BioImpacts. 7(2):115-133.
doi: 10.15171/bi.2017.15
Review
Molecular machineries of pH dysregulation in tumor microenvironment: potential targets for cancer therapy
Mohammad Reza Asgharzadeh 1, 2, Jaleh Barar 3, 4, Mohammad M. Pourseif 3, Morteza Eskandani 3, Mojtaba Jafari Niya 1, 2, Mohammad Reza Mashayekhi 5, Yadollah Omidi 3, 4, *
Author information:
1Department of Biology, Fars Science and Research Branch, Islamic Azad University, Marvdasht, Iran
2Department of Biology, Marvdasht Branch, Islamic Azad University, Marvdasht, Iran
3Research Center for Pharmaceutical Nanotechnology, Biomedicine Institute, Tabriz University of Medical Sciences, Tabriz, Iran
4Department of Pharmaceutics, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
5Department of Genetic, Tabriz Branch, Islamic Azad University, Tabriz, Iran
Abstract
Introduction:
Cancer is an intricate disorder/dysfunction of cells that can be defined as a genetic heterogeneity in human disease. Therefore, it is characterized by several adaptive complex hallmarks. Among them, the pH dysregulation appears as a symbol of aberrant functions within the tumor microenvironment (TME). In comparison with normal tissues, in the solid tumors, we face with an irregular acidification and alkalinization of the extracellular and intracellular fluids.
Methods:
In this study, we comprehensively discussed the most recent reports on the hallmarks of solid tumors to provide deep insights upon the molecular machineries involved in the pH dysregulation of solid tumors and their impacts on the initiation and progression of cancer.
Results:
The dysregulation of pH in solid tumors is fundamentally related to the Warburg effect and hypoxia, leading to expression of a number of molecular machineries, including: NHE1, H+ pump V-ATPase, CA-9, CA-12, MCT-1, GLUT-1. Activation of proton exchangers and transporters (PETs) gives rise to formation of TME. This condition favors the cancer cells to evade from the anoikis and apoptosis, granting them aggressive and metastasis phenotype, as well as resistance to chemotherapy and radiation therapy. This review aimed to discuss the key molecular changes of tumor cells in terms of bio-energetics and cancer metabolism in relation with pH dysregulation. During this phenomenon, the intra- and extracellular metabolites are altered and/or disrupted. Such molecular alterations provide molecular hallmarks for direct targeting of the PETs by potent relevant inhibitors in combination with conventional cancer therapies as ultimate therapy against solid tumors.
Conclusion:
Taken all, along with other treatment strategies, targeting the key molecular machineries related to intra- and extracellular metabolisms within the TME is proposed as a novel strategy to inhibit or block PETs that are involved in the pH dysregulation of solid tumors.
Keywords: Cancer, Carbonic anhydrases, Hypoxia, pH dysregulation, Sodium-hydrogen exchanger, Tumor microenvironment, Vacuolar-type H+-ATPase, Targeted therapy of cancer, Synthetic lethality
Copyright and License Information
© 2017 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Solid tumors, as a class of complex diseases, are characterized by their unique hallmarks, including: anomalous initiation and development through altered genotype and phenotype, self-organization and adaptation, collective specific behavior with molecular networks and irregular pattern formation and dynamism within tumor microenvironment (TME). Such traits result in an uncontrolled process of cell growth, division, metastasis and progression.
The cancer cells, in comparison with normal cells, exhibit distinctive physiopathology, including: (a) autonomous mechanisms of cell growth, (b) divergence from the factors involved in growth inhibition, (c) evasion from anoikis, immunosurveillance and apoptosis, (d) evolutionary regulation of growth, (e) invasiveness and metastatic colonization. These characteristics are manifested through genetic and epigenetic changes.
1,2
The initiation of cancer is deemed to be linked with the hypoxia and irregular metabolism in energetic pathway, in particular glucose, resulting in production of acidic byproducts. Such dysregulation of pH within the cancer cells and TME, in comparison with the normal cells, seems to be one of the main causes for the metastasis, drug resistance, and the recurrence of the disease after the course(s) of treatment. The normal cells have intracellular pH (pHi) of ~ 7.2, but in cancer cells, it is about 7.4. Further, in the normal cells extracellular pH (pHe) is approximately 7.4, while in cancer cells it reduces to 6.7-7.1.
3
Therefore, any changes in the activity and/or expression of membrane ion pumps and proton transfers may lead to a lower pHe and higher pHi. All these occur because the cancer cells can significantly increase the functional expression of several key molecular machineries, including: Na+–H+ exchanger (NHE1), H+-ATPases, carbonic anhydrase IX (CA IX) and carbonic anhydrase XII (CA XII).
1,4-7
Based on scientific praxis, a number of theories have been coined to explain the unique behavior of cancer cells. Some of studies have shown that the hypoxia, Warburg effect and aberrant metabolism play central roles in the pH dysregulation and also progression and metastasis of cancer cells.
8-10
In 1880s, Steven Paget articulated the "seed and soil" notion. Based on this theory, environment (soil) is necessary for the tumor cells (seed).
11,12
In this concept, however, there still remain some key questions. How exactly cancer cells can escape from the immune system and apoptosis? What are the main causes of their resistance to chemotherapy? In fact, answering all these questions can improve the missing link(s) between the actual mechanisms of cancer progression and pharmacotherapy strategies. The right answers to these questions may be entombed within the TME as a crucial part of the cancer puzzle. In this review, we will discuss the TME and its role in cancer development and the factors involved in the pH dysregulation. Further, the relationship between the pH dysregulation and some key molecular machineries such as NHE1, H+-ATPases, CA9 and CA12 are articulated in terms of their association with invasion, metastasis, and drug resistance. At the end of each section, we will focus on some genetic changes that can be occurred in the TME on the molecular machineries such as NHE1, H+-ATPases, CA9 and CA12.
Tumor microenvironment
Intricate interacting network between the cancerous and non-cancerous cells often lead to formation of a permissive milieu called tumor microenvironment. From pathological perspective, the TME include blood vessels, bone marrow-derived inflammatory cell, signaling molecules, extracellular matrix (ECM) and lymphocytes.
13
As shown in Fig. 1, the cells involved with TME are recognized by various cell-specific molecular markers often expressed on the cell surface.
14
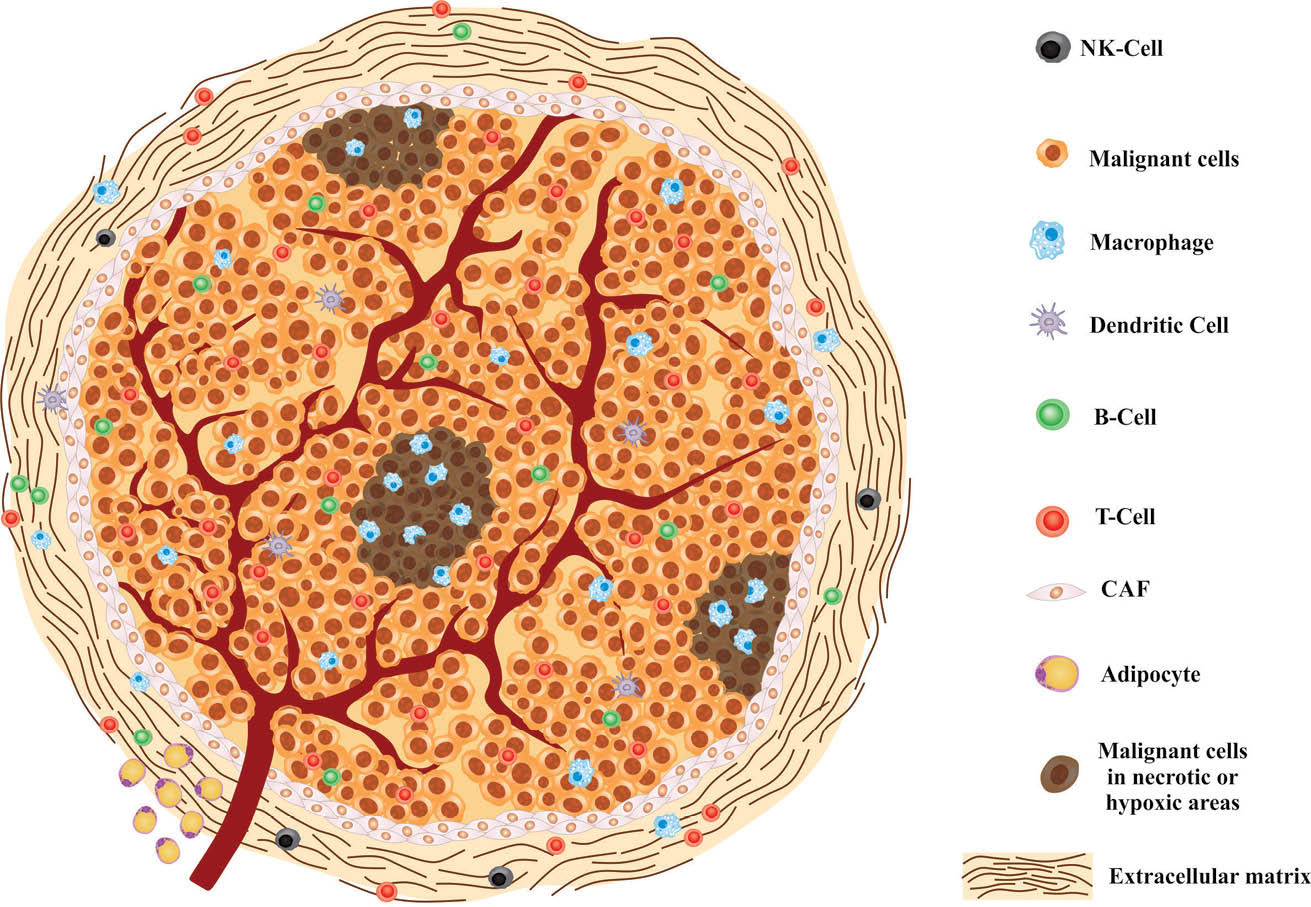
Fig. 1.
Schematic representation of tumor microenvironment. NK-cell: natural killer cell ,CAF: cancer associated-fibroblast.
.
Schematic representation of tumor microenvironment. NK-cell: natural killer cell ,CAF: cancer associated-fibroblast.
Based upon the Warburg effect, tumor cells tend to be anaerobic glycolytic pathway even in the presence of oxygen, and hence produce lactate. This is important in the acidic TME, as well as contribution to the homeostasis and the immune defense system. From a holistic point of view, lactate production may contribute in several functions, including: (a) evasion of cancer cells from immunosurveillance of the immune system, (b) alteration of the metabolism processes in the stromal cells function such as T cells, (c) increase of the inflammation mediated IL-17 production by T cells and macrophages, (d) inhibition of the activity of dendritic cells (DCs), (e) enhanced mobility of tumor cells, (f) prevention of the monocyte migration and cytokine extrication, and (g) initiation of angiogenesis and tumor vascularization by induction of factors such as IL-8, NF-κB and VEGF/VEGF-A via HIF-1.
15
Tumor microenvironment facilitates tumor invasion
Metastasis of cancer cells is a complex process that is associated with key serial events, including: (i) epithelial -mesenchymal transition (EMT), (ii) excitation of angiogenesis, (iii) invasion of cancer cells from the basal membrane into the lymphatic or blood vessel, (iv) extravasation of cancer cells from the blood or lymphatic vessel ,and finally (v) formation of new growing colonies in a far-away site.
1
Inflammation appears to be a main factor in tumorigenesis. Surprisingly, the markers inflammatory mediators that promote cancer development are cancer fighting T cells, DCs, macrophages, natural killer (NK) cells and tumor-infiltrating cells.
8,16,17
Previous studies have shown that the high level of cytokines such as interleukins (Ils), in particular Il-1, Il-6 and Il-8, are correlated with tumor progression, invasion, mobility and metastasis. Tumor-infiltrated immune cells produce several cytokines in favor of MET, including: TGF-β, TNF-α and Il-1B.
18
Cancer-associated fibroblasts (CAFs) are part of TME, that is the cause of tumor progression through production of Il-6.
19
Several cell survival signaling pathways mediated through Il-6 have so far been identified. Some of these pathways include PI3K/AKT, Ras/Raf/MEK-ERK1/2, JAK/STAT3, SHP2/RAFTK, and Src-family tyrosine kinase pathways.
20
Further, the TME is composed of various types of cells whose interactions with TGF- β appear to be diverse. This complex environment is created during cancer initiation, progression and metastasis within the TME. The TGF-β can inhibit the angiogenesis in the early-stage of cancer, but not very effective at the cancer progression stages. Efficiency of TGF-β based responses is highly dependent on the cell type and potential genetic changes on the TGF-β receptors and/or ligands. The TGF-β signaling pathways are related to activation of downstream targets such as TAK1, MAPK, Ras, Rho A, PP2A and SMAD.
21
It has been found that the TNF-α, not only involved in the MET induction in cancer cells, but also associated with an invasive phenotype of mammary epithelial cells and finally cancer metastasis. In the epithelial cells, TNF-α binds to its receptor with high efficiency. This connection can activate NF-κB transcription factor, and subsequently regulates the expression of some genes involved in MET and invasion. The NF-κB indirectly affects the Snail-like proteins. These proteins act as a key inhibitor in E-cadherin transcription. Besides, the NF-κB increases the expression of hepatocyte growth factor (HGF) ligand/receptor. The HGF secrets from the primary mesenchymal cells, such as cancer associated fibroblasts, and it is involved in planning and regulating cancer cell invasive growth.
22
Metastatic cancer cells are the main cause of cancer death. These can simply occur during the pH dysregulation, and GTPase CDC42 is one of the key requirements for the increase of pHi. Although CDC42 activity is not dependent on the physiological pH changes, several guanine nucleotide exchange factors (GEFs), such as GEF Dbl's Big Sister, are concerned in CDC42 motivation. These proteins are activated and released across the plasma membrane at pHi over 7.2. Moreover, higher pHi and lower pHe act as an enhancer in invasion and metastasis via other mechanisms. For instance, due to the opening of aquaporin-type water channels, which can lead to amoeboid movement, the invadopodia is formed and then induce the protease activities that lead to degradation of ECM by enzymes such as matrix metalloproteinase.
2
Recently Martin and colleagues mathematically predicted that the rate of tumor invasiveness does not increase consistently with pHe decrease. In this work, several parameters were considered such as the interaction between cancer cells, stromal cells, cell death and decomposition processes of ECM. This simulation model represented that the onset of the formation of acidic extracellular milieu, can initially increase the cell invasion and then lead to stromal cells death.
23
As reported previously, the pH dysregulation in TME induces a set of critical functions which can in return trigger progress and invasion of tumor cells. Warburg effect and aberrant glucose metabolism in cancer cells can result in production of protons and the other acidic metabolites that these conditions increase intracellular pH. After this change, the cancer cells use plasma membrane ionic pumps to maintain the intracellular pH. Such pH change is one of the main pathological characteristics of solid tumors that can lead to intracellular and extracellular fluids alkalinization and acidification, respectively.
1
These changes can be considered from several aspects, including:
simple
-
(i) pH changes that can modulate numerous protein regulating complex cell functions,
-
(ii) despite existence of several proteins involved in such phenomena, only a limited number of these proteins could only be used as a pH sensor. Therefore, regulatory proteins are selected based on the physiological changes in pHi and pHe, and
-
(iii) the pH dysregulation, along with the other regulatory mechanisms, can be effectively involved in post-translational protein modifications.
2
An increase in the pHi can result in induction of positive signals for cellular proliferation, and promotion of cellular survival by controlling apoptosis. Cell proliferation is associated with several pH-dependent mechanisms. Intracellular pH higher than 7.2 can stimulate growth factors and transit the cell cycle toward the S phase. This seems to be the main cause for progression of the cell cycle through G2/M phase. In contrast, pHi lower than 7.2 can transit the cells into the G2/M phase by the cyclin-dependent kinase 1-cyclin B. Therefore, pH-dependent regulating activity of CDK1-cyclin B could be a conservative mechanism for entering cellular division into the mitosis and meiosis.
24,25
It should be stated that the acidic pH imposes important structural changes in the pro-apoptotic protein BAX to improve its insertion into the peripheral mitochondrial membrane to make the holes that can enhance the membrane permeability and releases cytochrome-c and various pro-apoptotic proteins into the cytosol. Hereafter, activation of caspase by cytochrome-c in the cytosol is more efficient at a pHi of about 6.8.
24
Tumor hypoxia is important for TME, because the tumor-associated macrophages (TAMs) are part of the TME accumulation in the necrosis and hypoxia segments. As a result, it releases hypoxia-induced chemo-attractants such as endothelin(s), endothelial monocyte activating polypeptide II (EMAP2) and vascular endothelial growth factor (VEGF). TAMs contribute to the migration, invasion and metastasis of malignant cells, in large part through Ils such as Il-10 low and Il-12. Further, adipocytes within the TME provide fatty acids as a fuel source for the cancer cells, and as a result contribute to metastasis.
14
It should be also noted that the non-cellular parts of TME play important regulatory roles in the progression of cancer. These parts include biomolecules involved in the formation of ECM as well as physical and chemical parameters such as oxygen tension, pH, interstitial pressure, and fluid flux.
26,27
The ECM density is an important factor in tumor cells aggressive phenotype. For example, the density of the breast cancer tissue is associated with the phenotype of cancer more than 30%, while BCR1 and BCR2 mutations have only a 5% role in the breast cancer.
28,29
An increase in tissue stiffness appears to enhance cancer development through altering focal adhesions, integrin signaling, Rho/Rho-associated protein kinase (ROCK) pathway activation, and actomyosin- and cytoskeletal-dependent cell contractility.
30
Within the TME, all cellular secretions can be considered as non-cellular entities. The ECM-related biomolecules (e.g., collagens, elastin, proteoglycans, hyaluronan, glycoproteins), degrading enzyme and soluble factors (e.g., cytokines, growth factor and chemokines) are involved in the formation of TME.
31,32
It seems that the overexpression of ECM-degrading enzymes such as matrix metalloproteinases (MMPs) can increase the tumorigenesis. For example, in ovarian cancer, MMP-2 and MMP-9 have been shown to associate with cancer metastasis.
33,34
In addition to MMPs, other ECM proteins, such as "a disintegrin and metalloproteinase" (ADAM), are important in the development of tumors. Of these proteins, ADAM with thrombospondin motifs (ADAMTS) were shown to be involved in the remodeling of ECM in tumourigenesis.
35-37
Further, the lysyl oxidase (LOX) enzymes and transglutaminases are main molecules in tumor progression.
38,39
Integrins are cell membrane adhesion molecules that are highly contributed in the development of tumors. For example, in prostate cancer, the overexpression of metastatic αvβ6 integrin was shown to upregulate MMP-2, which in return can promote cancer cell-mediated program of osteolysis through degradation of matrix.
40
Studies have shown that hyaluronan plays an imperative role in the development of various types of cancers, such as prostate, breast and ovarian cancers. This biomolecule, by activating CD44 and RHAMM (receptor for hyaluronan-mediated motility), was shown to mediate some important signaling pathways to favor the cancer progression, including: NFκB and mitogen-activated protein kinase (MAPK) pathways.
41-43
A group of ECM-related biomolecules, so-called matricellular proteins, were shown to contribute in the modulation of ECM, including: tenascin C (TN-C), SPARC (secreted pro-protein, acidic and rich in cysteine), thrombospondin-1 and−2 and osteopontin.
44,45
Although these biomolecules do not directly involved in the formation of cell construction substances, they often participate in modulation of cell-matrix interactions. Of these, TN-C was shown to play a key role in invasiveness of tumor cells.
46
Now, in fact, there exist compelling evidence upon specific interactions between ECM molecules and cancer cells within the TME in service or disservice of cancer progression.
47,48
Drug resistance related to TME
Emergence of resistance to chemotherapy by cancer cells is major issue in clinical oncology, resulting in failure of treatment and possible relapse of disease. Various factors are involved in attaining such resistance against chemotherapy, immunotherapy and radiotherapy, including: epigenetic changes, genetic mutations, aberrant functions within TME, structural components of the stroma, the inter- and/or cross-communication of cancer cells with each other and other elements of TME, hypoxia condition and tumor cells pH.
49-54
The interaction between tumor cells and their microenvironment is very important in emergence of drug-resistance. It has been shown that insulin-like growth factor I (IGF-I) protects colon cancer cells against several cytotoxic factors.
55
The apoptotic response in small-cell lung cancer chemotherapy was shown by the inhibition of integrins through detrimental impacts on DNA.
52
Under hypoxia tumor condition, hypoxia-inducible factors (HIFs) as a transcription factor was shown to elicit the expression of several genes involved in the survival and angiogenesis of solid tumor.
53,56
Tumor hypoxia conditions can also disrupt the protein foldings in the endoplasmic reticulum,
57
which may favor cancer cells in attaining drug-resistance by targeting elements such astopoisomerase ΙΙ.
58
The traits of TME such as irregularly non-integrated tumor microvasculature, highly pressurized tumor interstitial fluid, and dysregulated pH appear to be involved in drug-resistance properties of cancer cells.
1,59,60
In fact, the penetration and absorption of anticancer drugs (e.g., vinblastine, doxorubicin, vincristine, mitoxantrone, paclitaxel) were shown to be altered in the acidic TME.
61,62
TME-mediated resistance can be initiated by several cells and some key structural components of the stroma, which are not limited to endothelia, fibroblasts, pericytes, neutrophils, macrophages, integrin and collagen.
63,64
Moreover, there exist several other extracellular factors that are involved in drug resistance, including: cytokines, proteases and growth factors.
65,66
While the drug resistance in cancer cells is regulated by various mechanisms, this phenomenon may increase the EMT as a direct effect through an array of molecules derived from TME. This mechanism occurs by activation of intracellular networks linked with epidermal growth factor (EGF)/EGF receptor (EGFR), hepatocyte growth factor/c-met, Wnt/beta-catenin axes and multiple cytokine/chemokine-mediated pathways.
67-69
Besides, solid tumors possess different strategies to utilize cancer stem cells (CSCs) and mesenchymal stem cells (MSCs). For example, breast tumors release monocyte chemotactic protein-1 and prostate tumors secrete CXCL16.
70,71
Likewise, MSCs can release polyunsaturated fatty acids, 12-oxo-5,8,10-heptadecatrienoic acid and hexadeca-4,7,10,13-tetraenoic acid which stimulate the resistance to many chemotherapeutic agents.
72
Recently, studies have shown that cancer-associated fibroblasts (CAFs) are able to secrete the collagen type-2, and hence reduce the absorption of chemotherapy, resulting in inevitable drug resistance.
73
Genetic and epigenetic changes in TME
To date, some de novo information has been obtained by genomics, epigenetics, transcriptomics, proteomics and metabolomics, which can be used in our combat against initiation and development of solid tumors.
74
DNA methylation is a chemical modification in the structure of DNA that can lead to chromatin structural remodeling/transformation, gene availability and expression. In mammals, DNA methylation happens at 70% of cytosine-guanine (CpG) rich regions. Recently, it has been found that DNA methylation profile may involve with initiation of the TME and progression of cancer. DNA methylation can influence the performance of different areas of the DNA such as promoter, silencer, enhancer and non-coding RNAs. Bisulfite sequencing and methyl-CpG-binding domain (MBD, or MethylCap-Seq) can be used to identify the methylated areas of DNA. Several studies have shown that, in the breast cancer, the promoter of some key genes are hypomethylated, including: insulin-like growth factor (IGF), multidrug resistance (MDR1) and metastasis promoting protease genes. By contrast, genes prompters involved in the DNA repair, apoptosis, metastasis, control of proliferation and angiogenesis are hypermethylated in cancer. For example, cyclin-dependent kinase inhibitor 2A (p16), O6-methylguanine DNA methyltransferase (MGMT), MutL homolog 1 (MLH1), retinoic acid receptor beta (RAR-β), Ras association domain-containing protein 1(RASSF1A) and phosphatase and tensin homolog (PTEN) epitomize these elements.
75
Histone modifications and DNA methylation have been observed in CAFs in various types of cancers, including: gastric and pancreatic cancer. It has been found that CYP19 and CXorf12 genes are hypermethylated in breast adipose fibroblasts (BAFs) and CAFs, respectively. Histone H3K27 methylation in breast cancer is associated with aggressive phenotype.
18
Alteration in microRNAs in TME
Micro RNAs are considered as biomarkers of different cancer types. New studies indicated that miRNAs influence the metastasis phenomena, in large part through interactions with different elements of TME. Especially, miR-210 is secreted by metastatic breast cancer cells and transferred to epithelial cells which lead to increase in cells migration and angiogenesis. The miRNAs can be transmitted between cells. For example, miR-223 released by TAMs are activated by IL-4 and transferred to the breast cancer cells, which are able to promote the tumor invasion and metastasis. The transmission mechanisms of miRNAs between cell types seems to be one of the mechanisms involved in metastasis within TME.
76
In the breast cancer, the miR-21 is overexpressed, resulting in upregulation of TGF-β. While the upregulation of miR-31 in the CAFs breast cancer disturbs the ability of CAFs to stimulate the cancer cell invasion and migration, the downregulation of miR-15 and miR-16 in prostate cancer CAFs lead to an enhanced tumor growth and progression. This latter effect seems to be mediated by reduced post-transcriptional modification of Fgf-2, resulting in promotion of metastasis.
19
Thus, microRNAs are considered as small non-coding RNA molecules that can control the inhibition or progression of cancer as well as many other pathogenic diseases. Of these systems, TGF-β interacts with SMADs, and hence, mutations and deletions in these areas are important in cancer. In prostate and colon cancer, 18q21 gene locus which encode SMADs 2 and 4 are most often mutated or deleted. SMAD4 mutations are rare, but can be found in breast cancer. Further, SMAD2 mutation has been recognized in lung, colon and head and neck cancer. Lack of SMAD3 expression in choriocarcinoma cells is associated with the downregulation of tissue inhibitor of metalloproteinases (TIMP-1), which may increase the activity of protein metalloproteinase - important for tumor invasion.
21
Na+/H+exchanger 1 (NHE1)
The pH change in the cancerous cells, in comparison with the normal tissue, seems to be common in most solid tumors. In fact, they show some similar phenotypic properties in terms of pH dysregulation, which includes some common key metabolic changes. Such alterations seem (i) to promote acid-producing pathways, (ii) to activate the oncogenic signals, (iii) to induce and develop the hypoxia, and (iv) to alter the functional expression of some key molecular transporters involved in the regulation of pH. Despite these changes, most of studies have focused on the clinical importance of proton transporters, in particular NHE1 and V-ATPase. Factors that promote the cellular pH or enhance the functional expression of NHE1 leading to an increased pHi and carcinogenicity include viruses (e.g., human papilloma virus), persistent hypoxia and HIF, oncogenes and viral proteins, p53 defect, growth factors, hormones, and chemicals carcinogenic and genomic products.
77
Structure and function of NHE1
Changes in the functional expression of plasma membrane ionic pumps and also transmitters which facilitates the distribution of H+ in the extracellular fluid may result in stabilized increased the pHi and decreases the pHe. One of the plasma membrane ionic pumps with enhanced functional expression is NHE1. The human NHE1 gene is a member of a gene family that is also called solute carrier 9 (SLC9). The NHE gene family is pH regulator of the cytoplasm and intracellular organelles. There are 9 different genes locus (NHE1-9 or /SLC9A1-9) in the human genome for the NHE gene family while some related pseudogenes have also been identified in human genome. These genes encode proteins that have 25 to 75% similarity with each other.
78
The human NHE1 (Fig. 2) is an integral membrane protein and encoded by SLC9A1 gene, which is composed of 815 amino acids, and comprises two domains:
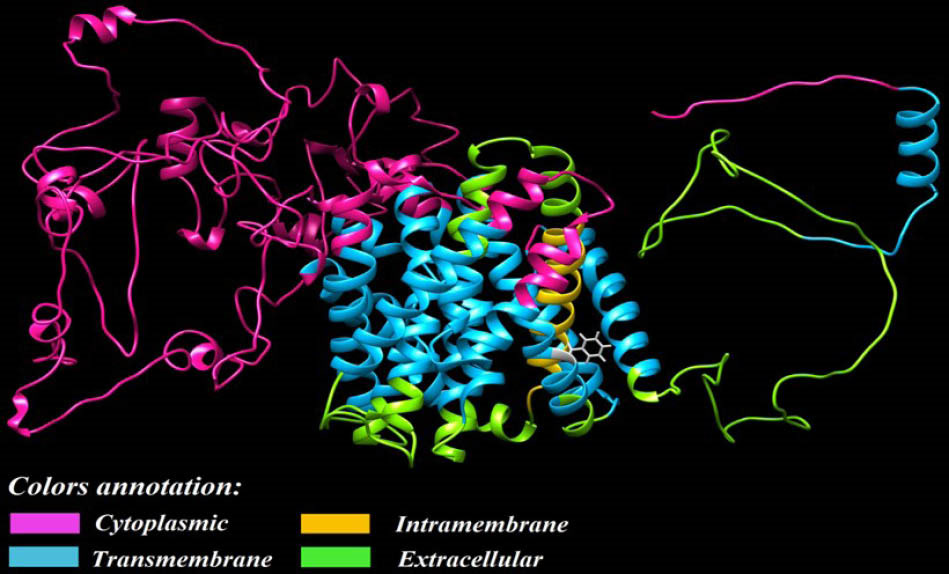
Fig. 2.
Three-dimensional representation of the modeled human
NHE1. The membrane-spanning protein, NHE1, was modeled by
means of Chimera software. Subcellular localization of different
domains of the protein is predicted via Uniprot database
.
Three-dimensional representation of the modeled human
NHE1. The membrane-spanning protein, NHE1, was modeled by
means of Chimera software. Subcellular localization of different
domains of the protein is predicted via Uniprot database
simple
-
(i) a hydrophobic N-terminal membrane domain which contains amino acids 1 to 500 and show a secondary structure with 12 transmembrane spans, to which the NHE1 transport activity is attributed, and
-
(ii) a domain embracing 501 to 815 amino acids constituting the cytoplasmic domain (C-terminus tail) that is fundamental for the NHE1 regulation.
The N-terminal domain of NHE1 exchanges intracellular H+ with extracellular Na+. This molecular exchanger protects cells from the acidification and regulates pHi.
79
Moreover, human NHE1 is involved in some important biological and physiological activities of cells, including: regulation of intracellular pH, acid-base electrolyte control, homeostasis, adhesion, migration and proliferation.
78
Studies have shown that early stage transformation by oncogene(s) in normal cells occurs along with NHE1 activity, and then the cytosolic alkalinization happens in consistent correlation with the occurrence of glycolysis. In addition, it has been shown that such alkalinization stimulates a set of transformation markers such as an increase in the growth rate, substrate-independent growth, and growth factor(s)-independent growth.
The intracellular pH determines that how cells obtain their energy, in which an alkalinized pHi stimulates an anaerobic glycolysis, while an acidic pH provokes an oxidative phosphorylation process. Such molecular storms caused by the activation of NHE1 decrease with its inhibition leading to suppression of tumor progression, in large part by acidifying the intracellular fluid of cancer cells.
77
Further, it should be noted that the tissue distribution and intracellular position of NHEs varry markedly. Recent studies have identified a variety of plasma membrane NHEs and several organelle-based isoforms with similar functions in mammalian physiology. NHE1 exists abundantly in various types of cells, which is found as an extended member. Although it is placed on the cell surface, its functional presences at different domains in various cell types reflects its diverse functions. For example, NHE1 in fibroblasts is localized at the boundary of lamellipodia, the basolateral membrane of epithelia, and the disks and t-tubules of cardiac.
80-82
It is believed that NHE1 performs two important functions. First, it is the chief alkalinizing mechanism in various types of cells to protect cells against the harmful effects of increased acidity. If the production of acidic metabolites increases, the H+-derived from the related metabolic pathways via different secretory routes are intensified and remain unchecked. Hence, to maintain the acid-base balance in the cytoplasm, it plays a central role along with bicarbonate-transporting systems such as, Na+-HCO3- co-transporters, Na+-dependent HCO3-/Cl- exchangers and Cl-/HCO3-exchangers. Second, it provides a channel for the traverse of Na+, which is paired with Cl¯ and absorbs H2O necessary to maintain the cell volume at a steady state level. As a result, the cytoplasmic contents is modulated by a sharp rise in extracellular osmolality. In many specialized secretive cell types, for example the acinar cells of the parotid and sublingual glands, the activation of NHE1 is also necessary for the secretion of derived fluids. Change in the activity of NHEs is related to the pathogenesis of several diseases, such as hypertension, congenital secretive diarrhea, diabetes, and tissue harm affected by ischemia/reperfusion.
78
The phosphorylation alteration of basic amino acids in C-terminal and the reaction with intracellular lipids and proteins can regulate the NHE1 activities (Fig. 3).
83
Transport activity of NHE1 is changed by regulators through altering affinity to intracellular H+, in which it is activated in alkaline pHi.
84
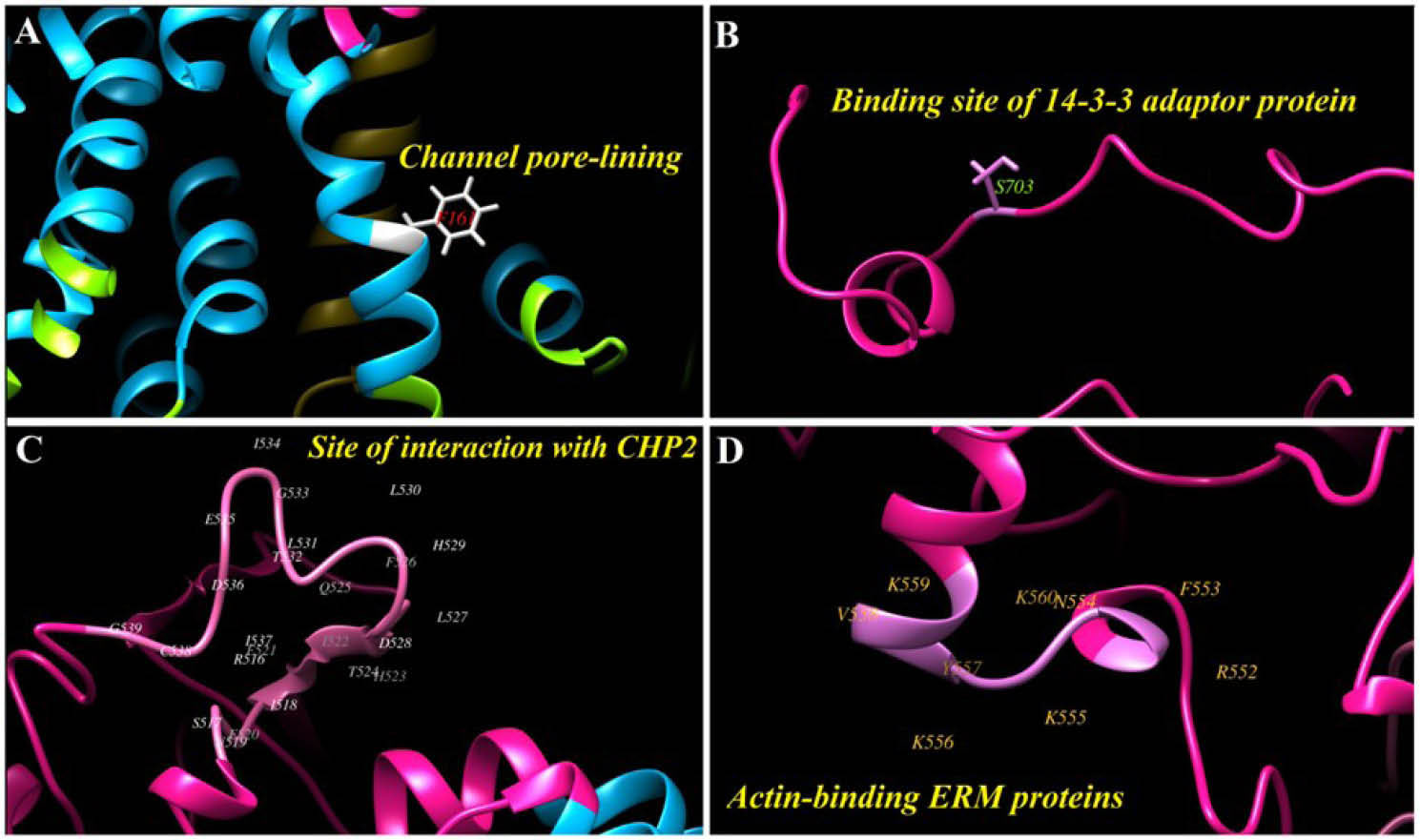
Fig. 3.
3. The main residues at the human NHE1 3D structure. A) Phenylalanine (F) 161 has a main role for covering the channel pore. B)
Serine (S) 703 can be modified to phosphoserine, in this form it is ready for binding to 14-3-3 adaptor protein. C) The site of interaction
between NHE1 and calcineurin homologous protein II (CHP2). D) The actin-binding site between ERM (ezrin, radixin and moesin) proteins
and NHE1.
.
3. The main residues at the human NHE1 3D structure. A) Phenylalanine (F) 161 has a main role for covering the channel pore. B)
Serine (S) 703 can be modified to phosphoserine, in this form it is ready for binding to 14-3-3 adaptor protein. C) The site of interaction
between NHE1 and calcineurin homologous protein II (CHP2). D) The actin-binding site between ERM (ezrin, radixin and moesin) proteins
and NHE1.
The approximate binding sites of regulatory proteins and cofactors which bind to NHE1 in breast cancer cells are shown in Table 1. A number of protein kinases are involved in the regulation of NHE1, protein kinase p160ROCK facilitates the assembly of actin stress fibers through RhoA which leads to the activation of fibroblastic NHE1.
85
Angiotensin II stimulates the NHE1 through ERK phosphorylation, which depends on p38MAPK. Therefore, the phosphorylation of NHE1 at residues T718, S723, S726, and S729 seem to regulate the apoptosis as reported previously for the pro-B cells.
86
Protein kinase B (Akt) plays a key role in phosphorylation of S648, and inhibits the activity of NHE1 in myocardial cells, perhaps by interfering with CaM binding. In other cell types, although Akt phosphorylation leads to the activation of NHE1, and plays a key role in the survival, growth, and perhaps metastasis. In the phosphorylation of NHE, some other protein kinases are involved through various mechanisms such as Ca2+/calmodulin-dependent kinase (CaMKII) and Nck-interacting kinase (NIK) mechanisms.
4
Table 1.
Summarizing NHE1 regulation by proteins, cofactors, and protein kinases
|
Regulators
|
Mechanism of action
|
Biding site(s)
|
Ref.
|
| Calmodulin (CaM) |
Binding to a high- and low-affinity, blocks an auto-inhibitory site, consequently activating NHE1 |
From AAs 636 to 700 |
79
|
| Calcineurin homologous protein (CHP) |
(i) CHP1: NHE1 activity and its stabilization and localization to the plasma membrane
(ii) CHP2: overexpressed in tumor cells; it is protective against serum deprivation–induced cell death by enhancing pHi and promoted proliferation of tumor cells
(iii) CHP3: promote maturation and cell surface stability
|
518 to 537 |
87-89,90
|
phosphatidylinositol 4,5-
biphosphate (PIP2)
|
Depletion of PIP2 results
in ATP-dependent inhibition of NHE1
|
513, 520, 556 and 564 |
91
|
| Carbonic anhydrase II (CAII) |
Catalyzing the production of HCO3- and H+ from the hydration of CO2
|
790 to 802 |
92
|
| Actin-binding ERM |
Proper localization of NHE1 to the plasma membrane and maintaining cell shape |
552 and 560 |
93
|
| Heat shock protein 70 (Hsp70) |
Binding to NHE1 and possible play a role in protein folding |
Unknown site |
94
|
| 14–3-3 adaptor protein |
Binding to NHE1 when it is phosphorylated at S703. Thereby NHE1 has activate by protecting S703 from dephosphorylation
|
703 |
95
|
| Several protein kinases |
NHE1 stimulation and activation |
|
85
|
Studies have shown that the activity of NHE1 in the breast cancer leads to acidification of extracellular microenvironment. This process facilitates the degradation ECM by protease and increases the invasion and metastasis.4 Fibroblast cells with mutated NHE1 show lower pHi as compared to the NHE1 of wild type cells. In such circumstances, the expression of enzymes related to the glycogenesis reduces 3-4 times, which include fructose 1,6 bisphosphatase, galactokinase and phosphorylase kinase. Besides, the concentration of lactate dehydrogenase (LDH) is reduced, which is an enzyme crucial for the Warburg effect.1 There are several key inhibitors and protein kinases that justify the activity of NHE1. These regulators can be considered as interesting molecular target for pharmacotherapy of cancer, (e.g., ErbB2).96 The NHE1 has a critical role in chronic and acute myeloid leukemia differentiation. The simultaneous pharmacological inhibition of P38 MAPK and NHE1 under hypoxia significantly suppresses the expression of C/EBP. These results show the hypoxia-induced K562 differentiation by inhibiting NHE1, in large part due to upregulation of C/EBPα through p38 MAPK signaling pathway. This suggests that the inhibition of NHE1 in hypoxic microenvironment could be considered as possible treatment for the leukaemic disease.
97
NHE1 inhibitor compounds
The progress and maintenance of converting pH gradient is directly caused by the ability of tumor cells in withdrawal of proton. Such proton emission depends on cell buffer size and is stimulated through the transporters and exchangers of the proton attached to the membrane, mainly by NHEs. This gene family has its specific inhibitors.
77
The NHE gene family can be inhibited by various groups of pharmacological compounds, including: amiloride and its analogues, benzyolguanidinium-based formatives, cimetidine, clonidine and harmaline. It might be inhibited by imidazoline or guanidinium groups. The sensitivity of these compounds for NHEs are largely dependent upon the type of NHE, where NHE1 display the greatest sensitivity as compared to others with the following trend (NHE1≥NHE2>NHE5>NHE3>NHE4).
78
Clinical inhibition of NHE1 with potent and much more selective inhibitors should be taken into consideration to attain highly selective and potential targeting in cancer therapy. For example, cariporide, compound 9 T, 2-aminophenoxazine-3-one (Phx-3) could be used.
98
The first inhibitor of NHE was amiloride, which was shown to decrease VEGF that is an activator of urokinase-type plasminogen (μPA), MMPs and other proteases. All these molecular machineries are in favor of activation of the metastasis. In recent years, many researchers have aimed to produce potent and specific compounds to inhibit the activity of NHEs. In this regard, potential amiloride analogues have been made, including: ethylisopropylamiloride (EIPA), hexamethylamiloride (HMA) and dimethylamiloride (DMA).
In addition to amiloride-based compounds, other compounds such as cariporide and eniporide have been used in several experiments to inhibit the activity of NHE1. These tests were not against cancer, but rather in the field of cardiological to ischaemic-reperfusion disservice. Finally, it has been shown that cariporide is beneficial to overcome drug resistance and metastasis process.
77
Studies have shown that cariporide diminishes the hypoxia-dependent tumor invasion and migration in human tongue squamous cell carcinoma and Tca8113 cells by inhibiting NHE1. Also, pharmacological inhibition of p38 mitogen-activated protein kinase (MAPK) specifically inhibits the expression of C/EBPα under hypoxia conditions, of course, which happens after the inhibition of NHE1.
97,99
The genetic variation and diversity
The NHEs have been detected generally in organisms from prokaryotes to eukaryotes such as plants, fungi and animals. In prokaryotes like the Escherichia coli, there are two types of NHEs (NhaA and NhaB) that are employed in H+ gradient produced by H+-ATPase to export Na+ or Li+. In terms of stoichiometry, the NhaA and NhaB transported 1Na+:2H+ and 2Na+:3H+, respectively. Various eukaryotes encoding different types of NHEs, for example, Arabidopsis thaliana contains at least several NHE proteins so-called AtSOS1 or AtNHX1–6 and the nematode Caenorhabditis elegans encodes nine NHE-like proteins such as CeNHX1–9.
78
Wakabayashi et al identified mutations of Tyr454 or Arg458 in transmembrane domain 11 (TM11) that disrupt the plasma membrane domain expression of NHE1 and possibly play a role in protein folding. Their studies showed that mutation of Arg440 in intracellular loop 5 (IL5) could reduce the sensitivity of NHE1 to pHi sensitivity, even though, mutation in Gly amino acid in Gly-rich area could increase its in TM11 domain.100 Genomic analyses of NHE1 display mutation at Phe162 amino acid of human NHE1 that extremely decrease the affinity of M4 domain to Na+. In return, the mutations of Glu350 and Gly356 in TM9 considerably reduce the catalytic efficiency of the transporter without affecting other subsections (Fig. 4).
78
Genetic change in human NHE1 is related to hypertension, which is clearly related to NHE1 phosphorylation that increases the activation of MAPK pathway. It is suggested that the change in protein kinase pathway possibility results in cation homeostasis, hence leading to hypertension.
101
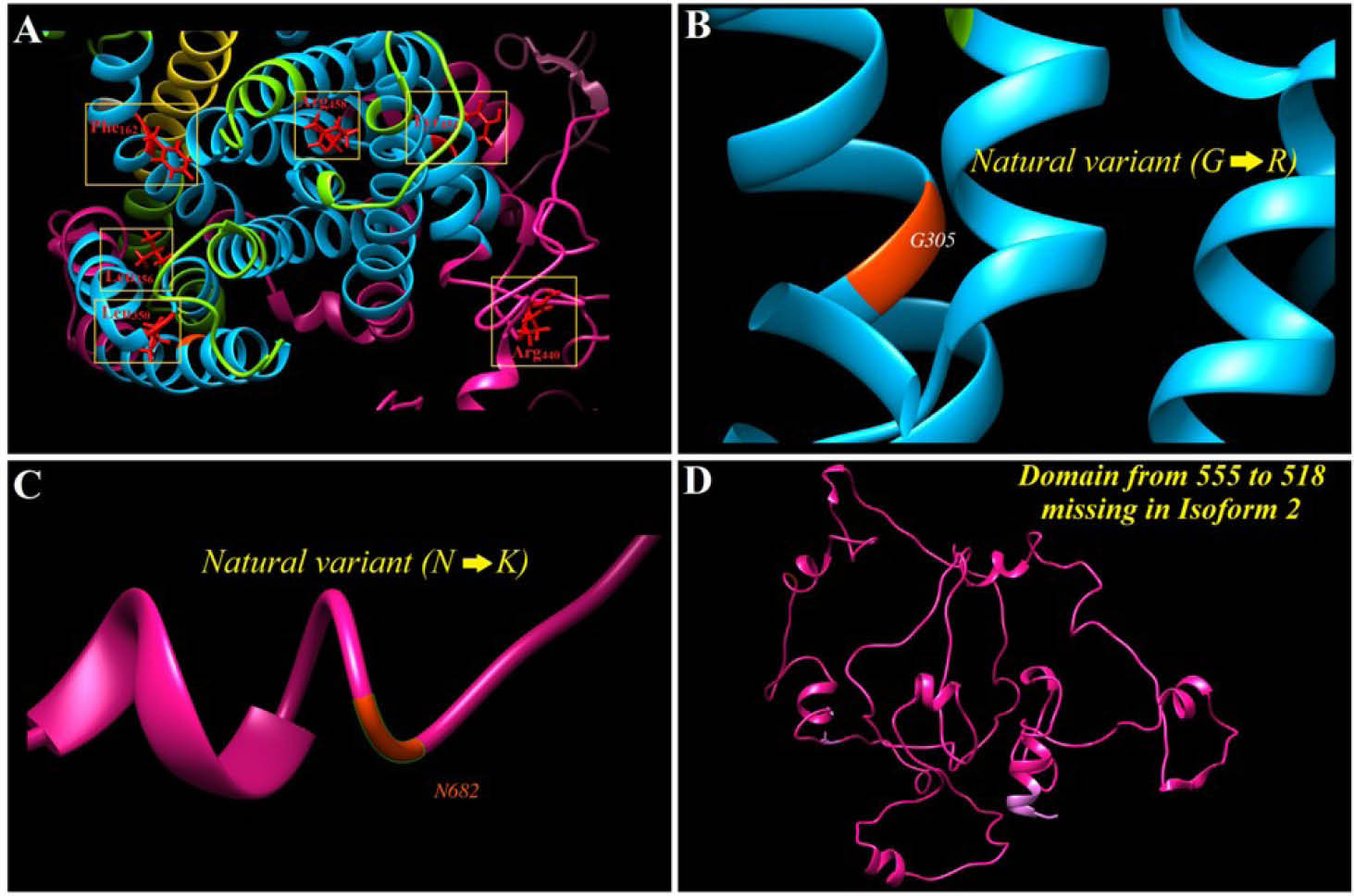
Fig. 4.
NHE1 sequence analysis in terms of probability variant(s). A and B) Some of main natural variants, C) Polymorphism at the position 682 lead to change from asparagine (N) to lysine (K). D) The 261-length domain that is missed in NHE1 isoform 2.
.
NHE1 sequence analysis in terms of probability variant(s). A and B) Some of main natural variants, C) Polymorphism at the position 682 lead to change from asparagine (N) to lysine (K). D) The 261-length domain that is missed in NHE1 isoform 2.
Vacuolar H+ ATPases (V-ATPases)
The V-ATPases are ATP-dependent H+ pumps that show a great variation in the cell membrane. The proteins are composed of several subunits (up to 14), a transmembrane domain is called V0 complex and V1 complex embedded in the cytoplasm. The V-ATPase functions includ pH homeostasis, contribution in endocytosis, participation in activation of proteases and invasion of tumor cells. Furthermore, it is involved in several cellular activities such as angiogenesis, autophagy and also interplay with mTOR for amino acids sensing.
98,102
Regulation and function of the V-ATPase
The V-ATPases have a wide functional expression in the membranes and intracellular organelles (e.g., endosomes, lysosomes and secretory vesicles) of numerous cells such as neutrophils, macrophages and sperm and tumor cells.
103,104
The V0 complex is proton pumpers across the membrane and is formed by a, e, c and c'' subunits. The V1 complex subunits are A, B, C, D, E, F and H, that organized into three subdomains, including:
simple
-
(i) the A3B3 cylinder (subunits A and B), which is involved in the hydrolysis of ATP,
-
(ii) the central stalk (subunits D, F and d) that returns energy from ATP to the complex, because the V-ATPase pumps use a rotary mechanism for the transfer of H+ in the cell membrane, and
-
(iii) the peripheral stalk (subunits E, G, C and H) binds to the A3B3 subdomain and prevents rotation of the stator during ATP hydrolysis.
102
Assembly regulation of V-ATPase domains is related to various stimuli. In yeast, glucose depletion causes rapid and reversible decay of V-ATPase, which may help to protect the cell from ATP reserves. It is suggested assembly of V-ATPase is controlled by pHi, thus an increase in pHi results in the promoted assembly of V-ATPase. In mammalian cells, the V-ATPase pump assembly is stimulated by the activation of phosphoinositide 3-kinase (PI3K) enzyme and mechanistic target of rapamycin complex 1 (mTORC1) in dendritic cells.
102
The trafficking regulation is the second main control system of V-ATPase pump activity. This is very important in regulation of proton secretion in epithelial cells in kidney and epididymis. In this regard, high levels of cAMP and an increase in the activity PKA have a positive effect, while AMP kinase has a negative impact. Such effects occur despite the fact that both positive and negative mechanisms use A-subunit for phosphorylation.
102
Other regulatory systems in mammalian cells can be referred to as heme-binding protein HRG-1 that, without involvement in the assembly of this pump, increases its activity.
105
Transcription factor EB (TFEB) controls the expression of V-ATPase genes, that is under the control of mTORC1.
106
The V-ATPase has an important role in intracellular signaling, especially in wingless/int (Wnt) and Notch pathways. In Wnt signaling, if V-ATPase is prohibited either pharmacologically or genetically, Wnt receptor on the cell surface (i.e., LRP6 receptor) cannot be activated. Further, in Notch signal, performance of the V-ATPase is suitable for the activation of Notch receptor. In Notch pathway, when the ligand binds to a cell surface receptor, the receptor breaks structurally, then Notch intracellular domain (NICD) is released and enters into the nucleus. However, if V-ATPase is inactivated, Notch cleavage through γ-secretase occurs in endosomes, and the liberation of NICD can be extremely reduced.
102
V-ATPase and cancer
In TME, aerobic and anaerobic metabolisms of glucose lead to the accumulation of acid products that can strongly affect the cancer cells and other neighboring cells. As a result, the pH dysregulation in both pHi and pHe within TME can influence cancer cells responses such as their response to cytotoxic drugs. It seems that V-ATPase pumps contribute to the reduction of pHe and a number of studies suggest that V-ATPase pumps play a key role in tumor invasion and cancer cells response to drugs in breast cancer, esophageal carcinoma, lung carcinoma, hepatocellular and pancreatic carcinoma, oral squamous cell carcinoma, sarcoma and other solid tumors.
98
Studies have shown that the inhibition of V-ATPase could increase the pHi in breast cancer cells, and led to the expression of BNIP3 pre-apoptotic protein and cell death.
107
Even short-term inhibition of V-ATPase leads to the activation of cellular stress response and autophagy.
108
In mammalian cells, the V-ATPase has four isoforms displayed as a1–a4. The a1 and a2 isoforms, which are present in the membrane of intracellular organelles, have been diagnosed in synaptic vesicles and endosomes, respectively. The a3 and a4 are located in particular cells membrane such as osteoclast cells and renal intercalated cells, respectively.
109,110
Recently, it has been shown that V-ATPase contributes to aggressive phenotype of breast cancer cells. The V-ATPase inhibition by specific inhibitors (i.e., bafilomycin and concanamycin A) was shown to reduce invasive phenotype of breast cancer MDA-MB231 cells.
111
It was also shown to contribute to the invasion of other types of tumor cells. For instance, in human pancreatic, increased expression of V-ATPase is associated with an increase in cancer degree.
112
Some studies have shown a relationship between rearrangement and arrangement of this pump in plasma membrane and cancer cell invasion.
One limitation on studying this pump is that V-ATPase inhibitors operate specifically and strongly, an hence they prohibit all the intracellular and plasma membrane V-ATPases. This is an important phenomenon because the potential role of intracellular V-ATPase is not considered in the discharge of the invasive factors being mainly growth factors and proteases.
113,114
Although the exact mechanism of V-ATPase activity is not clear in the invasion of cancer cells, it is proposed to contribute to the acidification of pHi that is an important factor in the activation of proteins such as cathepsins and matrix metalloproteases.
115,116
In colon cancer cells, TM9SF4 protein, as a member of the transmembrane-9 superfamily, was shown to interact with V-ATPase. The function of this protein has been reported in adhesion, phagocytosis and innate immunity. Further, it plays an important role in resistance to the 5-fluorouracil (5-FU) drug and invasive phenotype of cancer cells. The TM9SF4 interacts with the V-ATPase most likely through V1H subunit. It was also reported that the TM9SF4 inhibition could lead to a change in the activity of cancer cells in association with V-ATPase activity reduction in protons outflow. As a result of pHi reduction, the extracellular pH and the pH in intracellular vesicles may be increased.
117
It has been proven that V-ATPase increase in the membrane of tumor cells is associated with metastasis. The V-ATPase inhibition by archazolid is related to anoikis pathways in the invasive cancer cells. Probably, archazolid inhibits V-ATPase activity with binding to the c-subunit in the V0 domain. Anoikis is a form of programed cell death which was shown to occur in the cells isolated from the ECM, while metastatic cancer cells are resistant to such phenomenon. The anoikis begins with two pathways, including: (i) extrinsic pathway, where cell death receptors are involved and caspase-8 and Bcl-2 are the main players, and (ii) the intrinsic pathway that starts from mitochondria and is related to cytochrome C.
As mentioned, V-ATPase plays an important role in autophagy, in which the cells detached from ECM stimulate autophagy as a process to evade anoikis. Hence, the V-ATPase inhibition can be effective in stimulating anoikis. Recently, a number of studies have suggested that endolysosomal V-ATPase are attractive targets for inducing anti-tumor and anti-metastasis effects, since its inhibition could prevent the endocytotic traffic of migratory signaling molecules such as Rac1 and EGF receptor.
118
It should be stated that the increased activity of V-ATPase in metastatic cells could lead to an enhanced autophagy, in which process lysosomes play a fundamental role(s). In addition, it has been shown that the inhibition of mTOR signaling pathway reduces autophagy. Accordingly, it can be an alternative mechanism to proton pump inhibitors because V-ATPase is part of a regulation system that modulates mTOR.
98
The expression of E2F1 transcription factor is increased in lung, breast and hepatocellular carcinomas, which is strongly associated with the development of the invasive breast and bladder cancer cells. Accordingly, there have been plenty of evidences that show E2F1 is associated with biological processes such as regulation of cell growth, modulation of autophagy, invasion and metastasis of tumor cells, even though the exact molecular mechanisms are yet to be fully understood. Nathalie et al discovered that E2F1 could contribute to the lysosomal trafficking and mTORC1 signaling, leading to V-ATPase regulation. Their studies showed that E2F1 was able to stimulate the movement of lysosomes in the surrounding cells, and this procedure is necessary for the motivation of mTORC1 which could inhibit autophagy. E2F1 was shown to control the V-ATPase activity through promoting the assembly of V0 and V1 domain in V-ATPase complex. It was shown that the B-subunit of V-ATPase protein could be a suitable target for E2F1 transcription factor, therefore ectopic expression of B-subunit might enhance the activities of V-ATPase and mTORC1.
119
Archazolid treatment or silencing of V-ATPase in human breast cancer SKBR3 cells was shown to inhibit the Rac1 activity, and reduce the metastasis of breast cancer by inhibiting the activity of EGFR and Rho-GTPase Rac1 that is important for cell motility.
120
Further, a correlative expression between homo sapiens longevity assurance (LASS2), Kruppel-like factor 4 (KLF4) and 1-acylglycerol-3-phosphate O-acy ltransferase 9 (AGPAT9) were shown, in which the LASS2 could bind to V-ATPase C-subunit and hence play a role in chemoresistance. The KLF4 is a transcription factor and contributes in the inhibition of tumor formation and oncogenesis. The AGPAT9 is a critical enzyme, which is involved in the conversion of glycerol- 3-phosphate to lysophosphatidic acid in the triacylglycerol synthesis pathway, was shown to specifically inhibit the migration, invasion and metastasis of breast cancer cells. Meanwhile, it has been recently shown that the AGPAT9 functional expression may lead to an increased expression of specific KLF4 gene, perhaps via its direct binding to the promoter region of the LASS2 gene that could induce its expression. Eventually, LASS2 can inhibit the functionality of V-ATPase by binding to its c-subunit.
121
V-ATPase inhibitors
The acidic extracellular environment is one of the main features of solid tumors and the V-ATPase has an important role in this process. This milieu provides the necessary condition for tissue damage, elicits the activation of enzymes involved in degradation of ECM, creates a metastatic cell phenotype and is involved in many other unknown biologic processes. Therefore, the employment of V-ATPase inhibitors can suppress all these events and increase the survival rate of cancer patients. The first V-ATPase inhibitor was bafilomycin that has a microbial origin isolated from Streptomyces griseus. Another inhibitor is concanamycin that has been isolated from the Streptomyces neyagawaensis.
122,123
Archazolid inhibitor is similar to bafilomycin or concanamycin because it is able to attach to the c-subunit of V-ATPases. Furthermore, it has been shown that this compound has an important anti-tumor role both in vitro and in vivo.
120
Archazolid is produced by Cystobacter violaceus and Archangium gephyra.
124
Benzolactone enamides are another group of V-ATPases inhibitors, which are normally isolated from sea sponges, gram-negative bacteria, and ascidians. Key members of the family of V-ATPase inhibitors include salicylihalamide A, apicularen A, lobatamide A, oximidine I, and cruentaren. In addition to the listed cases, new V-ATPases inhibitors continue to be discovered and added to the list, including: NiK12192, PPI SB 242784 and FR202126.
125
The breast tumor cells treated with bafilomycin A1 were shown to significantly lose their migration and metastatic phenotype. These results might be related to the findings that cancer cells with low metastatic potential use HCO3- and NHE1 transporter to regulate the pH, while the high-metastatic cancer cells use of V-ATPase proton pump for this purpose.
126
It was shown that V-ATPase inhibition by bafilomycin or concanamycin could induce apoptosis. In this context, human gastric cancer cells were evaluated and lysosomal pH factor was studied, with results showing some changes could occur in lysosomal pH. Further, it was found that an increase in the existence of bafilomycin might lead to an increase in the activity of caspase-3.
127,128
To date, several related factors between lysosomes and cancer have been identified.
In short, the lysosomes have an acidic miliu which is controlled by V-ATPase, hence it can be considered as a potential therapeutic targets for cancer therapy. The pH in the normal cell lysosomes is about 5. However, in cancer cells, the pH in lysosome may be changed (e.g., 5.8 in MCF-7 cancer cells and 6.4 in promyelocytic leukemia HL-60 cancer cells). The reasons for such alterations could be due to (i) malignancy-related changes or (ii) treatment with drugs (e.g., V-ATPase inhibition with bafilomycin A).
Elipticin dsplays anti-tumor effects, and it is topoisomerase II inhibitor by DNA intercalation. Recently, it has been observed that treatment of the UKF-NB-4 cells with this inhibitor might increase the expression of V-ATPase. Therefore, the V-ATPase activity could result in lysosome alkalinization and resistance of UKF-NB-4ELLI cells to ellipticine-induced apoptosis. In this regard, the V-ATPase inhibition by concanamycin A might reduce the acidity of lysosomes in the PKSV-PR cells.
129
Carbonic anhydrases
The aberrant metabolism of cancer cells may lead to the production of proton and carbon dioxide (CO2), which can be converted into carbonic acids by the carbonic anhydrases (CAs) - a process that occurs due to high consumption of glucose. Many proteins are involved in the metabolism of glucose such as glucose transporters and pH regulatory proteins. Of these, carbonic anhydrase family plays an important role, especially CAIX and CAXII. It should be noted that little change (even 0.1 pH units) in the intracellular and extracellular pH could disrupt the molecular functions of solid tumor cells, including: ATP synthesis, enzyme functionalities, migration, invasiveness and metastasis. Also, different isoforms of extracellular proteins (e.g., fibronectin and tenascin) might be generated by alternative splicing that may not occur in the normal cells.
130
The carbonic anhydrases are zinc metalloproteinase enzymes which can be found among all organisms, including in microbes, fungi, plants, and animals0.13 Five groups of carbonic anhydrases (α, β, γ, δ, and ζ) have evolved independently, among which alpha-carbonic anhydrases were found in the human. There are 16 isoforms of alpha-carbonic anhydrase in primates, 15 isoforms of which are common and also shared in human. They are classified based on their location in the cell, catalytic activity and response to several groups of inhibitors, as follows: (i) some are placed in the cell membrane (CAI, CAII, CAIII, CAVII and CAXIII), (ii) several are attached to the membrane (CAIV, CAIX, CAXII and CAXIV), (iii) two of them are found in the mitochondria (CAVA and CAVB) and (iv) secretory isoforms (CAVI), three of them are catalytic isoforms, so-called CA-related proteins (CARPs) which include CAVIII, CAX and CAXI.
130-132
The carbonic anhydrases perform a vital reaction in cells, that is: “CO2+ + H2O↔HCO3- + H+”. Also, CA isoforms are involved in many process and functions in mammals. Among the most important ones, we can point out the acid-base balance, release of electrolytes, bone resorption, calcification, lipogenesis, gluconeogenesis and ureagenesis in human. Further, this enzyme appears to contribute in photosynthesis and carbon dioxide fixation reaction in bacteria and plants.
132,133
In human, CAs are present in a very wide range of tissues. In fact, they are present almost in most tissues, including: digestive, nervous and genital systems, renal, lungs, skin and others organs.
133
In 1992, Pastoreková et al showed that CAs are associated with cancer. Thus, it became clear that CAIX and CAXII increase simultaneously in many tumor tissues and normal tissue could be related to the cancer development and progression.
134
Sulphonamides, sulphamates and sulphamides are the main compounds for inhibition of carbonic anhydrases, which are able to bind to zinc ion and active site of the enzyme resulting in their inactivation.135 It has been over 50 years that sulphonamides have been used as diuretics and antiglaucoma medicine. Now, this component is proposed as anticonvulsant, anti-obesity, anticancer and anti-pain agent.
136
Various compounds have been designed for the inhibition of carbonic anhydrases, and some of them are mentioned in the following context:
simple
-
(i) Fluorescent sulphonamides are used for imaging purposes and CAs are involved in tumor acidity.
137
-
(ii)
Addition of positive and negative charges to sulphonamide and sulphamate enable them more likely to cross the cell membrane and influence the intracellular CAs such as CAIX and CAXII.
130
-
(iii)
Acetazolamide inhibits all CA isoforms with a high capability. To date, new acetazolamide derivatives have been produced with much better and efficient functional properties than the parent drug acetazolamide.
130
-
(iv)
Sugar-containing sulfonamides-sulfamates-sulfamides are extremely hydrophilic compounds that cannot easily cross the cell membrane. Thus, they influence the extracellular CAs, including CAIX and CAXII.
135
-
(v)
Coumarins, thiocoumarins and polyamines are new chemotypes that have CAs inhibitory properties. These inhibitors bind to CAs active site and do not have interaction with zinc ions of this enzyme.
138
-
(vi)
Protein tyrosine kinase (PTK) inhibitors (e.g., imatinib and nilotinib) are also the CAs inhibitors, however, they are not as effective as sulphonamides and coumarins.
139,140
-
(vii)
Monoclonal antibodies such as M75 and WX-G250 have been used.
141,142
-
(viii)
CA inhibitors coated gold nanoparticles have also been used.
143
Structure and function of CAIX in tumor cells
CAIX seems to be more complex than the other isoforms of CAs. Unlike other isoforms that contain a polypeptide chain with just one domain, CAIX is a multidomain protein (Fig. 5). The CAIX isoform is comprised of (i) a short intracytosolic sector with an unknown role, (ii) a small transmembrane section, (iii) an extracellular catalytic domain, (iv) a proteoglycan like domain that exclusively belongs to CAIX - not seen in other isoforms - and is involved in cell adhesion processes, and (v) a stocky peptide signal.
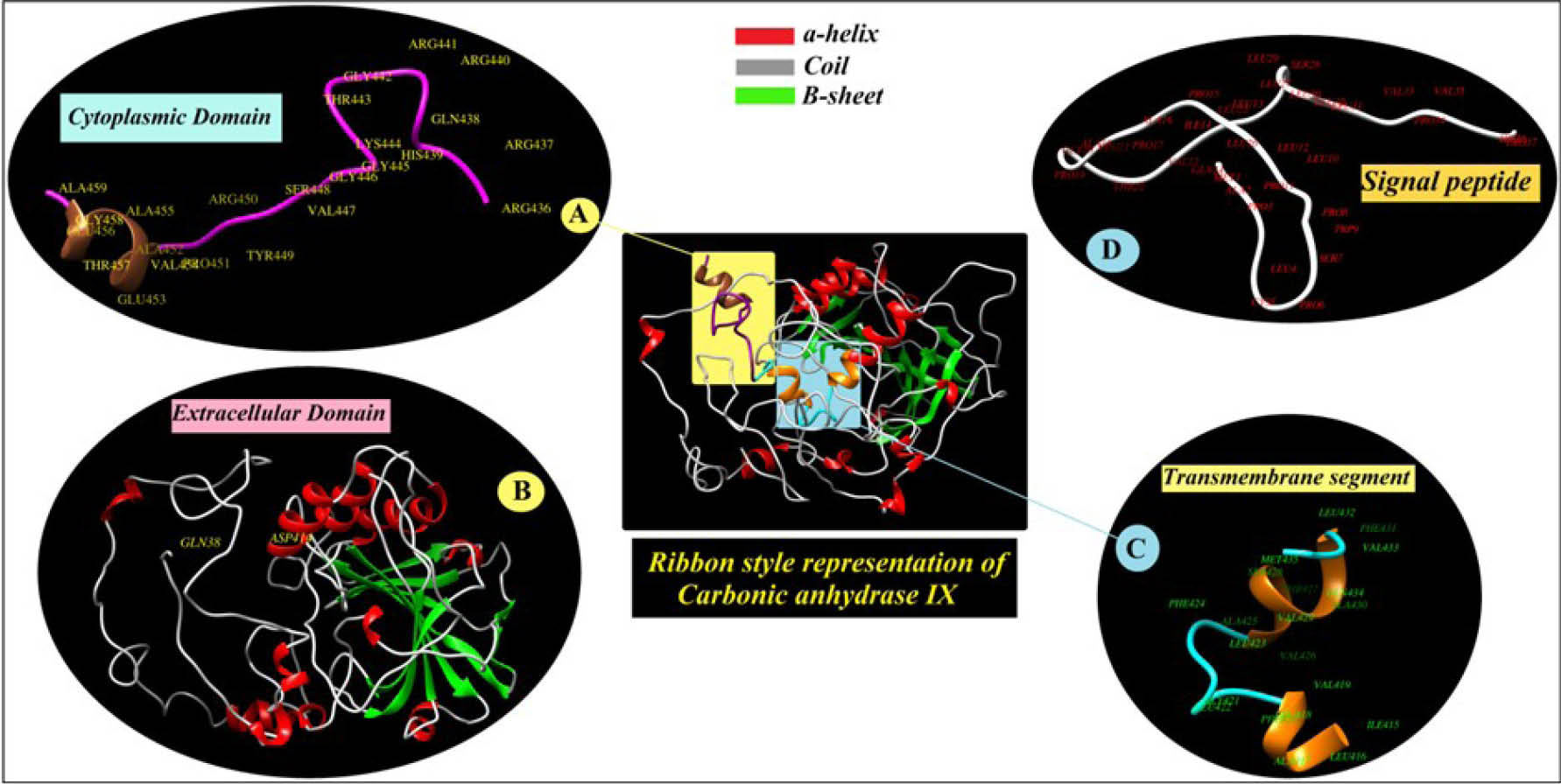
Fig. 5.
Subcellular localization, transmembrane topology and sequence processing of carbonic anhydrase IX (CAIX). A) The residues from
436 to 459 that is localized in cytoplasm. B) The residues from 38 to 414 that known as extracellular domain. C) Transmembrane segment
of the CA9 between amino acids 415 – 435. D) The N-terminal signal peptide at the first 37 residues are denoted as signal peptide.
.
Subcellular localization, transmembrane topology and sequence processing of carbonic anhydrase IX (CAIX). A) The residues from
436 to 459 that is localized in cytoplasm. B) The residues from 38 to 414 that known as extracellular domain. C) Transmembrane segment
of the CA9 between amino acids 415 – 435. D) The N-terminal signal peptide at the first 37 residues are denoted as signal peptide.
CAIX may be seen in the form of a dimmer, in which they may be connected to each other through disulfide bonds. Among CAs protein families, CAIX has a high catalytic activity. The catalytic domain of this protein contains zinc (Zn2+) ion as well as three histidine amino acids and a water molecule (Fig. 6).
131
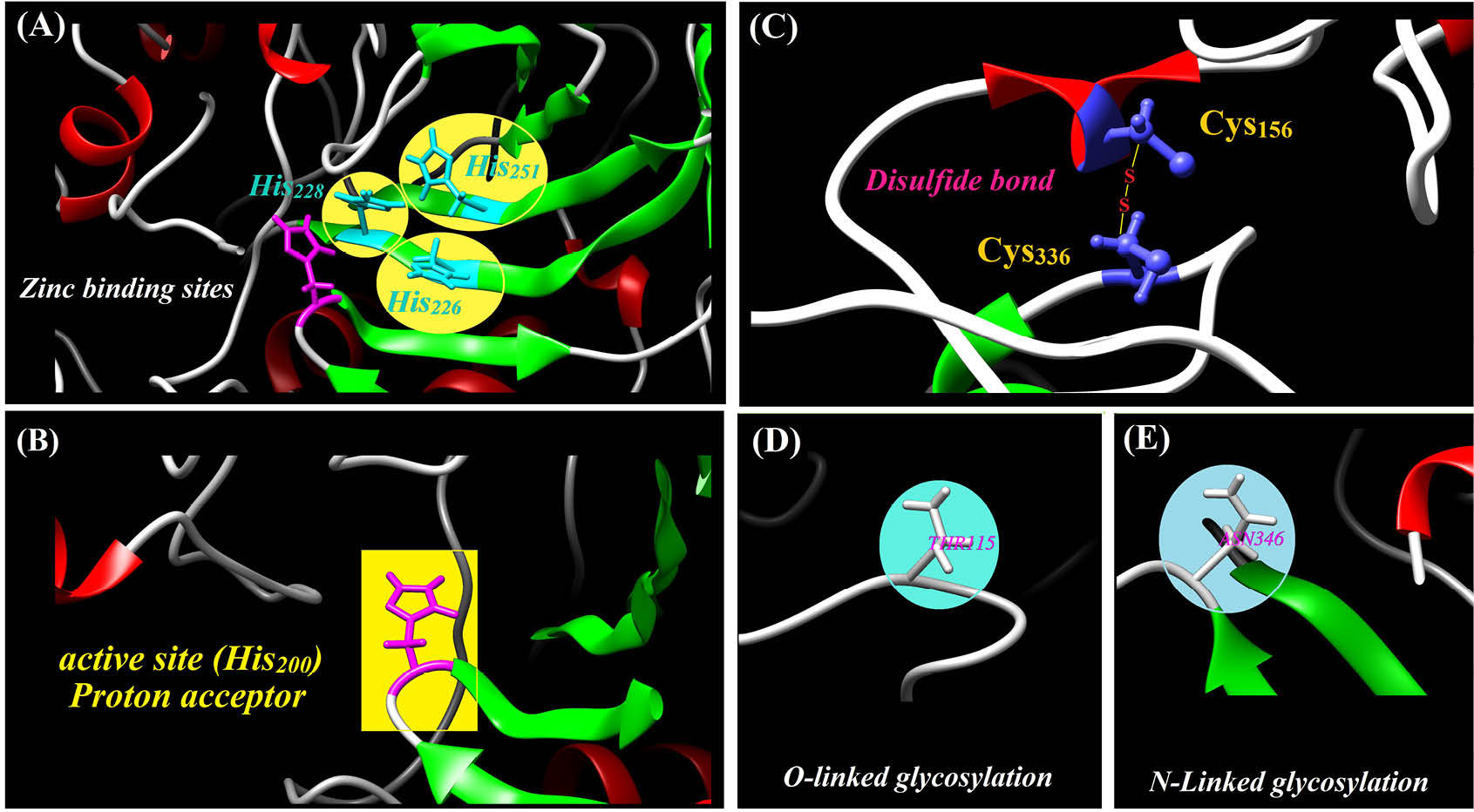
Fig. 6.
Functional sites, disulfide bond and post-translational modifications of the carbonic anhydrase IX. A) Zinc binding to the histidine
226, 228 and 251 require a catalytically activity for the CA9 protein. B) Histidine 200 act as a proton acceptor that is crucial for the enzyme
catalysis activity. C) The positions of cysteine 156 and 336 that are participated in formation of disulfide bond. D and E) the O- and N-linked
glycosylation relating to threonine 115 and asparagine 346
.
Functional sites, disulfide bond and post-translational modifications of the carbonic anhydrase IX. A) Zinc binding to the histidine
226, 228 and 251 require a catalytically activity for the CA9 protein. B) Histidine 200 act as a proton acceptor that is crucial for the enzyme
catalysis activity. C) The positions of cysteine 156 and 336 that are participated in formation of disulfide bond. D and E) the O- and N-linked
glycosylation relating to threonine 115 and asparagine 346
CAIX is highly expressed in a variety of different types of cancers, whose expression is regulated by the transcription factor HIF-1 and is highly inducted in the hypoxic condition, and also associate with the cellular responses to radiotherapy and chemotherapy.
132
The transcription factor HIF-1 consists of two subunits, i.e., alpha (α) and beta (β). In normoxia, the unit α is connected to pVHL (von Hippel–Lindau tumor suppressor protein), and therefore prevents the assembly of two subunits of α and β. However, under hypoxia, pVHL is separated from the unit α and leads to the connection of two α and β subunits to each other, at which point an active form of HIF-1 is created. Such phenomena result in the functional expression of the hypoxia responsive elements (HREs) such as glucose transporters (GLUT-1 and GLUT-3), vascular endothelial growth factor (VEGF) and CAIX.
56,134,144
Of these, CAIX contributes to the acidification of TME. It may also lead to the phenotype of metastasis. Moreover, this isoform is involved in the pH regulation and cellular connection in association with ion exchanger and Na+-HCO3- co-transporters.
145-147
Studies have shown that CAIX expression is under the control of promoter methylation in the kidney cells, which is an epigenetic mechanism.
146
Reports have shown that inactivation of CAIX gene with short hairpin RNA or inhibition of catalytic domain activity by drug could lead to a decrease in xenograft tumor size. Further, CAXII mRNA levels are upregulated in some cancer cells such as LS174Tr and HT29. Overall, suppression of both CAIX and CAXII appears to be the reason for 85% reduction in tumor growth. Intriguingly, CAIX expression in cancer cells could be a compensatory response to reduce the damaging effects of acidosis and hypoxia.
148-150
The use of CAIX inhibitors reduces proliferation of cancer cells and increases apoptosis in hypoxic conditions, sensitizing cancer cells to chemotherapy and radiotherapy.
151
The membrane transporters, pH regulation machineries (e.g., CAIX) and hypoxia in cancer cells are mostly located in invadopodia areas (i.e., membranous actin-rich protrusios of the cells). Using the import mechanism of HCO3- and export mechanism of H+, lactate and CO2 are able to facilitate the cell movement.
152
Special conditions of solid tumors (i.e., low pHe, hypoxia and its molecular responses) allow cancer cells to show stem cell-like phenotype.
153,154
In esophageal cancer model, it was found that the expression of CAIX and some other genes (i.e., Sox-2, Nanog and Lin28) are related to an increase in stem cell-like phenotype.
155
Studies about breast cancer have shown that the expression of CAIX with stem cell-related factors is increased markedly.
156,157
The expression of CAIX is related to the expression of CD44, which is a stem cell marker. Besides, CAIX contributes to the activation of EMT phenomenon in epithelial cancer cells.
155,158
It has been shown that the overexpression CAIX is involved in the formation of focal adhesion in the cells.
159
It associate with β-catenin and the separation of E-cadherin from cytoskeletal - a key process for the separation of cell-cell junction.
160
Further, the activations of PI3K/Akt and FAK/PI3K/mTOR/p70S6K pathways seem to be in association with CAIX.
161,162
Present and future prospects
Solid tumors display complex hallmarks. They are able to form a permissive microenvironment and show adaptation and evolution ed through functional expression of a number of different molecular machineries. Such traits make the treatment of solid tumors very hard. Although acid-base balance in cells is one of the main parameters to cellular homeostasis, unfortunately, this balance is disturbed in solid tumors. This is a unique characteristic of solid tumors, which seems to be initiated by hypoxia and aberrant metabolism of glucose resulting in production of acidic byproducts in cancerous cells. Such phenomena give rise to functional expression of array of functional proteins including transcription factors, enzymes and transporters. These biological elements are considered as potential targets, inhibition of which may provide effective pharmacotherapy in combination with chemotherapy and/or immunotherapy. Use of the proton transporter inhibitors as a new treatment for cancer were proposed by several research groups. Pouysségur group showed that use of these this inhibitor in combination with anti-angiogenic inhibitors could effectively abolish the solid tumors.
77
One advantage of this treatment modality seems to be the induction of less toxicity in comparison with the traditional chemotherapy strategies. In fact, the main purpose of this method was based on acid-base balance, employing the forces involved in controlling pH dysregulation in cancer cells and tissue, resulting in regression of tumor growth, prohibition of invasion and suppression of metastasis. This has been shown in many different human solid tumors. This could be beneficial because such treatment may lead to the inhibition of metastasis process and neutralization of drug resistance, resulting in much more sensitization of cancer cells to chemotherapy, radiotherapy and immunotherapy. Cancer cells in different tissues show distinctive behaviors because they have different genetic pattern and mutations or expression . For instance, lower metastatic cells in breast cancer often use NHEs and HCO3-- and H+- based transporting system to transfer proton, whereas extremely metastatic cells employ plasma membrane V-ATPases.
163
This issue is also true about CAs, for example, CAII mRNA level was shown to be decreased in the lung and colon cancers, while the level of CAIX mRNA was reported to be extremely increased in basal and triple-negative breast cancers. Further, CAXII mRNA levels was shown to be substantially enhanced in all types of breast cancer. In accordance with these, CAIX and CAXII mRNA levels were reported to be increased in squamous cell carcinoma of the lung, but not in the lung adenocarcinomas.
164
A variety of mechanisms are involved in pH-dependent cancer cells' behaviors such as proliferation and survival, metabolic adaptation, metastatic and invasion. Detection of the molecular basis of this phenomenon is very important in understanding of how pH dysregulation can influence the progression and metastasis of solid tumors. It is now well-known that an increased pHi is in favor of cancer cells proliferation, and it also increases the cell survival through inhibition of apoptosis. In such circumstances, cancer cells proliferation seems to be somewhat independent from the influence of growth factors. Further, cancer cells need high energy and nucleotid biosynthesis for growth and proliferation in comparison with normal cells. As a result, metabolic program changes in cancer cells, which is known as the Warburg effect or ‘aerobic’ glycolysis. In solid tumors, cancerous cells are able to modulate/remodel the ECM through pH dysregulation resulting in metastasis. The pHi increase seems to necessary for the migration of cells, while the pHe decrease appears to interfere with the degradation of ECM that favors further invasion of cancer. Researchers who attempt to discover pH dysregulation in solid tumors have focused on proton transport, while other ions may be involved in the cancer, including HCO3-. However, many fundamental questions remain unanswered. We need to address how cancer cells get capability to escape from the immune system surveillance within TME? What are the holistic roles of solute transporters? How the vesicular enzymes behave in lower pHe? Is pH dysregulation involved in anoikis? How single cancer cells communicate with neighboring cells during metastasis? How different types of cells communicate with TME? And, many other questions.
We have previously capitalized on development of immunotherapies and various multifunctional nanomedicines to combat different types of solid tumors.
165-182
However, it seems that ultimate therapy of solid tumors might need use of robust synthetic lethality through combination therapy via targeting different key molecules involved in the dysregulation of pH, formation of TME, tumor vascularization, and even tumor metabolism. he main aim of this review was to bring about the importance of molecular machineries related to the pH dysregulation within TME and that the use of relevant inhibitors against key regulators of pH might suppress tumor progression, metastasis and further invasion. It can be suggested that targeting the key elements involved in formation of TME and pH along with other conventional therapies can sensitize cancer cells to combinational therapy and hence reduce drug resistances. This may improve the survival rate of cancer patients. It should be also noted that the cancerous cells in different part of solid tumors may present distinct molecular patterns in terms of pH dysregulation, and hence some proof-of-concept conforming studies on epigenetic/genetic and proteomic/metabolomic aspects may help us to design much more effective treatment modalities to improve the currently used chemotherapy and immunotherapy.
Acknowledgment
The authors are very grateful for the financial support (grant No: RCPN-94012) provided by the Research Center for Pharmaceutical Nanotechnology (RCPN), BioMedicine Instiute, Tabriz University of Medical Sciences.
Conflict of interest
Authors declare no conflict of interest in this study.
Ethical approval
Not applicable.
Review Highlights
What is current knowledge?
simple
-
√ Cancer is characterized by several adaptive hallmarks.
-
√ Cancer cells are able to form permissive tumor microenvironment (TME).
-
√ Hypoxia, glycolysis and Warburg effects are the main causes for the formation of TME.
-
√ The pH dysregulation is resultant from aberrant metabolism of glucose and production of acidic byproducts. ancer cells migration.
What is new here?
simple
-
√ Dysregulated pH appears to be part of cancer hallmarks.
-
√ The pH dysregulation favors migration, metastasis and invasion of cancer cells.
-
√ Combined formulation of cytotoxic agents with different inhibitors of pH modulators in TME may provide a robust synthetic lethality against solid tumors.
-
√ The pH dysregulation machineries in TME may be considered as tumor molecular markers and used for cancer diagnosis.
-
√ Targeted therapy of cancer can be done by targeting pH modulators.
References
- Barar J, Omidi Y. Dysregulated pH in Tumor Microenvironment Checkmates Cancer Therapy. Bioimpacts 2013; 3:149-62. doi: 10.5681/bi.2013.036 [Crossref] [ Google Scholar]
- Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 2011; 11:671-7. doi: 10.1038/nrc3110 [Crossref] [ Google Scholar]
- Busco G, Cardone RA, Greco MR, Bellizzi A, Colella M, Antelmi E. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. Faseb j 2010; 24:3903-15. doi: 10.1096/fj.09-149518 [Crossref] [ Google Scholar]
- Amith SR, Fliegel L. Regulation of the Na+/H+ Exchanger (NHE1) in Breast Cancer Metastasis. Cancer Res 2013; 73:1259-64. doi: 10.1158/0008-5472.can-12-4031 [Crossref] [ Google Scholar]
- Lin Y, Chang G, Wang J, Jin W, Wang L, Li H. NHE1 mediates MDA-MB-231 cells invasion through the regulation of MT1-MMP. Exp Cell Res 2011; 317:2031-40. doi: 10.1016/j.yexcr.2011.05.026 [Crossref] [ Google Scholar]
- Daniel C, Bell C, Burton C, Harguindey S, Reshkin SJ, Rauch C. The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochim Biophys Acta 2013; 1832:606-17. doi: 10.1016/j.bbadis.2013.01.020 [Crossref] [ Google Scholar]
- Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 2005; 202(3):654-62. doi: 10.1002/jcp.20166 [Crossref] [ Google Scholar]
- Heinrich EL, Walser TC, Krysan K, Liclican EL, Grant JL, Rodriguez NL. The inflammatory tumor microenvironment, epithelial mesenchymal transition and lung carcinogenesis. Cancer Microenviron 2012; 5:5-18. doi: 10.1007/s12307-011-0089-0 [Crossref] [ Google Scholar]
- Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell 2008; 13:472-82. doi: 10.1016/j.ccr.2008.05.005 [Crossref] [ Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029-33. doi: 10.1126/science.1160809 [Crossref] [ Google Scholar]
- Paget S. The distribution of secondary growths in cancer of the breast 1889. Cancer Metastasis Rev 1989; 8:98-101. [ Google Scholar]
- Fidler IJ. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer 2003; 3:453-8. doi: 10.1038/nrc1098 [Crossref] [ Google Scholar]
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015; 348:74-80. doi: 10.1126/science.aaa6204 [Crossref] [ Google Scholar]
- Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci 2012; 125:5591-6. doi: 10.1242/jcs.116392 [Crossref] [ Google Scholar]
- Romero-Garcia S, Moreno-Altamirano MMB, Prado-Garcia H, Sánchez-García FJ. Lactate contribution to the tumor microenvironment: Mechanisms, effects on immune cells and therapeutic relevance. Frontiers in Immunology 2016; 7. doi: 10.3389/fimmu.2016.00052 [Crossref]
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 2009; 30:1073-81. doi: 10.1093/carcin/bgp127 [Crossref] [ Google Scholar]
- Cramer DW, Finn OJ. Epidemiologic perspective on immune-surveillance in cancer. Curr Opin Immunol 2011; 23:265-71. doi: 10.1016/j.coi.2011.01.002 [Crossref] [ Google Scholar]
- Tsai MJ, Chang WA, Huang MS, Kuo PL. Tumor microenvironment: a new treatment target for cancer. ISRN Biochem 2014; 2014:351959. doi: 10.1155/2014/351959 [Crossref] [ Google Scholar]
- Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev 2013; 32:303-15. doi: 10.1007/s10555-012-9415-3 [Crossref] [ Google Scholar]
- Shain KH, Dalton WS. Environmental-mediated drug resistance: a target for multiple myeloma therapy. Expert Rev Hematol 2009; 2:649-62. doi: 10.1586/ehm.09.55 [Crossref] [ Google Scholar]
- Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev 2006; 17:29-40. doi: 10.1016/j.cytogfr.2005.09.006 [Crossref] [ Google Scholar]
- Bigatto V, De Bacco F, Casanova E, Reato G, Lanzetti L, Isella C. TNF-alpha promotes invasive growth through the MET signaling pathway. Mol Oncol 2015; 9:377-88. doi: 10.1016/j.molonc.2014.09.002 [Crossref] [ Google Scholar]
- Martin NK, Gaffney EA, Gatenby RA, Maini PK. Tumour-stromal interactions in acid-mediated invasion: a mathematical model. J Theor Biol 2010; 267:461-70. doi: 10.1016/j.jtbi.2010.08.028 [Crossref] [ Google Scholar]
- Matsuyama S, Llopis J, Deveraux QL, Tsien RY, Reed JC. Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat Cell Biol 2000; 2:318-25. doi: 10.1038/35014006 [Crossref] [ Google Scholar]
- Putney LK, Barber DL. Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J Biol Chem 2003; 278:44645-9. doi: 10.1074/jbc.M308099200 [Crossref] [ Google Scholar]
- Detry B, Erpicum C, Paupert J, Blacher S, Maillard C, Bruyere F. Matrix metalloproteinase-2 governs lymphatic vessel formation as an interstitial collagenase. Blood 2012; 119:5048-56. doi: 10.1182/blood-2011-12-400267 [Crossref] [ Google Scholar]
- Noel A, Gutierrez-Fernandez A, Sounni NE, Behrendt N, Maquoi E, Lund IK. New and paradoxical roles of matrix metalloproteinases in the tumor microenvironment. Front Pharmacol 2012; 3:140. doi: 10.3389/fphar.2012.00140 [Crossref] [ Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009; 139:891-906. doi: 10.1016/j.cell.2009.10.027 [Crossref] [ Google Scholar]
- Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E. Mammographic density and the risk and detection of breast cancer. N Engl J Med 2007; 356:227-36. doi: 10.1056/NEJMoa062790 [Crossref] [ Google Scholar]
- Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011; 19:776-91. doi: 10.1016/j.ccr.2011.05.008 [Crossref] [ Google Scholar]
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science 2009; 326:1216-9. doi: 10.1126/science.1176009 [Crossref] [ Google Scholar]
- Jarvelainen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol Rev 2009; 61:198-223. doi: 10.1124/pr.109.001289 [Crossref] [ Google Scholar]
- Davidson B, Goldberg I, Gotlieb WH, Kopolovic J, Ben-Baruch G, Nesland JM. High levels of MMP-2, MMP-9, MT1-MMP and TIMP-2 mRNA correlate with poor survival in ovarian carcinoma. Clin Exp Metastasis 1999; 17:799-808. [ Google Scholar]
- Davidson B, Goldberg I, Gotlieb WH, Kopolovic J, Ben-Baruch G, Nesland JM. The prognostic value of metalloproteinases and angiogenic factors in ovarian carcinoma. Mol Cell Endocrinol 2002; 187:39-45. [ Google Scholar]
- Georgiadis D, Yiotakis A. Specific targeting of metzincin family members with small-molecule inhibitors: progress toward a multifarious challenge. Bioorg Med Chem 2008; 16:8781-94. doi: 10.1016/j.bmc.2008.08.058 [Crossref] [ Google Scholar]
- Lu X, Lu D, Scully M, Kakkar V. ADAM proteins - therapeutic potential in cancer. Curr Cancer Drug Targets 2008; 8:720-32. doi: 10.2174/156800908786733478 [Crossref] [ Google Scholar]
- Fontanil T, Rua S, Llamazares M, Moncada-Pazos A, Quiros PM, Garcia-Suarez O. Interaction between the ADAMTS-12 metalloprotease and fibulin-2 induces tumor-suppressive effects in breast cancer cells. Oncotarget 2014; 5:1253-64. doi: 10.18632/oncotarget.1690 [Crossref] [ Google Scholar]
- Mayorca-Guiliani A, Erler JT. The potential for targeting extracellular LOX proteins in human malignancy. Onco Targets Ther 2013; 6:1729-35. doi: 10.2147/ott.s38110 [Crossref] [ Google Scholar]
- Lentini A, Abbruzzese A, Provenzano B, Tabolacci C, Beninati S. Transglutaminases: key regulators of cancer metastasis. Amino Acids 2013; 44:25-32. doi: 10.1007/s00726-012-1229-7 [Crossref] [ Google Scholar]
- Dutta A, Li J, Lu H, Akech J, Pratap J, Wang T. Integrin alphavbeta6 promotes an osteolytic program in cancer cells by upregulating MMP2. Cancer Res 2014; 74:1598-608. doi: 10.1158/0008-5472.can-13-1796 [Crossref] [ Google Scholar]
- Craig EA, Parker P, Camenisch TD. Size-dependent regulation of Snail2 by hyaluronan: its role in cellular invasion. Glycobiology 2009; 19:890-8. doi: 10.1093/glycob/cwp064 [Crossref] [ Google Scholar]
- Telmer PG, Tolg C, McCarthy JB, Turley EA. How does a protein with dual mitotic spindle and extracellular matrix receptor functions affect tumor susceptibility and progression?. Commun Integr Biol 2011; 4:182-5. doi: 10.4161/cib.4.2.14270 [Crossref] [ Google Scholar]
- Dicker KT, Gurski LA, Pradhan-Bhatt S, Witt RL, Farach-Carson MC, Jia X. Hyaluronan: a simple polysaccharide with diverse biological functions. Acta Biomater 2014; 10:1558-70. doi: 10.1016/j.actbio.2013.12.019 [Crossref] [ Google Scholar]
- Bornstein P. Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol 1995; 130:503-6. doi: 10.1083/jcb.130.3.503 [Crossref] [ Google Scholar]
- Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 2002; 14:608-16. doi: 10.1016/S0955-0674(02)00361-7 [Crossref] [ Google Scholar]
- Varga I, Hutoczki G, Szemcsak CD, Zahuczky G, Toth J, Adamecz Z. Brevican, neurocan, tenascin-C and versican are mainly responsible for the invasiveness of low-grade astrocytoma. Pathol Oncol Res 2012; 18:413-20. doi: 10.1007/s12253-011-9461-0 [Crossref] [ Google Scholar]
- Ye J, Wu D, Wu P, Chen Z, Huang J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumour Biol 2014; 35:3945-51. doi: 10.1007/s13277-013-1561-x [Crossref] [ Google Scholar]
- S Skandalis S, J Aletras A, Gialeli C, D Theocharis A, Afratis N, N Tzanakakis G. Targeting the tumor proteasome as a mechanism to control the synthesis and bioactivity of matrix macromolecules. Current molecular medicine 2012; 12:1068-82. doi: 10.2174/156652412802480943 [Crossref] [ Google Scholar]
- Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol 2004; 14:198-206. doi: 10.1016/j.semradonc.2004.04.008 [Crossref] [ Google Scholar]
- Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res 2000; 60:2497-503. [ Google Scholar]
- Olive PL, Durand RE. Drug and radiation resistance in spheroids: cell contact and kinetics. Cancer Metastasis Rev 1994; 13:121-38. [ Google Scholar]
- Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med 1999; 5:662-8. doi: 10.1038/9511 [Crossref] [ Google Scholar]
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230-7. [ Google Scholar]
- Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res 1989; 49:4373-84. [ Google Scholar]
- Guo YS, Jin GF, Houston CW, Thompson JC, Townsend CM, Jr Jr.
Insulin-like growth factor-I promotes multidrug resistance
in MCLM colon cancer cells
. J Cell Physiol 1998; 175:141-8. doi: 10.1002/(sici)1097-4652(199805)175:2>141::aidjcp3<3.0.co;2-o [Crossref] [ Google Scholar]
- Eskandani M, Vandghanooni S, Barar J, Nazemiyeh H, Omidi Y. Cell physiology regulation by hypoxia inducible factor-1: Targeting oxygen-related nanomachineries of hypoxic cells. Int J Biol Macromol 2017; 99:46-62. doi: 10.1016/j.ijbiomac.2016.10.113 [Crossref] [ Google Scholar]
- Tomida A, Tsuruo T. Drug resistance mediated by cellular stress response to the microenvironment of solid tumors. Anticancer Drug Des 1999; 14:169-77. [ Google Scholar]
- Gray MD, Mann M, Nitiss JL, Hendershot LM. Activation of the unfolded protein response is necessary and sufficient for reducing topoisomerase IIalpha protein levels and decreasing sensitivity to topoisomerase-targeted drugs. Mol Pharmacol 2005; 68:1699-707. doi: 10.1124/mol.105.014753 [Crossref] [ Google Scholar]
- Omidi Y, Barar J. Targeting tumor microenvironment: crossing tumor interstitial fluid by multifunctional nanomedicines. Bioimpacts 2014; 4:55-67. doi: 10.5681/bi.2014.021 [Crossref] [ Google Scholar]
- Rafi MA, Omidi Y. A prospective highlight on exosomal nanoshuttles and cancer immunotherapy and vaccination. Bioimpacts 2015; 5:117-22. doi: 10.15171/bi.2015.22 [Crossref] [ Google Scholar]
- Cowan DS, Tannock IF. Factors that influence the penetration of methotrexate through solid tissue. Int J Cancer 2001; 91:120-5. [ Google Scholar]
- Gerweck LE, Vijayappa S, Kozin S. Tumor pH controls the in vivo efficacy of weak acid and base chemotherapeutics. Mol Cancer Ther 2006; 5:1275-9. doi: 10.1158/1535-7163.mct-06-0024 [Crossref] [ Google Scholar]
- Sun Y. Translational horizons in the tumor microenvironment: harnessing breakthroughs and targeting cures. Med Res Rev 2015; 35:408-36. doi: 10.1002/med.21338 [Crossref] [ Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013; 13:714-26. doi: 10.1038/nrc3599 [Crossref] [ Google Scholar]
- Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev 2011; 25:2465-79. doi: 10.1101/gad.180331.111 [Crossref] [ Google Scholar]
- Williams RT, den Besten W, Sherr CJ. Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev 2007; 21:2283-7. doi: 10.1101/gad.1588607 [Crossref] [ Google Scholar]
- Lee JK, Joo KM, Lee J, Yoon Y, Nam DH. Targeting the epithelial to mesenchymal transition in glioblastoma: the emerging role of MET signaling. Onco Targets Ther 2014; 7:1933-44. doi: 10.2147/ott.s36582 [Crossref] [ Google Scholar]
- Shostak K, Zhang X, Hubert P, Goktuna SI, Jiang Z, Klevernic I. NF-kappaB-induced KIAA1199 promotes survival through EGFR signalling. Nat Commun 2014; 5:5232. doi: 10.1038/ncomms6232 [Crossref] [ Google Scholar]
- Rosano L, Cianfrocca R, Tocci P, Spinella F, Di Castro V, Caprara V. Endothelin A receptor/beta-arrestin signaling to the Wnt pathway renders ovarian cancer cells resistant to chemotherapy. Cancer Res 2014; 74:7453-64. doi: 10.1158/0008-5472.can-13-3133 [Crossref] [ Google Scholar]
- Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012; 487:505-9. doi: 10.1038/nature11249 [Crossref] [ Google Scholar]
- Dwyer RM, Potter-Beirne SM, Harrington KA, Lowery AJ, Hennessy E, Murphy JM. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res 2007; 13:5020-7. doi: 10.1158/1078-0432.ccr-07-0731 [Crossref] [ Google Scholar]
- Roodhart JM, Daenen LG, Stigter EC, Prins HJ, Gerrits J, Houthuijzen JM. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell 2011; 20:370-83. doi: 10.1016/j.ccr.2011.08.010 [Crossref] [ Google Scholar]
- Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest 2006; 116:1955-62. doi: 10.1172/jci26532 [Crossref] [ Google Scholar]
- Sounni NE, Noel A. Targeting the tumor microenvironment for cancer therapy. Clin Chem 2013; 59:85-93. doi: 10.1373/clinchem.2012.185363 [Crossref] [ Google Scholar]
- Jeschke J, Collignon E, Fuks F. DNA methylome profiling beyond promoters - taking an epigenetic snapshot of the breast tumor microenvironment. Febs J 2015; 282:1801-14. doi: 10.1111/febs.13125 [Crossref] [ Google Scholar]
- Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med 2015; 13:45. doi: 10.1186/s12916-015-0278-7 [Crossref] [ Google Scholar]
- Harguindey S, Arranz JL, Polo Orozco JD, Rauch C, Fais S, Cardone RA. Cariporide and other new and powerful NHE1 inhibitors as potentially selective anticancer drugs--an integral molecular/biochemical/metabolic/clinical approach after one hundred years of cancer research. J Transl Med 2013; 11:282. doi: 10.1186/1479-5876-11-282 [Crossref] [ Google Scholar]
- Orlowski J, Grinstein S. Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch 2004; 447:549-65. doi: 10.1007/s00424-003-1110-3 [Crossref] [ Google Scholar]
- Wakabayashi S, Ikeda T, Iwamoto T, Pouyssegur J, Shigekawa M. Calmodulin-binding autoinhibitory domain controls "pH-sensing" in the Na+/H+ exchanger NHE1 through sequence-specific interaction. Biochemistry 1997; 36:12854-61. doi: 10.1021/bi9715472 [Crossref] [ Google Scholar]
- Grinstein S, Woodside M, Waddell TK, Downey GP, Orlowski J, Pouyssegur J. Focal localization of the NHE-1 isoform of the Na+/H+ antiport: assessment of effects on intracellular pH. Embo j 1993; 12:5209-18. [ Google Scholar]
- Biemesderfer D, Reilly RF, Exner M, Igarashi P, Aronson PS. Immunocytochemical characterization of Na(+)-H+ exchanger isoform NHE-1 in rabbit kidney. Am J Physiol 1992; 263:F833-40. [ Google Scholar]
- Petrecca K, Atanasiu R, Grinstein S, Orlowski J, Shrier A. Subcellular localization of the Na+/H+ exchanger NHE1 in rat myocardium. Am J Physiol 1999; 276:H709-17. [ Google Scholar]
- Malo ME, Fliegel L. Physiological role and regulation of the Na+/H+ exchanger. Can J Physiol Pharmacol 2006; 84:1081-95. doi: 10.1139/y06-065 [Crossref] [ Google Scholar]
- Putney LK, Denker SP, Barber DL. The changing face of the Na+/H+ exchanger, NHE1: structure, regulation, and cellular actions. Annu Rev Pharmacol Toxicol 2002; 42:527-52. doi: 10.1146/annurev.pharmtox.42.092001.143801 [Crossref] [ Google Scholar]
- Tominaga T, Ishizaki T, Narumiya S, Barber DL. p160ROCK mediates RhoA activation of Na-H exchange. Embo j 1998; 17:4712-22. doi: 10.1093/emboj/17.16.4712 [Crossref] [ Google Scholar]
- Khaled AR, Moor AN, Li A, Kim K, Ferris DK, Muegge K. Trophic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1, which induces intracellular alkalinization. Mol Cell Biol 2001; 21:7545-57. doi: 10.1128/mcb.21.22.7545-7557.2001 [Crossref] [ Google Scholar]
- Matsushita M, Tanaka H, Mitsui K, Kanazawa H. Dual functional significance of calcineurin homologous protein 1 binding to Na(+)/H(+) exchanger isoform 1. Am J Physiol Cell Physiol 2011; 301:C280-8. doi: 10.1152/ajpcell.00404.2010 [Crossref] [ Google Scholar]
- Pang T, Wakabayashi S, Shigekawa M. Expression of calcineurin B homologous protein 2 protects serum deprivation-induced cell death by serum-independent activation of Na+/H+ exchanger. J Biol Chem 2002; 277:43771-7. doi: 10.1074/jbc.M208313200 [Crossref] [ Google Scholar]
- Li QH, Wang LH, Lin YN, Chang GQ, Li HW, Jin WN. Nuclear accumulation of calcineurin B homologous protein 2 (CHP2) results in enhanced proliferation of tumor cells. Genes Cells 2011; 16:416-26. doi: 10.1111/j.1365-2443.2011.01497.x [Crossref] [ Google Scholar]
- Li X, Liu Y, Kay CM, Muller-Esterl W, Fliegel L. The Na+/H+ exchanger cytoplasmic tail: structure, function, and interactions with tescalcin. Biochemistry 2003; 42:7448-56. doi: 10.1021/bi027143d [Crossref] [ Google Scholar]
- Aharonovitz O, Zaun HC, Balla T, York JD, Orlowski J, Grinstein S. Intracellular pH regulation by Na(+)/H(+) exchange requires phosphatidylinositol 4,5-bisphosphate. J Cell Biol 2000; 150:213-24. [ Google Scholar]
- Li X, Liu Y, Alvarez BV, Casey JR, Fliegel L. A novel carbonic anhydrase II binding site regulates NHE1 activity. Biochemistry 2006; 45:2414-24. doi: 10.1021/bi051132d [Crossref] [ Google Scholar]
- Denker SP, Huang DC, Orlowski J, Furthmayr H, Barber DL. Direct binding of the Na--H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H(+) translocation. Mol Cell 2000; 6:1425-36. doi: 10.1016/S1097-2765(00)00139-8 [Crossref] [ Google Scholar]
- Silva NL, Haworth RS, Singh D, Fliegel L. The carboxyl-terminal region of the Na+/H+ exchanger interacts with mammalian heat shock protein. Biochemistry 1995; 34:10412-20. [ Google Scholar]
- Lehoux S, Abe J, Florian JA, Berk BC. 14-3-3 Binding to Na+/H+ exchanger isoform-1 is associated with serum-dependent activation of Na+/H+ exchange. J Biol Chem 2001; 276:15794-800. doi: 10.1074/jbc.M100410200 [Crossref] [ Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344:783-92. doi: 10.1056/nejm200103153441101 [Crossref] [ Google Scholar]
- Jin W, Li Q, Wang J, Chang G, Lin Y, Li H. Na+/H+ exchanger 1 inhibition contributes to K562 leukaemic cell differentiation. Cell Biol Int 2012; 36:739-45. doi: 10.1042/cbi20100919 [Crossref] [ Google Scholar]
-
Spugnini EP, Sonveaux P, Stock C, Perez-Sayans M, De Milito A, Avnet S, et al. Proton channels and exchangers in cancer. Biochimica et Biophysica Acta (BBA)-Biomembranes 2014.
- Lv C, Yang X, Yu B, Ma Q, Liu B, Liu Y. Blocking the Na+/H+ exchanger 1 with cariporide (HOE642) reduces the hypoxia-induced invasion of human tongue squamous cell carcinoma. Int J Oral Maxillofac Surg 2012; 41:1206-10. doi: 10.1016/j.ijom.2012.03.001 [Crossref] [ Google Scholar]
- Wakabayashi S, Hisamitsu T, Pang T, Shigekawa M. Mutations of Arg440 and Gly455/Gly456 oppositely change pH sensing of Na+/H+ exchanger 1. J Biol Chem 2003; 278:11828-35. doi: 10.1074/jbc.M213243200 [Crossref] [ Google Scholar]
- Lifton RP, Hunt SC, Williams RR, Pouyssegur J, Lalouel JM. Exclusion of the Na(+)-H+ antiporter as a candidate gene in human essential hypertension. Hypertension 1991; 17:8-14. [ Google Scholar]
- Cotter K, Stransky L, McGuire C, Forgac M. Recent Insights into the Structure, Regulation, and Function of the V-ATPases. Trends Biochem Sci 2015; 40:611-22. doi: 10.1016/j.tibs.2015.08.005 [Crossref] [ Google Scholar]
- Nelson N.
Structure and function of V-ATPases in endocytic and
secretory organelles
. J Exp Biol1992; 172:149-53. [ Google Scholar]
- Izumi H, Torigoe T, Ishiguchi H, Uramoto H, Yoshida Y, Tanabe M. Cellular pH regulators: potentially promising molecular targets for cancer chemotherapy. Cancer Treat Rev 2003; 29:541-9. doi: 10.1016/S0305-7372(03)00106-3 [Crossref] [ Google Scholar]
- O'Callaghan KM, Ayllon V, O'Keeffe J, Wang Y, Cox OT, Loughran G. Heme-binding protein HRG-1 is induced by insulin-like growth factor I and associates with the vacuolar H+-ATPase to control endosomal pH and receptor trafficking. J Biol Chem 2010; 285:381-91. doi: 10.1074/jbc.M109.063248 [Crossref] [ Google Scholar]
- Peña‐Llopis S, Vega‐Rubin‐de‐Celis S, Schwartz JC, Wolff NC, Tran TAT, Zou L. Regulation of TFEB and V‐ATPases by mTORC1. EMBO J 2011; 30:3242-58. doi: 10.1038/emboj.2011.257 [Crossref] [ Google Scholar]
- Graham RM, Thompson JW, Webster KA. Inhibition of the vacuolar ATPase induces Bnip3-dependent death of cancer cells and a reduction in tumor burden and metastasis. Oncotarget 2014; 5:1162. doi: 10.18632/oncotarget.1699 [Crossref] [ Google Scholar]
- von Schwarzenberg K, Wiedmann RM, Oak P, Schulz S, Zischka H, Wanner G. Mode of cell death induction by pharmacological vacuolar H+-ATPase (V-ATPase) inhibition. J Biol Chem 2013; 288:1385-96. doi: 10.1074/jbc.M112.412007 [Crossref] [ Google Scholar]
- Toyomura T, Oka T, Yamaguchi C, Wada Y, Futai M. Three subunit a isoforms of mouse vacuolar H(+)-ATPase Preferential expression of the a3 isoform during osteoclast differentiation. J Biol Chem 2000; 275:8760-5. doi: 10.1074/jbc.275.12.8760 [Crossref] [ Google Scholar]
- Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet 2000; 26:71-5. doi: 10.1038/79208 [Crossref] [ Google Scholar]
- Cotter K, Capecci J, Sennoune S, Huss M, Maier M, Martinez-Zaguilan R. Activity of Plasma Membrane V-ATPases Is Critical for the Invasion of MDA-MB231 Breast Cancer Cells. J Biol Chem 2015; 290(6):3680-92. doi: 10.1074/jbc.M114.611210 [Crossref] [ Google Scholar]
- Chung C, Mader CC, Schmitz JC, Atladottir J, Fitchev P, Cornwell ML. The vacuolar-ATPase modulates matrix metalloproteinase isoforms in human pancreatic cancer. Lab Invest 2011; 91(5):732-43. doi: 10.1038/labinvest.2011.8 [Crossref] [ Google Scholar]
- Nishisho T, Hata K, Nakanishi M, Morita Y, Sun-Wada G-H, Wada Y. The a3 isoform vacuolar type H+-ATPase promotes distant metastasis in the mouse B16 melanoma cells. Mol Cancer Res 2011; 9:845-55. doi: 10.1158/1541-7786.MCR-10-0449 [Crossref] [ Google Scholar]
- Hendrix A, Sormunen R, Westbroek W, Lambein K, Denys H, Sys G.
Vacuolar H+ ATPase expression and activity is required
for Rab27B‐dependent invasive growth and metastasis of breast
cancer
. Int J Cancer 2013; 133:843-54. doi: 10.1002/ijc.28079 [Crossref] [ Google Scholar]
- Gocheva V, Joyce JA. Cysteine cathepsins and the cutting edge of cancer invasion. Cell cycle 2007; 6:60-4. doi: 10.4161/cc.6.1.3669 [Crossref] [ Google Scholar]
- Gondi CS, Rao JS. Cathepsin B as a cancer target. Expert Opin Ther Targets 2013; 17:281-91. doi: 10.1517/14728222.2013.740461 [Crossref] [ Google Scholar]
- Lozupone F, Borghi M, Marzoli F, Azzarito T, Matarrese P, Iessi E. TM9SF4 is a novel V-ATPase-interacting protein that modulates tumor pH alterations associated with drug resistance and invasiveness of colon cancer cells. Oncogene 2015; 34(40):5163-74. doi: 10.1038/onc.2014.437 [Crossref] [ Google Scholar]
- Schempp CM, von Schwarzenberg K, Schreiner L, Kubisch R, Müller R, Wagner E. V-ATPase inhibition regulates anoikis resistance and metastasis of cancer cells. Mol Cancer Ther 2014; 13:926-37. doi: 10.1158/1535-7163 [Crossref] [ Google Scholar]
- Meo-Evoli N, Almacellas E, Massucci FA, Gentilella A, Ambrosio S, Kozma SC. V-ATPase: a master effector of E2F1-mediated lysosomal trafficking, mTORC1 activation and autophagy. Oncotarget 2015; 6:28057-70. doi: 10.18632/oncotarget.4812 [Crossref] [ Google Scholar]
- Wiedmann RM, von Schwarzenberg K, Palamidessi A, Schreiner L, Kubisch R, Liebl J. The V-ATPase-inhibitor archazolid abrogates tumor metastasis via inhibition of endocytic activation of the Rho-GTPase Rac1. Cancer res 2012; 72:5976-87. doi: 10.1158/0008-5472.CAN-12-1772 [Crossref] [ Google Scholar]
- Fan S-h, Wang Y-y, Wu Z-y, Zhang Z-f, Lu J, Li M-q. AGPAT9 suppresses cell growth, invasion and metastasis by counteracting acidic tumor microenvironment through KLF4/LASS2/V-ATPase signaling pathway in breast cancer. Oncotarget 2015; 6:18406. doi: 10.18632/oncotarget.4074 [Crossref] [ Google Scholar]
- Pérez-Sayáns M, García-García A, Reboiras-López MD MD, GándaraVila P.
Role of V-ATPases in solid tumors: importance of the
subunit C (Review)
. Int J Oncol 2009; 34:1513-20. doi: 10.3892/ijo_00000280 [Crossref] [ Google Scholar]
- Muroi M, Shiragami N, Nagao K, Yamasaki M, Takatsuki A. Folimycin (concanamycin A), a specific inhibitor of V-ATPase, blocks intracellular translocation of the glycoprotein of vesicular stomatitis virus before arrival to the Golgi apparatus. Cell Struct Funct 1993; 18:139-49. doi: 10.1247/csf.18.139 [Crossref] [ Google Scholar]
- Menche D, Hassfeld J, Sasse F, Huss M, Wieczorek H. Design, synthesis, and biological evaluation of novel analogues of archazolid: a highly potent simplified V-ATPase inhibitor. Bioorg Med Chem Lett 2007; 17:1732-5. doi: 10.1016/j.bmcl.2006.12.073 [Crossref] [ Google Scholar]
- Pérez-Sayáns M, Somoza-Martín JM, Barros-Angueira F, Rey JMG, García-García A. V-ATPase inhibitors and implication in cancer treatment. Cancer Treat Rev 2009; 35:707-13. doi: 10.1016/j.ctrv.2009.08.003 [Crossref] [ Google Scholar]
- Sennoune SR, Bakunts K, Martinez GM, Chua-Tuan JL, Kebir Y, Attaya MN. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am J Physiol Cell Physiol 2004; 286:C1443-C52. doi: 10.1152/ajpcell.00407.2003 [Crossref] [ Google Scholar]
- Nakashima S, Hiraku Y, Tada-Oikawa S, Hishita T, Gabazza EC, Tamaki S. Vacuolar H+-ATPase inhibitor induces apoptosis via lysosomal dysfunction in the human gastric cancer cell line MKN-1. J Biochem 2003; 134:359-64. doi: 10.1093/jb/mvg153 [Crossref] [ Google Scholar]
- Hishita T, Tada-Oikawa S, Tohyama K, Miura Y, Nishihara T, Tohyama Y. Caspase-3 activation by lysosomal enzymes in cytochrome c-independent apoptosis in myelodysplastic syndrome-derived cell line P39. Cancer Res 2001; 61:2878-84. [ Google Scholar]
- Zhitomirsky B, Assaraf YG. Lysosomes as mediators of drug resistance in cancer. Drug Resist Updat 2016; 24:23-33. doi: 10.1016/j.drup.2015.11.004 [Crossref] [ Google Scholar]
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011; 10:767-77. doi: 10.1038/nrd3554 [Crossref] [ Google Scholar]
- Ferry JG. The γ class of carbonic anhydrases. Biochim Biophys Acta 2010; 1804:374-81. doi: 10.1016/j.bbapap.2009.08.026 [Crossref] [ Google Scholar]
- Thiry A, Dogne J-M, Masereel B, Supuran CT. Targeting tumor-associated carbonic anhydrase IX in cancer therapy. Trends Pharmacol Sci 2006; 27:566-73. doi: 10.1016/j.tips.2006.09.002 [Crossref] [ Google Scholar]
-
Supuran C. Carbonic Anhydrases: Catalytic And Inhibition Mechanisms, Distribuion and Physiological Roles. In: Supuran C, Scozzafava A, Conway J, eds. Carbonic Anhydrase: Its Inhibitors and Activators. Boca Raton: CRC Press; 2004. p. 1-23.
-
Pastoreková S, Pastorek J. Cancer-related carbonic anhydrase isozymes and their inhibition. In: upuran C, Scozzafava A, Conway J, eds. Carbonic Anhydrase—Its Inhibitors and Activators. Boca Raton: CRC Press; 2004. p. 253-80.
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010; 20:3467-74. doi: 10.1016/j.bmcl.2010.05.009 [Crossref] [ Google Scholar]
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011; 3:1165-80. doi: 10.4155/fmc.11.69 [Crossref] [ Google Scholar]
- Švastová E, Hulıiková A, Rafajová M, Zat'ovičová M, Gibadulinová A, Casini A. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS let 2004; 577:439-45. doi: 10.1016/j.febslet.2004.10.043 [Crossref] [ Google Scholar]
- Maresca A, Temperini C, Pochet L, Masereel B, Scozzafava A, Supuran CT. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2009; 53:335-44. doi: 10.1021/jm901287j [Crossref] [ Google Scholar]
- Pearson MA, Fabbro D. Targeting protein kinases in cancer therapy: a success?. Expert Rev Anticancer Ther 2004; 4(6):1113-24. doi: 10.1586/14737140.4.6.1113 [Crossref] [ Google Scholar]
- Parkkila S, Innocenti A, Kallio H, Hilvo M, Scozzafava A, Supuran CT. The protein tyrosine kinase inhibitors imatinib and nilotinib strongly inhibit several mammalian α-carbonic anhydrase isoforms. Bioorg Med Chem Lett 2009; 19:4102-6. doi: 10.1016/j.bmcl.2009.06.002 [Crossref] [ Google Scholar]
- Chrastina A, Závada J, Parkkila S, Kaluz Š, Kaluzová M, Rajčáni J. Biodistribution and pharmacokinetics of 125I‐labeled monoclonal antibody M75 specific for carbonic anhydrase IX, an intrinsic marker of hypoxia, in nude mice xenografted with human colorectal carcinoma. Int J Cancer 2003; 105:873-81. doi: 10.1002/ijc.11142 [Crossref] [ Google Scholar]
- Siebels M, Rohrmann K, Oberneder R, Stahler M, Haseke N, Beck J. A clinical phase I/II trial with the monoclonal antibody cG250 (RENCAREX®) and interferon-alpha-2a in metastatic renal cell carcinoma patients. World J Urol 2011; 29:121-6. doi: 10.1007/s00345-010-0570-2 [Crossref] [ Google Scholar]
- Stiti M, Cecchi A, Rami M, Abdaoui M, Barragan-Montero V, Scozzafava A. Carbonic anhydrase inhibitor coated gold nanoparticles selectively inhibit the tumor-associated isoform IX over the cytosolic isozymes I and II. J Am Chem Soc 2008; 130:16130-1. doi: 10.1021/ja805558k [Crossref] [ Google Scholar]
- Höckel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 2001; 93:266-76. [ Google Scholar]
- Helmlinger G, Sckell A, Dellian M, Forbes NS, Jain RK. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin Cancer Res 2002; 8:1284-91. [ Google Scholar]
- Ashida S, Nishimori I, Tanimura M, Onishi S, Shuin T. Effects of von Hippel-Lindau gene mutation and methylation status on expression of transmembrane carbonic anhydrases in renal cell carcinoma. J Cancer Res Clin Oncol 2002; 128:561-8. doi: 10.1007/s00432-002-0374-x [Crossref] [ Google Scholar]
- Alvarez BV, Vilas GL, Casey JR. Metabolon disruption: a mechanism that regulates bicarbonate transport. EMBO J 2005; 24:2499-511. doi: 10.1038/sj.emboj.7600736 [Crossref] [ Google Scholar]
- Chiche J, Ilc K, Laferrière J, Trottier E, Dayan F, Mazure NM. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer res 2009; 69:358-68. doi: 10.1158/0008-5472.CAN-08-2470 [Crossref] [ Google Scholar]
- Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer res 2011; 71:3364-76. doi: 10.1158/0008-5472.CAN-10-4261 [Crossref] [ Google Scholar]
- McIntyre A, Patiar S, Wigfield S, Li J-l, Ledaki I, Turley H. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin Cancer Res 2012; 18:3100-11. doi: 10.1158/1078-0432 [Crossref] [ Google Scholar]
- Dubois L, Peeters S, Lieuwes NG, Geusens N, Thiry A, Wigfield S. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011; 99:424-31. doi: 10.1016/j.radonc.2011.05.045 [Crossref] [ Google Scholar]
- Stock C, Schwab A. Protons make tumor cells move like clockwork. Pflugers Arch 2009; 458:981-92. doi: 10.1007/s00424-009-0677-8 [Crossref] [ Google Scholar]
- Axelson H, Fredlund E, Ovenberger M, Landberg G, Påhlman S, editors editors. Hypoxia-induced dedifferentiation of tumor cells–a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin Cell Dev Biol 2005; 16(4-5):554-63. doi: 10.1016/j.semcdb.2005.03.007 [Crossref] [ Google Scholar]
- Hjelmeland AB, Wu Q, Heddleston J, Choudhary G, MacSwords J, Lathia J. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ 2011; 18:829-40. doi: 10.1038/cdd.2010.150 [Crossref] [ Google Scholar]
- Fujiwara D, Kato K, Nohara S, Iwanuma Y, Kajiyama Y. The usefulness of three-dimensional cell culture in induction of cancer stem cells from esophageal squamous cell carcinoma cell lines. Biochem Biophys Res Commun 2013; 434:773-8. doi: 10.1016/j.bbrc.2013.04.008 [Crossref] [ Google Scholar]
- Sansone P, Storci G, Giovannini C, Pandolfi S, Pianetti S, Taffurelli M. p66Shc/Notch‐3 Interplay Controls Self‐Renewal and Hypoxia Survival in Human Stem/Progenitor Cells of the Mammary Gland Expanded In Vitro as Mammospheres. Stem Cells 2007; 25:807-15. doi: 10.1634/stemcells.2006-0442 [Crossref] [ Google Scholar]
- Storci G, Sansone P, Trere D, Tavolari S, Taffurelli M, Ceccarelli C. The basal‐like breast carcinoma phenotype is regulated by SLUG gene expression. J Pathol 2008; 214:25-37. doi: 10.1002/path.2254 [Crossref] [ Google Scholar]
- Fiaschi T, Giannoni E, Taddei L, Cirri P, Marini A, Pintus G. Carbonic anhydrase IX from cancer-associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle 2013; 12:1791-801. doi: 10.4161/cc.24902 [Crossref] [ Google Scholar]
- Csaderova L, Debreova M, Radvak P, Stano M, Vrestiakova M, Kopacek J. The effect of carbonic anhydrase IX on focal contacts during cell spreading and migration. Front Physiol 2013; 4:10 .3389. doi: 10.3389/fphys.2013.00271 [Crossref] [ Google Scholar]
- Švastová E, Žilka N, Zat'ovičová M, Gibadulinová A, Čiampor F, Pastorek J. Carbonic anhydrase IX reduces E-cadherin-mediated adhesion of MDCK cells via interaction with β-catenin. Exp Cell Res 2003; 290:332-45. doi: 10.1016/S0014-4827(03)00351-3 [Crossref] [ Google Scholar]
- Dorai T, Sawczuk IS, Pastorek J, Wiernik PH, Dutcher JP.
The role
of carbonic anhydrase IX overexpression in kidney cancer
.
Exp Cell
Res
2005; 41:2935-47. doi: 10.1016/S0014-4827(03)00351-3 [Crossref] [ Google Scholar]
- Wiesener MS, Münchenhagen PM, Berger I, Morgan NV, Roigas J, Schwiertz A. Constitutive activation of hypoxia-inducible genes related to overexpression of hypoxia-inducible factor-1α in clear cell renal carcinomas. Cancer res 2001; 61:5215-22. [ Google Scholar]
- Sennoune SR, Bakunts K, Martínez GM, Chua-Tuan JL, Kebir Y, Attaya MN. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am J Physiol Cell Physiol 2004; 286:C1443-C52. doi: 10.1152/ajpcell.00407.2003 [Crossref] [ Google Scholar]
- Gorbatenko A, Olesen CW, Boedtkjer E, Pedersen SF. Regulation and roles of bicarbonate transporters in cancer. Front Physiol 2013; 5:130. doi: 10.3389/fphys.2014.00130 [Crossref] [ Google Scholar]
- Shakoori Z, Ghanbari H, Omidi Y, Pashaiasl M, Akbarzadeh A, Jomeh Farsangi Z. Fluorescent multi-responsive cross-linked P(N-isopropylacrylamide)-based nanocomposites for cisplatin delivery. Drug Dev Ind Pharm 2017; 43:1283-91. doi: 10.1080/03639045.2017.1313859 [Crossref] [ Google Scholar]
- Rahmanian N, Eskandani M, Barar J, Omidi Y. Recent trends in targeted therapy of cancer using graphene oxide-modified multifunctional nanomedicines. J Drug Target 2017; 25:202-15. doi: 10.1080/1061186X.2016.1238475 [Crossref] [ Google Scholar]
- Bakhtiary Z, Barar J, Aghanejad A, Saei AA, Nemati E, Ezzati Nazhad Dolatabadi J. Microparticles containing erlotinib-loaded solid lipid nanoparticles for treatment of non-small cell lung cancer. Drug Dev Ind Pharm 2017; 43:1244-53. doi: 10.1080/03639045.2017.1310223 [Crossref] [ Google Scholar]
- Same S, Aghanejad A, Akbari Nakhjavani S, Barar J, Omidi Y. Radiolabeled theranostics: magnetic and gold nanoparticles. Bioimpacts 2016; 6:169-81. doi: 10.15171/bi.2016.23 [Crossref] [ Google Scholar]
- Johari-Ahar M, Barar J, Alizadeh AM, Davaran S, Omidi Y, Rashidi MR. Methotrexate-conjugated quantum dots: synthesis, characterisation and cytotoxicity in drug resistant cancer cells. J Drug Target 2016; 24:120-33. doi: 10.3109/1061186X.2015.1058801 [Crossref] [ Google Scholar]
- Azhdarzadeh M, Atyabi F, Saei AA, Varnamkhasti BS, Omidi Y, Fateh M. Theranostic MUC-1 aptamer targeted gold coated superparamagnetic iron oxide nanoparticles for magnetic resonance imaging and photothermal therapy of colon cancer. Colloids Surf B Biointerfaces 2016; 143:224-32. doi: 10.1016/j.colsurfb.2016.02.058 [Crossref] [ Google Scholar]
- Barar J, Kafil V, Majd MH, Barzegari A, Khani S, Johari-Ahar M. Multifunctional mitoxantrone-conjugated magnetic nanosystem for targeted therapy of folate receptor-overexpressing malignant cells. J Nanobiotechnology 2015; 13:26. doi: 10.1186/s12951-015-0083-7 [Crossref] [ Google Scholar]
- Matthaiou EI, Barar J, Sandaltzopoulos R, Li C, Coukos G, Omidi Y. Shikonin-loaded antibody-armed nanoparticles for targeted therapy of ovarian cancer. Int J Nanomedicine 2014; 9:1855-70. doi: 10.2147/IJN.S51880 [Crossref] [ Google Scholar]
- Mashinchian O, Johari-Ahar M, Ghaemi B, Rashidi M, Barar J, Omidi Y. Impacts of quantum dots in molecular detection and bioimaging of cancer. Bioimpacts 2014; 4:149-66. doi: 10.15171/bi.2014.008 [Crossref] [ Google Scholar]
- Barar J, Omidi Y. Surface modified multifunctional nanomedicines for simultaneous imaging and therapy of cancer. Bioimpacts 2014; 4:3-14. doi: 10.5681/bi.2014.011 [Crossref] [ Google Scholar]
- Tohidkia MR, Asadi F, Barar J, Omidi Y. Selection of potential therapeutic human single-chain Fv antibodies against cholecystokinin-B/gastrin receptor by phage display technology. BioDrugs 2013; 27:55-67. doi: 10.1007/s40259-012-0007-0 [Crossref] [ Google Scholar]
- Najar AG, Pashaei-Asl R, Omidi Y, Farajnia S, Nourazarian AR. EGFR antisense oligonucleotides encapsulated with nanoparticles decrease EGFR, MAPK1 and STAT5 expression in a human colon cancer cell line. Asian Pac J Cancer Prev 2013; 14:495-8. [ Google Scholar]
- Heidari Majd M, Asgari D, Barar J, Valizadeh H, Kafil V, Abadpour A. Tamoxifen loaded folic acid armed PEGylated magnetic nanoparticles for targeted imaging and therapy of cancer. Colloids Surf B Biointerfaces 2013; 106:117-25. doi: 10.1016/j.colsurfb.2013.01.051 [Crossref] [ Google Scholar]
- Barar J, Omidi Y. Targeted Gene Therapy of Cancer: Second Amendment toward Holistic Therapy. Bioimpacts 2013; 3:49-51. doi: 10.5681/bi.2013.014 [Crossref] [ Google Scholar]
- Khosroushahi AY, Naderi-Manesh H, Yeganeh H, Barar J, Omidi Y. Novel water-soluble polyurethane nanomicelles for cancer chemotherapy: physicochemical characterization and cellular activities. J Nanobiotechnology 2012; 10:2. doi: 10.1186/1477-3155-10-2 [Crossref] [ Google Scholar]
- Omidi Y. Smart multifunctional theranostics: simultaneous diagnosis and therapy of cancer. Bioimpacts 2011; 1:145-7. doi: 10.5681/bi.2011.019 [Crossref] [ Google Scholar]
- Nakhlband A, Barar J, Bidmeshkipour A, Heidari HR, Omidi Y. Bioimpacts of anti epidermal growth factor receptor antisense complexed with polyamidoamine dendrimers in human lung epithelial adenocarcinoma cells. J Biomed Nanotechnol 2010; 6:360-9. [ Google Scholar]
- Majidi J, Barar J, Baradaran B, Abdolalizadeh J, Omidi Y. Target therapy of cancer: implementation of monoclonal antibodies and nanobodies. Hum Antibodies 2009; 18:81-100. doi: 10.3233/HAB-2009-0204 [Crossref] [ Google Scholar]