Bioimpacts. 2025;15:31226.
doi: 10.34172/bi.31226
Original Article
Hybrid deep learning models for text-based identification of gene-disease associations
Noor Fadhil Jumaa Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Visualization, Writing – original draft, 1 
Jafar Razmara Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Supervision, Validation, Writing – review & editing, 1, *
Sepideh Parvizpour Validation, Writing – review & editing, 2
Jaber Karimpour Validation, Writing – review & editing, 1
Author information:
1Department of Computer Science, Faculty of Mathematics, Statistics, and Computer Science, University of Tabriz, Tabriz, Iran
2Research Center for Pharmaceutical Nanotechnology, Biomedicine Institute, Tabriz University of Medical Sciences, Tabriz, Iran
Abstract
Introduction:
Identifying gene-disease associations is crucial for advancing medical research and improving clinical outcomes. Nevertheless, the rapid expansion of biomedical literature poses significant obstacles to extracting meaningful relationships from extensive text collections.
Methods:
This study uses deep learning techniques to automate this process, using publicly available datasets (EU-ADR, GAD, and SNPPhenA) to classify these associations accurately. Each dataset underwent rigorous pre-processing, including entity identification and preparation, word embedding using pre-trained Word2Vec and fastText models, and position embedding to capture semantic and contextual relationships within the text. In this research, three deep learning-based hybrid models have been implemented and contrasted, including CNN-LSTM, CNN-GRU, and CNN-GRU-LSTM. Each model has been equipped with attentional mechanisms to enhance its performance.
Results:
Our findings reveal that the CNN-GRU model achieved the highest accuracy of 91.23% on the SNPPhenA dataset, while the CNN-GRU-LSTM model attained an accuracy of 90.14% on the EU-ADR dataset. Meanwhile, the CNN-LSTM model demonstrated superior performance on the GAD dataset, achieving an accuracy of 84.90%. Compared to previous state-of-the-art methods, such as BioBERT-based models, our hybrid approach demonstrates superior classification performance by effectively capturing local and sequential features without relying on heavy pre-training.
Conclusion:
The developed models and their evaluation data are available at https://github.com/NoorFadhil/Deep-GDAE.
Keywords: Gene-disease association extraction, Deep learning, Attention mechanism, Feature extraction
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
None.
Introduction
The discovery of gene-disease associations is crucial for advancing medical research and improving clinical outcomes, especially in preventing, diagnosing, and treating genetic illnesses. However, deriving significant links from the rapidly expanding biomedical literature has become more difficult.1 The MEDLINE database adds around 500 000 new biomedical abstracts each year, resulting in an enormous amount of data practically impossible for academics to evaluate manually. This difficulty emphasizes the critical need for computational tools that may reveal hidden insights within large amounts of textual data.1 Text mining has emerged as a critical method for assessing massive amounts of biomedical literature. It allows researchers to automatically identify noteworthy patterns, correlations, and links in unstructured data. Using text mining tools, researchers may easily find gene-disease connections, keeping up with the field's fast progress. These techniques have considerably improved the speed and accuracy of information retrieval, revealing important insights into complicated biological systems.2,3
Despite these advancements, typical traditional techniques for relation extraction (RE) remain difficult and time-consuming. Due to the large amount of data available, experimental approaches for discovering gene-disease associations are sometimes impractical. Therefore, automated systems combining machine learning, deep learning, and natural language processing (NLP) have been created to solve this issue.4 Most recent developments in biomedical research have changed the emphasis from researching individual genes and proteins to examining whole genomes and biosystems. Advanced methods that can reveal hidden correlations in huge, unstructured datasets are required for this progress. Computational approaches that automate the extraction of these correlations may save researchers substantial time, effort, and money while allowing them to prioritize preventive, diagnostic, and treatment initiatives.4,5
The existing computational techniques for gene-disease association extraction range from basic co-occurrence algorithms to complex machine learning and deep learning models. However, many solutions have shortcomings, such as poor context management, accuracy, and data imbalance. These difficulties call for creative approaches that successfully convey the intricate relationships and subtleties of context present in biomedical texts.6,7
In this paper, we have developed three deep-learning classification models: CNN-LSTM, CNN-GRU, and CNN-GRU-LSTM. Each model combines convolutional and recurrent neural networks to classify gene-disease correlations accurately. Our classifiers outperformed the other state-of-the-art methods in determining gene-disease relationships without needing biological features. The main contributions of this paper include the following:
-
The development of hybrid neural network architectures, specifically the CNN-LSTM, CNN-GRU, and CNN-GRU-LSTM models, designed for classifying gene-disease associations,
-
Adding attention mechanisms to these models to focus on essential parts of the input sequence, making classification tasks more accurate and efficient,
-
Thorough testing of the suggested models using three public datasets: EU-ADR, GAD, and SNPPhenA, to evaluate their performance for identifying gene-disease associations,
-
Employing pre-trained word embedding models like word2vec and fastText to capture semantic relationships in the text, enhancing the feature representation for the classifiers, and
-
Employing a position embedding technique to capture the relative positions of words within sentences, helping to understand contextual relationships in the text.
Literature review
Biomedical text mining is crucial for extracting significant connections from the vast and expanding collection of biomedical literature. Relation extraction aims explicitly to uncover and describe connections between biomedical entities, such as genes, illnesses, and single nucleotide polymorphisms (SNPs). In recent years, several studies have suggested novel machine learning, deep learning, and Transformer-Based Approaches to improve the accuracy and efficacy of extracting these relationships. A comparative summary of the reviewed studies, highlighting their approaches, proposed methods, and identified limitations, is presented in Table S1.
Machine learning-based approaches
Several early studies applied traditional machine learning techniques to the problem of gene-disease association extraction. Bhasuran et al8 suggested a supervised machine learning approach for extracting gene-disease associations, wherein an ensemble support vector machine was trained with an extensive feature set, including conceptual, syntactic, and semantic features concurrently acquired using the Word2Vec word embedding technique. The system achieved a F1-score of 83.93% on the GAD corpus, 87.39% on the CoMAGC corpus, 85.55% on the EU-ADR corpus, and 85.57% on the PolySearch dataset. However, difficulties developed when dealing with long and complex texts, leading to inaccuracies in identifying associations. In future work, the authors addressed simplifying intricate sentences and enhancing the classifier's effectiveness in identifying negative instances to improve significantly the extraction process's accuracy. However, Bokharaeian et al9 suggested a novel method for extracting SNP-phenotype associations from biomedical literature. The focus is on identifying negation signals and neutral candidates to enhance the accuracy of relation extraction. Utilizing features such as negation scope and neutral candidate detection, they implemented a linguistic-based approach that was assessed on the SNPPhenA corpus. With an F1-score of 75.60%, their method outperformed kernel-based approaches in capturing SNP-phenotype associations with consideration of confidence levels. However, the scarcity of neutral candidates in the corpus impacted the model's robustness. Future work suggested improving the model by incorporating supplementary linguistic features and investigating larger datasets to generalize the results across biomedical texts. While, Wang et al10 developed HNEEM and HNEEM-PLUS using graph embedding and ensemble learning to predict gene-disease associations. They implemented six graph embedding approaches to create a heterogeneous network integrating genes, diseases, chemicals, and chemicals and learned representative vectors of genes and diseases. The ensemble model HNEEM was created by averaging prediction models for each graph embedding representation with random forest classifiers. In contrast, the HNEEM-PLUS model was created by adding a multilayer perceptron classifier to increase base predictor diversity. Their HNEEM-PLUS model outperformed the other previous methods with an F1-score of 82.60 in the collected dataset from CTD. However, the intricacy of ensemble approaches limits scalability and overfitting, according to the study. Therefore, the authors suggest applying more advanced ensemble approaches and biological data sources to improve prediction accuracy. Xiang et al11 proposed a hybrid method for improving disease-gene prediction by integrating multiscale module structures under the name HyMM. They applied three algorithms for multiscale module decomposition: modularity optimization, asymptotic surprise, and fast hierarchical clustering on biological networks to extract multiscale modules. These modules were then used for ranking genes according to their disease association, and the integration of multiple rankings was performed under a theoretical framework based on naïve Bayes theory. 5-fold cross-validation and independent tests showed that HyMM worked better than traditional methods. The authors point out that, for future work, there is a need for more flexible and robust methods for detecting modules capable of incorporating more types of biological data to predict disease-causing genes better.
Deep learning-based approaches
Recent advances in deep learning have significantly improved gene-disease relation extraction by leveraging complex neural architectures. Wu et al12 proposed a deep learning-based RENET model, which was trained on a collected dataset of 30,192 abstracts from MEDLINE fetched through DisGeNet to extract gene-disease associations. Their model consisted of two main stages; the first stage is a representation of words and sentences using convolutional neural networks (CNNs), while in the second stage, the relationships between each sentence were captured in a document with RNN, such as GRU/LSTM. Their approach outperformed tools like BeFree and DTMiner regarding precision and recall values while improving the F-Score by more than 20%. Nonetheless, their technique required better NER and a deeper contextual understanding of sentence boundaries. Future work includes exploring more efficient models for handling document-level semantics and incorporating domain-specific knowledge bases to improve the accuracy of association extraction further. Nourani et al13 presented Deep-GDAE, a new deep-learning algorithm for extracting gene-disease associations from biological texts. It used a sentence-level attention-based neural network architecture that combined transfer learning with pre-trained embeddings from PubMed and PMC. Deep-GDAE uses CNNs and bidirectional LSTMs (BiLSTMs) to analyze and categorize sentences using an attention mechanism. Their model outperformed previous methods, with F1-scores of 85.80, 71.63, and 73.97 for the EU-ADR, GAD, and SNPPhenA corpora, respectively. However, researchers note constraints such as potential performance differences due to the pre-trained embeddings' quality and the biomedical texts' unique structure. For future work, the authors recommend further investigating more advanced attention mechanisms and incorporating supplementary contextual information to improve the model's robustness and accuracy. On the other hand, Su et al14 proposed RENET2, a deep learning-based strategy for extracting relationships in discovering gene-disease associations from abstract and full-text articles. Section filtering and modeling of ambiguous relationships were incorporated into this approach. Since the annotated full-text datasets are rare, the authors developed an iterative strategy for augmenting the training data. Their model structure incorporates the CNN layer for extracting local semantic features and the RNN layer, which captures sequential dependencies and contextual information about the sentences. RENET2 outperformed some existing tools, such as BeFree, DTMiner, and BioBERT, with an F1-score of 72.13 % in extracting gene-disease relationships from annotated full-text datasets. However, their strategy demonstrated poor processing efficacy for one gene-disease pair at a time and needed to integrate advanced deep language models like ELMo and BERT for enhancing full-text relation extraction. Bokharaeian et al15 introduced a method for extracting and ranking SNP-phenotype associations from biomedical literature using machine learning and deep learning models. In their study, several machine learning techniques were developed, including traditional methods such as random forest, logistic regression, SVM, KNN, GaussianNB, GradientBoosting, and DecisionTree, as well as advanced deep learning models like CNN-LSTM, BERT-LSTM, and PubMedBERT-LSTM for extracting associations with a focus on the degree of certainty. In testing models on SNPPhenA and the EU-ADR dataset, the PubMedBERT-LSTM model gave an F1-score of 83.9 and 80.7, respectively, compared to prior methods. In future work, the authors suggested focusing on treating complicated linguistic forms, structures, and integrations of more biomedical databases for model validation and refinement. However, Dehghani et al16 proposed a deep learning method for extracting SNP-phenotype associations from biomedical literature using BioBERT-GRU. The model used pre-trained BioBERT for feature representation, with a CNN layer over it for feature extraction, followed by a bidirectional GRU for capturing the dependencies of the sequence data. Results showed that BioBERT-GRU outperformed the prior models and reached an F1-score of 88.30 at the sentence level and 64.50 at the abstract level when evaluated in the SNPPhenA dataset. Future work involved determining the type of relationship, direct or indicative of a pathway effect, and possibly the consideration of the use of fuzzy relations instead of crisp relations.
Transformer-Based Approaches
Transformer-based models have recently emerged as powerful tools for biomedical text mining, offering enhanced performance through contextualized embeddings. Lee et al17 introduced BioBERT, a specialized language representation model that enhances BERT by pre-training it with extensive biological corpora, such as PubMed abstracts and PMC full-text articles. The BioBERT model achieved superior performance compared to BERT and other advanced models in three critical tasks of biomedical text analysis: named entity recognition, relation extraction, and question answering, where the model demonstrated an improvement of 0.62% in F1-score for named entity recognition, 2.80% in F1-score for relation extraction, and 12.24% in MRR for question answering. One of the drawbacks highlighted was the computational expense and time requirements associated with pre-training. A recommendation was offered to improve the efficiency of BioBERT in future research and to explore its potential for a broader range of applications in biomedical text mining. However, Deng et al18 contributed an approach for extracting gene-disease associations from biomedical literature using BioBERT, a version pre-trained on large-scale biomedical corpora like BERT. After downloading relevant literature from PubMed, the text was processed through a tokenizer. Afterward, BioBERT is used in two successive steps: named entity recognition for identifying genes and diseases, and then relation extraction to find associations between the identified entities. Their model evaluation achieved an F1-score of 79.98% on the EU-ADR dataset. However, the limitations of this method include that setting up a local BioBERT environment requires high computational resources and expertise, and it ignores abstracts without either gene or disease mentions, which might exclude real associations. In future work, the authors suggested improving the efficiency and accuracy of the model by integrating advanced models or using other biomedical datasets for better performance.
As shown in Table S1, many prior studies have made considerable advancements in gene-disease relationship extraction. However, there are still constraints, such as scalability issues, high processing costs, inadequate handling of complicated sentence structures, and difficulties with negation and speculative language. To bridge these gaps, the current study suggests using lightweight hybrid models (CNN-GRU, CNN-LSTM, and CNN-GRU-LSTM) that mix convolutional and recurrent architectures with attention mechanisms. These models aim to capture both local and long-range dependencies more efficiently, improve contextual information handling, and reduce computational demands when compared to transformer-based approaches such as BioBERT, all while maintaining high classification performance across diverse biomedical datasets.
Methods
The methodology of the proposed method is shown in Fig. 1. It consists of two main phases: (i) the preprocessing phase, and (ii) the classification phase. The datasets were obtained from diverse biological libraries including EU-ADR,19 GAD,20 and SNPPhenA21 and used for predicting gene-disease associations. Each dataset undergoes the preprocessing phase, where all sentences are preprocessed to be represented by pre-trained embedding vectors, thereby rendering them suitable for further processing by the classifier. Following the preprocessing stage, the prepared datasets proceed to the classification phase. In this phase, deep-learning-based models are utilized to categorize gene-disease associations. Specifically, a multi-class classification task is performed on the GAD dataset to classify gene-disease associations into positive (Y), negative (N), and irrelevant (F) categories. Conversely, the EUADR and SNPs datasets are subjected to binary classification, categorizing gene-disease associations as either positive (Y) or negative (N). To improve model performance, an attention mechanism at the word level was applied to each proposed model during the classification phase, enabling the models to concentrate on the most relevant words within each sentence throughout the classification process.
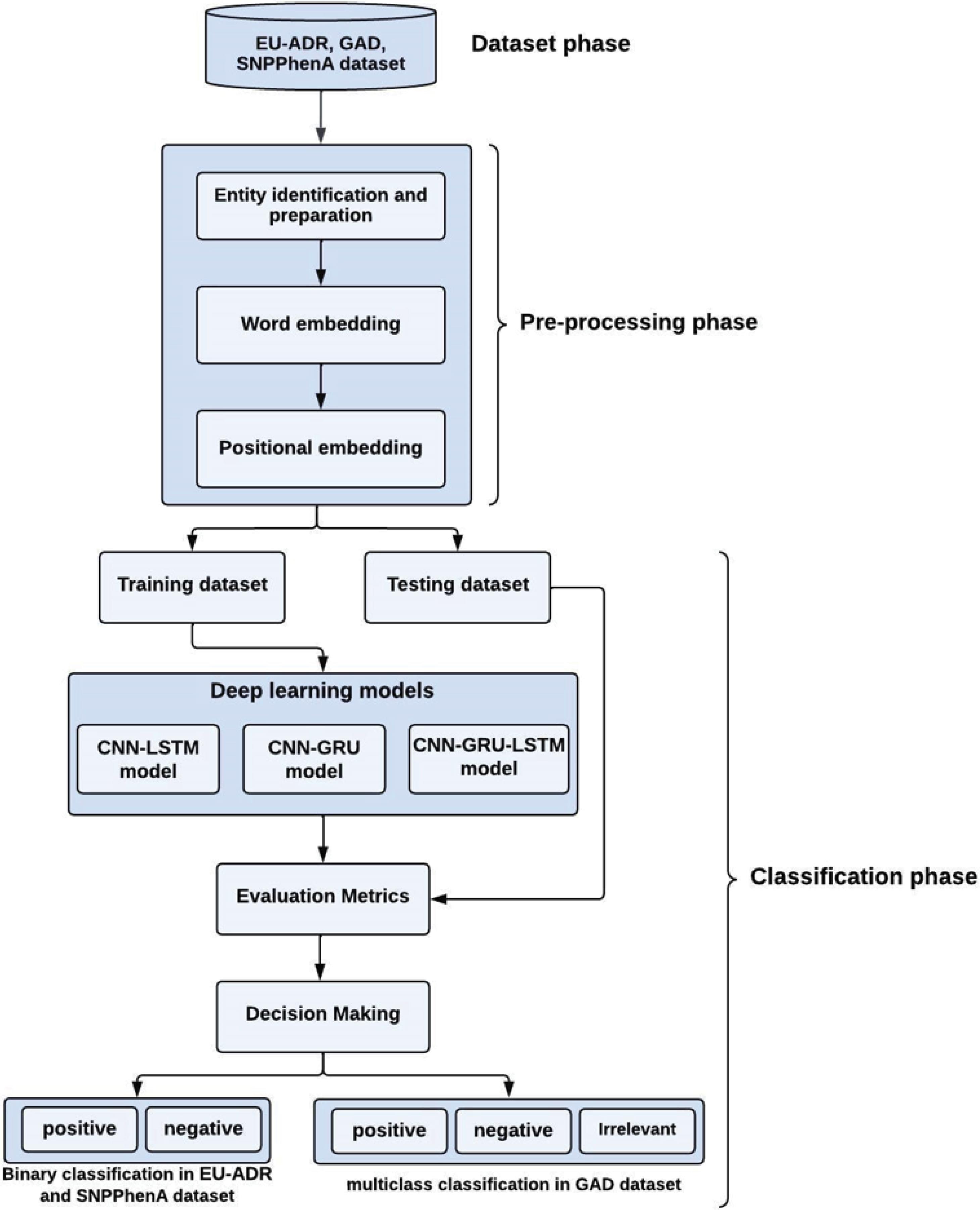
Fig. 1.
The proposed methodology.
.
The proposed methodology.
Dataset description
Different gene-disease association datasets were collected from three diverse publicly available comprehensive resources, including EU-ADR,19 GAD,20 and SNPPhenA.21 The purpose of utilizing this diverse set of datasets is to evaluate the performance of deep learning models in classifying disease-gene associations across different benchmark datasets. We used datasets that fell into the gene-disease or SNP-phenotype association extraction categories. Several recent studies have used these standard biomedical relation extraction datasets, such as Bhasuran et al,8 Nourani et al,13 Bokharaeian et al,9 Lee et al,17 Deng et al,18 and Bokharaeian et al.15 These datasets are described in detail below.
EU-ADR dataset
EU-ADR19 stands for European adverse drug reaction terminology, which consists of annotated scientific literature from Medline that focuses on drugs, diseases, targets (such as proteins, genes, and gene variations), and relationships between these entities. In this study, we used only GDAs to assess the proposed models. The EU-ADR dataset consists of 355 disease-gene associations, 218 genes, and 118 diseases. These associations are categorized into positive associations (PA), negative associations (NA), speculative associations (SA), and false associations (FA) based on the degree of certainty that they possess. Table S2 presents the statistics of the EU-ADR dataset.
Genetic Association Database (GAD)
GAD20 is a comprehensive database of human genetic associations, specifically consisting of data collected from published association and genome-wide association studies. The GAD dataset contains 5,330 disease-gene associations between 1131 genes and 535 diseases. These associations are classified into three classes: positive association (Y), negative association (N), and no semantic association (F). Table S3 represents the GAD dataset in summary.
SNPPhenA dataset
SNPPhenA21 demonstrates ranked relationships between SNPs and phenotypes identified in the literature. It was generated in three steps: collecting relevant abstracts, automatic named entity recognition (NER), and determining SNP-phenotype associations, negations, method flags, and confidence level. The SNP dataset contains 1300 SNP-phenotype associations that involve 417 SNPs (genes) and 360 phenotypes (diseases), separated into training (935 samples) and testing (365 samples) data files. These associations are divided into three classes: positive, negative, and neutral associations. Table S4 illustrates the statistics for the SNPPhenA dataset.
Table S5 provides a comparative summary of the number of instances per class better to illustrate the distribution of association types across datasets. A class imbalance exists in all datasets, with positive associations generally outnumbering negative or uncertain associations. This study merged speculative, neutral, and false associations into the main positive or negative classes where appropriate. Specifically, in the EU-ADR dataset, false associations (FA) and negative associations (NA) were merged into a single negative class. In contrast, positive associations (PA) and speculative associations (SA) were merged into a single positive class. In the SNPPhenA dataset, positive and neutral associations were combined into a positive class. However, no merging was performed in the GAD dataset, and three distinct classes were retained. The decision to merge certain uncertain classes was made to focus on the extraction of definitive gene-disease relationships (i.e., positive and negative associations), as uncertain instances could introduce ambiguity and decrease the reliability of the models. Furthermore, modeling speculative or neutral associations requires specialized techniques, such as uncertainty modeling or fuzzy logic, which are beyond the scope of the current study. Future work could consider including such associations to enrich the relation extraction process.
Preprocessing phase
Each dataset was subjected to a series of operations during the preprocessing phase to enhance its quality and facilitate the subsequent analysis process when fed into the classifiers. The initial step is to identify and prepare entities within each dataset, which includes tasks such as sentence preprocessing, entity identification, padding, and label mapping. The following step involves applying the technique of word embedding to represent each sentence as a dense vector. Finally, positional embedding is employed to encode the sequential or positional order of words within each sentence. Fig. 2 illustrates the workflow of the preprocessing phase.
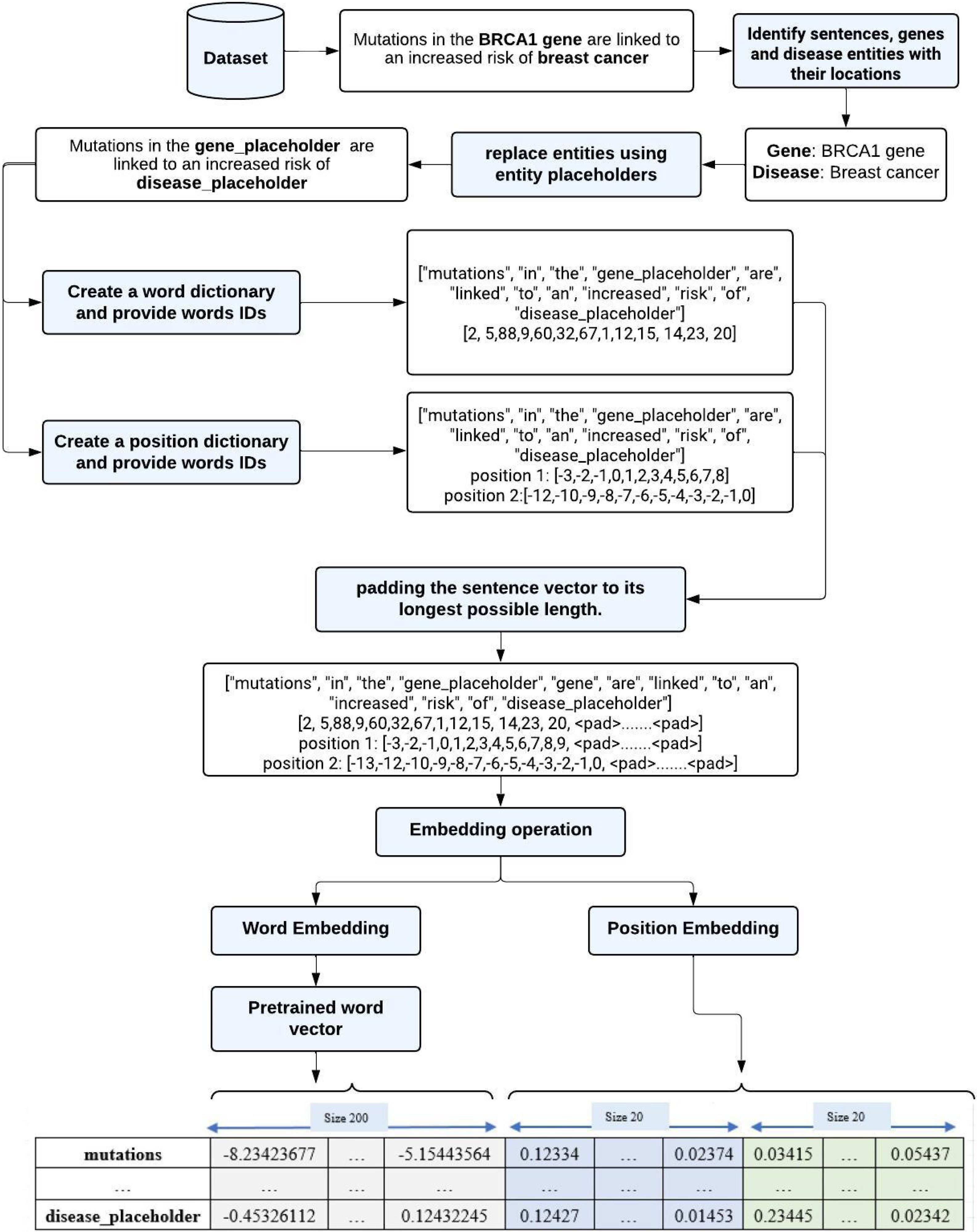
Fig. 2.
The workflow of the preprocessing phase for a sample sentence.
.
The workflow of the preprocessing phase for a sample sentence.
Entity identification and preparation
The essential entities within a dataset are identified: the sentence, the disease and gene names, their respective locations, and association labels. The names of diseases and genes in each sentence are then replaced with placeholders to ensure the task's generality. Subsequently, each sentence goes through processing procedures to improve its analyzability. These steps involve converting the text to lowercase, removing punctuation marks, replacing "/" characters with spaces, and tokenizing the sentence to generate a list of individual words. For convenience and standardization, a dictionary was created by leveraging pre-existing vocabularies and assigning a unique identifier to each word (e.g., '351' was assigned to the word 'humans'). Following this, each sentence vector was padded to the maximum length using a special token represented as < PAD > . This maximum length, determined empirically through dataset analysis, was set to 100. Padding sentence vectors to the same length are necessary to ensure uniform input lengths, facilitating efficient processing and compatibility with models requiring fixed-size inputs. Finally, the labels associated with each sentence are mapped to numeric IDs. In the EU-ADR dataset, false and negative associations were merged into a single negative class (label 0), while positive and speculative associations were incorporated into a positive class (label 1). In the SNPPhenA dataset, positive and neutral associations were merged and assigned as the positive class (label 1), while negative associations were labeled as 0. In the GAD dataset, the positive, negative, and no semantic associations were assigned three distinct labels: 0, 1, and 2, respectively. Subsequently, these mapped labels undergo conversion into one-hot encoded.
Word embedding
Word embedding is a method for representing words as dense vectors of actual numerical values in a continuous vector space. Recently, word embedding in deep learning models has demonstrated outstanding effectiveness in capturing semantic associations, allowing dimensionality reduction, and enabling transfer learning, enhancing various natural language processing tasks. To speed up training and improve outcomes, two popular pre-trained word embedding models were used: the word2vec model, trained on PubMed and PMC datasets, and the fastText model, trained on a large corpus of text data gathered through web crawling. While general-purpose embeddings such as word2vec and fastText may have limitations in capturing domain-specific nuances of biomedical texts, our use of Entity Relation (ER) preprocessing mitigates these limitations. By replacing medical terms (e.g., gene and disease names) with general placeholders, the biomedical text becomes more standardized and comparable to general text, allowing word2vec and fastText to capture relationships between entities effectively. Nonetheless, domain-specific embeddings like BioBERT and PubMedBERT remain promising for capturing complex semantic relationships inherent in biomedical literature and will be considered for future work. The word2vec model has been applied to the EU-ADR and GAD datasets, while the fastText model was applied to the SNPs Dataset to embed words. During the embedding process, each word is fed through a pre-trained model that weights the words depending on their context and relationships learned during training. These weights are expressed as vectors, and each word is assigned a 200-dimensional embedding.
Position embedding
Position embedding is a method employed in NLP tasks to present information regarding the order or position of words in a sentence. The relative distance between words and target entities is an important component in determining the association between entities. Therefore, to capture this positional information, we trained a position embedding model on our training dataset, with each relative position embedding having a dimension of 20. This form of embedding allows the neural network to keep track of the distance of each word to a gene or disease entity. For example, in the sentence “Mutations in the BRCA1 gene are linked to an increased risk of breast cancer”; the relative distance from “breast cancer” to the gene “BRCA1 gene” is 8. Finally, we merged all word and position vectors into a unified vector to create the final representation of each sentence, which is then passed as inputs to the deep learning models.
Classification phase
Three hybrid deep learning models (CNN-LSTM, CNN-GRU, and CNN-GRU-LSTM) were designed to extract gene-disease associations during the classification phase. These architectures were chosen to combine the benefits of convolutional and recurrent models. CNNs are excellent at capturing local n-gram characteristics and short-range dependencies inside sentences, essential for recognizing patterns, including gene and disease names. Meanwhile, recurrent neural networks (RNNs) can simulate sequential information and long-distance dependencies, which are required for comprehending complex biomedical assertions. GRUs were used for the CNN-GRU model because they are computationally simpler and train faster than LSTMs while maintaining high performance. LSTMs, which are employed in the CNN-LSTM model, were chosen for their greater capacity to model long-term dependencies, which is especially relevant in biological phrases that frequently span extensive semantic links between clauses. The hybrid CNN-GRU-LSTM model was created to combine the strengths of GRUs and LSTMs and improve performance on texts that display short- and long-range dependencies. Furthermore, attention methods were built into each model to allow for dynamic weighting of words within sentences. This allowed the models to focus more on essential sections of the input and improved the extraction of relevant gene-disease connections. Prior research findings demonstrated that hybrid architectures and attention significantly improve relation extraction tasks, particularly in complex biological domains, influencing these design decisions. Although the models were not explicitly designed with dedicated negation and speculation detection modules, combining pre-trained embedding and attention mechanisms helps capture some contextual clues related to negated or uncertain expressions.
Models architectures
Three hybrid neural network architectures were developed for gene-disease association classification: CNN-LSTM, CNN-GRU, and CNN-GRU-LSTM. The model architecture consists of one embedding layer, convolutional layers (three layers with the GAD dataset and two layers with the EUADR and SNPPhenA datasets), a bidirectional RNN layer (LSTM or GRU), and a fully connected layer, as shown in Fig. 3. Each model receives preprocessed sentences as input to start the model training process, where the training process is done by adjusting the connected model weights to minimize the error value between the target and the estimated value of the model. The binary cross-entropy loss function was used for binary classification to measure the difference between the actual and expected distribution. On the other hand, the cross-entropy loss function was used for multi-class classification to measure the difference between the actual and expected distributions. The training phase of the models consists of two stages: the feed-forward stage and the backpropagation stage. The feed-forward phase of the models includes the following steps:
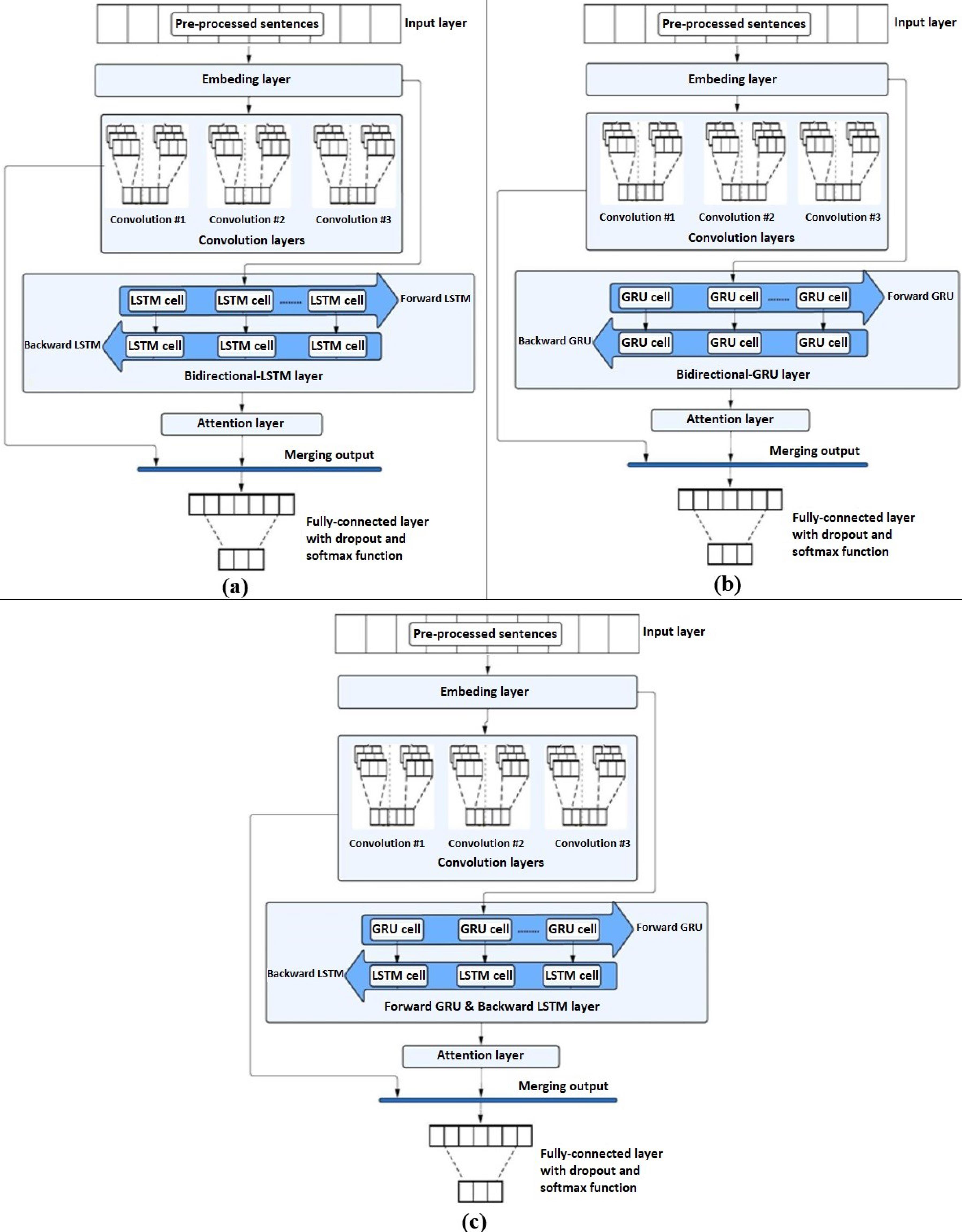
Fig. 3.
The architecture of deep models for gene-disease classification. (a) The CNN-LSTM model. (b) The CNN-GRU model. (c) The CNN-GRU-LSTM model.
.
The architecture of deep models for gene-disease classification. (a) The CNN-LSTM model. (b) The CNN-GRU model. (c) The CNN-GRU-LSTM model.
Step (1): Initially, the embedding layer receives an input layer consisting of sequences of integer-encoded sentences from the pre-processed dataset, where it converts sequences into dense vectors of fixed dimension size to capture semantic meanings and contextual relationships inherent in the sentences.
Step (2): The embedded sequences are then passed into two different paths, described in the following:
-
In the first path, the embedded sequences are input to a CNN layer with a series of 1-D convolution layers for extracting the local features of every sentence. The model used two convolutional layers for the EU-ADR dataset, with 256 filters in the first Conv1D layer using kernel size 5 and 128 filters in the second Conv1D layer using kernel size 3. In the SNPPhenA dataset, two convolutional layers were used with 256 filters of kernel size 7 in the first Conv1D layer and 128 filters of kernel size 5 in the second Conv1D layer. In the GAD dataset, three convolutional layers have been used, with 64 filters in Conv1D layer one of kernel size 5, 128 filters in Conv1D layer two of kernel size 3, and 256 filters in Conv1D layer three of kernel size 3. Each filter was applied with the ReLU activation function to capture different features in sentences and represent them in feature maps. Max pooling and dropout were then employed after each convolutional layer, where max pooling was used for down-sampling to reduce the spatial dimensions of the feature maps. At the same time, dropout prevented overfitting by randomly ignoring neurons during training. The output of the last convolutional layer is subjected to global max pooling to reduce the dimensionality by taking the maximum value in each feature map, resulting in a fixed-size vector regardless of the input sequence length.
-
In the second path, the embedded sequences enter bidirectional RNN layers, with the specific type of RNN layer differing in each of the three models. The CNN-LSTM model feeds the embedded sequences into a bidirectional LSTM layer, which has 100 cells for each forward and backward direction. Each LSTM cell contains input, output, and forget gates, essential for determining information retention. These gates enable the LSTM to adjust cell states based on current and prior inputs, guiding the decision-making process at each time step. The LSTM cells process embedded sequences in both forward and backward directions, thereby capturing contextual information from the beginning to the end and vice versa. In contrast, the CNN-GRU model feeds the embedded sequences to a bidirectional GRU layer with 100 cells. Each GRU cell contains update and reset gates to regulate information flow. Like LSTM, the GRU processes sequences directionally, enabling it to extract contextual information from both directions of the sentence. Finally, the CNN-GRU-LSTM model transfers the embedded sequences to a hybrid RNN layer. This layer consists of a GRU for forward processing and an LSTM for backward processing, with cells set to 100 for both layers. At each time step, these cells make decisions regarding information retention and adjust their internal states in tandem. The hybrid approach enabled the model to develop a more sophisticated comprehension of the contextual information in the sentence by utilizing the capabilities of both GRUs and LSTMs. The outputs from the forward and backward RNN layers were concatenated and subsequently passed through an attention mechanism, enabling the model to focus on the most impactful words for classification automatically. The attention-enhanced feature vectors were then pooled to reduce the sequence dimensionality and retain only the most significant information.
Step (3): The outputs from the two paths are merged into unified vectors and passed to a fully connected layer, which contains two dense layers. The first dense layer performs the ReLU activation function and L2 regularization to combine features from earlier layers. The outputs from the two paths are merged into unified vectors and passed to a fully connected layer, which contains two dense layers. The first dense layer performs the ReLU activation function and L2 regularization to combine features from earlier layers and reduce the dimensionality of the features and overfitting. As for the last dense layer, it will take the previous dense layer's output and perform a softmax activation function to produce the model's final output with two classes (positive and negative) with the EU-ADR & SNPPhenA datasets or three classes (positive, negative, irrelevant) with the GAD dataset. Finally, in the backpropagation stage, the error between the predicted and actual output is calculated, and then, the Adam optimization algorithm is used to update the weight values.
All models' hyperparameter settings were standardized to provide uniform training and equitable assessment, as shown in Table 1. Epochs (70) were chosen based on initial experiments without overfitting, but converged well. A batch size of 32 was selected to balance memory usage against the convergence rate. The dimensionality of embedding was set to 200 for the EU-ADR and SNPPhenA datasets and 300 for the GAD dataset, considering the heterogeneous vocabularies and complexities of the datasets, but a balance between richness in representation and complexity of computation. A drop rate of 0.4 following convolutional layers and dense layers was utilized for preventing overfitting, which is of great importance in biomedical text with relatively smaller amounts of labeled data. The capacity of RNN cells was set to a fixed value of 100 for GRU and LSTM units to allow sufficient model capacity for learning complex dependencies without exposing the model to the risk of extensive models and overfitting. The Adam optimizer with a learning rate of 0.001 was used because it is stable and adaptive learning for different datasets.
Table 1.
The hyper-parameter settings used for the proposed models
|
Hyperparameter
|
Value (All Models)
|
| Number of epochs |
70 |
| Batch size |
32 |
| Training samples size |
284 (EU-ADR), 935 (SNPPhenA), 4264 (GAD) |
| Maximum sentence length |
102 (EU-ADR), 91 (SNPPhenA), 81 (GAD) |
| Embedding dimension |
200 (EU-ADR & SNPPhenA), 300 (GAD) |
| Number of convolutions |
2 (EU-ADR, SNPPhenA), 3 (GAD) |
| Number of filters |
256, 128 (EU-ADR, SNPPhenA); 64, 128, 256 (GAD) |
| Kernel sizes |
(5,3) EU-ADR, (7,5) SNPPhenA, (5,3,3) GAD |
| Max pooling size |
3 |
| Dropout (CNN layers) |
0.4 |
| RNN cells |
100 GRU (CNN-GRU), 100 LSTM (CNN-LSTM), 100 GRU + 100 LSTM (CNN-GRU-LSTM) |
| Optimizer |
Adam |
| Learning rate |
0.001 |
| Bias |
0 |
| L2 regularizer (Dense Layers) |
0.05 |
| Dropout (Dense Layers) |
0.4 |
Attention mechanism
Attention mechanisms in neural networks have demonstrated exceptional efficacy and adaptability across various NLP tasks. Their impact was especially noticeable in fundamental tasks like machine translation,22 sentiment analysis,23 and profiling users on social networks.24 Furthermore, attention mechanisms have been demonstrated to be effective in more complicated applications such as question answering,25 drug repurposing,26 disease classification,27 and thermal prediction.28 In this study, we incorporated an attention mechanism into each of the three proposed models for relation classification using the RNN output (either LSTM or GRU) at the word level. After extracting word-level feature vectors from the embedded sequence inputs in both forward and backward directions, the attention layer assigns different weights to words within each sentence, allowing the model to focus on the most informative parts of the input sequence. This layer generated a vector of weights and was then used to scale the word-level features across each time step. Let H be a matrix of LSTM output vectors [h1, h2, …, hn], where n is the sentence length. A weighted sum of LSTM output vectors composes the representation r of the sentence.
Where
, dw is word embedding size, w is a vector of trainable weights, and wT is a transpose. The dimension of w, α, and r are dw, n, and dw, respectively. The sentence-pair representation for classification is attained by:
Experimental Results
In this section, we evaluated the performance of our models across three separate datasets, using precision, F1-score, and recall metrics to demonstrate their effectiveness in classifying gene-disease associations. To validate our evaluation, we employed a 5-fold cross-validation approach on the GAD and EU-ADR datasets to ensure the maintenance of consistent class distribution across the folds, where the models were trained on four folds and tested on the remaining fold of the dataset. For the EU-ADR dataset, class imbalance was addressed by applying class weight balancing during training to ensure more equitable model performance across different classes. For SNPPhenA, the dataset was already partitioned into training and testing files when it was obtained.
Evaluation matrices
The performance measurement matrix for gene-disease associations (positive or negative) extraction systems comprises four performance measures. These are the Accuracy (Acc), precision (P), recall (R), and F1-score (F). These measures can be used to assess the proposed system's accuracy in extracting association types. The matrix identifies two categories of errors in the results produced by the proposed system, which will assist us in determining the performance level of the underlying implementation of the gene-disease relations extraction model. Type I errors, which are caused by false positives (FP), and type II errors, which are caused by false negatives (FN), can be distinguished by precision and recall, respectively. The F-score is a comprehensive performance measure that considers both the precision and recall scores. The harmonic mean of precision and recall is referred to as the F1-score. Accuracy, on the other hand, measures the overall correctness of the system by considering both true positives (TP) and true negatives (TN) among all evaluated cases. The evaluation metrics are:
Where a correct positive association was counted as TP if the predicted association matched the true association, and an incorrect positive association was counted as FP if it did not match the corresponding true association. Moreover, for negative associations, a correct negative association was counted as a TN, and an incorrect association was counted as an FN.
Evaluations using the EU-ADR dataset
The evaluation of the proposed models on the EU-ADR dataset highlights the strengths and limitations of each architecture, as shown in Table 2. The CNN-GRU-LSTM model achieved the highest accuracy (90.14%), followed by CNN-GRU (88.73%) and CNN-LSTM (85.92%). The superior performance of CNN-GRU-LSTM is attributed to its ability to capture both short- and long-range dependencies effectively, while CNN-GRU offered competitive performance with fewer parameters, reducing the risk of overfitting on the relatively small dataset. All models demonstrated stronger performance on the positive class than the negative class, due to class imbalance, where negative associations were underrepresented. For example, CNN-GRU-LSTM achieved a precision of 92.59% and a recall of 94.34% for the positive class, but only 82.35% precision and 77.78% recall for the negative class. Although class weight balancing was applied to mitigate this issue, it only partially improved minority class performance, suggesting that more advanced techniques like oversampling or synthetic data generation could further address the imbalance. To assess model robustness, additional experiments with noisy and missing data were conducted. Under 10% perturbation, CNN-GRU experienced a 5.3% accuracy drop. In contrast, CNN-GRU-LSTM exhibited greater resilience with only a 3.2% reduction, highlighting the benefit of combining GRU and LSTM units in handling noise and variability.
Table 2.
Performance comparison of deep learning models on different datasets
|
Dataset
|
Model
|
|
Precision (%)
|
Recall (%)
|
F1-score (%)
|
Support (%)
|
Accuracy (%)
|
| EU-ADR |
CNN-GRU-LSTM |
Negative |
82.35 |
77.78 |
80.00 |
18 |
90.14 |
| Positive |
92.59 |
94.34 |
93.46 |
53 |
| Macro avg. |
87.47 |
86.06 |
86.73 |
71 |
| Weighted avg. |
90.00 |
90.14 |
90.05 |
71 |
| CNN-GRU |
Negative |
81.25 |
72.22 |
76.47 |
18 |
88.73 |
| Positive |
90.91 |
94.34 |
92.59 |
53 |
| Macro avg. |
86.08 |
83.28 |
84.53 |
71 |
| Weighted avg. |
88.46 |
88.73 |
88.51 |
71 |
| CNN-LSTM |
Negative |
72.22 |
72.22 |
72.22 |
18 |
85.92 |
| Positive |
90.57 |
90.57 |
90.57 |
53 |
| Macro avg. |
81.39 |
81.39 |
81.39 |
71 |
| Weighted avg. |
85.92 |
85.92 |
85.92 |
71 |
| GAD |
CNN-GRU-LSTM |
Irrelevant |
86.28 |
84.27 |
85.27 |
515 |
84.80 |
| Positive |
81.23 |
84.8 |
82.98 |
342 |
| Negative |
87.38 |
86.12 |
86.75 |
209 |
| Macro avg. |
84.96 |
85.06 |
85.00 |
1066 |
| Weighted avg. |
84.88 |
84.8 |
84.82 |
1066 |
| CNN-GRU |
Irrelevant |
88.58 |
81.36 |
84.82 |
515 |
84.52 |
| Positive |
81.28 |
85.09 |
83.14 |
342 |
| Negative |
81.28 |
91.39 |
86.04 |
209 |
| Macro avg. |
83.72 |
85.94 |
84.67 |
1066 |
| Weighted avg. |
84.81 |
84.52 |
84.52 |
1066 |
| CNN-LSTM |
Irrelevant |
89.01 |
81.75 |
85.22 |
515 |
84.90
|
| Positive |
82.34 |
84.50 |
83.41 |
342 |
| Negative |
80.58 |
93.30 |
86.47 |
209 |
| macro avg. |
83.97 |
86.52 |
85.03 |
1066 |
| Weighted avg. |
85.21 |
84.90 |
84.89 |
1066 |
| SNPPhenA |
CNN-GRU-LSTM |
Negative |
96.10 |
69.16 |
80.43 |
107 |
90.14 |
| Positive |
88.54 |
98.84 |
93.41 |
258 |
| Macro avg. |
92.32 |
84.00 |
86.92 |
365 |
| Weighted avg. |
90.76 |
90.14 |
89.6 |
365 |
| CNN-GRU |
Negative |
93.10 |
75.70 |
83.51 |
107 |
91.23
|
| Positive |
90.65 |
97.67 |
94.03 |
258 |
| Macro avg. |
91.88 |
86.69 |
88.77 |
365 |
| Weighted avg. |
91.37 |
91.23 |
90.94 |
365 |
| CNN-LSTM |
Negative |
94.12 |
74.77 |
83.33 |
107 |
91.23
|
| Positive |
90.36 |
98.06 |
94.05 |
258 |
| Macro avg. |
92.24 |
86.41 |
88.69 |
365 |
| Weighted avg. |
91.46 |
91.23 |
90.91 |
365 |
Evaluations using the GAD dataset
Evaluation of the models on the GAD dataset for multi-class classification of gene-disease associations demonstrated strong and relatively consistent performance across different architectures, as shown in Table 2. The CNN-LSTM model achieved the highest accuracy at 84.90%, followed closely by the CNN-GRU-LSTM model (84.80%) and the CNN-GRU model (84.52%). The superior performance of CNN-LSTM can be attributed to its strong ability to capture long-term dependencies, which are essential in the GAD dataset where complex sentence structures are common. Although the CNN-GRU-LSTM model also performed competitively, its slightly lower accuracy may be due to its increased complexity, which could introduce minor overfitting on large but diverse datasets. Meanwhile, CNN-GRU showed robust generalization across classes due to its efficiency and fewer parameters but slightly underperformed compared to CNN-LSTM in capturing longer contextual dependencies. Overall, the results indicate that all three models could identify gene-disease associations effectively, with CNN-LSTM slightly outperforming others in modeling the nuanced relationships inherent in the GAD dataset.
Evaluations using the SNPPhenA dataset
The performance of the proposed models was evaluated using the SNPPhenA dataset, which is known for its complex structure and diverse association patterns. Among the models, the CNN-GRU achieved an accuracy of 91.23% with a slightly higher Macro average F1-score of 88.77%, indicating a better balance and improved recall for the negative class (75.70%), as shown in Table 2. The CNN-GRU model's superior performance is attributed to the GRU's efficiency in modeling short to medium-range dependencies while avoiding overfitting. This is particularly beneficial given the relatively small and complex nature of the SNPPhenA dataset. The GRU architecture's simplified gating mechanisms enable quicker learning and greater generalization, capturing fundamental relationship patterns with minimal complexity. Similarly, the CNN-LSTM model also attained an accuracy of 91.23% with a Macro average F1-score of 88.69%, although its recall for the negative class was lower (74.77%), suggesting occasional difficulty in identifying true negatives. In contrast, the CNN-GRU-LSTM model recorded the highest Macro average precision (92.32%) but had the lowest recall for the negative class (69.16%), resulting in a Macro average F1-score of 86.92% and a slightly reduced accuracy of 90.14%. While the CNN-GRU-LSTM model excelled in detecting true positives, its weaker performance in capturing true negatives affected its overall balance. Among the three models, the CNN-GRU demonstrated the best trade-off between precision, recall, and F1-score, establishing it as the most effective model for analyzing the SNPPhenA dataset.
Error analysis
Even though the proposed models achieved strong overall performance in classifying gene-disease associations, an analysis of misclassified samples revealed several recurring challenges. Errors were widespread in sentences with complex syntactic structures, especially when there were long-distance dependencies between the mentions of genes and diseases. Furthermore, the models made slightly inaccurate predictions when dealing with speculative statements (such as "Gene X can be held responsible for Disease Y") and negated relationships (such as "Gene X is not related to Disease Y"). Class imbalance was another primary source of error, which was mainly observed in the EU-ADR dataset, where the negative relationships were nowhere near being represented to a satisfactory degree compared to positive relationships. This distortion made it more challenging for the models to make accurate predictions of less frequent classes. These findings suggest that even with the incorporation of attention mechanisms, the models still struggle to detect fine-grained linguistic patterns, subtle semantic cues, and underrepresented class distributions. Subsequent research can overcome these constraints by exploring more advanced architectures, such as transformer-based models, and by adding specialized negation and speculation detection modules to enhance the extraction of complex biomedical relations further.
Performance evaluation compared to previous studies
The performance of the proposed models was compared to that of previous models that addressed the task of gene-disease association classification using the same datasets (EU-ADR, GAD, and SNPPhenA) as shown in Tables S2, S3, and S4. For the EU-ADR dataset, our CNN-GRU-LSTM model outperformed all prior techniques, with an F1-score of 93.46%, as shown in Table 3. Bhasuran et al offered an Ensemble SVM with an F1-score of 85.34%, whereas Nourani et al showed a minor improvement with a CNN-BiLSTM model at 85.80%. Despite using domain-specific pre-trained models, BioBERT-based techniques by Lee et al and Deng et al produced F1-scores of 84.83% and 79.98%, respectively, falling short of our model's performance. The CNN-GRU-LSTM model's ability to efficiently combine convolutional and sequential operations for feature extraction and dependency modeling enables it to handle the dataset's complexity more effectively than previous methods, setting a new benchmark for EU-ADR classification.
Table 3.
Evaluation results on the EU-ADR, GAD, and SNPPhenA datasets
|
Dataset
|
Study
|
Method
|
Precision (%)
|
Recall (%)
|
F1-Score (%)
|
| EU-ADR |
Bhasuran et al8 |
EnsembleSVM |
76.43 |
98.01 |
85.34 |
| Nourani et al13 |
CNN-BiLSTM |
78.10 |
97.0 |
85.80 |
| Lee et al17 |
BioBERT |
80.92 |
90.81 |
84.83 |
| Deng et al18 |
BioBERT |
75.03 |
76.17 |
79.98 |
| Proposed method |
CNN-GRU-LSTM |
92.59
|
94.34
|
93.46
|
| GAD |
Nourani et al13 |
CNN-BiLSTM |
71.62 |
72.64 |
71.63 |
| Lee et al17 |
BioBERT |
75.95 |
88.08
|
81.52 |
| Proposed method |
CNN-LSTM |
83.97
|
86.52 |
85.03
|
| SNPPhenA |
Bokharaeian et al9 |
NNB |
75.40 |
79.60 |
75.60 |
| Nourani et al13 |
CNN-BiLSTM |
75.99 |
74.69 |
73.97 |
| Bokharaeian et al15 |
PubMedBERT–LSTM |
84.00 |
84.40 |
83.90 |
| Proposed method |
CNN-GRU |
91.88
|
86.69
|
88.77
|
On the GAD dataset, the CNN-LSTM model achieved an F1-score of 85.03%, as shown in Table 3. In terms of capturing the complex characteristics of the dataset, our model outperforms the CNN-BiLSTM model of Nourani et al, which achieved an F1-score of 71.63%. Similarly, the BioBERT-based model of Lee et al achieved an F1-score of 81.52%, somewhat lower than our CNN-LSTM model. The comparison indicates that our hybrid CNN-LSTM architecture is more successful than previous techniques in handling the problems of the GAD dataset.
For the SNPPhenA dataset, our CNN-GRU model outperformed previous techniques with an F1-score of 88.77%, as shown in Table 3. The negation-neutral-based technique (NNB) by Bokharaeian et al produced an F1-score of 75.60%, while the CNN-BiLSTM model by Nourani et al scored 73.97%, despite the dataset's intrinsic problems. More recently, Bokharaeian et al used PubMedBERT-LSTM and produced an F1-score of 83.90%, which demonstrated improvements but fell short of our suggested model. Our model's use of convolutional and GRU layers allows it to effectively capture spatial and temporal dependencies within the data, resulting in higher performance.
Discussion
This study examined the reliability of hybrid deep learning-based models in classifying gene-disease associations from biomedical literature, augmented by attention mechanisms. By utilizing three distinct publicly accessible datasets (EU-ADR, GAD, and SNPPhenA), the models demonstrated their ability to uncover significant associations in extensive text collections. A comprehensive evaluation was conducted on the proposed models, which include CNN-LSTM, CNN-GRU, and CNN-GRU-LSTM. The CNN-GRU model demonstrated outstanding performance on the SNPPhenA dataset, achieving an accuracy of 91.23%. Likewise, the CNN-GRU-LSTM model showed strong results on the EU-ADR dataset, with an accuracy of 90.14%.
Meanwhile, the CNN-LSTM model achieved an accuracy of 84.90% on the GAD dataset. These results indicate that the hybrid approach, which combines convolutional neural networks with recurrent neural networks (specifically the integration of LSTM and GRU layers), effectively captures subtle and contextual details essential for accurate gene-disease association extraction. Attention mechanisms enhanced model performance by enabling a focus on the most relevant information within the input sequences, helping to overcome challenges posed by imbalanced datasets. Notably, all models faced performance limitations on underrepresented classes, particularly in the EU-ADR dataset, highlighting the need for future methods to handle class imbalance more effectively.
Regarding computational cost, the CNN-GRU model exhibited the fewest parameters among all models, owing to its simpler structure, making it the most computationally efficient in terms of model size. However, the training time differences across the datasets were relatively minor and more influenced by dataset size than model complexity. Specifically, training on the EU-ADR dataset took approximately 1 minute and 10 seconds, while training on the GAD dataset took about 15 minutes, and about 4 minutes for the SNPPhenA dataset. These variations primarily reflect differences in data volume rather than significant computational disparities between models. As for hardware requirements, all training experiments were conducted on a system equipped with an Intel Core i5-12400 CPU and an NVIDIA RTX 3060 Ti GPU, running Windows 10 and using Python version 3.7 within the Anaconda environment. Furthermore, although transformer-based models such as BioBERT and PubMedBERT have achieved remarkable results in biomedical NLP tasks, they were not employed in this study primarily due to their high computational demands, which may not be accessible to all researchers or practitioners. Instead, emphasis was placed on developing lighter hybrid models that balance strong performance with practical computational requirements.
Conclusion
The findings of this study demonstrate that lightweight hybrid deep learning models, when enhanced with attention mechanisms, can effectively extract gene-disease associations from biomedical literature with high accuracy and computational efficiency. Each model showcased strengths depending on the dataset characteristics, with CNN-GRU achieving superior results on SNPPhenA, CNN-GRU-LSTM excelling on EU-ADR, and CNN-LSTM performing best on GAD. Regarding real-world applicability, although the models were initially developed and validated for research applications, their relatively low computational demand and strong predictive performance suggest that, with further tuning and domain-specific validation, they could be adapted for use within clinical decision-support systems or biomedical research pipelines. Future research is encouraged to explore adaptive architectures that dynamically adjust to varying data characteristics, integrate advanced transformer-based models to capture deeper semantic relationships, and develop enhanced preprocessing strategies to improve model robustness and generalizability for clinical deployment. Future work should also focus on explicitly addressing negations and speculative language by integrating specialized detection modules or enhancing attention mechanisms to better distinguish uncertain and negated associations in biomedical texts.
Research Highlights
What is the current knowledge?
-
Rapidly growing repositories of biomedical literature
-
Different techniques for text preprocessing and encoding to be used in machine learning
-
Different deep learning models, including CNN, LSTM, and GRU
What is new here?
-
An efficient model for text-based gene-disease association identification is proposed.
-
Combining deep learning models, including CNN, LSTM, and GRU, yields highly accurate results.
Competing Interests
The authors declare that they have no conflict of interest.
Data Availability Statement
The primary data and materials used in this research are available at https://github.com/NoorFadhil/Deep-GDAE. The GitHub repository includes the full code for preprocessing, model training, and evaluation to ensure complete reproducibility of the experiments. Additionally, the word embeddings used in this study were obtained from publicly available sources on Kaggle, specifically the Word2Vec embeddings and FastText embeddings.
Ethical Approval
The authors declare no ethical issue to be considered.
Supplementary files
Supplementary file 1 contains Tables S1-S5.
(pdf)
References
- Kim J, Kim JJ, Lee H. An analysis of disease-gene relationship from Medline abstracts by DigSee. Sci Rep 2017; 7:40154. doi: 10.1038/srep40154 [Crossref] [ Google Scholar]
- Adnan K, Akbar R. Limitations of information extraction methods and techniques for heterogeneous unstructured big data. Int J Eng Bus Manag 2019; 11:1847979019890771. doi: 10.1177/1847979019890771 [Crossref] [ Google Scholar]
- Chen L, Xu S, Zhu L, Zhang J, Lei X, Yang G. A deep learning-based method for extracting semantic information from patent documents. Scientometrics 2020; 125:289-312. doi: 10.1007/s11192-020-03634-y [Crossref] [ Google Scholar]
- Callahan TJ, Tripodi IJ, Pielke-Lombardo H, Hunter LE. Knowledge-based biomedical data science. Annu Rev Biomed Data Sci 2020; 3:23-41. doi: 10.1146/annurev-biodatasci-010820-091627 [Crossref] [ Google Scholar]
- Graw S, Chappell K, Washam CL, Gies A, Bird J, Robeson MS 2nd. Multi-omics data integration considerations and study design for biological systems and disease. Mol Omics 2021; 17:170-85. doi: 10.1039/d0mo00041h [Crossref] [ Google Scholar]
- Zhou J, Hong X, Jin P. Information fusion for multi-source material data: progress and challenges. Appl Sci 2019; 9:3473. doi: 10.3390/app9173473 [Crossref] [ Google Scholar]
- Xing R, Luo J, Song T. BioRel: towards large-scale biomedical relation extraction. BMC Bioinformatics 2020; 21:543. doi: 10.1186/s12859-020-03889-5 [Crossref] [ Google Scholar]
- Bhasuran B, Natarajan J. Automatic extraction of gene-disease associations from literature using joint ensemble learning. PLoS One 2018; 13:e0200699. doi: 10.1371/journal.pone.0200699 [Crossref] [ Google Scholar]
- Bokharaeian B, Diaz A. Automatic Extraction of Ranked SNP-Phenotype Associations from Literature through Detecting Neural Candidates, Negation and Modality Markers. ArXiv [Preprint]. December 2, 2020. Available from: https://arxiv.org/abs/2012.00902.
- Wang H, Wang X, Yu Z, Zhang W. Graph embedding and ensemble learning for predicting gene-disease associations. Int J Data Min Bioinform 2020; 23:360-79. doi: 10.1504/ijdmb.2020.108704 [Crossref] [ Google Scholar]
- Xiang J, Meng X, Zhao Y, Wu FX, Li M. HyMM: hybrid method for disease-gene prediction by integrating multiscale module structure. Brief Bioinform 2022; 23:bbac072. doi: 10.1093/bib/bbac072 [Crossref] [ Google Scholar]
-
Wu Y, Luo R, Leung HC, Ting HF, Lam TW. RENET: a deep learning approach for extracting gene-disease associations from literature. In: Cowen L, ed. Research in Computational Molecular Biology. Cham: Springer; 2019. p. 272-84. doi: 10.1007/978-3-030-17083-7_17.
- Nourani E, Reshadat V. Association extraction from biomedical literature based on representation and transfer learning. J Theor Biol 2020; 488:110112. doi: 10.1016/j.jtbi.2019.110112 [Crossref] [ Google Scholar]
- Su J, Wu Y, Ting HF, Lam TW, Luo R. RENET2: high-performance full-text gene-disease relation extraction with iterative training data expansion. NAR GenomBioinform 2021; 3:lqab062. doi: 10.1093/nargab/lqab062 [Crossref] [ Google Scholar]
- Bokharaeian B, Dehghani M, Diaz A. Automatic extraction of ranked SNP-phenotype associations from text using a BERT-LSTM-based method. BMC Bioinformatics 2023; 24:144. doi: 10.1186/s12859-023-05236-w [Crossref] [ Google Scholar]
-
Dehghani M, Bokharaeian B, Yazdanparast Z. BioBERT-based SNP-trait associations extraction from biomedical literature. In: 2023 13th International Conference on Computer and Knowledge Engineering (ICCKE). Mashhad, Iran: IEEE; 2023. p. 216-21. doi: 10.1109/iccke60553.2023.10326231.
- Lee J, Yoon W, Kim S, Kim D, Kim S, So CH. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 2020; 36:1234-40. doi: 10.1093/bioinformatics/btz682 [Crossref] [ Google Scholar]
- Deng C, Zou J, Deng J, Bai M. Extraction of gene-disease association from literature using BioBERT. In: The 2nd International Conference on Computing and Data Science. Association for Computing Machinery; 2021.
- van Mulligen EM, Fourrier-Reglat A, Gurwitz D, Molokhia M, Nieto A, Trifiro G. The EU-ADR corpus: annotated drugs, diseases, targets, and their relationships. J Biomed Inform 2012; 45:879-84. doi: 10.1016/j.jbi.2012.04.004 [Crossref] [ Google Scholar]
- Becker KG, Barnes KC, Bright TJ, Wang SA. The genetic association database. Nat Genet 2004; 36:431-2. doi: 10.1038/ng0504-431 [Crossref] [ Google Scholar]
- Bokharaeian B, Diaz A, Taghizadeh N, Chitsaz H, Chavoshinejad R. SNPPhenA: a corpus for extracting ranked associations of single-nucleotide polymorphisms and phenotypes from literature. J Biomed Semantics 2017; 8:14. doi: 10.1186/s13326-017-0116-2 [Crossref] [ Google Scholar]
-
He W, Wu Y, Li X. Attention mechanism for neural machine translation: a survey. In: 2021 IEEE 5th Information Technology, Networking, Electronic and Automation Control Conference (ITNEC). Xi'an, China: IEEE; 2021. p. 1485-9. doi: 10.1109/itnec52019.2021.9586824.
- Vazan M, Razmara J. Jointly modeling aspect and polarity for aspect-based sentiment analysis in Persian reviews. ArXiv [Preprint]. September 16, 2021. Available from: https://arxiv.org/abs/2109.07680.
- Babaei Giglou H, Rahgooy T, Razmara J, Rahgouy M, Rahgooy Z. Profiling Haters on Twitter using Statistical and Contextualized Embeddings. CLEF (Working Notes); 2021.
- Lu S, Liu M, Yin L, Yin Z, Liu X, Zheng W. The multi-modal fusion in visual question answering: a review of attention mechanisms. PeerJComput Sci 2023; 9:e1400. doi: 10.7717/peerj-cs.1400 [Crossref] [ Google Scholar]
- Amiri R, Razmara J, Parvizpour S, Izadkhah H. A novel efficient drug repurposing framework through drug-disease association data integration using convolutional neural networks. BMC Bioinformatics 2023; 24:442. doi: 10.1186/s12859-023-05572-x [Crossref] [ Google Scholar]
- Guan Q, Huang Y, Zhong Z, Zheng Z, Zheng L, Yang Y. Thorax disease classification with attention guided convolutional neural network. Pattern Recognit Lett 2020; 131:38-45. doi: 10.1016/j.patrec.2019.11.040 [Crossref] [ Google Scholar]
- Tabrizchi H, Razmara J, Mosavi A. Thermal prediction for energy management of clouds using a hybrid model based on CNN and stacking multi-layer bi-directional LSTM. Energy Rep 2023; 9:2253-68. doi: 10.1016/j.egyr.2023.01.032 [Crossref] [ Google Scholar]