BioImpacts. 9(1):15-23.
doi: 10.15171/bi.2019.03
Original Research
Highly efficient novel recombinant L-asparaginase with no glutaminase activity from a new halo-thermotolerant Bacillus strain
Azam Safary 1  , Rezvan Moniri 2, 3, Maryam Hamzeh-Mivehroud 4, 5, Siavoush Dastmalchi 4, 5, 6, *
, Rezvan Moniri 2, 3, Maryam Hamzeh-Mivehroud 4, 5, Siavoush Dastmalchi 4, 5, 6, *
Author information:
1 Connective Tissue Diseases Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
2 Anatomical Sciences Research Center, Kashan University of Medical Sciences, Kashan, Iran
3 Department of Microbiology and Immunology, Faculty of Medicine, Kashan University of Medical Sciences, Kashan, Iran
4 Biotechnology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
5 School of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
6 Faculty of Pharmacy, Near East University, POBOX:99138, Nicosia, North Cyprus, Mersin 10, Turkey
Abstract
Introduction:
The bacterial enzyme has gained more attention in therapeutic application because of the higher substrate specificity and longer half-life. L-asparaginase is an important enzyme with known antineoplastic effect against acute lymphoblastic leukemia (ALL).
Methods:
Novel L-asparaginase genes were identified from a locally isolated halo-thermotolerant Bacillus strain and the recombinant enzymes were overexpressed in modified E. coli strains, OrigamiTM B and BL21. In addition, the biochemical properties of the purified enzymes were characterized, and the enzyme activity was evaluated at different temperatures, pH, and substrate concentrations.
Results:
The concentration of pure soluble enzyme obtained from Origami strain was ~30 mg/L of bacterial culture, which indicates the significant improvement compared to L-asparaginase produced by E. coli BL21 strain. The catalytic activity assay on the identified L-asparaginases (ansA1 and ansA3 genes) from Bacillus sp. SL-1 demonstrated that only ansA1 gene codes an active and stable homologue (ASPase A1) with high substrate affinity toward L-asparagine. The Kcat and Km values for the purified ASPase A1 enzyme were 23.96s-1 and 10.66 µM, respectively. In addition, the recombinant ASPase A1 enzyme from Bacillus sp. SL-1 possessed higher specificity to L-asparagine than L-glutamine. The ASPase A1 enzyme was highly thermostable and resistant to the wide range of pH 4.5–10.
Conclusion:
The biochemical properties of the novel ASPase A1 derived from Bacillus sp. SL-l indicated a great potential for the identified enzyme in pharmaceutical and industrial applications.
Keywords: Recombinant L-asparaginase,
Bacillus sp. SL-1
, Origami, Cloning, Soluble overexpression
Copyright and License Information
© 2019 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
L-asparaginase (EC 3.5.1.1; L-asparagine amidohydrolase) an important enzyme with known antineoplastic effect since 1967
1
catalyzes the conversion of L-asparagine amino acid (Asn) to L-aspartic acid and ammonia.
2
This therapeutic enzyme is applied primarily for the management of acute lymphoblastic leukemia (ALL). ALL is a case of leukemia affecting mainly children, contributing to approximately 80% of childhood and 20% of adult leukemias, respectively.
3,4
L-asparaginase is found in various organisms including animals, plant cells, yeast, fungi, and bacteria,
5
however, the bacterial enzyme has gained more attention in therapeutic application because of the higher substrate specificity and longer half-life.
1,2
Currently, Erwinia chrysanthemi and Escherichia coli are widely used in the pharmaceutical industry to produce L-asparaginase for the treatment of leukemia.
6
In the United States, 3 asparaginase formulations are widely used against ALL including native E. coli asparaginase (Elspar®), its pegylated form (Oncaspar®) and Erwinase from cultures of E. chrysanthemi.
3
In recent years, the PEGylation has widely been used in order to improve the blood-circulation half-life, to minimize the immunological clearances via reticuloendothelial system (RES) and to maximize the intratumoral penetration and biodistribution of the administered drugs.
7-9
The anti-leukemia activity of L-asparaginase is due to its depleting effect on the concentration of Asn in the extracellular fluid. Unlike normal cells, tumor cells, more specifically leukemia cells, have little or no asparagine synthetase enzyme and require exogenous asparagine to keep their rapid malignant growth.
10,11
Diminished level of L-asparagine and inability of leukemic cells to synthesize their own L-asparagine leads to the inhibition of protein synthesis, cell cycle arrest in the G1-phase, and ultimately apoptosis in susceptible leukemic cells.
12
In addition, L-asparaginase is enzymatically active against L-glutamine (Gln) but with a significantly lower affinity than that for L-asparagine.
10
This property leads to L-glutaminase activity associated side effects of L-asparaginase treatment. However, this activity contributes to the depletion of both Asn and Gln amino acids which have shown in some studies leads to the suppression of malignant cell growth in certain cancers.
3
Despite significant advantages of L-asparaginase development, clinical application of this bacterial enzyme is limited by immune system response and antibody production against foreign L-asparaginase.
5,13
Anti-asparaginase antibodies are responsible for major toxicity and resistance to asparaginase therapy and also reduce the therapeutic efficacy of asparaginase in some cancer cases.
3
Rapid plasma clearance, shorter duration of activity, frequent injections to maintain the required therapeutic level, and development of immune responses, as well as the anaphylactic shock, are the main restrictions of current commercial L-asparaginases.
3,12
Low substrate specificity is another problem associated with the commercial L-asparaginases, which can cause liver dysfunction, pancreatitis, leucopenia, neurological seizures, and coagulation abnormalities leading to intracranial thrombosis or hemorrhage.
6
It has been demonstrated that L-asparaginases derived from various microorganisms show different properties and do not share antigenic cross-reactivity. Therefore, when patients develop hypersensitivity to L-asparaginase from E. coli, enzyme from E. chrysanthemi can be replaced to avoid risk of allergic reactions.
5,14,15
This condition attracted scientist for discovering this activity in many other microorganisms. Hence there is a request for the novel and robust L-asparaginase from alternative GRAS (generally recognized as safe) microorganisms. Researchers are studying new microbial sources of L-asparaginase which have improved stability, lower glutaminase activity with high substrate affinity, and sufficient half-life under physiological conditions.
3,4
Halo-thermotolerant or halo-thermophile organisms, living under extreme physicochemical conditions, are a valuable source of stable and resistant enzymes that can potentially lead to advances in biocatalysis production.
16
Thermostable enzymes can be stored at room temperature for long periods of time without significant loss of activities. Another advantage of these enzymes is their typical resistance to the variable concentration of sodium chloride.
17,18
In the current study, a new halo-thermotolerant Bacillus sp. SL-1, isolated from Saline Lake of Iran
16
was investigated as the source of novel L-asparaginases.
19
Two ansA1 and ansA3 L-asparaginase genes from Bacillus sp. SL-1 were cloned and overexpressed in Origami(modified E. coli expression strain trxB2/gor2 double mutant) and E. coli BL21. The biochemical properties of the purified enzymes were characterized, and the enzyme activity was evaluated at different temperatures, pH, and substrate concentrations. The results of the present study revealed that only ansA1 gene codes for a novel catalytically active L-asparaginase, with high catalytic activity, whereas ansA3 gene was an inactive protein without any L-asparaginase activity.
Materials and Methods
Reagents
In the present study, all of the mentioned reagents were analytical grade. DNA extraction kit was obtained from Qiagen (Germany). Other kits such as Gel extraction, plasmid purification, and BM chemiluminescence western blotting kits were purchased from Roche Company (Germany). Micro BCA protein assay kit was obtained from Thermo Scientific (USA). DNA ladders, NdeI, XhoI, XbaI, SalI and T4 DNA ligase were acquired from Fermentas (Russia). Ni Sepharose 6 Fast flow medium was from GE Healthcare Life Sciences (Sweden). His-Tag monoclonal primary antibody and goat anti-mouse IgG-HRP conjugated secondary antibody were obtained from GE Healthcare and Santa Cruz companies, respectively. Tryptone and NaCl were from Scharlau (Barcelona, Spain). Finally, yeast extract, agar, and glycerol were provided from Applichem (Darmstadt, Germany). Clinically approved E. coli L-asparaginase (Leunase®) was purchased from Kyowa Hakko Kirin Company (Japan).
Cells and strains conditions
According to the authors previous study,
16
halo-thermotolerant Bacillus sp. SL-1 was isolated from the Aran-Bidgol salt lake located in the central region of Iran. Later on, this strain was donated into Iranian Biological Resource Center (IBRC-M 11052), recognized as an international culture collection, and is currently available for all scientists and investigators. In the current study, all of the utilized E. coli strains (i.e., Origami, DH5α, and BL21) were obtained from Novagen (Darmstadt, Germany).
Cloning and sub-cloning of L-asparaginase genes from Bacillus sp. SL-1
According to the manufacturer’s instructions, QIAamp DNA kit (Qiagen, Germany) was utilized for Genomic DNA extraction from Bacillus sp. SL-1. The principles, which were followed for primer design were based on the high similarity of 16S rDNA sequences derived from Bacillus sp. SL-1 (GenBank accession no. JQ996502) and Bacillus licheniformis (strain ATCC 14580). Polymerase chain reactions were carried out for amplification of the L-asparaginase genes (ansA1 and ansA3) by utilizing the ansA1 forward (5′-TCTTCATATGAATAAAAAA GTAGCTCTCATTACAACG-3′) and ansA1 reverse (5′- AACAGTCGACCTAATAGCA GAATTTGTCTTTTATGCCTT-3′) and ansA3 forward (5′- TATACATATGAAAAAGTTAC TGCTGTTGACCACC-3′) and ansA3 reverse (5′- CAAGGTCGACTTATATGATGATATCGTCTGCAATC-3′)(Bioron, Germany, ordered via FAZA Biotech, Iran). It should be noticed that the italic scripts in the primers are the recognition sites for NdeI and SalI endonucleases. Thereupon, NdeI and SalI endonucleases cleaved the PCR products and those products were subsequently ligated into the pET28a+ expression vector at 16°C overnight. In a process known as bacterial transformation, the produced constructs were transferred into E. coli DH5α for plasmid amplification and extraction by utilizing plasmid purification kit. Subsequently, for examining the presence of ansA1 and ansA3 coding sequences, the purified plasmid constructs had to be tested. The aforementioned examination was conducted by PCR reactions utilizing the previously mentioned primers and universal primers for the pET vectors. Additionally, XbaI and SalI endonucleases were utilized in order to sub-cloning the ansA1 and ansA3 genes into the pET22b+ expression vector. The sequencing of the generated constructs are required in order to confirm the sequence of the ansA1 and ansA3 genes, therefore the accuracy of the generated constructs was verified by Sanger sequencing
20
at Sequetech, USA.
Sequence analysis of L-asparaginase genes from Bacillus sp. SL-1
After analyzing the nucleotide sequences of L-asparaginase genes, they were also analyzed by BLASTn database search method provided by NCBI (http://www.ncbi.nlm.nih.gov). In addition, the translation tool provided by the ExPASy server (http://web.expasy.org/translate/) was utilized to translate the nucleotide sequence in order to obtain the amino acid sequences of Bacillus sp. SL-1 L-asparaginases. ClustalW and Jalview programs were also utilized for alignment. ProtParam (http://www.expasy.org/tools/protparam.html) was used for predicting the L-asparaginase related molecular mass and theoretical pI (isoelectric point) values.
Expression and extraction of recombinant L-asparaginase enzymes
For investigating the expression profile of the recombinant L-asparaginases, E. coli BL21 and Origami prokaryotic expression systems were utilized in aerobic condition in Luria-Bertani (LB) medium. The recombinant constructs pET22-ansA1 and pET22-ansA3 were transformed into chemically competent E. coli cells (BL21 and Origami). The transformed cells were plated on LB ampicilin plates and incubated at 37°C overnight. A single well isolated colony was used to inoculate 10 mL LB+ampicilin medium followed by overnight incubation at 37°C. The starter cultures were diluted 1:100 in fresh LB medium supplemented with ampicillin in the scale of 100 μg/mL and incubated at 37°C with shaking at 180 rpm until optical density of 0.6. Then, the expression was induced by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) in 0.4 mM and overnight incubation at 20°C with shaking at 140 rpm. Bacterial cells were harvested by centrifugation at 5000×g for 15 minutes. After discarding the supernatant, the pellet was resuspended in the lysis buffer (50 mM Tris-HCl pH 8, 100 mM NaCl, 1% Triton X-100, 1.4 mM protease inhibitor (PMSF), 0.1% β-mercaptoethanol and 0.1 mg/mL lysozyme). For efficient disruption of the cells, three rounds of freezing and thawing steps were utilized using liquid nitrogen. Subsequently, the lysate was sonicated 5 times on ice at 60% amplitude for 30 seconds with 1 minute interval. Bacterial debris was removed by centrifugation at 13 000×g for 10 minutes at 4°C and then the soluble fraction was filtered through a 0.22 µm filter and subjected to purification step.
Purification of recombinant L-asparaginase, SDS-PAGE and western blot analysis
Purification of the recombinant L-asparaginase enzymes was performed by exploiting affinity chromatography in certain conditions (bath and gravity-flow conditions), as described previously.17 Briefly, the filtered soluble fraction was applied to Ni Sepharose resin pre-equilibration with lysis buffer and incubated at 4°C for one hour. After removal of the flow through, the affinity resin was washed (Tris 50 mM, NaCl 150 mM, β-mercaptoethanol 0.1%, and imidazole 20 mM) three times and the bound 6×His-tagged proteins were eluted by imidazole solution (imidazole 500 mM, sodium phosphate 20 mM, NaCl 500 mM, pH 7). To remove imidazole, the purified protein samples were dialyzed against the buffer containing 100 mM NaCl and 50 mM Tris-HCl adjusted at pH 8. The prepared purified recombinant protein concentration was determined by micro BCA protein assay method using bovine serum albumin (BSA) standard solutions and the obtained proteins named L-asparaginase A1 (ASPase A1) and L-asparaginase A3 (ASPase A3). Protein analysis at different steps of expression and purification was carried out on 12% polyacrylamide gel (SDS-PAGE) stained by Coomassie brilliant blue G-250. Immediately after SDS-PAGE, the proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane for western blotting. The membrane was blocked at 4°C overnight in 5% BSA dissolved in TBST (20 mM Tris–HCl, containing 150 mM NaCl and 0.05% Tween-20 at pH 7.5). Subsequently, the blocked membrane was incubated with mouse anti-His monoclonal primary antibody (1:3,000) at room temperature for 1.5 hours. The membrane was washed three times with TBST for 5 min each at room temperature. Then, the membrane was incubated with goat anti-mouse IgG-HRP in 3% BSA at 1:8,000 dilution for one hour at room temperature and finally the protein bands were detected by chemiluminescence.
Enzyme activity assay of ASPase A1 and ASPase A3
The enzyme activities were determined by measuring the released ammonia during L-asparagine hydrolysis using Nessler’s reagent.
21
In order to reach temperature equilibration the reaction mixture (2 mL), consisting of 50 mM Tris-HCl (pH 8.6) and 10 mM L-asparagine was incubated at 37°C for 5-6 minutes. After adding 100 µL diluted enzyme at zero time, the reaction proceeded at 37°C for precisely 10 minutes and paused by adding 100 µL of 1.5 M trichloroacetic acid (TCA) only to "Test" tubes. Prior to incubation, the blank tube was terminated by adding 100 µL of 1.5 M TCA. After centrifugation of the reaction mixture, 500 µL of clarified supernatant was mixed with 7.0 mL of ultrapure water and 1.0 mL of Nessler's reagent and incubated at room temperature for 10 minutes. All the measurements were done in triplicates by using spectrophotometer at 480 nm. Using the ammonium sulfate standard curve the micromoles of the released ammonia were determined. An amount of enzyme that under the specified conditions such as the temperature of 37°C and the pH of 8.6 is required to produce one micromole of the ammonia per minute is defined as one unit of the enzyme. According to the manufacturer’s instructions, a micro BCA protein assay kit utilized in order to determine the protein concentration. Finally, the results of the recombinant ASPase A1 activity were compared with the clinically approved E. coli L-asparaginase (Leunase®) activity that named ASPase E in this study.
Biochemical characterization of recombinant ASPase A1 and substrate specificity assay
According to the spectroscopic method mentioned above, we used various concentrations of L-asparagine as a substrate in 50 mM Tris-HCl (pH 8.6) in order to determine the kinetic parameters Km and Kcat of the purified recombinant ASPase A1. The data were fitted to the Michaelis–Menten equation by nonlinear regression using Prism software (version 6.01, GraphPad Software Inc.). The Michaelis-Menten constant Km indicates the concentration of the substrate when the reaction rate is one-half of the maximal velocity (Vmax). The constant Kcat (catalytic rate) shows the number of substrate molecules turned over into the product by enzyme per second.
22
In order to determine the effect of pH on the activity and stability of ASPase A1, L-asparagine hydrolysis by the enzyme in 50 mM Tris-HCl (pH 8.6) was considered. At -20, 4, 25, 37 and 50°C and by using L-asparagine in 50 mM Tris-HCl and pH 8.6 during 120-hour incubation, the thermostability of ASPase A1 was investigated. In different intervals, samples were taken and immediately assayed for residual enzymatic activity by utilizing the L-asparagine hydrolysis. L-glutaminase activity of ASPase A1 was determined by 10 mM L-glutamine as a substrate in 50 mM Tris-HCl at 37°C and pH 8.6 using Nessler’s method.
Results
Characterization of L-asparaginase sequences (ansA1 and ansA3) from Bacillus sp. SL-1
In this study, based on 97.0% similarity between the16S rDNA gene sequences of Bacillus sp. SL-1 and B. licheniformis (strain ATCC 14580),
16
the sequences for L-asparaginase enzyme from the B. licheniformis were used as a template for designing appropriate primers for identifying the corresponding genes from the genome of the Bacillus sp. SL-1. This approach led to the amplification of the coding sequences of ansA1 (969 bp) and ansA3 (990 bp) of Bacillus sp. SL-1. The DNA sequence analysis of the cloned ansA3(SL-1) showed the high degree of identity 99.90% to the same gene from B. licheniformis with just a silent T522A substitution (Tables 1 and 2). The ansA1(SL-1) gene (second homologous enzyme) was also identified from Bacillus sp. SL-1 with 99.07% similarity to ansA1 from B. licheniformis consists of 969 bp corresponding to 322 residues with five nucleotide substitutions (A177T, C184G, G229A, C462T, A946T) leading to tow silence and 3 residue changes in the protein sequence (None, Q62 to E, D77 to N, None, I316 to L), respectively (Tables 1 and 2). From the primary sequence alignment of ASPase A1 and A3, as shown in Fig. 1, it was clear that the identified L-asparaginases from Bacillus sp. SL-1 have conserved amino acids compared to the L-asparaginases from B. licheniformis, Bacillus sp. SL-1, B. subtilis 168, E. chrysanthemi and E. coli, represented in the dark blue columns. The primary structure of Bacillus sp. SL-1 ASPase A3 enzyme shows high sequence identity to B. subtilis 168 (79.0%) and B. licheniformis (ATCC14580) (100.0%) L-asparaginases. ASPase A1 sequence from Bacillus sp. SL-1 was more similar only to L-asparaginase from B. licheniformis (ATCC14580) (99.21%).
Table 1.
Comparison of the corresponding genes for L-asparaginase enzyme from the genome of Bacillus sp. SL-1 with B. licheniformis ATCC14580, B. subtilis 168, E. coli K-12, and Erwinia chrysanthemi
|
Genes
|
GenBank Accession numbers
|
Coding sequence alignment (Identity %)
|
B. licheniformis
ATCC
|
B. subtilis
168
|
E. coli
k-12
|
Erwinia
chrysanthemi
|
|
ansA3
|
KX129701 |
99.90 |
72.04 |
44.37 |
42.66 |
|
ansA1
|
KX681674 |
99.48 |
46.35 |
41.99 |
45.20 |
Table 2.
Pairwise alignment of nucleotide and amino acid sequences of L-asparaginase enzymes from Bacillus sp. SL-1 with B. licheniformis
(ATCC14580) and analysis of their modifications
|
Enzymes
|
Protein sequence alignment
|
|
Nucleotide modification
|
Amino acid modification
|
Identity (%)
|
| ASPase A3 (ansA3) |
T522A |
None |
100.0 |
| ASPase A1 (ansA1) |
A177T
C184G
G229A
C462T
A946T
|
None
Q62 to E
D77 to N
None
I316 to L
|
99.07 |

Fig. 1.
Alignment of the amino acid sequence of ASPase A1 and A3 from Bacillus sp. SL-1 with identified L-asparaginase from closely related microorganisms. P00805, E. coli K12; AAU41311, ASPase A1 B. licheniformis ATCC; AQA28589, ASPase A1 Bacillus sp. SL-1; APT68285, ASPase A3 Bacillus sp. SL-1; AAU42953, ASPase A3 B. licheniformis ATCC; AQR86584, B. subtilis 168; P06608, Erwinia chrysanthemi. Highly conserved amino acid represented in the dark blue columns.
.
Alignment of the amino acid sequence of ASPase A1 and A3 from Bacillus sp. SL-1 with identified L-asparaginase from closely related microorganisms. P00805, E. coli K12; AAU41311, ASPase A1 B. licheniformis ATCC; AQA28589, ASPase A1 Bacillus sp. SL-1; APT68285, ASPase A3 Bacillus sp. SL-1; AAU42953, ASPase A3 B. licheniformis ATCC; AQR86584, B. subtilis 168; P06608, Erwinia chrysanthemi. Highly conserved amino acid represented in the dark blue columns.
Characterization of the produced recombinant ASPase A1 and A3 from Bacillus sp. SL-1
In the present study, the L-asparaginase corresponding genes from Bacillus sp. SL-1 were successfully cloned and expressed in E. coli cells (BL21 and Origami) expression system under aerobic condition. Using Origami expression system under aerobic conditions, ASPase A1 and A3 were expressed significantly in the soluble fraction by the appearance of a strong band with a molecular weight around ∼37 kDa as shown in the SDS-PAGE analysis (Fig. 2) corresponding to the molecular weight of L-asparaginase monomers from B. licheniformis and B. subtilis. The result of the western-blot analysis was in agreement with that of the SDS-PAGE experiment. The production of enzymes in E. coli BL21 expression system led to precipitation and formation of insoluble aggregates as inclusion bodies in cytoplasmic space. After expression of 6×His-tagged ansA1(SL-1) and ansA3(SL-1) L-asparaginases, the clear soluble fractions were subjected to the Ni Sepharose resin for purification (Fig. 3). The concentration of soluble purified L-asparaginase in Origami expression system was ~30 mg per one-liter bacterial culture that showed improvement over the produced enzyme in E. coli BL21 strain under the same condition.
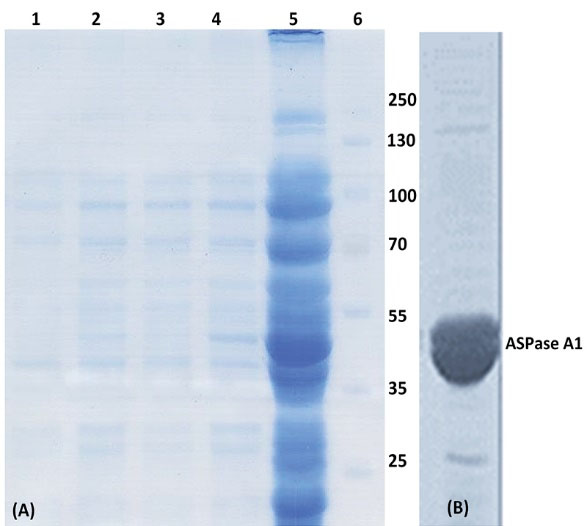
Fig. 2.
The SDS-PAGE and western blot analysis of 6×His-tagged ASPase A1. (A) SDS-PAGE analysis of samples prepared from the soluble fraction of cell lysate prepared from Origami transformed with the plasmid harboring 6×His-tagged ASPase A1 gene. Lane 1, immediately after induction; lane 2, 1 h after induction; lane 3, 2 h after induction; lane 4, 3 h after induction; lane 5, 16 h after induction and lane 6, protein size markers. (B) Western blot analysis of 6×His-tagged ASPase A1 production in Origami expression system.
.
The SDS-PAGE and western blot analysis of 6×His-tagged ASPase A1. (A) SDS-PAGE analysis of samples prepared from the soluble fraction of cell lysate prepared from Origami transformed with the plasmid harboring 6×His-tagged ASPase A1 gene. Lane 1, immediately after induction; lane 2, 1 h after induction; lane 3, 2 h after induction; lane 4, 3 h after induction; lane 5, 16 h after induction and lane 6, protein size markers. (B) Western blot analysis of 6×His-tagged ASPase A1 production in Origami expression system.
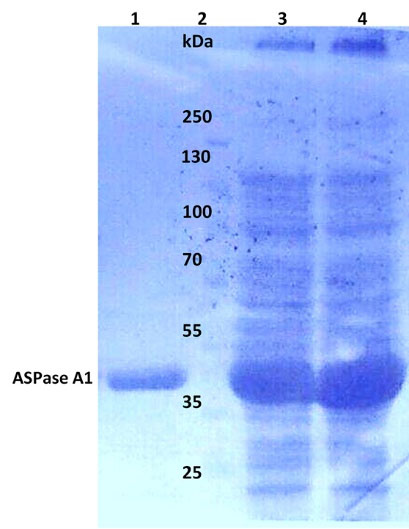
Fig. 3.
The SDS-PAGE analysis of 6×His-tagged ASPase A1 prepared from the soluble fraction of the Origami lysate. Lane 1, purified ASPase A1 with the size of ~37 kDa; lane 2, protein size markers; lane 3, soluble fraction (total protein) of the Origami lysate for ASPase A1; lane 4, soluble fraction (total protein) of the Origami lysate for ASPase A3.
.
The SDS-PAGE analysis of 6×His-tagged ASPase A1 prepared from the soluble fraction of the Origami lysate. Lane 1, purified ASPase A1 with the size of ~37 kDa; lane 2, protein size markers; lane 3, soluble fraction (total protein) of the Origami lysate for ASPase A1; lane 4, soluble fraction (total protein) of the Origami lysate for ASPase A3.
Activity assay of recombinant ASPase A1 and ASPase A3 from Bacillus sp. SL-1
The preliminary hydrolytic activity assay of recombinant ASPase A1 and A3 from Bacillus sp. SL-1 using L-asparagine as substrate was showed that the identified ASPase A3 is completely inactive regardless of the presence of 6×His-tags in either N- or C-terminal of the target protein. However, the second homologous enzyme ASPase A1 from Bacillus sp. SL-1 was produced in high purity and characterized with excellent enzymatic activity. To investigate the ASPase A3 inactivity and its relationship with polyhistidine-tag position, the 6×His-tag was moved from C- terminal to N-terminal of the L-asparaginase by sub-cloning of the ansA3 coding sequence into pET22b (+). The lack of activity for ASPase A3, in either case, shows that the presence of 6×His-tag was not the reason for the observed inactivity.
Biochemical characterization and kinetic parameters of recombinant ASPase A1 compared to ASPase E
The ASPase A1 expressed in Origamiunder aerobic condition was investigated for its kinetic and biochemical properties. As exhibited in Fig. 4, the Km value of ASPase A1 was almost two times higher than the determined value for ASPase E, while the Kcat value of 7.727 s-1 for ASPase E was revealed to be 3 times lower than the Kcat of 23.96s-1 for ASPase A1 (Table 3). Purified ASPase A1 from Bacillus sp. SL-1 was stable in a wide range of pH values ranging from 4.5–10 over a period of 24 hours with maximum residual activity (100%). Temperature effect on enzyme stability and activity was studied between -20°C until 50°C at pH 8.6 during 24 until 120 hours. As shown in Fig. 5, the enzyme exhibited high stability at -20°C where its residual activity was about 100% after one-year storage at this temperature. In addition, the ASPase A1 retained about 40% of its residual activity after 72 hours incubation at 37 °C. The ansA1(SL-1) L-asparaginase showed maximum activity (~100%) after 24 hours incubation in room temperature (~ 25°C).
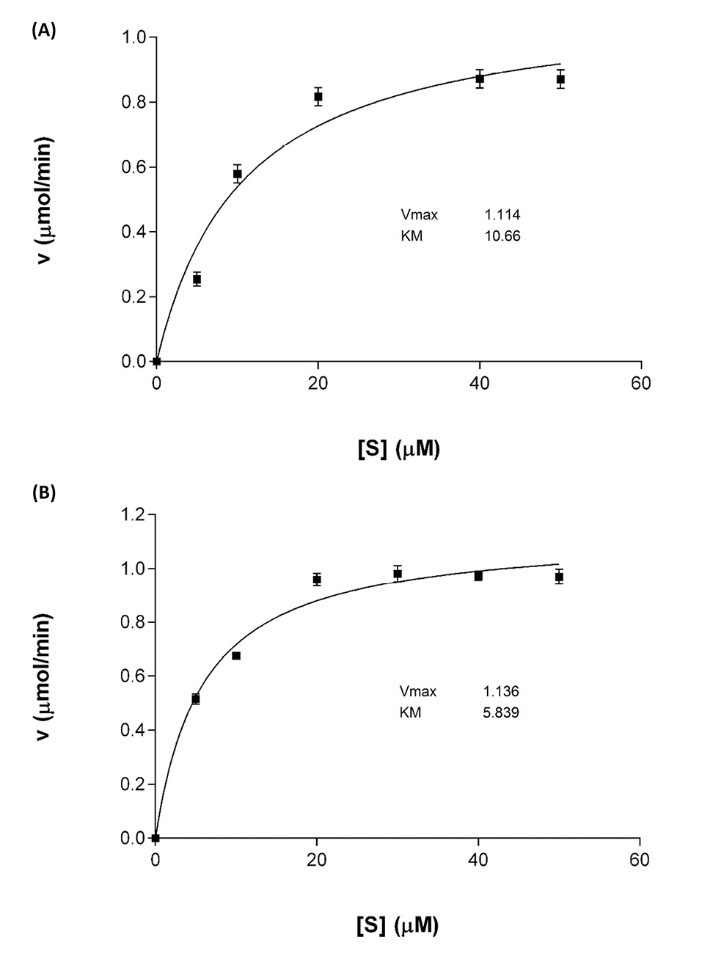
Fig. 4.
Kinetic parameters analysis for ASPase A1 and ASPase E (A) Michaelis–Menten plot for ASPase A1 and (B) Michaelis–Menten plot for ASPase E.
.
Kinetic parameters analysis for ASPase A1 and ASPase E (A) Michaelis–Menten plot for ASPase A1 and (B) Michaelis–Menten plot for ASPase E.
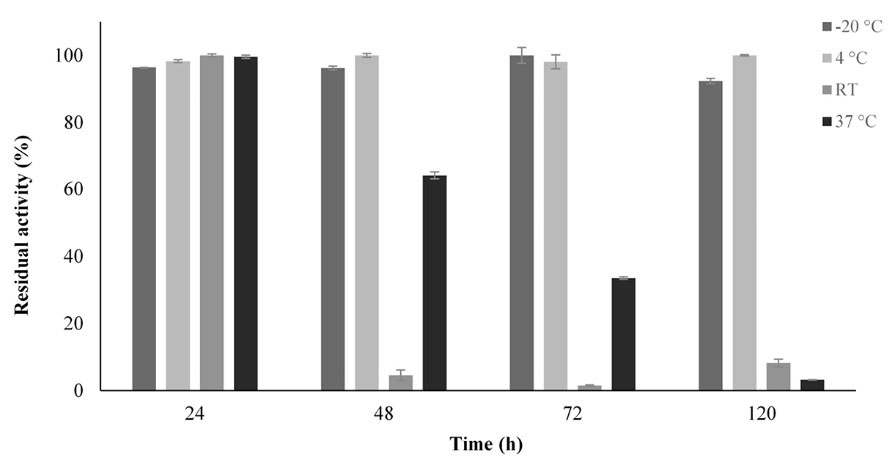
Fig. 5.
Thermostability assessment of the recombinant ASPase A1. The residual activity of the enzyme was determined after incubation for 24, 48, 72 and 120 hours at different temperatures.
.
Thermostability assessment of the recombinant ASPase A1. The residual activity of the enzyme was determined after incubation for 24, 48, 72 and 120 hours at different temperatures.
Table 3.
Kinetic properties of recombinant ASPase A1 from Bacillus sp. SL-1 in comparison with the L-asparaginase activity from other closely
related organisms
|
Source of L-Asparginase
|
K
m
(µM)
|
k
cat
(s
-1
)
|
k
cat
/K
m
|
References
|
|
B. licheniformis SL-1
|
10.30 |
23.96 |
2.326 |
Present study |
|
B. licheniformis MTCC 429
|
670 |
- |
- |
4
|
|
B. subtilis B11−06
|
430 |
- |
- |
5
|
|
E. coli k-12
|
5.788 |
7.727 |
1.339 |
Present study |
Discussion
To reach the main goal of the current work in developing the new recombinant efficient L-asparaginase with improved therapeutic properties, the focus of this study was the production of a soluble and functional enzyme with high-concentration and low L-glutaminase activity within reasonable time and cost. Recently L-asparaginase from B. licheniformis with low-glutaminase activity has been considered as a key therapeutic agent in the treatment of ALL.
12
The glutaminase activity of L-asparaginase is probably related to the structural similarity between L-asparagine and L-glutamine amino acids although the latter has an extra methylene (-CH2-) group.
23
In accordance to our results, previous studies have shown that recombinant L-asparaginase from different bacterial and fungal sources overexpressed in various strains of E. coli BL21 (DE3), E. coli BL21 (DE3) pLysS, E. coli JM109, E. coli BLR (DE3) can lead to different enzyme production profiles of soluble or insoluble forms.
4,5,24,25
More recently, Saeed et al reported the overexpression of L-asparaginase from Aspergillus terreus in E. coli BL21 (DE3) pLysS that precipitated in the form of amorphous inclusion bodies upon induction with 2 mM IPTG for 18 h at 37°C.
26
In general E. coli is considered as a proper host for heterologous protein production in research and biotechnology industry.
27
However, the inability of E. coli to support the complex post-translational modifications is a serious drawback which can lead to misfolding, aggregation and lack of functionality of the heterologously expressed proteins in the cytoplasm of this expression system.
28
Solubilizing and refolding the proteins from inclusion bodies is considered as a common strategy to resolve this problem, because of the presence of a high concentration of target recombinant proteins in such aggregates.
29
However, the cost and time of the whole process must be considered if the goal is to produce a large-scale manufactured product. The soluble expression of recombinant proteins often leads to the production of properly folded, functional, and easy to purify products. Moreover, the refolding process of proteins from the aggregated inclusion bodies is not complete and can fail in many cases.
30
Therefore, in this study, the solubility is considered as a key factor for the production of recombinant L-asparaginase in heterologous expression systems. Based on previous experiences regarding the expression profiles for the other enzymes from B. licheniformis in different E. coli strains, in the current study, the modified E. coli strain (Origami) was used to increase expression concentration and appropriate folding of L-asparaginase in soluble form. Moreover, it is noteworthy to mention that ansA1 from Bacillus sp. SL-1 compared to B. licheniformis ATCC 14580 have 3 residue variations (Table 3) which may be the reason for the observed dissimilar expression profile. It has been demonstrated that such limited alterations only change the structure of the enzyme very slightly, but may still be enough to make a different subunit interaction of the produced recombinant proteins leading to aggregation.
31
Due to native variations in Origami, which leads to its oxidative cytoplasmic environment, the soluble overexpression of L-asparaginases A1 and A3 have been facilitated leading to correct folding of the enzymes in this expression system.
According to the hydrolytic activity assay of recombinant ASPase A1 and A3, the identified ASPase A3 was completely inactive in the presence of 6×His-tags in either N- or C-terminal of the target protein. In contrast to our results, Sudhir et al have shown that the coding sequence ansA3 from another strain of B. licheniformis MTCC 429 produces an active and stable enzyme, while the enzyme from ansA1 is highly unstable.
4
The molecular basis of functional differences observed for the homologous enzymes is strongly related to their 3-dimensional structures.
32
The first possibility for enzymatic inactivity of ASPase A3 was the 6×Histidine residue-tag in C-terminal of protein that may interfere with protein folding, oligomerization and enzyme functionality.
3,33-35
, However, this tag is present in ASPase A1and hence rules out its adverse effect on the activity. The more possible reason for the observed activity difference can be attributed to the high sequence difference (44% similarity) between these two homologs due to the sequence divergence during the evolution may have rendered one of the proteins inactive while the other preserved the activity.
36
Kinetic parameters of the purified enzyme were measured by assessing the hydrolysis of L-asparagine amino acid as a substrate at different concentrations (Fig. 4). The ASPase A1 from Bacillus sp. SL-1 showed a Michaelis–Menten profile typical for such enzymes. The Km value of ASPase A1 was approximately 43 and 67 times lower than the reported value for B. subtilis B11−06 and B. licheniformis MTCC 429, but the value was close to that of B. licheniformis RAM-8 L-asparaginase as shown in Table 3.
4,12
Interestingly, the Kcat value of ASPase A1 was comparatively higher than the calculated value for therapeutical ASPase E from E.coli, which unraveled the high potential of the ASPase A1 for being applied in the therapeutical and pharmaceutical fields.
The purified ASPase A1 from Bacillus sp. SL-1 possessed higher specificity for L-asparagine than L-glutamine amino acid similar to isolated L-asparaginase from B. licheniformis RAM-8 (soil isolate).
12
Purified ASPase A1 from Bacillus sp. SL-1 was stable in wide range of pH values ranging from 4.5–10 over period of 24 hours with maximum residual activity (100%) similar to the reported pH range 7.0-9.0 for L-asparaginase from B. licheniformis RAM-8.
12
L-asparaginase is one of the amidases that are commonly active and stable at neutral to alkaline pH, whereas, pH range 5.0-9.0 has also been reported to be optimum for amidase activity.
37
The thermostability of ansA1(SL-1) at -20 °C was similar to the stability of L-asparaginase from B. licheniformis RAM-8.
12
In another study, the L-asparaginase from B. subtilis B11−06 retains 80% and 75% of the initial activity after 10 h incubation at 20°C and 30°C, respectively. The ASPase A1 lost completely its hydrolytic activity after 24 hours incubation at 50°C, while the L-asparaginase reported from B. licheniformis RAM-8 retained 20% of its hydrolytic activity after 24 hours incubation at 60°C.
12
These biochemical and kinetic variations are attributed to the genetic nature of the microbial strain sources.
37
These results indicated that the thermostability profile of ASPase A1 from Bacillus sp. SL-1 was almost comparable with L-asparaginase from B. licheniformis RAM-8, E. coli K12 (ASPase E) and was higher than that reported for L-asparaginase from B. subtilis B11−06.
12
Conclusion
In summary, the current study introduces a new and efficient recombinant L-asparaginase enzyme from locally newly isolated halo-thermotolerant Bacillus strain with high-level soluble overexpression in the modified E. coli strain (Origami). The cloning, expression and activity assay of recombinant ansA1 and ansA3 showed that only ansA1 gene produced an active and stable homologues enzyme with high substrate affinity toward L-asparagine. The concentration of soluble and functional recombinant L-asparaginase production was optimized in the modified E. coli expression system. The purified recombinant ASPase A1 was highly thermostable and resistant to the wide range of pH values. The L-glutaminase-free L-asparaginase activity and higher Kcat value for ASPase A1 from Bacillus sp. SL-1 in compared to other studied bacterial L-asparaginases reveals the potential of this enzyme for pharmaceutical and industrial application.
Acknowledgment
The authors would like to thank the Biotechnology Research Center, Tabriz University of Medical Sciences for providing all necessary research facilitates.
Funding sources
The aid of Anatomical Science Research Center, Kashan University of Medical Science is highly appreciated for providing financial support (grant no. 9171).
Ethical statement
None to be declared.
Competing interests
The authors declare that they have no conflict of interest.
Authors contribution
AS was the PhD student and conducted the experiments, gathered the data and drafted the manuscript. MHM was advisor in this study. SD and RM designed the experiments and finalized the manuscript and supervised the overall study.
Research Highlights
What is the current knowledge?
simple
-
√ L-asparaginase is an important anti-cancer enzyme against
ALL.
-
√ Currently, Erwinia chrysanthemi and Escherichia coli
are used in the pharmaceutical industry to production of
L-asparaginase.
What is new here?
simple
-
√ L-asparaginase enzymes from newly isolated Bacillus sp.SL-1 were investigated.
-
√ Cloned L-asparaginases were overexpressed in solubleform in Origami strain.
-
√ Only one of the two produced recombinant enzymes
showed catalytic activity.
-
√ Novel active enzyme (A1) showed high Kcat value with no
L-glutaminase activity.
-
√ Enzyme A1 was highly thermo-stable and resistant to wide
range of pH values.
References
- Sajitha S, Vidya J, Binod P, Pandey A. Cloning and expression of l-asparaginase from E coli in eukaryotic expression system. Biochem Eng J 2015; 102:14-7. doi: 10.1016/j.bej.2015.02.027 [Crossref] [ Google Scholar]
- Zuo S, Xue D, Zhang T, Jiang B, Mu W. Biochemical characterization of an extremely thermostable l-asparaginase from Thermococcus gammatolerans EJ3. J Mol Catal B Enzym 2014; 109:122-9. doi: 10.1016/j.molcatb.2014.08.021 [Crossref] [ Google Scholar]
- Shrivastava A, Khan AA, Khurshid M, Kalam MA, Jain SK, Singhal PK. Recent developments in l-asparaginase discovery and its potential as anticancer agent. Crit Rev Oncol Hematol 2016; 100:1-10. doi: 10.1016/j.critrevonc.2015.01.002 [Crossref] [ Google Scholar]
- Sudhir AP, Dave BR, Prajapati AS, Panchal K, Patel D, Subramanian R. Characterization of a recombinant glutaminase-free l-asparaginase (ansa3) enzyme with high catalytic activity from bacillus licheniformis. Appl Biochem Biotechnol 2014; 174:2504-15. doi: 10.1007/s12010-014-1200-z [Crossref] [ Google Scholar]
- Jia M, Xu M, He B, Rao Z. Cloning, expression, and characterization of L-asparaginase from a newly isolated Bacillus subtilis B11–06. J Agric Food Chem 2013; 61:9428-34. doi: 10.1021/jf402636w [Crossref] [ Google Scholar]
- Duval M, Suciu S, Ferster A, Rialland X, Nelken B, Lutz P. Comparison of Escherichia coli–asparaginase withErwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment of Cancer—Children's Leukemia Group phase 3 trial. Blood 2002; 99:2734-9. doi: 10.1182/blood.V99.8.2734 [Crossref] [ Google Scholar]
- Omidi Y, Barar J. Targeting tumor microenvironment: crossing tumor interstitial fluid by multifunctional nanomedicines. Bioimpacts 2014; 4:55-67. doi: 10.5681/bi.2014.021 [Crossref] [ Google Scholar]
- Nischan N, Hackenberger CP. Site-specific PEGylation of proteins: recent developments. J Org Chem 2014; 79:10727-33. doi: 10.1021/jo502136n [Crossref] [ Google Scholar]
- Barar J, Omidi Y. Translational approaches towards cancer gene therapy: hurdles and hopes. Bioimpacts 2012; 2:127-43. doi: 10.5681/bi.2012.025 [Crossref] [ Google Scholar]
- Song P, Ye L, Fan J, Li Y, Zeng X, Wang Z. Asparaginase induces apoptosis and cytoprotective autophagy in chronic myeloid leukemia cells. Oncotarget 2015; 6:3861. doi: 10.18632/oncotarget.2869 [Crossref] [ Google Scholar]
- Chen S-H. Asparaginase therapy in pediatric acute lymphoblastic leukemia: a focus on the mode of drug resistance. Pediatr Neonatol 2015; 56:287-93. doi: 10.1016/j.pedneo.2014.10.006 [Crossref] [ Google Scholar]
- Mahajan RV, Kumar V, Rajendran V, Saran S, Ghosh PC, Saxena RK. Purification and characterization of a novel and robust L-asparaginase having low-glutaminase activity from Bacillus licheniformis: in vitro evaluation of anti-cancerous properties. PLoS One 2014; 9:e99037. doi: 10.1371/journal.pone.0099037 [Crossref] [ Google Scholar]
- Safary A, Akbarzadeh Khiavi M, Mousavi R, Barar J, Rafi MA. Enzyme replacement therapies: what is the best option?. Bioimpacts 2018; 8:153-7. doi: 10.15171/bi.2018.17 [Crossref] [ Google Scholar]
- Frank BH, Pekar AH, Veros AJ, Ho PP. Crystalline L-Asparaginase from Escherichia coli B II Physical Properties, Subunits, And Reconstitution Behavior. J Biol Chem 1970; 245:3716-24. [ Google Scholar]
- Yun M-K, Nourse A, White SW, Rock CO, Heath RJ. Crystal structure and allosteric regulation of the cytoplasmic Escherichia coli L-asparaginase I. J Mol Biol 2007; 369:794-811. doi: 10.1016/j.jmb.2007.03.061 [Crossref] [ Google Scholar]
- Safary A, Moniri R, Mirhashemi SM, Nikzad H, Khiavi MA. Phylogenetic and biochemical characterization of a new halo-thermotolerant, biofilm-forming Bacillus from Saline Lake of Iran. Polish J Microbiol 2013; 62:419-25. [ Google Scholar]
- Safary A, Moniri R, Hamzeh-Mivehroud M, Dastmalchi S. A strategy for soluble overexpression and biochemical characterization of halo-thermotolerant Bacillus laccase in modified E coli. J Biotechnol 2016; 227:56-63. doi: 10.1016/j.jbiotec.2016.04.006 [Crossref] [ Google Scholar]
- Giugliani R, Federhen A, Rojas MV, Vieira T, Artigalas O, Pinto LL. Mucopolysaccharidosis I, II, and VI: Brief review and guidelines for treatment. Genet Mol Biol 2010; 33:589-604. doi: 10.1590/s1415-47572010005000093 [Crossref] [ Google Scholar]
- Safary A, Moniri R, Hamzeh-Mivehroud M, Dastmalchi S. Identification and Molecular Characterization of Genes Coding Pharmaceutically Important Enzymes from Halo-Thermo Tolerant Bacillus. Adv Pharm Bull 2016; 6(4):551-561. doi: 10.15171/apb.2016.069 [Crossref] [ Google Scholar]
- Moghaddas Sani H, Hamzeh-Mivehroud M, Silva AP, Walshe JL, Mohammadi SA, Rahbar-Shahrouziasl M. Expression, purification and DNA-binding properties of zinc finger domains of DOF proteins from Arabidopsis thaliana. Bioimpacts 2018; 8:167-76. doi: 10.15171/bi.2018.19 [Crossref] [ Google Scholar]
- Imada A, Igarasi S, Nakahama K, Isono M. Asparaginase and glutaminase activities of micro-organisms. Microbiology 1973; 76:85-99. [ Google Scholar]
- Bisswanger H. Enzyme assays. Perspect Sci (Neth) 2014; 1:41-55. doi: 10.1016/j.pisc.2014.02.005 [Crossref] [ Google Scholar]
- Nagarethinam S, Nagappa A, Udupa N, Rao V, Vanathi M. Microbial L-Asparaginase and its future prospects. Asian J Med Res 2012; 1:159-68. [ Google Scholar]
- Hong SJ, Lee YH, Khan AR, Ullah I, Lee C, Park CK. Cloning, expression, and characterization of thermophilic L‐asparaginase from Thermococcus kodakarensis KOD1. J Basic Microbiol 2014; 54:500-8. doi: 10.1002/jobm.201300741 [Crossref] [ Google Scholar]
- Sindhu R, Manonmani H. Expression and characterization of recombinant l-asparaginase from Pseudomonas fluorescens. Protein Expr Purif 2018; 143:83-91. doi: 10.1016/j.pep.2017.09.009 [Crossref] [ Google Scholar]
- Saeed H, Ali H, Soudan H, Embaby A, El-Sharkawy A, Farag A. Molecular cloning, structural modeling and production of recombinant Aspergillus terreus L asparaginase in Escherichia coli. Int J Biol Macromol 2018; 106:1041-51. doi: 10.1016/j.ijbiomac.2017.08.110 [Crossref] [ Google Scholar]
- Peti W, Page R. Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Expr Purif 2007; 51:1-10. doi: 10.1016/j.pep.2006.06.024 [Crossref] [ Google Scholar]
- Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol 2014; 5:172. doi: 10.3389/fmicb.2014.00172 [Crossref] [ Google Scholar]
- Singh A, Upadhyay V, Upadhyay AK, Singh SM, Panda AK. Protein recovery from inclusion bodies of Escherichia coli using mild solubilization process. Microb Cell Fact 2015; 14:41. doi: 10.1186/s12934-015-0222-8 [Crossref] [ Google Scholar]
- Sørensen HP, Mortensen KK. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb Cell Fact 2005; 4:1. doi: 10.1186/1475-2859-4-1 [Crossref] [ Google Scholar]
-
Patel AK, Singhania RR, Pandey A. Production, purification, and application of microbial enzymes. In: Brahmachari G, ed. Biotechnology of Microbial Enzymes: Elsevier; 2017. p. 13-41.
- Todd AE, Orengo CA, Thornton JM. Sequence and structural differences between enzyme and nonenzyme homologs. Structure 2002; 10:1435-51. doi: 10.1016/S0969-2126(02)00861-4 [Crossref] [ Google Scholar]
- Pradita A, Schweiger R, Schwenkert S, Soll J. N-Terminal His-Tagged AtTPR7 Interactions with Hsp70 and Hsp90 Proteins. HAYATI Journal of Biosciences 2014; 21:197-200. doi: 10.4308/hjb.21.4.197 [Crossref] [ Google Scholar]
- Park WJ, You SH, Choi HA, Chu YJ, Kim GJ. Over-expression of recombinant proteins with N-terminal His-tag via subcellular uneven distribution in Escherichia coli. Acta Biochim Biophys Sin (Shanghai) 2015; 47:661. doi: 10.1093/abbs/gmv069 [Crossref] [ Google Scholar]
- Schalk AM, Nguyen HA, Rigouin C, Lavie A. Identification and structural analysis of an L-asparaginase enzyme from guinea pig with putative tumor cell killing properties. J Biol Chem 2014; 289:33175-86. doi: 10.1074/jbc.M114.609552 [Crossref] [ Google Scholar]
- Todd AE, Orengo CA, Thornton JM. Sequence and structural differences between enzyme and nonenzyme homologs. Structure 2002; 10:1435-51. doi: 10.1016/S0969-2126(02)00861-4 [Crossref] [ Google Scholar]
- El-Naggar NE-A, Deraz SF, Soliman HM, El-Deeb NM, El-Ewasy SM. Purification, characterization, cytotoxicity and anticancer activities of L-asparaginase, anti-colon cancer protein, from the newly isolated alkaliphilic Streptomyces fradiae NEAE-82. Sci Rep 2016; 6:32926. doi: 10.1038/srep32926 [Crossref] [ Google Scholar]