BioImpacts. 8(1):39-52.
doi: 10.15171/bi.2018.06
Original Research
A novel B- and helper T-cell epitopes-based prophylactic vaccine against Echinococcus granulosus
Mohammad M. Pourseif 1, 2, Gholamali Moghaddam 1, *, Hossein Daghighkia 1, Ahmad Nematollahi 3, Yadollah Omidi 2, 4, * 
Author information:
1 Department of Animal Sciences, Faculty of Agriculture, University of Tabriz, Tabriz, Iran
2 Research Center for Pharmaceutical Nanotechnology, Biomedicine Institute, Tabriz University of Medical Sciences, Tabriz, Iran
3 Department of Pathobiology, Veterinary Collage, University of Tabriz, Tabriz, Iran
4 Department of Pharmaceutics, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
Abstract
Introduction:
In this study, we targeted the worm stage of Echinococcus granulosus to design a novel multi-epitope B- and helper T-cell based vaccine construct for immunization of dogs against this multi-host parasite.
Methods:
The vaccine was designed based on the local Eg14-3-3 antigen (Ag). DNA samples were extracted from the protoscoleces of the infected sheep’s liver, and then subjected to the polymerase chain reaction (PCR) with 14-3-3 specific forward and reverse primers. For the vaccine designing, several in silico steps were undertaken. Three-dimensional (3D) structure of the local Eg14-3-3 Ag was modeled by EasyModeller software. The protein modeling accuracy was then analyzed via various validation assays. Potential transmembrane helix, signal peptide, post-translational modifications and allergenicity of Eg14-3-3 were evaluated as the preliminary measures of B-cell epitopes (BEs ) prediction. Having used many web-servers, a well-designed process was carried out for improved prediction of BEs. High ranked linear and conformational BEs were utilized for engineering the final vaccine construct. Possible T-helper epitopes (TEs) were identified by the molecular docking between 13-mer fragments of the Eg14-3-3 Ag and two high frequent dog class II MHC alleles (i.e., DLA-DRB1*01101 and DRB1*01501). The epitopes coverage was evaluated by Shannon’s variability plot.
Results:
The final designed construct was analyzed based on different physicochemical properties, which was then codon optimized for high-level expression in Escherichia coli k12. This minigene construct is the first dog-specific epitopic vaccine construct that is established based on TEs with high-binding affinity to canine MHC alleles.
Conclusion:
This in silico study is the first part of a multi-antigenic vaccine designing work that represents as a novel dog-specific vaccine against E. granulosus. Here, we present key data on the step-by-step methodologies used for designing this de novo vaccine, which is under comprehensive in vivo investigations.
Keywords: B-cell epitope,
Echinococcus granulosus
, Eg14-3-3 antigen, Leukocyte antigen, T-helper epitope, Vaccine
Copyright and License Information
© 2018 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
The hydatid disease is one of the most important zoonotic parasitic infections worldwide. The main part of Echinococcus granulosus life cycle depends upon the growth and differentiation of protoscoleces (PSCs) within the intestine of definitive hosts.
1
The survival of an organism is mainly dependent on its indirect transmission cycle from the definitive hosts (particularly dogs) to the intermediate hosts, including sheep and human. According to the latest updated report of the World Health Organization (WHO) in March 2017, there may be over one million infected cases with echinococcosis at any time.
2
A hydatid cyst (HC) encompasses numerous PSCs and cyst fluid and is formed in visceral organs (liver and lung) of the infected intermediate hosts.
3
Morphogenesis of PSCs from the cystic viscera into worm’s scolex (or head) occurs on the surface of the small intestine of the definitive host, and then the head-like structure attaches to the gut epithelial lining and develops into an adult worm within 32-80 days post-infection.
4
The currently used treatment modalities against echinococcosis, as multi-stage parasitic infection, are based on the anthelmintic drugs such as albendazole, praziquantel, and ivermectin. However, these medicaments are often associated with some issues such as inadvertent side effects.
5-7
Therefore, to control this parasitic infection, it is necessary to improve preventive interventions such as vaccination in addition to hygiene practices. Several constraining factors may influence the vaccine development against such multi-stage pathogens, including economic, socio-cultural/political issues and technical hurdles.
8
Thus, to overcome the issues, it is necessary to implement a rationalized approach towards the construction of multipotent vaccines through targeting a different aspect of the infection. In silico modeling of vaccines provides a cost- and time-effective approach that can improve such endeavor towards designing and developing effective vaccines.
9
Further, such approach favors the identification and selection of key vaccine candidate antigens (VCAs) with high immunogenicity. Ideally, a vaccine construct, to be highly effective, should encompass several parts, including epitopes of one/more VCAs, B-cells epitope and T-cells epitopes (BEs and TEs, respectively).
10
Nevertheless, even a well-designed vaccine may fail to elicit the immune system's responses because of the complexity of various cascades involved in the immunity against parasitic infections. Altogether, epitope-based vaccines (EVs) seem to be one of the most effective vaccines. However, despite being a tedious and laborious approach, designing effective EV type vaccines depends upon several key parameters, including the selection of the best epitopes of a right VCA among other factors.
11
Of various VCAs involved in echinococcosis, the E. granulosus (Eg) 14-3-3 as a secretory Ag plays key roles in parasite’s survival, cell cycle, growth and differentiation regulation and apoptotic cell death, as well as the interaction between the adult worm and definitive host.
12,13
The main immune response to the echinococcus infection is based on the activation of humoral immunity, mainly through production of the specific antibodies (Abs).
14,15
In this study, to design an effective prophylactic EV, we capitalized on the protein sequence of Eg14-3-3 isolated from the infected cases in Iran and used it for 3D structure homology modeling and prediction of BEs and TEs. This dog-specific vaccine construct, to the best of our knowledge, is the first EV designed for the immunization of definitive hosts against the adult worm of E. granulosus.
Materials and Methods
Echinococcus granulosus parasites and genomic DNA isolation from PSCs
The PSCs were aspirated from the lungs and livers of 15 sheep infected with hydatid cysts (Tabriz slaughterhouse, north-west Iran). Then, DNA samples were extracted using a standard protocol as follows: (i) lysing with the lysis buffer (NaCl 150 mM, EDTA 2 mM, Triton 0.5%, Tris-HCL 50 mM), (ii) processing through freeze-thaw-vortex cycles (×3), (iii) suspending with phenolchloroform-isoamylalcohol, and (iv) centrifuging at 9000 ×g for 10 minutes. The concentration and purity of the isolated DNA were analyzed using NanoDropTM spectrophotometer ND-1000 (Thermo Fisher Scientific, Waltham, USA) and 1% agarose gel electrophoresis.
DNA amplification
Eg14-3-3 gene was amplified by polymerase chain reaction (PCR). The PCR reaction performed in a 25 µL reaction volume as follows: 9.5 µL DNase free deionized water, 1 µL template DNA (1017.6 ng/µL), 1 µL each primer (100 pmoles/µL) and 12.5 µL PCR master mix (2x Master Mix RED, Taq DNA polymerase, 0.5 µM of dNTPs and 1.5 mM MgCl2) obtained from Ampliqon (Odense, Denmark). Based on NCBI’s Eg14-3-3 gene sequence (GenBank accession No: AY942149), the forward and reverse primers were designed using OLIGO 7 software.
16
The primers were as follows: the forward primer, 5'-ATGTCTTCTCTCAGTAAGCGCGA-3' and the reverse primer, 5'-ATCGGCTTTCGGCGGTTCAG-3'. The gene amplification was conducted using following condition: (i) one cycle at 94°C for 4 minutes as primary denaturation, (ii) 32 cycles at 94 °C for 1 minute (denaturation), 52°C for 1 minute (annealing) and 72°Cfor 1 minute (extension), and (iii) three cycles as final extension (72°C for 10 minutes). The PCR products were visualized by electrophoresis on a 1% agarose gel and size of the product analyzed with 1 kb DNA ladder (GeneRulerTM 1 kb Plus DNA ladder), obtained from Fermentase (Thermo Fisher Scientific, Waltham, USA). The PCR-amplified product was sequenced, and its mRNA sequence was registered in NCBI GenBank as Iran Eg14-3-3 isolate (GenBank: KU739136).
In silico analysis
Prediction of transmembrane topology and signal peptide
Possible transmembrane helix (TMH) was predicted using three web-servers, including TOPCONS,
17
TMHMM server v2.0 (http://www.cbs.dtu.dk/services/TMHMM/) and TMpred.
18
Potential secretory signal peptide (SigP) was predicted by means of TOPCONS and SignalP v4.1 online tools.
19
The D-cutoff value in the SignalP server was 0.45 and eukaryotic protein datasets were selected for SigP prediction.
Computing post-translational modifications
Neural network-based algorithms, NetNGlyc v1.0
20
and NetOGlyc v4.0
21
were used respectively for the prediction of N-linked and mucin-type CalNAc O-linked glycosylation sites. NetPhos v2.0 web-server was served for the prediction of serine, threonine, and tyrosine phosphorylation sites.
22
Allergenicity prediction
The whole Eg14-3-3 amino acid sequence was aligned against databases of two online servers, including AlgPred and SDAP (Structural Database of Allergenic Proteins). These web servers aid us to predict potential cross-reactivity between the Eg14-3-3 protein and known allergens by means of FAO/WHO allergenicity rules. The algpred server was used to serve the hybrid method consisted of four different algorithms (SVMc + known IgE epitopes + ARPs BLAST + MAST).
23
The Eg14-3-3 sequence was compared to the FASTA-formatted protein sequences based on two main parameters, including (i) the E-value cutoff of 0.01 for the full-length sequence similarity >35% with a sliding window alignment of 80 amino acids, and (ii) short sequences of six contiguous amino acids that match to those of known allergenic proteins.
24
Identification of surface accessible and hydrophobic residues
It should be pointed out that the hydrophilic regions of Ags interact with the binding sites of Abs and the basis of the current vaccine is BEs. Thus, the predicted BEs should be located in the highly hydrophilic region with a lot of accessible residues. Accordingly, the structure and plot of the hydrophobicity profile of Eg14-3-3 Ag were analyzed respectively through UCSF Chimera software and the algorithm of Kyte and Doolittle (KD).
25
The surface accessible residues were predicted by means of the ProtScale online server.
26
The secondary structure prediction
The secondary structure of Eg14-3-3 was predicted using the PSIPRED v3.3 web-server.
27
In this regard, we predicted three states of the secondary structure, including α-helix (H), β-strand (S), and coil (C).
The tertiary structure prediction
The NCBI blastp algorithm (protein-protein BLAST) was utilized to compare the Eg14-3-3 sequence with the crystallized protein structures in Protein Data Bank (PDB). For the homology modeling, we capitalized on EasyModeller v4.0 software and five high rank homologous PDB structures as template structures, including 4WRQ (chain B), 3RDH (chain A), 4HKC (chain A), 2V7D (chain A) and 1A38 (chain A).
28
The most similarity percentage in this protein-protein blast was about 65%. Therefore, the 3D structure of Eg14-3-3 was modeled with acceptable accuracy. The predicted structure was visualized through the UCSF Chimera v1.11 standalone software.
29
The model refinement
The model was automatically refined via the loop model module of the EasyModeller software. The Swiss-Pdb Viewer v4.1.0 software was used for normalization of the internal aberrant interactions within the model.
30
This step was performed using the energy minimization tool in a vacuum with the GROMOS96 43B1 parameters set and without the reaction field.
The model validation assays
The model quality was assessed based on the GA341 score and DOPE (Discrete Optimized Protein Energy) scores of EasyModeller program. The ProSA web-server implemented for the prediction of the local and overall model quality.
31
The Verify3D server was exploited to assess the quality of the refined model.
32
The homology modeling stereochemical accuracy was also evaluated based on ϕ and ψ Torsion angles by means of Ramachandran’s map obtained from RAMPAGE online server.
33
Linear and conformational B-cell epitope prediction
The protein sequence of Iran Eg14-3-3 isolate (GenBank: AMX81438) has been served with the prediction of BE and TE. In order to improve the accuracy of computational epitope mapping, we used different online predictor servers, including ElliPro
34
LBTope,
35
BepiPred v1.0,
36
BcePred,
37
ABCpred,
38
and Immune Epitope Database (IEDB) server (window size 7, threshold 1.0).
39-42
The BepiPred can predict potential epitopes based on a combination of a hidden Markov model and a propensity scoring method with the values above 0.35 as the threshold. In BcePred server, epitopes were predicted via the physicochemical properties and default thresholds. The linear BEs in ABCpred server were predicted based on the artificial neural network algorithm (windows size 14, threshold 0.51). The conformational epitopes were selected based on the high-score outcomes of SEPPA v2.0,
43
ElliPro,
34
and DiscoTope v2.0 (threshold: -3.7) online servers.
44
Shannon entropy analysis for predicted epitopes
The protein sequence of local Eg14-3-3 was aligned with five NCBI’s complete sequences of other reported Eg14-3-3 isolate (GenBank accessions: CDS22750, CDS15498, AAX73175, AAM94863, AAF19966). Afterward, imprecise regions at both ends of sequences were trimmed using BioEdit package v7.1.3.
45
The variability metrics for these sequences were then characterized based on Shannon’s entropy (Hx) plot obtained by means of BioEdit software. Shannon’s entropy data were imported into Excel program to make the Shannon variability plot.
CD4+ T-helper epitope prediction using docking method
Two highly frequent Dog Leukocyte Antigen (DLA)-DRB1 alleles were selected (01101 and 01501) for docking. The simulation and binding prediction were performed by using Hex software v8.0.0 and between the binding groove of the DLA-class II alleles and the 13-mer fragments of the Eg14-3-3 antigen.
Modeling the 3D structure of MHC alleles and validation
Amino acid sequences of DLA-DRB1*01501 and DRB1*01101 were retrieved from IPD-MHC database (https://www.ebi.ac.uk/ipd/mhc/). The tertiary structures of MHC alleles were modeled using I-TASSER online server.
46
The DLA-DRB1*01501 model was energy minimized via Swiss-PdbViewer software.
30
The modeling validation tests were analyzed based on I-TASSER scores [C-score, TM-score, and root mean square deviation (RMSD)], ProSA z-scores and Ramachandran plots obtained from Chimera software.
MHC-peptide molecular docking
The Eg14-3-3 model and the MHC alleles were initially processed using Dock Prep tool of Chimera software. The key changes were automatically applied upon the molecular docking, including (i) deletion of solvent molecules, (ii) removal of alternate locations keeping only the highest occupying positions, (iii) replacement of incomplete side chains by Dunbrack rotamer library, and (iv) addition of hydrogen atoms and charge.
47
The AMBER ff14SB force field was computed for the standard residues and the AM1-BCC force fields for other residues. Several 13-mer fragments of Eg14-3-3 protein sequence were separately docked with the 3D structure of both MHC alleles (01501 and 01101). The MHC-peptide docking process was performed using Hex v8.0.0 software.
28
The Hex docking orientation control was set for the default display. For reduction of the number of “false-positive” orientations, the “Number of Solutions” was set at 5000. The most negative E-total values, as the stable system, were selected as final TEs.
Visualization of epitope-MHC interactions
The hydrogen and hydrophobic interactions between MHC alleles (DLA-DRB1*01501 and 01101) and Eg14-3-3 peptide fragments were surveyed using LigPlot+ v1.4.5 program.
11
Engineering of the designed fusion construct
The consensus B- and T-cell epitopes were bound together through the appropriate amino acid spacers. The 3D structure of vaccine construct was modeled via I-TASSER server based on the threading and ab initio modeling approaches.
48
The modeling validation assays (e.g., Z-score, C-score, TM-score, and RMSD) were performed for evaluation of the model stability.
Computing the physicochemical and 3D properties
The physicochemical and structural properties of the formulated fusion vaccine construct were characterized by means of Expasy’s ProtParam tool.
49
The calculated parameters were as follows: peptide length, molecular weight, theoretical isoelectric point (pI), in vitro and in vivo based estimated half-life, stability index, aliphatic index, extinction coefficient and “grand average of hydropathicity” (GRAVY). Furthermore, the net charge at pH 7.0 was computed via protein calculator v3.4 (http://protcalc.sourceforge.net/).
Back translation, codon optimization, and mRNA stability
Species-specific codon bias is a crucial factor for the protein expression in different host’s cells. Further, a large number of G/C content and also the formation of an unstable mRNA folding may cause negative impact(s) on the translation initiation and elongation steps. Thus, the Codon Adaptation Index (CAI), G/C content, mRNA secondary structure and Gibbs free energy (∆G) of the optimized mRNA structure were performed using Visual Gene Developer v1.7 program.
50
The codon frequency table of E. coli K12, which is postulated for codon and mRNA analysis, was directly imported from CUTG (Codon Usage Tabulated from GenBank) website (http://www.kazusa.or.jp/codon/).
Results
PCR-based DNA amplification
The double-stranded PCR product of Eg14-3-3 (approximately 744 bp) was visualized on a cyber-green stained agarose gel electrophoresis (Fig. 1a).
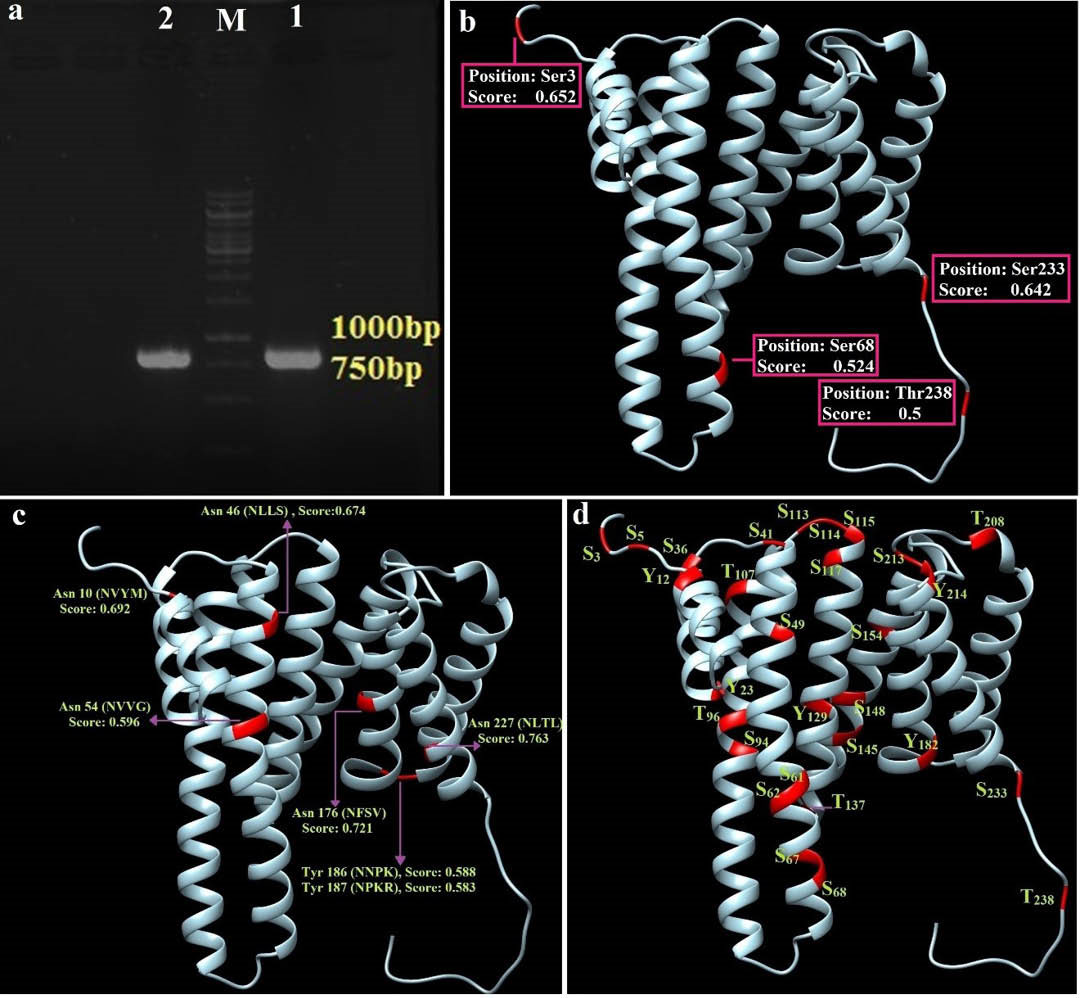
Fig. 1.
Amplification of genomic DNA of E. granulosus protoscoleces and structure-based representation of the predicted post-translational modifications. a) The gene was amplified by polymerase chain reaction (PCR) in two repeats (1 & 2). M: 1 kb DNA marker. b) Shows four potential mucin type O-glycosylation regions and their prediction scores. c) The N-glycolysation residues and prediction values. d) The potential phosphorylation of serine, threonine and tyrosine residues as the 3D structure of the Eg14-3-3 protein.
.
Amplification of genomic DNA of E. granulosus protoscoleces and structure-based representation of the predicted post-translational modifications. a) The gene was amplified by polymerase chain reaction (PCR) in two repeats (1 & 2). M: 1 kb DNA marker. b) Shows four potential mucin type O-glycosylation regions and their prediction scores. c) The N-glycolysation residues and prediction values. d) The potential phosphorylation of serine, threonine and tyrosine residues as the 3D structure of the Eg14-3-3 protein.
Transmembrane topology and signal peptide prediction
The results of TOPCONS, TMpred, TMHMM v2.0, and SignalP web-servers showed that there was no significant TM-helix and signal peptide in the Eg14-3-3 protein sequence (Figs S1, S2, S3 and S4, Electronic Supplementary Data). Therefore, the whole protein sequence was used for prediction of the BE(s).
Post-translational modifications
The neural network-based NetOGlyc v4.0 online server showed that there were four potential glycosylation sites, where the glycosylation score was higher than 0.5 (Fig. 1b). The N-linked glycosylation sites in the Eg14-3-3 appear to be seven asparagine residues consisting of Asn10, 46, 54, 176, 186, 187 and 227 (Fig. 1c). The output of NetPhos v2.0 server indicated that there were 18 phosphorylated serine residues (S), 4 threonines (T) and 5 tyrosines (Y) phosphorylation sites in the Eg14-3-3 protein sequence (Fig. 1d). The results of N-glycosylation and phosphorylation were also plotted (Fig. S5, Electronic Supplementary Data).
Potential allergenic cross-reactivity
In AlgPred server, the prediction score of SVMc method based on amino acid composition (threshold= -0.4) and dipeptide composition (threshold= -0.2) was found to be about -0.696 and -0.419, respectively. Based on the SDAP prediction algorithm, the similarities (%) between Eg14-3-3 sequence and the most similar templates were ranged from 5.67 to 12.96, which are less than the threshold (35%). Moreover, in these alignments, no contiguous amino acids were observed. Based on the results of the SDAP and hybrid method of AlgPred, the Eg14-3-3 was not detected as an allergenic protein.
Accessibility and hydrophobicity
The accessibility and hydrophobicity plots of residues in the Eg14-3-3 protein and their positions within the 3D structure are shown in Fig. 2a and 2c . The residues between aa169-182 were predicted as a highly hydrophobic segment (Fig. 2b). In five amino acid fragments (amino acids 16-23, 132-134, 166-171 and 181-184 and 221-224), the surface accessibility scores were less than 5.0. Hence, they were predicted as highly non-accessible regions (Fig. 2d). Such hydrophobic and non-accessible residues will be ignored during the BE prediction because they are unlikely to interact with the binding site of specific Abs.
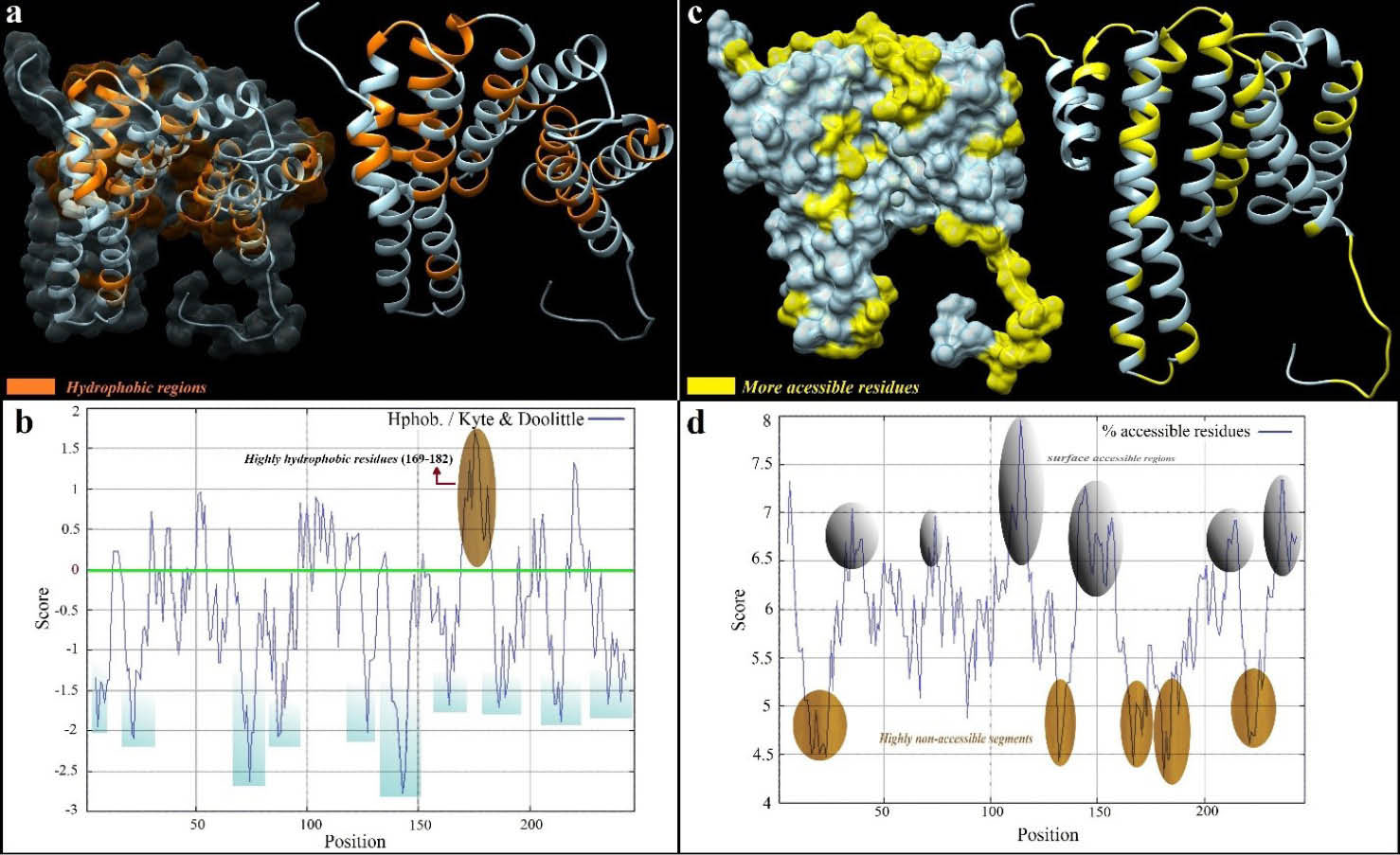
Fig. 2.
Solvent accessible and hydrophilic regions of the Eg14-3-3 protein. a) The orange residues show the hydrophilic regions of Eg14-3-3 protein as 3D structure. b) Indicates ProtScale hydrophobicity plot. One highly hydrophobic region (aa169-182) was predicted as brown. c) The surface-accessible regions are illustrated as 3D structure. d) Accessible residues are illustrated as the ProtScale plot. The residues above the threshold (6.0) are highlighted as surface accessible residues. The 3D structure of Eg14-3-3 was visualized as transparency and ribbon style via UCSF Chimera software (freely available at: http://www.rbvi.ucsf.edu/chimera).
.
Solvent accessible and hydrophilic regions of the Eg14-3-3 protein. a) The orange residues show the hydrophilic regions of Eg14-3-3 protein as 3D structure. b) Indicates ProtScale hydrophobicity plot. One highly hydrophobic region (aa169-182) was predicted as brown. c) The surface-accessible regions are illustrated as 3D structure. d) Accessible residues are illustrated as the ProtScale plot. The residues above the threshold (6.0) are highlighted as surface accessible residues. The 3D structure of Eg14-3-3 was visualized as transparency and ribbon style via UCSF Chimera software (freely available at: http://www.rbvi.ucsf.edu/chimera).
Evaluation of the predicted secondary structure
Based on our analysis, there were 11 α-helix and 12 coil regions within the secondary structure of the Eg14-3-3 (Fig. S6, Electronic Supplementary Data).
Assessment of Eg14-3-3 homology modeling quality
The 3D structure of modeled Eg14-3-3 is shown in Fig. 3. The EasyModeller software provided two types of validation scores, including the GA341 score that is ranged from zero to 1.0, and the normalized DOPE score as an energy profile for each residue. In this regard, the model with correct folding showed GA341 scores near 1.0 and the model with more negative DOPE values counted as a stable model. The energetic values of the GA341 score and DOPE score relevant to the modeled Eg14-3-3 were 1.0, and -19088.43 kcal/mole, respectively. The DOPE score was altered to -26746.53 after loop refinement. Therefore, the relative conformational stability of the predicted model was increased. The DOPE score profile for the initially generated model and the loop-refined model are displayed in Fig. 3a . The Eg14-3-3 model was re-arranged after energy minimization (EM) and the EM value was measured -12650.81 kJ/mol. Two main structural rearrangements occurred at the Gly138 and Cys20 amino acids (Fig. 3c).
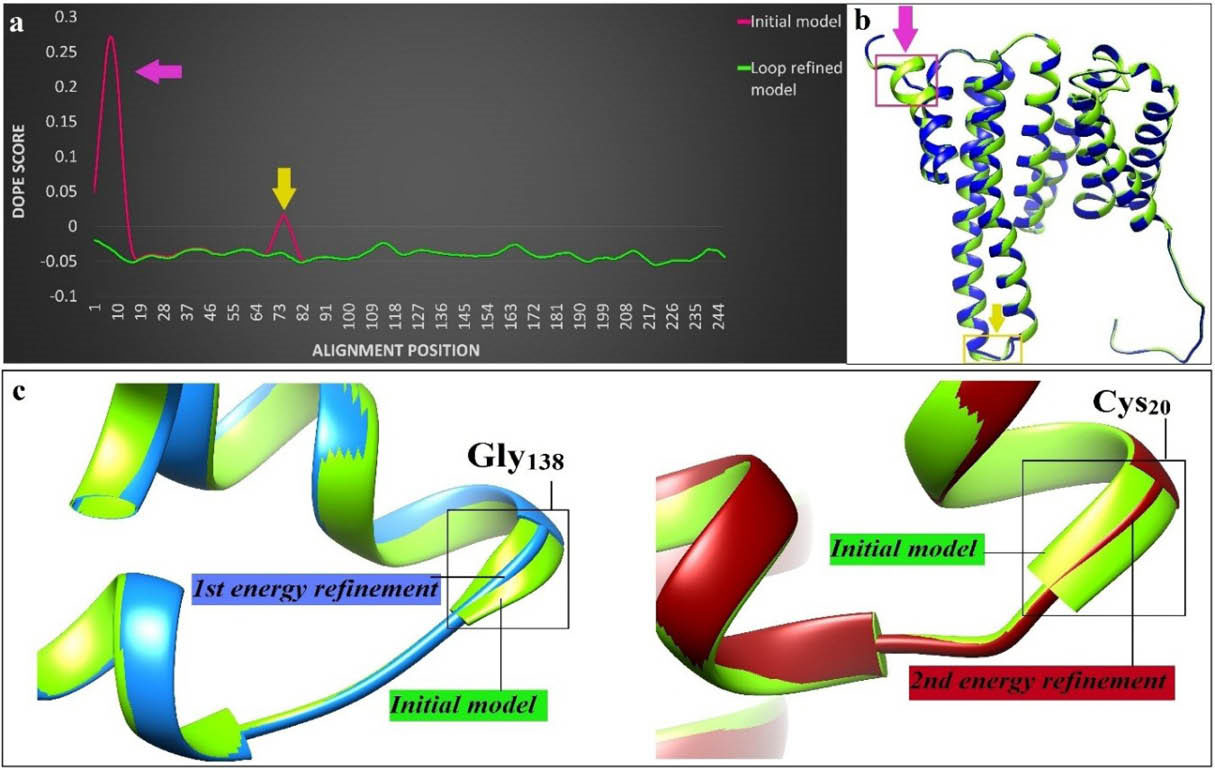
Fig. 3.
The model loop-refinement and energy minimization. a) The DOPE score values of each residue were imported to Excel program and their related profiles before (red) and after (green) loop refinement were plotted. b) The 3D structure of the loop-refined model (green structure) and the newly built model (blue structure) were compared through MatchMaker tool of Chimera software. Two main changes after loop-refinement are marked as arrows. c) The structural superimposed image of the initially predicted model (green color) and geometrically minimized model. Alpha helix of glycine 138 (left) and cysteine 20 (right) in initial model were converted to strand after the first and second energy refinements, respectively.
.
The model loop-refinement and energy minimization. a) The DOPE score values of each residue were imported to Excel program and their related profiles before (red) and after (green) loop refinement were plotted. b) The 3D structure of the loop-refined model (green structure) and the newly built model (blue structure) were compared through MatchMaker tool of Chimera software. Two main changes after loop-refinement are marked as arrows. c) The structural superimposed image of the initially predicted model (green color) and geometrically minimized model. Alpha helix of glycine 138 (left) and cysteine 20 (right) in initial model were converted to strand after the first and second energy refinements, respectively.
C-alpha atoms of the Eg14-3-3 were analyzed by ProSA-web server and exhibited the overall model quality by the z-score value of -7.21 (Fig. S7, Electronic Supplementary Data). The Verify3D-1D profile showed that 83.81% of the model residues has an averaged 3D-1D score over 0.2 (Fig. S8, Electronic Supplementary Data).
Ramachandran plot of the predicted model before and after energy minimization was also assessed (Fig. S9, Electronic Supplementary Data). After structure optimization, the number of residues within the favored region was increased from 94.7% (232 amino acids) to 98.8% (242 amino acids). Before energy minimization, seven residues (2.9%) were within the outlier region, however, after energy optimization, all of these residues were distributed to the favored and/or allowed regions.
B-cell epitope prediction and analysis of their mutative/conservative
We used two types of algorithms for increasing accuracy of the BE prediction, including (i) the physicochemical-based algorithms (e.g., Bcepred and IEDB-based linear epitope prediction) and (ii) the machine learning based algorithms (e.g., ABCpred, BepiPred, and LBtope). The residue-based scores for different predictor web-servers were imported into the Microsoft Excel for plotting and selection of the best BEs (Figs. 4 and 5).
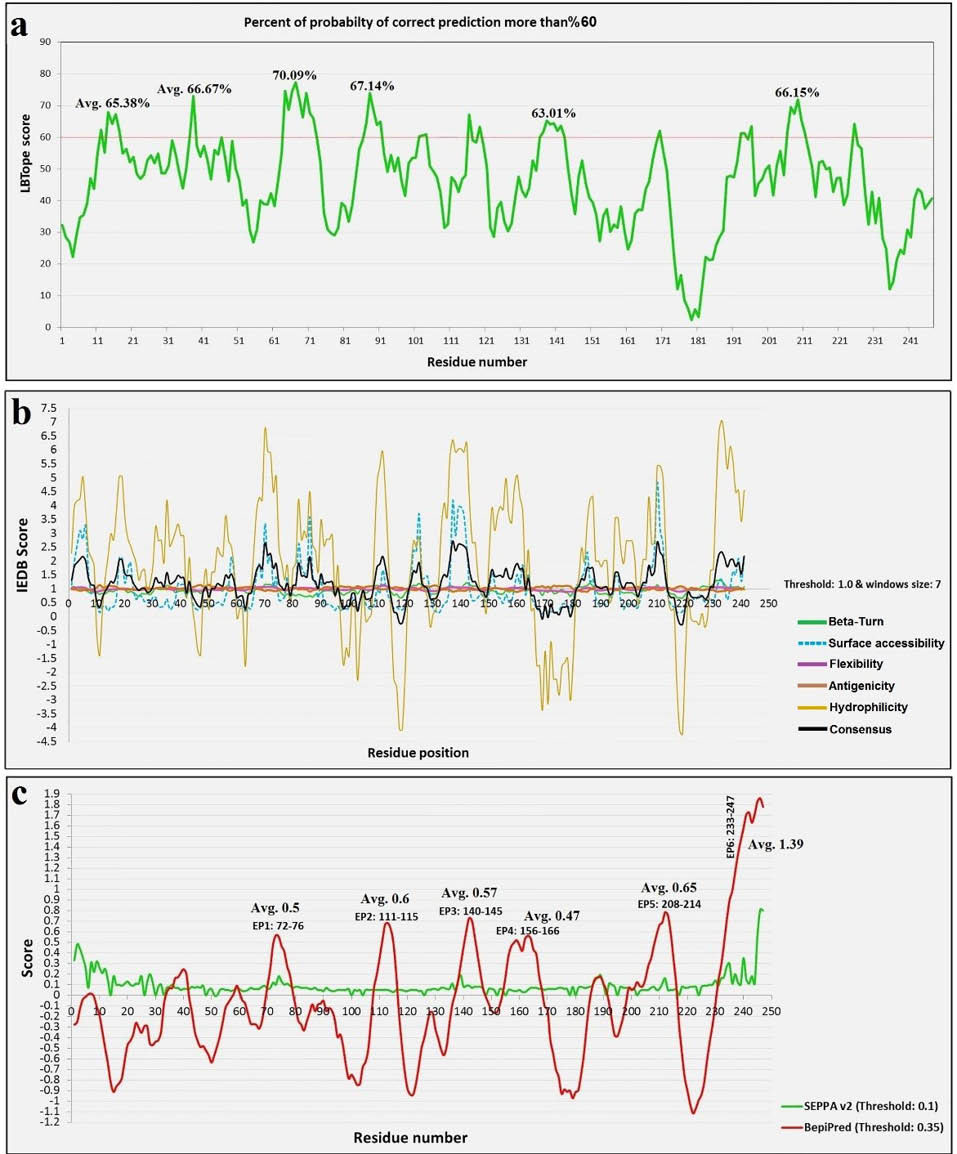
Fig. 4.
The BE prediction scores for LBTope, IEDB, SEPPA and BepiPred web-servers. a) The plot obtained by the scores of LBtope server. In six positions the values are more than the threshold (60%). b) The result of five different physicochemical algorithms of IEDB online server (flexibility, antigenicity, surface accessibility, beta-turn, and hydrophilicity) and the consensus result (black curve) plotted for prediction of linear BEs. c) The residue-based scores of the SEPPA and BepiPred online tools. Six high-score peptides and their average values that were predicted by means of BepiPred server were exhibited top of the red curve.
.
The BE prediction scores for LBTope, IEDB, SEPPA and BepiPred web-servers. a) The plot obtained by the scores of LBtope server. In six positions the values are more than the threshold (60%). b) The result of five different physicochemical algorithms of IEDB online server (flexibility, antigenicity, surface accessibility, beta-turn, and hydrophilicity) and the consensus result (black curve) plotted for prediction of linear BEs. c) The residue-based scores of the SEPPA and BepiPred online tools. Six high-score peptides and their average values that were predicted by means of BepiPred server were exhibited top of the red curve.
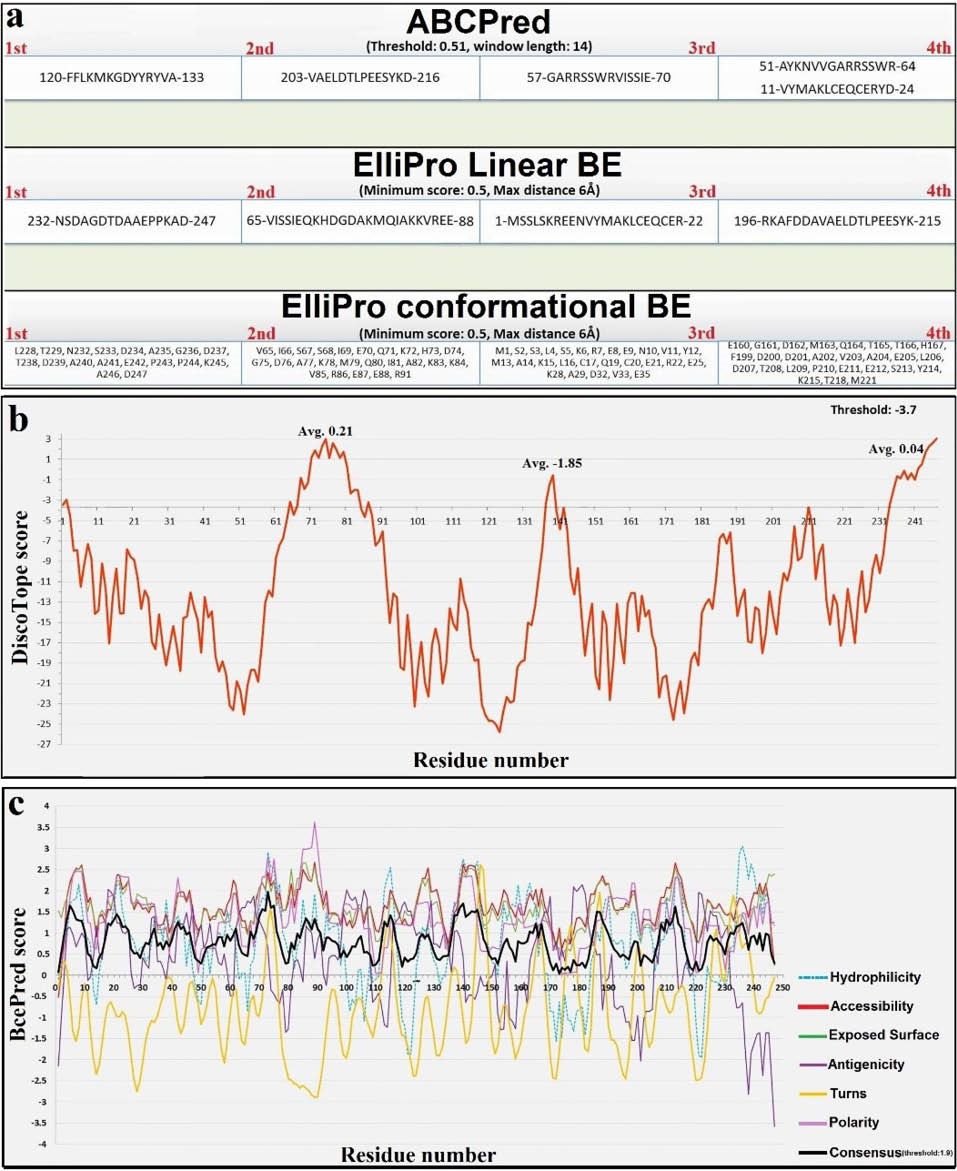
Fig. 5.
The residue-based scores for different BE predictor web-servers. a) The graphical panel of BcePred prediction result. The curve of six different methods and result of the consensus method (black curve) are indicated simultaneously. b) The sequence of four high-rank epitopes that were predicted by means of ABCPred and ElliPro servers. c) The residue-based values of DiscoTope web-server. Three positions are observed as possible discontinuous BEs (threshold: -3.7). Avg: Average.
.
The residue-based scores for different BE predictor web-servers. a) The graphical panel of BcePred prediction result. The curve of six different methods and result of the consensus method (black curve) are indicated simultaneously. b) The sequence of four high-rank epitopes that were predicted by means of ABCPred and ElliPro servers. c) The residue-based values of DiscoTope web-server. Three positions are observed as possible discontinuous BEs (threshold: -3.7). Avg: Average.
Mean Shannon’s entropy value for Eg14-3-3 isolates was 0.584, implying that the Eg14-3-3 isolates are semi-conserved among different E. granulosus strains (Fig. S10, Electronic Supplementary Data). The entropy values at each residue position in the alignment were ranged from zero (high conservation) to 1.242 (hyper-variable).
The computationally mapped BEs and their mean entropy (Hx) values were ranked based on their scores (Table 1). These data reflect the fact that the predicted epitopes were conserved in some, but not all, of the isolated of E. granulosus. The mean entropy values for each predicted epitope provide some information in terms of the vaccine coverage. The epitopes 1 (LTLWNSDAGDTDAAEPPKAD) and 4 (FCTGDERKQASDNS) exhibited a lower sequence variability, thus can cover more variety of E. granulosus strains.
Table 1.
The consensus predicted B-cell epitopes
| No. |
Consensus B-cell Epitope* |
Position |
Length |
Mean entropy values |
| 1 |
LTLWNSDAGDTDAAEPPKAD |
228–247 |
20 |
0.376 |
| 2 |
MSSLSKREENVYMAKLCEQCERYDE |
1–25 |
25 |
0.845 |
| 3 |
RKAFDDAVAELDTLPEESYKD |
196–216 |
21 |
0.508 |
| 4 |
FCTGDERKQASDNS |
135–148 |
14 |
0.372 |
| 5 |
GARRSSWRVISSIEQKHDGDAKMQIAKKVREE |
57–88 |
32 |
0.427 |
*The predicted epitopes were sorted based on their score.
Validation of homology modeling of the MHC alleles and energy minimization
The modeling validation parameters, including RMSD, C-score, TM-score (template modeling score) and ProSA Z-score is shown in Table S1. The zero and negative values in ProSA plots were found to be related to the stabilized models. Therefore, the 3D predicted models were found to be structurally acceptable (Fig. S11, Electronic Supplementary Data). The topological stereochemistry of DLA-DRB1*01501 model after energy minimization was -7270.35 kJ/mol. The energy minimization for DRB1*01101 model resulted in some residual dislocations, while the percentage of favored residues in Ramachandran plot was decreased. Therefore, the prepared DRB1*01501 model and initially generated DRB1*01101 model were used for the subsequent docking simulation (Fig. 6).
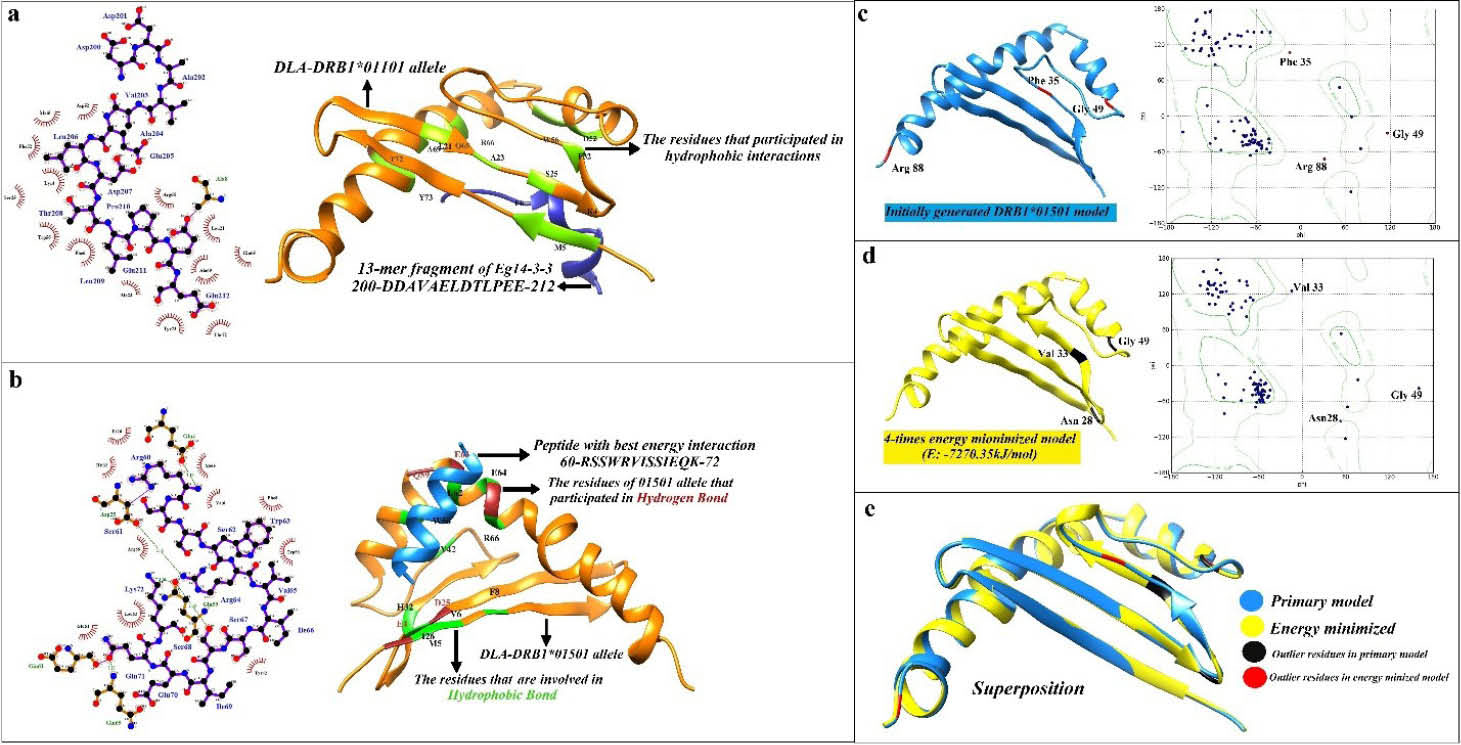
Fig. 6.
Visualization of the 3D structure of DLA-DRB1*01501 allele, and the DLA-epitope docking. a) The docking analysis of DLA-DRB1*01101 and the peptide fragment of Eg14-3-3 from 200 to 212. The LigPlot software was used to plot the left image in the panel a, showing a two-dimensional diagram of the intra-molecular hydrophobic (red spline curves) and hydrogen (green dotted lines) binding interactions. The UCSF Chimera program was used to plot the right image in the panel a. In two-dimensional diagrams, the peptide bonds, as well as MHC allele bonds, are shown as purple and brown lines, respectively. b) The docking between DLA-DRB1*01501 and the peptide fragment of Eg14-3-3 from 60 to 72. c) Initially modeled DLA-DRB1*01501 allele and its Ramachandran plot. Phe35, Gly49 and Arg88 were located into the outlier region of the plot. d) Tertiary structure and Ramachandran plot of DRB1*01501 allele after energy minimization. E-total for the energy-minimized model was -7270.35 kJ/mol. e) The structural superposition of tertiary structure of DRB1*01501 allele before and after geometry optimization. MatchMaker tool of Chimera (Needleman-Wunsch algorithm, BLOSUM-62 matrix) was conducted for structural comparison. Iterative pruning of atom pairs exceeding 2.0 Å RMSD was used to obtain the best-matching core regions.
.
Visualization of the 3D structure of DLA-DRB1*01501 allele, and the DLA-epitope docking. a) The docking analysis of DLA-DRB1*01101 and the peptide fragment of Eg14-3-3 from 200 to 212. The LigPlot software was used to plot the left image in the panel a, showing a two-dimensional diagram of the intra-molecular hydrophobic (red spline curves) and hydrogen (green dotted lines) binding interactions. The UCSF Chimera program was used to plot the right image in the panel a. In two-dimensional diagrams, the peptide bonds, as well as MHC allele bonds, are shown as purple and brown lines, respectively. b) The docking between DLA-DRB1*01501 and the peptide fragment of Eg14-3-3 from 60 to 72. c) Initially modeled DLA-DRB1*01501 allele and its Ramachandran plot. Phe35, Gly49 and Arg88 were located into the outlier region of the plot. d) Tertiary structure and Ramachandran plot of DRB1*01501 allele after energy minimization. E-total for the energy-minimized model was -7270.35 kJ/mol. e) The structural superposition of tertiary structure of DRB1*01501 allele before and after geometry optimization. MatchMaker tool of Chimera (Needleman-Wunsch algorithm, BLOSUM-62 matrix) was conducted for structural comparison. Iterative pruning of atom pairs exceeding 2.0 Å RMSD was used to obtain the best-matching core regions.
Molecular docking for CD4+ T-cell epitope prediction
The CD4+ T-cells can recognize usually peptides with a length of 12 to 16 amino acids. Therefore, the Eg14-3-3 protein was cleaved into 48 peptides with 13 amino acids in length. These 13-mer peptide fragments were docked separately with the 3D structure of the modeled DRB1*01501 and DRB1*01101 alleles. The results of docking energy (E-Total) between each peptide and active site of the MHC-alleles were calculated (Table 2). The residues from 60 to 72 exhibited the lowest Edocked value (-630.88 kcal/mol) with DRB1*01501, and the fragment from 200 to 212 showed the best interaction with DRB1*01101. Therefore, these peptides were selected as the most desired and suitable TEs.
Table 2.
Interaction energy obtained of docking between 13-mer fragments of Eg14-3-3 and DLA-DRB1*01501 and 01101 alleles
|
Peptides
|
IE
a
with 01501
|
IE with 01101
|
| 1-MSSLSKREENVYM-13 |
-467.12 |
-393.37 |
| 5-SKREENVYMAKLC-17 |
-464.66 |
-377.41 |
| 10-NVYMAKLCEQCER-22 |
-476.41 |
-416.64 |
| 15-KLCEQCERYDEMV-27 |
-413.18 |
-432.72 |
| 20-CERYDEMVKAMKD-32 |
-457.45 |
-422.64 |
| 25-EMVKAMKDVLESG-37 |
-430.89 |
-426.05 |
| 30-MKDVLESGADLSV-42 |
-424.83 |
-454.77 |
| 35-ESGADLSVEERNL-47 |
-391.16 |
-480.62 |
| 40-LSVEERNLLSVAY-52 |
-401.08 |
-405.71 |
| 45-RNLLSVAYKNVVG-57 |
-546.14 |
-366.23 |
| 50-VAYKNVVGARRSS-62 |
-599.53 |
-323.88 |
| 55-VVGARRSSWRVIS-67 |
-626.77 |
-326.02 |
| 60-RSSWRVISSIEQK-72 |
-630.88b
|
-357.23 |
| 65-VISSIEQKHDGDA-77 |
-421.34 |
-475.67 |
| 70-EQKHDGDAKMQIA-82 |
-443.58 |
-418.82 |
| 75-GDAKMQIAKKVRE-87 |
-566.51 |
-364.02 |
| 80-QIAKKVREEIERE-92 |
-534.85 |
-449.85 |
| 85-VREEIERELSATC-97 |
-383.64 |
-468.54 |
| 90-ERELSATCKEILD-102 |
-410.01 |
-501.15 |
| 95-ATCKEILDLLDKT-107 |
-405.22 |
-431.04 |
| 100-ILDLLDKTLLPAA-112 |
-403.44 |
-416.42 |
| 105-DKTLLPAASSSES-117 |
-432.24 |
-425.85 |
| 110-PAASSSESKIFFL-122 |
-441.83 |
-383.09 |
| 115-SESKIFFLKMKGD-127 |
-517.40 |
-441.31 |
| 120-FFLKMKGDYYRYV-132 |
-593.50 |
-350.43 |
| 125-KGDYYRYVAEFCT-137 |
-473.07 |
-432.97 |
| 130-RYVAEFCTGDERK-142 |
-501.14 |
-383.05 |
| 135-FCTGDERKQASDN-147 |
-407.47 |
-394.59 |
| 140-ERKQASDNSLMAY-152 |
-531.54 |
-436.38 |
| 145-SDNSLMAYKSATE-157 |
-387.36 |
-403.49 |
| 150-MAYKSATEVAEGD-162 |
-480.35 |
-490.82 |
| 155-ATEVAEGDMQTTH-167 |
-395.45 |
-506.01 |
| 160-EGDMQTTHPIRLG-172 |
-421.58 |
-403.32 |
| 165-TTHPIRLGLALNF-177 |
-553.50 |
-395.60 |
| 170-RLGLALNFSVFYY-182 |
-530.83 |
-420.93 |
| 175-LNFSVFYYEIMNN-187 |
-464.83 |
-422.16 |
| 180-FYYEIMNNPKRAC-192 |
-538.72 |
-344.73 |
| 185-MNNPKRACELARK-197 |
-530.96 |
-328.38 |
| 190-RACELARKAFDDA-202 |
-524.07 |
-390.61 |
| 195-ARKAFDDAVAELD-207 |
-418.53 |
-518.86 |
| 200-DDAVAELDTLPEE-212 |
-378.88 |
-670.52b
|
| 205-ELDTLPEESYKDA-217 |
-362.19 |
-551.86 |
| 210-PEESYKDATLIMQ-222 |
-367.47 |
-441.26 |
| 215-KDATLIMQLLRDN-227 |
-452.84 |
-409.63 |
| 220-IMQLLRDNLTLWN-232 |
-534.29 |
-382.89 |
| 225-RDNLTLWNSDAGD-237 |
-460.99 |
-503.78 |
| 230-LWNSDAGDTDAAE-242 |
-398.08 |
-611.17 |
| 235-AGDTDAAEPPKAD-247 |
-447.34 |
-563.94 |
aIE: Interaction energy (kJ/mol). b The lowest interaction energy between peptide fragments of Eg14-3-3 and both MHC alleles (01501 and 01101), that are selected as CD4+ T-helper epitopes.
The interactions between the binding core of the MHC alleles and the top docked peptides were visualized using LigPlot+ and UCSF Chimera program (Fig. 6). Fourteen residues of DLA-DRB1*01101 (i.e., Lys4, Met5, Phe6, Leu21, Ala23, Ser25, Phe32, Asp52, Trp56, Gln65, Arg66, Ala69, Thr72, Tyr73) participated in the hydrophobic interaction with the peptide fragment of Eg14-3-3 (residue from 200 to 212). The DRB1*01501 allele indicated ten hydrophobic and five hydrogen binding interactions with the peptide fragment (residue from 200 to 212). The residues engaged in hydrophobic interactions were Met5, Val6, Phe8, Ile26, His32, Tyr42, Trp56, Leu62, Glu64 and Arg66. The hydrogen bonds were variable in length, about 2.10 – 3.16 Å (Fig. 6).
Based on the MHC-peptide docking results, we selected two TEs with a binding ability to two types of DLA-DRB1 alleles. These two 13-mer peptide fragments, including the residues from 60 to 72 (RSSWRVISSIEQK) and residues from 200 to 212 (DDAVAELDTLPEE), were found to associate with the highest binding affinity to the active site of DLA-DRB1*01501 and DLA-DRB1*01101, respectively.
Computational epitope-scaffold designing
To enhance the bioactivity of vaccine construct, it is so important to assemble the epitopes together via appropriate linker amino acids based on their antigenicity and immune-dominancy. Some important epitopes can be intentionally repeated in the vaccine construct to obtain maximal immune system responses. For this reason, the selected TEs were inserted into the vaccine construct with two repeats, and HEYGAEALERAG sequence was added between the repeated TEs. We used KFERQ linker sequence at the both NH2– and –COOH termini. This specific linker can enzymatically and/or chemically be cleaved by proteasomal degradation machinery, making a rational distance between the minigene construct and expression plasmid. Additionally, due to their sensitivity to proteasomes, the linker can promote the peptides cleavage efficiency.
51,52
A four-residue spacer, Gly-Pro-Ser-Leu (GPSL), was served between the T and B cell epitopes. This type of linkers acts as a flexible spacer and can provide a free folding potential for T and B cell epitopes independently.
53
A short linker peptide, GGSSGG, was used between the BEs to minimize the interference between the adjacent epitopes and prevent formation of the new epitope. A six-glycine linker was conjugated upstream of six histidine-tag in order to minimize the possible effect of the tag on other epitopes.
54,55
The Shine-Dalgarno sequence (AGGAGG) was added to the upstream of the start codon to improve the mRNA stability, ribosomal orientation, and translation initiation. The scaffold of the vaccine construct is displayed in Fig. 7. The C-score, TM-score and estimated RMSD of the construct 3D structure were -2.30, 0.44±0.14 and 10.9±4.6 Å, respectively.
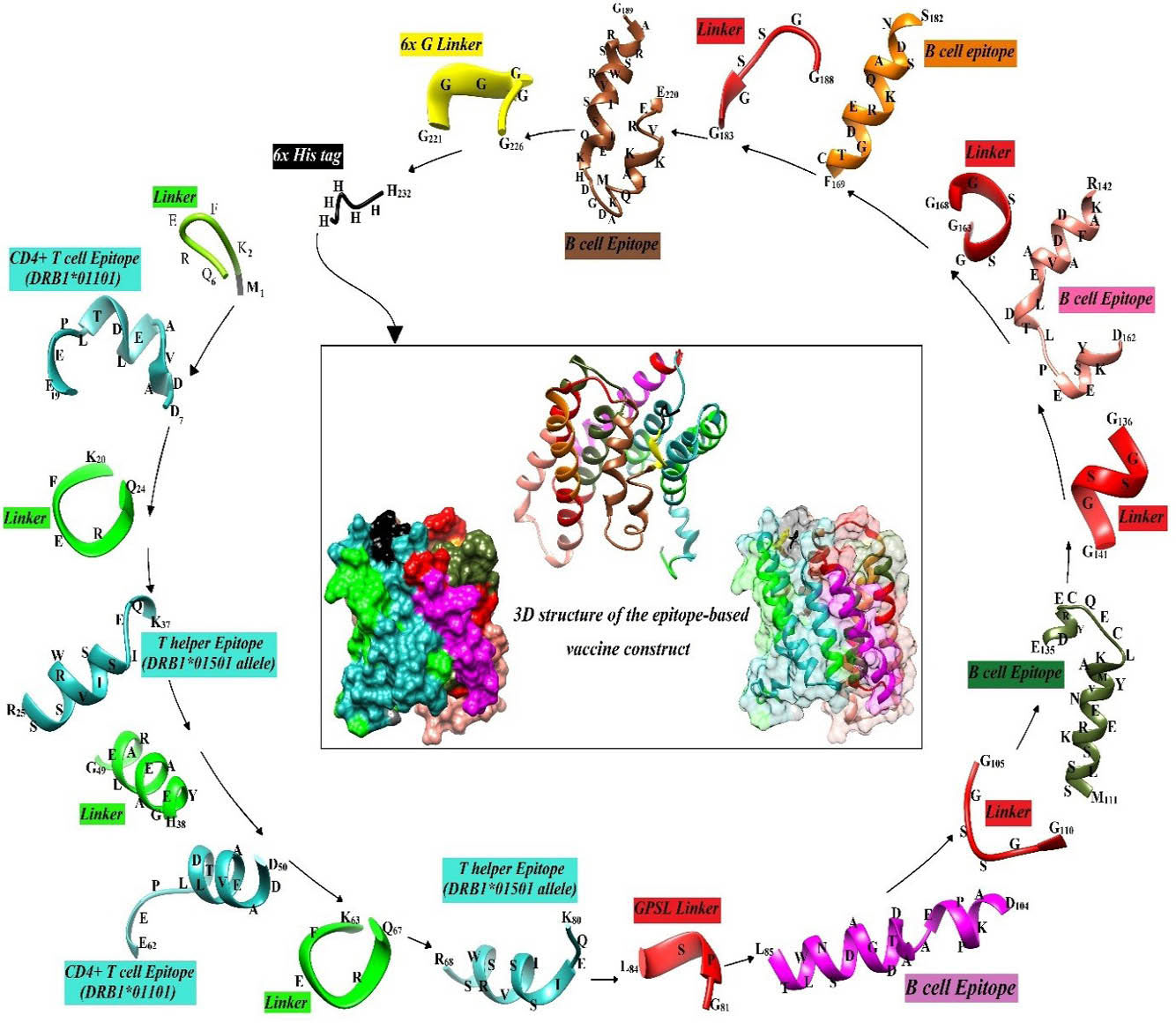
Fig. 7.
Flowchart for scaffolding the vaccine construct and 3D structure of the minigene construct. The helper T- and B-cell epitopes are separated by the specific molecular linkers (KFERQ, HEYGAEALERAG, GPSL, GGSSGG, and GGGGGG). The amino acid sequence corresponding to each peptide was written on the 3D structure of the peptides.
.
Flowchart for scaffolding the vaccine construct and 3D structure of the minigene construct. The helper T- and B-cell epitopes are separated by the specific molecular linkers (KFERQ, HEYGAEALERAG, GPSL, GGSSGG, and GGGGGG). The amino acid sequence corresponding to each peptide was written on the 3D structure of the peptides.
Computing the physicochemical properties of the construct
The full-length sequence of the designed vaccine construct in consideration with six histidine tag in C-terminal was 232 aa. The molecular weight of the protein vaccine was predicted to be ~ 25 kDa. Three most abundant residues in the minigene construct were Gly (12.5%), Ser (12.1%) and Gln (11.6%). Theoretical isoelectric point (pI) was calculated to be 5.01, thus it was classified as an acidic protein. The half-life of the construct in mammalian reticulocytes, yeast, and E. coli was estimated 30 h, >20 h, and >10 h, respectively. The instability index (II) was calculated to be 67.94 that was more than 40 as a threshold. Aliphatic index (AI), which can explain the thermal stability of the target protein, was estimated to be 50.13. The AI values are ranged from 65.36 to 138.39, where the proteins with more values can be stable in a wide temperature range. The extinction coefficient at 280 nm wavelength was 28085 M-1 cm-1. This index elucidates the protein-protein and protein-ligand interactions in the solution condition. Sum of the hydropathy values of all the amino acids was reported as “grand average hydropathy” (GRAVY) index, which was computed to be -1.042. The negative values of GRAVY indicate the protein hydrophilicity and its tendency for interaction with the surrounding water molecules. The net charge of the vaccine construct was predicted to be -13.2 at pH 7.0 based on Protein Calculator v3.4 web-server. The estimated net charge cannot be verifiable for folded proteins; however, it may be useful for the protein purification planning.
Back-translation, codon usage, and mRNA secondary structure optimization
One of the approaches for improvement in the gene expression level is the optimization of codons and the secondary structure of mRNA. In this sense, the amino acid sequence of the multi-epitope construct was reversely translated to the DNA sequence using Visual Gene Developer v1.7. Further, E. coli K12 codon frequency table was used to replace each amino acid with the corresponding and most frequently occurring codon. Table S2 represents several key parameters, including the overall G/C content, G/C content at the first, second and third places, tRNA adaptation index (tAI), the effective number of codons (Nc) and CAI values before and after the optimization. Synonymous codons substitution reduced the percentage of G/C content at the third codon site. The part of unfavorable hairpin stem-loops at the beginning of the mRNA was optimized to stabilize the mRNA structure and ribosomal binding site. The mRNA secondary structure, their overall Gibbs free energy and their residue-based plots before and after the optimization are shown in Fig. 8 and Fig. S12 (Electronic Supplementary Data). The Gibbs free energy (ΔG) of mRNA secondary structure before, and after the optimization was -279.0 and -285.30 kcal/mol, respectively. The overall negative Gibbs free energy indicates favorable mRNA folding process. The stop codon (TAA) was finally added at the 3ꞌ-end of the optimized construct.
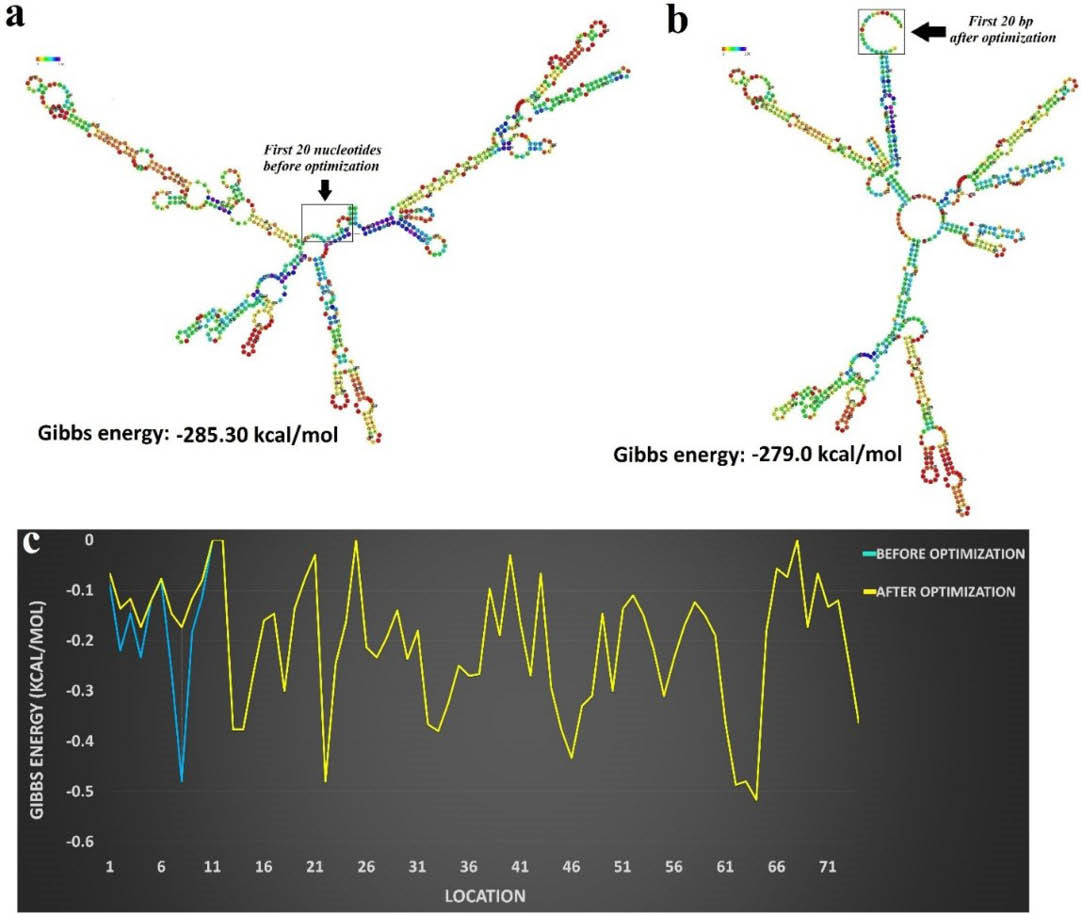
Fig. 8.
The mRNA secondary structure before and after optimization. a) The secondary structure of mRNA before energy optimization. b) The effects of energy optimization on the secondary structure of mRNA, in particular on the beginning of the sequence. c) The diagram of Gibbs energy profile for each nucleotide sequence, before (blue curve) and after (yellow curve) optimization.
.
The mRNA secondary structure before and after optimization. a) The secondary structure of mRNA before energy optimization. b) The effects of energy optimization on the secondary structure of mRNA, in particular on the beginning of the sequence. c) The diagram of Gibbs energy profile for each nucleotide sequence, before (blue curve) and after (yellow curve) optimization.
Discussion
The human health is often related to the animals’ health, in large part, because there are several human diseases (e.g., hydatid cyst) that are derived from the domestic and/or wild animals – categorized as the zoonotic diseases. For instance, dogs with echinococcus infection are the main cause of the human hydatidosis. Therefore, the aim of this study was to design an effective dog-specific prophylactic vaccine construct considering the 3D structure, post-translational modifications (PTMs) and hydrophobic/hydrophilic regions of the Eg14-3-3 used as an Ag isolated from the infected cases in Iran (Figs. 1 and 2). This designed minigene construct was comprised of BEs and TEs that were predicted based on Eg14-3-3 Ag. In this minigene vaccine, two most frequent DLA class II alleles were included in order to increase the probability of major histocompatibility (MHC) epitope binding in various dog breeds. We believe that such design, based on DLA class II alleles together with the multilateral computational assays (Figs. 3-6), might improve the coverage rate of the vaccination among different dog population.
E. granulosus infection in dogs is mainly mediated by the production of Ag-specific B-cell Abs.
14,15,56
However, differentiation of B-lymphocytes usually requires activation of naive helper T-cells. Therefore, we inserted the predicted CD4+ T-helper epitopes at the 3ꞌ-end of the construct. It should be noted that, in some bioinformatics investigations, vaccines were simply concentrated based upon the prediction of BEs from different VCAs, regardless of TEs.
57,58
In some other studies, TEs were predicted based on the major MHC alleles of animal models,
59,60
while these cannot thoroughly be substituted for the natural hosts of the parasite. All of these issues were considered for designing of the current multi-epitopic vaccine (Table 1; Figs. 6 and 7). Additionally, sequence-based TE prediction for the vaccine design against HC disease is associated with some technical limitations. Unfortunately, little is known about the canine MHC alleles. Therefore, the sequence-based TE prediction algorithms cannot be used in the prediction of specific TEs for dog vaccination. Given such facts, the structure-based approaches, especially computational molecular docking techniques, might be recruited for the prediction of either the helper or the cytotoxic T-cell epitopes.
61
In the face of extracellularly associated parasites such as nematodes and cestodes, MHC class II molecules are mainly involved in the binding and the epitope presenting to the CD4+ T-helper cells.
62
As reported previously, the DLA-DRB1 genotype is mainly associated with levels of anti-disease Abs in some of the canine diseases such as chronic hepatitis,
63
visceral leishmaniasis,
64
and rheumatoid arthritis.
65
MHC-DRB1 in canine has been suggested as a key factor for the resistance or susceptibility to infectious diseases. Moreover, among different studies on dog populations, DRB1 allele is a highly frequent allele than the DLA-DQA1 and DLA-DQB1 alleles.
64
Generally, two points have been considered in the selection of the most suitable MHC alleles for the TE prediction, including (i) the frequency of DLA-DRB1 alleles between diverse dog breeds
63,66-69
and (ii) the allele(s) associated with the parasitic diseases. Thereby, two high frequent DLA class II alleles (DRB1*01501 and DRB1*01101) were served for the peptide-MHC docking and finally for the prediction of MHC class II-restricted TEs (Table S1; Figs. 6 and S11).
The Eg14-3-3 is a secretory component from the rostellar glands of E. granulosus adult worms. Immunolocalization studies have revealed some localized areas of Eg14-3-3 in 35-day-old worms. In this developmental stage, the Eg14-3-3 was reported to be discernible within the periphery of testicle cells, apical of rostellum and around tegument.
13
Further, the capability of Eg14-3-3 as an immunotherapeutic Ag has been recently studied,
70
in which the recombinant Eg14-3-3 was extracted and purified from E. coli BL21, and subcutaneously administered to the ICR mice. This vaccination trial exhibited a high binding potential (84.7%) to the anti-Eg14-3-3 IgG subclasses (IgG1, IgG2a, and IgG2b), and significant enhancement in the production of IL-2 and INFγ against E. granulosus. Given that the parasite secretory products play key roles in the survival of the worms within the definitive host's intestine; therefore, it might count as an ideal Ag for the vaccination against E. granulosus.
70
As a part of this study, for the first time, we modeled the secondary and 3D structure of Eg14-3-3 Ag (Figs. 1 and S6). In this regard, we validated the accuracy of the protein modeling by means of different computational tools such as Verify3D, ProSA z-score and ProSA energy plot (Figs. S7 and S8). Given that the zero and negative values of z-score are related to the stabilized models, the predicted 3D model appeared to be structurally favorable. The ProSA energy plot represents the local model quality (Fig. S7). The Verify3D was used to calculate the compatibility of the 3D structure of the predicted model with its own amino acid sequence (1D), and also to evaluate various structural regions of the model. This server gives 3D-1D structural profile to assess the compatibility of each residue with their local environment.
32,71
Shannon's entropy analysis demonstrated that BEs of the designed vaccine are conserved and semi-conserved and can cover the majority of E. granulosus strains. Further, the Eg14-3-3 isolates were found to be semi-conserved among different E. granulosus strains (Fig. S10), and hence it is not necessary to consider all the isolate of this Ag for the vaccine design. The CD4+ T helper epitopes were predicted based on the molecular docking between the 13-mer peptide residues of the whole Ag sequence and the high frequent dog MHC (Canine Leukocyte Antigen, CLA/DLA) class II alleles. The physicochemical and immunological properties of the designed minigene vaccine revealed that this immunogenic peptide was not sufficiently stable at the different temperatures. Furthermore, we used the protease specificity prediction server (PROSPER) as an on line web-server used for the prediction of potential endogenous proteases activity of E. coli. The potential cleavage sites into the minigene construct were found (Fig. S13 and Table S3, Electronic Supplementary Data). It should be stated that such results upon the assessment of proteases activities would be favorable for the extraction and purification of this immunogenic peptide from E. coli.
As reported previously, an ideal vaccine should be safe, effective and broad-spectrum. That is why we recruited several steps for designing this rational EV (Fig. 7), including (i) identifying and selecting a key immunogenic Ag, (ii) using bioinformatics tools for analysis of different parameters of the selected Ag, Eg14-3-3, (iii) predicting BEs and TEs computationally, and (iv) assembling the selected epitopes with each other using the suitable molecular linkers.
41
To revalidate such approaches, we also looked at the mRNA secondary structure before and after optimization of the construct (Table S2; Figs. 8 and S12).
The expression level of many of E. granulosus VCAs are dependent upon the stage of the parasite life cycle. Therefore, for designing a strong poly-epitopic vaccine, we need to pay a close attention to several indices of the selected Ag, including the role of Ag in the parasite survival, antigenicity, immunogenicity and allergenicity, stage of Ag expression, and its localization in the parasite body [e.g., intra- and/or extracellular, somatic and/or excretory-secretory].
72
The hydrophobic interactions are the main parameters for the Ag-Ab binding energy in most of Ags.
73
Because of some known correlations between BEs and surface exposed regions of Ags,
74
we focused on the surface-exposed and accessible regions of Eg14-3-3 where there are more possibilities for interaction with the Ab binding sites.
75
Transmembrane helices need several continuous hydrophobic residues to spin accurately within the cell membrane. Accordingly, the hydrophobic transmembrane domains of the Ag cannot be detected by the immune system’s Abs. Moreover, signal sequences act as key players in terms of protein secretion onto the extracellular membrane as well as periplasmic space. Signal peptidases can cleave the signal peptide off (as co-translationally or post-translationally) to produce a mature protein.
76
Therefore, the signal peptide cannot also be as an appropriate segment for the epitope-paratope interaction (Figs. S1, S2, S3 and S4). The minigene protein processing during post-translational modifications (PTM), especially N-glycosylation, can block the accessibility of some epitopes for neutralizing Abs and affect the proteolytic process by specific immunoproteasome components.
77
It should be highlighted that the PTMs (e.g., phosphorylated and glycosylated regions) of the epitopes used for the selection of BEs need to be taken into consideration (Fig. S5), in large part because such epitopes are usually located on the unexposed regions.
78
In fact, some of the PTMs can partially/fully mask epitopes, in particular, those that are linear.
79
To the best of our knowledge, conclusively, there is no similar in silico Eg14-3-3 based multi-epitope vaccine, and this is the first report on the design of a multimodal dog-specific vaccine. In addition, for designing of this multi-epitope construct, we capitalized on various computational software and web-servers to address all issues related to the improved functionality of such multi-epitope vaccine, which is envisioned to be effective for a diverse population of dog breeds. It should be noted that the predicted epitopes of Eg14-3-3 can be used in combination with the conserved epitopes of some other Ags to produce a universal vaccine with high-rate successful vaccination.
Conclusion
The current in silico study is the first part of the multi-antigenic vaccine designing project that resulted in promising outcomes for immunization of dogs against E. granulosus. In this current work, we reported on a rationalized in silico methodology to design this novel prophylactic vaccine. The undertaken in silico approach may provide vaccinologists a robust tool for designing host-specific multi-epitopic and/or multi-antigenic vaccines against different pathogens. Our vaccine construct designed by use of the local isolate of E. granulosus from the infected cases in Iran. The TEs parts of the vaccine were predicted based on two highly frequent canine MHC alleles through several MHC-peptide docking procedures. Notwithstanding, for more accurate in silico BE identification, we modeled 3D structure of Eg14-3-3 using homology modeling methods. Taken all, the designed vaccine construct comprised of four CD4+ MHC-II epitopes, which can elicit protective MHC class II, restricted responses based on DLA-DRB1*01101 and 01501 alleles, and five BEs from E. granulosus 14-3-3 antigen. Codon usage of the construct was comprehensively optimized in terms of codon usage table of Escherichia coli k-12. Based on our findings, conclusively, we envision that the constructed dog-specific multi-epitope vaccine against echinococcosis might provide an improved treatment modality. These data may encourage the vaccinologists to focus on such multi-epitopic vaccines to fight against multi-host and genetically variable pathogens.
Ethical approval
There is none to be declared.
Conflict of interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article (doi: 10.15171/bi.2018.06) contains supplementary material, which is available to authorized users.
(pdf)
Research Highlights
What is current knowledge?
simple
-
√ The Eg14-3-3 a key antigen in the host-parasite interactions was served for designing a creative platform for vaccination of dogs against E. granulosus.
What is new here?
simple
-
√ Iran 14-3-3 antigen has been isolated from the local E. granulosus.
-
√ The homology modeling approaches were conducted for the prediction of the 3D structure of Eg14-3-3 protein.
-
√ The molecular docking methods were performed for the identification of the best MHC-CD4+ T cell epitope binding.
-
√ A multi-modal methodology for in silico epitope mapping was presented for more precise B-cell epitope prediction.
Acknowledgements
This work is part of the Ph.D. thesis project that was financially supported by the University of Tabriz and the Research Center for Pharmaceutical Nanotechnology (RCPN) at Tabriz University of Medical Sciences (grant No.: RCPN-95002).
References
- Carmena D, Cardona GA. Echinococcosis in wild carnivorous species: epidemiology, genotypic diversity, and implications for veterinary public health. Vet Parasitol 2014; 202:69-94. doi: 10.1016/j.vetpar.2014.03.009 [Crossref] [ Google Scholar]
-
Anonymous. World Health Organization (WHO). Echinococcosis. Available from: http://wwwwhoint/mediacentre/factsheets/fs377/en/2015.
- Bingham GM, Budke CM, Larrieu E, Del Carpio M, Mujica G, Slater MR. A community-based study to examine the epidemiology of human cystic echinococcosis in Rio Negro Province, Argentina. Acta Trop 2014; 136:81-8. doi: 10.1016/j.actatropica.2014.04.005 [Crossref] [ Google Scholar]
- Moro P, Schantz PM. Echinococcosis: a review. Int J Infect Dis 2009; 13:125-33. doi: 10.1016/j.ijid.2008.03.037 [Crossref] [ Google Scholar]
- Kern P. Echinococcus granulosus infection: clinical presentation, medical treatment, and outcome. Langenbecks Arch Surg 2003; 388:413-20. doi: 10.1007/s00423-003-0418-y [Crossref] [ Google Scholar]
- Vercruysse J, Knox DP, Schetters TP, Willadsen P. Veterinary parasitic vaccines: pitfalls and future directions. Trends Parasitol 2004; 20:488-92. doi: 10.1016/j.pt.2004.07.009 [Crossref] [ Google Scholar]
- Schellenberg D, Abdulla S, Roper C. Current issues for anti-malarial drugs to control P falciparum malaria. Curr Mol Med 2006; 6:253-60. doi: 10.2174/156652406776055168 [Crossref] [ Google Scholar]
- Bethony JM, Cole RN, Guo X, Kamhawi S, Lightowlers MW, Loukas A. Vaccines to combat the neglected tropical diseases. Immunol Rev 2011; 239:237-70. doi: 10.1111/j.1600-065X.2010.00976.x [Crossref] [ Google Scholar]
- Gori A, Longhi R, Peri C, Colombo G. Peptides for immunological purposes: design, strategies and applications. Amino Acids 2013; 45:257-68. doi: 10.1007/s00726-013-1526-9 [Crossref] [ Google Scholar]
- Dhanda SK, Usmani SS, Agrawal P, Nagpal G, Gautam A, Raghava GP. A novel in silico tools for designing peptide-based subunit vaccines and immunotherapeutics. Brief Bioinform 2016. doi: 10.1093/bib/bbw025 [Crossref]
- Pourseif MM, Moghaddam G, Saeedi N, Barzegari A, Dehghani J, Omidi Y. Current status and future prospective of vaccine development against Echinococcus granulosus. Biologicals 2017. doi: 10.1016/j.biologicals.2017.10.003 [Crossref]
- Siles-Lucas M, Felleisen RS, Hemphill A, Wilson W, Gottstein B. Stage-specific expression of the 14-3-3 gene in Echinococcus multilocularis. Mol Biochem Parasitol 1998; 91:281-93. doi: 10.1016/S0166-6851(97)00208-9 [Crossref] [ Google Scholar]
- Siles-Lucas M, Nunes CP, Zaha A, Breijo M. The 14-3-3 protein is secreted by the adult worm of Echinococcus granulosus. Parasite Immunol 2000; 22:521-8. doi: 10.1046/j.1365-3024.2000.00334.x [Crossref] [ Google Scholar]
- Zhang W, Ross AG, McManus DP. Mechanisms of immunity in hydatid disease: implications for vaccine development. J Immunol 2008; 181:6679-85. doi: 10.4049/jimmunol.181.10.6679 [Crossref] [ Google Scholar]
- Zhang W, Li J, McManus DP. Concepts in immunology and diagnosis of hydatid disease. Clin Microbiol Rev 2003; 16:18-36. doi: 10.1128/CMR.16.1.18-36.2003 [Crossref] [ Google Scholar]
- Rychlik W. OLIGO 7 primer analysis software. Methods Mol Biol 2007; 402:35-60. doi: 10.1007/978-1-59745-528-2_2 [Crossref] [ Google Scholar]
- Tsirigos KD, Peters C, Shu N, Kall L, Elofsson A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res 2015; 43:W401-7. doi: 10.1093/nar/gkv485 [Crossref] [ Google Scholar]
-
Hofmann K, Stoffel W. TMbase-A database of membrane spanning protein segments. 1993.
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 40: discriminating signal peptides from transmembrane regions. Nat Methods 2011; 8:785-6. doi: 10.1038/nmeth.1701 [Crossref] [ Google Scholar]
-
Gupta R, Jung E, Brubak S. NetNGlyc: Prediction of N-glycosylation sites in human proteins. software2005.
- Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J 2013; 32:1478-88. doi: 10.1038/emboj.2013.79 [Crossref] [ Google Scholar]
- Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 1999; 294:1351-62. doi: 10.1006/jmbi.1999.3310 [Crossref] [ Google Scholar]
- Saha S, Raghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res 2006; 34:W202-9. doi: 10.1093/nar/gkl343 [Crossref] [ Google Scholar]
- Ivanciuc O, Schein CH, Braun W. SDAP: database and computational tools for allergenic proteins. Nucleic Acids Res 2003; 31:359-62. doi: 10.1093/nar/gkg010 [Crossref] [ Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982; 157:105-32. doi: 10.1016/0022-2836(82)90515-0 [Crossref] [ Google Scholar]
-
Gasteiger E, Hoogland C, Gattiker A, Duvaud Se, Wilkins MR, Appel RD, et al. Protein identification and analysis tools on the ExPASy server: Springer; 2005.
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics 2000; 16:404-5. doi: 10.1093/bioinformatics/16.4.404 [Crossref] [ Google Scholar]
- Ritchie DW. Recent progress and future directions in protein-protein docking. Curr Protein Pept Sci 2008; 9:1-15. doi: 10.2174/138920308783565741 [Crossref] [ Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605-12. doi: 10.1002/jcc.20084 [Crossref] [ Google Scholar]
- Johansson MU, Zoete V, Michielin O, Guex N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC bioinformatics 2012; 13:1. doi: 10.1186/1471-2105-13-173 [Crossref] [ Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 2007; 35:W407-10. doi: 10.1093/nar/gkm290 [Crossref] [ Google Scholar]
- Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature 1992; 356:83-5. doi: 10.1038/356083a0 [Crossref] [ Google Scholar]
- Lovell SC, Davis IW, Arendall WB, 3rd 3rd, de Bakker PI, Word JM, Prisant MG. Structure validation by Calpha geometry: phi,psi, and Cbeta deviation. Proteins 2003; 50:437-50. doi: 10.1002/prot.10286 [Crossref] [ Google Scholar]
- Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 2008; 9:514. doi: 10.1186/1471-2105-9-514 [Crossref] [ Google Scholar]
- Singh H, Ansari HR, Raghava GP. An improved method for linear B-cell epitope prediction using antigen's primary sequence. PLoS One 2013; 8:e62216. doi: 10.1371/journal.pone.0062216 [Crossref] [ Google Scholar]
- Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res 2006; 2:2. doi: 10.1186/1745-7580-2-2 [Crossref] [ Google Scholar]
-
Saha S, Raghava G, editors. BcePred: Prediction of continuous
B-cell epitopes in antigenic sequences using physicochemical
properties. International Conference on Artificial Immune Systems;
2004: Springer.
- Saha S, Raghava GP. Prediction of continuous B-cell epitopes in an antigen using the recurrent neural network. Proteins 2006; 65:40-8. doi: 10.1002/prot.21078 [Crossref] [ Google Scholar]
- Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 1986; 25:5425-32. doi: 10.1021/bi00367a013 [Crossref] [ Google Scholar]
- Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 1990; 276:172-4. doi: 10.1016/0014-5793(90)80535-Q [Crossref] [ Google Scholar]
- Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol 1978; 47:45-148. doi: 10.1002/9780470122921.ch2 [Crossref] [ Google Scholar]
- Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol 1985; 55:836-9. [ Google Scholar]
- Qi T, Qiu T, Zhang Q, Tang K, Fan Y, Qiu J. SEPPA 20--more refined server to predict spatial epitope considering species of immune host and subcellular localization of protein antigen. Nucleic Acids Res 2014; 42:W59-63. doi: 10.1093/nar/gku395 [Crossref] [ Google Scholar]
- Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol 2012; 8:e1002829. doi: 10.1371/journal.pcbi.1002829 [Crossref] [ Google Scholar]
-
Hall TA, editor. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic acids symposium series; 1999: [London]: Information Retrieval Ltd., c1979-c2000.
- Gopal V, Guruprasad K. Structure prediction, and validation of an affibody engineered for cell-specific nucleic acid targeting. Syst Synth Biol 2010; 4:293-7. doi: 10.1007/s11693-011-9074-7 [Crossref] [ Google Scholar]
- Dunbrack RL, Jr Jr. Rotamer libraries in the 21st century. Curr Opin Struct Biol 2002; 12:431-40. [ Google Scholar]
- Zhang W, Yang J, He B, Walker SE, Zhang H, Govindarajoo B. Integration of QUARK and I-TASSER for Ab Initio Protein Structure Prediction in CASP11. Proteins 2016; 84 Suppl 1:76-86. doi: 10.1002/prot.24930 [Crossref] [ Google Scholar]
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 1999; 112:531-52. [ Google Scholar]
- Jung SK, McDonald K. Visual gene developer: a fully programmable bioinformatics software for synthetic gene optimization. BMC Bioinformatics 2011; 12:340. doi: 10.1186/1471-2105-12-340 [Crossref] [ Google Scholar]
- Amani J, Mousavi SL, Rafati S, Salmanian AH. In silico analysis of chimeric espA, eae and tir fragments of Escherichia coli O157:H7 for oral immunogenic applications. Theor Biol Med Model 2009; 6:28. doi: 10.1186/1742-4682-6-28 [Crossref] [ Google Scholar]
- Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng 2001; 14:529-32. [ Google Scholar]
-
Kaumaya PT, Kobs-Conrad S, DiGeorge AM, Stevens VC. “De novo” engineering of peptide immunogenic and antigenic determinants as potential vaccines. Peptides: Springer; 1994. p. 133-64.
- Shao JJ, Wong CK, Lin T, Lee SK, Cong GZ, Sin FW. Promising multiple-epitope recombinant vaccine against foot-and-mouth disease virus type O in swine. Clin Vaccine Immunol 2011; 18:143-9. doi: 10.1128/CVI.00236-10 [Crossref] [ Google Scholar]
- Funakoshi M, Hochstrasser M. Small epitope-linker modules for PCR-based C-terminal tagging in Saccharomyces cerevisiae. Yeast 2009; 26:185-92. doi: 10.1002/yea.1658 [Crossref] [ Google Scholar]
- Deplazes P, Thompson RC, Constantine CC, Penhale WJ. Primary infection of dogs with Echinococcus granulosus: systemic and local (Peyer's patches) immune responses. Vet Immunol Immunopathol 1994; 40:171-84. doi: 10.1016/0165-2427(94)90032-9 [Crossref] [ Google Scholar]
- Li YJ, Yang J, Zhao H, Jia HY, Zhang LN, Liu XX. Bioinformatics prediction of egA31 recombinant antigen epitopes of Echinococcus granulosus. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 2012; 30:78-80. [ Google Scholar]
- Lu G, Lu Y, Li L, Wu L, Fan Z, Shi D. Identification and bioinformatics analysis of lactate dehydrogenase genes from Echinococcus granulosus. Asian Pacific Journal of Tropical Medicine 2010; 3:757-61. doi: 10.1016/S1995-7645(10)60182-4 [Crossref] [ Google Scholar]
- Ma X, Zhou X, Zhu Y, Li Y, Wang H, Mamuti W. The prediction of T- and B-combined epitope and tertiary structure of the Eg95 antigen of Echinococcus granulosus. Exp Ther Med 2013; 6:657-62. doi: 10.3892/etm.2013.1187 [Crossref] [ Google Scholar]
- Gan W, Zhao G, Xu H, Wu W, Du W, Huang J. Reverse vaccinology approach identify an Echinococcus granulosus tegumental membrane protein enolase as a vaccine candidate. Parasitol Res 2010; 106:873-82. doi: 10.1007/s00436-010-1729-x [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Naghili B, Saeedi N, Parvizpour S, Nematollahi A. A novel in silico minigene vaccine based on CD4+ T-helper and B-cell epitopes of EG95 isolates for vaccination against cystic echinococcosis. Computational Biology and Chemistry 2017. doi: 10.1016/j.compbiolchem.2017.11.008 [Crossref]
- Schwensow N, Fietz J, Dausmann KH, Sommer S. Neutral versus adaptive genetic variation in parasite resistance: the importance of major histocompatibility complex supertypes in a free-ranging primate. Heredity (Edinb) 2007; 99:265-77. doi: 10.1038/sj.hdy.6800993 [Crossref] [ Google Scholar]
- Bexfield NH, Watson PJ, Aguirre-Hernandez J, Sargan DR, Tiley L, Heeney JL. DLA class II alleles and haplotypes are associated with risk for and protection from chronic hepatitis in the English Springer spaniel. PLoS One 2012; 7:e42584. doi: 10.1371/journal.pone.0042584 [Crossref] [ Google Scholar]
- Quinnell RJ, Kennedy LJ, Barnes A, Courtenay O, Dye C, Garcez LM. Susceptibility to visceral leishmaniasis in the domestic dog is associated with MHC class II polymorphism. Immunogenetics 2003; 55:23-8. doi: 10.1007/s00251-003-0545-1 [Crossref] [ Google Scholar]
- Ollier WE, Kennedy LJ, Thomson W, Barnes AN, Bell SC, Bennett D. Dog MHC alleles containing the human RA shared epitope confer susceptibility to canine rheumatoid arthritis. Immunogenetics 2001; 53:669-73. doi: 10.1007/s002510100372 [Crossref] [ Google Scholar]
- Wilbe M, Ziener ML, Aronsson A, Harlos C, Sundberg K, Norberg E. DLA class II alleles are associated with risk for canine symmetrical lupoid onychodystrophy [corrected](SLO). PLoS One 2010; 5:e12332. doi: 10.1371/journal.pone.0012332 [Crossref] [ Google Scholar]
- Runstadler JA, Angles JM, Pedersen NC. Dog leucocyte antigen class II diversity and relationships among indigenous dogs of the island nations of Indonesia (Bali), Australia and New Guinea. Tissue Antigens 2006; 68:418-26. doi: 10.1111/j.1399-0039.2006.00696.x [Crossref] [ Google Scholar]
- Kennedy LJ, Barnes A, Short A, Brown JJ, Lester S, Seddon J. Canine DLA diversity: 1 New alleles and haplotypes. Tissue Antigens 2007; 69 Suppl 1:272-88. doi: 10.1111/j.1399-0039.2006.00779.x [Crossref] [ Google Scholar]
- Tsai KL, Starr-Moss AN, Venkataraman GM, Robinson C, Kennedy LJ, Steiner JM. Alleles of the major histocompatibility complex play a role in the pathogenesis of pancreatic acinar atrophy in dogs. Immunogenetics 2013; 65:501-9. doi: 10.1007/s00251-013-0704-y [Crossref] [ Google Scholar]
- Li ZJ, Wang YN, Wang Q, Zhao W. Echinococcus granulosus 14-3-3 protein: a potential vaccine candidate against challenge with Echinococcus granulosus in mice. Biomed Environ Sci 2012; 25:352-8. doi: 10.3967/0895-3988.2012.03.014 [Crossref] [ Google Scholar]
- Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 1997; 277:396-404. [ Google Scholar]
- Monterrubio-Lopez GP, Gonzalez YMJA, Ribas-Aparicio RM. Identification of Novel Potential Vaccine Candidates against Tuberculosis Based on Reverse Vaccinology. Biomed Res Int 2015; 2015:483150. doi: 10.1155/2015/483150 [Crossref] [ Google Scholar]
- Murphy K, Weaver C. Janeway's immunobiology. Garland Science 2016. doi: 10.1002/bmb.20272 [Crossref]
- Thornton JM, Edwards MS, Taylor WR, Barlow DJ. Location of 'continuous' antigenic determinants in the protruding regions of proteins. EMBO J 1986; 5:409-13. doi: 10.1002/j.1460-2075.1986.tb04226.x [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Saeedi N, Barzegari A, Dehghani J, Omidi Y. Current status and future prospective of vaccine development against Echinococcus granulosus. Biologicals 2018; 51:1-11. doi: 10.1016/j.biologicals.2017.10.003 [Crossref] [ Google Scholar]
-
Dalbey R, von Heijne G. Protein targeting, transport, and translocation: Academic Press; 2002.
- Wolfert MA, Boons GJ. Adaptive immune activation: glycosylation does matter. Nat Chem Biol 2013; 9:776-84. doi: 10.1038/nchembio.1403 [Crossref] [ Google Scholar]
- Dowling W, Thompson E, Badger C, Mellquist JL, Garrison AR, Smith JM. Influences of glycosylation on antigenicity, immunogenicity, and protective efficacy of ebola virus GP DNA vaccines. J Virol 2007; 81:1821-37. doi: 10.1128/JVI.02098-06 [Crossref] [ Google Scholar]
- Caoili SE. Benchmarking B-cell epitope prediction for the design of peptide-based vaccines: problems and prospects. J Biomed Biotechnol 2010; 2010:910524. doi: 10.1155/2010/910524 [Crossref] [ Google Scholar]