Bioimpacts. 2025;15:30378.
doi: 10.34172/bi.30378
Review
Deciphering the role of classical oestrogen receptor in insulin resistance and type 2 diabetes mellitus: From molecular mechanism to clinical evidence
Haryati Ahmad Hairi Conceptualization, Data curation, Investigation, Methodology, Visualization, Writing – original draft, 1 
Nurul Izzah Ibrahim Data curation, Investigation, Writing – original draft, 2
Muhammad Zulfiqah Sadikan Formal analysis, Writing – original draft, 3
Putri Ayu Jayusman Conceptualization, Data curation, Investigation, Validation, Visualization, Writing – original draft, 4
Ahmad Nazrun Shuid Formal analysis, Methodology, Validation, Visualization, Writing – review & editing, 5, *
Author information:
1Department of Biochemistry, Faculty of Medicine, Manipal University College Malaysia, Jalan Batu Hampar, Bukit Baru, 75150 Melaka, Malaysia
2Department of Pharmacology, Faculty of Medicine, Universiti Kebangsaan Malaysia, Jalan Yaacob Latif, Bandar Tun Razak, Cheras, 56000 Kuala Lumpur, Malaysia
3Department of Pharmacology, Faculty of Medicine, Manipal University College Malaysia, Jalan Batu Hampar, Bukit Baru, 75150 Melaka, Malaysia
4Department of Craniofacial Diagnostics and Biosciences, Faculty of Dentistry, Universiti Kebangsaan Malaysia, 50300 Kuala Lumpur, Malaysia
5Department of Pharmacology, Faculty of Medicine, Universiti Teknologi Mara (UITM), Jalan Hospital, 47000 Sungai Buloh, Selangor, Malaysia
Abstract
The biological actions of oestrogen are mediated by the oestrogen receptor α or β (ERα or ERβ), which are members of a broad nuclear receptor superfamily. Numerous in vivo and in vitro studies have demonstrated that loss of circulating oestrogen modulated by classical ERα and ERβ led to rapid changes in pancreatic β-cell and islet function, GLUT4 expression, insulin sensitivity and glucose tolerance, dysfunctional lipid homeostasis, oxidative stress, and inflammatory cascades. Remarkably, 17β-oestradiol (E2) can completely reverse these effects. This review evaluates the current understanding of the protective role of classical ER in critical pathways and molecular mechanisms associated with insulin resistance and type 2 diabetes mellitus (T2DM). It also examines the effectiveness of menopausal hormone therapy (MHT) in reducing the risk of developing T2DM in menopausal women. Clinical trials have shown the protective effects of MHT on glucose metabolism, which may be useful to treat T2DM in perimenopausal women. However, there are concerns about E2's potential side effects of obesity and hyperlipidaemia in menopausal women. Further studies are warranted to gain understanding and find other oestrogen alternatives for treatment of insulin resistance and T2DM in postmenopausal women.
Keywords: oestrogen receptor, Insulin resistance, Type 2 diabetes mellitus, Menopausal hormone therapy
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
No funds, grants, or other support was received.
Introduction
Diabetes mellitus is among the most rapidly rising global epidemics, with a complication that poses a major health threat. The main finding of diabetes mellitus is chronic hyperglycaemia caused by either impaired insulin secretion, impaired insulin effect, or both.1 The two most common forms of diabetes are type 1 diabetes (T1DM) resulting from pancreatic cell destruction that causes an absolute insulin deficiency, and type 2 diabetes (T2DM), which is caused by insufficient insulin secretion and impaired response to insulin.2 Gestational diabetes, in addition to T1DM and T2DM, is another common form of diabetes. However, other less common specific types exist.3 Leastways, T2DM is the type that is mainly responsible for the global health challenges of this disease. Globally, the burden of T2DM is on the rise in terms of incidence, prevalence, death and disability-adjusted life-years (DALYs).4 Approximately 1 in 11 adults has been reported to have diabetes mellitus, with 90% of them having T2DM. Additionally, it is worth noting that Asia is the primary region affected by this global T2DM epidemic.5 The inadequate action of insulin, the major glucose-lowering hormone, is the pathogenic driver of T2DM.6
Physiologically, the elevation of postprandial blood glucose concentration is sensed by pancreatic β-cells, which eventually release insulin into the circulation and inhibit glucose production in the liver. The circulating insulin binds to insulin receptors in almost all mammalian cells. In muscle and adipose tissues, glucose uptake is dependent on the insulin-responsive glucose transporter GLUT4, demonstrating its fundamental role in plasma glucose clearance and glucose homeostasis.7 Insulin resistance is defined as a state of inappropriate or reduced responsiveness of peripheral tissues to high insulin levels, causing ineffective glucose homeostasis.8 Insulin resistance contributes to increased glucose production in the liver and decreased glucose uptake in muscle, liver and adipose tissue.9 In a prediabetic state, the level of insulin increases to meet normal insulin requirements, which leads to chronic hyperinsulinemia, hyperglycaemia-induced β-cell dysfunction and subsequently leading to T2DM.5,10 In essence, malfunction of the feedback loops between insulin action and insulin secretion results in a hyperglycaemic state in T2DM.11
Prolonged exposure to hyperglycaemia is considered the main causal factor in the pathogenesis of diabetes complications. In fact, the global burden of diabetes mellitus contributes to its morbidity and mortality from complications of the disease.12 The incidence of diabetic complications escalates with the duration of diabetes mellitus, followed by ageing-related disability and reduced life expectancy.13 Hence, clinical trials have suggested that menopausal hormone therapy (MHT) has a favourable effect on glucose homeostasis and could delay the onset of T2DM in women. Although this protective mechanism is still unclear, the action of oestrogens on glucose homeostasis and insulin sensitivity observed in human and animal models suggested its role in preventing diabetes.14,15
Oestrogens boost hepatic insulin sensitivity by lowering gluconeogenesis and glycogenolysis and increasing insulin release in the Langerhans islets. 17β-oestradiol (E2) stimulates insulin production and release in cells of in vitro and in vivo via oestrogen receptors, which should counteract insulin resistance.16 Furthermore, oestrogen inhibits β-cell apoptosis and pro-inflammatory signalling, as well as improves insulin function. As a result, the greater quantity of visceral adipose tissue observed in men, along with lower endogenous oestrogen levels, may be connected to higher insulin resistance compared to that of premenopausal women.17 On the other hand, Choi et al demonstrated that weight loss due to limited diets increased first-phase insulin secretion in ovariectomised rats, but that decreased second-phase insulin secretion at 120 minutes under a hyperglycaemia clamp was not overcome. Nevertheless, the replacement of oestrogen corrected the reduced second-phase and first-phase insulin secretion.18 Therefore, this present review evaluates the current knowledge and understanding of the role of oestrogen and its classical receptors in critical pathways and molecular mechanisms involved in insulin resistance and T2DM. Additionally, it aims to examine the beneficial effect of MHT in menopausal women who are at risk of T2DM.
Classical oestrogen receptor: structure and expression
Oestrogens, renowned as one of the most important hormones, are involved in female physiology, specifically in the reproductive and non-reproductive systems. Estrone, estriol and 17β‐oestradiol (also known as oestradiol or E2) constitute a group of human oestrogens. Chemically, oestrogen belongs to the steroid hormone group, whose core structure comprises 17 carbon-carbon bonds. These bonds are arranged as four fused rings consisting of three cyclohexane rings and one cyclopentane ring. All four oestrogens contain 18 carbons and a phenolic hydroxyl group. They also have one benzene ring with a ketone group for estrone and a varying number of hydroxyl groups for the other three oestrogens.19 Among all, oestradiol possesses the most potent oestrogenic properties and is commonly utilised to symbolise oestrogen owing to its physiological significance and predominance.20,21
Oestrogen is involved in the development and homeostasis of several tissues in females and males. In females, oestrogen is primarily produced by the ovaries and in smaller amounts by other tissues, including the liver, pancreas, adipose tissue, adrenal glands and breast.22 In males, the production of local oestrogen occurs by aromatisation of testosterone in reproductive tract cells, including Sertoli cells, Leydig cells and mature spermatocytes. In certain conditions, including pregnancy, oestrogen is also produced by the placenta. Oestrogen exerts its actions by binding to the oestrogen receptors (ERs), which comprise two distinct types: ERα, which is mostly expressed in the uterus, ovary, mammary glands, liver, bone, male reproductive organs and adipose tissue, and ERβ, which is mainly expressed in the uterus, ovary, lung, immune system, prostate and colon20,23 ERα and ERβ are approximately 95% homologous in their DNA-binding domains.24 Oestrogen could activate both receptors, but the biological effects exerted may be different due to the structural differences in their ligand-binding domains and amino acid sequence identity, which differentiates their ligand affinities.25,26
The binding of oestrogen to their receptors activates cytoplasmic signalling pathways and nucleus transcription in the nucleus that are essential in the regulation of gene expression, metabolism, cell growth and proliferation.21 These actions are basically mediated via genomic and non-genomic pathways. The genomic effect (or nuclear) involves the direct binding of ER complexes to specific sequences in gene promoters and works as transcription factors in the nucleus. The non-genomic effect (or cytoplasmic) does not involve direct binding to DNA but via cytosolic signal transduction pathway that takes place at the plasma membrane.19,21 All four oestrogens could bind to nuclear and membrane oestrogen receptors but with varying affinity and strength.27
It is also known that the role of oestrogen is much broader than preceding thought. Ultimately, under normal circumstances, oestrogen controls cell autophagy, proliferation, differentiation, apoptosis and survival, but their action can be dysregulated in diseases.28 Derangement or dysregulation of oestrogen activity has been shown to be involved in the pathogenesis and progression of many diseases, including diabetes.21 Findings from human and animal studies suggested that oestrogen has protective effects against diabetes owing to the role of oestrogen in glucose homeostasis, insulin sensitivity and secretion.14,29,30
Oestrogen receptor in insulin resistance and type 2 diabetes mellitus (T2DM): preclinical evidence
Insulin resistance is a condition described as the inability of circulating insulin to effectively regulate the uptake and/or utilisation of glucose by insulin-sensitive tissues and organs. When blood glucose levels rise, the secretion of insulin from pancreatic β-cells increases while the synthesis of glucose in the liver decreases.31 On the other hand, insulin-resistant people are unresponsive to this signalling pathway and, as a result, do not experience increased insulin secretion and continue with hepatic glucose synthesis. This could lead to the development or worsening of hyperglycaemia.32 Insulin resistance during the prediabetic stage causes β-cell to hypersecrete insulin to maintain normal blood glucose levels, resulting in hyperinsulinemia. Prolonged insulin release exhausts pancreatic β-cells and causes apoptosis, which is a risk factor for the onset of T2DM that can result in decreased insulin secretion.33 Besides, impaired oestrogen signalling is associated with dysfunctional pancreatic β-cells, dysregulation of GLUT4 expression, glucose intolerance, insulin insensitivity and dysfunctional lipid homeostasis, all of which contribute to the development of insulin resistance, T2DM and obesity (Fig. 1).
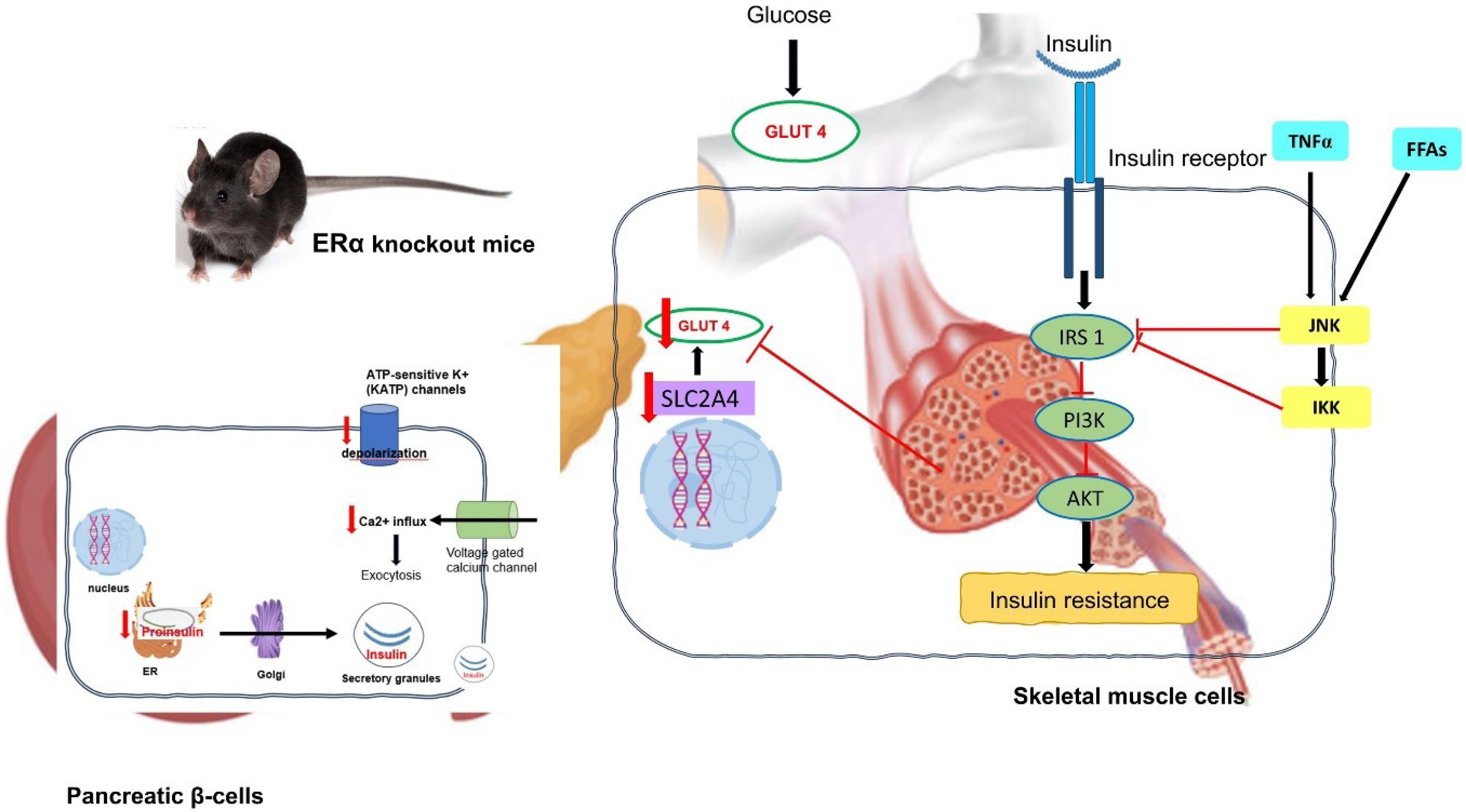
Fig. 1.
The effect of ERα deletion in pancreatic β-cells and skeletal muscle on insulin sensitivity and glucose tolerance in the pancreatic β-cells and skeletal muscle of ERαKO mice. Abbreviation: ERα- oestrogen receptor alpha; ER- endoplasmic reticulum; GLUT4- glucose transporter-4; IRS-1- insulin receptor substrate 1; PI3K- Phosphatidylinositol 3 kinase; AKT- protein kinase B; TNF- Tumor necrosis alpha; FFA: free fatty acid; JNK – c Jun N-terminal kinase; IKK- IκB kinase.
.
The effect of ERα deletion in pancreatic β-cells and skeletal muscle on insulin sensitivity and glucose tolerance in the pancreatic β-cells and skeletal muscle of ERαKO mice. Abbreviation: ERα- oestrogen receptor alpha; ER- endoplasmic reticulum; GLUT4- glucose transporter-4; IRS-1- insulin receptor substrate 1; PI3K- Phosphatidylinositol 3 kinase; AKT- protein kinase B; TNF- Tumor necrosis alpha; FFA: free fatty acid; JNK – c Jun N-terminal kinase; IKK- IκB kinase.
Modulation of oestrogen receptor in pancreatic β-cells and islet function
Oestrogen is known to regulate pancreatic β-cell activity. The effect on β-cells is immediate, as it controls membrane depolarisation, Ca2+ influx and insulin secretion.34 Previous studies indicated that 17β-oestradiol (E2) has favourable effects on insulin action and β-cell function by maintaining insulin sensitivity. For example, a study indicated that in vivo treatment of male mice with E2 at the dose of 100 µg/kg/d for 4 days increased insulin content and insulin secretion in response to high blood glucose.35 Treatment with pure antioestrogen, ICI182780 inhibited this action, indicating a classic ER-mediated effect.36 ER- knockout (ERKO) mouse is vulnerable to oxidative stress, which causes pancreatic β-cell death and insulin-deficient diabetes. This model exhibited insulin resistance and reduced glucose tolerance. It was also evident that the most common receptor isoform for regulating insulin levels in the pancreas is ERα.37
The blocking of ATP-sensitive potassium (KATP) channels, which control the β-cell resting membrane potential, is an important event in the insulin release pathway. This blockage can cause a specific electrical activity pattern that comprises bursts of action potentials by the opening of voltage-gated Ca2+, Na+ and K+ channels, as well as an increase in intracellular Ca2+, culminating in the release of insulin after the channel is closed.38,39 Changes in the expression and/or function of these ion channels influence insulin secretion and are a major risk factor for T2DM.40 Oestrogen controls the KATP channels in β-cells.41 Bisphenol-induced insulin hypersecretion might represent one of the changed pathways causing insulin resistance, which is a risk factor for T2DM and obesity.42 Bisphenol analogues are xenoestrogen capable of interacting with ERα and ERβ.43 Bisphenol S (BPS) or Bisphenol F (BPF) have been shown to boost insulin release while decreasing KATP channel activity. Additionally, 48-hour treatment with BPS or BPF increased insulin release and lowered the expression of many ion channel subunits in wild-type (WT)-cells but had no effect in ERβ-knockout (ERβKO) cells.44
Since the 1980s, it has been shown that oestrogen has a direct effect on the pancreatic islet cells to modulate insulin release.45 Islet dysfunction and hyperinsulinemia are ensued from the absence of ERα. For example, 17β-oestradiol (E2) increased insulin content in cultured islets of WT mice but not islets from ERα-knockout (ERαKO) mice.46,47 Furthermore, anti-apoptotic effects of oestrogen were largely mediated by ERα. E2 treatment reduced STZ-induced β-cell death while increasing insulin production and decreasing insulin resistance and glucose intolerance.48 E2's protective effects were abolished in ERαKO female mice.
Classical ERα and ERβ are nuclear transcription factors implicated in regulating various physiological processes in humans. Physiological functions of oestrogenic compounds are modulated in the cell nucleus regulating the transcription of specific target genes by binding to associated DNA regulatory sequences. In a study by Gay et al, it was demonstrated that the functions of the oestrogen receptor are modulated by the LIM/HD proteins, which belong to a subset of homeodomain (HD)-containing transcription factors. The insulin gene enhancer protein, Islet-1 (ISL1), is the most extensively studied among the LIM/HD transcription factors. It is located in discrete brain areas, which is important in motoneuron differentiation and is responsible for ER expression.It has been demonstrated that ISL1 could directly interact with oestrogen receptors, as shown by the dual immunohistochemistry of ISL1-ER co-expression by the same neuronal subpopulation within the rat hypothalamic arcuate nucleus. The interaction between ISL1 via its N-terminal LIM domains and ER inhibited oestrogen receptor dimerisation, thus leading to a strong and specific inhibition of oestrogen receptor DNA binding activity.Therefore, this study indicated that ISL1, via its N-terminal LIM domains, could specifically inhibit the oestrogen receptor-driven transcriptional activation, while the ER could serve as a coactivator for ISL1.49
In the study by Alonso et al to determine the pancreatic insulin content regulation by oestrogen receptors, ERα selective agonist, propyl pyrazole triol (PPT) and ERβ selective agonist, diarylpropionitrile (DPN) were supplemented to isolated pancreatic β-cells of genetically modified mice (ERαKO and ERβKO). Treatment with oestrogen receptor agonists demonstrated that only ERα had increased insulin content of the islets following a 48-hour culture. This action replicated the response produced by oestrogen, specifically the E2 treatment. The study also demonstrated an upregulation of pancreatic insulin content following the activation of ERα by its agonist, which rapidly activated the mitogen-activated protein kinases/extracellular signal-regulated kinase (MAPK/ERK) 1/2 pathway.50 In the study by Longuet et al using a MIN6 β-cell line and isolated rat islets of Langerhans, it was discovered that MAPK/ERK 1/2 pathway was associated with glucose-dependent actions in β-cells and played a role in the overall glucose-induced insulin secretion. The ERK1/2 cascade activation may induce the phosphorylation of cytoplasmic proteins implicated in the exocytosis of insulin granules.51 Hence, it was suggested that the participation of ERα in endocrine pancreatic function and blood glucose homeostasis took place through the MAPK/ERK 1/2 pathway.
Modulation of oestrogen receptor in GLUT4 expression, insulin sensitivity and glucose tolerance
ERα is far more abundant than ERβ in β-cell of the pancreas.52 In ERKO mice, homozygous ERβ deletion did not result in insulin resistance, but homozygous deletion of ERα causes substantial skeletal muscle insulin resistance.53 Bryzgalova et al suggested reduced total glucose transporter type 4 (GLUT4) levels in muscle as the underlying cause of the ERαKO insulin resistance phenotype.54 Meanwhile, Ribas et al reported that it was due to defective insulin signal transduction.29 ERα has previously been implicated in the regulation of proximal insulin signal transduction, as E2 administration to insulin-resistant rodents increases insulin receptor substrate (IRS)-1 abundance and insulin-stimulated tyrosine phosphorylation, as well as Akt phosphorylation at the activation site.55 Additionally, ERαKO caused obesity at the expense of white adipose tissue growth, decreased energy expenditure, insulin resistance and glucose intolerance.56 In comparison to wild-type mice, ERβKO mice showed normal glucose tolerance and insulin release, and it was proposed that ERβKO protected against diet-induced insulin resistance and glucose intolerance. Other than that, Manrique et al also discovered that the insulin receptor substrate 1/phosphatidylinositol 3-kinase association and protein kinase B activation were decreased in ERαKO mice, resulting in impaired skeletal-muscle insulin signalling and glucose uptake.57 The findings on the effect of ERα on systemic and skeletal muscle insulin sensitivity may help to better understand the mechanisms involved in oestrogen protection against insulin resistance.
Identification of oestrogen receptors 1 and 2 (ESR1 and ESR2), as well as insulin-sensitive GLUT4, provided an opportunity to shed light on oestrogen regulation of glycaemic homeostasis.58 It was also observed that ESR1 deficiency may increase the possibility of insulin resistance and T2DM.59 Besides, ESR1 could decrease lipogenesis in white adipose and liver tissue, improve insulin sensitivity and aid the maintenance of pancreatic cells.60 It has been recorded that selective deletion of ESR1/ ERα from skeletal muscle in female mice and cultured myotubes caused marked insulin resistance.61 The solute carrier family 2 member 4 (SLC2A4) gene encodes the insulin-responsive glucose transporter isoform GLUT4, which is essential for glucose uptake in muscle and adipose tissues, playing a critical role in clearing plasma glucose.62 Additionally, the inhibition of SLC2A4 gene expression and eventual reduction of GLUT4 protein in chronic insulin resistance has been linked to T2DM.63 Moreover, mRNA SLC2A4 expression was reduced in the subcutaneous and visceral adipose tissues of ESR1 knockout female mice.64 SLC2A4 deletion produced insulin resistance, whereas SLC2A4 overexpression improved glycaemic control, even in diabetic mice.58,65 While decreased SLC2A4 expression is associated with insulin resistance, increased SLC2A4 expression is considered to be related to improved glycaemic management.
Skeletal muscle is responsible for approximately 75% of glucose elimination in response to insulin release.66 Insulin receptor, insulin receptor substrate (IRS), phosphatidylinositol-3 (PI3K) and AKT kinase are components of the insulin signalling system governing glucose absorption.67,68 When this pathway is activated, the cytoplasmic GLUT4 translocates to the cell membrane, where it enhances glucose transport into the cell. GLUT4 is abundant in muscle and is a rate-limiting step in insulin-induced glucose absorption.69,70 E2 influences glucose homeostasis in the muscle mostly through its impacts on important proteins in the insulin signalling system, such as GLUT4 expression and translocation.71 Insulin signalling pathway is required for GLUT4 transport to the cell surface, while insulin resistance has been well known to lower GLUT4 expression.72,73 Both ER receptors play a role in the regulation of insulin sensitivity in the liver and the expression of GLUT4 in skeletal muscle, among other functions. It is suggested that increasing ERα expression may enhance the sensitivity of skeletal muscle cells to insulin, ultimately leading to glucose uptake.74 Further, in ERα knockout male mice, tamoxifen, an ER antagonist, improved insulin sensitivity with fasting hypoglycaemia, increased GLUT4 expression in skeletal muscle and improved pancreatic islet function.75 Transgenic mice lacking or overexpressing GLUT4 have lower or higher whole-body insulin sensitivity, respectively, highlighting its role in maintaining glucose homeostasis.76
E2 administration to insulin-resistant rodents increased insulin receptor substrate (IRS)-1 abundance and insulin-stimulated tyrosine phosphorylation, as well as phosphorylation of Akt at the activation site Ser473.77 Furthermore, ERα-mediated tyrosine phosphorylation of the IRS protein contributed to insulin-stimulated glucose uptake, as demonstrated in 3T3-L1 adipocytes.78 Remarkably, it was demonstrated that oestrogen/ER-induced adenosine monophosphate protein kinase (AMPK) activation acted upstream of IRS-1/Akt signalling and that E2 modulated the expression of genes associated with glucose metabolism in 3T3-L1 adipocytes by activating ER-AMPK in the absence of insulin-induced signalling activation.79
The Akita strain is a monogenic mouse model for type 1 diabetes characteristics. A spontaneous mutation in the insulin 2 (Ins2) gene caused improper folding of the insulin protein, resulting in a decreased number of β-cells and decreased insulin output, which led to dysfunctional β-cells. By 3 to 4 weeks, heterozygous Ins2 Akita mice developed insulin-dependent diabetes, which included hyperglycaemia, hypoinsulinemia, polydipsia and polyuria. Intriguingly, conjugated oestrogen was shown to promote the degradation of misfolded Akita proinsulin proteasomal in pancreatic cells, preventing pancreatic cell death and diabetes.80
Modulation of oestrogen receptor in dysfunctional lipid homeostasis
Dysfunctional lipid homeostasis within pancreatic β-cells has been associated with metabolic and oxidative stress. This may lead to β-cell damage and progression from insulin resistance to T2DM.81 The suppression of fatty acids and lipids has been associated with protection against β cell failure and T2DM.82,83 In a study by Tiano et al, a slow-release 17β-oestradiol (E2) pellet subcutaneously implanted behind the neck of male Zucker diabetic fatty (ZDF) rats was able to inhibit the synthesis and accumulation of fatty acids and glycerolipids in pancreatic islets. Studies using rat β cell line, cultured ZDF rat, mouse and human islets reported the association between the anti-lipogenic action of E2 with the activation of ERα, ERβ and G protein-coupled ER (GPER). It was also demonstrated that activating oestrogen receptors inhibited β-cell lipid synthesis by suppressing the activity of fatty acid synthase (FAS) via a nonclassical pathway dependent on activated signal transducer and activator of transcription 3 (STAT3). Accordingly, pancreas-specific deletion of STAT3in mice inhibited ER-mediated suppression of lipid synthesis. The data suggested that extranuclear ERs may be promising therapeutic targets for preventing β-cell failure in T2DM. The study conducted by Tiano et al suggested that oestrogen receptors, specifically ERα, ERβ and GPER, modulated the expression of FAS through the activated STAT3 pathway. This finding could potentially have therapeutic implications for addressing β cell dysfunction in obesity-associated T2DM.83
The expression of this transporter gene, which is directly related to the development of insulin resistance, decreases with obesity. Obesity causes an abundance of free fatty acids (FFAs), which in turn induce cellular malfunction, particularly in the mitochondria.84 When adipocytes are subjected to high levels of either glucose or FFA, they exhibit elevated levels of mitochondrial fission. This is accomplished by manipulating the insulin transduction pathway as well as endogenous ROS generation and release. Mitochondrial fission boosts p38 MAPK expression while suppressing IRS-1 and protein kinase B (Akt) activity.85,86 According to a study conducted in patients with varying degrees of obesity, this mitochondrial malfunction appeared to alter transcriptional levels.87 As adipose tissue grows, mitochondrial transcription levels fall, resulting in reduced glucose utilisation in this tissue.88 E2 therapy improved insulin sensitivity and glucose tolerance in ovariectomised mice fed a high-fat diet (HFD) but not in ER-deficient mice.54,89 Furthermore, this study found that oestrogen improved insulin sensitivity and glucose tolerance in HFD-fed rats via ERα activation.54 E2 therapy, on the other hand, reduced HFD-induced insulin resistance by 50% and increased insulin signalling (Akt phosphorylation) in insulin-stimulated skeletal muscles during hyperinsulinemic-euglycemic clamp trials.90
Adiposity is a global epidemic that raises the risk of diabetes and cardiometabolic disorders. According to epidemiological research, postmenopausal women have low oestrogen levels, gain weight in general and redistribute adipose tissue, resulting in increased abdominal fat accumulation. Oestrogen significantly increases the activity of the enzyme 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, which is the rate-limiting enzyme in hepatic cholesterol biosynthesis under high dietary cholesterol loads, implying that de novo synthesis of cholesterol in the liver may be increased.91 The presence of ERs in the nucleus and mitochondria suggests a mechanism for oestrogen coordination of mitochondrial DNA (mtDNA) and nuclear-encoded mitochondrial respiratory complex genes. Mitochondria ERα and ERβ engage in the coordination of the cell's energy requirements as well as in OXPHOS biosynthesis, influencing ROS production and apoptosis induction.92 For instance, Hamilton et al revealed that selective ERα activation with the ER agonist, 4,40,4′′-(4-Propyl-[1H]-pyrazole-1,3,5-triyl) (PPT) enhanced systemic oxidative metabolism, mitochondrial function, and insulin resistance in low-density lipoprotein receptor-deficient mice fed a western high-fat diet.93
Phosphoenolpyruvate carboxykinase (PEPCK) has been identified as a crucial enzyme in gluconeogenesis, particularly in the liver and kidney.94 Evidence suggested that increased PEPCK activity led to higher glucose output and diabetes aggravation.95 Previous studies discovered that E2 suppressed hepatic gluconeogenic genes like PEPCK-1 and glucose 6-phosphatase (G6Pase); however, this action was missing in mice lacking the liver oestrogen receptor (LERKO mice). Male LERKO mice had increased hepatic gluconeogenic activity as well as fasting hyperglycaemia. Additionally, enhanced liver lipid deposits and triglyceride levels were also seen in male LERKO mice owing to enhanced hepatic lipogenesis, as evidenced by higher mRNA levels of fatty acid synthase (FAS) and acetyl-CoA carboxylase (Acc1). Intriguingly, E2 promoted ESR1 binding to the promoters of PEPCK-1, glucose-6-phosphatase (G6Pase), FAS and Acc1.96 Thus, it is apparent that obesity and metabolic disorders are linked to impaired ESR1 function.
Modulation of oestrogen receptor in oxidative stress and inflammatory signalling cascade
Hyperglycaemia increases reactive oxygen species (ROS) generation while decreasing mitochondrial biogenesis, resulting in mitochondrial dysfunction. Excessive ROS formation activated important pathways that produced oxidative stress in the cellular environment, causing β-cell function degradation and increasing insulin resistance in T2DM patients.97,98 ROS, such as hydrogen peroxide (H2O2), could cause muscle insulin resistance by activating c-Jun amino-terminal kinases (JNK), which block the insulin signalling cascade at the IRS level.98 E2 suppresses oxidative stress through nongenomic and genomic actions by activating pathways that prevent the generation of ROS and increasing the efficiency with which ROS are scavenged. The mechanism by which E2 affects mitochondrial activity appeared to be via transcription by nuclear translocation of dimerised, E2-bound ERs or transcriptional activation of mitochondrial genes by ERs localised inside the mitochondria.99 A study reported ER-containing mitochondrial extracts bound to putative mitochondrial EREs (mtEREs), with E2 increasing the binding and ER-deficient cells missing the binding.100 Meanwhile, tumour necrosis factor-α (TNF-α) and interleukin-6 (IL6) cytokines released by adipose tissue were linked to decreased GLUT4 expression, decreased glucose absorption by muscle and compensatory hyperinsulinemia.101 Activated inflammatory signalling networks caused JNK and IκB kinase (IKK) activation, leading to insulin resistance via Protein kinase C (PKC).102 This was also shown by Ribas et al, which reported that stimulation of inflammatory signalling kinases and cascades such as JNK, AMPK, IKK and TNF-α reduced insulin action by limiting IRS activity in ERα knockout mice.29 Also, when JNK1 deletion mice were subjected to a high-fat diet, they experienced long-term metabolic protection from diet-induced obesity and demonstrated significantly higher insulin sensitivity, while maintaining a normal life span.103
The prevalence of cardiovascular disease in diabetic menopausal women has been shown to be higher. T2DM was associated with high levels of triglycerides, total cholesterol, low-density lipoprotein and fasting blood glucose, all of which are major risk factors for cardiovascular diseases, including coronary artery disease in menopausal women.104 Moreover, In vivo studies revealed that T2DM induces cardiovascular disease by modifying glycaemic and lipid profiles and pro-inflammatory cytokine levels. These studies also found a relationship between TNF-α activation and lipoprotein buildup in rat artery walls, as well as response to cardiac ischemia and reperfusion. Recent research by Khaksari et al investigated the beneficial effects of administrating selective oestrogen receptor modulators (SERMs) and oestrogen (E2), alone or in combination, in postmenopausal diabetic cardiovascular disease. E2 or SERMs appeared to help the cardiovascular system by lowering pro-inflammatory cytokines such as TNF-α, IL-10 and Angiotensin II (Ang II) levels in ovariectomised diabetic rats.105 Subsequent studies from Ebrahami et al and Azizian et al showed that a combination of Tamoxifen (SERM) and E2 is more effective than either tamoxifen or E2 alone in reducing diabetes mellitus and cardiovascular dysfunction by improving lipid and glycaemic profiles, pro-inflammatory cytokines and atherogenic index.106,107
Table 1 summarizes in vivo and in vitro findings on the role of ER.
Table 1.
In vivo and in vitro findings on the role of ER in pancreas β-cells and islet functions, GLUT4 expression, glucose tolerance, insulin sensitivity, lipid homeostasis, oxidative stress and inflammation signalling cascade
|
Type of study/Model used
|
Summary of findings
|
Comments/Outcomes
|
References
|
|
Role of ER in pancreas β-cells and islet function
|
| In vivo/wild-type mice |
Bisphenol S (BPS) and Bisphenol F (BPF) have been demonstrated to increase glucose-stimulated insulin secretion (GSIS) while lowering KATP channel activity via an ERβ-mediated mechanism. |
ERβ activation changed three critical cellular events in β-cell, namely ion channel expression and activity, as well as insulin release. |
42
|
| In vivo/Estrogen-induced adult Wistar strain female rats. |
Insulin gene enhancer protein Islet-1 (ISL1) and ERα and ERβ were co-expressed by the same neuronal subpopulation within the rat hypothalamic arcuate nucleus. |
The inhibition of ER dimerisation led to a strong and specific inhibition of ER-DNA binding activity. |
47
|
| In vitro/ Transient transfection of Chinese hamster ovary cells with ERE-SV-Luciferase |
In vitro interaction between ISL1-ER inhibited ER dimerisation. |
| In vivo/ Adult wild type, ERαKO and ERβKO of Swiss albino OF1 mice |
The levels of activated ERK1/2 in the isolated β-cells were increased with treatment using ERα. |
ERα is involved in pancreatic function and glucose homeostasis via MAPK/ERK 1/2 pathway |
48
|
|
Roles of ER in GLUT4 expression, insulin sensitivity and glucose tolerance
|
| In vivo/ ERαKO and ERβKO mice |
ERKO mice were found to lack skeletal muscle glucose absorption via GLUT 4, resulting in significant hepatic insulin resistance. |
ERαKO mice exhibited severe hepatic insulin resistance together with impairment in skeletal muscle glucose absorption. |
52
|
| In vivo/ ERαKO mice |
ERαKO mice had reduced insulin receptor substrate 1/phosphatidylinositol 3-kinase association and protein kinase B activation, and immunostaining for 3-nitrotyrosine was increased, resulting in impaired skeletal-muscle insulin signalling and glucose absorption. |
ERαKO had poor glucose tolerance and significant insulin resistance in their skeletal muscles. |
55
|
| In vivo/ ERαKO of C57Bl6 mice |
The insulin receptor substrate (IRS)-1 on serine residues was observed to be decreased in ERαKO mice, which led to impaired insulin signalling and glucose transport. The increased fat weight gain and overall body fat percentage in ERKO mice resulted from the buildup of bioactive lipid intermediates and the decreased levels of PPARα, PPARδ and UCP2 mRNA. |
59
|
| In vivo/ Esr1 knockout (Esr1KO) mice |
SLC2A4 gene expression, which encodes GLUT4, was increased in female Esr1KO mice treated with E2 in subcutaneous and visceral adipose tissues. |
E2 induced adipocyte development and SLC2A4/GLUT4 expression via an ESR1-mediated mechanism. |
62
|
| In vivo/ ERαKO male mice |
In ERαKO male mice, tamoxifen improved insulin sensitivity with fasting hypoglycemia, as determined by tolerance tests for insulin (ITT) and glucose (GTT), increased GLUT4 expression in skeletal muscle, and improved pancreatic islet function. |
Tamoxifen improved glucose tolerance in skeletal muscle by increasing GLUT4 expression. |
73
|
| In vitro/ 3T3-L1 adipocytes. |
ERα-mediated tyrosine phosphorylation of the IRS protein was associated with insulin-stimulated glucose absorption in 3T3-L1 adipocytes. |
ERα promoted insulin-stimulated glucose absorption via regulation of tyrosine phosphorylation of the IRS-1 protein. |
76
|
| In vivo/Akita mice |
Conjugated oestrogen (CE) activated nuclear and membrane ER, increased transcriptional repression and proteasomal degradation of UBC6e, a ubiquitin-conjugating enzyme and ER-associated protein degradation (ERAD) degrader. |
CE promoted misfolded proinsulin degradation, suppressed endoplasmic
reticulum stress and protected insulin secretion in Akita mice |
78
|
|
Roles of ER in lipid homeostasis
|
| In vivo/Non-diabetic Zucker fatty (ZF) rats, Zucker diabetic fatty (ZDF) rats, Zucker lean (ZL) rats/ Pancreas-specific null deletion of ERα/mice |
Suppression of synthesis and accumulation of fatty acids and glycerolipids in islets of the rats.
Pancreas-specific null deletion of ERα in mice (PERα–/–) inhibited lipid synthesis induced by E2. |
ER activation inhibited β-cell lipid synthesis by suppressing the activity of fatty acid synthase via a nonclassical pathway dependent on activated STAT3. |
81
|
| In vivo/ ERαKO and ERβKO mice |
In ERKO mice, the plasma leptin level was increased, while the adiponectin concentration and Lepr expression were decreased. Genes involved in hepatic lipid production were increased in ERKO mice, while genes involved in lipid transport were downregulated. |
Increment in lipogenic genes resulted in insulin resistance in the liver, which was caused by a decrease in Lepr expression. |
52
|
| In vivo/ mice lacking liver estrogen receptor α (Esr1) (LERKO mice) |
The LERKO mice demonstrated higher levels of hepatic triglyceride as well as elevated FAS and Acc1 expression. E2 inhibited the expression of hepatic gluconeogenic genes such as phosphoenolpyruvate carboxykinase 1 (PEPCK-1) and glucose 6-phosphatase (G6Pase). |
The ESR1 signalling system may be a promising therapeutic target to prevent and correct lipid and glucose metabolic disorders. |
91
|
| In vivo/Western high-fat diet ovariectomised low-density lipoprotein receptor-deficient mice |
In WHFD-fed OVX mice, selective ERa activation with ERα agonist, 4,40,4′′-(4-Propyl-[1H]-pyrazole-1,3,5-triyl) (PPT) avoided weight gain, enhanced insulin action and reduced visceral fat formation.
PPT treatment increased systemic metabolism, oxygen consumption and core body temperature and induced the expression of several metabolic genes in the heart, liver, skeletal muscle, and adipose tissue, including peroxisome proliferator-activated receptor gamma (PPARγ), coactivator 1 alpha, and nuclear respiratory factor 1. |
Selective ER activation with PPT improves metabolic effects such as insulin resistance, whole-body energy metabolism, and mitochondrial function in OVX mice with metabolic syndrome. |
96
|
| In vivo/ ovariectomised diabetic (high-fat diet and streptozotocin) female Wistar rats |
Combination of E2 and tamoxifen treatment lowered triglyceride, total cholesterol, low-density lipoprotein, and fasting blood glucose in ovariectomised diabetic rats. |
Combination of E2 and tamoxifen treatment reduced lipid profile and atherogenic index. |
109
|
|
Role of ER in oxidative stress and inflammation signalling cascade
|
| In vivo/ ERαKO of C57Bl6 mice |
The activation of inflammatory signalling cascades such as JNK, AMPK, IKK, and TNFα inhibited insulin action by reducing IRS activity in adipose tissue of ERαKO mice. |
ERα is a regulator of the inflammatory signalling cascade in adipose tissue and has been linked to insulin resistance. |
59
|
| In vivo/ ovariectomised diabetic (high-fat diet and streptozotocin) female Wistar rats |
Combination treatment of E2 and tamoxifen decreased serum Angiotensin II (Ang II), TNF-α, and the TNF- α to IL-10 ratio, indicating an improvement in inflammatory balance. |
E2 and Tamoxifen therapy can reduce Ang I) production, which leads to enhanced insulin sensitivity and cardioprotection by lowering inflammatory cytokines. |
108
|
| In vivo/ ovariectomised diabetic (high-fat diet and streptozotocin) female Wistar rats |
Tamoxifen reduced mean arterial blood pressure, fasting blood glucose, and inflammatory cytokine levels. IL-10. Tamoxifen boosted the protein expression of ERα and ERβ |
Tamoxifen protects against diabetic cardiovascular impairment and is a promising E2 replacement candidate. |
110
|
Menopause and T2DM: clinical evidence
Menopause is the end of a woman's reproductive life caused by ovarian ageing.108 It is characterised by a significant decrease in endogenous oestrogen concentrations, changes in body weight, adipose tissue distribution and energy expenditure, as well as insulin secretion and activity. All these can predispose to the development of T2DM, both independently and additively, to ageing.109 Menopause occurs at an average age of 50 to 52 years.110 However, roughly 10 per cent of the female population experiences menopause before the age of 45, a condition known as "early" or "premature" menopause.111 Given the protective impact of oestrogen on pancreatic β-cell function and insulin resistance, one plausible mechanism of T2DM is reduced exposure to endogenous oestrogen.112 Tyler et al discovered a link between reduced ovarian function and reduced pancreatic β-cell function, which has an impact on glucose metabolism and diabetes risk.113 Postmenopausal women experience a significant decrease in sex hormones, which leads to a further decline and eventual cessation of ovarian function. Weight gain, increased visceral fat and impaired glucose homeostasis are all related to the menopause transition, which are key risk factors for T2DM.114
Based on the clinical data, certain studies have indicated a correlation between early menopause and a heightened risk of T2DM. Conversely, other studies reported that late menopause is also a significant risk factor for T2DM. According to a Dutch study, postmenopausal women had a higher incidence of T2DM.115 This suggested that postmenopausal status is related to T2DM. However, in a Vietnam study, a significant connection between menopausal state and T2DM was lost when age, occupation, BMI, physical activity, parental history of T2DM, residential status and hypertension were taken into account.116 In a Chinese study on women from Henan Province, the risk of T2DM rose with postmenopausal versus premenopausal states. The highest correlation between postmenopausal status and T2DM was recorded among women with a BMI of less than 24.0 kg/m2.117 Moreover, T2DM risk was enhanced by postmenopausal status, which interacted with BMI, hypertension, triglyceride level and waist circumference.
Brand and colleagues reported a higher incidence of T2DM among individuals experiencing early menopause after conducting an 11-year prospective follow-up study as part of the EPIC (European Prospective Investigation into Cancer)-InterAct trial. Furthermore, the study by Brand et al revealed a correlation between premature ovarian insufficiency (menopause before the age of 40) and a 32 per cent higher risk for T2DM.118 After adjusting for multiple variables, a prospective case-cohort study conducted in Europe revealed that women aged 40 and above exhibit a higher risk of developing T2DM.119 This finding is consistent with a study from Latin America, which showed the association between diabetes with early menopause in women under the age of 45.120 This is also supported by a previous study's meta-analysis concluding that women with early menopause (40 to 45 years of age) or premature ovarian insufficiency (40 years of age) have an increased risk of T2DM.121 Similarly, another Chinese observational study of 16,299 women identified that early menopause (before 45 years) was related to a 20 per cent increased incidence of T2DM.122 A similar finding was also obtained in a Beijing longitudinal study, which discovered that early menopausal women (age ≤ 45 years) with postmenopausal obesity had the highest risk of developing T2DM compared to premenopausal women.123
Abdominal obesity is associated with a higher risk of developing T2DM than peripheral obesity as visceral fat stores have more impact on insulin metabolism by releasing free fatty acids into the portal circulation, which may impair hepatic clearance of insulin and lead to insulin resistance and hyperinsulinemia.124 A study from South India discovered that the average age of menopause in diabetic women is 44.7 years, much younger than the age of menopause in non-diabetic women (48.2 years). The study also discovered that diabetic women have higher BMIs than non-diabetic women, which could be attributed to changes in body composition and an increase in abdominal fat following menopause. These changes were more pronounced in diabetics due to altered insulin sensitivity and glucose metabolism.125 Consistently, findings from South Korea showed that significant obesity, severe insulin secretion abnormalities and severe insulin resistance resulted in early hyperglycaemia symptoms.126 Another study presented that visceral fats reduced insulin sensitivity in Chinese T2DM patients regardless of BMI or subcutaneous fat level with a positive correlation with homeostatic model assessment-insulin resistance (HOMA2-IR).127 All these study findings have confirmed T2DM risk as a postmenopausal complication.
Diabetes has been causally linked to early menopause. Previous studies have found that T2DM women appeared to enter menopause at an earlier age than non-diabetic women. According to the Women's Health Across the Nation (SWAN) study (n = 2171), women with diabetes have their last menstrual period (FMP) at a significantly younger age than women without diabetes.128 The cross-sectional study of 6079 women aged 40 to 59 years from 11 Latin American countries found that the risk of being postmenopausal in women aged 40 to 44 years is nearly three times higher in those with diabetes than in those without diabetes, even after adjustment for confounding factors like obesity and hypertension.129 This is also consistent with data from Brand et al, revealing that women with diabetes before the age of 20 have an earlier menopause than non-diabetic women, whereas women with diabetes after the age of 50 have later menopause.130 A recent Canadian data revealed an association between early T2DM diagnosis (30-39 years) and earlier menopause in comparison to non-diabetics.131 In addition, Wellons et al reported an observational finding that women with type II diabetes are more likely to experience ovulation difficulties and early menopause.131
The global studies above agreed on the existence of a relationship between the timing and duration of menopause with T2DM. Any disparities in study outcomes were most likely attributable to changes in model adjustment, sample population, and/or genetic traits among women in different nations. It was also probable that the relationship between T2DM and menopausal age varied by country and was influenced by a variety of factors, including nutrition and lifestyle choices.
Menopausal hormone therapy and T2DM
Menopausal hormone treatment (MHT) is commonly recommended for menopausal women who experience vasomotor symptoms, such as night sweats and hot flashes.132 The potential relationship between T2DM and vasomotor symptoms has been evaluated in 150,007 women who participated in the Women’s Health Initiative (WHI) trial from 1993 to 2014. The study found that these vasomotor symptoms were associated with an 18 per cent increase in the incidence of T2DM, and the association was independent of obesity.133 In addition, menopausal women who utilised MHT tend to experience benefits from the metabolic effects of oestrogen replacement. They became more energetic, experienced improved glucose metabolism, as well as having better weight control and improved bone density.134 The favourable effect of MHT on glucose metabolism appeared to entail several physiological mechanisms, including increased lipid oxidation and increased energy expenditure, which help to reduce the trend towards central obesity and belly fat accumulation.135 According to Garcia et al, animal studies have shown that oestradiol and ERα-specific agonists enhanced energy homeostasis and enhanced body fat distribution, besides decreasing insulin resistance, β-cell dysfunction and inflammation. To extrapolate these reported benefits to humans, additional studies would be necessary to develop targeted oestrogen mimics with metabolic benefits without having the adverse effects of MHT.56
Postmenopausal women have faster development of visceral adiposity, insulin resistance, and T2DM. Oestrogen deficiency during menopause is related to an increased risk of developing T2DM, which could be alleviated with MHT. Several clinical trials on postmenopausal women receiving MHT found a lower incidence of T2DM, lower glucose plasma levels and increased systemic insulin sensitivity. According to most randomised clinical trials, MHT is related to a reduction in central adiposity and an increase in insulin sensitivity. In the WHI intervention trials, both conjugated equine oestrogen (CEE) alone and CEE plus medroxyprogesterone acetate (MPA) were associated with a lower risk of developing T2DM compared to placebo.136 The T2DM incidence was reduced by 35% in the MHT arm of the Heart and Oestrogen/Progestin Replacement Study (HERS), by 21% in the WHI study, and by 25% in a French cohort study of women on MHT.137-139
In the randomised controlled clinical trial of HERS (women with known CVD), oestrogen treatment prevented the rise of blood glucose levels over time and resulted in fewer women with impaired fasting glucose at study onset progressing to overt diabetes when compared to controls.140 Consistently, from the year 1999 to 2007, several forms of MHT were reported to improve glucose control in women with T2DM, which included CEE alone,141 or in combination with MPA,142,143 E2 alone,144 or in combination with cyclical,145 or continuous combined norethisterone acetate.146,147 Furthermore, a meta-analysis of 107 studies (49,973 patient-years) followed for an average of 1.5 years revealed that MHT reduced abdominal obesity, insulin resistance and new T2DM incidence.148 However, MHT has been linked to an increase in the incidence of thrombotic events in older women with T2DM ( > 60 years or > 10 years in menopause).149
Therefore, the 'timing theory' has been investigated in terms of oestrogen effects on glucose metabolism. Effects on insulin activity may be one mechanism by which MHT decreased the incidence of T2DM in early postmenopausal women. For example, Pereira et al used the hyperinsulinemic-euglycemic clamp to compare the effect of one-week administration of transdermal E2 vs placebo in early postmenopausal women (six years since last menstruation) versus late postmenopausal women (more than ten years since last menstruation). E2 was found to boost insulin activity in early postmenopausal women while decreasing it in late postmenopausal women.15 This is consistent with the finding of an observational trial conducted in Japan, which found age-stratified findings of HbA1C reduction in response to oral standard-dose continuous combination CEE/MPA in women aged 40 to 49 years but not in those aged more than 50 years.150 Besides, transdermal E2 has more favourable effects on inflammatory markers and triglycerides than oral E2 and does not increase the risk of venous thromboembolism. Recently, Speksnijder et al published a meta-analysis in 2023 on the impact of postmenopausal hormone therapy on glucose regulation in women with type 1 or type 2 diabetes. This study found that MHT had a positive impact on glucose regulation in women with type 2 diabetes. MHT significantly reduced fasting glucose and HbA1c (mean difference -0.56% [95% CI -0.80, -0.31], -6.08 mmol/mol [95% CI -8.80, -3.36), as well as fasting glucose (mean difference -1.15 mmol/L [95% CI -1.78, -0.51]). When MHT is used to treat menopausal symptoms in women with type 2 diabetes, it is likely to have a neutral to favourable effect on glucose regulation.151 In addition, MHT has a positive effect on glucose metabolism since it increases lipid oxidation and energy expenditure, followed by the reduction of fat storage in the abdomen area with a decreased risk of central obesity.135
Most clinical trials have included women receiving CEE in combination with MPA. Oral oestrogen has been demonstrated to be more effective than transdermal oestrogen (patch HRT). However, oral oestrogen has some drawbacks. For instance, it enhances triglyceride and clotting factors formation by the liver and raises inflammatory indicators more than transdermal products.152
SERM and T2DM
SERMs are a class of compounds that interact with ERs and generate a specific receptor conformation that correlates with distinct behaviours in oestrogen-responsive tissues.153 SERMs exert agonist or antagonist activity on ERs in a tissue-specific manner, which is determined by the complexity of ER signalling, such as ER tissue distribution, ligand binding specificity and various interactions with coactivators or corepressors.154 SERM has been shown to reduce the risk of osteoporosis and cardiovascular disease while also inhibiting oestrogenic activation in the breast and uterus in postmenopausal women.108 SERM, on the other hand, has no effect in lowering blood glucose levels or increasing insulin sensitivity. As a result, it becomes ineffective for menopausal women with T2DM.155 Raloxifene is equally effective in lowering the risk of invasive breast cancer and preventing postmenopausal osteoporosis. However, short-term raloxifene treatment (12 weeks or 6 months) did not affect fasting blood glucose or insulin levels, nor glucose tolerance or insulin sensitivity in postmenopausal women with or without T2DM.156,157 Raloxifene significantly reduced LDL cholesterol levels and increased serum leptin levels in postmenopausal women.158 However, tamoxifen was reported to improve glycaemic control and provide cardioprotective benefits in ovariectomised diabetic rats by decreasing lipid profiles and pro-inflammatory cytokines.105-107 Therefore, further prospective clinical trials are warranted to highlight the potential relevance of SERM in the management of menopausal diabetic women.
Bazedoxifene (BZA) is a novel SERM with oestrogen antagonistic activity in the breast and uterus but oestrogen agonistic activity in bone, preventing osteoporosis while protecting the breast and uterus from oestrogenic stimulation.159 Tissue selective oestrogen complexes (TSECs), which combine conjugated oestrogen (CE) and bazedoxifene in a single pill, become a new therapy for menopausal symptoms.160 The main innovation of TSEC is that it provides all the benefits of CE treatment plus breast and uterine protection without using progestin.161 It is noteworthy that TSEC enhances glucose and insulin homeostasis in OVX mice,162 but it seemed to not affect postmenopausal women.163 Therefore, further studies are required to investigate the effects of TSEC on glucose and insulin homeostasis in postmenopausal women.
Evidence showed that hyperglycaemia can lead to an increase in angiotensin 2 (Ang II) production, which in turn leads to a decrease in insulin sensitivity; nevertheless, the renin-angiotensin system (RAS) inhibition improves insulin sensitivity.164 SERMs alone may not have the same impact on type 2 diabetes as oestrogen, but when combined with oestrogen, they can reduce trophic effects, thrombosis and CVD events. An in vivo study by Khaksari et al recently discovered that combining SERMs and oestrogen can reduce oestrogen's deleterious effects (carcinogenesis in breast and uterine tissues) while retaining its favourable benefits on cardiovascular, glycaemic and inflammatory markers.105 Nonetheless, more human trials are required to determine the therapeutic target of combining SERM and oestrogen for the treatment of postmenopausal disorders such as osteoporosis and cardiovascular diseases. Furthermore, a retrospective cohort analysis has been carried out involving female patients with early-stage breast cancer who were treated with or without SERMs at a Korean tertiary care hospital from 2008 to 2020. According to the analysis, SERMs, notably tamoxifen in general, were not related to an increased risk of diabetes for long-term use ( ≥ 1500 days).155
Concluding remarks
Numerous in-vitro and in-vivo studies have identified the oestrogen receptor as the key player in insulin resistance and T2DM. The comprehensive review findings demonstrated that ER is critical in β-cell function, insulin action, glucose homeostasis and the aetiology of insulin resistance, diabetes and its consequences.
The discoveries of ERα and ERβ as modulators of GLUT4 expression, as well as the identification of E2 as a crucial regulator of glucose homeostasis, have opened new avenues for developing T2DM coadjuvant therapy. The idea of selectively activating ERα and inhibiting ERβ to increase GLUT4 expression and glucose absorption appears very promising.
Menopause is associated with a poor metabolic profile and an increased risk of T2DM. MHT has been shown to enhance β-cell insulin secretion, glucose effectiveness and insulin sensitivity. These improvements have been evaluated using clinically relevant indices, such as the HOMA/IR and IVGTT/OGTT. MHT improves glycaemic control in young women with T2DM but should be individually commenced after careful consideration of other risk factors in older women (60 years of age or more than 10 years post menopause onset). The issue with MHT is its adverse effects on obesity and hyperlipidemia in menopausal women, exposing them to thromboembolic events. On the other hand, SERMs that deliver estrogenic benefits in selected tissues do not influence insulin sensitivity and glucose homeostasis. TSEC is another potential alternative but requires further studies to determine its role in T2DM therapy in postmenopausal women.
In conclusion, although the study findings are intriguing, additional research is demanded to gain a comprehensive understanding of the mechanism and optimal clinical strategy for ER regulation as a potential therapy for insulin resistance and T2DM, especially in postmenopausal women.
Review Highlights
What is the current knowledge?
-
Clinical trials have shown that MHT improves glucose homeostasis and may delay the onset of type 2 diabetes in women. But the underlying protective mechanism remains unknown. However, several studies have demonstrated that oestrogens have an influence on glucose homeostasis and insulin sensitivity in animal and in-vitro models, suggesting a potential role in diabetes prevention.
What is new here?
-
We have written a comprehensive review on the involvement of the classical oestrogen receptor in insulin resistance and T2DM, including molecular mechanisms and clinical data. Numerous in-vitro and in-vivo studies have confirmed oestrogen receptor to be the key player in both insulin resistance and T2DM. The classical ER pathway demonstrated that ER is critical for pancreatic β-cells and islet function, GLUT4 expression, insulin sensitivity and glucose tolerance, dysfunctional lipid homeostasis, oxidative stress, and inflammatory signalling. There is compendium of clinical findings for MHT and SERM till the year of 2023. Therefore, the goal of this review to investigate the beneficial effects of MHT in menopausal women who at risk of T2DM.
Competing Interests
The authors have no relevant financial or non-financial interests to disclose.
Data Availability Statement
Not applicable.
Ethical Statement
Not applicable.
References
- Petersmann A, Müller-Wieland D, Müller UA, Landgraf R, Nauck M, Freckmann G. Definition, classification and diagnosis of diabetes mellitus. Exp Clin Endocrinol Diabetes 2019; 127:S1-7. doi: 10.1055/a-1018-9078 [Crossref] [ Google Scholar]
- American Diabetes Association. Anonymous Standards of medical care in diabetes-2015 abridged for primary care providers. Clin Diabetes 2015; 33:97-111. doi: 10.2337/diaclin.33.2.97 [Crossref] [ Google Scholar]
- Tuomi T, Santoro N, Caprio S, Cai M, Weng J, Groop L. The many faces of diabetes: a disease with increasing heterogeneity. Lancet 2014; 383:1084-94. doi: 10.1016/s0140-6736(13)62219-9 [Crossref] [ Google Scholar]
- Lin X, Xu Y, Pan X, Xu J, Ding Y, Sun X. Global, regional, and national burden and trend of diabetes in 195 countries and territories: an analysis from 1990 to 2025. Sci Rep 2020; 10:14790. doi: 10.1038/s41598-020-71908-9 [Crossref] [ Google Scholar]
- Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol 2018; 14:88-98. doi: 10.1038/nrendo.2017.151 [Crossref] [ Google Scholar]
- Lee SH, Park SY, Choi CS. Insulin resistance: from mechanisms to therapeutic strategies. Diabetes Metab J 2022; 46:15-37. doi: 10.4093/dmj.2021.0280 [Crossref] [ Google Scholar]
- Klip A, McGraw TE, James DE. Thirty sweet years of GLUT4. J Biol Chem 2019; 294:11369-81. doi: 10.1074/jbc.REV119.008351 [Crossref] [ Google Scholar]
- Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev 2018; 98:2133-223. doi: 10.1152/physrev.00063.2017 [Crossref] [ Google Scholar]
- Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci 2020; 21:6275. doi: 10.3390/ijms21176275 [Crossref] [ Google Scholar]
- Cerf ME. Beta cell dysfunction and insulin resistance. Front Endocrinol (Lausanne) 2013; 4:37. doi: 10.3389/fendo.2013.00037 [Crossref] [ Google Scholar]
- Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005; 365:1333-46. doi: 10.1016/s0140-6736(05)61032-x [Crossref] [ Google Scholar]
- Zimmet P, Alberti KG, Magliano DJ, Bennett PH. Diabetes mellitus statistics on prevalence and mortality: facts and fallacies. Nat Rev Endocrinol 2016; 12:616-22. doi: 10.1038/nrendo.2016.105 [Crossref] [ Google Scholar]
- Gregg EW, Sattar N, Ali MK. The changing face of diabetes complications. Lancet Diabetes Endocrinol 2016; 4:537-47. doi: 10.1016/s2213-8587(16)30010-9 [Crossref] [ Google Scholar]
- Vogel H, Mirhashemi F, Liehl B, Taugner F, Kluth O, Kluge R. Estrogen deficiency aggravates insulin resistance and induces β-cell loss and diabetes in female New Zealand obese mice. Horm Metab Res 2013; 45:430-5. doi: 10.1055/s-0032-1331700 [Crossref] [ Google Scholar]
- Pereira RI, Casey BA, Swibas TA, Erickson CB, Wolfe P, Van Pelt RE. Timing of estradiol treatment after menopause may determine benefit or harm to insulin action. J Clin Endocrinol Metab 2015; 100:4456-62. doi: 10.1210/jc.2015-3084 [Crossref] [ Google Scholar]
- Zhu J, Zhou Y, Jin B, Shu J. Role of estrogen in the regulation of central and peripheral energy homeostasis: from a menopausal perspective. Ther Adv Endocrinol Metab 2023; 14:20420188231199359. doi: 10.1177/20420188231199359 [Crossref] [ Google Scholar]
- Ciarambino T, Crispino P, Guarisco G, Giordano M. Gender differences in insulin resistance: new knowledge and perspectives. Curr Issues Mol Biol 2023; 45:7845-61. doi: 10.3390/cimb45100496 [Crossref] [ Google Scholar]
- Choi SB, Jang JS, Park S. Estrogen and exercise may enhance beta-cell function and mass via insulin receptor substrate 2 induction in ovariectomized diabetic rats. Endocrinology 2005; 146:4786-94. doi: 10.1210/en.2004-1653 [Crossref] [ Google Scholar]
- Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol 2019; 116:135-70. doi: 10.1016/bs.apcsb.2019.01.001 [Crossref] [ Google Scholar]
- Hairi HA, Mustafa NH, Jayusman PA, Shuid AN. The potential effects of isoflavones on nuclear receptor modulation in bone remodeling: a review. J Appl Pharm Sci 2023; 13:73-84. doi: 10.7324/japs.2023.126975 [Crossref] [ Google Scholar]
- Faltas CL, LeBron KA, Holz MK. Unconventional estrogen signaling in health and disease. Endocrinology 2020; 161:bqaa030. doi: 10.1210/endocr/bqaa030 [Crossref] [ Google Scholar]
- Barakat R, Oakley O, Kim H, Jin J, Ko CJ. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep 2016; 49:488-96. doi: 10.5483/bmbrep.2016.49.9.141 [Crossref] [ Google Scholar]
- Paterni I, Granchi C, Katzenellenbogen JA, Minutolo F. Estrogen receptors alpha (ERα) and beta (ERβ): subtype-selective ligands and clinical potential. Steroids 2014; 90:13-29. doi: 10.1016/j.steroids.2014.06.012 [Crossref] [ Google Scholar]
- Al-Bader MD, Malatiali SA, Redzic ZB. Expression of estrogen receptor α and β in rat astrocytes in primary culture: effects of hypoxia and glucose deprivation. Physiol Res 2011; 60:951-60. doi: 10.33549/physiolres.932167 [Crossref] [ Google Scholar]
- Lee HR, Kim TH, Choi KC. Functions and physiological roles of two types of estrogen receptors, ERα and ERβ, identified by estrogen receptor knockout mouse. Lab Anim Res 2012; 28:71-6. doi: 10.5625/lar.2012.28.2.71 [Crossref] [ Google Scholar]
- Menazza S, Murphy E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res 2016; 118:994-1007. doi: 10.1161/circresaha.115.305376 [Crossref] [ Google Scholar]
- Watson PJ, Fairall L, Schwabe JW. Nuclear hormone receptor co-repressors: structure and function. Mol Cell Endocrinol 2012; 348:440-9. doi: 10.1016/j.mce.2011.08.033 [Crossref] [ Google Scholar]
- Wilkenfeld SR, Lin C, Frigo DE. Communication between genomic and non-genomic signaling events coordinate steroid hormone actions. Steroids 2018; 133:2-7. doi: 10.1016/j.steroids.2017.11.005 [Crossref] [ Google Scholar]
- Ribas V, Nguyen MT, Henstridge DC, Nguyen AK, Beaven SW, Watt MJ. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERα-deficient mice. Am J Physiol Endocrinol Metab 2010; 298:E304-19. doi: 10.1152/ajpendo.00504.2009 [Crossref] [ Google Scholar]
- Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 2013; 34:309-38. doi: 10.1210/er.2012-1055 [Crossref] [ Google Scholar]
- James DE, Stöckli J, Birnbaum MJ. The aetiology and molecular landscape of insulin resistance. Nat Rev Mol Cell Biol 2021; 22:751-71. doi: 10.1038/s41580-021-00390-6 [Crossref] [ Google Scholar]
- Aedh AI, Alshahrani MS, Huneif MA, Pryme IF, Oruch R. A glimpse into milestones of insulin resistance and an updated review of its management. Nutrients 2023; 15:921. doi: 10.3390/nu15040921 [Crossref] [ Google Scholar]
- Vaidya RA, Desai S, Moitra P, Salis S, Agashe S, Battalwar R. Hyperinsulinemia: an early biomarker of metabolic dysfunction. Front Clin Diabetes Healthc 2023; 4:1159664. doi: 10.3389/fcdhc.2023.1159664 [Crossref] [ Google Scholar]
- Li K, Bian J, Xiao Y, Wang D, Han L, He C. Changes in pancreatic senescence mediate pancreatic diseases. Int J Mol Sci 2023; 24:3513. doi: 10.3390/ijms24043513 [Crossref] [ Google Scholar]
- Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environ Health Perspect 2006; 114:106-12. doi: 10.1289/ehp.8451 [Crossref] [ Google Scholar]
- Kumar R, Balhuizen A, Amisten S, Lundquist I, Salehi A. Insulinotropic and antidiabetic effects of 17β-estradiol and the GPR30 agonist G-1 on human pancreatic islets. Endocrinology 2011; 152:2568-79. doi: 10.1210/en.2010-1361 [Crossref] [ Google Scholar]
- Kooptiwut S, Kaewin S, Semprasert N, Sujjitjoon J, Junking M, Suksri K. Estradiol prevents high glucose-induced β-cell apoptosis by decreased BTG2 expression. Sci Rep 2018; 8:12256. doi: 10.1038/s41598-018-30698-x [Crossref] [ Google Scholar]
- Ashcroft FM. KATP channels and the metabolic regulation of insulin secretion in health and disease: the 2022 banting medal for scientific achievement award lecture. Diabetes 2023; 72:693-702. doi: 10.2337/dbi22-0030 [Crossref] [ Google Scholar]
- Abdul Mu-U-Min RB, Diane A, Allouch A, Al-Siddiqi HH. Ca(2 + )-mediated signaling pathways: a promising target for the successful generation of mature and functional stem cell-derived pancreatic beta cells in vitro. Biomedicines 2023; 11:1577. doi: 10.3390/biomedicines11061577 [Crossref] [ Google Scholar]
- Thompson B, Satin LS. Beta-cell ion channels and their role in regulating insulin secretion. Compr Physiol 2021; 11:1-21. doi: 10.1002/cphy.c210004 [Crossref] [ Google Scholar]
- Gil-Rivera M, Medina-Gali RM, Martínez-Pinna J, Soriano S. Physiology of pancreatic β-cells: Ion channels and molecular mechanisms implicated in stimulus-secretion coupling. Int Rev Cell Mol Biol 2021; 359:287-323. doi: 10.1016/bs.ircmb.2021.02.006 [Crossref] [ Google Scholar]
- Farrugia F, Aquilina A, Vassallo J, Pace NP. Bisphenol A and type 2 diabetes mellitus: a review of epidemiologic, functional, and early life factors. Int J Environ Res Public Health 2021; 18:716. doi: 10.3390/ijerph18020716 [Crossref] [ Google Scholar]
- Pérez-Bermejo M, Mas-Pérez I, Murillo-Llorente MT. The role of the bisphenol A in diabetes and obesity. Biomedicines 2021; 9:666. doi: 10.3390/biomedicines9060666 [Crossref] [ Google Scholar]
- Marroqui L, Martinez-Pinna J, Castellano-Muñoz M, Dos Santos RS, Medina-Gali RM, Soriano S. bisphenol-S and bisphenol-F alter mouse pancreatic β-cell ion channel expression and activity and insulin release through an estrogen receptor ERβ mediated pathway. Chemosphere 2021; 265:129051. doi: 10.1016/j.chemosphere.2020.129051 [Crossref] [ Google Scholar]
- Winborn WB, Sheridan PJ, McGill HC. Estrogen receptors in the islets of Langerhans of baboons. Cell Tissue Res 1983; 230:219-23. doi: 10.1007/bf00216041 [Crossref] [ Google Scholar]
- Chu C, Gao X, Li X, Zhang X, Ma R, Jia Y. Involvement of estrogen receptor-α in the activation of Nrf2-antioxidative signaling pathways by silibinin in pancreatic β-cells. Biomol Ther (Seoul) 2020; 28:163-71. doi: 10.4062/biomolther.2019.071 [Crossref] [ Google Scholar]
- Allard C, Morford JJ, Xu B, Salwen B, Xu W, Desmoulins L. Loss of nuclear and membrane estrogen receptor-α differentially impairs insulin secretion and action in male and female mice. Diabetes 2019; 68:490-501. doi: 10.2337/db18-0293 [Crossref] [ Google Scholar]
- Zuo Z, Wu T, Lin M, Zhang S, Yan F, Yang Z. Chronic exposure to tributyltin chloride induces pancreatic islet cell apoptosis and disrupts glucose homeostasis in male mice. Environ Sci Technol 2014; 48:5179-86. doi: 10.1021/es404729p [Crossref] [ Google Scholar]
- Gay F, Anglade I, Gong Z, Salbert G. The LIM/homeodomain protein islet-1 modulates estrogen receptor functions. Mol Endocrinol 2000; 14:1627-48. doi: 10.1210/mend.14.10.0538 [Crossref] [ Google Scholar]
- Alonso-Magdalena P, Ropero AB, Carrera MP, Cederroth CR, Baquié M, Gauthier BR. Pancreatic insulin content regulation by the estrogen receptor ERα. PLoS One 2008; 3:e2069. doi: 10.1371/journal.pone.0002069 [Crossref] [ Google Scholar]
- Longuet C, Broca C, Costes S, Hani EH, Bataille D, Dalle S. Extracellularly regulated kinases 1/2 (p44/42 mitogen-activated protein kinases) phosphorylate synapsin I and regulate insulin secretion in the MIN6 beta-cell line and islets of Langerhans. Endocrinology 2005; 146:643-54. doi: 10.1210/en.2004-0841 [Crossref] [ Google Scholar]
- Merino B, García-Arévalo M. Sexual hormones and diabetes: the impact of estradiol in pancreatic β cell. Int Rev Cell Mol Biol 2021; 359:81-138. doi: 10.1016/bs.ircmb.2021.02.004 [Crossref] [ Google Scholar]
- Hevener AL, Ribas V, Moore TM, Zhou Z. The impact of skeletal muscle ERα on mitochondrial function and metabolic health. Endocrinology 2020; 161:bqz017. doi: 10.1210/endocr/bqz017 [Crossref] [ Google Scholar]
- Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL. Evidence that oestrogen receptor-alpha plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia 2006; 49:588-97. doi: 10.1007/s00125-005-0105-3 [Crossref] [ Google Scholar]
- Sakurai Y, Kubota N, Yamauchi T, Kadowaki T. Role of insulin resistance in MAFLD. Int J Mol Sci 2021; 22:4156. doi: 10.3390/ijms22084156 [Crossref] [ Google Scholar]
- Garcia-Arevalo M, Lorza-Gil E, Leite N, Brunetto S, Boschero AC, Carneiro EM. The 17-beta-estradiol improves insulin sensitivity in a rapid estrogen receptor alpha-dependent manner in an animal model of malnourishment. J Endocrinol Metab 2019; 9:133-46. doi: 10.14740/jem612 [Crossref] [ Google Scholar]
- Manrique C, Lastra G, Habibi J, Mugerfeld I, Garro M, Sowers JR. Loss of estrogen receptor α signaling leads to insulin resistance and obesity in young and adult female mice. Cardiorenal Med 2012; 2:200-10. doi: 10.1159/000339563 [Crossref] [ Google Scholar]
- Gregorio KC, Laurindo CP, Machado UF. Estrogen and glycemic homeostasis: the fundamental role of nuclear estrogen receptors ESR1/ESR2 in glucose transporter GLUT4 regulation. Cells 2021; 10:99. doi: 10.3390/cells10010099 [Crossref] [ Google Scholar]
- Clapauch R, Mourão AF, Mecenas AS, Maranhão PA, Rossini A, Bouskela E. Endothelial function and insulin resistance in early postmenopausal women with cardiovascular risk factors: importance of ESR1 and NOS3 polymorphisms. PLoS One 2014; 9:e103444. doi: 10.1371/journal.pone.0103444 [Crossref] [ Google Scholar]
- Xu H, Du X, Xu J, Zhang Y, Tian Y, Liu G. Pancreatic β cell microRNA-26a alleviates type 2 diabetes by improving peripheral insulin sensitivity and preserving β cell function. PLoS Biol 2020; 18:e3000603. doi: 10.1371/journal.pbio.3000603 [Crossref] [ Google Scholar]
- Ribas V, Drew BG, Zhou Z, Phun J, Kalajian NY, Soleymani T. Skeletal muscle action of estrogen receptor α is critical for the maintenance of mitochondrial function and metabolic homeostasis in females. Sci Transl Med 2016; 8:334ra54. doi: 10.1126/scitranslmed.aad3815 [Crossref] [ Google Scholar]
- Passarelli M, Machado UF. AGEs-induced and endoplasmic reticulum stress/inflammation-mediated regulation of GLUT4 expression and atherogenesis in diabetes mellitus. Cells 2021; 11:104. doi: 10.3390/cells11010104 [Crossref] [ Google Scholar]
- Aldahish A, Balaji P, Vasudevan R, Kandasamy G, James JP, Prabahar K. Elucidating the potential inhibitor against type 2 diabetes mellitus associated gene of GLUT4. J Pers Med 2023; 13:660. doi: 10.3390/jpm13040660 [Crossref] [ Google Scholar]
- Fatima LA, Campello RS, Barreto-Andrade JN, Passarelli M, Santos RS, Clegg DJ. Estradiol stimulates adipogenesis and Slc2a4/GLUT4 expression via ESR1-mediated activation of CEBPA. Mol Cell Endocrinol 2019; 498:110447. doi: 10.1016/j.mce.2019.05.006 [Crossref] [ Google Scholar]
- Yonamine CY, Alves-Wagner AB, Esteves JV, Okamoto MM, Correa-Giannella ML, Giannella-Neto D. Diabetes induces tri-methylation at lysine 9 of histone 3 at Slc2a4 gene in skeletal muscle: a new target to improve glycemic control. Mol Cell Endocrinol 2019; 481:26-34. doi: 10.1016/j.mce.2018.11.006 [Crossref] [ Google Scholar]
- Chadt A, Al-Hasani H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflugers Arch 2020; 472:1273-98. doi: 10.1007/s00424-020-02417-x [Crossref] [ Google Scholar]
- Savova MS, Mihaylova LV, Tews D, Wabitsch M, Georgiev MI. Targeting PI3K/AKT signaling pathway in obesity. Biomed Pharmacother 2023; 159:114244. doi: 10.1016/j.biopha.2023.114244 [Crossref] [ Google Scholar]
- Landis J, Shaw LM. Insulin receptor substrate 2-mediated phosphatidylinositol 3-kinase signaling selectively inhibits glycogen synthase kinase 3β to regulate aerobic glycolysis. J Biol Chem 2014; 289:18603-13. doi: 10.1074/jbc.M114.564070 [Crossref] [ Google Scholar]
- Chang YC, Chan MH, Yang YF, Li CH, Hsiao M. Glucose transporter 4: insulin response mastermind, glycolysis catalyst and treatment direction for cancer progression. Cancer Lett 2023; 563:216179. doi: 10.1016/j.canlet.2023.216179 [Crossref] [ Google Scholar]
- Jaldin-Fincati JR, Pavarotti M, Frendo-Cumbo S, Bilan PJ, Klip A. Update on GLUT4 vesicle traffic: a cornerstone of insulin action. Trends Endocrinol Metab 2017; 28:597-611. doi: 10.1016/j.tem.2017.05.002 [Crossref] [ Google Scholar]
- Tao Z, Cheng Z. Hormonal regulation of metabolism-recent lessons learned from insulin and estrogen. Clin Sci (Lond) 2023; 137:415-34. doi: 10.1042/cs20210519 [Crossref] [ Google Scholar]
- van Gerwen J, Shun-Shion AS, Fazakerley DJ. Insulin signalling and GLUT4 trafficking in insulin resistance. Biochem Soc Trans 2023; 51:1057-69. doi: 10.1042/bst20221066 [Crossref] [ Google Scholar]
- Suren Garg S, Kushwaha K, Dubey R, Gupta J. Association between obesity, inflammation and insulin resistance: insights into signaling pathways and therapeutic interventions. Diabetes Res Clin Pract 2023; 200:110691. doi: 10.1016/j.diabres.2023.110691 [Crossref] [ Google Scholar]
- Fu Q, Li T, Zhang C, Ma X, Meng L, Liu L. Butyrate mitigates metabolic dysfunctions via the ERα-AMPK pathway in muscle in OVX mice with diet-induced obesity. Cell Commun Signal 2023; 21:95. doi: 10.1186/s12964-023-01119-y [Crossref] [ Google Scholar]
- Barros RP, Gabbi C, Morani A, Warner M, Gustafsson JA. Participation of ERalpha and ERbeta in glucose homeostasis in skeletal muscle and white adipose tissue. Am J Physiol Endocrinol Metab 2009; 297:E124-33. doi: 10.1152/ajpendo.00189.2009 [Crossref] [ Google Scholar]
- Carvalho E, Kotani K, Peroni OD, Kahn BB. Adipose-specific overexpression of GLUT4 reverses insulin resistance and diabetes in mice lacking GLUT4 selectively in muscle. Am J Physiol Endocrinol Metab 2005; 289:E551-61. doi: 10.1152/ajpendo.00116.2005 [Crossref] [ Google Scholar]
- Dominici FP, Hauck S, Argentino DP, Bartke A, Turyn D. Increased insulin sensitivity and upregulation of insulin receptor, insulin receptor substrate (IRS)-1 and IRS-2 in liver of Ames dwarf mice. J Endocrinol 2002; 173:81-94. doi: 10.1677/joe.0.1730081 [Crossref] [ Google Scholar]
- Muraki K, Okuya S, Tanizawa Y. Estrogen receptor alpha regulates insulin sensitivity through IRS-1 tyrosine phosphorylation in mature 3T3-L1 adipocytes. Endocr J 2006; 53:841-51. doi: 10.1507/endocrj.k06-005 [Crossref] [ Google Scholar]
- Kim JY, Jo KJ, Kim BJ, Baik HW, Lee SK. 17β-estradiol induces an interaction between adenosine monophosphate-activated protein kinase and the insulin signaling pathway in 3T3-L1 adipocytes. Int J Mol Med 2012; 30:979-85. doi: 10.3892/ijmm.2012.1070 [Crossref] [ Google Scholar]
- Xu B, Allard C, Alvarez-Mercado AI, Fuselier T, Kim JH, Coons LA. Estrogens promote misfolded proinsulin degradation to protect insulin production and delay diabetes. Cell Rep 2018; 24:181-96. doi: 10.1016/j.celrep.2018.06.019 [Crossref] [ Google Scholar]
- Benito-Vicente A, Jebari-Benslaiman S, Galicia-Garcia U, Larrea-Sebal A, Uribe KB, Martin C. Molecular mechanisms of lipotoxicity-induced pancreatic β-cell dysfunction. Int Rev Cell Mol Biol 2021; 359:357-402. doi: 10.1016/bs.ircmb.2021.02.013 [Crossref] [ Google Scholar]
- Oh YS, Bae GD, Baek DJ, Park EY, Jun HS. Fatty acid-induced lipotoxicity in pancreatic beta-cells during development of type 2 diabetes. Front Endocrinol (Lausanne) 2018; 9:384. doi: 10.3389/fendo.2018.00384 [Crossref] [ Google Scholar]
- Tiano JP, Delghingaro-Augusto V, Le May C, Liu S, Kaw MK, Khuder SS. Estrogen receptor activation reduces lipid synthesis in pancreatic islets and prevents β cell failure in rodent models of type 2 diabetes. J Clin Invest 2011; 121:3331-42. doi: 10.1172/jci44564 [Crossref] [ Google Scholar]
- Santoro A, Kahn BB. Adipocyte regulation of insulin sensitivity and the risk of type 2 diabetes. N Engl J Med 2023; 388:2071-85. doi: 10.1056/NEJMra2216691 [Crossref] [ Google Scholar]
- Van Huynh T, Rethi L, Rethi L, Chen CH, Chen YJ, Kao YH. The complex interplay between imbalanced mitochondrial dynamics and metabolic disorders in type 2 diabetes. Cells 2023; 12:1223. doi: 10.3390/cells12091223 [Crossref] [ Google Scholar]
- Xega V, Alami T, Liu JL. Recent progress on the role of cellular communication network factors (CCN) 3, 4 and 6 in regulating adiposity, liver fibrosis and pancreatic islets. J Cell Commun Signal 2023; 17:297-306. doi: 10.1007/s12079-023-00765-8 [Crossref] [ Google Scholar]
- Kwon EJ, You YA, Park B, Ha EH, Kim HS, Park H. Association between the DNA methylations of POMC, MC4R, and HNF4A and metabolic profiles in the blood of children aged 7-9 years. BMC Pediatr 2018; 18:121. doi: 10.1186/s12887-018-1104-0 [Crossref] [ Google Scholar]
- Kuryłowicz A. Estrogens in adipose tissue physiology and obesity-related dysfunction. Biomedicines 2023; 11:690. doi: 10.3390/biomedicines11030690 [Crossref] [ Google Scholar]
- Yokota-Nakagi N, Omoto S, Tazumi S, Kawakami M, Takamata A, Morimoto K. Estradiol replacement improves high-fat diet-induced insulin resistance in ovariectomized rats. Physiol Rep 2022; 10:e15193. doi: 10.14814/phy2.15193 [Crossref] [ Google Scholar]
- Ramos PA, Lytle KA, Delivanis D, Nielsen S, LeBrasseur NK, Jensen MD. Insulin-stimulated muscle glucose uptake and insulin signaling in lean and obese humans. J Clin Endocrinol Metab 2021; 106:e1631-46. doi: 10.1210/clinem/dgaa919 [Crossref] [ Google Scholar]
- Song R, Hu M, Qin X, Qiu L, Wang P, Zhang X. The roles of lipid metabolism in the pathogenesis of chronic diseases in the elderly. Nutrients 2023; 15:3433. doi: 10.3390/nu15153433 [Crossref] [ Google Scholar]
- Almikhlafi MA, Karami MM, Jana A, Alqurashi TM, Majrashi M, Alghamdi BS. Mitochondrial medicine: a promising therapeutic option against various neurodegenerative disorders. Curr Neuropharmacol 2023; 21:1165-83. doi: 10.2174/1570159x20666220830112408 [Crossref] [ Google Scholar]
- Hamilton DJ, Minze LJ, Kumar T, Cao TN, Lyon CJ, Geiger PC. Estrogen receptor alpha activation enhances mitochondrial function and systemic metabolism in high-fat-fed ovariectomized mice. Physiol Rep 2016; 4:e12913. doi: 10.14814/phy2.12913 [Crossref] [ Google Scholar]
- Yu S, Meng S, Xiang M, Ma H. Phosphoenolpyruvate carboxykinase in cell metabolism: roles and mechanisms beyond gluconeogenesis. Mol Metab 2021; 53:101257. doi: 10.1016/j.molmet.2021.101257 [Crossref] [ Google Scholar]
- Franckhauser S, Muñoz S, Elias I, Ferre T, Bosch F. Adipose overexpression of phosphoenolpyruvate carboxykinase leads to high susceptibility to diet-induced insulin resistance and obesity. Diabetes 2006; 55:273-80. doi: 10.2337/diabetes.55.02.06.db05-0482 [Crossref] [ Google Scholar]
- Qiu S, Vazquez JT, Boulger E, Liu H, Xue P, Hussain MA. Hepatic estrogen receptor α is critical for regulation of gluconeogenesis and lipid metabolism in males. Sci Rep 2017; 7:1661. doi: 10.1038/s41598-017-01937-4 [Crossref] [ Google Scholar]
- Lima JE, Moreira NC, Sakamoto-Hojo ET. Mechanisms underlying the pathophysiology of type 2 diabetes: from risk factors to oxidative stress, metabolic dysfunction, and hyperglycemia. Mutat Res Genet Toxicol Environ Mutagen 2022; 874-875:503437. doi: 10.1016/j.mrgentox.2021.503437 [Crossref] [ Google Scholar]
- Singh A, Kukreti R, Saso L, Kukreti S. Mechanistic insight into oxidative stress-triggered signaling pathways and type 2 diabetes. Molecules 2022; 27:950. doi: 10.3390/molecules27030950 [Crossref] [ Google Scholar]
- Yaşar P, Ayaz G, User SD, Güpür G, Muyan M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol 2017; 16:4-20. doi: 10.1002/rmb2.12006 [Crossref] [ Google Scholar]
- Ince BA. Dominant Negative Mutants of the Human Estrogen Receptor. University of Illinois Urbana-Champaign; 1995.
- Khodabandehloo H, Gorgani-Firuzjaee S, Panahi G, Meshkani R. Molecular and cellular mechanisms linking inflammation to insulin resistance and β-cell dysfunction. Transl Res 2016; 167:228-56. doi: 10.1016/j.trsl.2015.08.011 [Crossref] [ Google Scholar]
- Nandipati KC, Subramanian S, Agrawal DK. Protein kinases: mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol Cell Biochem 2017; 426:27-45. doi: 10.1007/s11010-016-2878-8 [Crossref] [ Google Scholar]
- Sabio G, Davis RJ. cJun NH2-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance. Trends Biochem Sci 2010; 35:490-6. doi: 10.1016/j.tibs.2010.04.004 [Crossref] [ Google Scholar]
- Liu Q, Si F, Liu Z, Wu Y, Yu J. Association between triglyceride-glucose index and risk of cardiovascular disease among postmenopausal women. Cardiovasc Diabetol 2023; 22:21. doi: 10.1186/s12933-023-01753-3 [Crossref] [ Google Scholar]
- Khaksari M, Raji-Amirhasani A, Bashiri H, Ebrahimi MN, Azizian H. Protective effects of combining SERMs with estrogen on metabolic parameters in postmenopausal diabetic cardiovascular dysfunction: the role of cytokines and angiotensin II. Steroids 2022; 183:109023. doi: 10.1016/j.steroids.2022.109023 [Crossref] [ Google Scholar]
- Ebrahimi MN, Khaksari M, Sepehri G, Asadi Karam G, Raji-Amirhasani A, Azizian H. The effects of alone and combination tamoxifen, raloxifene and estrogen on lipid profile and atherogenic index of ovariectomized type 2 diabetic rats. Life Sci 2020; 263:118573. doi: 10.1016/j.lfs.2020.118573 [Crossref] [ Google Scholar]
- Azizian H, Khaksari M, Asadikaram G, Sepehri G, Najafipour H. Therapeutic effects of tamoxifen on metabolic parameters and cytokines modulation in rat model of postmenopausal diabetic cardiovascular dysfunction: role of classic estrogen receptors. Int Immunopharmacol 2018; 65:190-8. doi: 10.1016/j.intimp.2018.10.009 [Crossref] [ Google Scholar]
- Das A, Destouni A. Novel insights into reproductive ageing and menopause from genomics. Hum Reprod 2023; 38:195-203. doi: 10.1093/humrep/deac256 [Crossref] [ Google Scholar]
- Smith-Ryan AE, Hirsch KR, Cabre HE. Physiology of menopause. In: Hackney AC, ed. Sex Hormones, Exercise and Women: Scientific and Clinical Aspects. Cham: Springer; 2023. p. 351-67. 10.1007/978-3-031-21881-1_15.
- Moir ME, Corkery AT, Senese KA, Miller KB, Pearson AG, Loggie NA. Age at natural menopause impacts cerebrovascular reactivity and brain structure. Am J Physiol Regul Integr Comp Physiol 2023; 324:R207-15. doi: 10.1152/ajpregu.00228.2022 [Crossref] [ Google Scholar]
- Sochocka M, Karska J, Pszczołowska M, Ochnik M, Fułek M, Fułek K. Cognitive decline in early and premature menopause. Int J Mol Sci 2023; 24:6566. doi: 10.3390/ijms24076566 [Crossref] [ Google Scholar]
- Yoh K, Ikeda K, Horie K, Inoue S. Roles of estrogen, estrogen receptors, and estrogen-related receptors in skeletal muscle: regulation of mitochondrial function. Int J Mol Sci 2023; 24:1853. doi: 10.3390/ijms24031853 [Crossref] [ Google Scholar]
- Tyler KA, Habermehl TL, Mason JB. Manipulation of ovarian function influenced glucose metabolism in CBA/J mice. Exp Gerontol 2019; 126:110686. doi: 10.1016/j.exger.2019.110686 [Crossref] [ Google Scholar]
- Marlatt KL, Pitynski-Miller DR, Gavin KM, Moreau KL, Melanson EL, Santoro N. Body composition and cardiometabolic health across the menopause transition. Obesity (Silver Spring) 2022; 30:14-27. doi: 10.1002/oby.23289 [Crossref] [ Google Scholar]
- Muka T, Asllanaj E, Avazverdi N, Jaspers L, Stringa N, Milic J. Age at natural menopause and risk of type 2 diabetes: a prospective cohort study. Diabetologia 2017; 60:1951-60. doi: 10.1007/s00125-017-4346-8 [Crossref] [ Google Scholar]
- Nguyen CT, Pham NM, Nguyen QV, Nguyen VQ, La QN, Lee AH. Menopausal status and type 2 diabetes: a nationwide epidemiological survey in Vietnam. Public Health 2016; 138:168-9. doi: 10.1016/j.puhe.2016.04.010 [Crossref] [ Google Scholar]
- Ren Y, Zhang M, Liu Y, Sun X, Wang B, Zhao Y. Association of menopause and type 2 diabetes mellitus. Menopause 2019; 26:325-30. doi: 10.1097/gme.0000000000001200 [Crossref] [ Google Scholar]
- Brand JS, van der Schouw YT, Onland-Moret NC, Sharp SJ, Ong KK, Khaw KT. Age at menopause, reproductive life span, and type 2 diabetes risk: results from the EPIC-InterAct study. Diabetes Care 2013; 36:1012-9. doi: 10.2337/dc12-1020 [Crossref] [ Google Scholar]
- Karvonen-Gutierrez CA, Park SK, Kim C. Diabetes and menopause. Curr Diab Rep 2016; 16:20. doi: 10.1007/s11892-016-0714-x [Crossref] [ Google Scholar]
- Monterrosa-Castro A, Blümel JE, Portela-Buelvas K, Mezones-Holguín E, Barón G, Bencosme A. Type II diabetes mellitus and menopause: a multinational study. Climacteric 2013; 16:663-72. doi: 10.3109/13697137.2013.798272 [Crossref] [ Google Scholar]
- Anagnostis P, Christou K, Artzouchaltzi AM, Gkekas NK, Kosmidou N, Siolos P. Early menopause and premature ovarian insufficiency are associated with increased risk of type 2 diabetes: a systematic review and meta-analysis. Eur J Endocrinol 2019; 180:41-50. doi: 10.1530/eje-18-0602 [Crossref] [ Google Scholar]
- Shen L, Song L, Li H, Liu B, Zheng X, Zhang L. Association between earlier age at natural menopause and risk of diabetes in middle-aged and older Chinese women: the Dongfeng-Tongji cohort study. Diabetes Metab 2017; 43:345-50. doi: 10.1016/j.diabet.2016.12.011 [Crossref] [ Google Scholar]
- Jiang J, Cui J, Wang A, Mu Y, Yan Y, Liu F. Association between age at natural menopause and risk of type 2 diabetes in postmenopausal women with and without obesity. J Clin Endocrinol Metab 2019; 104:3039-48. doi: 10.1210/jc.2018-02310 [Crossref] [ Google Scholar]
- Ruze R, Liu T, Zou X, Song J, Chen Y, Xu R. Obesity and type 2 diabetes mellitus: connections in epidemiology, pathogenesis, and treatments. Front Endocrinol (Lausanne) 2023; 14:1161521. doi: 10.3389/fendo.2023.1161521 [Crossref] [ Google Scholar]
- Sekhar TV, Medarametla S, Rahman A, Adapa SS. Early menopause in type 2 diabetes - a study from a south Indian tertiary care centre. J Clin Diagn Res 2015; 9:Oc08-10. doi: 10.7860/jcdr/2015/14181.6628 [Crossref] [ Google Scholar]
- Ha KH, Park CY, Jeong IK, Kim HJ, Kim SY, Kim WJ. Clinical characteristics of people with newly diagnosed type 2 diabetes between 2015 and 2016: difference by age and body mass index. Diabetes Metab J 2018; 42:137-46. doi: 10.4093/dmj.2018.42.2.137 [Crossref] [ Google Scholar]
- Huang H, Zheng X, Wen X, Zhong J, Zhou Y, Xu L. Visceral fat correlates with insulin secretion and sensitivity independent of BMI and subcutaneous fat in Chinese with type 2 diabetes. Front Endocrinol (Lausanne) 2023; 14:1144834. doi: 10.3389/fendo.2023.1144834 [Crossref] [ Google Scholar]
- Khalil N, Sutton-Tyrrell K, Strotmeyer ES, Greendale GA, Vuga M, Selzer F. Menopausal bone changes and incident fractures in diabetic women: a cohort study. Osteoporos Int 2011; 22:1367-76. doi: 10.1007/s00198-010-1357-4 [Crossref] [ Google Scholar]
- Mayor S. Type 2 diabetes triples risk of early menopause, study shows. BMJ 2013; 347:f7676. doi: 10.1136/bmj.f7676 [Crossref] [ Google Scholar]
- Brand JS, Onland-Moret NC, Eijkemans MJ, Tjønneland A, Roswall N, Overvad K. Diabetes and onset of natural menopause: results from the European Prospective Investigation into Cancer and Nutrition. Hum Reprod 2015; 30:1491-8. doi: 10.1093/humrep/dev054 [Crossref] [ Google Scholar]
- Wellons MF, Matthews JJ, Kim C. Ovarian aging in women with diabetes: an overview. Maturitas 2017; 96:109-13. doi: 10.1016/j.maturitas.2016.11.019 [Crossref] [ Google Scholar]
- Stuenkel CA, Davis SR, Gompel A, Lumsden MA, Murad MH, Pinkerton JV. Treatment of symptoms of the menopause: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2015; 100:3975-4011. doi: 10.1210/jc.2015-2236 [Crossref] [ Google Scholar]
- Gray KE, Katon JG, LeBlanc ES, Woods NF, Bastian LA, Reiber GE. Vasomotor symptom characteristics: are they risk factors for incident diabetes?. Menopause 2018; 25:520-30. doi: 10.1097/gme.0000000000001033 [Crossref] [ Google Scholar]
- Genazzani AR, Monteleone P, Giannini A, Simoncini T. Hormone therapy in the postmenopausal years: considering benefits and risks in clinical practice. Hum Reprod Update 2021; 27:1115-50. doi: 10.1093/humupd/dmab026 [Crossref] [ Google Scholar]
- Paschou SA, Papanas N. Type 2 diabetes mellitus and menopausal hormone therapy: an update. Diabetes Ther 2019; 10:2313-20. doi: 10.1007/s13300-019-00695-y [Crossref] [ Google Scholar]
- Manson JE, Chlebowski RT, Stefanick ML, Aragaki AK, Rossouw JE, Prentice RL. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women's Health Initiative randomized trials. JAMA 2013; 310:1353-68. doi: 10.1001/jama.2013.278040 [Crossref] [ Google Scholar]
- Kanaya AM, Herrington D, Vittinghoff E, Lin F, Grady D, Bittner V. Glycemic effects of postmenopausal hormone therapy: the heart and estrogen/progestin replacement study A randomized, double-blind, placebo-controlled trial. Ann Intern Med 2003; 138:1-9. doi: 10.7326/0003-4819-138-1-200301070-00005 [Crossref] [ Google Scholar]
- Margolis KL, Bonds DE, Rodabough RJ, Tinker L, Phillips LS, Allen C. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women's Health Initiative Hormone Trial. Diabetologia 2004; 47:1175-87. doi: 10.1007/s00125-004-1448-x [Crossref] [ Google Scholar]
- de Lauzon-Guillain B, Fournier A, Fabre A, Simon N, Mesrine S, Boutron-Ruault MC. Menopausal hormone therapy and new-onset diabetes in the French Etude Epidemiologique de Femmes de la Mutuelle Générale de l'Education Nationale (E3N) cohort. Diabetologia 2009; 52:2092-100. doi: 10.1007/s00125-009-1456-y [Crossref] [ Google Scholar]
- Cobin RH, Goodman NF. American Association of Clinical Endocrinologists and American College of Endocrinology position statement on menopause–2017 update. Endocr Pract 2017; 23:869-80. doi: 10.4158/ep171828.ps [Crossref] [ Google Scholar]
- Friday KE, Dong C, Fontenot RU. Conjugated equine estrogen improves glycemic control and blood lipoproteins in postmenopausal women with type 2 diabetes. J Clin Endocrinol Metab 2001; 86:48-52. doi: 10.1210/jcem.86.1.7094 [Crossref] [ Google Scholar]
- Manning PJ, Allum A, Jones S, Sutherland WH, Williams SM. The effect of hormone replacement therapy on cardiovascular risk factors in type 2 diabetes: a randomized controlled trial. Arch Intern Med 2001; 161:1772-6. doi: 10.1001/archinte.161.14.1772 [Crossref] [ Google Scholar]
- Samaras K, Hayward CS, Sullivan D, Kelly RP, Campbell LV. Effects of postmenopausal hormone replacement therapy on central abdominal fat, glycemic control, lipid metabolism, and vascular factors in type 2 diabetes: a prospective study. Diabetes Care 1999; 22:1401-7. doi: 10.2337/diacare.22.9.1401 [Crossref] [ Google Scholar]
- Brussaard HE, Gevers Leuven JA, Frölich M, Kluft C, Krans HM. Short-term oestrogen replacement therapy improves insulin resistance, lipids and fibrinolysis in postmenopausal women with NIDDM. Diabetologia 1997; 40:843-9. doi: 10.1007/s001250050758 [Crossref] [ Google Scholar]
- Andersson B, Mattsson LA, Hahn L, Mårin P, Lapidus L, Holm G. Estrogen replacement therapy decreases hyperandrogenicity and improves glucose homeostasis and plasma lipids in postmenopausal women with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 1997; 82:638-43. doi: 10.1210/jcem.82.2.3746 [Crossref] [ Google Scholar]
- McKenzie J, Jaap AJ, Gallacher S, Kelly A, Crawford L, Greer IA. Metabolic, inflammatory and haemostatic effects of a low-dose continuous combined HRT in women with type 2 diabetes: potentially safer with respect to vascular risk?. Clin Endocrinol (Oxf) 2003; 59:682-9. doi: 10.1046/j.1365-2265.2003.01906.x [Crossref] [ Google Scholar]
- Kernohan AF, Sattar N, Hilditch T, Cleland SJ, Small M, Lumsden MA. Effects of low-dose continuous combined hormone replacement therapy on glucose homeostasis and markers of cardiovascular risk in women with type 2 diabetes. Clin Endocrinol (Oxf) 2007; 66:27-34. doi: 10.1111/j.1365-2265.2006.02679.x [Crossref] [ Google Scholar]
- Salpeter SR, Walsh JM, Ormiston TM, Greyber E, Buckley NS, Salpeter EE. Meta-analysis: effect of hormone-replacement therapy on components of the metabolic syndrome in postmenopausal women. Diabetes Obes Metab 2006; 8:538-54. doi: 10.1111/j.1463-1326.2005.00545.x [Crossref] [ Google Scholar]
- Stuenkel CA. Managing menopausal vasomotor symptoms in older women. Maturitas 2021; 143:36-40. doi: 10.1016/j.maturitas.2020.08.005 [Crossref] [ Google Scholar]
- Okada M, Nomura S, Ikoma Y, Yamamoto E, Ito T, Mitsui T. Effects of postmenopausal hormone replacement therapy on HbA(1c) levels. Diabetes Care 2003; 26:1088-92. doi: 10.2337/diacare.26.4.1088 [Crossref] [ Google Scholar]
- Speksnijder EM, Ten Noever de Brauw GV, Malekzadeh A, Bisschop PH, Stenvers DJ, Siegelaar SE. Effect of postmenopausal hormone therapy on glucose regulation in women with type 1 or type 2 diabetes: a systematic review and meta-analysis. Diabetes Care 2023; 46:1866-75. doi: 10.2337/dc23-0451 [Crossref] [ Google Scholar]
- Davis SR, Baber RJ. Treating menopause - MHT and beyond. Nat Rev Endocrinol 2022; 18:490-502. doi: 10.1038/s41574-022-00685-4 [Crossref] [ Google Scholar]
- Das A, Lavanya KJ, Nandini Nandini, Kaur K, Jaitak V. Effectiveness of selective estrogen receptor modulators in breast cancer therapy: an update. Curr Med Chem 2023; 30:3287-314. doi: 10.2174/0929867329666221006110528 [Crossref] [ Google Scholar]
- Acconcia F, Fiocchetti M, Busonero C, Fernandez VS, Montalesi E, Cipolletti M. The extra-nuclear interactome of the estrogen receptors: implications for physiological functions. Mol Cell Endocrinol 2021; 538:111452. doi: 10.1016/j.mce.2021.111452 [Crossref] [ Google Scholar]
- Choi YJ, Bak K, Yeo Y, Choi Y, Shin S. Incident type 2 diabetes risk of selective estrogen receptor modulators in female patients with breast cancer. Pharmaceuticals (Basel) 2021; 14:925. doi: 10.3390/ph14090925 [Crossref] [ Google Scholar]
- Cagnacci A, Paoletti AM, Zanni A, Arangino S, Ibba G, Orrù M. Raloxifene does not modify insulin sensitivity and glucose metabolism in postmenopausal women. J Clin Endocrinol Metab 2002; 87:4117-21. doi: 10.1210/jc.2002-020120 [Crossref] [ Google Scholar]
- Nagamani M, Szymajda A, Sepilian V, Urban RJ, Gilkison C. Effects of raloxifene on insulin sensitivity, beta-cell function, and hepatic insulin extraction in normal postmenopausal women. Fertil Steril 2008; 89:614-9. doi: 10.1016/j.fertnstert.2007.03.083 [Crossref] [ Google Scholar]
- Xu B, Lovre D, Mauvais-Jarvis F. Effect of selective estrogen receptor modulators on metabolic homeostasis. Biochimie 2016; 124:92-7. doi: 10.1016/j.biochi.2015.06.018 [Crossref] [ Google Scholar]
- Lello S, Capozzi A, Scambia G. The tissue-selective estrogen complex (bazedoxifene/conjugated estrogens) for the treatment of menopause. Int J Endocrinol 2017; 2017:5064725. doi: 10.1155/2017/5064725 [Crossref] [ Google Scholar]
- Komm BS, Mirkin S, Jenkins SN. Development of conjugated estrogens/bazedoxifene, the first tissue selective estrogen complex (TSEC) for management of menopausal hot flashes and postmenopausal bone loss. Steroids 2014; 90:71-81. doi: 10.1016/j.steroids.2014.06.004 [Crossref] [ Google Scholar]
- Baquedano L, Sánchez-Borrego R, Abad P, Jurado AR, Manubens M, Mendoza N. Innovative treatment in menopause: tissue-selective estrogen complex (TSEC). Reprod Med Int 2018; 1:1-5. doi: 10.23937/rmi-2017/1710004 [Crossref] [ Google Scholar]
- Kim JH, Lee JH, Kim YJ. Retraction: protective effects of bazedoxifene paired with conjugated estrogens on pancreatic β-cell dysfunction. Biol Pharm Bull 2016. 10.1248/bpb.b15-00585.
- Marlatt KL, Lovre D, Beyl RA, Tate CR, Hayes EK, Burant CF. Effect of conjugated estrogens and bazedoxifene on glucose, energy and lipid metabolism in obese postmenopausal women. Eur J Endocrinol 2020; 183:439-52. doi: 10.1530/eje-20-0619 [Crossref] [ Google Scholar]
- Al-Kouh A, Babiker F, Al-Bader M. Renin-angiotensin system antagonism protects the diabetic heart from ischemia/reperfusion injury in variable hyperglycemia duration settings by a glucose transporter type 4-mediated pathway. Pharmaceuticals (Basel) 2023; 16:238. doi: 10.3390/ph16020238 [Crossref] [ Google Scholar]