Bioimpacts. 2025;15:30335.
doi: 10.34172/bi.30335
Original Article
In-silico based discovery of potential Keap1 inhibitors using the strategies of pharmacophore screening, molecular docking, and MD simulation studies
Ekta Singh Conceptualization, Data curation, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing, 1, 2 
Gurubasavaraja Swamy Purawarga Matada Formal analysis, Project administration, Resources, Supervision, Validation, Visualization, 1, *
Prasad Sanjay Dhiwar Data curation, Investigation, 1
Rajesh B. Patil Data curation, Investigation, 3
Rohit Pal Writing – original draft, Writing – review & editing, 1
Author information:
1Integrated Drug Discovery Centre, Department of Pharmaceutical Chemistry, Acharya & BM Reddy College of Pharmacy, Bengaluru 560107, Karnataka, India
2Aditya Bangalore Institute of Pharmacy Education and Research, Department of Pharmaceutical Chemistry, 560064, Karnataka, India
3Sinhgad Technical Education Society’s, Sinhgad College of Pharmacy, Department of Pharmaceutical Chemistry, Off Sinhgad Road, Vadgaon (Bk), Pune 411041, Maharashtra, India
Abstract
Introduction:
The main objective of this research is to identify potential leads for developing potent Keap1 inhibitors.
Methods:
In the current research article, in-silico methods have been employed to discover potential Keap1 inhibitors. 3D-QSAR was generated using the ChemBL database of Keap1 inhibitors with IC50. The best pharmacophore was selected for the screening of three different libraries namely Asinex, MiniMaybridge, and Zinc. The molecules screened from the databases were filtered through druggability rules and molecular docking studies. The best binding molecules obtained after docking studies were subjected to physicochemical properties toxicity determination by in-silico methods. The best hits were studied for stability in the cavity of Keap1 by molecular dynamic simulations.
Results:
The virtual screening of different databases was carried out separately and three leads, were obtained. These lead molecules ASINEX 508, MiniMaybridgeHTS_01719, and ZINC 0000952883 showed the best binding in the Keap1 cavity. The molecular dynamic simulations of the binding complexes of the leads support the docking analysis. The leads (ASINEX 508, MiniMaybridgeHTS_01719, and ZINC 0000952883) were stabilized in the Keap1 binding cavity throughout 100 ns simulation, with average RMSD values of 0.100, 0.114, and 0.106 nm, respectively.
Conclusion:
This research proposes three lead molecules as potential Keap1 inhibitors based on high throughput screening, docking, and MD simulation studies. These hit molecules can be used for further design and development of Keap1 inhibitors. This research provides the preliminary data for discovering novel Keap1 inhibitors. It opens new avenues for medicinal chemists to explore antioxidant-stimulating molecules targeting the Keap1-Nrf2 pathway.
Keywords: Keap1 inhibitors, Oxidative stress, Virtual screening, 3DQSAR, Docking, Molecular dynamics
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This work is not funded by any organization.
Introduction
The Keap1-Nrf2 system is composed of the Kelch-like erythroid cell-derived protein with Cap and collar homology [ECH]-associated protein 1 (Keap1) and Nuclear factor erythroid 2 related factor 2 (Nrf2). This system is directly involved in the production of antioxidants1 and gets activated by the increase in the oxidative stress inside the cell. Oxidative stress is a condition in which there is an increase in the amount of pro-oxidants. These pro-oxidants usually are charged species (positively charged species or negatively charged species) or free radicals which are generated during biochemical reactions.2,3 So, antioxidants are needed to neutralize these pro-oxidants to prevent oxidative damages to cell organelles. The cellular antioxidants produced such as glutathione (GSH), NAD(P)H quinone oxidoreductase 1 (NQO1), glutathione peroxidase (GPx), heme oxygenase (OH), superoxide dismutase (SOD), and peroxidases defend the oxidative stress. The production of these antioxidant molecules is regulated through Nrf2 as shown in Fig. 1.4,5 In non-oxidative conditions Nrf2 is held in a complex by Keap1 which allows degradation of Nrf2 by ubiquitination.6 The inhibition of Keap1 causes detachment of Nrf2 from the complex. The free Nrf2 plies to the nucleus7 and activates the antioxidant response element (ARE) present in antioxidant genes leading to the production of the antioxidant bio-molecules.8
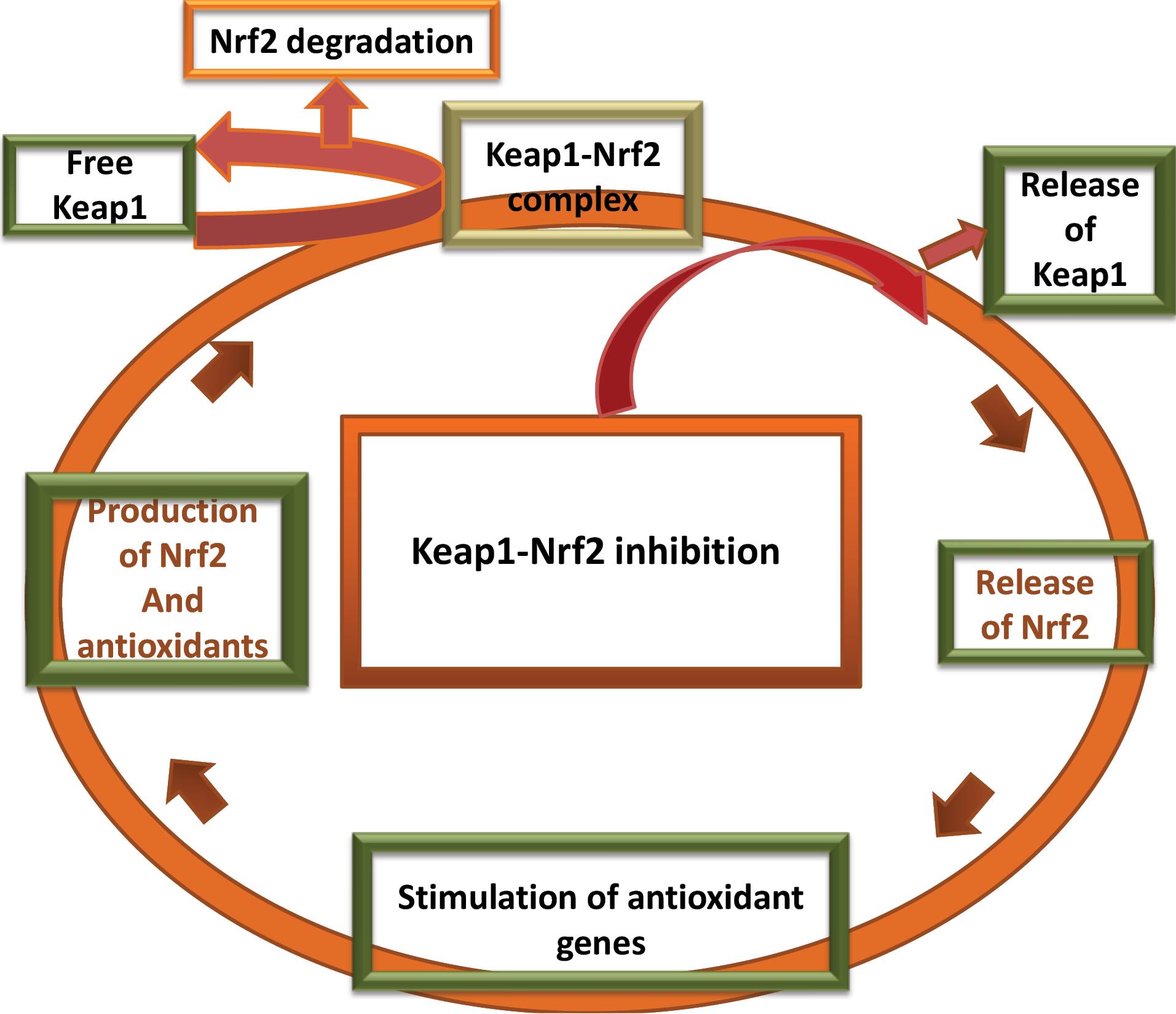
Fig. 1.
Schematic diagram of the Keap1-Nrf2 pathway.
.
Schematic diagram of the Keap1-Nrf2 pathway.
Keap1 undergoes a conformational change when some oxidants bind to one or more cysteine residues present on the extruded sides of the protein.9 The cysteine residues especially CYS151 are susceptible towards the pro-oxidants. These cysteine subunits present on Keap1-Nrf2 are referred to as cysteine sensors.10-12 The release of Nrf2 from Keap1 follows a hinge-latch mechanism and is controlled by multiple factors.13
The increased oxidative stress is responsible for cellular damage. The pro-oxidants interact with the cellular organelles and impart detrimental effects. This leads to the initiation and propagation of various diseases like cancer,14 diabetes,15 inflammatory conditions,16-18 and neurodegeneration.19,20 The reason behind low levels of antioxidants in the body has provided several research questions. Scientists are exploring the field of antioxidant response and its relation to diseases. To counterpoise oxidative stress Keap1 inhibition or Nrf2 activation is targeted. The possibility of decreased antioxidant levels cannot be ruled out in the pathogenesis of several diseases. Hence Keap1 inhibitors are the desirable therapeutic agents for diseases related to oxidative stress.
The recently reported in-vitro, in-vivo, and clinical trial studies on Keap1 inhibitors are mentioned below separately. The molecules reported in in-vitro and in-vivo studies are depicted in Fig. 2.
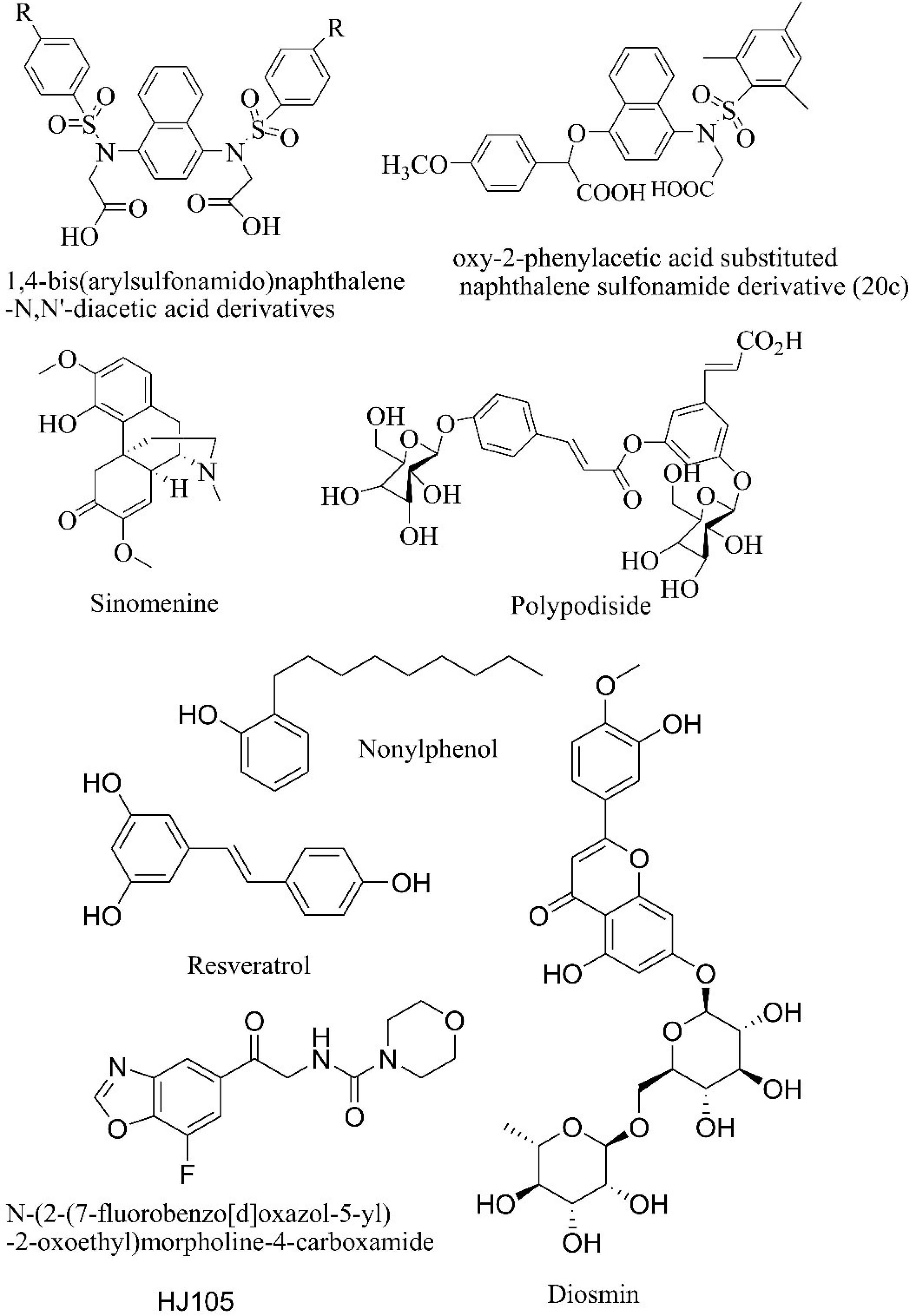
Fig. 2.
Molecules reported in recent in-vitro studies and preclinical studies.
.
Molecules reported in recent in-vitro studies and preclinical studies.
Recently reported in-vitro studies on Keap1 inhibitors
The 1,4-bis(arylsulfonamido)naphthalene-N, N'-diacetic acid derivatives were reported as to disrupt the Keap1-Nrf2 interaction. A fluorescence polarization assay was carried out to find out whether they could directly inhibit Keap1-Nrf2 interaction. The derivatives showed IC50 in the range of 7.2 and 31.3 nM.21 The compound 20c which is a derivative of 2-oxy-2-phenylacetic acid substituted naphthalene sulfonamide was reported to have IC50 of 75 nM in disrupting the Keap1-Nrf2 interaction. Its dissociation constant (Kd) from Keap1 was found to be 24 nM. The supporting studies on mouse cell models and in-vivo models show that 20c leads to Nrf2 target gene expression.22 Sinomenine increases the expression of heme oxygenase through Keap1-Nrf2 system. The reported concentration for Keap1-Nrf2 inhibition was 100 μM. This effect is reduced in in-vivo studies on Nrf2 knock-out mice.23 Polypodiside in 10 μМ concentration increases the concentration of Nrf2 thereby increasing the gene expression involved in the production of antioxidants.24
Recently reported in-vivo studies on Keap1
A study on rat models showed that Nonylphenol activated the Keap1-Nrf2 system in low doses thereby increasing the production of antioxidants. But, the effect of a high dose of Nonylphenol was just the opposite. Nonylphenol inhibited the Keap1-Nrf2 system in high doses and in prolonged exposure time.25 Resveratrol study on C57BL/6 wild-type mice with thoracic blast exposure-induced brain injury showed that it showed that resveratrol has a protective action against reactive oxygen species. The level of Keap increased and the level of Nrf2 decreased indicating that resveratrol shows its activity through the Keap1-Nrf2 system along with other inflammatory pathways.26 HJ105 was studied for its neuroprotective effect on a rat model of Alzheimer’s disease. It increased the levels of Nrf2 and biological antioxidants like superoxide dismutase, catalase, and glutathione peroxidase.27 Gentamycin is an antibiotic that induces nephrotoxicity, antioxidant imbalance, and decrease in Nrf2 level. Diosmin increased the levels of Nrf2, hemeoxygenase, and superoxide dismutase 3 along with other protective effects.28
Reported phase 2 clinical trial on Keap1 inhibitor
Clinical trials are being conducted (NCT04698681, NCT04265534, and NCT02417701) to find out the possibility of managing different types of cancers with Keap1 mutation. One of the most studied Keap1 inhibitors is sulforaphane. Several studies show sulforaphane is effective in decreasing oxidative stress in in-vitro models.29-31 Sulforaphane was studied in the phase 2 clinical trial for its effectiveness in chronic obstructive pulmonary disease (NCT01335971).32 But, clinical trials failed to establish the effectiveness of sulforaphane.33 So, the discovery of novel Keap1 inhibitors is warranted.
Materials and Methods
Data and software availability
ChemDraw Ultra 8.0 was used for drawing 2D structures of ligands. BIOVIA Discovery Studio (DS) (BIOVIA, Dassault Systèmes, Discovery Studio Visualizer, 20.1.0, San Diego, California, USA) was used for in-silico studies. Keap1 protein structure (PDB: 4N1B)34 was downloaded from the web page: https://www.rcsb.org/structure/4n1b of the Research Collaboratory for Structural Bioinformatics (RCSB) website. The protein was prepared for docking by the Protein Preparation module. Both protein and ligands were exposed to CHARMM (Chemistry at HARvard Molecular Mechanics) force field. The ligand preparation module and protein preparation module of Biovia DS 3.5 were used. 3DQSAR module of DS was used for pharmacophore. CDOCKER module of Biovia DS 3.5 was used for docking the ligands in the PDB site of 4N1B. Results were analyzed in DS visualize v.2021. The virtual toxicity prediction module used in this study was TOxicity Prediction by Komputer Assisted Technology (TOPKAT). In-silico calculations of Ames mutagenicity, lethal dose, skin irritation, skin sensitization, and ADMET (absorption, distribution, metabolism, excretion, and toxicity) were computed. CheMBL database was searched for Keap1 inhibitors. The assay results of Keap1 were used to obtain the molecules with IC50 values. Online Swiss ADME was used for computing physicochemical parameters and CYP enzyme inhibition. Molecular dynamics investigations were carried out using the GROMACS 2020.4 programme on a remote computer at the Bioinformatics Resources and Applications Facility (BRAF), C-DAC, Pune. The initial approach was developed by utilizing the CHARMM-36 force field to produce the protein topology, and the CGenFF server's CHARMM General Force Field to build the topologies of each ligand.
Selection of protein
Keap1 structures available on the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) website were evaluated for admissibility for in-silico investigations. The term “Keap1 inhibitor” was searched on rcsb.org. Numerous structures were displayed in the search results. A filter of Homo sapiens was created to screen protein structures. By using subsequent filters, NMR structures, and apoproteins were removed. Finally, a PDB ID with X-ray crystallographic structure, co-crystallized with a ligand and having E. coli expression with a resolution of 2.55 Å was selected (4N1B) for this in-silico research work. Hydrogen was added to the protein and 4N1B was energy minimized using CHARMM force field. The protein was prepared and viewed for any missing loop. Ramchandran’s plot was generated and the alignment of amino acids in the binding cavity was assured. The prepared protein had no breaks in the chain of amino acids. The amino acids were found to be in the binding cavity as shown in the plot. Both protein and ligands were prepared using suitable modules of DS. These structures were exposed to the CHARMM force field used for molecular dynamics. This force field simulation included the calculation of geometries, interaction and conformation energies, local minima, barriers to rotation, time-dependent dynamic behavior, and free energy. The binding site of the protein was selected from its PDB site. The radius of the binding site was 8.7 A◦ and XYZ coordinates were: 16.133, -21.95, and -36.218. The prepared protein was desolvated as retention of water in the binding cavity was not emphasized in the published details of the protein. The in-built ligand (1S,2R)-2-{[(1S)-1-[(1-oxo-1,3-dihydro-2H-isoindol-2-yl)methyl]-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}cyclohexane carboxylic acid was removed from the binding cavity before docking.
Methodology
The discovery of potential Keap1 inhibitors was schematically done using in-silico methods. The schematic representation of the workflow is depicted in Fig. 3. A 3D-QSAR model was generated using reported Keap1 inhibitors. This pharmacophore was used for screening three molecular libraries namely the Zinc database, Asinex, and MiniMaybridge. The resultant molecules from each library were screened using Lipinski’s and Verber’s rules. The screened molecules were subjected to energy minimization and ligand preparation. The number of conformation generation during ligand preparation was restricted to 3. Ligand preparation led to an increase in the number of ligands because conformers were generated in the output file of each library. These molecules were screened using predicted AlogP values in DS. Molecules with values ≥ 3 were selected for docking. The best-docked molecule in each library was selected for the MD simulation study. The details of the methods have been discussed below.
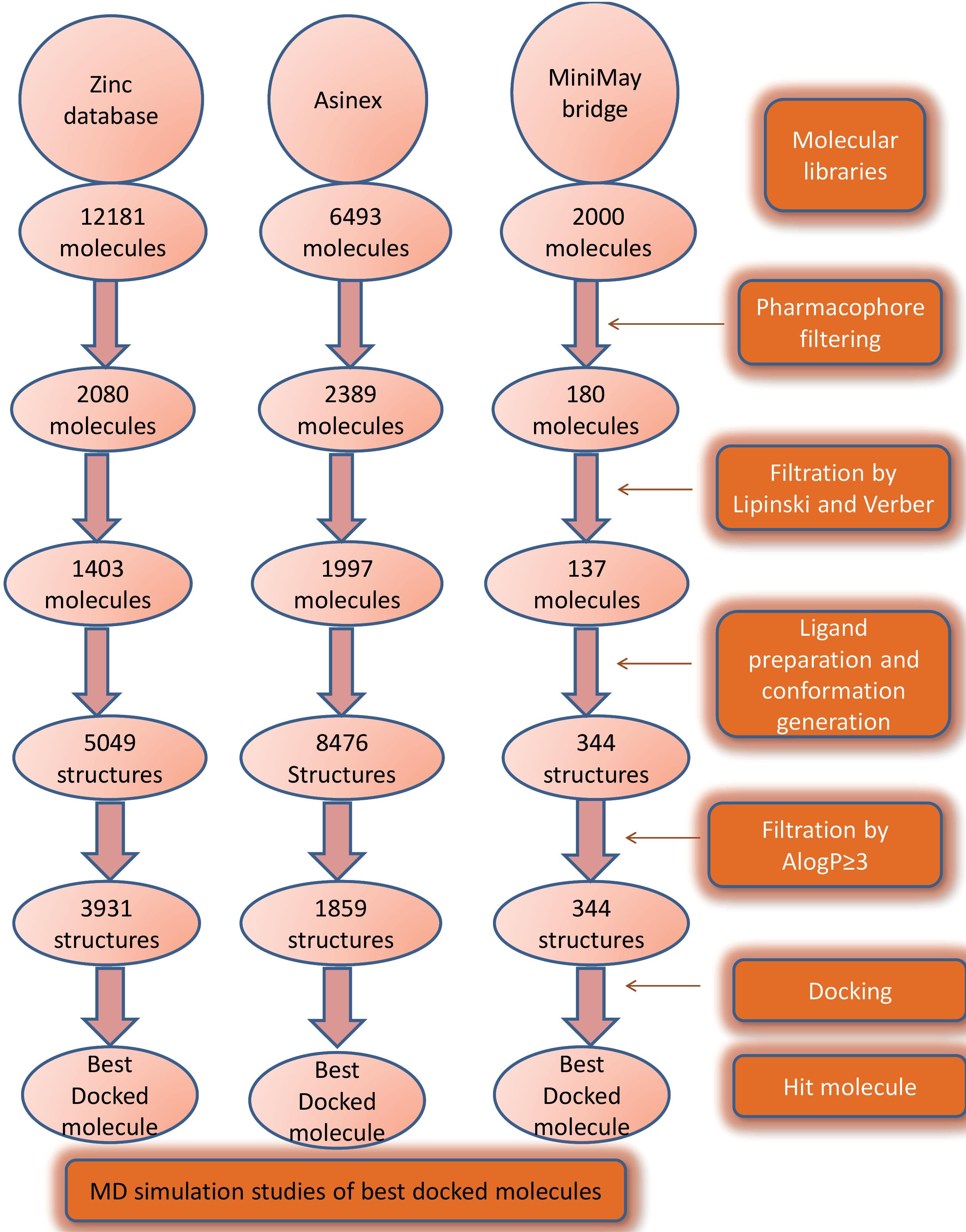
Fig. 3.
Steps involved in screening and discovery of potential Keap1 inhibitors.
.
Steps involved in screening and discovery of potential Keap1 inhibitors.
Collection of molecules with IC50 for 3D QSAR pharmacophore
CheMBL database was searched for Keap1 inhibitors. The assay results of Keap1 were used to obtain the molecules with reported activity. A collection of 60 molecules with IC50 values was used to generate a pharmacophore. 60 molecules were randomly distributed into training and test sets. The details of training set molecules are provided in Table 1.
Table 1.
Experimental and estimated activity of individual training set compounds
|
Compound No.
|
IC50
value (μM)
|
Errors
|
Fit value
|
Activity scale
|
|
Experimental
|
Estimated
|
Estimated
|
Experimental
|
Molecule-16
|
9.2 |
10 |
-1.1 |
8.51 |
+ + + + |
+ + + + |
Molecule-5
|
18 |
10 |
+ 1.8 |
8.22 |
+ + + |
+ + + + |
Molecule-4
|
64 |
13 |
+ 4.8 |
7.67 |
+ + + |
+ + + |
Molecule-8
|
40 |
25 |
+ 1.6 |
7.87 |
+ + + |
+ + + |
Molecule-14
|
40 |
31 |
+ 1.3 |
7.87 |
+ + + |
+ + + |
Molecule-7
|
32 |
34 |
-1.1 |
7.97 |
+ + + |
+ + + |
Molecule-22
|
63 |
42 |
+ 1.5 |
7.67 |
+ + + |
+ + + |
Molecule-17
|
68 |
47 |
+ 1.4 |
7.64 |
+ + + |
+ + + |
Molecule
|
51 |
54 |
-1.1 |
7.77 |
+ + + |
+ + + |
Molecule-19
|
34 |
66 |
-2 |
7.95 |
+ + + |
+ + + |
Molecule-9
|
120 |
85 |
+ 1.4 |
7.40 |
+ + |
+ + + |
Molecule-3
|
750 |
630 |
+ 1.2 |
6.59 |
+ + |
+ + |
Molecule-24
|
790 |
660 |
+ 1.2 |
6.58 |
+ + |
+ + |
Molecule-6
|
880 |
720 |
+ 1.2 |
6.53 |
+ + |
+ + |
Molecule-12
|
2600 |
750 |
+ 3.4 |
6.06 |
+ |
+ + |
Molecule-2
|
690 |
890 |
-1.3 |
6.63 |
+ + |
+ + |
Molecule-20
|
1000 |
940 |
+ 1.1 |
6.46 |
+ + |
+ + |
Molecule-18
|
1000 |
940 |
+ 1.1 |
6.46 |
+ + |
+ + |
Molecule-13
|
340 |
1100 |
-3.3 |
6.94 |
+ + + |
+ |
Molecule-23
|
290 |
1300 |
-4.5 |
7.01 |
+ + + |
+ |
Molecule-10
|
740 |
1300 |
-1.7 |
6.60 |
+ |
+ |
Molecule-15
|
1200 |
1700 |
-1.4 |
6.39 |
+ |
+ |
Molecule-11
|
1000 |
1800 |
-1.7 |
6.46 |
+ |
+ |
Molecule-21
|
1200 |
2300 |
-1.9 |
6.39 |
+ |
+ |
Generation of pharmacophore
3D QSAR was generated using Keap1 inhibitors obtained from the CheMBL database. The investigation considered three pharmacophoric features: H-bond donors, H-bond acceptors, and ring aromatic. Hypotheses were computer-generated by DS with default names 1 to 10. Different hypotheses of pharmacophore considered different feature distances and considered different ratios of contribution by these features. The result of pharmacophore shows the maximum value of fitment of the molecules in the pharmacophore. The cost of the hypothesis should be high. The value away from the null hypothesis is the best cost. The lower value of RMSD indicates the best hypothesis. The correlation coefficient along with RMSD was also considered for evaluating the hypotheses. In the description box, the features contributing to pharmacophore are listed and their respective contribution in ratio was mentioned. The contribution of each feature may or may not vary. A regressed graph was automatically generated by DS based on reported IC50 provided for during the making of 3D QSAR models and the estimated activity is calculated based on pharmacophore. The regressed graph shows green dots as active molecules. Black dots as moderately active and red dots as inactive molecules. The mapping table shows the name of the molecule, fit value in the pharmacophore (better active molecules have better-fit value), activity, and mapping score. The mapping score shows how well each feature has been mapped in that particular molecule. In some cases, the feature has no value against it. The Asterisk shows that the molecule does not have the contribution of that particular feature as calculated by the pharmacophore algorithm. Validation of all the hypotheses was done by mapping with active and inactive molecules.35 In some cases moderately active molecules have also been used. All the hypotheses confirmed the validation criteria. A comparison of pharmacophores was done based on the RMSD and null cost. Fig. 4 shows the distance between the pharmacophoric features of hypothesis 1. The details of all the pharmacophores are provided in Supplementary file 1.
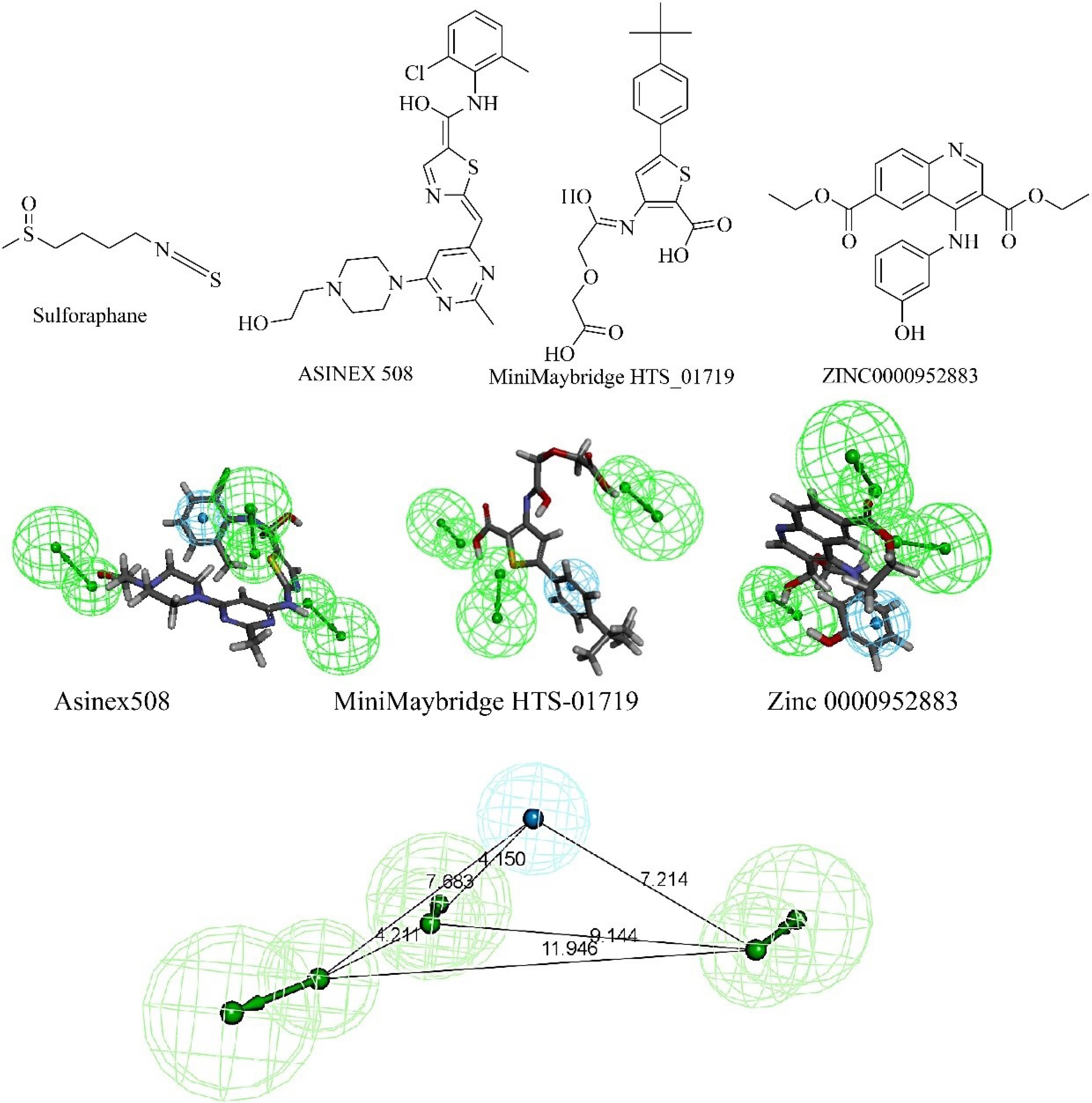
Fig. 4.
The structure of sulforaphane and the best hits from three different libraries are shown. The best hits were obtained from screening through pharmacophore hypothesis 1. The mapping diagram is shown with the pharmacophore. The pharmacophoric features and distance between the features of hypothesis 1 are shown. Cyan color shows hydrophobic features and green region shows hydrogen bond acceptor.
.
The structure of sulforaphane and the best hits from three different libraries are shown. The best hits were obtained from screening through pharmacophore hypothesis 1. The mapping diagram is shown with the pharmacophore. The pharmacophoric features and distance between the features of hypothesis 1 are shown. Cyan color shows hydrophobic features and green region shows hydrogen bond acceptor.
Screening of database using pharmacophore
The zinc database (comprising 12182 molecules) Asinex library (comprising of 6493 molecules) MiniMaybridge library (comprising of 2000 molecules) were screened using the pharmacophore (hypothesis 1). In Fig. 3 stepwise screening procedure of three libraries has been mentioned. In the last step of screening, molecular docking was done and the best-docked molecules from each library were selected as a hit. Fig. 4 shows the alignment of best hits with the pharmacophore.
Docking of hit molecules with 4N1B
The molecular docking was performed for the hits from the zinc database (Zinc 0000952883), Asinex library (508), and MiniMaybridge library (HTS-01719) using Discovery Studio 3. The preparation of Keap1 protein (PDB ID: 4N1B) and ligands performed using DS. Co-crystal ligand-bound Keap1 (PDB ID: 4N1B) was obtained from a protein data bank (www.rcsb.org). The protein was prepared by adding hydrogen atoms followed by the removal of water molecules. During docking investigations, the negative CDOCKER interaction energy of the best poses from all derivatives was determined. The grid used for docking had XYZ values 16.133, -21.95, and -36.218 with an 8.7 radius. Force-field-based scoring functions were used to analyze the binding energies of the compounds in terms of Kcal/mol. Sulforaphane was used as the standard. The results were analyzed using the Discovery Studio 4.0. Fig. 4 shows the structure of hit molecules from different libraries and the structure of sulforaphane.35,36
Molecular dynamics simulation and MM-PBSA calculations
The binding interactions and possible binding affinities of the top 3 hits (Hit_Asinex, Hit_MiniMaybridge, and Hit_Zinc) were investigated through molecular dynamics simulations (MDS) by using the similar protocol as demonstrated by Matada et al,35,36 each equilibrated system was screened against MDS using Gromacs 2020.4 programs, run on a remote computer at the Bioinformatics Resources and Applications Facility (BRAF), C-DAC, Pune.37-46 Radius of gyration (Rg), root mean square fluctuations (RMSF), and root mean square deviations (RMSD) in the ligand and backbone atoms have been determined from the different trajectories. Additionally, a hydrogen bond analysis was conducted, and Poisson Boltzmann surface area continuum solvation (MM-PBSA) computations were applied to the collected trajectories at 100 ps intervals.47-49
Results and Discussion
Fischer's randomization test (95% confidence interval), test set analysis, and cost analysis were used in the pharmacophore investigation. As per the graph shown in Fig. 5, hypothesis 1 has a low cost and is considered the best hypothesis amongst all that were generated. The cost analysis values are included in Supplementary file 1.
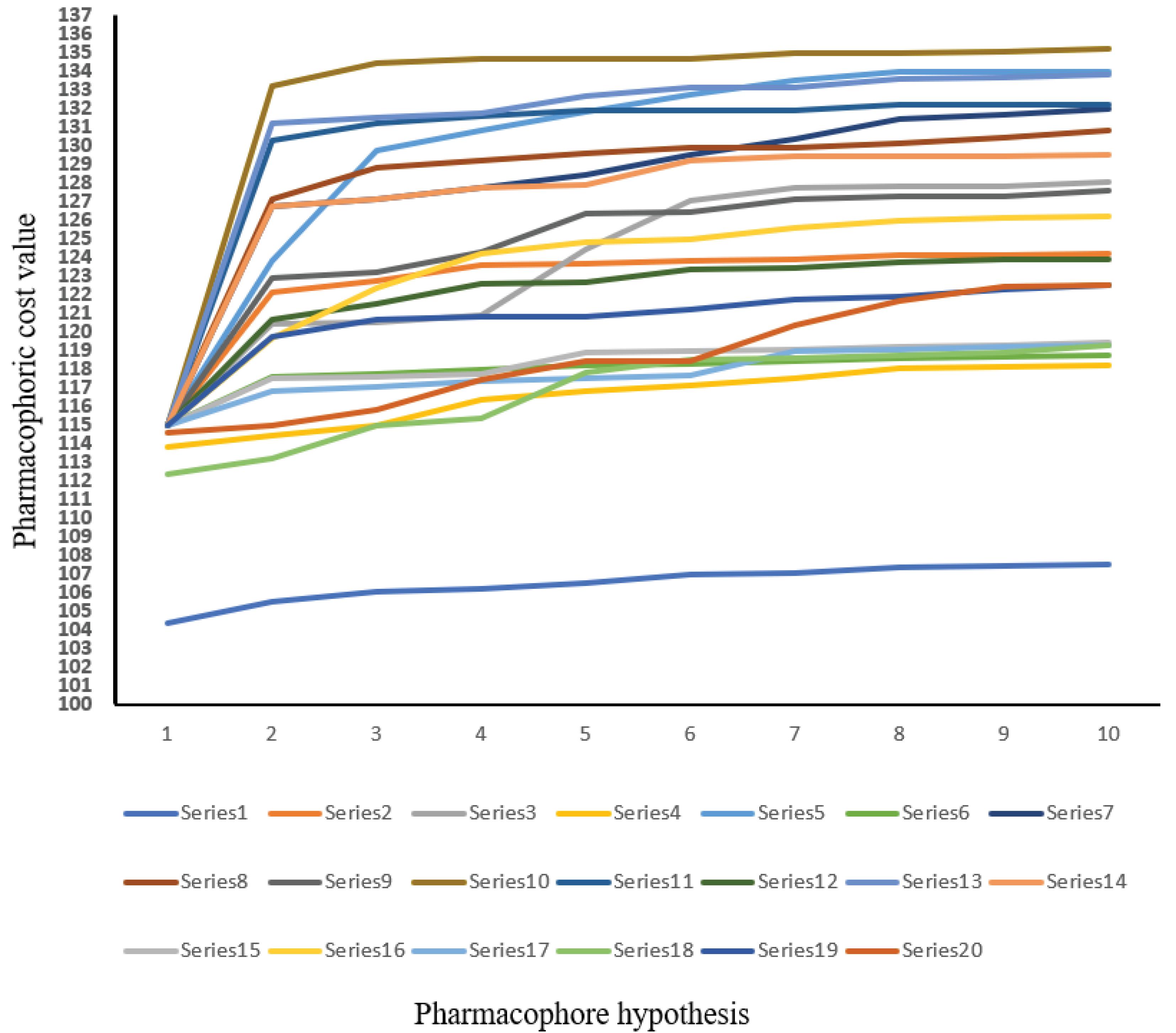
Fig. 5.
Fischer’s randomization tests 95% confidence level. A graph between the pharmacophore hypothesis and 20 scrambled runs. The series represents the HypoGen algorithms used to generate the top 10 pharmacophores.
.
Fischer’s randomization tests 95% confidence level. A graph between the pharmacophore hypothesis and 20 scrambled runs. The series represents the HypoGen algorithms used to generate the top 10 pharmacophores.
Fischer validation of pharmacophore
The Discovery Studio 3DQSAR module generated twenty arbitrary hypotheses and fisher Validation findings were acquired in a graphical style. The confidence interval selected was 95%. The costs of the entire hypotheses were represented in the graph by different points. From the graphical representation, it can be concluded that hypothesis 1 i.e., ‘series 1’ qualifies to be the best hypothesis when compared with other randomly generated 19 hypotheses. Fig. 6 represents Fischer’s randomization test with a 95% confidence level. It shows the graph between the pharmacophore hypothesis and pharmacophoric cost value.
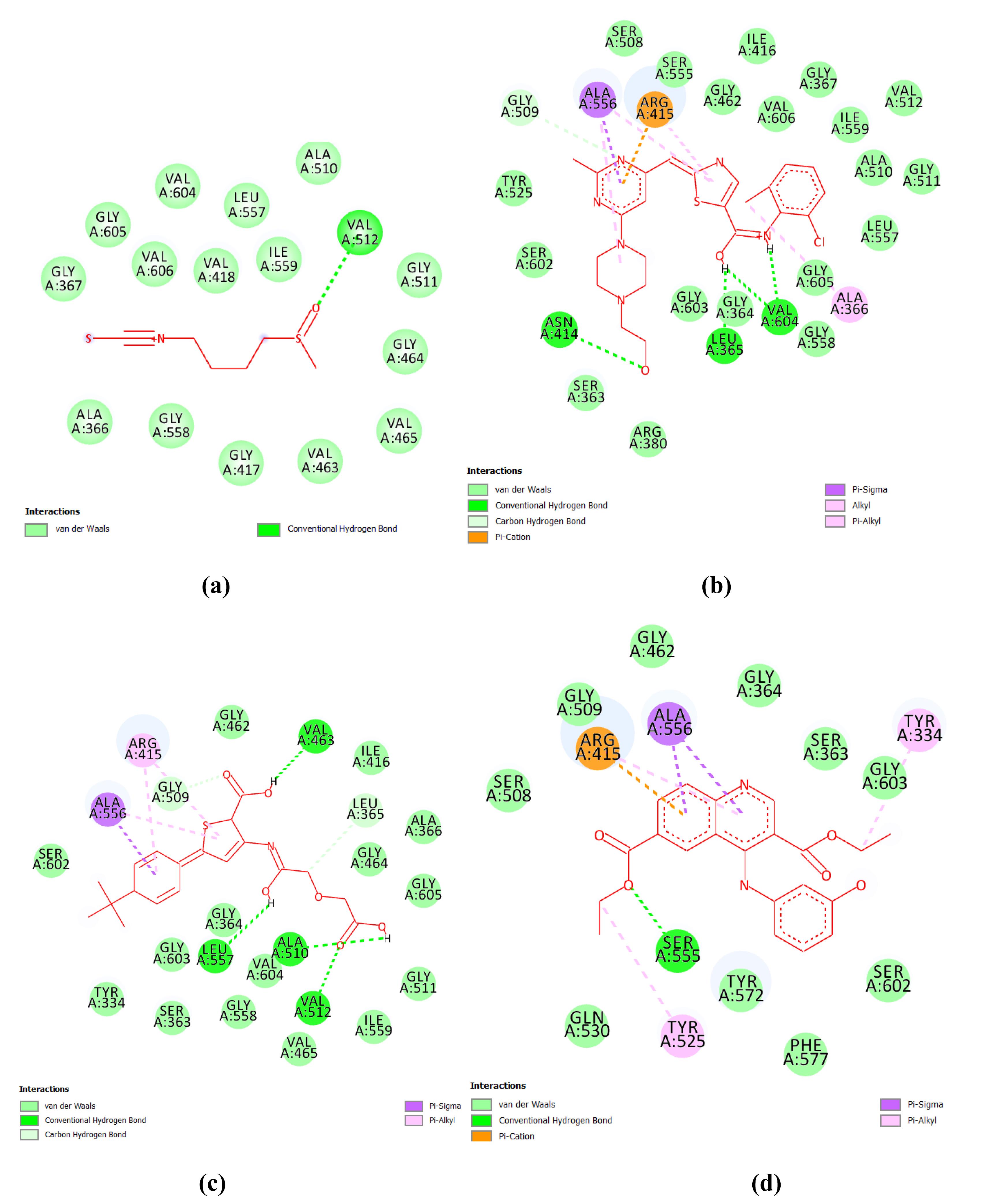
Fig. 6.
2D Representation of docking results of best hits from three databases and sulforaphane (standard) (a) standard: sulforaphane (b) Hit_Asinex (c) Hit_MiniMaybridge and (d) Hit_zinc database.
.
2D Representation of docking results of best hits from three databases and sulforaphane (standard) (a) standard: sulforaphane (b) Hit_Asinex (c) Hit_MiniMaybridge and (d) Hit_zinc database.
The best pharmacophore was used for screening the libraries under study. The pharmacophore screening filtered out the best-mapped molecules. Based on the mapping score, hits were selected for further in-silico study.
Test set validation
The ligands in test set were validated using the Pharmacophore 1 hypothesis where the biological activity was mapped with predicted activity.The regression plot of hypothesis 1 shows the log activity and the predicted activity values for the test set. Details are included in Fig. S1.
Predicted ADMET properties and drug-likeness properties
Discovery Studio 3.5 was employed for in-silico ADMET parameter computation, which helped prevent the drug from failing in the last phases of the discovery process. All 60 generated compounds were analyzed, and it was discovered that they had optimal levels of absorption (ranging from 0 to 1), indicating that the compounds were easily absorbed. The aqueous solubility and blood-brain barrier (BBB) levels were both within the ideal range of 0-2, suggesting acceptable solubility. Based on this data, it was recommended that the generated compounds be used in drug development, and they were subsequently analyzed for docking studies. The ADMET characteristics of the hit molecules are presented in Supplementary file 1.
Toxicity prediction
Toxicity prediction studies are utilized as a preclinical evaluation to assist minimize the duration and cost of clinical trials. The Ames test determines a drug's mutagenicity, which is its capacity to cause mutation in cells. The skin irritation test determines the suitable dose of chemicals for topical use. To evaluate toxicity, a calculated probability is used: if it falls between 0 and 0.29, the chemical is regarded non-toxic; if it falls between 0.3 and 0.69, the result is equivocal; and if it falls between 0.7 and 1, the compound is hazardous. The mutagenicity, toxicity, and irritability were determined utilizing in-silico techniques. The results show that all of the compounds analysed were inconclusive. Supplementary file 1 contain in-silico prediction results.
Drug likeness
The structures of hit molecules were studied for their ability to make hydrogen bonds. The details of a number of hydrogen bond acceptors, hydrogen bond donor, lipophilicity, molecular polar surface area, and molar refractivity are provided in Supplementary file 1. These values correspond to the ability of the molecules to cross the cell membrane and act as a drug.
Molecular docking
The in-silico interaction of the hit molecules indicates that the ligand is binding with the amino acids as shown in Table 2. Fig. 6 shows 2D interactions between 4N1B and ligands. The hit molecules showed good interaction with amino acids in the target through hydrogen bonds. The amino acid backbone of the binding pocket with the ligand is shown in Fig. S2 in Supplementary file 1. The ligands with good 4N1B binding interactions potentially be effective Keap1 inhibitors. Surface diagrams of the docked complexes are shown in Fig. S3 in Supplementary file. Sulforaphane is taken as the standard because this is the only reported Keap1 inhibitor for which several clinical trials were conducted. The best binding pose of the molecules with 4N1B was selected for MD simulation.
Table 2.
Docking analysis of the hit molecules from three databases
|
Compounds
|
Binding energy (kcal/mol)
|
Interacting amino acid residues
|
| Sulforaphane (Std) |
-19.058 |
SER602, SER363, TYR334, ARG380 |
| Hit_Asinex |
-55.761 |
ALA556, ARG415,SER363,GLY603, VAL606 |
| Hit_MiniMaybridge |
-63.279 |
ALA556, TYR572, ARG380, ASN414, ARG415 |
| Hit_Zinc |
-37.18 |
ARG380, TRY334, ARG415, ALA556, TYR572, GLY603, GLY364, ILE416 |
Molecular dynamics studies and MM-PBSA calculations of the best hits
Molecular simulations were analyzed for top compounds to study an in-depth understanding of the dynamic behavior of proteins and ligands-proteins complex in a simulated biological environment with constant volume, temperature, and pressure conditions. The simulation was conducted for 100 nanoseconds, and several trajectories (RMSD, Rg, RMSF, and H-bonding) were computed. The RMSD assesses the protein-ligand complex's stability during the MD run's equilibration period, demonstrating dynamic changes in protein and ligand at various temperatures and pressures across the simulation's duration. The lower the RMSD value, the greater the conformational stability of the protein and ligand. The average RMSD of protein backbone atoms for complexes (Hit_Asinex, Hit_MiniMaybridge, and Hit_Zinc) were found to be 0.100, 0.114, and 0.106 nm respectively as compared to the standard drug (0.082 nm). Initially there (5-15 ns) for all three complexes, thereafter they were stable throughout the simulation. RMSD variations in ligand atoms were almost similar in protein backbone atoms. Among the ligands, Hit_MiniMaybridge was the most stable with no deviation or confirmational change throughout the simulation. Standard drug displayed the lowest RMSD in ligand atoms with the value of 0.133 nm; while the RMSDs in the atoms of Hit_Asinex, Hit_MiniMaybridge, and Hit_Zinc were 0.193, 0.152, and 0.146 respectively. Interestingly, a few spikes in RMSD plots at around 22- 40 ns and 69-78 ns show few major conformational changes in the ligand structures. The Hit_Asinex is undergoing a major conformational change at these simulation periods. The RMSD analysis suggests that all the hits and standards are in the binding site and are dynamically stable within the pocket of protein with few deviations in starting structures.
In isothermic and isobaric processes, the root mean square fluctuation (RMSF) governs fluctuations in protein-ligand complex residues or local changes. The analysis of root mean square fluctuations (RMSF) can also provide evidence about the stability of the protein-ligand complex and elasticity of the side chains of binding site residues and residues in loop regions. In the case of complex with Hit_Asinex, the proteins side chain residues Val604, Ser363, and Tyr334 seems to undergo major conformational changes (Fig. S4 in Supplementary file 1). The hydrogen bonds formed with these residues in trajectories extracted at different time intervals revealed the involvement of the aforementioned residues in the stabilization of this complex. Hit_MiniMaybridge forms a hydrogen bond interaction with Arg415, Asn414, and Ser363 residues in the equilibrated complex (Fig. S5 in Supplementary file 1). None of these hydrogen bonds are stable however as at around 25 ns a new hydrogen bond forms with residue Ser602, at 50 ns with Gly574, and 100 ns with Gln530 and Arg483. Thus, there would be fluctuations in the side chains of these residues. In the case of Hit_Zinc, the residue Arg415 forms a consistent hydrogen bond till 50 ns which breaks during 75 ns and again forms at 100 ns (Fig. S6 in Supplementary file 1). While standard drug forms a hydrogen bond with Phe546 at 25 ns, Asn414 at 50 ns, and Gln372 at 100 ns (Fig. S7 in Supplementary file 1). The Hit_Zinc and standard drug do not show any hydrogen bonds in the equilibrated systems. In line with these results, the RMSF analysis also suggests the fluctuations in the side chains of many of these residues (Fig. 7). In the case of Hit_Asinex, the major fluctuations were observed in residues ranging from 430-440, while for Hit_MiniMaybridge and Hit_Zinc the fluctuations were observed in residues ranging from 380-390 and 440-450.
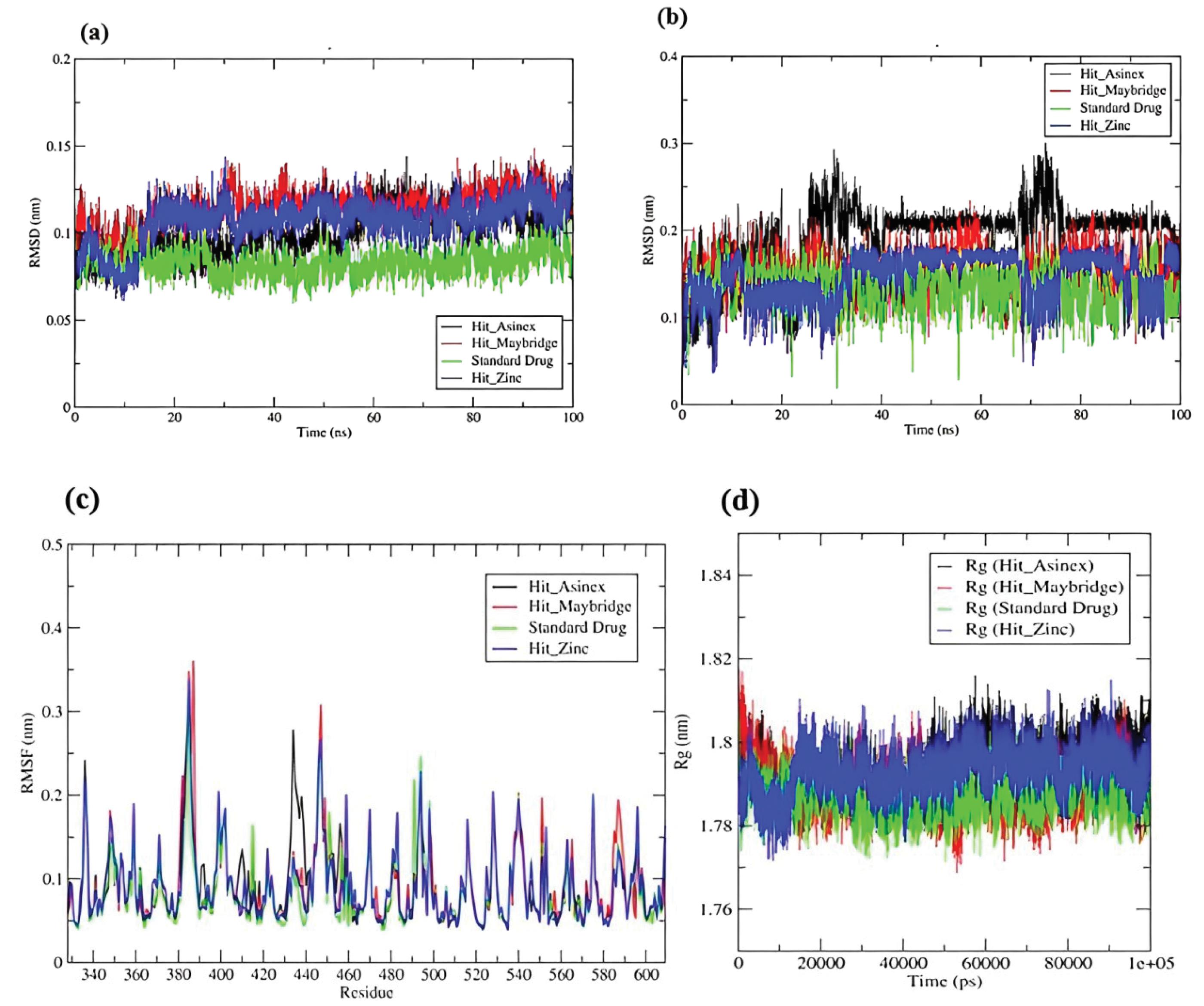
Fig. 7.
Root mean square deviation (RMSD) in (a) Backbone atoms of protein and (b) Atoms of hit compounds and standard drug (c) RMSF analysis (d) Radius of gyration of Cα atoms of protein in all systems understudy.
.
Root mean square deviation (RMSD) in (a) Backbone atoms of protein and (b) Atoms of hit compounds and standard drug (c) RMSF analysis (d) Radius of gyration of Cα atoms of protein in all systems understudy.
The radius of gyration (Rg) analysis calculates the root mean square distance of a group of atoms from their common center of mass, as well as acquires insight into the system's overall compactness. Till around 10 ns in all the systems, the Rg seems to be largely deviating pointing to the conformational changes in the loop regions (Fig. 8). However, thereafter almost all the systems have almost constant total Rg values within a narrow range of 1.78 to 1.8 nm. This suggests the stability of all the systems.
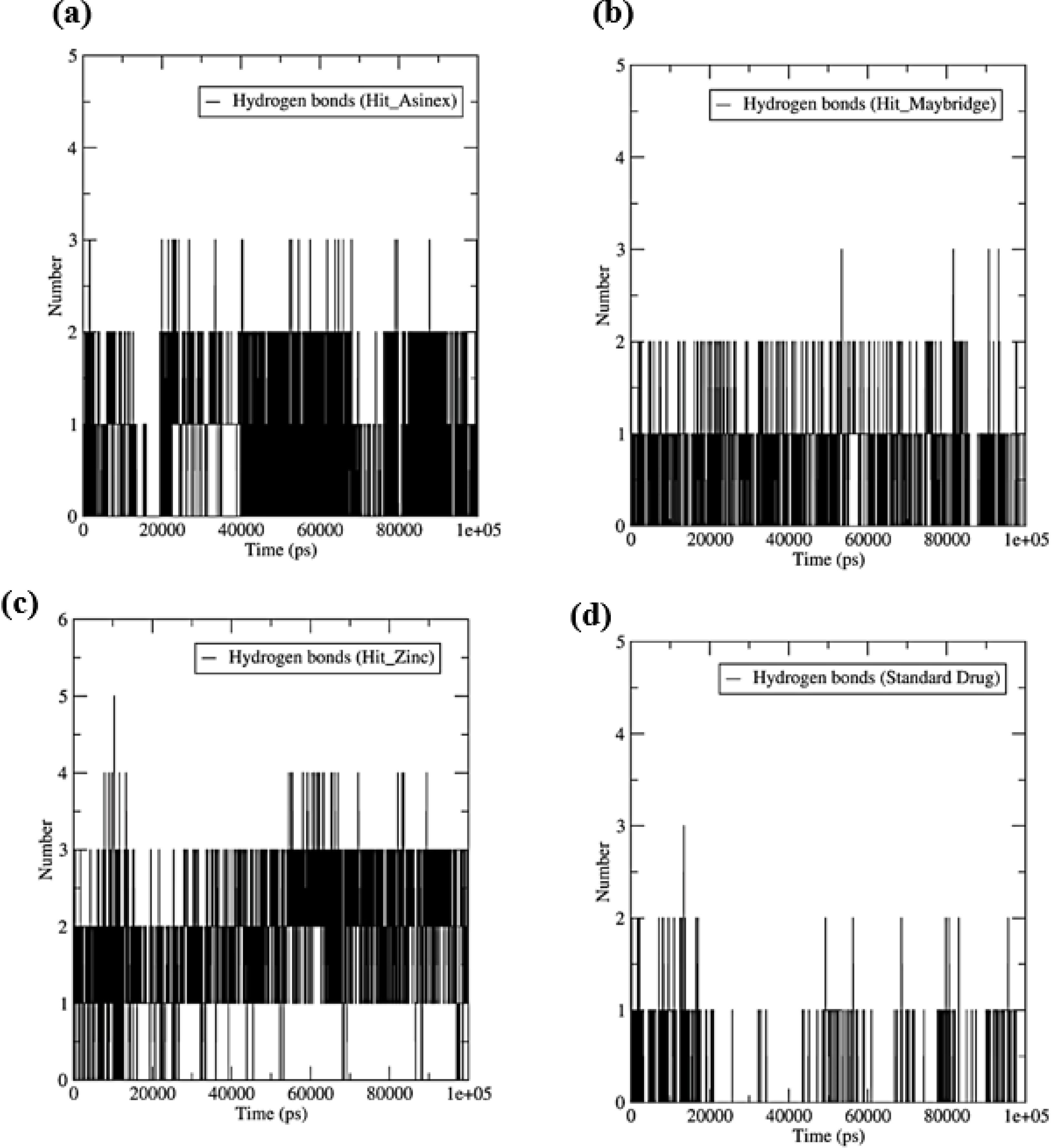
Fig. 8.
The number of hydrogen bonds formed between the ligand and binding site residues. (a) HIT_ASINEX, (b) Hit_MiniMaybridge, (c) Hit_Zinc, and (d) Standard drug.
.
The number of hydrogen bonds formed between the ligand and binding site residues. (a) HIT_ASINEX, (b) Hit_MiniMaybridge, (c) Hit_Zinc, and (d) Standard drug.
Hydrogen bond interactions signify the interactions in the stabilization of protein-ligand complexes. The more the number of hydrogen bonds formed better the stability of the resulting protein-ligand complex. Three hydrogen bonds were formed with Hit_Asinex, where a minimum of two hydrogen bonds are formed consistently throughout the MDS (Fig. 8). Similarly, in Hit_MiniMaybridge, a maximum of three hydrogen bonds could form but less frequently, and one hydrogen bond is found forming consistently. Interestingly, a maximum of five hydrogen bonds were found forming with Hit_Zinc, but only two of them are found to form consistently. The standard drug however forms only one hydrogen bond occasionally during MDS.
The binding free energy may be computed more precisely using the MM-PBSA technique. The free energy calculation analysis provides a quantitative estimate of binding free energy, which indicates the stability of the protein-ligand complex. The binding free energies were computed using the final 75-100 ns of MD trajectories, as shown in Table 3. The results revealed that Hit_Asinex had the lowest binding energy, -260.398 kJ.mol-1, and hence the highest binding affinity when compared to other ligands. Hit_Asinex has higher favorable electrostatic and polar solvation energies than other ligands. The Hit_Zinc has slightly higher binding free energy compared to other ligands.
Table 3.
Results of free energy calculation by molecular mechanics generalized Born surface area (MM-PBSA)
|
Compound
|
van der Waal energy
(kJ.mol-1) |
Electrostatic energy
(kJ.mol-1) |
Polar solvation energy
(kJ.mol-1) |
SASA energy
(kJ.mol-1) |
Binding energy
(kJ.mol-1) |
| Hit_Asinex |
-197.540 (1.754) |
-282.812 (2.425) |
241.625 (3.258) |
-21.957 (0.180) |
-260.398 (2.835) |
| Hit_MiniMaybridge |
-146.472 (2.005) |
-24.195 (2.248) |
115.649 (4.135) |
-16.638 (0.247) |
-71.635 (2.198) |
| Hit_Zinc |
-72.330 (1.554) |
-13.507 (2.967) |
50.356 (3.850) |
-9.218 (0.160) |
-45.073 (2.030) |
| Standard drug |
-131.653 (1.710) |
-64.973 (2.552) |
152.739 (3.029) |
-16.341 (0.175) |
-60.435 (1.955) |
Standard deviations are given in parentheses.
Conclusion
The top molecules from the screening of three libraries namely Asinex library, MiniMaybridge library, and Zinc database were selected. The top hit molecule from each library abides by Lipinski’s rule of five and Verber’s rule. All the three hit molecules have in acceptable values of predicted ADMET parameters and predicted toxicity. Docking results of the hit molecules indicate the interaction with the amino acids of the Kelch domain. The Kelch domain is that part of Keap1 which holds Nrf2 through its c-terminal. Hence these interactions are considered significant and might lead to a conformational change in the protein which in turn would release Nrf2. Hit_Asinex (2-(4-(6-(-5-((2-chloro-6-methyl phenylamino) (hydroxy)methylene)thiazol-2(5H)-ylidene)methyl)-2-methylpyrimidin-4-yl) piperazin-1-yl)ethanol) and Hit_MiniMaybridge (HTS_01719) show interactions with hydrogen bonding and stability in molecular dynamics. In terms of stability in the Keap1 pocket, Hit_Asinex has comparatively better electrostatic and binding energy than the other two hits. Prediction of the probable active molecules is an integral part of rational drug design. So, the results of these hit molecules can be used for the discovery of novel Keap1 inhibitors. There is a huge research gap in the field of Keap1 inhibitors as no Keap1 inhibitor has cleared all the phases of clinical trials. This research work can further be extended to designing and synthesizing more potent Keap1 inhibitors.
Research Highlights
What is the current knowledge?
-
Keap1 inhibitors are still in the clinical trial studies
-
No Keap1 inhibitor has cleared the clinical trials
-
There is no Keap1 inhibitor being used for therapeutic use
What is new here?
-
The screening of molecular libraries has been done in a systematic way to filter the best hits from each of the three libraries.
-
The process of drug discovery mentioned in this article can be used for different targets.
-
The results of the screening have provided three hits. Binding interactions of these with the Keap1 cavity have been studied using docking and molecular dynamic simulations.
-
This work can be extended to designing and developing of more potent Keap1 inhibitors.
Competing Interests
Authors declare no conflict of interest.
Ethical Statement
None to be declared.
Supplementary files
The supplementary file 1 contains Figs. S1-S7.
(pdf)
Acknowledgements
The authors acknowledge the support of Acharya & BM Reddy College of Pharmacy for providing the facilities to carry out this research work.
References
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999; 13:76-86. doi: 10.1101/gad.13.1.76 [Crossref] [ Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003; 552:335-44. doi: 10.1113/jphysiol.2003.049478 [Crossref] [ Google Scholar]
- Lushchak VI. Classification of oxidative stress based on its intensity. EXCLI J 2014; 13:922-37. [ Google Scholar]
- Kovac S, Angelova PR, Holmström KM, Zhang Y, Dinkova-Kostova AT, Abramov AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta 2015; 1850:794-801. doi: 10.1016/j.bbagen.2014.11.021 [Crossref] [ Google Scholar]
- Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol 2004; 37:139-43. doi: 10.5483/bmbrep.2004.37.2.139 [Crossref] [ Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003; 8:379-91. doi: 10.1046/j.1365-2443.2003.00640.x [Crossref] [ Google Scholar]
- Ganner A, Pfeiffer ZC, Wingendorf L, Kreis S, Klein M, Walz G. The acetyltransferase p300 regulates NRF2 stability and localization. Biochem Biophys Res Commun 2020; 524:895-902. doi: 10.1016/j.bbrc.2020.02.006 [Crossref] [ Google Scholar]
- Wang X, Yu JY, Sun Y, Wang H, Shan H, Wang S. Baicalin protects LPS-induced blood-brain barrier damage and activates Nrf2-mediated antioxidant stress pathway. Int Immunopharmacol 2021; 96:107725. doi: 10.1016/j.intimp.2021.107725 [Crossref] [ Google Scholar]
- Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 2018; 98:1169-203. doi: 10.1152/physrev.00023.2017 [Crossref] [ Google Scholar]
- Dayalan Naidu S, Muramatsu A, Saito R, Asami S, Honda T, Hosoya T. C151 in KEAP1 is the main cysteine sensor for the cyanoenone class of NRF2 activators, irrespective of molecular size or shape. Sci Rep 2018; 8:8037. doi: 10.1038/s41598-018-26269-9 [Crossref] [ Google Scholar]
- He X, Ma Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol Pharmacol 2009; 76:1265-78. doi: 10.1124/mol.109.058453 [Crossref] [ Google Scholar]
- Dinkova-Kostova AT, Kostov RV, Canning P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch Biochem Biophys 2017; 617:84-93. doi: 10.1016/j.abb.2016.08.005 [Crossref] [ Google Scholar]
- Horie Y, Suzuki T, Inoue J, Iso T, Wells G, Moore TW. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun Biol 2021; 4:576. doi: 10.1038/s42003-021-02100-6 [Crossref] [ Google Scholar]
- Senyuk V, Eskandari N, Jiang Y, Garcia-Varela R, Sundstrom R, Leanza L. Compensatory expression of NRF2-dependent antioxidant genes is required to overcome the lethal effects of Kv111 activation in breast cancer cells and PDOs. Redox Biol 2021; 45:102030. doi: 10.1016/j.redox.2021.102030 [Crossref] [ Google Scholar]
- Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol 2013; 1:45-9. doi: 10.1016/j.redox.2012.10.001 [Crossref] [ Google Scholar]
- Behl T, Kaur I, Sehgal A, Sharma E, Kumar A, Grover M. Unfolding Nrf2 in diabetes mellitus. Mol Biol Rep 2021; 48:927-39. doi: 10.1007/s11033-020-06081-3 [Crossref] [ Google Scholar]
- Singh E, Purawarga Matada GS, Abbas N, Dhiwar PS, Ghara A, Das A. Management of COVID-19-induced cytokine storm by Keap1-Nrf2 system: a review. Inflammopharmacology 2021; 29:1347-55. doi: 10.1007/s10787-021-00860-5 [Crossref] [ Google Scholar]
- Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun 2016; 7:11624. doi: 10.1038/ncomms11624 [Crossref] [ Google Scholar]
- Pajares M, Jiménez-Moreno N, García-Yagüe Á J, Escoll M, de Ceballos ML, Van Leuven F. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016; 12:1902-16. doi: 10.1080/15548627.2016.1208889 [Crossref] [ Google Scholar]
- Singh E, Devasahayam G. Neurodegeneration by oxidative stress: a review on prospective use of small molecules for neuroprotection. Mol Biol Rep 2020; 47:3133-40. doi: 10.1007/s11033-020-05354-1 [Crossref] [ Google Scholar]
- Abed DA, Lee S, Wen X, Ali AR, Mangipudy V, Aleksunes LM. Optimization of 1,4-bis(arylsulfonamido)naphthalene-N,N'-diacetic acids as inhibitors of Keap1-Nrf2 protein-protein interaction to suppress neuroinflammation. Bioorg Med Chem 2021; 44:116300. doi: 10.1016/j.bmc.2021.116300 [Crossref] [ Google Scholar]
- Lu MC, Shao HL, Liu T, You QD, Jiang ZY. Discovery of 2-oxy-2-phenylacetic acid substituted naphthalene sulfonamide derivatives as potent KEAP1-NRF2 protein-protein interaction inhibitors for inflammatory conditions. Eur J Med Chem 2020; 207:112734. doi: 10.1016/j.ejmech.2020.112734 [Crossref] [ Google Scholar]
- Liao K, Su X, Lei K, Liu Z, Lu L, Wu Q. Sinomenine protects bone from destruction to ameliorate arthritis via activating p62(Thr269/Ser272)-Keap1-Nrf2 feedback loop. Biomed Pharmacother 2021; 135:111195. doi: 10.1016/j.biopha.2020.111195 [Crossref] [ Google Scholar]
- Yao H, Zhang N, Zhang W, Li J, Hua H, Li Y. Discovery of polypodiside as a Keap1-dependent Nrf2 activator attenuating oxidative stress and accumulation of extracellular matrix in glomerular mesangial cells under high glucose. Bioorg Med Chem 2020; 28:115833. doi: 10.1016/j.bmc.2020.115833 [Crossref] [ Google Scholar]
- Ke Q, Yang J, Liu H, Huang Z, Bu L, Jin D. Dose- and time-effects responses of nonylphenol on oxidative stress in rat through the Keap1-Nrf2 signaling pathway. Ecotoxicol Environ Saf 2021; 216:112185. doi: 10.1016/j.ecoenv.2021.112185 [Crossref] [ Google Scholar]
- Cong P, Wang T, Tong C, Liu Y, Shi L, Mao S. Resveratrol ameliorates thoracic blast exposure-induced inflammation, endoplasmic reticulum stress and apoptosis in the brain through the Nrf2/Keap1 and NF-κB signaling pathway. Injury 2021; 52:2795-802. doi: 10.1016/j.injury.2021.08.019 [Crossref] [ Google Scholar]
- Yang X, Zhi J, Leng H, Chen Y, Gao H, Ma J. The piperine derivative HJ105 inhibits Aβ(1-42)-induced neuroinflammation and oxidative damage via the Keap1-Nrf2-TXNIP axis. Phytomedicine 2021; 87:153571. doi: 10.1016/j.phymed.2021.153571 [Crossref] [ Google Scholar]
- Ali FE, Sayed AM, El-Bahrawy AH, Omar ZM, Hassanein EH. Targeting KEAP1/Nrf2, AKT, and PPAR-γ signals as a potential protective mechanism of diosmin against gentamicin-induced nephrotoxicity. Life Sci 2021; 275:119349. doi: 10.1016/j.lfs.2021.119349 [Crossref] [ Google Scholar]
- Dinkova-Kostova AT, Fahey JW, Wade KL, Jenkins SN, Shapiro TA, Fuchs EJ. Induction of the phase 2 response in mouse and human skin by sulforaphane-containing broccoli sprout extracts. Cancer Epidemiol Biomarkers Prev 2007; 16:847-51. doi: 10.1158/1055-9965.epi-06-0934 [Crossref] [ Google Scholar]
- Cox AG, Gurusinghe S, Abd Rahman R, Leaw B, Chan ST, Mockler JC. Sulforaphane improves endothelial function and reduces placental oxidative stress in vitro. Pregnancy Hypertens 2019; 16:1-10. doi: 10.1016/j.preghy.2019.02.002 [Crossref] [ Google Scholar]
- Qin Y, Zhang H, Liu Q, Jiang B, Chen J, Zhang T. Sulforaphane attenuates oxidative stress and inflammation induced by fine particulate matter in human bronchial epithelial cells. J Funct Foods 2021; 81:104460. doi: 10.1016/j.jff.2021.104460 [Crossref] [ Google Scholar]
- Wise RA, Holbrook JT, Criner G, Sethi S, Rayapudi S, Sudini KR. Lack of effect of oral sulforaphane administration on Nrf2 expression in COPD: a randomized, double-blind, placebo-controlled trial. PLoS One 2016; 11:e0163716. doi: 10.1371/journal.pone.0163716 [Crossref] [ Google Scholar]
- Alumkal JJ, Slottke R, Schwartzman J, Cherala G, Munar M, Graff JN. A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Invest New Drugs 2015; 33:480-9. doi: 10.1007/s10637-014-0189-z [Crossref] [ Google Scholar]
- Jnoff E, Albrecht C, Barker JJ, Barker O, Beaumont E, Bromidge S. Binding mode and structure-activity relationships around direct inhibitors of the Nrf2-Keap1 complex. ChemMedChem 2014; 9:699-705. doi: 10.1002/cmdc.201300525 [Crossref] [ Google Scholar]
- Purawarga Matada GS, Dhiwar PS, Abbas N, Singh E, Ghara A, Das A. Molecular docking and molecular dynamic studies: screening of phytochemicals against EGFR, HER2, estrogen and NF-KB receptors for their potential use in breast cancer. J Biomol Struct Dyn 2022; 40:6183-92. doi: 10.1080/07391102.2021.1877823 [Crossref] [ Google Scholar]
- Pal R, Teli G, Sengupta S, Maji L, Purawarga Matada GS. An outlook of docking analysis and structure-activity relationship of pyrimidine-based analogues as EGFR inhibitors against non-small cell lung cancer (NSCLC). J Biomol Struct Dyn 2023: 1-17. 10.1080/07391102.2023.2252082.
- Berendsen HJ, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun 1995; 91:43-56. doi: 10.1016/0010-4655(95)00042-e [Crossref] [ Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J Chem Theory Comput 2012; 8:3257-73. doi: 10.1021/ct300400x [Crossref] [ Google Scholar]
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 2010; 31:671-90. doi: 10.1002/jcc.21367 [Crossref] [ Google Scholar]
- Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A 2001; 105:9954-60. doi: 10.1021/jp003020w [Crossref] [ Google Scholar]
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys 2007; 126:014101. doi: 10.1063/1.2408420 [Crossref] [ Google Scholar]
- Berendsen HJ, Postma JP, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984; 81:3684-90. doi: 10.1063/1.448118 [Crossref] [ Google Scholar]
- Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput 2008; 4:116-22. doi: 10.1021/ct700200b [Crossref] [ Google Scholar]
- Pal R, Jawaid Akhtar M, Raj K, Singh S, Sharma P, Kalra S. Design, synthesis and evaluation of piperazine clubbed 1,2,4-triazine derivatives as potent anticonvulsant agents. J Mol Struct 2022; 1257:132587. doi: 10.1016/j.molstruc.2022.132587 [Crossref] [ Google Scholar]
- Petersen HG. Accuracy and efficiency of the particle mesh Ewald method. J Chem Phys 1995; 103:3668-79. doi: 10.1063/1.470043 [Crossref] [ Google Scholar]
- Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 1981; 52:7182-90. doi: 10.1063/1.328693 [Crossref] [ Google Scholar]
- Kumari R, Kumar R, Lynn A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 2014; 54:1951-62. doi: 10.1021/ci500020m [Crossref] [ Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 2001; 98:10037-41. doi: 10.1073/pnas.181342398 [Crossref] [ Google Scholar]
- Pal R, Kumar B, Swamy P, Chawla PA. Design, synthesis of 1,2,4-triazine derivatives as antidepressant and antioxidant agents: In vitro, in vivo and in silico studies. Bioorg Chem 2023; 131:106284. doi: 10.1016/j.bioorg.2022.106284 [Crossref] [ Google Scholar]