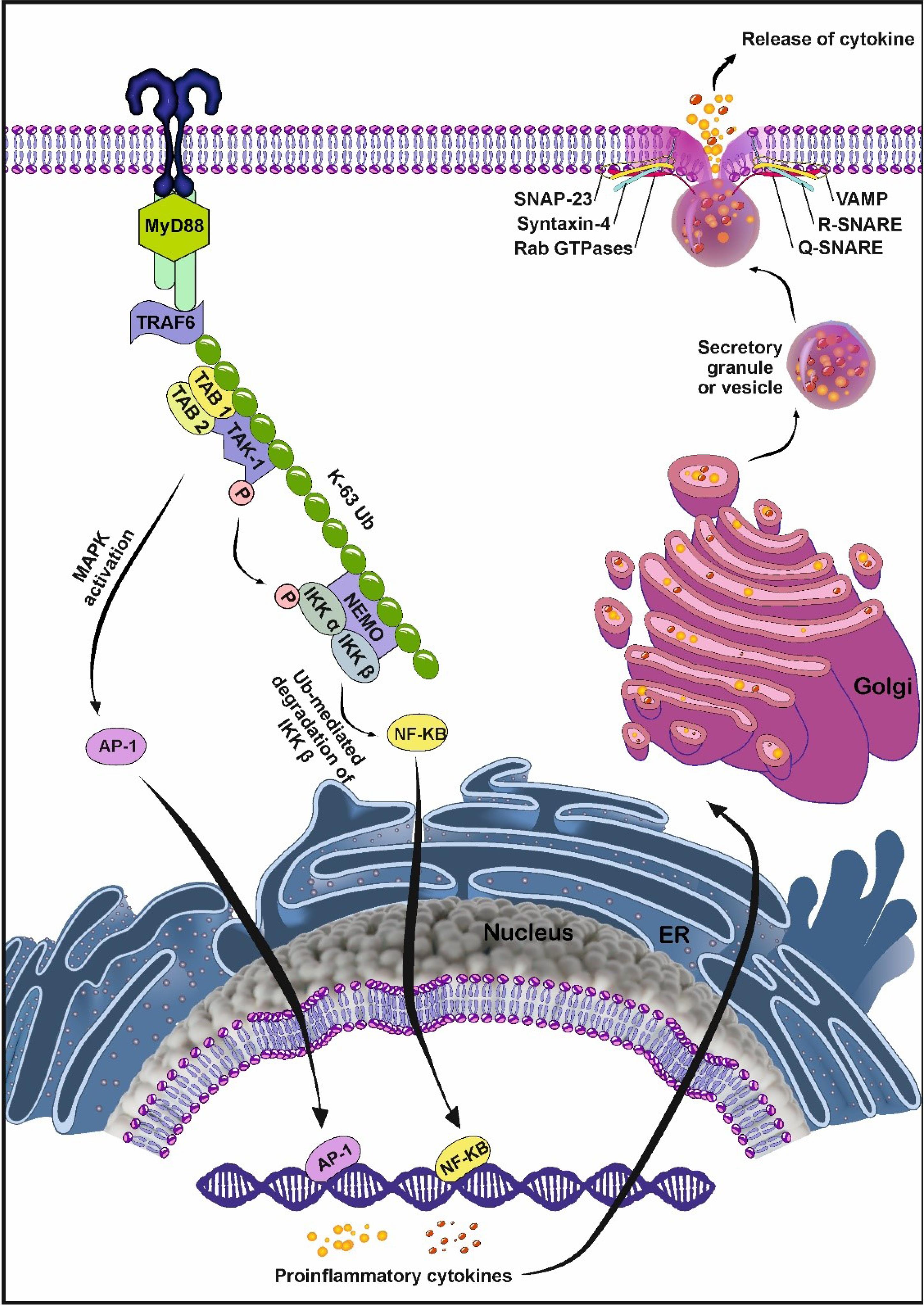
Fig. 3.
Polyubiquitination systems can connect TLRs with exocytosis. After ligand-TLR interaction, TRAF6 is activated via association between IRAK4/1 and TRAF6 in a MyD88-dependent manner. TRAF6 functions as a ubiquitin ligase and stimulates the synthesis of K63-linked polyubiquitin. The physical attachment of TRAF6 to the ubiquitin lysine residue activates protein kinase TAK1. TAK1 activates IκB kinases (IKKα, IKKβ, IKKγ) and phosphorylates IkB-α. Phosphorylation and ubiquitination of the protein inhibitor IkB lead to its degradation. Afterward, NF-κB is activated by the degradation of IkB through the ubiquitination pathway with the translocation of NF-κB into the nucleus. NF-κB in the nucleus promotes the synthesis of numerous pro-inflammatory cytokines. In the next step, pro-inflammatory cytokines synthesized in the ER and Golgi complex, are transported to the cell surface. A set of SNAREs is required for the binding of secretory granules to the plasma membrane and the release of cytokines from the granules. An R-SNARE in the membrane of the secretory organelle associates with a Q-SNARE-like complex (SNAP-23 and syntaxin-4) in the target membrane, forming a 4-helix coil structure which is necessary for the release of cytokines. Created by CorelDRAW Graphics Suite Ver. 250.0.0.230.
.
Polyubiquitination systems can connect TLRs with exocytosis. After ligand-TLR interaction, TRAF6 is activated via association between IRAK4/1 and TRAF6 in a MyD88-dependent manner. TRAF6 functions as a ubiquitin ligase and stimulates the synthesis of K63-linked polyubiquitin. The physical attachment of TRAF6 to the ubiquitin lysine residue activates protein kinase TAK1. TAK1 activates IκB kinases (IKKα, IKKβ, IKKγ) and phosphorylates IkB-α. Phosphorylation and ubiquitination of the protein inhibitor IkB lead to its degradation. Afterward, NF-κB is activated by the degradation of IkB through the ubiquitination pathway with the translocation of NF-κB into the nucleus. NF-κB in the nucleus promotes the synthesis of numerous pro-inflammatory cytokines. In the next step, pro-inflammatory cytokines synthesized in the ER and Golgi complex, are transported to the cell surface. A set of SNAREs is required for the binding of secretory granules to the plasma membrane and the release of cytokines from the granules. An R-SNARE in the membrane of the secretory organelle associates with a Q-SNARE-like complex (SNAP-23 and syntaxin-4) in the target membrane, forming a 4-helix coil structure which is necessary for the release of cytokines. Created by CorelDRAW Graphics Suite Ver. 250.0.0.230.
Several studies have shown that secretory nanovesicles are cytokine-transporting vesicles.60 Upon the production of secretory cytokines in the endoplasmic reticulum (ER), they are transferred to the Golgi apparatus and packed. After that, cytokines are released from the cell by secretory vesicles via exocytosis.61,62 In this pathway, small GTPases belonging to the Rab family are involved in the formation, movement, attachment, and fusion of vesicles to target membranes.63 Some studies indicate that secretory granules are physically connected to the plasma membrane in response to TLR stimulation via the activation of SNAREs as intracellular proteins.61,64
SNAREs are a family of proteins with conserved structures and form a complex in combination with membrane proteins that fuse docked bilayer lipids.65 Depending on the type of amino acid in the central core, SNAREs fall into two subfamilies: Q-SNARE with glutamine (Q) residue and R-SNARE with arginine (R) residue as a central functional residue in motif SNAREs.66 A Q-SNARE complex contains syntaxin, synaptosomal-associated protein (SNAP), and R-SNARE contains VAMP. To form four helical bundles for membrane fusion, one R-SNARE on the secretory vesicle and two or three Q-SNAREs on the plasma membrane are essential.67,68 Q- and R-SNAREs pairing is important in trafficking cytokines from the host cells.69 By interacting subfamily of SNAREs from the N-terminus to the C-terminus of the opposite membrane, the α-helix bundle (zipper) is formed, which is called trans-SNARE. The Zipper complex provides the energy required to keep hydrophobic lipids in the plasma membrane and overcome hydrostatic pressure.70
Effect of TLRs on mitochondria in myocardial infraction
During myocardial infarction, DAMPs are released with the death of cardiac cells. DAMPs stimulate the resident immune cells by binding to cardiomyocyte pattern recognition receptors (PRRs).71 Recruitment of immune cells followed by the release of inflammatory cytokines leads to inflammation in the infarcted area.72 The heart as an energy-demanding organ has a large number of mitochondria in each cardiomyocyte. Necrotic cardiomyocytes release mitochondrial DAMPs (mtDAMPs) into the cytosol or extracellular space.73 Studies have shown that mtDAMPs can induce inflammatory factors through multiple signaling pathways, including Toll-like receptor (TLR) 9, cGAS- (cyclic guanosine monophosphate-adenosine monophosphate synthase-) STING, and pyrin domain-containing protein 3 (NLRP3). Released mtDAMPs are oxidized when exposed to reactive oxygen species (ROS) and oxidized mtDAMPs can trigger TLRs.74 During prolonged ischemic and hypoxic conditions, large amounts of free radicals are produced, and high amounts of cardiomyocyte mtDNA are recognized by PRRs.75,76 Research shows that endogenous DAMPs released from damaged cardiomyocytes bind to TLR receptors in intact cardiomyocytes. TLR-9 has been identified as the most important mtDNA receptor through the identification of an unmethylated CPG motif in mtDNA structure.77 The activation of endosomal TLR-9 by mtDAMPs stimulates NF-κB in the MyD88-dependent pathway. As the main regulator of inflammation, NF-κB plays a role in the increased expression of pro-inflammatory cytokines, tissue remodeling, and immune responses.76 In addition, the elevation of ROS after mitochondrial injury causes oxidative stress and the secretion of chemokines and pro-inflammatory cytokines (IL-6, and -8).78 Besides, the increase of mtDNA and mitochondrial ROS cardiomyocyte cytoplasm contributes to the formation of NLRP3 inflammasomes. The inflammasomes are the main source of interleukin 1β (IL-1β) and (IL-18) and play an important role in the recruitment of inflammatory cells and the promotion of cardiomyocyte death.81,82 In this pathway, caspase-1 regulates the inflammatory factors such as IL-1β and IL-18. Activation of signaling pathways by mtDAMPs causes more damage to mitochondria and thus more mtDAMPs are released.83 Another inflammatory pathway activated by mtDNA is cyclic GMP-AMP (cGAMP) synthase-(cGAS-) STING pathway (cGAS-STING). The mtDNA released in the cytoplasm recognizes the cGAS-STING pathway, after that it leads to STING homodimerization and activation. Further phosphorylation of IκB activates the transcription factor NF-κB.83,84 Previous data have shown that circulating mtDNA plays a significant role in the development of heart diseases by activating inflammatory pathways.85
Impact of Inflammation on exosome production and release
The findings are consistent with the fact that several inflammatory stimuli such as metabolic disease and pathological conditions can affect the Exo biogenesis and secretion.86,87 On the other hand, exosomal cargo can regulate the extent and duration of inflammatory response under physiological and pathological conditions.88 In some inflammatory diseases, Exos induce pro-inflammatory cytokines like IL-6, IL-1β, and TNF-α through PAMP receptors such as TLRs.89 For example, IL-1β cytokine secretion is responsible for fever and leukocyte recruitment after the initiation of inflammation.90 In response to intracellular damage and endogenous pathogens, the formation of protein complexes namely NLRP3 inflammasomes is initiated via canonical and non-canonical pathways.91 In the canonical axis, the activation of IL-1β, Caspase 1, and IL-18 are the main fundamental factors that lead to the inflammasome-associated inflammatory response.92 Recent works have indicated the importance of Caspase 1 activation on intracellular trafficking of vesicles via the regulation of Rab-interacting lysosomal protein (RILP).93 In an experiment conducted by Wozniak and colleagues, THP-1 macrophages priming with bacterial LPS (inflammasome inducer) and ATP (inflammasome activator) can increase RILP cleavage, leading to the activation of Exo cargo sorting and secretion.93 Based on the data, these effects were blunted when the non-cleavable form of RILP was expressed in the host cells.93 The activation of RILP is an appropriate strategy to close the MVBs' direction toward the cell membrane. Along with these changes, fragile X mental retardation protein (FMRP) with KH domain can sort miRNAs with AAUGC motif onto the ILVs inside the MVBs via the collaboration with ESCRT complex after Caspase 1 cleavage.93 Of note, circulating Exos harbors high contents of FMRP and IL-1β in patients with non-alcoholic steatohepatitis compared to healthy counterparts.93 To support the existence of a direct relationship between Exo secretion and inflammation, Chen et al found that pre-treatment of mouse RAW264.7 macrophages with dexamethasone and incubation with bacterial LPS reduced Exo release compared to the control LPS group. These data coincided with the reduction of exosomal inflammatory miRNA-155.94 Under metabolic diseases, the stimulation of the endosomal system and dysregulation of lysosomal activity can affect the host cell's capacity to release the Exos.95 In this regard, Huang and co-workers examined the effect of homocysteine on Exo biogenesis and the secretion of podocytes in a mouse model.95 They claimed that in response to hyperhomocysteinemia, NLRP3 inflammasomes are activated and sequestrated onto the ILVs in later endosomes and MVBs. It was suggested that simultaneous activation of lysosomal acid sphingomyelinase increases the intracellular levels of sphingolipids and ceramides, resulting in the reduction of MVB-lysosome interaction and an increase of Exo release from podocytes. The inhibition of acidic and neutral sphingomyelinase activity using Amitriptyline and GW4869 can stimulates MVB-lysosome interaction and Exo biogenesis, respectively.95 Simultaneous treatment with mTORC1 inhibitor, rapamycin, triggers the fusion of MVBs with lysosomes and cargo degradation.95 Data indicated that regulated inhibition of inflammatory Exo secretion can alleviate glomerular inflammation under pathological conditions. In line with these changes, the inflammatory response can expedite the process of Exo biogenesis and release into the ECM space. Due to changes in cargo sorting into Exos and secretion of certain inflammatory factors, the induction of intracellular lysosomal degradation is touted as an effective approach to the expansion of inflammation and pathological changes to juxtaposed sites. Likewise, Ye and co-workers indicated that the induction of albuminuric nephropathy in mice using Adriamycin led to enhanced Exo secretion by Rab27a activity.96 The prolonged exposure of tubular epithelial cells to albumin increased the transcription of interferon regulatory factor 1, a factor that regulates the expression of GTPase Rab27a.97 Based on the data, Rab27a silencing increased lysosomal degradation of Exo and intracellular albumin and diminished the production of cellular inflammatory cytokines.96 It is postulated that phagocytes and inflammatory cells can exert their effective role during pathological conditions via the regulation of Exo biogenesis. In support of this notion, systemic levels of TNF-α, IL-1β, IL-6, and cardiovascular pathologies were significantly reduced after injection of endotoxin in mice pre-treated with Exo biogenesis blocker GW4869.98 It should not be forgotten that the release of Exos can also occur in response to the intracellular accumulation of noxious and undegraded materials to prevent cell atresia.99 Guix and co-workers declared that the progressed aging process in neurons can distort the normal activity of Exo biogenesis and release.100 By the progression of aging changes, the number of large-sized MVBs, and lysosome-associated multilamellar bodies increased along with the lysosome heterogeneity.100 Also, the number of Exos with small diameter sizes increased in cell culture supernatant, reflecting the enlargement of MVBs inside the cytosol. These effects are associated with reduction of cholesterol-transporter NPC1 protein, resulting in the accumulation of cholesterol and GM2 in endosomal vesicles and lysosomes. Under such conditions, the reduced activity of Akt-mTOR axis and suppression of autophagosomes with lysosomes are evident inside the affected cells.100 Of note, Exos can also from in dying apoptotic cells and cells exposed to severe pathological conditions.101 Immunofluorescence staining indicated the formation of CD63+ microvesicle in apoptotic HeLa cells induced by staurosporine in a caspase-dependent manner. Based on data obtained from a study conducted by Park and colleagues, the activated sphingosine-1-phosphate/sphingosine-1-phopshate receptors are involved in the production and release of ILVs in apoptotic HeLa cells.101
Whether the contact of immune cells with normal and/or cancer Exos can lead to different inflammatory responses is at the center of the subject. It was suggested that palmitoylated proteins on breast cancer cell Exos can stimulate NF-κB and increase the release of pro-inflammatory cytokines such as TNF-α, IL-6, chemokine ligand 2 (CCL2), granulocyte colony-stimulating factor (GCSF).102,103 In this regard, the entry of cancer Exos to leukemic monocytes (THP-1 cells) increases the TNF-α. The attachment of this factor to TLRs (2 and 4) promotes NF-κB and STAT3. It has been well-known that the reciprocal interaction of NF-κB and STAT3 regulates the inflammatory response during neoplastic changes.104,105 These features unveil the potent paracrine activity of Exos between the tumor cells and tumor-associated macrophages (TAMs). Exos released by breast and gastric tumors triggers the phenotype acquisition of macrophages toward M1.102 Certain molecular signatures such as Annexin A2 in the exosomal membrane directly activate the p38MAPK/NFκB/STAT-3 axis and production of IL-6 and TNF-α.106 In addition to the regulatory effect of Exo on the inflammatory response, the attachment of LPS or angiotensin II can force cardiac macrophages to stimulate inflammatory status in fibroblasts via miR-155-bearing Exos.107 Other genetic materials such as exosomal circ_0075932 increase Aurora A kinase in dermal keratinocytes after binding to PUM2 protein, resulting in inflammatory responses. Under these conditions, the secretion of MMP-2, and -9 is induced following the activation of the NF-κB signaling pathway. It is thought that these factors can increase tumor cell resistance to therapeutic agents.108 The release of EVs from hypoxic endothelial cells (ECs) and cardiomyocytes led to in situ accumulation of IL-6 and chemokines CCK2 and CCK7.109 Unexpectedly, the induction of inflammation via TLRs in the tumor niche can lead to the inhibition of tumoricidal effects. For instance, exosomal miR21 and mir290 from cancer cells increase the migration of cancer cells after binding to TLR-7 and -8.110 Within the pulmonary niche, EC Exos intensify inflammation in COVID-19, COPD, and asthma subjects via local accumulation of IL-6 (Fig. 4).111,112
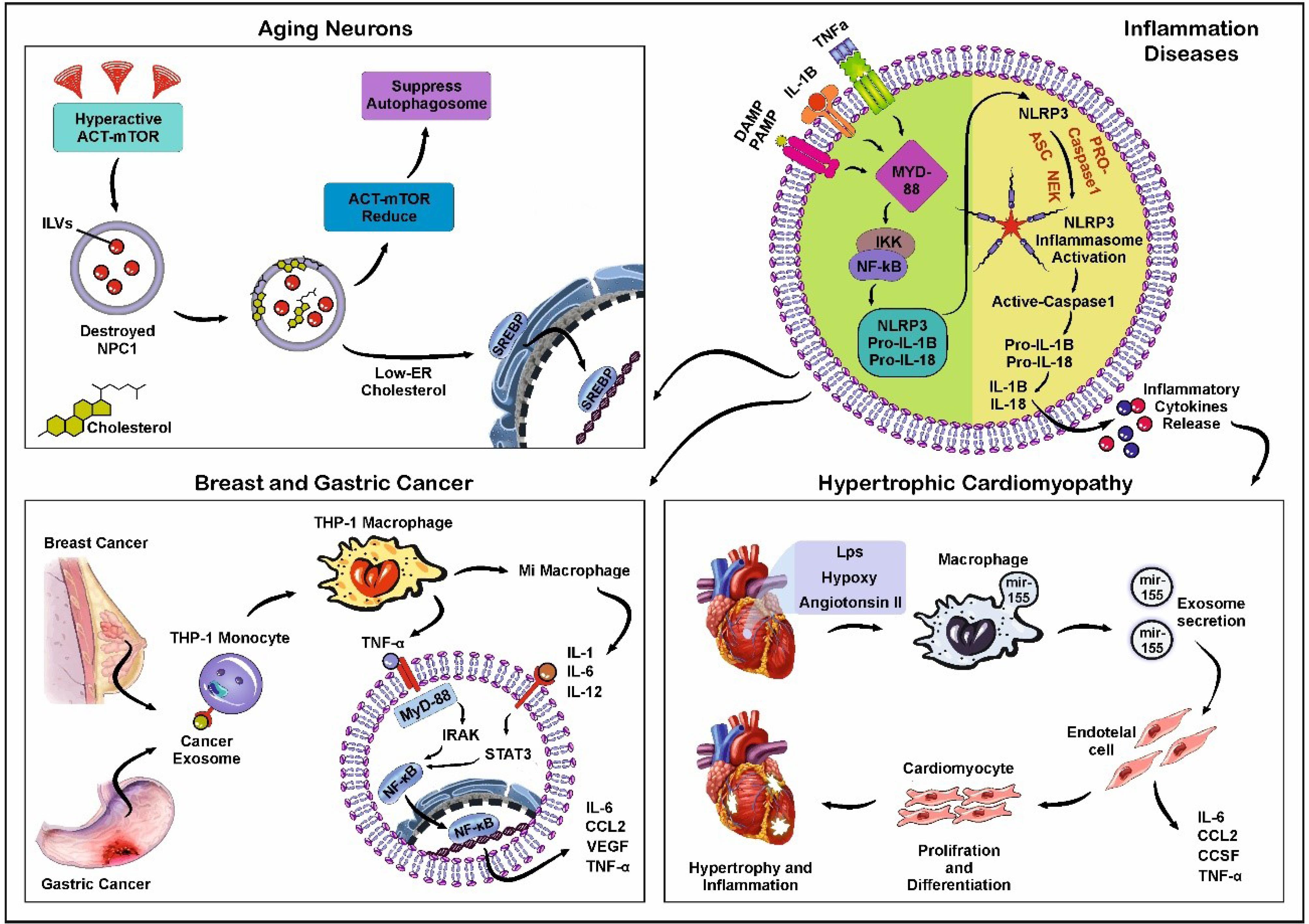
Fig. 4.
Schematic illustration of inflammatory diseases. The activation of NLRP3 Inflammasomes (Upper right). The binding of DAMPs, PAMPs, IL-1B, and TNF-α to cognate cell surface receptors leads to the up-regulation of NLRP3, pro-IL-1, and pro-IL-18 through the activation of NF-κB signaling axis. Some factors can activate NLRP3 inflammasomes independently. After activation of the NLRP3 inflammasomes, Caspase-1 can process pro-IL-1 and pro-IL-18 into their mature forms. Scheme of the alterations found in aging neurons (Upper left). Hyperactivation of the AKT/mTOR pathway during neuronal aging leads to the degeneration of type C protein (NPC1) and accumulation of cholesterol in the endosomal membrane. This phenomenon, in turn, increases the number of ILVs inside MVBs. Decreased NPC1 triggers a low-rate endosome-to-ER cholesterol transport. Also, the down-regulation of the AKT/mTOR pathway suppresses the formation of autophagolysosomes. Schematic of monocyte activation, NF-κB accumulation, and secretion of proinflammatory cytokine in breast and gastric cancers (Lower left). Exos released from breast and gastric tumor cells increases THP-1 monocytes differentiating to mature macrophages. Proinflammatory cytokines such as TNF-a can activate STAT3 and TLRs in a MyD88-dependent manner. In inflamed cardiac tissue (Lower right), some factors such as angiotensin II, LPS, and hypoxic conditions increase the transfer of miR-155 from macrophage into Exos. Secretory Exos enter endothelial cells (ECs) and activate NF-κB, which is a key molecule between inflammation and cardiovascular disease. Synthesis and secretion of pro-inflammatory cytokines by NF-κB lead to hypertrophy and inflammation in the affected cardiac tissue. Created using CorelDRAW Graphics Suite Ver. 250.0.0.230.
.
Schematic illustration of inflammatory diseases. The activation of NLRP3 Inflammasomes (Upper right). The binding of DAMPs, PAMPs, IL-1B, and TNF-α to cognate cell surface receptors leads to the up-regulation of NLRP3, pro-IL-1, and pro-IL-18 through the activation of NF-κB signaling axis. Some factors can activate NLRP3 inflammasomes independently. After activation of the NLRP3 inflammasomes, Caspase-1 can process pro-IL-1 and pro-IL-18 into their mature forms. Scheme of the alterations found in aging neurons (Upper left). Hyperactivation of the AKT/mTOR pathway during neuronal aging leads to the degeneration of type C protein (NPC1) and accumulation of cholesterol in the endosomal membrane. This phenomenon, in turn, increases the number of ILVs inside MVBs. Decreased NPC1 triggers a low-rate endosome-to-ER cholesterol transport. Also, the down-regulation of the AKT/mTOR pathway suppresses the formation of autophagolysosomes. Schematic of monocyte activation, NF-κB accumulation, and secretion of proinflammatory cytokine in breast and gastric cancers (Lower left). Exos released from breast and gastric tumor cells increases THP-1 monocytes differentiating to mature macrophages. Proinflammatory cytokines such as TNF-a can activate STAT3 and TLRs in a MyD88-dependent manner. In inflamed cardiac tissue (Lower right), some factors such as angiotensin II, LPS, and hypoxic conditions increase the transfer of miR-155 from macrophage into Exos. Secretory Exos enter endothelial cells (ECs) and activate NF-κB, which is a key molecule between inflammation and cardiovascular disease. Synthesis and secretion of pro-inflammatory cytokines by NF-κB lead to hypertrophy and inflammation in the affected cardiac tissue. Created using CorelDRAW Graphics Suite Ver. 250.0.0.230.
TLR and angiogenesis potential of ECs
Angiogenesis is a vital biological process in multicellular creatures under physiological and pathological conditions.113 The term angiogenesis points to the formation of new blood vessels from the preexisting vascular networks in response to hypoxia and ischemic conditions.114 Compared to angiogenesis, the term vascularization refers to the generation of de novo blood vessels with the participation of endothelial progenitor cells (EPCs).115 It is thought that the angiogenic switch coincides with the activation of ECs and the formation of neovessel sprouts toward cytokine gradients.116 Whether and how the TLR signaling pathway can regulate the activity of ECs under pathological conditions is at the center of debate. The role of TLR in terms of angiogenesis can be studied in two different aspects; the modulation of TLRs in ECs, and ECs response to non-ECs after inhibition/stimulation of TLRs. Under pro-inflammatory conditions, the activation of TLRs especially TLR-4 in the recruited immune cells can promote inflammation-induced angiogenesis.117 The activation of TLR-4 leads to the activation and homing of certain immune cells such as lymphocytes into injured sites. Upon reaching the target sites, these cells can release several angiogenesis factors and inflammatory cytokines.118 Notably, ECs like several cell types can express TLRs and these receptors are activated in response to damage signals and pathogens.119 It was suggested that the activation of endothelial TLRs can be done in a paracrine manner via the interchange of Exos between the ECs and other cell types such as immune cells.120 For instance, Dalvi et al claimed that LPS- and interferon alpha-primed monocytes secret Exos with distinct miRNA signatures that can promote the expression of adhesion molecules such as CCL2, ICAM-1, VCAM-1 on brain microvascular ECs via the activation of TLR-4 in a MyD88-dependent manner.120 The suppression of Exo secretion in activated monocytes using GW4869 can reduce the inflammatory response in ECs (IL-1β↓, and IL-6↓). Thus, one could hypothesize that the TLR signaling axis has a critical role in the angiogenesis potential of ECs under pathological conditions. It should be noted that the activation of all TLR types does not yield similar angiogenesis potential. In an experiment conducted by Liotti and co-workers, they declared that the activation of TLR-7 in non-small cell lung cancer by imiquimod, a TLR-7 agonist, can reduce the release of VEGF-A, CXCL1, Ang-1, leading to the suppression of angiogenesis response.121 Previously, it was indicated that the incubation of EPCs with LPS leads to the increase of Exo secretion via the elevation of intracellular Ca2+ in P3R-dependent endoplasmic reticulum manner and alteration of PMCA 1/4 expression after stimulation of TLR-4.122 Heidarzadeh et al indicated that the incubation with LPS can affect the EPC survival rate in a dose-dependent manner in in vitro condition.123 In LPS-treated EPCs, the levels of p-ERK1/2 and Erk1/2, NF-kB and TRIF were increased and led to the expression of Exo-related factors. Along with these changes, non-toxic levels of LPS can promote the angiogenesis potential of EPCs in mouse model of subcutaneously injected Matrigel plug.123 Besides the critical role of the TLR signaling pathway in the production and release of Exos from ECs, Exos in turn promotes this signaling cascade in the recipient ECs. In an interesting study, Migneault and co-workers found that the exposure of ECs to serum-depleted conditions promotes apoptotic changes with simultaneous apoptotic Exos production. The attachment of apoptotic Exos to neighboring ECs induced cell migration and wound closure capacity while the angiogenesis potential was significantly diminished.124 Molecular analysis showed that apoptotic Exo lumen contains specific genetic cargo with the potential to activate the NF-kB pathway via engaging TLRs, and RIG-I-like receptors.124 In a similar study, it was shown that the treatment of rat aortic ECs with 100 ng/ml LPS promotes the generation and abscission of Exos compared to the non-treated ECs.125 The uptake of Exos released from LPS-treated ECs was significantly higher in NR8383 alveolar macrophages compared to normal EC Exos. The internalization of pro-inflammatory Exos in macrophages induces M1-type phenotypes with the increase of CD86, and inducible nitric oxide synthase (iNOS). These features were orchestrated via the activation of TLR-4/NF-ĸB and phosphorylation of subunit p65.125 It is also noteworthy to mention that human CD54 + monocyte microparticles harbor higher levels of pro-inflammatory cytokines like TNF-α, IL-6, and -8.126 These particles can per se stimulates the production of Exos in brain hCMEC/D3 ECs.126 Likewise, Treponema pallidum-treated dendritic cell Exos yielded similar outcomes on HUVECs via the activation of the TLR4/MyD88/NF-κB axis.127 HUVECs exposed to dendritic cell Exos exhibited inflammatory phenotypes indicated with IL-1β, -6, and TNF-α. The inhibition of NF-κB with chemical inhibitor BAY11-7082 reduced these effects.127 Emerging data have revealed the close relationship between the TLR signaling pathway and Exo release in endothelial lineage.128 In mouse cardiac ECs, the activation of TLR-4 with bacterial LPS produced Exos with higher levels of Lamp-2, and αvβ6. Further uptake of αvβ6-loaded Exos by dendritic cells and macrophages can contribute to production of VEGF and angiogenesis response.129 The phenomenon of Exo secretion and release is associated with the activation of several intracellular factors. In an experiment, it was shown that the release of Exo is dependent on certain miRNAs, especially miR-214. ECs needs miR-214 to release the Exos in which the inhibition of this factor blunt the angiogenesis capacity.130 In several cancer cells, the activation of TLRs can induce specific miRNA types such as miR-214.131 These data show the possible cross-talk between the TLR signaling pathway and Exo biogenesis mechanisms.
Conclusion
TLR signaling pathway can affect several bioactivities in the host cells after being exposed to different danger signals. Based on emerging data, it was suggested that the TLR signaling pathway can affect the paracrine activity of cells via the release of Exos into the ECM. It is thought that several shared molecular mechanisms exist between the TLR signaling pathway and endosomal machinery. Thus, the modulation of TLRs can affect the Exo biogenesis and abscission in different cell types such as stem cells. Meanwhile, Exos can also mediate their effects on the target cells in part via the modulation of TLRs especially in immune cells. To be specific, these nano-sized particles can harbor several inflammatory cytokines that target PAMP receptors and TLR, leading to the stimulation of downstream molecules. Overall, continued and extensive research is needed to investigate the relationship between exosomes, inflammation, and tumors and how they mediate the inflammatory process. In the context of angiogenesis, the close relationship between the Exo, and TLRs is integral to EC activity. Thus, using certain therapeutic strategies, i.e., inhibition or stimulation of distinct TLR types, it is possible to control the blood supply to the affected sites.
Review Highlights
What is the current knowledge?
-
Exos participate in different cell functions and bioactivities related to several lineages.
-
How and by which mechanisms, Exos exert their biological effects remain unknown.
What is new here?
Competing Interests
The authors declare that they have no competing interests.
Ethical Statement
This study was approved by the local ethics committee of Tabriz University of Medical Sciences under an ethical code of IR.TBZMED.VCR.REC.1399.477.
Acknowledgements
Authors wish to thank the personnel of the Faculty of Advanced Medical Sciences, Faculty of Medicine, and Stem Cell Research Center for help and guidance.
References
- Rodrigues CR, Balachandran Y, Aulakh GK, Singh B. TLR10: an intriguing toll-like receptor with many unanswered questions. J Innate Immun 2024; 16:96-104. doi: 10.1159/000535523 [Crossref] [ Google Scholar]
- Zhou Y, Little PJ, Downey L, Afroz R, Wu Y, Ta HT. The role of toll-like receptors in atherothrombotic cardiovascular disease. ACS Pharmacol Transl Sci 2020; 3:457-71. doi: 10.1021/acsptsci.9b00100 [Crossref] [ Google Scholar]
- Duan T, Du Y, Xing C, Wang HY, Wang RF. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol 2022; 13:812774. doi: 10.3389/fimmu.2022.812774 [Crossref] [ Google Scholar]
- Parizadeh SM, Ghandehari M, Heydari-Majd M, Seifi S, Mardani R, Parizadeh SM. Toll-like receptors signaling pathways as a potential therapeutic target in cardiovascular disease. Curr Pharm Des 2018; 24:1887-98. doi: 10.2174/1381612824666180614090224 [Crossref] [ Google Scholar]
- Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll-like receptors: significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol 2018; 37:20-36. doi: 10.1080/08830185.2017.1380200 [Crossref] [ Google Scholar]
- Lannoy V, Côté-Biron A, Asselin C, Rivard N. TIRAP, TRAM, and toll-like receptors: the untold story. Mediators Inflamm 2023; 2023:2899271. doi: 10.1155/2023/2899271 [Crossref] [ Google Scholar]
- Yang M, Zhang C. The role of innate immunity in diabetic nephropathy and their therapeutic consequences. J Pharm Anal 2024; 14:39-51. doi: 10.1016/j.jpha.2023.09.003 [Crossref] [ Google Scholar]
- Wu Z, Zhang Z, Lei Z, Lei P. CD14: biology and role in the pathogenesis of disease. Cytokine Growth Factor Rev 2019; 48:24-31. doi: 10.1016/j.cytogfr.2019.06.003 [Crossref] [ Google Scholar]
- Goulopoulou S, McCarthy CG, Webb RC. Toll-like receptors in the vascular system: sensing the dangers within. Pharmacol Rev 2016; 68:142-67. doi: 10.1124/pr.114.010090 [Crossref] [ Google Scholar]
- Fitzpatrick JM, Minogue E, Curham L, Tyrrell H, Gavigan P, Hind W. MyD88-dependent and -independent signalling via TLR3 and TLR4 are differentially modulated by Δ9-tetrahydrocannabinol and cannabidiol in human macrophages. J Neuroimmunol 2020; 343:577217. doi: 10.1016/j.jneuroim.2020.577217 [Crossref] [ Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011; 34:637-50. doi: 10.1016/j.immuni.2011.05.006 [Crossref] [ Google Scholar]
- Gao W, Xiong Y, Li Q, Yang H. Inhibition of toll-like receptor signaling as a promising therapy for inflammatory diseases: a journey from molecular to nano therapeutics. Front Physiol 2017; 8:508. doi: 10.3389/fphys.2017.00508 [Crossref] [ Google Scholar]
- Crews FT, Qin L, Sheedy D, Vetreno RP, Zou J. High mobility group box 1/toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry 2013; 73:602-12. doi: 10.1016/j.biopsych.2012.09.030 [Crossref] [ Google Scholar]
- Yuk JM, Jo EK. Toll-like receptors and innate immunity. J Bacteriol Virol 2011; 41:225-35. doi: 10.4167/jbv.2011.41.4.225 [Crossref] [ Google Scholar]
- Ajibade AA, Wang HY, Wang RF. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol 2013; 34:307-16. doi: 10.1016/j.it.2013.03.007 [Crossref] [ Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol 2010; 11:373-84. doi: 10.1038/ni.1863 [Crossref] [ Google Scholar]
- Brennan RS, Healy TM, Bryant HJ, La MV, Schulte PM, Whitehead A. Integrative population and physiological genomics reveals mechanisms of adaptation in killifish. Mol Biol Evol 2018; 35:2639-53. doi: 10.1093/molbev/msy154 [Crossref] [ Google Scholar]
- Fakhraei Lahiji S, Jang Y, Ma Y, Dangol M, Yang H, Jang M. Effects of dissolving microneedle fabrication parameters on the activity of encapsulated lysozyme. Eur J Pharm Sci 2018; 117:290-6. doi: 10.1016/j.ejps.2018.03.003 [Crossref] [ Google Scholar]
- Ha T, Hua F, Li Y, Ma J, Gao X, Kelley J. Blockade of MyD88 attenuates cardiac hypertrophy and decreases cardiac myocyte apoptosis in pressure overload-induced cardiac hypertrophy in vivo. Am J Physiol Heart Circ Physiol 2006; 290:H985-94. doi: 10.1152/ajpheart.00720.2005 [Crossref] [ Google Scholar]
- Leulier F, Lemaitre B. Toll-like receptors--taking an evolutionary approach. Nat Rev Genet 2008; 9:165-78. doi: 10.1038/nrg2303 [Crossref] [ Google Scholar]
- Aheget H, Mazini L, Martin F, Belqat B, Marchal JA, Benabdellah K. Exosomes: their role in pathogenesis, diagnosis and treatment of diseases. Cancers (Basel) 2020; 13:84. doi: 10.3390/cancers13010084 [Crossref] [ Google Scholar]
- Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell 2016; 164:1226-32. doi: 10.1016/j.cell.2016.01.043 [Crossref] [ Google Scholar]
- Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 2018; 7:1535750. doi: 10.1080/20013078.2018.1535750 [Crossref] [ Google Scholar]
- Johnstone RM. Revisiting the road to the discovery of exosomes. Blood Cells Mol Dis 2005; 34:214-9. doi: 10.1016/j.bcmd.2005.03.002 [Crossref] [ Google Scholar]
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 2013; 200:373-83. doi: 10.1083/jcb.201211138 [Crossref] [ Google Scholar]
- Moghadasi S, Elveny M, Rahman HS, Suksatan W, Jalil AT, Abdelbasset WK. A paradigm shift in cell-free approach: the emerging role of MSCs-derived exosomes in regenerative medicine. J Transl Med 2021; 19:302. doi: 10.1186/s12967-021-02980-6 [Crossref] [ Google Scholar]
- Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol 2015; 40:41-51. doi: 10.1016/j.semcdb.2015.02.010 [Crossref] [ Google Scholar]
- Abbasi-Malati Z, Azizi SG, Milani SZ, Serej ZA, Mardi N, Amiri Z. Tumorigenic and tumoricidal properties of exosomes in cancers; a forward look. Cell Commun Signal 2024; 22:130. doi: 10.1186/s12964-024-01510-3 [Crossref] [ Google Scholar]
- Kenific CM, Zhang H, Lyden D. An exosome pathway without an ESCRT. Cell Res 2021; 31:105-6. doi: 10.1038/s41422-020-00418-0 [Crossref] [ Google Scholar]
- Hurley JH. ESCRTs are everywhere. EMBO J 2015; 34:2398-407. doi: 10.15252/embj.201592484 [Crossref] [ Google Scholar]
- Kalra H, Drummen GP, Mathivanan S. Focus on extracellular vesicles: introducing the next small big thing. Int J Mol Sci 2016; 17:170. doi: 10.3390/ijms17020170 [Crossref] [ Google Scholar]
- Kowal J, Tkach M, Théry C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol 2014; 29:116-25. doi: 10.1016/j.ceb.2014.05.004 [Crossref] [ Google Scholar]
- Villarroya-Beltri C, Baixauli F, Gutiérrez-Vázquez C, Sánchez-Madrid F, Mittelbrunn M. Sorting it out: regulation of exosome loading. Semin Cancer Biol 2014; 28:3-13. doi: 10.1016/j.semcancer.2014.04.009 [Crossref] [ Google Scholar]
- Hanson PI, Cashikar A. Multivesicular body morphogenesis. Annu Rev Cell Dev Biol 2012; 28:337-62. doi: 10.1146/annurev-cellbio-092910-154152 [Crossref] [ Google Scholar]
- Airola MV, Hannun YA. Sphingolipid metabolism and neutral sphingomyelinases. Handb Exp Pharmacol 2013; 57-76. 10.1007/978-3-7091-1368-4_3.
- Stuffers S, Sem Wegner C, Stenmark H, Brech A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009; 10:925-37. doi: 10.1111/j.1600-0854.2009.00920.x [Crossref] [ Google Scholar]
- Meckes DG Jr, Raab-Traub N. Microvesicles and viral infection. J Virol 2011; 85:12844-54. doi: 10.1128/jvi.05853-11 [Crossref] [ Google Scholar]
- Stipp CS, Kolesnikova TV, Hemler ME. Functional domains in tetraspanin proteins. Trends Biochem Sci 2003; 28:106-12. doi: 10.1016/s0968-0004(02)00014-2 [Crossref] [ Google Scholar]
- Subra C, Grand D, Laulagnier K, Stella A, Lambeau G, Paillasse M. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J Lipid Res 2010; 51:2105-20. doi: 10.1194/jlr.M003657 [Crossref] [ Google Scholar]
- Andreu Z, Yáñez-Mó M. Tetraspanins in extracellular vesicle formation and function. Front Immunol 2014; 5:442. doi: 10.3389/fimmu.2014.00442 [Crossref] [ Google Scholar]
- Charrin S, Jouannet S, Boucheix C, Rubinstein E. Tetraspanins at a glance. J Cell Sci 2014; 127:3641-8. doi: 10.1242/jcs.154906 [Crossref] [ Google Scholar]
- Short B. β-Catenin gets an honorable discharge. J Cell Biol 2010; 190:945. doi: 10.1083/jcb.1906if [Crossref] [ Google Scholar]
- Edgar JR, Eden ER, Futter CE. Hrs- and CD63-dependent competing mechanisms make different sized endosomal intraluminal vesicles. Traffic 2014; 15:197-211. doi: 10.1111/tra.12139 [Crossref] [ Google Scholar]
- Perez-Hernandez D, Gutiérrez-Vázquez C, Jorge I, López-Martín S, Ursa A, Sánchez-Madrid F. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. J Biol Chem 2013; 288:11649-61. doi: 10.1074/jbc.M112.445304 [Crossref] [ Google Scholar]
- Beach A, Zhang HG, Ratajczak MZ, Kakar SS. Exosomes: an overview of biogenesis, composition and role in ovarian cancer. J Ovarian Res 2014; 7:14. doi: 10.1186/1757-2215-7-14 [Crossref] [ Google Scholar]
- Jadli AS, Ballasy N, Edalat P, Patel VB. Inside(sight) of tiny communicator: exosome biogenesis, secretion, and uptake. Mol Cell Biochem 2020; 467:77-94. doi: 10.1007/s11010-020-03703-z [Crossref] [ Google Scholar]
- Hsu C, Morohashi Y, Yoshimura S, Manrique-Hoyos N, Jung S, Lauterbach MA. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J Cell Biol 2010; 189:223-32. doi: 10.1083/jcb.200911018 [Crossref] [ Google Scholar]
- Short B, Barr FA. Membrane traffic: exocyst III--makes a family. Curr Biol 2002; 12:R18-20. doi: 10.1016/s0960-9822(01)00641-8 [Crossref] [ Google Scholar]
- Stenmark H, Olkkonen VM. The Rab GTPase family. Genome Biol 2001; 2:Reviews3007. doi: 10.1186/gb-2001-2-5-reviews3007 [Crossref] [ Google Scholar]
- Garcia NA, Ontoria-Oviedo I, González-King H, Diez-Juan A, Sepúlveda P. Glucose starvation in cardiomyocytes enhances exosome secretion and promotes angiogenesis in endothelial cells. PLoS One 2015; 10:e0138849. doi: 10.1371/journal.pone.0138849 [Crossref] [ Google Scholar]
- Fukuda M. Rab27 effectors, pleiotropic regulators in secretory pathways. Traffic 2013; 14:949-63. doi: 10.1111/tra.12083 [Crossref] [ Google Scholar]
- Yue B, Yang H, Wang J, Ru W, Wu J, Huang Y. Exosome biogenesis, secretion and function of exosomal miRNAs in skeletal muscle myogenesis. Cell Prolif 2020; 53:e12857. doi: 10.1111/cpr.12857 [Crossref] [ Google Scholar]
- Dingjan I, Linders PTA, Verboogen DRJ, Revelo NH, Ter Beest M, van den Bogaart G. Endosomal and phagosomal SNAREs. Physiol Rev 2018; 98:1465-92. doi: 10.1152/physrev.00037.2017 [Crossref] [ Google Scholar]
- Collins CA, Brown EJ. Cytosol as battleground: ubiquitin as a weapon for both host and pathogen. Trends Cell Biol 2010; 20:205-13. doi: 10.1016/j.tcb.2010.01.002 [Crossref] [ Google Scholar]
- Loureiro J, Ploegh HL. Antigen presentation and the ubiquitin-proteasome system in host-pathogen interactions. Adv Immunol 2006; 92:225-305. doi: 10.1016/s0065-2776(06)92006-9 [Crossref] [ Google Scholar]
- Sun SC, Ley SC. New insights into NF-kappaB regulation and function. Trends Immunol 2008; 29:469-78. doi: 10.1016/j.it.2008.07.003 [Crossref] [ Google Scholar]
- Courtois G. Tumor suppressor CYLD: negative regulation of NF-kappaB signaling and more. Cell Mol Life Sci 2008; 65:1123-32. doi: 10.1007/s00018-007-7465-4 [Crossref] [ Google Scholar]
- Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004; 430:694-9. doi: 10.1038/nature02794 [Crossref] [ Google Scholar]
- Lilley BN, Ploegh HL. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A 2005; 102:14296-301. doi: 10.1073/pnas.0505014102 [Crossref] [ Google Scholar]
- Carta S, Lavieri R, Rubartelli A. Different members of the IL-1 family come out in different ways: DAMPs vs. cytokines? Front Immunol 2013; 4:123. doi: 10.3389/fimmu.2013.00123 [Crossref] [ Google Scholar]
- Stanley AC, Lacy P. Pathways for cytokine secretion. Physiology (Bethesda) 2010; 25:218-29. doi: 10.1152/physiol.00017.2010 [Crossref] [ Google Scholar]
- Lacy P. The role of Rho GTPases and SNAREs in mediator release from granulocytes. Pharmacol Ther 2005; 107:358-76. doi: 10.1016/j.pharmthera.2005.03.008 [Crossref] [ Google Scholar]
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 2009; 10:513-25. doi: 10.1038/nrm2728 [Crossref] [ Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science 2009; 323:474-7. doi: 10.1126/science.1161748 [Crossref] [ Google Scholar]
- Stow JL, Manderson AP, Murray RZ. SNAREing immunity: the role of SNAREs in the immune system. Nat Rev Immunol 2006; 6:919-29. doi: 10.1038/nri1980 [Crossref] [ Google Scholar]
- Collins LE, DeCourcey J, Soledad di Luca M, Rochfort KD, Loscher CE. An emerging role for SNARE proteins in dendritic cell function. Front Immunol 2015; 6:133. doi: 10.3389/fimmu.2015.00133 [Crossref] [ Google Scholar]
- Maringer K, Fernandez-Sesma A. Message in a bottle: lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor Rev 2014; 25:669-79. doi: 10.1016/j.cytogfr.2014.08.004 [Crossref] [ Google Scholar]
- Stow JL, Manderson AP, Murray RZ. SNAREing immunity: the role of SNAREs in the immune system. Nat Rev Immunol 2006; 6:919-29. doi: 10.1038/nri1980 [Crossref] [ Google Scholar]
- Lacy P, Stow JL. Cytokine release from innate immune cells: association with diverse membrane trafficking pathways. Blood 2011; 118:9-18. doi: 10.1182/blood-2010-08-265892 [Crossref] [ Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 24 A resolution. Nature 1998; 395:347-53. doi: 10.1038/26412 [Crossref] [ Google Scholar]
- Torp MK, Vaage J, Stensløkken KO. Mitochondria-derived damage-associated molecular patterns and inflammation in the ischemic-reperfused heart. Acta Physiol (Oxf) 2023; 237:e13920. doi: 10.1111/apha.13920 [Crossref] [ Google Scholar]
- Nakayama H, Otsu K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem J 2018; 475:839-52. doi: 10.1042/bcj20170714 [Crossref] [ Google Scholar]
- Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res 2016; 119:91-112. doi: 10.1161/circresaha.116.303577 [Crossref] [ Google Scholar]
- Rutkovskiy A, Bliksøen M, Hillestad V, Amin M, Czibik G, Valen G. Aquaporin-1 in cardiac endothelial cells is downregulated in ischemia, hypoxia and cardioplegia. J Mol Cell Cardiol 2013; 56:22-33. doi: 10.1016/j.yjmcc.2012.12.002 [Crossref] [ Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013; 339:826-30. doi: 10.1126/science.1229963 [Crossref] [ Google Scholar]
- Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 2012; 26:203-34. doi: 10.1101/gad.183434.111 [Crossref] [ Google Scholar]
- Kato K, Omura H, Ishitani R, Nureki O. Cyclic GMP-AMP as an endogenous second messenger in innate immune signaling by cytosolic DNA. Annu Rev Biochem 2017; 86:541-66. doi: 10.1146/annurev-biochem-061516-044813 [Crossref] [ Google Scholar]
- Apostolakis S, Vogiatzi K, Amanatidou V, Spandidos DA. Interleukin 8 and cardiovascular disease. Cardiovasc Res 2009; 84:353-60. doi: 10.1093/cvr/cvp241 [Crossref] [ Google Scholar]
- Song M, Meng L, Liu X, Yang Y. Feprazone prevents free fatty acid (FFA)-induced endothelial inflammation by mitigating the activation of the TLR4/MyD88/NF-κB pathway. ACS Omega 2021; 6:4850-6. doi: 10.1021/acsomega.0c05826 [Crossref] [ Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417-26. doi: 10.1016/s1097-2765(02)00599-3 [Crossref] [ Google Scholar]
- Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A 2011; 108:19725-30. doi: 10.1073/pnas.1108586108 [Crossref] [ Google Scholar]
- Li S, Hu Q, Huang J, Wu X, Ren J. Mitochondria-derived damage-associated molecular patterns in sepsis: from bench to bedside. Oxid Med Cell Longev 2019; 2019:6914849. doi: 10.1155/2019/6914849 [Crossref] [ Google Scholar]
- Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol 2004; 75:995-1000. doi: 10.1189/jlb.0703328 [Crossref] [ Google Scholar]
- Donnino MW, Liu X, Andersen LW, Rittenberger JC, Abella BS, Gaieski DF. Characterization of mitochondrial injury after cardiac arrest (COMICA). Resuscitation 2017; 113:56-62. doi: 10.1016/j.resuscitation.2016.12.029 [Crossref] [ Google Scholar]
- Galluzzi L, Vanpouille-Box C, Bakhoum SF, Demaria S. SnapShot: CGAS-STING signaling. Cell 2018; 173: 276-276.e1. 10.1016/j.cell.2018.03.015.
- Rezaie J, Hosseinpour H, Rahbarghazi R, Keyhanmanesh R, Khanzadeh S, Mahdipour M. Type 2 diabetes mellitus stimulated pulmonary vascular inflammation and exosome biogenesis in rats. Cell Biochem Funct 2023; 41:78-85. doi: 10.1002/cbf.3764 [Crossref] [ Google Scholar]
- Matikainen S, Nyman TA, Cypryk W. Inflammasomes: exosomal miRNAs loaded for action. J Cell Biol 2020; 219:e202008130. doi: 10.1083/jcb.202008130 [Crossref] [ Google Scholar]
- Chan BD, Wong WY, Lee MM, Cho WC, Yee BK, Kwan YW. Exosomes in inflammation and inflammatory disease. Proteomics 2019; 19:e1800149. doi: 10.1002/pmic.201800149 [Crossref] [ Google Scholar]
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 2009; 1:a001651. doi: 10.1101/cshperspect.a001651 [Crossref] [ Google Scholar]
- Das S, Halushka MK. Extracellular vesicle microRNA transfer in cardiovascular disease. Cardiovasc Pathol 2015; 24:199-206. doi: 10.1016/j.carpath.2015.04.007 [Crossref] [ Google Scholar]
- Elizagaray ML, Gomes MT, Guimaraes ES, Rumbo M, Hozbor DF, Oliveira SC. Canonical and non-canonical inflammasome activation by outer membrane vesicles derived from Bordetella pertussis. Front Immunol 2020; 11:1879. doi: 10.3389/fimmu.2020.01879 [Crossref] [ Google Scholar]
- Vijayaraj SL, Feltham R, Rashidi M, Frank D, Liu Z, Simpson DS. The ubiquitylation of IL-1β limits its cleavage by caspase-1 and targets it for proteasomal degradation. Nat Commun 2021; 12:2713. doi: 10.1038/s41467-021-22979-3 [Crossref] [ Google Scholar]
- Wozniak AL, Adams A, King KE, Dunn W, Christenson LK, Hung WT, et al. The RNA binding protein FMR1 controls selective exosomal miRNA cargo loading during inflammation. J Cell Biol 2020; 219. 10.1083/jcb.201912074.
- Chen Y, Zhang M, Zheng Y. Glucocorticoids inhibit production of exosomes containing inflammatory microRNA-155 in lipopolysaccharide-induced macrophage inflammatory responses. Int J Clin Exp Pathol 2018; 11:3391-7. [ Google Scholar]
- Huang D, Li G, Bhat OM, Zou Y, Li N, Ritter JK. Exosome biogenesis and lysosome function determine podocyte exosome release and glomerular inflammatory response during hyperhomocysteinemia. Am J Pathol 2022; 192:43-55. doi: 10.1016/j.ajpath.2021.10.005 [Crossref] [ Google Scholar]
- Feng Y, Lv LL, Tang TT, Zhong X, Chen J, Liu BC. FO007Rab27a dependent exosome secretion from tubular epithelial cell mediates albumin handling and promotes tubulointerstitial inflammation in albuminuric nephropathy. Nephrol Dial Transplant 2019; 34: gfz096.FO007. 10.1093/ndt/gfz096.FO007.
- Feng Y, Zhong X, Tang TT, Wang C, Wang LT, Li ZL. Rab27a dependent exosome releasing participated in albumin handling as a coordinated approach to lysosome in kidney disease. Cell Death Dis 2020; 11:513. doi: 10.1038/s41419-020-2709-4 [Crossref] [ Google Scholar]
- Essandoh K, Yang L, Wang X, Huang W, Qin D, Hao J. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim Biophys Acta 2015; 1852:2362-71. doi: 10.1016/j.bbadis.2015.08.010 [Crossref] [ Google Scholar]
- Mathews PM, Levy E. Exosome production is key to neuronal endosomal pathway integrity in neurodegenerative diseases. Front Neurosci 2019; 13:1347. doi: 10.3389/fnins.2019.01347 [Crossref] [ Google Scholar]
- Guix FX, Capitán AM, Casadomé-Perales Á, Palomares-Pérez I, López Del Castillo I, Miguel V. Increased exosome secretion in neurons aging in vitro by NPC1-mediated endosomal cholesterol buildup. Life Sci Alliance 2021; 4:e202101055. doi: 10.26508/lsa.202101055 [Crossref] [ Google Scholar]
- Park SJ, Kim JM, Kim J, Hur J, Park S, Kim K. Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc Natl Acad Sci U S A 2018; 115:E11721-30. doi: 10.1073/pnas.1811432115 [Crossref] [ Google Scholar]
- Wu L, Zhang X, Zhang B, Shi H, Yuan X, Sun Y. Exosomes derived from gastric cancer cells activate NF-κB pathway in macrophages to promote cancer progression. Tumour Biol 2016; 37:12169-80. doi: 10.1007/s13277-016-5071-5 [Crossref] [ Google Scholar]
- Chow A, Zhou W, Liu L, Fong MY, Champer J, Van Haute D. Macrophage immunomodulation by breast cancer-derived exosomes requires toll-like receptor 2-mediated activation of NF-κB. Sci Rep 2014; 4:5750. doi: 10.1038/srep05750 [Crossref] [ Google Scholar]
- Bretz NP, Ridinger J, Rupp AK, Rimbach K, Keller S, Rupp C. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via toll-like receptor signaling. J Biol Chem 2013; 288:36691-702. doi: 10.1074/jbc.M113.512806 [Crossref] [ Google Scholar]
- Bromberg J, Darnell JE Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000; 19:2468-73. doi: 10.1038/sj.onc.1203476 [Crossref] [ Google Scholar]
- Maji S, Chaudhary P, Akopova I, Nguyen PM, Hare RJ, Gryczynski I. Exosomal annexin II promotes angiogenesis and breast cancer metastasis. Mol Cancer Res 2017; 15:93-105. doi: 10.1158/1541-7786.mcr-16-0163 [Crossref] [ Google Scholar]
- Wang C, Zhang C, Liu L, A X, Chen B, Li Y. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol Ther 2017; 25:192-204. doi: 10.1016/j.ymthe.2016.09.001 [Crossref] [ Google Scholar]
- Wei TW, Wu PY, Wu TJ, Hou HA, Chou WC, Teng CJ. Aurora A and NF-κB survival pathway drive chemoresistance in acute myeloid leukemia via the TRAF-interacting protein TIFA. Cancer Res 2017; 77:494-508. doi: 10.1158/0008-5472.can-16-1004 [Crossref] [ Google Scholar]
- Loyer X, Zlatanova I, Devue C, Yin M, Howangyin KY, Klaihmon P. Intra-cardiac release of extracellular vesicles shapes inflammation following myocardial infarction. Circ Res 2018; 123:100-6. doi: 10.1161/circresaha.117.311326 [Crossref] [ Google Scholar]
- Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R. MicroRNAs bind to toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 2012; 109:E2110-6. doi: 10.1073/pnas.1209414109 [Crossref] [ Google Scholar]
- Maremanda KP, Sundar IK, Rahman I. Protective role of mesenchymal stem cells and mesenchymal stem cell-derived exosomes in cigarette smoke-induced mitochondrial dysfunction in mice. Toxicol Appl Pharmacol 2019; 385:114788. doi: 10.1016/j.taap.2019.114788 [Crossref] [ Google Scholar]
- Benedikter BJ, Wouters EF, Savelkoul PH, Rohde GG, Stassen FR. Extracellular vesicles released in response to respiratory exposures: implications for chronic disease. J Toxicol Environ Health B Crit Rev 2018; 21:142-60. doi: 10.1080/10937404.2018.1466380 [Crossref] [ Google Scholar]
- Dudley AC, Griffioen AW. Pathological angiogenesis: mechanisms and therapeutic strategies. Angiogenesis 2023; 26:313-47. doi: 10.1007/s10456-023-09876-7 [Crossref] [ Google Scholar]
- Krattiger LA, Moser LO, Odabasi R, Odriozola A, Simona BR, Djonov V. Recovery of therapeutically ablated engineered blood-vessel networks on a plug-and-play platform. Adv Healthc Mater 2024; 13:e2301142. doi: 10.1002/adhm.202301142 [Crossref] [ Google Scholar]
- Zhao Y, Du L, Han L, Liu F, Chen S, Li Z. Exosomal hsa_circ_0093884 derived from endothelial progenitor cells promotes therapeutic neovascularization via miR-145/SIRT1 pathway. Biomed Pharmacother 2024; 173:116343. doi: 10.1016/j.biopha.2024.116343 [Crossref] [ Google Scholar]
- Hassanpour P, Sadeghsoltani F, Haiaty S, Zakeri Z, Saghebasl S, Izadpanah M. Mitochondria-loaded alginate-based hydrogel accelerated angiogenesis in a rat model of acute myocardial infarction. Int J Biol Macromol 2024; 260:129633. doi: 10.1016/j.ijbiomac.2024.129633 [Crossref] [ Google Scholar]
- Bhagwani A, Thompson AA, Farkas L. When innate immunity meets angiogenesis-the role of toll-like receptors in endothelial cells and pulmonary hypertension. Front Med (Lausanne) 2020; 7:352. doi: 10.3389/fmed.2020.00352 [Crossref] [ Google Scholar]
- Murad S. Toll-like receptor 4 in inflammation and angiogenesis: a double-edged sword. Front Immunol 2014; 5:313. doi: 10.3389/fimmu.2014.00313 [Crossref] [ Google Scholar]
- Salvador B, Arranz A, Francisco S, Córdoba L, Punzón C, Llamas M. Modulation of endothelial function by toll-like receptors. Pharmacol Res 2016; 108:46-56. doi: 10.1016/j.phrs.2016.03.038 [Crossref] [ Google Scholar]
- Dalvi P, Sun B, Tang N, Pulliam L. Immune activated monocyte exosomes alter microRNAs in brain endothelial cells and initiate an inflammatory response through the TLR4/MyD88 pathway. Sci Rep 2017; 7:9954. doi: 10.1038/s41598-017-10449-0 [Crossref] [ Google Scholar]
- Liotti F, Marotta M, Sorriento D, Pone E, Morra F, Melillo RM. Toll-like receptor 7 mediates inflammation resolution and inhibition of angiogenesis in non-small cell lung cancer. Cancers (Basel) 2021; 13:740. doi: 10.3390/cancers13040740 [Crossref] [ Google Scholar]
- Xia L, Wang X, Yao W, Wang M, Zhu J. Lipopolysaccharide increases exosomes secretion from endothelial progenitor cells by toll-like receptor 4 dependent mechanism. Biol Cell 2022; 114:127-37. doi: 10.1111/boc.202100086 [Crossref] [ Google Scholar]
- Heidarzadeh M, Biray Avci C, Saberianpour S, Ahmadi M, Hassanpour M, Saghaei Bagheri H. Activation of toll-like receptor signaling in endothelial progenitor cells dictates angiogenic potential: from hypothesis to actual state. Cell Tissue Res 2021; 384:389-401. doi: 10.1007/s00441-020-03405-4 [Crossref] [ Google Scholar]
- Migneault F, Dieudé M, Turgeon J, Beillevaire D, Hardy MP, Brodeur A. Apoptotic exosome-like vesicles regulate endothelial gene expression, inflammatory signaling, and function through the NF-κB signaling pathway. Sci Rep 2020; 10:12562. doi: 10.1038/s41598-020-69548-0 [Crossref] [ Google Scholar]
- He Z, Greven J, Shi Y, Qin K, Zhao Q, Zhang X. Extracellular vesicles derived from endothelial cells modulate macrophage phenotype in vitro. Eur J Med Res 2023; 28:506. doi: 10.1186/s40001-023-01427-6 [Crossref] [ Google Scholar]
- Wen B, Combes V, Bonhoure A, Weksler BB, Couraud PO, Grau GE. Endotoxin-induced monocytic microparticles have contrasting effects on endothelial inflammatory responses. PLoS One 2014; 9:e91597. doi: 10.1371/journal.pone.0091597 [Crossref] [ Google Scholar]
- Zhang R, Lian T, Liu J, Du F, Chen Z, Zhang R. Dendritic cell-derived exosomes stimulated by treponema pallidum induce endothelial cell inflammatory response through the TLR4/MyD88/NF-κB signaling pathway. ACS Infect Dis 2023; 9:2299-305. doi: 10.1021/acsinfecdis.3c00348 [Crossref] [ Google Scholar]
- Song J, Chen X, Wang M, Xing Y, Zheng Z, Hu S. Cardiac endothelial cell-derived exosomes induce specific regulatory B cells. Sci Rep 2014; 4:7583. doi: 10.1038/srep07583 [Crossref] [ Google Scholar]
- Nam EH, Park SR, Kim PH. TGF-beta1 induces mouse dendritic cells to express VEGF and its receptor (Flt-1) under hypoxic conditions. Exp Mol Med 2010; 42:606-13. doi: 10.3858/emm.2010.42.9.059 [Crossref] [ Google Scholar]
- van Balkom BW, de Jong OG, Smits M, Brummelman J, den Ouden K, de Bree PM. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 2013; 121:3997-4006. doi: 10.1182/blood-2013-02-478925 [Crossref] [ Google Scholar]
- Nahid MA, Satoh M, Chan EK. MicroRNA in TLR signaling and endotoxin tolerance. Cell Mol Immunol 2011; 8:388-403. doi: 10.1038/cmi.2011.26 [Crossref] [ Google Scholar]