Bioimpacts. 2025;15:30566.
doi: 10.34172/bi.30566
Original Article
The diagnostic and prognostic value of C1orf174 in colorectal cancer
Elham Nazari Conceptualization, Formal analysis, Methodology, Project administration, Validation, 1, 2, , * # 
Ghazaleh Khalili-Tanha Methodology, Validation, Writing – original draft, 2, 3
Ghazaleh Pourali Writing – original draft, 2
Fatemeh Khojasteh-Leylakoohi Methodology, Validation, 3
Hanieh Azari Methodology, 3
Mohammad Dashtiahangar Formal analysis, Methodology, 4
Hamid Fiuji Writing – review & editing, 5, #
Zahra Yousefli Writing – original draft, 2, 3
Alireza Asadnia Formal analysis, Methodology, 2, 3
Mina Maftooh Writing – review & editing, 2, 6
Hamed Akbarzade Writing – review & editing, 2
Mohammadreza Nassiri Writing – review & editing, 7
Seyed Mahdi Hassanian Writing – review & editing, 2
Gordon A Ferns Writing – review & editing, 8
Godefridus J Peters Writing – review & editing, 5, 9
Elisa Giovannetti Writing – review & editing, 5, 10
Jyotsna Batra Writing – review & editing, 11, 12
Majid Khazaei Supervision, Writing – review & editing, 2
Amir Avan Conceptualization, Project administration, Supervision, Writing – review & editing, 2, 12, *
Author information:
1Proteomics Research Center, Faculty of Paramedical Sciences, Shahid Beheshti University of Medical Sciences, Tehran, Iran
2Metabolic Syndrome Research Center, Mashhad University of Medical Sciences, Mashhad, Iran
3Medical Genetics Research Center, Mashhad University of Medical Sciences, Mashhad, Iran
4School of Medicine, Gonabad University of Medical Sciences, Gonabad, Iran
5Department of Medical Oncology, Cancer Center Amsterdam, Amsterdam U.M.C., VU. University Medical Center (VUMC), Amsterdam, The Netherlands
6College of Medicine, University of Warith Al-Anbiyaa, Karbala, Iraq
7Recombinant Proteins Research Group, The Research Institute of Biotechnology, Ferdowsi University of Mashhad, Mashhad, Iran
8Brighton & Sussex Medical School, Division of Medical Education, Falmer, Brighton, Sussex BN1 9PH, UK
9Professor In Biochemistry, Medical University of Gdansk,Gdansk, Poland
10Cancer Pharmacology Lab, AIRC Start up Unit, Fondazione Pisana per La Scienza, Pisa, Italy
11Centre for Genomics and Personalised Health, Queensland University of Technology, Brisbane 4059, Australia
12Faculty of Health, School of Biomedical Sciences, Queensland University of Technology, Brisbane 4059, Australia
#Equaly contributed as first author.
Abstract
Introduction:
Colorectal cancer (CRC) is among the lethal cancers, indicating the need for the identification of novel biomarkers for the detection of patients in earlier stages. RNA and microRNA sequencing were analyzed using bioinformatics and machine learning algorithms to identify differentially expressed genes (DEGs), followed by validation in CRC patients.
Methods:
The genome-wide RNA sequencing of 631 samples, comprising 398 patients and 233 normal cases was extracted from the Cancer Genome Atlas (TCGA). The DEGs were identified using DESeq package in R. Survival analysis was evaluated using Kaplan–Meier analysis to identify prognostic biomarkers. Predictive biomarkers were determined by machine learning algorithms such as Deep learning, Decision Tree, and Support Vector Machine. The biological pathways, protein-protein interaction (PPI), the co-expression of DEGs, and the correlation between DEGs and clinical data were evaluated. Additionally, the diagnostic markers were assessed with a combioROC package. Finally, the candidate tope score gene was validated by Real-time PCR in CRC patients.
Results:
The survival analysis revealed five novel prognostic genes, including KCNK13, C1orf174, CLEC18A, SRRM5, and GPR89A. Thirty-nine upregulated, 40 downregulated genes, and 20 miRNAs were detected by SVM with high accuracy and AUC. The upregulation of KRT20 and FAM118A genes and the downregulation of LRAT and PROZ genes had the highest coefficient in the advanced stage. Furthermore, our findings showed that three miRNAs (mir-19b-1, mir-326, and mir-330) upregulated in the advanced stage. C1orf174, as a novel gene, was validated using RT-PCR in CRC patients. The combineROC curve analysis indicated that the combination of C1orf174-AKAP4-DIRC1-SKIL-Scan29A4 can be considered as diagnostic markers with sensitivity, specificity, and AUC values of 0.90, 0.94, and 0.92, respectively.
Conclusion:
Machine learning algorithms can be used to Identify key dysregulated genes/miRNAs involved in the pathogenesis of diseases, leading to the detection of patients in earlier stages. Our data also demonstrated the prognostic value of C1orf174 in colorectal cancer.
Keywords: Colorectal cancer, TCGA, Machine learning, Prognostic biomarker, Diagnostic biomarker
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This study was supported by Mashhad University of Medical Sciences. Advance Queensland Industry Research Fellowship supports JB. Grant No. 401092.
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer, representing 10% of worldwide cancer incidence and 9.4% of cancer-related deaths.1 Early detection is crucial for improving patients' survival.2 RNA sequencing (RNA-seq) and microRNA profiling represent a new era of identifying biomarkers and there is growing attention on employing relevant bioinformatics technologies to study the potential role of RNAs and miRNAs in screening, determining progression-free survival, prognosis, and recurrence of colon adenocarcinoma.3-6
The exponential increase in biological data, driven by high-throughput sequencing and other advanced technologies, presents a significant challenge due to its complexity and the time required for analysis.7 Previous research has largely depended on bioinformatics alone for biomarker discovery, a process that, while useful, often requires manual data interpretation, making it time-consuming and less effective for handling large datasets. In contrast, integrates bioinformatics with machine learning techniques to improve biomarker discovery. The incorporation of machine learning algorithms facilitates more efficient analysis of complex datasets, uncovers patterns that traditional methods may miss, and enhances the accuracy and reliability of the findings.8
Machine learning,9 a new branch of artificial intelligence, is widely utilized to establish a signature for early cancer detection with high accuracy of prognosis prediction.10-12 Previous studies indicated the roles of differentially expressed RNAs and miRNAs as determinants of diagnosis and prognosis in various stages of CRC. Liu et al. have reported a 9-gene signature (NTRK2, DTNA, BTG2, COL11A1, Smad2, Smad4, PIK3R1, BCL2, and AXIN2) to have diagnostic significance in the early stages of CRC in a patient cohort.13 In Ghatak et al’s study, four upregulated (BDNF, PTGS2, GSK3B, and CTNNB1) and one downregulated gene (HPGD) were identified as diagnostic and prognostic biomarkers in 1850 primary CRC tissues.14 Furthermore, higher expression of let-7g, miR-21, miR140, miR143, miR-181, miR-192, and miR-215, in 200 CRC patients have been reported to be significantly associated with patients’ overall survival in stages III and IV of CRC. In addition, Jacob et al. identified a 16-miRNA panel (miR-143-5p, miR-27a-3p, miR-31-5p, miR-181a-5p, miR-30b-5p, miR-30d-5p, miR-146a-5p, miR-23a-3p, miR-150-5p, miR-210-3p, miR-25-3p, miR-196a-5p, miR-148a-3p, miR-222-3p, miR-30c-5p and miR-223-3p) as markers of poor survival in stage II and III colon cancer in a cohort of 111 CRC cases.5 Despite extensive efforts in preclinical and clinical phases to identify patients in earlier stages, the survival rate of patients in advanced stages remains poor. Several ML approaches, Deep Learning (DL), Decision Tree, and Support Vector Machine (SVM) are being used. In particular, DL is considered a core domain of ML and artificial intelligence (AI), which has a good learning capability from historical data by using multiple layers, DL can be employed for building intelligent systems and automation.15 Due to many parameters, training a model with DL is time-consuming. However, running during testing requires a short amount of time compared to other ML techniques.16 A decision tree is another algorithm of ML that provides a tool for building prediction models by classification and regression. It works efficiently toward extensive data with a tree-like structure in nodes, branches, and leaves representing tests, test outcomes, and class distributors, respectively.17 The trees vary based on the types of values (classes). A regression tree would suit a discrete set of values, a classification tree, and continuous values. The top-most node in a tree is the root node, and the path would be traced from the root to each leaf node holding a specific class distributor.18 A SVM is a wildly used algorithm supervised by ML, providing a classification tool. Numerous studies showed that ML methods have great potential in discovering biomarkers for various diseases, including cancer.19-21 Resmini et al proposed an ensemble method combining genetic algorithm and SVM for breast cancer diagnosis using thermographic data.22 Gupta et al showed how various ML algorithms could predict colon cancer stages by utilizing information from histopathology reports, intra-operative findings, history taking, and chart records. They used RF, AdaBoost, SVM, MLP, and kNN classifiers on the original dataset without augmentation during training. The results demonstrated that the SVM classifier outperformed the other algorithms in accurately predicting colon cancer stages. The study highlights the importance of integrating multiple sources of data for improved predictive accuracy in medical decision-making.23 In both the training and validation cohorts, a study demonstrated that the optimal SVM classification model accurately distinguished between colon and rectal cancer based on an accuracy of 82.1% and 82.2%, respectively, and an AUC of 0.87 and 0.91, respectively.24 A study utilized various ML methods on RNASeq data to discover new biomarkers in colorectal cancer, offering potential for early diagnosis, treatment, and prognosis improvements.25 Asadnia et al employed decision tree and deep learning techniques in an integrated bioinformatics approach on genomics and transcriptomics data. Their results revealed two novel biomarkers in colorectal cancer. These biomarkers were found to have a significant association with disease progression and patient prognosis, highlighting their potential as promising targets for future research and clinical applications in the field of colorectal cancer.26
The novelty of our work lies in conducting genome-wide RNA and microRNA profiling in CRC patients using advanced bioinformatics and machine learning techniques, including Deep Learning, Decision Trees, and Support Vector Machines. This analysis was subsequently validated in an independent cohort of CRC patients to discover prognostic biomarkers for CRC (Fig. 1A). By addressing these aspects, our work not only advances the methodological framework for biomarker discovery in CRC but also provides valuable prognostic tools that can enhance patient care. We believe that our approach represents a significant step forward in the field, offering both technical innovations and practical applications in oncology.
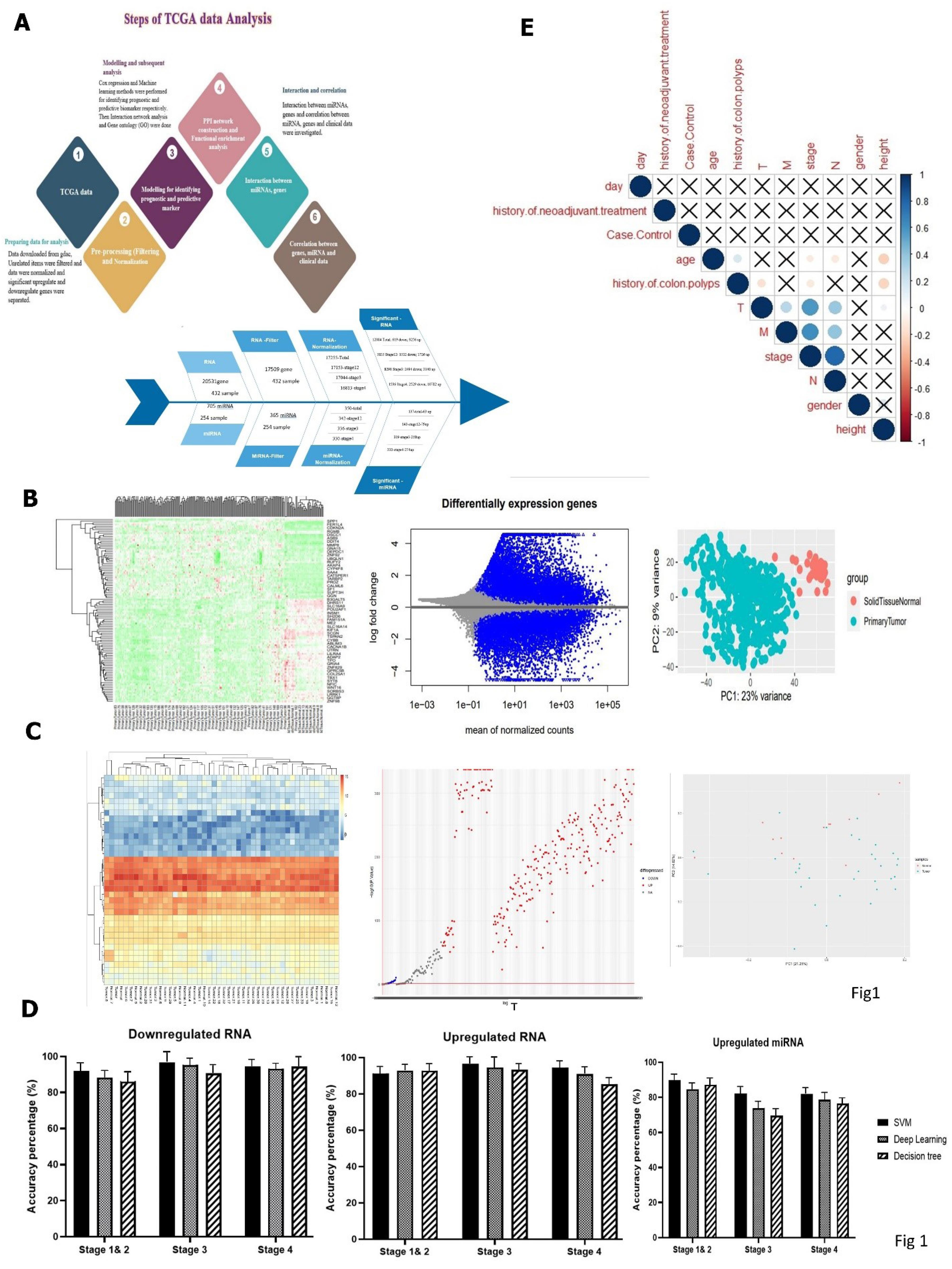
Fig. 1.
(A) The overall workflow, (B) heatmap of DEGs and (C) DEMs of COAD was drawn by R software, (D) comparison of five different machine learning algorithms, including Deep Learning, Decision Tree, Support Vector Machine (SVM), (E) correlation of upregulated and downregulated genes in CRC. A correlation of less than 0.3 is weak, between 0.3 and 0,6 was considered moderate, and more than 0.6 is strong.
.
(A) The overall workflow, (B) heatmap of DEGs and (C) DEMs of COAD was drawn by R software, (D) comparison of five different machine learning algorithms, including Deep Learning, Decision Tree, Support Vector Machine (SVM), (E) correlation of upregulated and downregulated genes in CRC. A correlation of less than 0.3 is weak, between 0.3 and 0,6 was considered moderate, and more than 0.6 is strong.
Materials and Methods
Data source and raw data
The gdac database (https://gdac.broadinstitute.org/) extracted Colorectal adenocarcinoma data from 631 samples, comprising 398 patients and 233 normal cases. 17509 RNA gene expression, 508 microRNA, and clinical data were downloaded. RNA-Seq data were collected from the TCGA database(https://portal.gdc.cancer.gov/). Voom and TMM normalization methods normalized The raw counts of RNA-Seq and miR-Seq data reads. All the analyses were conducted in R software. The DESeq2 package in R software was utilized to indicate the differentially expressed, and the concluded data were filtered based on the |LogFC| > 1, P-value <0.05 were considered as significant thresholds.
Differential expression analysis
The raw counts of RNA-Seq and miR-Seq data reads. were screened by filtering, that the zero expression and duplicate genes were eliminated, then data were normalized with limma and DESEQ2 packages in R 4.0.3 software. The adjusted P < 0.05 and |LogFC| > 1 were identified for upregulate and downregulate significant genes for subsequent analysis.
Identification of predictive biomarkers
Three machine learning techniques were used to identify essential genes and miRNAs, including deep learning, decision tree, and Support vector machine. The relief weight feature selection algorithm was implemented as a feature selection method in combination with three machine learning classifiers. The relief algorithm calculates a feature score for each feature, which is then used to rank and select the top-scoring features for inclusion in the classification model. Features with a score greater than 0.9 were selected for this purpose. In this approach, the method first evaluates the feature weights using ReliefF, sorts the features based on these weights, and then eliminates irrelevant genes according to a predefined threshold. Subsequently, three classifiers-support vector machine (SVM), decision tree, and deep learning-are employed to classify and identify the sample data after dimensionality reduction. This multi-step process ensures that only the most relevant features are used for classification, thereby improving the model's performance and generalization capabilities. The model was performed with Rapidminer 9.10. Model architecture design to ensure that the most relevant features are considered during training and parameters were set on learning rate = 0.01, activation function = Rectifier, hidden layer = 50, and epochs = 20 for deep learning model. Maximal depth = 10 and linear Kernel were provided for decision tree and SVM. Overall, the standard workflow of utilizing models involves splitting the data into two sets, training and test, training the model on the training set, evaluating its performance on the test set, and iterating on the model and data pre-processing techniques to optimize the performance and generalization ability of the model. This study divided the dataset into 70% training and 30% test sets. Further for considering generalizibility, external validation tests using various GEO datasets (Gene Expression Omnibus datasets) such as GSE4045, GSE4107, GSE5851, GSE44861, and GSE113513 were performed. R2, auc_curve, accuracy, confusion matrix were considered for the performance of machine learning methods. The high-performance technique (with high R2 and ROC (receiver operating characteristic)) was selected as the final classifier for necessary gene identifiers.
Correlation between case/control and clinicopathological factors
The binary correlation of variables such as age, sex, cancer stages, and case/control were examined using a correlation matrix. R4.1.0 was selected for analysis.
Enrichment analysis
For in-silico functional enrichment analysis, Gene Ontology (GO) was used to annotate biological processes (BPs), molecular functions (MFs), and cellular components (CCs) of genes. The Kyoto Encyclopedia of Genes and Genomes (KEGG) annotated the gene pathways.
PPI network construction and functional enrichment analysis
STRING v11.5 database (http://string-db.org/) was employed for Interaction network analysis, and GO and KEGG enrichment analysis was used to identify significant pathways. Results with enrichment score > 1, and adjust P < 0.05 were determined as statistically significant results.
Identification of significant targets for miRNA and RNA-miRNA integration
The miRNA-targeted genes were predicted by datasets, including miRWalk, miRDB, and TargetScan, and then visualized by Cytoscape software. The Venn diagram demonstrates the relationships among miRNA-targeted and DEGs genes.
Identification prognostic biomarker
The Kaplan–Meier analysis was performed on DEGs to estimate overall survival (OS) between the two groups (upregulate and downregulate). Candidate genes were screened with log-rank P < 0.05 as prognostic-related genes.
Identification diagnostic biomarker
CombioROC package in R was used in selecting the optimal combination(s) of diagnostic biomarkers through a simple analytical method biomarker detection. In this package, GLM model is used basically for determining the most important coefficient in biomarker combinations. Finally, the best combinations can be chosen in accordance with model criteria. Also, the best biomarkers are introduced by sensitivity, specificity, and AUC. Combined receiver operating characteristic (ROC) curve analysis was used to evaluate diagnostic performance. Sensitivity, specificity, cut-off value, positive predictive value, negative predictive value, and area under the ROC curve were evaluated to assess the discrimination of individual or combined biomarkers. All procedures were performed using the CombioROC R package.
Quantitative real-time PCR
Total RNA was extracted after paraffinization from thirty FFPE (Formalin-Fixed Paraffin-Embedded) tissue samples using a Parstous kit (Parstous, Tehran, Iran) according to the manufacturer's protocol. The local Hospital Ethics Committee of Mashhad University of Medical Sciences approved all procedures. The quality and quantity of extractions were evaluated by a Nanodrop 2000 spectrophotometer (BioTek, USA EPOCH). The cDNA was synthesized by performing a cDNA synthesis kit (Parstous, Tehran, Iran). Quantitative real-time PCR was performed using specific primers for the C1orf174 gene (Betagene, Mashhad, Iran) and the SYBR green master mix (Parstous Co. Tehran, Iran) by an ABI-PRISM StepOne instrument (Applied Biosystems, Foster City, CA). Gene expression data were normalized to GAPDH using a standard curve of cDNAs purchased from Quantitative PCR Human Reference RNA (Stratagene, La Jolla, CA).
Results
Data description, DEGs genes/miRNA profiling based on machine learning method
Considering that all analyses were performed separately by stages, the descriptive characteristics of the population in three stages are shown in Tables S1 and S2 (Supplementary file 1). The information showed that 53.3% of patients were women aged 64 years, and 52% were in the primary stage. The critical features were extracted using the threshold for the correlation coefficient (set correlation > 0.8). Finally, 12084 DEGs of RNA and 137 differentially expressed miRNAs (DEMs) were identified based on the specific criteria and then visualized by the heat map (Figs. 1B and C).
Identifying the most effective method of machine learning algorithms
The key genes were analysed by three different machine learning algorithms, including deep learning, decision tree, and SVM. They were examined with five metrics and illustrated in Fig. 1D by accuracy in all stages. Finally, we chose the SVM as the suitable algorithm with the best accuracy = 96.67, R2 = 95, and AUC = 1. The confusion matrix was presented in Table S3.
Correlation between case/control and clinicopathological factors
Our result showed no correlation between case/control and clinical data, while a significant negative correlation was observed between stage and age (Fig. 1E).
Gene ontology, functional annotation, and pathway enrichment
Based on the R software, our findings showed that 39 upregulated and 40 downregulated genes and 40 upregulated miRNAs were detected in the advanced stage (stages 3 and 4). DEGs were enriched in upregulated genes detected by machine learning techniques; MFs of downregulated DEGs regarded amyloid-beta binding, acetylcholine receptor activity, neurotransmitter receptor activity, calcium channel activity, calcium ion transmembrane transporter activity, voltage-gated calcium channel activity, G protein-coupled amine receptor activity, peptide binding, divalent inorganic cation transmembrane transporter activity, serine-type endopeptidase activity, and cation channel activity. The upregulated DEGs regulated vinculin binding, monocarboxylic acid transmembrane transporter activity, and organic acid transmembrane transporter activity. GO analysis for downregulated genes in cellular components revealed that the differentially expressed genes predominantly contributed to synaptic vesicles, exocytic vesicles, synaptic membranes, transport vesicles, and voltage-gated calcium channel complexes. The outputs of KEGG pathway analysis indicated that downregulated genes participated in pathways involving calcium signalling pathways, metabolic pathways, neuroactive ligand-receptor interaction, cholinergic synapse, and pathways of neurodegeneration - multiple diseases, etc.; for upregulated genes, KEGG gene set metabolic pathways, MicroRNAs in cancer, PI3K-Akt signaling pathway was enriched (Fig. 2).
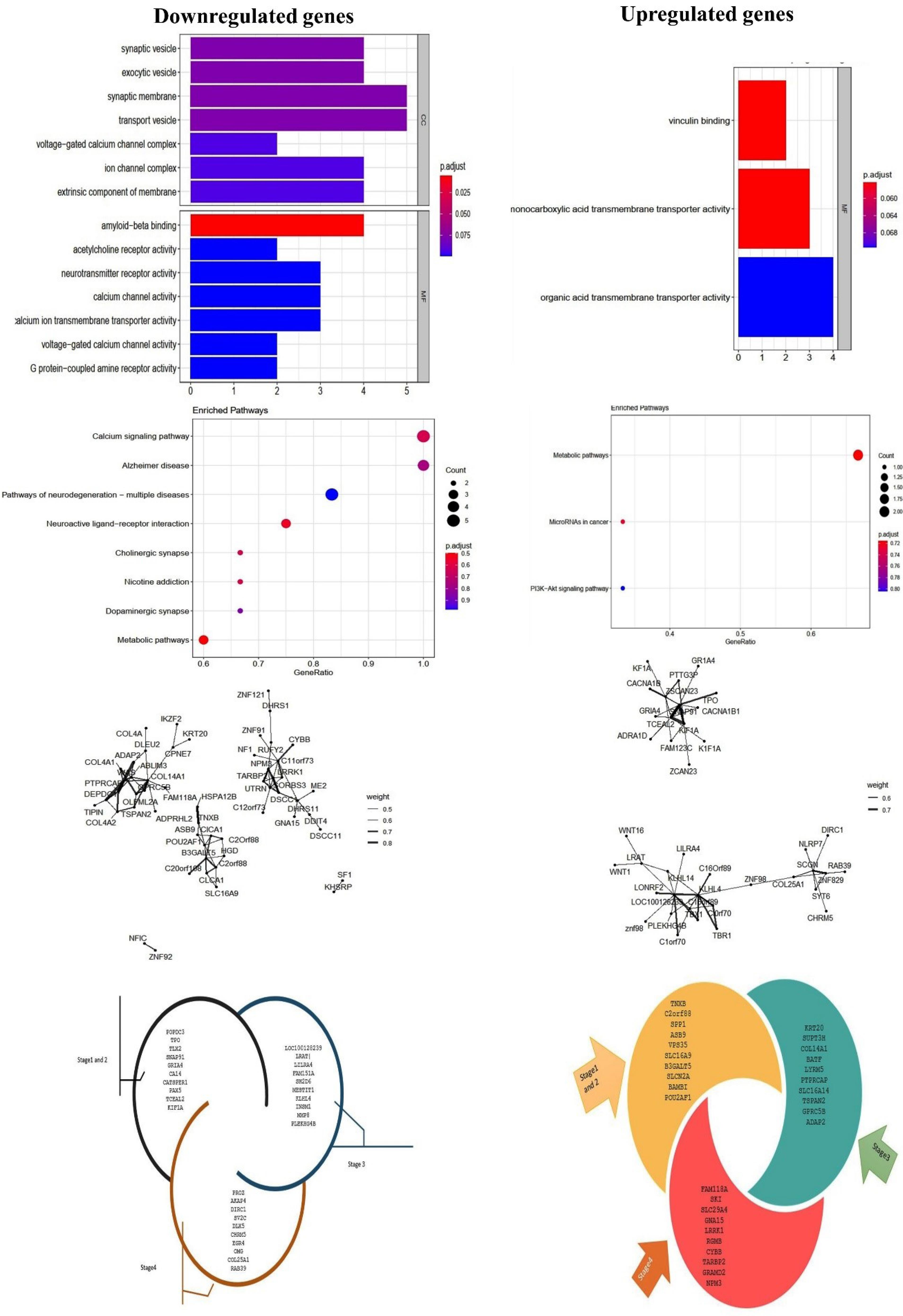
Fig. 2.
GO functional annotation and KEGG functional pathways of enrichment terms in CRC. The P-value is less than 0.05 and is shown by the colour.
.
GO functional annotation and KEGG functional pathways of enrichment terms in CRC. The P-value is less than 0.05 and is shown by the colour.
PPI network construction and identification of targets for miRNA and RNA-miRNA integration
The network of DEGs was analysed and depicted by Tidyverse and Igraph package of R software (Figs. 3A, B, and C).
Moreover, 68 mRNA were detected as targets of 40 DEGs in an advanced stage, of which three miRNAs (hsa-mir-19b-1, hsa-mir-326, and hsa-mir-330) upregulated in both stages 3 and 4. These miRNAs targeted eight mRNA, including OLFML2A, RGS16, SUPT3H, NFIC, DDIT4, GPRC5B, IKZF2, and COL14A1. Furthermore, the survival analysis revealed that dysregulation of hsa-miR-28, which targeted CASS4, KCNIP2, and ATP8B1 decreased OS (Fig. 4). Also, the candidate miRNAs were validated using dbDMEC (https://www.biosino.org/dbDEMC/index) which contains the demiRs in human cancers based on public repositories like ArrayExpress, Gene Expression Omnibus (GEO), Sequence Read Archive (SRA), and The Cancer Genome Atlas (TCGA). As shown in Fig. 4, the interaction of DEGs of RNA and miRNA was analyzed and visualized by String; the interaction score was 0.4.
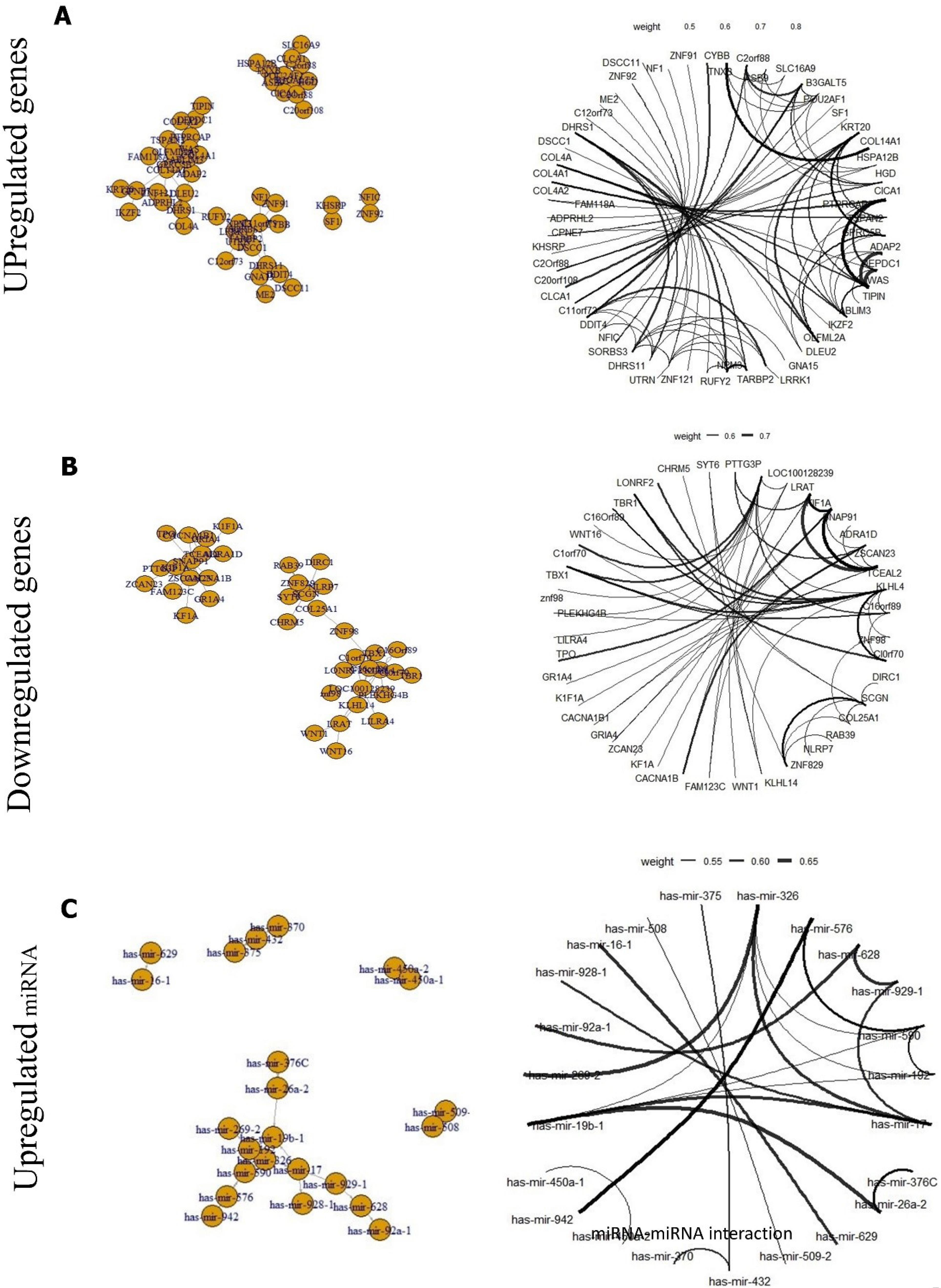
Fig. 3.
The network of DEGs was analysed and depicted by Tidyverse and Igraph package of R software. (A) Upregulated genes, (B) downregulated genes, and (C) miRNA.
.
The network of DEGs was analysed and depicted by Tidyverse and Igraph package of R software. (A) Upregulated genes, (B) downregulated genes, and (C) miRNA.
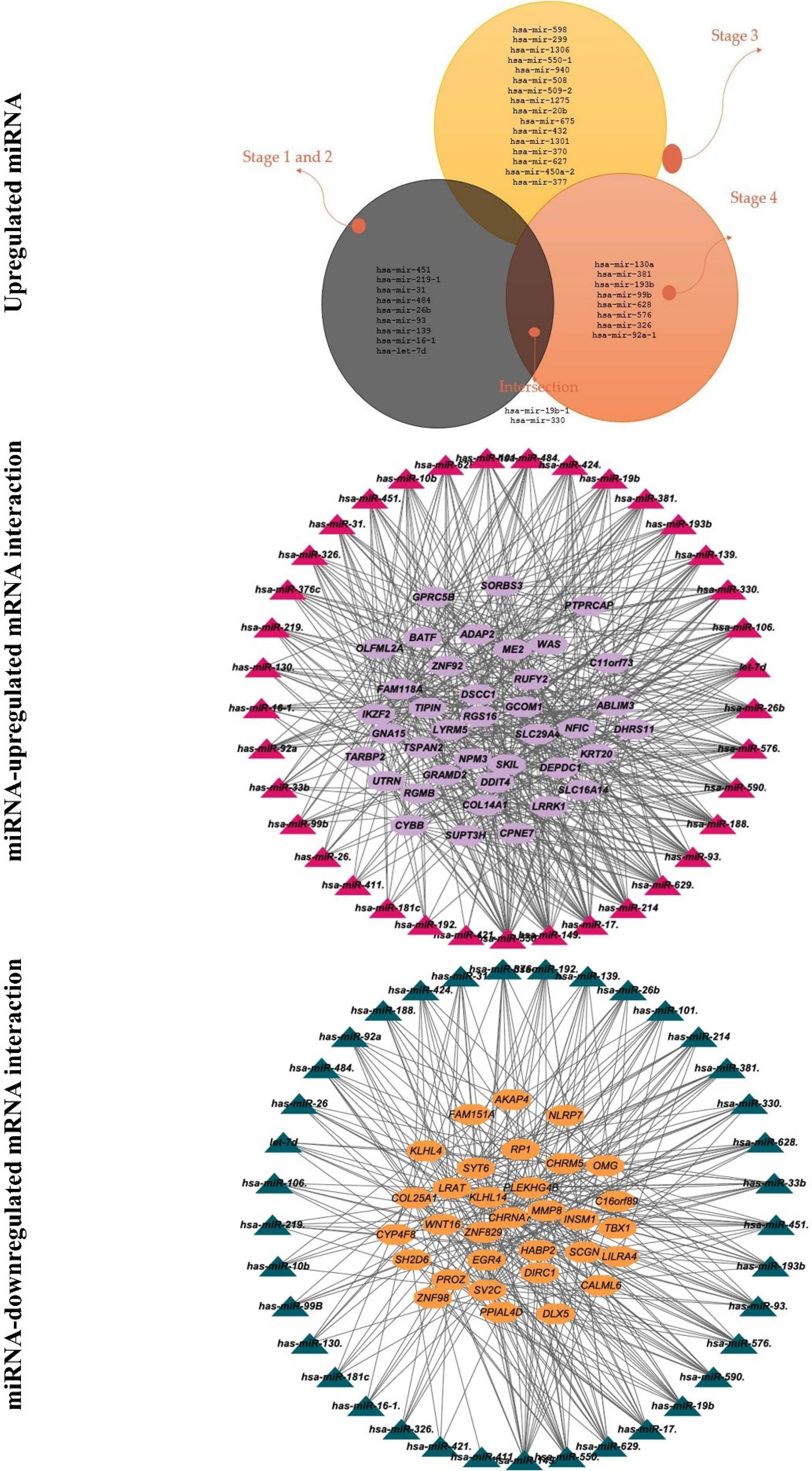
Fig. 4.
Identification of significant targets for miRNA and RNA-miRNA integration.
.
Identification of significant targets for miRNA and RNA-miRNA integration.
Identifying prognostic markers
The survival analysis revealed five novel prognostic genes of CRC, including KCNK13, C1orf174, CLEC18A, SRRM5, and GPR89A (Fig. 5). Additionally, we identified seven prognostic biomarkers mentioned in previous studies, including CASS4, KCNIP2, CLDN9, ATP8B1, RPIA, HPRT1, and ZNF805 (Figs. 5, 6 and 7A).
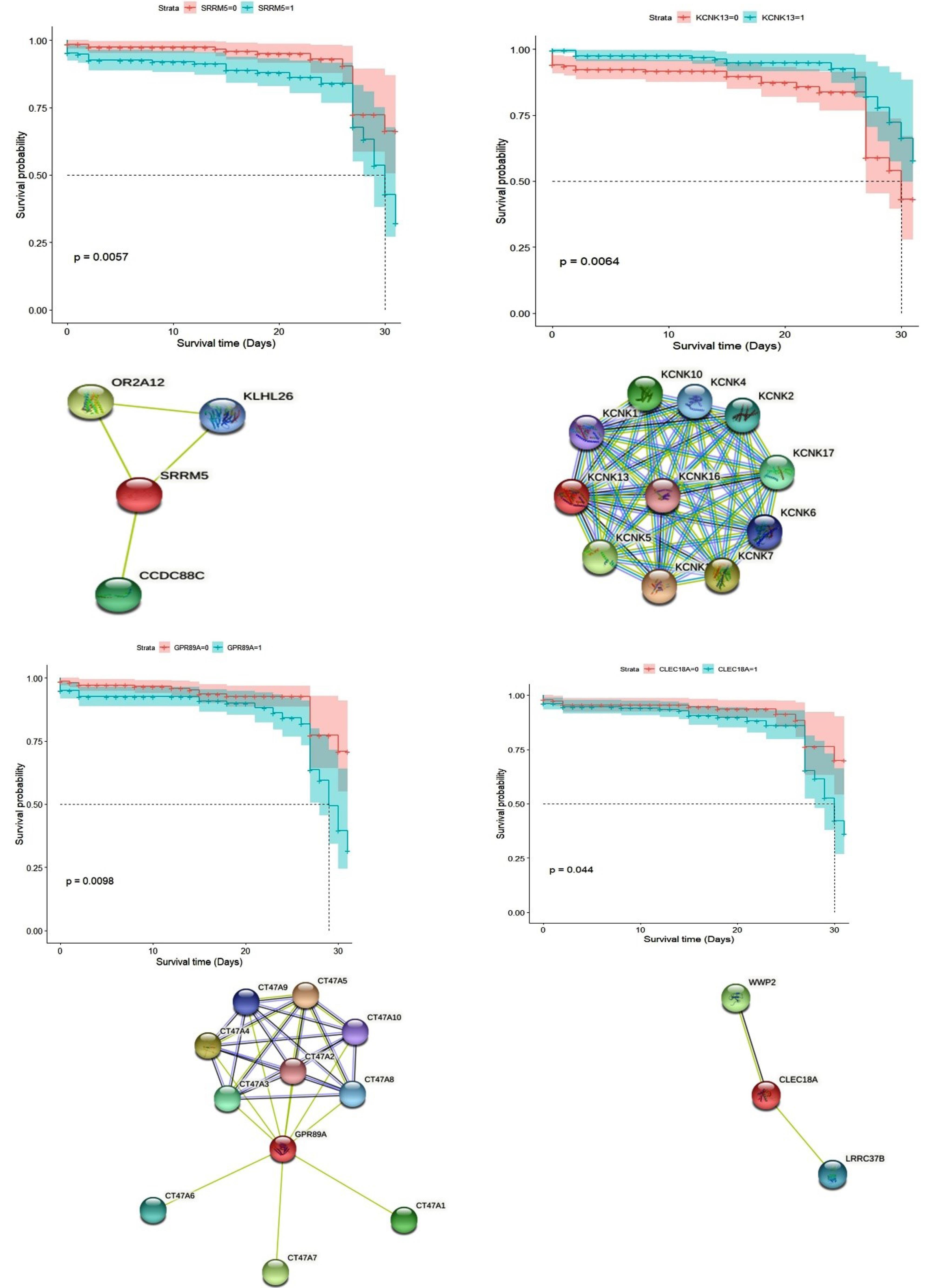
Fig. 5.
PPI network of novel prognostic genes from String, and Kaplan–Meier plot.
.
PPI network of novel prognostic genes from String, and Kaplan–Meier plot.
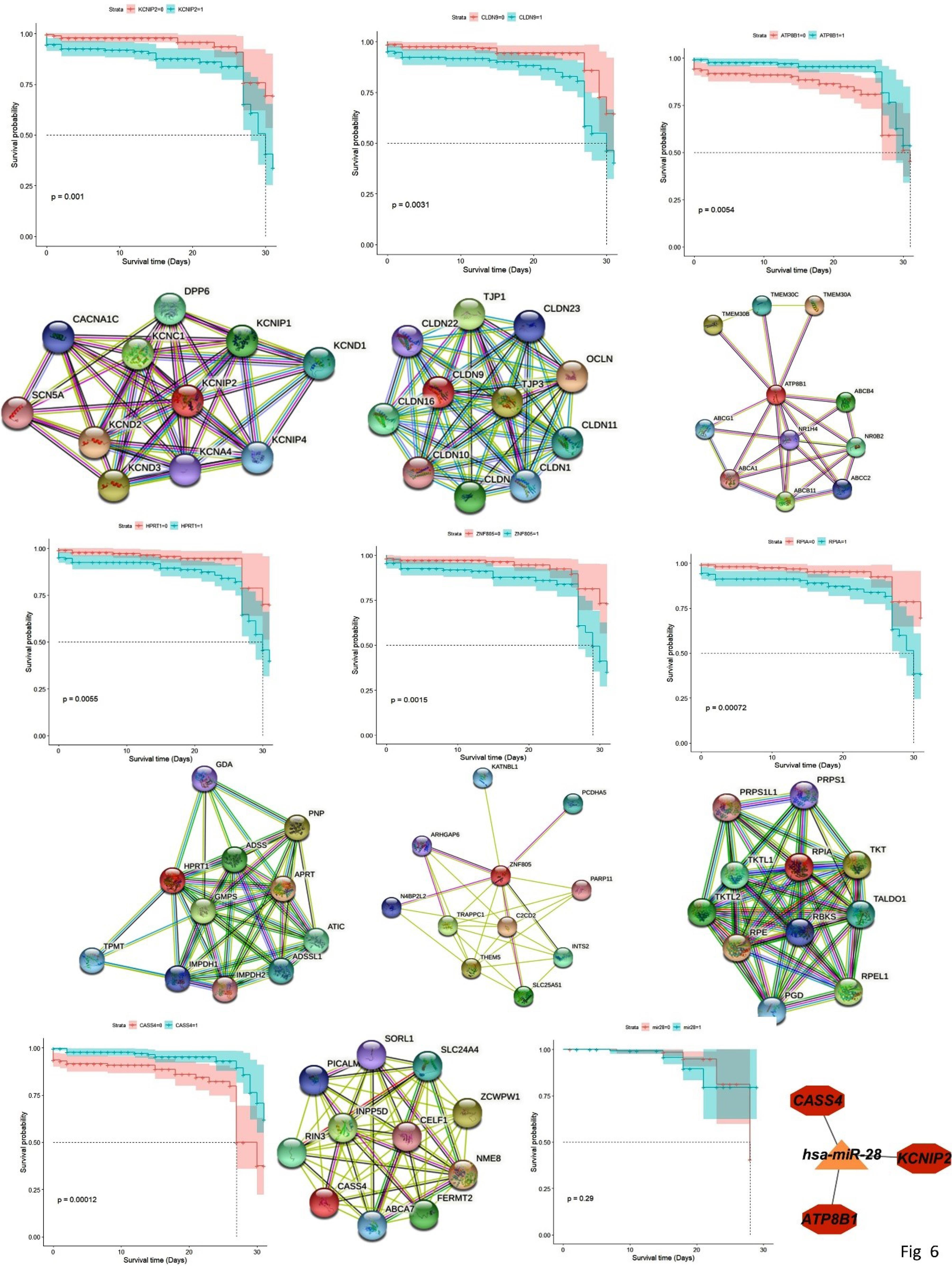
Fig. 6.
continued: PPI network of prognostic genes from String, and Kaplan–Meier plot.
.
continued: PPI network of prognostic genes from String, and Kaplan–Meier plot.
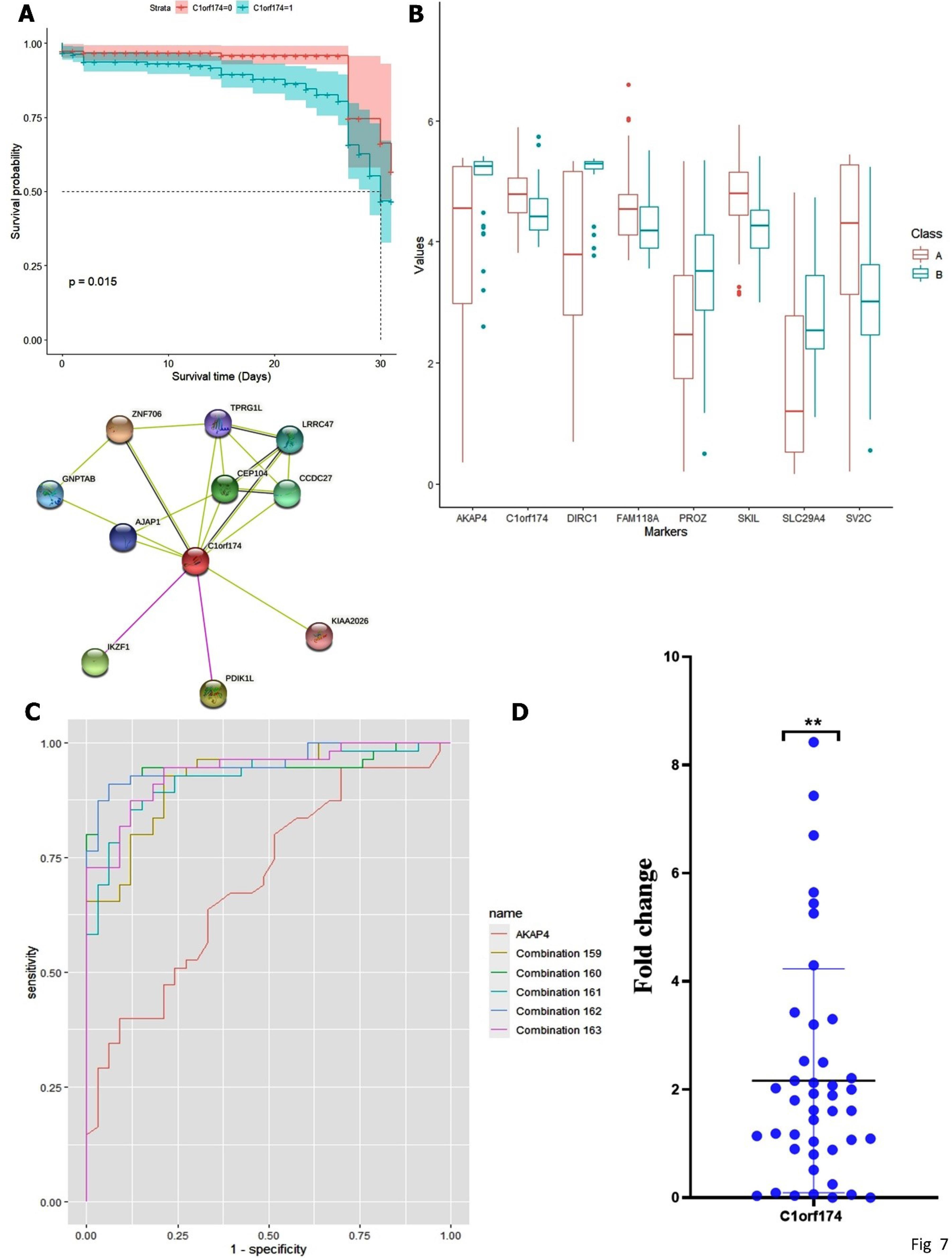
Fig. 7.
(A) PPI network from String, and Kaplan–Meier plot of C1orf174 gene, (B-C) combineROC curve of C1orf174 gene (combination159: AKAP4-C1orf174-DIRC1-PROZ-SKIL, combination160: AKAP4-C1orf174-DIRC1-PROZ-SLC29A4, combination 161: AKAP4-C1orf174-DIRC1-PROZ-SV2C, combination 162: AKAP4-C1orf174-DIRC1-SKIL-SLC29A4, and combination 163: AKAP4-C1orf174-DIRC1-SKIL-SV2C), (D) The expression level of C1orf174 in tumor tissue, as detected by RT-PCR.
.
(A) PPI network from String, and Kaplan–Meier plot of C1orf174 gene, (B-C) combineROC curve of C1orf174 gene (combination159: AKAP4-C1orf174-DIRC1-PROZ-SKIL, combination160: AKAP4-C1orf174-DIRC1-PROZ-SLC29A4, combination 161: AKAP4-C1orf174-DIRC1-PROZ-SV2C, combination 162: AKAP4-C1orf174-DIRC1-SKIL-SLC29A4, and combination 163: AKAP4-C1orf174-DIRC1-SKIL-SV2C), (D) The expression level of C1orf174 in tumor tissue, as detected by RT-PCR.
ROC curve for identification of diagnostic markers
For stage I–II, combination of CATSPER1-GRIA4-POPDC3-TLX2 had the highest rank (AUC of 0.91, 95% CI with sensitivity of 0.84 and specificity of 0.91), for stage 3, FAM151A-LILRA4-LRAT-SH2D6 among other individual biomarkers had the top value (AUC of 0.91,95%CI with sensitivity of 0.92 and specificity of 0.90). In stage 4, our finding showed that the AUC value for the AKAP4-C1orf174-DIRC1-SKIL-SLC29A4 combination was 0.95,95%CI with 0.90 sensitivity and 0.94 specificity. Among prognostic biomarkers, a combination of ATP8B1-C1orf174-CASS4-KCNK13 biomarkers with (AUC = 0.95, CI = 95%, sensitivity = 0.94, and Specificity = 0.88) was identified as an important combination for diagnosis of adenocarcinoma colon. GLM model analysis for ATP8B1-C1orf174-CASS4-RPIA combination in prognostic biomarkers resulted in superior diagnostic biomarkers with -3.0909, 1.0238, 0.2464, and 1.6263 coefficients and good AIC. These results suggest that C1orf174 has potential diagnostic value in combination with other genes such as AKAP4, DIRC1, and SLC29A4 to detect adenocarcinoma colon. Selected Diagnostic biomarkers, according to stages and prognostic biomarkers, are shown in Table S4. The results of GLM model are presented in Table S5 and Figs. 7B and 7C.
C1orf174 validation
Patient demographic and clinicopathological features are presented in the Table S6. The data showed that the mean expression of C1orf174 (Mean ± SD = 2.1 ± 2.02) was higher in tumor cells (P < 0.05). Furthermore, there was no correlation between dysregulation of the C1orf174 gene and demographic and clinicopathological characteristics (Fig. 7D).
Discussion
Colorectal cancer is the third most common cause of cancer-related mortality. To date, millions of people have died as a result of the disease worldwide.27 Researchers have been inspired to create novel diagnostic and prognostic biomarkers as a result of the rise in colorectal cancer morbidity and mortality. Similar to this, physicians are experimenting with various treatment plans to enhance patients' prognoses and minimize their suffering from colorectal cancer. Lowering the mortality rate requires uncovering biomarkers linked to mortality and survival utilizing patient datasets that are currently accessible.
Previous studies have primarily relied on bioinformatics analysis alone for biomarker discovery, which, although valuable, often involves manual data interpretation, making it time-consuming and less effective for large datasets. In contrast, our study combines bioinformatics and machine learning methods to enhance biomarker discovery. Integrating machine learning algorithms allows for more efficient analysis of complex datasets, identification of patterns not evident through traditional methods, and improved accuracy and reliability of findings.
Utilizing information from public databases, the quick development of bioinformatic techniques makes it possible to identify the characteristic genes of disorders.28-30 Machine learning9 methods have been used to analyse various kinds of biological datasets to predict the biomarkers for the categorization of samples and genes linked to a specific clinical condition.26,31-33 However, only a few researchers have simultaneously employed deep learning, SVM, and decision tree algorithms, three well-known machine learning methods, to uncover CRC biomarkers. DL may be used to create automated and intelligent systems.15 Training a model with DL requires a lot of time because of the numerous parameters. However, compared to other ML approaches, running while testing takes very little time.16 The decision tree is another ML approach that may be used to create classification and regression-based prediction models. It operates effectively on large amounts of data and has a tree-like structure with nodes, branches, and leaves that, respectively, represent tests, test results, and class distributors.17 A classification tree, a discrete collection of values, and continuous values would all work well with a regression tree. A tree's root node is the top node, and the route to each leaf node storing a particular class distributor would be traced from the root.18 SVMs are widely used algorithms supported by machine learning and offer a classification tool. It is frequently used to create a classification plane that divides samples into two categories. Additionally, it has distinct advantages, particularly when handling issues with high dimensions, a limited sample size, and nonlinearity.34
Due to the availability of public datasets, we employed a clinical dataset from TCGA to train ML algorithms in order to make use of the potential of the dataset for the diagnosis and prognosis of CRC patients. We created a pipeline to predict characteristics related to CRC patients' survival, such as genes and clinical factors.
We have identified five novel prognostic genes, including KCNK13, C1orf174, CLEC18A, SRRM5, and GPR89A, and seven prognostic biomarkers as described in previous studies, including CASS4, KCNIP2, CLDN9, ATP8B1, RPIA, HPRT1, and ZNF805. Our finding validated the prognostic value of C1orf174 in CRC. Machine learning was performed to identify the critical genes in the advanced stage of CRC. Our findings showed 39 upregulated genes and 40 downregulated genes, of which the upregulation of KRT20 and FAM118A genes and downregulation of LRAT and PROZ genes had the highest coefficient in the advanced stage.
In this study, the Machine Learning analysis detected 40 DEMs in the advanced stage of CRC, which targeted 68 genes. Three miRNAs (mir-19b-1, mir-326, and mir-330) were upregulated in stages 3 and 4. These miRNA targeted eight mRNA, including OLFML2A, RGS16, SUPT3H, NFIC, DDIT4, GPRC5B, IKZF2, and COL14A1, which are involved in the proliferation and metastasis by Wnt and MAPK, mTOR, NF-κB signaling.35-39 Furthermore, the survival analysis revealed that dysregulation of hsa-miR-28, which targeted CASS4, KCNIP2, and ATP8B1 decreased OS in CRC patients.
The potential role of the identified genes in different pathways, their native functions, their interactions with other genes, etc., are described in the following sections.
C1orf174, chromosome 1 open reading frame 174, is located in the 1p36.32 and consists of 6 exons. Our analysis revealed both the diagnostic and prognostic value of C1orf174 in CRC. The function of C1orf174 is still unclear, and according to the HPA RNA-seq project, the highest expression of this gene was reported in the placenta and appendix. During tumorigenesis, cancer cells produce a wide range of proteins originally produced by the placental and embryo. The inappropriate expression of these proteins in developed tissue is considered a tumor marker. Carcinoembryonic antigen (CEA) and alpha-fetoprotein (AFP) are the most common placental and foetus tumor markers. Min et al. reported the C1orf174 gene as a prognostic marker in thyroid papillary carcinoma with an AUC value of 0.79.40 The result of the string dataset showed that C1orf174 significantly correlates with AJAP1 and IKZF1 genes. AJAP1 modulated the adhesion and migration of cancer cells, including glioblastoma,9 breast cancer,41 hepatocellular carcinoma,42 and oesophageal squamous cell carcinoma,43 as well as serves as a potential prognostic biomarker. The IKZF1 gene belongs to the zinc finger 1 Faily and is known as the Ikaros family, which plays a pivotal role in developing and regulating the immune system. Recent evidence showed that IKZF1 could be considered a prognostic and predictive marker.44-47
CASS4 is a member of the Cas scaffold family, also known as HEPL, and regulates cell adhesion and invasion by colonizing focal molecules, such as FAK1 and paxillin.48,9 Li et al reported that CASS4 decreased the eexpression of E-cadherin by phosphorylating the AKT, promoting metastasis in non-small cell lung cancer (NSCLC).50 Bioinformatics analysis of TCGA data of lung squamous carcinoma tissues and lung adenocarcinoma showed the dysregulation of CASS4.49 KCNIP2 belongs to the family of potassium Voltage-Gated channels. The RNASeq analysis data of ovarian cancer demonstrated that KCNIP2 increases cancer incidence and OS.51 In glioblastomas, KCNIP2 Expression is associated with OS.52 Previous evidence suggests that CLDN9 expression promotes tumorigenesis, metastasis, and poor survival in gastric cancer,53,54 hepatocellular carcinoma (HCC),55 and cervical cancer.56 Claudin9 has two promoters: transcriptional factors, which bind to different sequences and activate cell growth, proliferation, and invasion.57 The high expression of CLDN9 increased metastasis in cervical cancer and HCC by activating the TyK2/Stat3 pathway.55,56 ATP8B1 belongs to the ATPase class I type 8b member 1, critical in translocating molecules such as phospholipids. Some studies suggested ATP8B1 is a tumor suppressor gene downregulated in the CRC.58-60 In agreement with our result, Qiu et al. showed that RPIA overexpression reduced OS in CRC patients.61 Overexpression of RPIA increases the level of reactive oxygen species, resulting in cell proliferation. An in-vivo model showed a high level of RPIA-induced Wnt signalling pathway and enhanced HCC proliferation, and the knockdown of a gene by shRNA reduced the cell growth in the xenograft model.62 The previous investigation reported that RPIA is upregulated in the mRNA and protein level and is an appropriate prognostic biomarker in HCC and CRC, promoting cell growth via modulating Erk signalling.61,63 HPRT1, Hypoxanthine phosphoribosyl transferase 1, a conversion enzyme that transfers 5-phosphoribosyl and produces inosine and guanosine, is recently known as a tumorigenesis factor.64,65 The RNASeq analysis of Uterine Corpus Endometrial Carcinoma (UCEC) data from TCGA showed the elevation of HPRT1 negatively associated with OS.66 The knockdown HPRT1 in breast cancer decreases cancer cell growth.66 ZNF805 is a zinc finger family member involved in binding transcription factors to DNA. The analysis of TCGA data revealed the upregulation of ZNF805 in the early stage of gastric cancer.67 The examine the blood of patients with small-cell lung cancer (SCLC) showed ZNF805 significantly upregulated, so it can be considered a potential and non-invasive marker.67
KRT20, also known as CK20, is a member of the cytokeratin family reported as a marker in different diseases, including gastrointestinal and Merkel cell carcinoma.68 Chan et al showed that KRT20 is considered a biomarker in 38 different types of CRC cell lines.69 In-silico analysis of data from the GEO dataset (GSE113513, GSE37182, GSE25070, and GSE10950) demonstrated 43 hub genes in CRC, including KRT20.70 The survival analysis of bladder cancer showed KRT20 overexpression is significantly associated with recurrence-free survival (RFS), progression-free survival (PFS), and cancer-specific survival (CSS), so it can be a predictive marker for re-occurrence and progress of cancer.71 Our findings showed that FAM118A (a family with sequence similarity 118 member A) was identified as a prognostic marker. A broad analysis of gene expression profiles using microarray, western blot, qPCR, and bioinformatic showed the high expression of FAM118A in glioblastoma.72 Our result showed downregulation of LRAT, which aligns with the previous reports. The LRAT expression decreased in various cancers, such as CRC, prostate, bladder, and renal.73-79 Cheng et al reported that LRAT was downregulated in the early stage of CRC due to hypermethylation of the promoter.74 PROZ gene encodes a vitamin K-dependent protein Z glycoprotein synthesized in the liver and is released in the blood. The previous data reported dysregulated expression of PROZ in pancreatic cancer and CRC.80-82
The results of this study suggest that ML-based prediction/classification models can effectively aid in the prognosis of CRC patients based on clinical and genetic indicators linked with CRC diagnosis/survival. In aggregate, our findings provide a novel insight into the prognostic and diagnostic value of C1orf174 in CRC. Further functional studies are warranted to investigate the molecular function of C1orf174 in CRC and validate other novel identified biomarkers in a larger multicentre setting population of colorectal cancer. Our study had several limitations. Validation of combination biomarkers was not performed, although they had high specificity and sensitivity due to our in-silico analyses. The RNA-sequencing analysis was not performed on our own patients' samples. Furthermore, the sensitivity and specificity of the identified prognostic and diagnostic biomarkers were not compared with those of gold-standard known markers. This should also be considered in further studies to help translate the novel findings of this research into the clinic.
Conclusion
Our findings underscore the utility of machine learning algorithms in identifying key dysregulated genes and miRNAs involved in the pathogenesis of CRC, which can facilitate early detection and improve patient outcomes. Moreover, the prognostic value of C1orf174 in CRC was demonstrated, providing a potential target for future research and therapeutic interventions. We recommend future research involving multi-omics analysis of C1orf174, further investigation into the protein's specific functions and conserved residues, and experimental validation of predicted protein interactions. Additionally, studying genetic variation in the C1orf174 gene and its expression across different cancer types could provide valuable insights for cancer therapy.
Research Highlights
What is the current knowledge?
-
CRC is the third most commonly diagnosed cancer, representing 10% of worldwide cancer incidence.
-
Identifying novel biomarkers for early detection of CRC patients is essential.
What is new here?
-
RNA sequencing (RNA-seq) and microRNA profiling herald a new era in biomarker identification, with increasing focus on relevant bioinformatics technologies.
-
Machine learning, a subset of artificial intelligence, is increasingly used for early cancer detection, offering high accuracy in prognosis prediction.
Competing Interests
The authors declare that they have no conflict of interest
Ethical Statement
The local Hospital Ethics Committee of Mashhad University of Medical Sciences approved all procedures (IR.MUMS.MEDICAL.REC.1401.404).
Supplementary files
Supplementary file 1 contains Tables S1-S6.
(pdf)
References
- Xi Y, Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Transl Oncol 2021; 14:101174. doi: 10.1016/j.tranon.2021.101174 [Crossref] [ Google Scholar]
- Brody H. Colorectal cancer. Nature 2015; 521:S1. doi: 10.1038/521S1a [Crossref] [ Google Scholar]
- Wei HT, Guo EN, Liao XW, Chen LS, Wang JL, Ni M. Genome-scale analysis to identify potential prognostic microRNA biomarkers for predicting overall survival in patients with colon adenocarcinoma. Oncol Rep 2018; 40:1947-58. doi: 10.3892/or.2018.6607 [Crossref] [ Google Scholar]
- Yang J, Ma D, Fesler A, Zhai H, Leamniramit A, Li W. Expression analysis of microRNA as prognostic biomarkers in colorectal cancer. Oncotarget 2017; 8:52403-12. doi: 10.18632/oncotarget.14175 [Crossref] [ Google Scholar]
- Jacob H, Stanisavljevic L, Storli KE, Hestetun KE, Dahl O, Myklebust MP. Identification of a sixteen-microRNA signature as prognostic biomarker for stage II and III colon cancer. Oncotarget 2017; 8:87837-47. doi: 10.18632/oncotarget.21237 [Crossref] [ Google Scholar]
- Liu JX, Li W, Li JT, Liu F, Zhou L. Screening key long non-coding RNAs in early-stage colon adenocarcinoma by RNA-sequencing. Epigenomics 2018; 10:1215-28. doi: 10.2217/epi-2017-0155 [Crossref] [ Google Scholar]
- Lightbody G, Haberland V, Browne F, Taggart L, Zheng H, Parkes E. Review of applications of high-throughput sequencing in personalized medicine: barriers and facilitators of future progress in research and clinical application. Brief Bioinform 2019; 20:1795-811. doi: 10.1093/bib/bby051 [Crossref] [ Google Scholar]
- Atas Guvenilir H, Doğan T. How to approach machine learning-based prediction of drug/compound-target interactions. J Cheminform 2023; 15:16. doi: 10.1186/s13321-023-00689-w [Crossref] [ Google Scholar]
- Di C, Mladkova N, Lin J, Fee B, Rivas M, Chunsheng K. AJAP1 expression modulates glioma cell motility and correlates with tumor growth and survival. Int J Oncol 2018; 52:47-54. doi: 10.3892/ijo.2017.4184 [Crossref] [ Google Scholar]
- Goldenberg SL, Nir G, Salcudean SE. A new era: artificial intelligence and machine learning in prostate cancer. Nat Rev Urol 2019; 16:391-403. doi: 10.1038/s41585-019-0193-3 [Crossref] [ Google Scholar]
- Kleppe A, Skrede OJ, De Raedt S, Liestøl K, Kerr DJ, Danielsen HE. Designing deep learning studies in cancer diagnostics. Nat Rev Cancer 2021; 21:199-211. doi: 10.1038/s41568-020-00327-9 [Crossref] [ Google Scholar]
- Yu C, Helwig EJ. The role of AI technology in prediction, diagnosis and treatment of colorectal cancer. Artif Intell Rev 2022; 55:323-43. doi: 10.1007/s10462-021-10034-y [Crossref] [ Google Scholar]
- Liu J, Liu F, Li X, Song X, Zhou L, Jie J. Screening key genes and miRNAs in early-stage colon adenocarcinoma by RNA-sequencing. Tumour Biol 2017; 39:1010428317714899. doi: 10.1177/1010428317714899 [Crossref] [ Google Scholar]
- Ghatak S, Mehrabi SF, Mehdawi LM, Satapathy SR, Sjölander A. Identification of a novel five-gene signature as a prognostic and diagnostic biomarker in colorectal cancers. Int J Mol Sci 2022; 23:793. doi: 10.3390/ijms23020793 [Crossref] [ Google Scholar]
- Sarker IH. Deep learning: a comprehensive overview on techniques, taxonomy, applications and research directions. SN Comput Sci 2021; 2:420. doi: 10.1007/s42979-021-00815-1 [Crossref] [ Google Scholar]
- Xin Y, Kong L, Liu Z, Chen Y, Li Y, Zhu H. Machine learning and deep learning methods for cybersecurity. IEEE Access 2018; 6:35365-81. doi: 10.1109/access.2018.2836950 [Crossref] [ Google Scholar]
- Han J, Kamber M, Pei J. Classification: basic concepts. In: Han J, Kamber M, Pei J, eds. Data Mining. 3rd ed. Boston: Morgan Kaufmann; 2012. p. 327-91.
- Singh M, Wadhwa PK, Sandhu PW. Human protein function prediction using decision tree induction. International Journal of Computer Science and Network Security 2007; 7:92-8. [ Google Scholar]
- Nazari E, Khalili-Tanha G, Asadnia A, Pourali G, Maftooh M, Khazaei M. Bioinformatics analysis and machine learning approach applied to the identification of novel key genes involved in non-alcoholic fatty liver disease. Sci Rep 2023; 13:20489. doi: 10.1038/s41598-023-46711-x [Crossref] [ Google Scholar]
- Azari H, Nazari E, Mohit R, Asadnia A, Maftooh M, Nassiri M. Machine learning algorithms reveal potential miRNAs biomarkers in gastric cancer. Sci Rep 2023; 13:6147. doi: 10.1038/s41598-023-32332-x [Crossref] [ Google Scholar]
- Nazari E, Pourali G, Khazaei M, Asadnia A, Dashtiahangar M, Mohit R. Identification of potential biomarkers in stomach adenocarcinoma using machine learning approaches. Curr Bioinform 2023; 18:320-33. doi: 10.2174/1574893618666230227103427 [Crossref] [ Google Scholar]
- Resmini R, Silva L, Araujo AS, Medeiros P, Muchaluat-Saade D, Conci A. Combining genetic algorithms and SVM for breast cancer diagnosis using infrared thermography. Sensors (Basel) 2021; 21:4802. doi: 10.3390/s21144802 [Crossref] [ Google Scholar]
- Gupta P, Chiang SF, Sahoo PK, Mohapatra SK, You JF, Onthoni DD. Prediction of colon cancer stages and survival period with machine learning approach. Cancers (Basel) 2019; 11:2007. doi: 10.3390/cancers11122007 [Crossref] [ Google Scholar]
- Zhang Y, Wu Y, Gong ZY, Ye HD, Zhao XK, Li JY. Distinguishing rectal cancer from colon cancer based on the support vector machine method and RNA-sequencing data. Curr Med Sci 2021; 41:368-74. doi: 10.1007/s11596-021-2356-8 [Crossref] [ Google Scholar]
- Khalili-Tanha G, Mohit R, Asadnia A, Khazaei M, Dashtiahangar M, Maftooh M. Identification of ZMYND19 as a novel biomarker of colorectal cancer: RNA-sequencing and machine learning analysis. J Cell Commun Signal 2023; 17:1469-85. doi: 10.1007/s12079-023-00779-2 [Crossref] [ Google Scholar]
- Asadnia A, Nazari E, Goshayeshi L, Zafari N, Moetamani-Ahmadi M, Goshayeshi L. The prognostic value of ASPHD1 and ZBTB12 in colorectal cancer: a machine learning-based integrated bioinformatics approach. Cancers (Basel) 2023; 15:4300. doi: 10.3390/cancers15174300 [Crossref] [ Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021; 71:209-49. doi: 10.3322/caac.21660 [Crossref] [ Google Scholar]
- Chen L, Lu D, Sun K, Xu Y, Hu P, Li X. Identification of biomarkers associated with diagnosis and prognosis of colorectal cancer patients based on integrated bioinformatics analysis. Gene 2019; 692:119-25. doi: 10.1016/j.gene.2019.01.001 [Crossref] [ Google Scholar]
- Jung Y, Lee S, Choi HS, Kim SN, Lee E, Shin Y. Clinical validation of colorectal cancer biomarkers identified from bioinformatics analysis of public expression data. Clin Cancer Res 2011; 17:700-9. doi: 10.1158/1078-0432.ccr-10-1300 [Crossref] [ Google Scholar]
- Yang G, Zhang Y, Yang J. A five-microRNA signature as prognostic biomarker in colorectal cancer by bioinformatics analysis. Front Oncol 2019; 9:1207. doi: 10.3389/fonc.2019.01207 [Crossref] [ Google Scholar]
- Maurya NS, Kushwaha S, Chawade A, Mani A. Transcriptome profiling by combined machine learning and statistical R analysis identifies TMEM236 as a potential novel diagnostic biomarker for colorectal cancer. Sci Rep 2021; 11:14304. doi: 10.1038/s41598-021-92692-0 [Crossref] [ Google Scholar]
- Koppad S, Basava A, Nash K, Gkoutos GV, Acharjee A. Machine learning-based identification of colon cancer candidate diagnostics genes. Biology (Basel) 2022; 11:365. doi: 10.3390/biology11030365 [Crossref] [ Google Scholar]
- Hossain MJ, Chowdhury UN, Islam MB, Uddin S, Ahmed MB, Quinn JM. Machine learning and network-based models to identify genetic risk factors to the progression and survival of colorectal cancer. Comput Biol Med 2021; 135:104539. doi: 10.1016/j.compbiomed.2021.104539 [Crossref] [ Google Scholar]
- Wang Y, Liu K, Ma Q, Tan Y, Du W, Lv Y. Pancreatic cancer biomarker detection by two support vector strategies for recursive feature elimination. Biomark Med 2019; 13:105-21. doi: 10.2217/bmm-2018-0273 [Crossref] [ Google Scholar]
- Ma S, Duan L, Dong H, Ma X, Guo X, Liu J. OLFML2A downregulation inhibits glioma proliferation through suppression of Wnt/β-catenin signaling. Front Oncol 2021; 11:717917. doi: 10.3389/fonc.2021.717917 [Crossref] [ Google Scholar]
- Liang X, Gao J, Wang Q, Hou S, Wu C. ECRG4 represses cell proliferation and invasiveness via NFIC/OGN/NF-κB signaling pathway in bladder cancer. Front Genet 2020; 11:846. doi: 10.3389/fgene.2020.00846 [Crossref] [ Google Scholar]
- Du F, Sun L, Chu Y, Li T, Lei C, Wang X. DDIT4 promotes gastric cancer proliferation and tumorigenesis through the p53 and MAPK pathways. Cancer Commun (Lond) 2018; 38:45. doi: 10.1186/s40880-018-0315-y [Crossref] [ Google Scholar]
- Kim YJ, Hirabayashi Y. Caveolin-1 prevents palmitate-induced NF-κB signaling by inhibiting GPRC5B-phosphorylation. Biochem Biophys Res Commun 2018; 503:2673-7. doi: 10.1016/j.bbrc.2018.08.022 [Crossref] [ Google Scholar]
- Wang Y, Han E, Xing Q, Yan J, Arrington A, Wang C. Baicalein upregulates DDIT4 expression which mediates mTOR inhibition and growth inhibition in cancer cells. Cancer Lett 2015; 358:170-9. doi: 10.1016/j.canlet.2014.12.033 [Crossref] [ Google Scholar]
- Min XS, Huang P, Liu X, Dong C, Jiang XL, Yuan ZT. Bioinformatics analyses of significant prognostic risk markers for thyroid papillary carcinoma. Tumour Biol 2015; 36:7457-63. doi: 10.1007/s13277-015-3410-6 [Crossref] [ Google Scholar]
- Xu C, Wang F, Hao L, Liu J, Shan B, Lv S. Expression patterns of Ezrin and AJAP1 and clinical significance in breast cancer. Front Oncol 2022; 12:831507. doi: 10.3389/fonc.2022.831507 [Crossref] [ Google Scholar]
- Qu W, Wen X, Su K, Gou W. MiR-552 promotes the proliferation, migration and EMT of hepatocellular carcinoma cells by inhibiting AJAP1 expression. J Cell Mol Med 2019; 23:1541-52. doi: 10.1111/jcmm.14062 [Crossref] [ Google Scholar]
- Tanaka H, Kanda M, Koike M, Iwata N, Shimizu D, Ezaka K. Adherens junctions associated protein 1 serves as a predictor of recurrence of squamous cell carcinoma of the esophagus. Int J Oncol 2015; 47:1811-8. doi: 10.3892/ijo.2015.3167 [Crossref] [ Google Scholar]
- Vrooman LM, Silverman LB. Treatment of childhood acute lymphoblastic leukemia: prognostic factors and clinical advances. Curr Hematol Malig Rep 2016; 11:385-94. doi: 10.1007/s11899-016-0337-y [Crossref] [ Google Scholar]
- Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009; 360:470-80. doi: 10.1056/NEJMoa0808253 [Crossref] [ Google Scholar]
- Morel G, Deau MC, Simand C, Caye-Eude A, Arfeuille C, Ittel A. Large deletions of the 5' region of IKZF1 lead to haploinsufficiency in B-cell precursor acute lymphoblastic leukaemia. Br J Haematol 2019; 186:e155-9. doi: 10.1111/bjh.15994 [Crossref] [ Google Scholar]
- van der Veer A, Waanders E, Pieters R, Willemse ME, Van Reijmersdal SV, Russell LJ. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 2013; 122:2622-9. doi: 10.1182/blood-2012-10-462358 [Crossref] [ Google Scholar]
- Singh MK, Dadke D, Nicolas E, Serebriiskii IG, Apostolou S, Canutescu A. A novel CAS family member, HEPL, regulates FAK and cell spreading. Mol Biol Cell 2008; 19:1627-36. doi: 10.1091/mbc.e07-09-0953 [Crossref] [ Google Scholar]
- Deneka A, Korobeynikov V, Golemis EA. Embryonal Fyn-associated substrate (EFS) and CASS4: the lesser-known CAS protein family members. Gene 2015; 570:25-35. doi: 10.1016/j.gene.2015.06.062 [Crossref] [ Google Scholar]
- Li A, Zhang W, Xia H, Miao Y, Zhou H, Zhang X. Overexpression of CASS4 promotes invasion in non-small cell lung cancer by activating the AKT signaling pathway and inhibiting E-cadherin expression. Tumour Biol 2016; 37:15157-64. doi: 10.1007/s13277-016-5411-5 [Crossref] [ Google Scholar]
- Gopalan L, Sebastian A, Praul CA, Albert I, Ramachandran R. Metformin affects the transcriptomic profile of chicken ovarian cancer cells. Genes (Basel) 2021; 13:30. doi: 10.3390/genes13010030 [Crossref] [ Google Scholar]
- Néant I, Haiech J, Kilhoffer MC, Aulestia FJ, Moreau M, Leclerc C. Ca2+-dependent transcriptional repressors KCNIP and regulation of prognosis genes in glioblastoma. Front Mol Neurosci 2018; 11:472. doi: 10.3389/fnmol.2018.00472 [Crossref] [ Google Scholar]
- Zavala-Zendejas VE, Torres-Martinez AC, Salas-Morales B, Fortoul TI, Montaño LF, Rendon-Huerta EP. Claudin-6, 7, or 9 overexpression in the human gastric adenocarcinoma cell line AGS increases its invasiveness, migration, and proliferation rate. Cancer Invest 2011; 29:1-11. doi: 10.3109/07357907.2010.512594 [Crossref] [ Google Scholar]
- Rendón-Huerta E, Teresa F, Teresa GM, Xochitl GS, Georgina AF, Veronica ZZ. Distribution and expression pattern of claudins 6, 7, and 9 in diffuse- and intestinal-type gastric adenocarcinomas. J Gastrointest Cancer 2010; 41:52-9. doi: 10.1007/s12029-009-9110-y [Crossref] [ Google Scholar]
- Liu H, Wang M, Liang N, Guan L. Claudin-9 enhances the metastatic potential of hepatocytes via Tyk2/Stat3 signaling. Turk J Gastroenterol 2019; 30:722-31. doi: 10.5152/tjg.2019.18513 [Crossref] [ Google Scholar]
- Zhu J, Wang R, Cao H, Zhang H, Xu S, Wang A. Expression of claudin-5, -7, -8 and -9 in cervical carcinoma tissues and adjacent non-neoplastic tissues. Int J Clin Exp Pathol 2015; 8:9479-86. [ Google Scholar]
- Lentjes MH, Niessen HE, Akiyama Y, de Bruïne AP, Melotte V, van Engeland M. The emerging role of GATA transcription factors in development and disease. Expert Rev Mol Med 2016; 18:e3. doi: 10.1017/erm.2016.2 [Crossref] [ Google Scholar]
- Deng L, Niu GM, Ren J, Ke CW. Identification of ATP8B1 as a tumor suppressor gene for colorectal cancer and its involvement in phospholipid homeostasis. Biomed Res Int 2020; 2020:2015648. doi: 10.1155/2020/2015648 [Crossref] [ Google Scholar]
- Althenayyan S, AlGhamdi A, AlMuhanna MH, Hawsa E, Aldeghaither D, Iqbal J. Modulation of ATP8B1 gene expression in colorectal cancer cells suggest its role as a tumor suppressor. Curr Cancer Drug Targets 2022; 22:577-90. doi: 10.2174/1568009622666220517092340 [Crossref] [ Google Scholar]
- Aziz MA, Periyasamy S, Al Yousef Z, AlAbdulkarim I, Al Otaibi M, Alfahed A. Integrated exon level expression analysis of driver genes explain their role in colorectal cancer. PLoS One 2014; 9:e110134. doi: 10.1371/journal.pone.0110134 [Crossref] [ Google Scholar]
- Qiu Z, Guo W, Wang Q, Chen Z, Huang S, Zhao F, et al. MicroRNA-124 reduces the pentose phosphate pathway and proliferation by targeting PRPS1 and RPIA mRNAs in human colorectal cancer cells. Gastroenterology 2015; 149: 1587-98.e11. 10.1053/j.gastro.2015.07.050.
- Chou YT, Chen LY, Tsai SL, Tu HC, Lu JW, Ciou SC. Ribose-5-phosphate isomerase A overexpression promotes liver cancer development in transgenic zebrafish via activation of ERK and β-catenin pathways. Carcinogenesis 2019; 40:461-73. doi: 10.1093/carcin/bgy155 [Crossref] [ Google Scholar]
- Ciou SC, Chou YT, Liu YL, Nieh YC, Lu JW, Huang SF. Ribose-5-phosphate isomerase A regulates hepatocarcinogenesis via PP2A and ERK signaling. Int J Cancer 2015; 137:104-15. doi: 10.1002/ijc.29361 [Crossref] [ Google Scholar]
- Wu T, Jiao Z, Li Y, Su X, Yao F, Peng J. HPRT1 promotes chemoresistance in oral squamous cell carcinoma via activating MMP1/PI3K/Akt signaling pathway. Cancers (Basel) 2022; 14:855. doi: 10.3390/cancers14040855 [Crossref] [ Google Scholar]
- Ohl F, Jung M, Xu C, Stephan C, Rabien A, Burkhardt M. Gene expression studies in prostate cancer tissue: which reference gene should be selected for normalization?. J Mol Med (Berl) 2005; 83:1014-24. doi: 10.1007/s00109-005-0703-z [Crossref] [ Google Scholar]
- Townsend MH, Ence ZE, Felsted AM, Parker AC, Piccolo SR, Robison RA. Potential new biomarkers for endometrial cancer. Cancer Cell Int 2019; 19:19. doi: 10.1186/s12935-019-0731-3 [Crossref] [ Google Scholar]
- Liu C, Wang J, Huang S, Chang J, Yu H, Zhu Z. P112-19 Identification and potential application of human blood exosomal RNA in small cell lung cancer. J Thorac Oncol 2019; 14:S541-2. doi: 10.1016/j.jtho.2019.08.1132 [Crossref] [ Google Scholar]
- Moll R, Schiller DL, Franke WW. Identification of protein IT of the intestinal cytoskeleton as a novel type I cytokeratin with unusual properties and expression patterns. J Cell Biol 1990; 111:567-80. doi: 10.1083/jcb.111.2.567 [Crossref] [ Google Scholar]
- Chan CW, Wong NA, Liu Y, Bicknell D, Turley H, Hollins L. Gastrointestinal differentiation marker cytokeratin 20 is regulated by homeobox gene CDX1. Proc Natl Acad Sci U S A 2009; 106:1936-41. doi: 10.1073/pnas.0812904106 [Crossref] [ Google Scholar]
- Ebadfardzadeh J, Kazemi M, Aghazadeh A, Rezaei M, Shirvaliloo M, Sheervalilou R. Employing bioinformatics analysis to identify hub genes and microRNAs involved in colorectal cancer. Med Oncol 2021; 38:114. doi: 10.1007/s12032-021-01543-5 [Crossref] [ Google Scholar]
- Ramírez-Backhaus M, Fernández-Serra A, Rubio-Briones J, Cruz Garcia P, Calatrava A, Garcia Casado Z. External validation of FXYD3 and KRT20 as predictive biomarkers for the presence of micrometastasis in muscle invasive bladder cancer lymph nodes. Actas Urol Esp 2015; 39:473-81. doi: 10.1016/j.acuro.2015.02.002 [Crossref] [ Google Scholar]
- Stangeland B, Mughal AA, Grieg Z, Sandberg CJ, Joel M, Nygård S. Combined expressional analysis, bioinformatics and targeted proteomics identify new potential therapeutic targets in glioblastoma stem cells. Oncotarget 2015; 6:26192-215. doi: 10.18632/oncotarget.4613 [Crossref] [ Google Scholar]
- Boorjian S, Tickoo SK, Mongan NP, Yu H, Bok D, Rando RR. Reduced lecithin:retinol acyltransferase expression correlates with increased pathologic tumor stage in bladder cancer. Clin Cancer Res 2004; 10:3429-37. doi: 10.1158/1078-0432.ccr-03-0756 [Crossref] [ Google Scholar]
- Cheng YW, Pincas H, Huang J, Zachariah E, Zeng Z, Notterman DA. High incidence of LRAT promoter hypermethylation in colorectal cancer correlates with tumor stage. Med Oncol 2014; 31:254. doi: 10.1007/s12032-014-0254-7 [Crossref] [ Google Scholar]
- Brown GT, Cash BG, Blihoghe D, Johansson P, Alnabulsi A, Murray GI. The expression and prognostic significance of retinoic acid metabolising enzymes in colorectal cancer. PLoS One 2014; 9:e90776. doi: 10.1371/journal.pone.0090776 [Crossref] [ Google Scholar]
- Guo X, Knudsen BS, Peehl DM, Ruiz A, Bok D, Rando RR. Retinol metabolism and lecithin:retinol acyltransferase levels are reduced in cultured human prostate cancer cells and tissue specimens. Cancer Res 2002; 62:1654-61. [ Google Scholar]
- Sheren-Manoff M, Shin SJ, Su D, Bok D, Rando RR, Gudas LJ. Reduced lecithin:retinol acyltransferase expression in human breast cancer. Int J Oncol 2006; 29:1193-9. [ Google Scholar]
- Zhan HC, Gudas LJ, Bok D, Rando R, Nanus DM, Tickoo SK. Differential expression of the enzyme that esterifies retinol, lecithin:retinol acyltransferase, in subtypes of human renal cancer and normal kidney. Clin Cancer Res 2003; 9:4897-905. [ Google Scholar]
- De Paoli V, Barany F, Cheng YW. LRAT promoter hypermethylation as a prognostic marker for colorectal cancer impairs retinol metabolism. J Clin Gastroenterol Treat 2017; 3:49. doi: 10.23937/2469-584X/1510049 [Crossref] [ Google Scholar]
- Li X, Peng S. Identification of metastasis-associated genes in colorectal cancer through an integrated genomic and transcriptomic analysis. Chin J Cancer Res 2013; 25:623-36. doi: 10.3978/j.issn.1000-9604.2013.11.01 [Crossref] [ Google Scholar]
- Wu X, Zhang ZX, Chen XY, Xu YL, Yin N, Yang J. A panel of three biomarkers identified by iTRAQ for the early diagnosis of pancreatic cancer. Proteomics Clin Appl 2019; 13:e1800195. doi: 10.1002/prca.201800195 [Crossref] [ Google Scholar]
- Sung MK, Bae YJ. Linking obesity to colorectal cancer: application of nutrigenomics. Biotechnol J 2010; 5:930-41. doi: 10.1002/biot.201000165 [Crossref] [ Google Scholar]