BioImpacts. 9(3):131-144.
doi: 10.15171/bi.2019.18
Original Research
A multi-method and structure-based in silico vaccine designing against Echinococcus granulosus through investigating enolase protein
Mohammad Mostafa Pourseif 1, 2, Mitra Yousefpour 1, *, Mohammad Aminianfar 2, Gholamali Moghaddam 3, Ahmad Nematollahi 4 
Author information:
1 Department of Physiology, Faculty of Medicine, AJA University of Medical Sciences, Tehran, Iran
2 Infectious Diseases and Tropical Medicine Research Center (IDTMRC), Department of Aerospace and Subaquatic Medicine, AJA University of Medical Sciences, Tehran, Iran
3 Department of Animal Sciences, Faculty of Agriculture, University of Tabriz, Tabriz, Iran
4 Department of Pathobiology, Veterinary College, University of Tabriz, Tabriz, Iran
Abstract
Introduction:
Hydatid disease is a ubiquitous parasitic zoonotic disease, which causes different medical, economic and serious public health problems in some parts of the world. The causal organism is a multi-stage parasite named Echinococcus granulosus whose life cycle is dependent on two types of mammalian hosts viz definitive and intermediate hosts.
Methods:
In this study, enolase, as a key functional enzyme in the metabolism of E. granulosus (EgEnolase), was targeted through a comprehensive in silico modeling analysis and designing a host-specific multi-epitope vaccine. Three-dimensional (3D) structure of enolase was modeled using MODELLER v9.18 software. The B-cell epitopes (BEs) were predicted based on the multi-method approach and via some authentic online predictors. ClusPro v2.0 server was used for docking-based T-helper epitope prediction. The 3D structure of the vaccine was modeled using the RaptorX server. The designed vaccine was evaluated for its immunogenicity, physicochemical properties, and allergenicity. The codon optimization of the vaccine sequence was performed based on the codon usage table of E. coli K12. Finally, the energy minimization and molecular docking were implemented for simulating the vaccine binding affinity to the TLR-2 and TLR-4 and the complex stability.
Results:
The designed multi-epitope vaccine was found to induce anti-EgEnolase immunity which may have the potential to prevent the survival and proliferation of E. granulosus into the definitive host.
Conclusion:
Based on the results, this step-by-step immunoinformatics approach could be considered as a rational platform for designing vaccines against such multi-stage parasites. Furthermore, it is proposed that this multi-epitope vaccine is served as a promising preventive anti-echinococcosis agent.
Keywords:
Echinococcus granulosus
, Enolase, In silico vaccinology, Molecular docking, Epitope
Copyright and License Information
© 2019 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Transmission dynamics of Echinococcus granulosus mainly depends on the dog-sheep cycle in several endemic areas. As such, adult worm’s eggs is transferred from definitive hosts feces (dog) to the body of intermediate hosts (sheep and human) during grazing. The eggs can penetrate into the small intestine’s mucosa and lead to the formation of hydatid cysts – comprising abundant protoscoleces (PSCs) and cyst fluid (CF) – in the internal organs of intermediate hosts (e.g., liver and lungs). The dog will be infected upon feeding the cystic viscera of the intermediate hosts and then PSCs are released and attached to the gut mucosa of the dog which grow and differentiate to the adult worm.
1,2
The last report of the World Health Organization (WHO) in terms of different aspects of echinococcosis has highlighted the main facts about the potential and actual risks of E. granulosus.
3
Vaccination acts through eliciting the host immune system – the natural barrier for fighting against infectious agents.
4,5
Along with the development of newer technologies, the informatics-based vaccines as next-generation of the vaccines were born from the multidisciplinary sciences.
6
The current challenges for vaccine development against such a multi-stage parasite is originated from major funding constraints, and socio-cultural and political problems.
7
Therefore, focusing on the multivalent and multistage vaccines seems to be a rational and time- and cost-effective approach.
8
In the past decade, the immunobiological data about the function of MHC molecules, host-pathogen interactions, and immunological signaling pathways have provided new avenues for the design and development of rational vaccines.
9
The vaccines that are designed by immunoinformatic analysis seem to be a promising vaccinology platform for the prevention of the parasite life cycle. Many candidate antigens have been used for the production of epitope-based recombinant vaccines against E. granulosus.
9
Nevertheless, in designing such vaccines, the preliminary in silico analyses using immunoinformatic tools have yet to be considered thouroughly. Some of the in silico-based investigations were only concentrated on the prediction of B-cell epitopes (BCEs) exploiting different vaccine candidate antigens regardless of T-cell epitopes.
10-12
However, it should be noted that there is not any web-server for the T-cell epitope prediction based on the canine MHC alleles. Therefore, for designing a host-specific epitope vaccine, one needs to switch from the sequence-based methods to the structure-based techniques.
13,14
For this reason, the docking tools can be recruited for the simulation of binding affinity and molecular interactions between T-cell epitope and MHC alleles.
As reported previously, the B lymphocyte cells have a central function in protective immune responses against the extracellular parasitic pathogen.
14
Despite such fact, B-cell provoking mechanisms are mainly dependent on the effectors of T-helper cells,
15
which implies that the B-cell based epitope vaccines, to be more efficient and immunogenic, need to be linked to the antigen-specific T helper cells.
16
In this study, we capitalized on the EgEnolase, a multifunctional glycolytic and excretory-secretory (ES) antigen, for designing an epitope-based vaccine construct. The EgEnolase has a significant expression and localization in the PSCs and adult worm stages of the parasite.
17
This enzyme is involved in the key processes such as motility, adhesion, invasion, growth, and differentiation of the parasite.
18-20
In this line, the 3D structure of the enolase was modeled and the T-helper epitopes were predicted using the molecular docking methods and based on the most frequent canine leukocyte antigen (DLA) alleles. To the best of our knowledge, this is one of the first studies for in silico epitope mapping against this pathogen, which might provide a creative platform for the construction of the host-specific and immunogenic recombinant, DNA, and edible-based vaccines against such parasitic pathogens.
21
Materials and Methods
Prerequisite analysis for designing the vaccine construct
The protein sequence retrieval and prediction of antigenicity
The previous reverse vaccinology assay showed that the tegumental membrane enzymes of E. granulosus have a strong immunogenicity and key abilities in the parasite pathogenicity.
22
Therefore, the seven E. granulosus enolase (EgEnolase) protein sequences were obtained from the national center for biotechnology information (NCBI) protein database and subjected to the antigenicity prediction. The ANTIGENpro tool of SCRATCH server and VaxiJen v2.0 server (threshold 0.4%) were used for the prediction of the proteins antigenicity.
23
Based on the antigenicity result, the sequence with the highest antigenic properties was selected for the next analysis.
Prediction of the transmembrane helix and signal peptide
The proteins with membrane localization have one or more transmembrane helices that cannot be detected by the humoral B-cells. Further, the signal sequence is a temporary part of mature proteins. Having considered these immunological facts, transmembrane helix (TMH) of EgEnolase was predicted using the following online web-servers; TOPCONS,
24
TMHMM server v2.0,
25
and TMPred.
26
The Signal-3L v2.0,
27
TOPCONS,
24
and SignalP v4.1 (D-cutoff: 0.45)
28
online tools were used for the prediction of the potential signal peptide (SigP) cleavage site.
Prediction of post-translational modifications
NetPhos v3.1 server was used for the identification of possible prediction of serine, threonine, and tyrosine phosphorylation sites.
29
NetNGlyc v1.0
30
and NetOGlyc v4.0
31
online predictors respectively served the identification of possible N-linked and mucin-type CalNAc O-linked glycosylation regions.
Prediction of surface accessibility and hydrophobicity
Basically, the surface accessible and hydrophilic residues of antigens are usually involved in the Ag-Ab interaction; therefore, the predicted BEs should not be located in the highly hydrophobic and non-accessible parts of the protein. Thus, we plotted the hydrophobicity profile of EgEnolase using the method of Kyte and Doolittle (KD).
32
The accessible amino acids were also predicted via the ProtScale online server.
33
Homology modeling, energy minimization, and validation
Three-dimensional (3D) structure of the EgEnolase, DLA-DRB1*01101, toll-like receptor 2 and 4 was modeled by MODELLER v9.18 software.
34
The amino acid sequence of the proteins was retrieved from the NCBI and UniProtKB databases (TLR-2: Q689D1, TLR-4: F1PDB9, EgEnolase: CDS19796, and DRB1*01101: BAU68163). The six template structures for each protein was achieved by protein-protein BLAST (blastp) algorithm of NCBI’s protein blast tool and against the Protein Data Bank.
The UCSF Chimera v1.13.1 standalone program was used for energy minimization of the models.
35
The 3D models were minimized energetically up to 400 steps steepest descent algorithm (i.e., steepest descent step size 0.02Å), 40 steps conjugate gradient algorithm (i.e., conjugate gradient step size 0.02Å), by applying AMBER ff14SB force field.
36
The GA341 and DOPE (Discrete Optimized Protein Energy) scores are calculated by MODELLER to choose the best model among the constructed structures. The overall quality of the chosen model can be assessed by ProSA,
37
Verify3D,
38
ERRAT
39
web-servers. The stereochemistry quality in the generated model was also analyzed based on the Ramachandran plot, which was obtained from the RAMPAGE online server.
40
B-cell epitope prediction and variability metrics analysis
Linear B-cell epitope prediction
The BCE prediction is an essential step in vaccine designing, especially against extracellular pathogens. In order to improve the accuracy of in silico epitope mapping, we applied a multimethod process from the Immune Epitope Database (IEDB) analysis resource,
41-44
LBtope,
45
BepiPred v1.0,
46
BCEPred,
47
and ABCpred,
48
and SVMTrip
49
online servers. Of these, the BepiPred can predict the linear BEs based on the combination of a Hidden Markov Model (HMM) and a propensity scoring method with the scores above the threshold of 0.35. Having used the BCEPred server, epitopes were predicted through the physicochemical features. The linear BEs in ABCpred tool were predicted based on the artificial neural network (ANN) method.
Conformational B-cell epitope prediction
The conformational epitopes were predicted by inputting the 3D structure of EgEnolase and then were selected based on the high-score outcomes of SEPPA v2.0,
50
ElliPro,
51
and DiscoTope v2.0 online web-servers.
Revealing mutative/conservative of the predicted B-cell epitopes
The variability metrics of the predicted BEs were characterized based on the data content measured by the Shannon’s entropy (Hx) plot and using the BioEdit program.
52
Structure-based CD4+ T-helper epitope prediction
The helper T-cell response plays a key role in cell-mediated immunity and especially helps in the clearance of extracellular pathogen through different cytokines and stimulation of humoral immune responses.
53
Therefore, the helper T-cell epitopes (HTEs) are definitely an essential part of the prophylactic and therapeutic vaccines.
9
Currently, there is no organized software or web-server for the sequence-based prediction of peptide binders to the cognate groove of dog MHC alleles I and II. In view of that, we used the structure-based method (peptide-protein docking) for prediction of HTEs of EgEnolase antigen.
14
Although the length of the peptides bound to the binding pocket of MHC class II molecules is not constrained, it has been reported that the longer antigenic peptides bound to MHC-II molecules are usually trimmed by peptidases to a length of 13–17 amino acids.
54
In this peptide-protein docking procedure, we used a highly frequent dog leukocyte antigen (DLA), so-called DLA-DRB1*01101 as a receptor, and 84 fragments of 13-mer peptides of the EgEnolase protein as a ligand. The ClusPro 2.0 online server was used to measure the binding free energy between the DLA allele and the 13-mer peptides.
55
The intramolecular interactions (e.g., hydrogen and hydrophobic) between DLA-DRB1*01101 and EgEnolase peptide fragments were analyzed by UCSF Chimera v1.13.1 and LigPlot+v1.4.5 programs.
56
Designing minigene vaccine construct
The high-immunogenic and efficient fusion vaccine constructs require two requisite elements: (i) epitope and adjuvant, and (ii) the linkers (or spacers).
Epitope and adjuvant
To plan a sufficient immunization against E. granulosus, the designed vaccine not only should have a high ability to trigger helper T-cell and humoral immune responses, but also it must effectively induce the innate immunity. On the other hand, the designed minigene vaccine must consist of helper T-cell and BCEs plus an immunogenic intramolecular adjuvant. E. granulosus is an extracellular helminth parasite, and the cell-surface Toll-like receptors (TLRs) are effectively involved in detection of surface echinococcus antigens. Of these, TLR-2 and TLR-4 are the best candidates to recognize the surface antigens of E. granulosus.
57
Thus, we used Leptospira surface adhesin (Lsa21),
58
a ligand for TLRs 2 and 4, and RS09 (Sequence: APPHALS),
59
a TLR-4 ligand.
Linker
Selecting an appropriate epitope-specific linker (e.g., flexible, rigid, cleavable) is an essential step in designing an immunogenic multi-epitope protein.
60
Conformational BCEs need a certain degree of free moving and folding to be more accessible to antibodies. Based on the length and rigidity/flexibility properties of the protein linkers, we used the (Gly4Ser)2 linker between the BCEs. The non-polar polyglycine residues plus a polar serine residue provide a flexible linker with minimum linker-epitope interactions and in total preserve the BE function. A four-amino-acid rigid and cleavable linker (FFRK), which contained both cathepsin and proteasomal cleavage sites, was added to the C-terminal of HTEs to optimize T-cell epitope processing. Intramolecular adjuvants were linked to each other by a (EAAAK)2 linker.
61
It can minimize the interference between the adjacent intramolecular adjuvants.
13
To provide an independent immunogenic function, GPSL spacer was used between the TH and BC epitopes. Intramolecular T helper epitopes were joined each other by the GPGPG linker.
Analysis of the minigene vaccine construct
Prediction of antigenicity, allergenicity, and autoimmunity
Prediction of the vaccine antigenicity represents a numerical criterion for the capability of the vaccine to bind to the B- and T-cell receptors. In this work, we used ANTIGENpro online web-server, as a pathogen independent and alignment-free predictor tool, to check the protein sequences in terms of antigenic behavior.
The protein allergenicity is due to the sensitization and allergic reactions associated with the IgE antibody response. The constructed vaccine must not show an allergic reaction into the body. The vaccine construct amino acid sequence was submitted to the AlgPred and AllerTOP web-servers to check potential cross-reactivity of the vaccine and known allergens based on the FAO/WHO allergenicity rules. In AlgPred we utilized the hybrid method consisted of 4 algorithms, including (i) SVMc, (ii) mapping of known IgE epitopes feature, (iii) BLAST search against 2890 allergen-representative peptides (ARPs), and (iv) searching among MEME/MAST allergen motifs using MAST. In this method, the FASTA-formatted protein sequences were compared with EgEnolase sequence on the basis of two standard indices, including (i) E-value cutoff of 0.01 for the full-length sequence similarity more than 35% with a sliding window size 80 amino acids, and (ii) contiguous 6-mer amino acids that match to those of the determined allergenic proteins.
62,63
Likewise, the vaccine protein sequence was blasted against non-redundant protein sequences of Canis lupus familiaris. Further, the blastp (protein-protein BLAST) algorithm of NCBI was recruited to check the similarity percentage between the minigene construct and the known endogenous proteins of Canis familiaris.
Physicochemical properties and efficiency of the vaccine construct
Computing the physicochemical properties
The post-injection behavior of the designed vaccine into the body is the main goal of vaccination. Therefore, the physicochemical features of the formulated minigene vaccine should be analyzed. Therefore, we used the ProtParam tool of ExPasy web-server.
33
In this web-server, various parameters were computed, including (i) molecular weight (kDa), (ii) theoretical isoelectric point (pI), (iii) in vitro and in vivo based estimated half-life, (iv) stability index, (v) aliphatic index, (vi) extinction coefficient, and (vii) grand average of hydropathicity (GRAVY).
Simulation of the vaccine binding affinity to the TLRs
The preferred orientation and binding affinity between the designed vaccine construct and Canis lupus familiaris TLR-2 and TLR-4 molecules in their stable complex form were predicted through the molecular docking method.
64
The tertiary structure of the minigene construct was modeled based on a template-based tertiary structure prediction web-server, RaptorX.
65
The protein sequences of Canis lupus familiaris TLR-2 and TLR-4 were retrieved from the UniProtKB database and their 3D structures were modeled by MODELLER v9.18.
66
The vaccine model and TLR-2 and TLR-4 were prepared for docking by use of the Dock Prep tool of Chimera software. The preparation steps were as follows: (a) solvent molecules were deleted, (b) alternate locations were removed, (c) incomplete side chains were replaced by Dunbrack rotamer library, and (d) hydrogen atoms and charge were added.
67
AMBER ff14SB force field was computed for the standard residues and AM1-BCC force fields for the other residues. The vaccine-TLR docking process was performed using ClusPro v2.0 web-server.
55
In this protein-protein docking procedure, the refined structure of the designed vaccine was used as a ligand for TLR-2 and TLR-4. In ClusPro docking, the orientation control was set at 70000. Visualization and more analysis of the docking result were implemented using the Chimera v1.13.1 and DIMPLOT program of the LigPlot+v1.4.5, respectively.
Codon adaptation and analysis of the mRNA stability
High expression rate of the foreign gene into the various protein expression systems need a main attention to the codon adaptation step. According to our established strategy for the vaccine development (e.g., DNA vaccine, edible vaccine, or recombinant protein vaccine), we conducted different host-specific codon bias procedures and selected different cloning systems. Therefore, back translation of the vaccine protein sequence and its codon optimization was carried out by the visual gene developer (VGD) v1.7 software and based on the codon frequency table of E. coli K12 that directly was imported from the Codon Usage Tabulated from GenBank (CUTG).
68
Besides, a large number of G/C content and creation of an unsteady folding in the mRNA structure showed a negative correlation with the gene expressivity into the host.
69
For this reason, the codon adaptation index (CAI), G/C content (%) of the optimized sequence, the mRNA secondary structure pre- and post-optimization, and it's Gibbs free energy (∆G) were evaluated by VGD program.
3D Modeling, refinement, energy minimization, and validation of the vaccine construct
The 3D structure of the designed vaccine was modeled using the RaptorX structure prediction online server.
65
The RaptorX generates a high-quality 3D model based on the ab initio web-based method. In the next step, the initially 3D-modeled vaccine was subjected to the GalaxyRefine web-server to be refined structurally.
70
The vaccine 3D structure was refined through rebuilding all side-chains, and using two mild and then aggressive relaxation steps that were followed by short MD simulation. Further, to resolve the model steric clashes and determine the stability of the initially created model at the microscopic level and during a timeframe, we applied the refined model to different structure analysis tools of the UCSF Chimera, which is an extensible software for interactive representation and molecular structure analysis, such as molecular dynamic trajectories, energy minimization, equilibration and so on.
35,71
Initially, the molecule charges were assigned by applying AMBER ff14SB force field. Then, the 3D model was centered into a cubic boundary box with a minimum distance of 2 Å from the edge of the box to any protein atom, and filled by water molecules using the simple point charge water model (SPCFWBOX), and chloride ion (Cl-) was used for the charge neutralization of the structure. The particle mesh Ewald method was used for the electrostatic interactions, and Lennard-Jones interaction method was set as default. The 3D model was then re-arranged and structurally optimized using 500 steepest descent minimization steps (steepest descent step size 0.02Å), 50 steps of conjugate gradient (conjugate gradient step size 0.02Å), and by applying AMBER ff14SB force field.
36
The overall model quality was checked using ProSA,
37
Verify3D,
38
ERRAT,
39
web-servers. The stereochemistry quality in the vaccine 3D model was analyzed based on the Ramachandran plot, which was obtained from the Chimera software.
35
Results
Antigenicity of EgEnolase protein sequences
In order to design an immunogenic vaccine, the sum of seven EgEnolase protein sequences was retrieved from the NCBI protein database. Of these, the ANTIGENpro and VaxiJen results showed the sequence (with NCBI accession no. CDS19796) with the highest antigenic scores of 0.5903 and 0.4814, respectively. The obtained scores indicated the antigenic nature of the selected EgEnolase protein sequence, therefore, the sequence was used for the multi-epitope vaccine designing. In Table S1 (Supplementary file 1), the antigenic score for all EgEnolase protein sequences is shown based on the method of prediction.
Prediction of immunological non-accessible residues
Membrane-spanning and signal peptide regions
The probability plot of TMHMM shows that there is no significant alpha-helix transmembrane (TM) topology in the EgEnolase protein sequence. In TMPred web-server only scores more than 500 were predicted as significant TM helix. In this case, two fragments 325–345 (Score: 529) and 358–377 (Score: 856) were predicted to be the strong inside-to-outside and outside-to-inside orientations of membrane spanning, respectively. Among five algorithms of the TOPCONS web-server, only one 20-amino-acid-segment (733–753) was predicted using the SPOCTOPUS method. According to the outputs of TOPCONS, SignalP, and Signal-3L online engines, there is no signal sequence at the beginning of the EgEnolase protein sequence (Fig. S1, Supplementary file 1).
Post-translational modifications
The NetOglyc web-server uses the neural network algorithms to predict the mucin-type GalNAc O-glycosylation sites in the mammalian proteins.
31
In this server, only residues with a prediction score of more than 0.5 are predicted as local O-glycosylated. The server output showed there was no O-glycosylation site in the EgEnolase protein sequence. In NetNGlyc server based on the threshold of 0.5, only one potential N-linked glycosylation site (position 17; sequence: NPTV; score: 0.8082) was observed (Fig. S2A, Supplementary file 1). The output of NetPhos v2.0 server indicated 20 phosphorylated serine residues (S), 6 threonine (T), and 4 tyrosine (Y) phosphorylation sites in the EgEnolase protein sequence (Fig. S2B, Supplementary file 1).
Identification of the non-accessible and hydrophobic residues
The non-accessible and hydrophobic areas of EgEnolase protein sequence are shown as plots in Figs S3A and B (Supplementary file 1). The non-accessible regions of the EgEnolase were considered during the prediction of conformational BCEs.
Homology modeling, energy minimization, and validation
The homologous PDB templates that were used for homology modeling are shown in Table S2 (Supplementary file 1). The 3D structures of the modeled EgEnolase, DLA-DRB1*01101, TLR-2 and TLR-4 proteins before and after energy minimization and their energy are shown in Fig. 1.
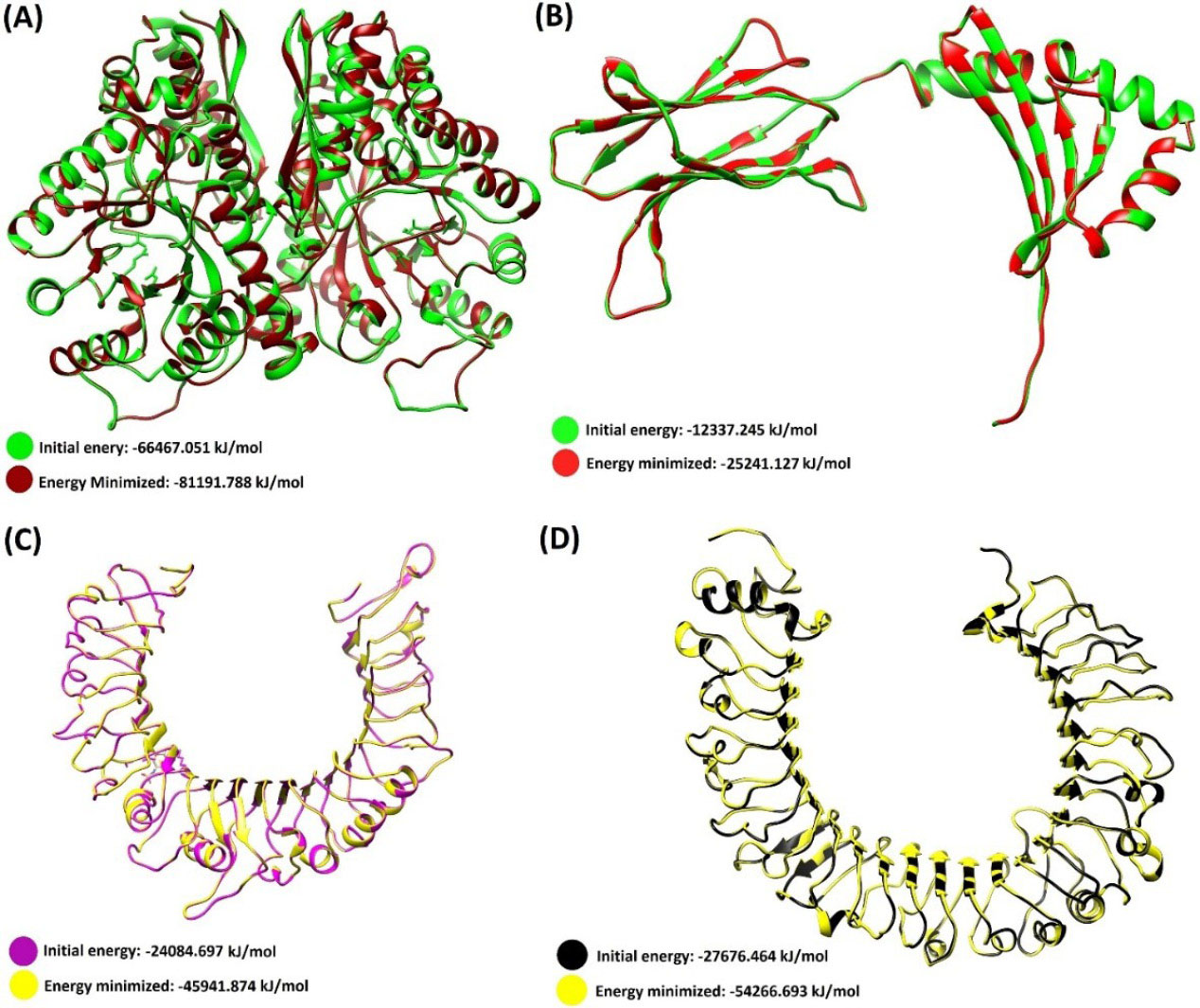
Fig. 1.
Three-dimensional structure of the initial and energy minimized modeled proteins. (A)E. granulosus Enolase protein. (B) DLA-DRB1*01101. (C)C. lupus Toll-like receptor 2. (D)C. lupus Toll-like receptor 4. The Chimera software (freely available at http://www.rbvi.ucsf.edu/chimera) was used for the energy minimization and ribbon style representation of the 3D models.
.
Three-dimensional structure of the initial and energy minimized modeled proteins. (A)E. granulosus Enolase protein. (B) DLA-DRB1*01101. (C)C. lupus Toll-like receptor 2. (D)C. lupus Toll-like receptor 4. The Chimera software (freely available at http://www.rbvi.ucsf.edu/chimera) was used for the energy minimization and ribbon style representation of the 3D models.
The models were re-arranged and structurally optimized after the energy minimization (Table 1). Afterward, the homology modeling procedure was validated, providing two modeling validation scores (GA341, and the normalized DOPE scores). The energetic value of the GA341 and DOPE scores for the 3D models are represented in Table 1. The alpha carbon (Cα) atoms of the modeled structures were analyzed by ProSA server Z-score plot. Herein, the Z-score values for each model showed the overall quality of the models is favorable (Table 1). The local quality of the models was checked by the energy plot of the ProSA-web. Their entropy plots are provided in Fig. S4 (Supplementary file 1). The ERRAT and Verify3D scores for the modeled structures are exhibited in Table 1.
Table 1.
Summary of energy minimization and structure validation for the 3D structures
|
Model
|
Energy minimization
|
MODELLER
|
Web-based tools
|
Ramachandran*
|
|
EM
0
|
EM
1
|
GA341
|
DOPE
¶
|
ProSA z-score
|
Verify3D
|
ERRAT
|
Favored
|
Allowed
|
| EgEnolase |
-66467.1 |
-81191.8 |
1.0 |
– 33668.3 |
– 10.5 |
90.1 |
98.6 |
97.0 |
2.6 |
| 01101 |
-12337.2 |
-25241.1 |
1.0 |
– 29341.6 |
– 5.9 |
86.8 |
97.1 |
96.3 |
3.2 |
| TLR-2 |
-24084. 7 |
-45941.9 |
1.0 |
– 26093.7 |
– 5.9 |
89.9 |
80.9 |
87.4 |
10.6 |
| TLR-4 |
-27676.5 |
-54266.7 |
1.0 |
– 30419.1 |
– 7.3 |
97.2 |
84.0 |
93.9 |
5.3 |
EM
0: Energy for the initial model (Kj/mol). EM1: Energy minimized model. * Totality of the residues within the favored regions of psi/phi Ramachandran plot is reported. ¶kcal/mole. ERRAT: A good 3D model should have a value > 50%. Verify3D: This criterion assigns a score to each residue between -1 to +1, and in good 3D models more than 80% of the amino acids should score >0.2.
B-cell epitope prediction and variability metrics for the predicted epitopes
We used both the physicochemical and the machine learning-based BE prediction tools to increase the accuracy of in silico BCE mapping. In this multimethod analysis, the high-rank epitopes of each predictor tool were schematically revealed under the EgEnolase protein sequence. Through this multimethod analysis a vivid overview was provided to discover the high immunogenic regions of EgEnolase protein, which facilitated and improved the accuracy of selecting the best BCEs (Fig. 2).
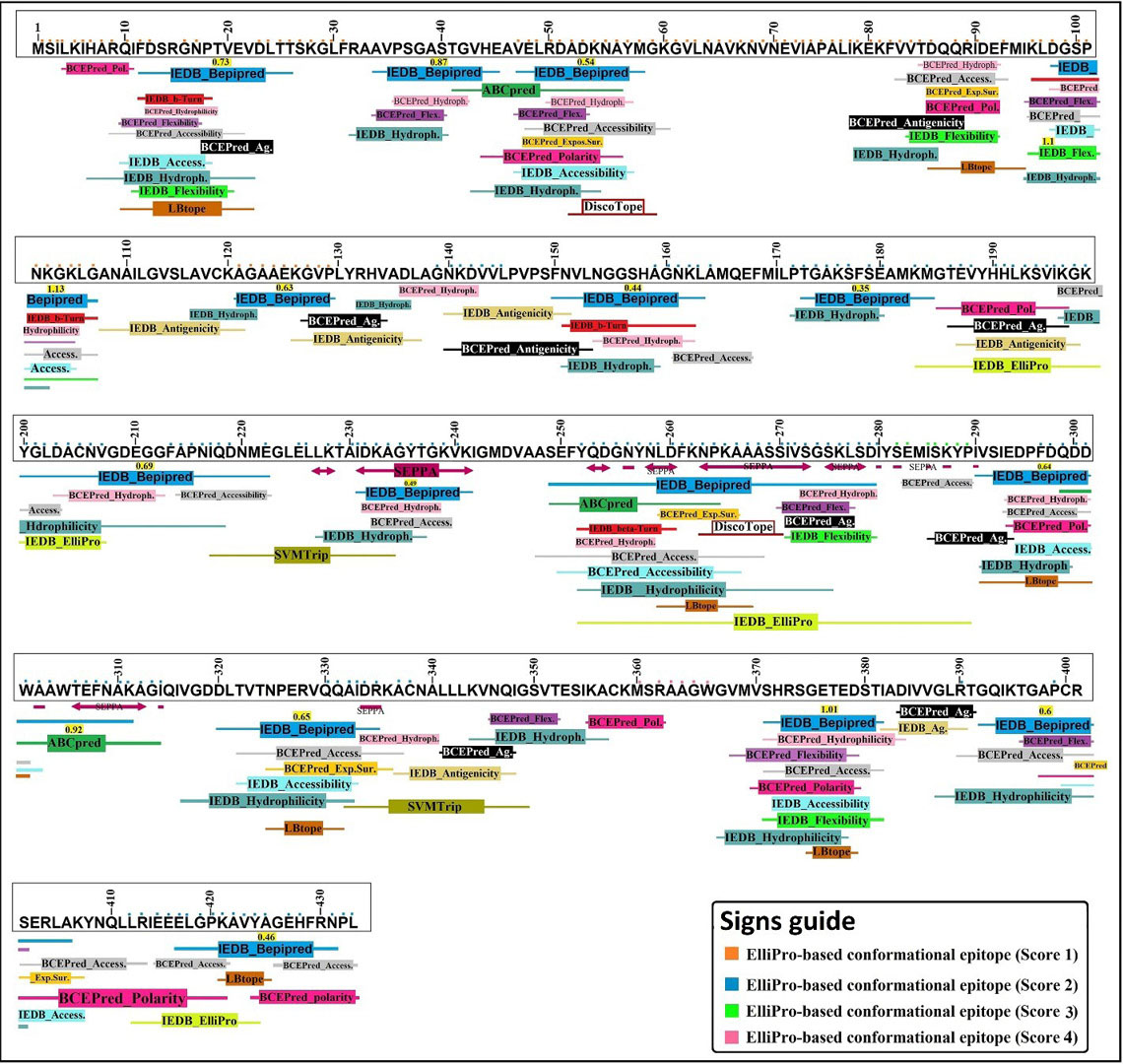
Fig. 2.
The result of high score predicted BEs based on the multimethod BCE prediction methods. The prediction method consists of 19 different BCE prediction algorithms. The position of high score epitopes is characterized by different colors. The conformational BCEs are marked by the use of the colored squares above each one-letter amino acids. As shown in the “signs guide”, the conformational BEs are differentiated from each other using different colored squares.
.
The result of high score predicted BEs based on the multimethod BCE prediction methods. The prediction method consists of 19 different BCE prediction algorithms. The position of high score epitopes is characterized by different colors. The conformational BCEs are marked by the use of the colored squares above each one-letter amino acids. As shown in the “signs guide”, the conformational BEs are differentiated from each other using different colored squares.
Total 12 BCEs with different lengths were predicted based on the multimethod BCE mapping analysis. Of these, 5 epitopes were finalized (Table 2) due to their high surface accessibility score and allergen analysis (Fig. S5, Supplementary file 1).
Table 2.
The predicted BCEs, their positions in the sequence, amino acid length, and variability score of each epitope
|
No.
|
Predicted B-cell Epitopes
|
Position
|
Length
|
Mean entropy value
|
|
1
|
MSILKIHARQIFDSRGNPTVEVDLTT |
1 – 26 |
26 |
0.236 |
|
2
|
PSGASTGVHEAVELRDADKNAYMGK* |
36 – 60 |
25 |
0.149 |
|
3
|
IKEKFVVTDQQRIDEFMIKLDGSPNKGKLG* |
78 – 107 |
30 |
0.393 |
|
4
|
KAGAAEKGVPL |
120 – 130 |
11 |
0.126 |
|
5
|
LAGNKDVVLP |
137 – 146 |
10 |
0.208 |
|
6
|
MGTEVYHHLKSVIKGKYGLDACNV* |
184 – 207 |
24 |
0.173 |
|
7
|
KTAIDKAGYTGKVK* |
228 – 241 |
14 |
0.245 |
|
8
|
SEFYQDGNYNLDFKNPKAAASSIVSGSKLSDI* |
249 – 280 |
32 |
0.335 |
|
9
|
PFDQDDWAAWTEFNAKAGI |
296 – 314 |
19 |
0.222 |
|
10
|
DLTVTNPERVQQAIDRKAC |
320 – 338 |
19 |
0.109 |
|
11
|
VMVSHRSGETEDSTIAD |
368 – 384 |
17 |
0.484 |
|
12
|
GQIKTGAPCRSERLAKYNQLLRIEEELGPKAVYAGEHFR |
388 – 430 |
42 |
0.458 |
* Indicates the selected B-cell epitopes.
Due to the comprehensive multimethod analysis, we guess our vaccine can effectively elicit humoral immunity. As shown in Table 2 , the mean entropy (Hx) values of the computationally predicted BEs are less than 1.0 (threshold). This implies the epitopes are conserved among different E. granulosus enolase isolates and can be used for the immunization against different strains of E. granulosus. The Shannon entropy (Hx) plot is represented in Fig. 3.
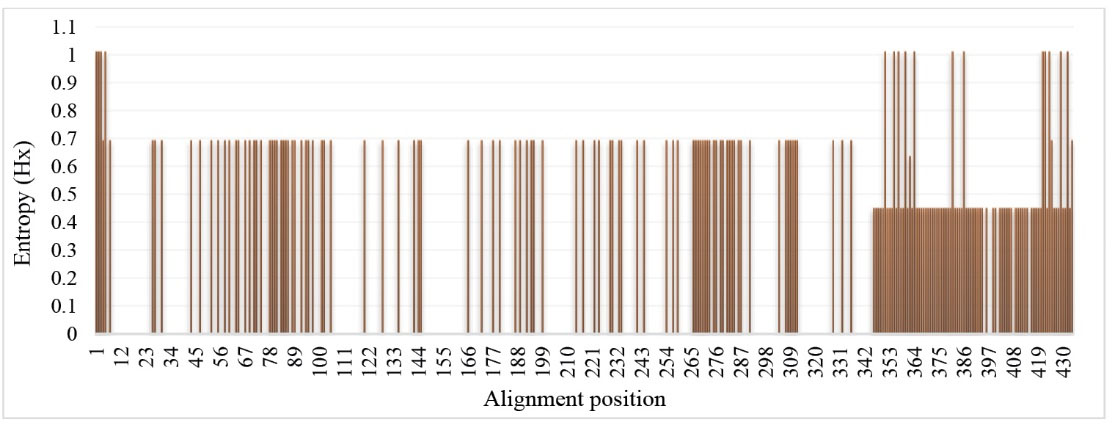
Fig. 3.
Entropy plot of deduced amino acid sequences of different EgEnolase protein sequences. The Shannon entropy plot was attained using the BioEdit program. Entropy (Hx) values varied from 0 (highly conserved) to 1.0 (hypervariable).
.
Entropy plot of deduced amino acid sequences of different EgEnolase protein sequences. The Shannon entropy plot was attained using the BioEdit program. Entropy (Hx) values varied from 0 (highly conserved) to 1.0 (hypervariable).
Helper T-lymphocyte (HTL) epitope prediction
The EgEnolase 3D structure was subjected to Chimera software to be cleaved manually into the eighty-four 13-mer peptides. In order to identify the highest immunogenic epitope(s), all 13-mer peptides were docked with a DRB1*01101 molecule, separately. The binding energy corresponding to each receptor-ligand complex is shown in Table S3. Of these, twelve 13-mer peptides with the lowest binding energy were selected for constructing the minigene vaccine scaffold (Table 3).
Table 3.
Final selected helper T-cell epitopes
|
No.
|
Sequence
|
Binding energy (kJ/mol)
|
|
Center
|
Lowest energy
|
| 1 |
KLAMQEFMILPTG |
-906.4 |
-1016.2 |
| 2 |
GALIIHARQIFDS |
-825.7 |
-999.1 |
| 3 |
AMQEFMILPTGAK |
-882.7 |
-983.0 |
| 4 |
EFMILPTGAKSFS |
-801.7 |
-946.9 |
| 5 |
LIIHARQIFDSR |
-793.3 |
-945.2 |
| 6 |
MSRAAGWGVMVSH |
-922.9 |
-922.9 |
| 7 |
AGWGVMVSHRSGE |
-819.7 |
-916.8 |
| 8 |
LRIEEELGPKAVY |
-710.7 |
-909.9 |
| 9 |
KAVYAGEHFRNPL |
-743.9 |
-903.3 |
| 10 |
YPIVSIEDPFDQD |
-876.3 |
-896.0 |
| 11 |
VLPVPSFNVLNGG |
-844.0 |
-896.6 |
| 12 |
GYTGKVKIGMDVA |
-859.7 |
-859.7 |
The arrangement of the minigene vaccine construct
The nucleotide and amino acid sequence of the minigene vaccine are shown in Fig. S6 (Supplementary file 1). The final vaccine construct of 601 amino acid residues was designed by means of 5 BEs and 12 helper T-cell epitopes, 2 intramolecular adjuvants, and 5 types of different peptide linkers. To design an adjuvant vaccine with the maximum immune response, TLR-2 and TLR-4 agonists (Lsa21 and RSO9, respectively) were linked to the N-terminal of the minigene construct as intramolecular adjuvants. Since the designed minigene vaccine construct consists of immunogenic BCEs and HTEs along with two immunogenic intramolecular adjuvant and suitable peptide spacers, it may have a high ability to induce the innate and adaptive anti-echinococcus immune responses within the host body.
13,72,73
The 3D structure prediction, refinement, energy minimization, and validation
The 3-dimensional structure of the vaccine construct represents good information about the arrangement of epitopes and important residues. The vaccine 3D structure modeling was used by the RaprorX server and the best template for this work was the crystal structure of Drosophila enolase (PDB ID: 5WRO). All residues were modeled as three domains with 39 positions (6%) disorder. The secondary structure of the 3D model displayed 21% helix, 23% β-sheet, and 54% coiled. The P-value of the homology modeling was 1.12e-06, which is a parameter of relative quality of the model. In this case, a small P-value refers to a good modeling quality.
74
The GalaxyRefine web-server performed some repeated structure perturbations and also structural relaxation by short molecular dynamics simulation to refine the vaccine 3D structure. In this procedure, the number of residues that were located in the favored region of the Ramachandran plot was increased from 87.3% to 91.3%. The refined structure was also energy minimized by the UCSF-Chimera and the system’s energy decreased to -15990.18 kJ/mol.
To validate the general and local quality of the vaccine construct, the Ramachandran plot was depicted after refinement and energy minimization of the model. As represented in Fig. 4 , the vaccine model showed that only 4.49% of the residues is placed in the outlier regions. The general quality of the 3D structure of the vaccine was evaluated by ERRAT, Verify3D, and ProSA web-servers (Fig. 4). The overall quality scores of the model obtained from the ERRAT and Verify3D servers were 77.37% and 71.71%, respectively. ProSA server resulted in the Z-score of -4.6 for the uploaded vaccine 3D model, which was laid close to the native protein structures.
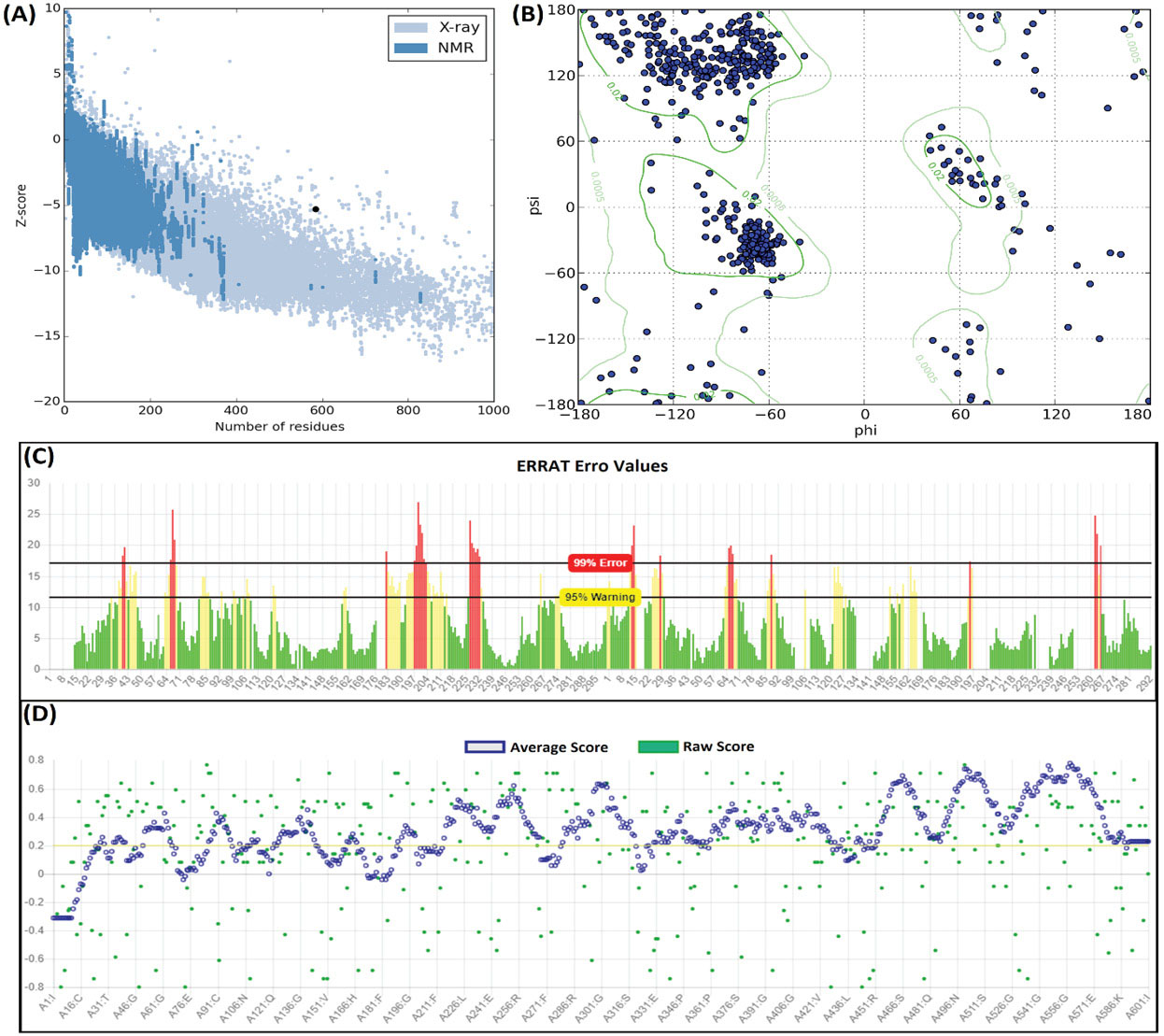
Fig. 4.
Structural validation of the modeled vaccine. (A) The ProSA Z-score of the vaccine 3D model. (B) Ramachandran map analysis of the modeled vaccine construct, showing 95.51% of residues in favored and allowed regions. (C) The ERRAT plot of the vaccine 3D model. (D) The Verify3D plot of the vaccine 3D model. The overall modeling quality values showed the quality of the modeled vaccine is relatively satisfactory.
.
Structural validation of the modeled vaccine. (A) The ProSA Z-score of the vaccine 3D model. (B) Ramachandran map analysis of the modeled vaccine construct, showing 95.51% of residues in favored and allowed regions. (C) The ERRAT plot of the vaccine 3D model. (D) The Verify3D plot of the vaccine 3D model. The overall modeling quality values showed the quality of the modeled vaccine is relatively satisfactory.
Evaluation of the vaccine efficiency
Prediction of antigenicity, allergenicity, and autoimmunity
The result of ANTIGENpro online server showed that the designed vaccine had an antigenicity probability of 0.847. This value represents the antigenic nature of the designed vaccine.
An allergic vaccine can cause the allergen-specific cross-reactive immune responses and symptoms in the host body such as skin rash and swelling of the mucosal membranes. Allergenicity of the designed vaccine was predicted by two web-servers (i.e., AlgePred and AllerTOP). The NCBI protein-protein BLAST against Canis lupus familiaris proteome was used for prediction of possible molecular mimicary between the vaccine sequence and endogenous proteins in dogs' body. The results showed the epitopic vaccine will not be associated with any allergic or autoimmune responses in vivo and is safe to be administrated to dogs (Fig. S5, Supplementary file 1).
Physicochemical properties of the vaccine
The ProtParam server was used for the assessment of nine important physicochemical parameters of the designed vaccine. The molecular weight (kDa) and theoretical pI of vaccine construct were predicted to be ~ 63 and 9.75, respectively. The total number of positively (Arg and Lys) and negatively (Asp and Glu) charged residues were 71 and 44, respectively. The estimated half-life of the vaccine construct in mammalian reticulocytes was 20 hours, in vitro; while 30 min and 10 hours in yeast and Escherichia coli, in vivo. The extinction coefficient at 280 nm wavelength was computed in water to be 36330 M-1 cM-1, assuming that all Cys residues are reduced. The instability index (II) of the vaccine construct was computed at 27.78, which indicates the stable nature of the minigene construct. The estimated values of the aliphatic index and the Grand average of hydropathicity (GRAVY) were 65.57 and −0.241, respectively.
The vaccine capability to interact with TLR-2 and TLR-4
The vaccine construct was engineered in such a way to be able for effective interaction with TLR-2 and TLR-4 receptors. In this step, we analyzed this protein-protein interaction using the ClusPro v2.0 server.
Vaccine–TLR-2 complex
Docking between the vaccine construct and TLR-2 leads to the generation of total 9 models. Of these, the model number 000.01 showed the lowest energy scores (-1193.2 kJ/mol), the highest binding affinity, which was properly located in interaction with the receptor, thus was selected as the best-docked complex (Fig. 5). The intramolecular interactions including hydrophobic and hydrogen bonds are represented in Fig. 5. The DIMPLOT analysis exhibited that nineteen residues of the vaccine construct were involved in hydrogen bond (H-bond) with TLR-2 and the length of H-bonds was ranged from 2.55 to 3.04 Å (Fig. 5).
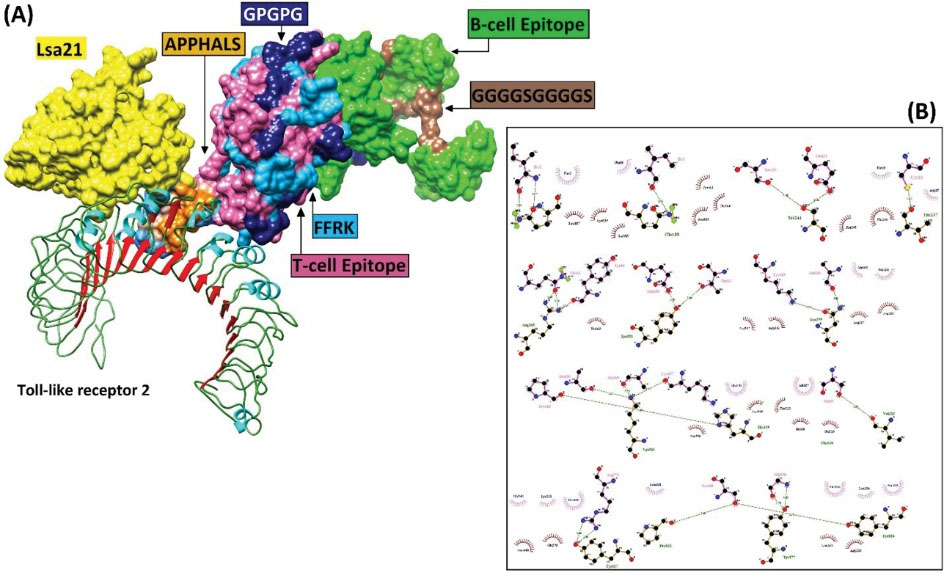
Fig. 5.
The docked complex of the minigene vaccine and the TLR-2 receptor. (A) Various components of the vaccine construct, including adjuvant (e.g., Lsa21, and APPHALS), linkers (e.g., EAAAK, FFRK, GPGPG, GPSL, and GGGGSGGGGS), and B- and T-cell epitopes are shown in contact with TLR-2 (B) The hydrophobic and hydrogen bonds between the vaccine construct and TLR-2.
.
The docked complex of the minigene vaccine and the TLR-2 receptor. (A) Various components of the vaccine construct, including adjuvant (e.g., Lsa21, and APPHALS), linkers (e.g., EAAAK, FFRK, GPGPG, GPSL, and GGGGSGGGGS), and B- and T-cell epitopes are shown in contact with TLR-2 (B) The hydrophobic and hydrogen bonds between the vaccine construct and TLR-2.
Vaccine–TLR-4 complex
Among total 9 generated complexes, the model number 000.00 showed the lowest binding energy values (-1018.3 kJ/mol) between the vaccine construct and TLR-4. This model was properly located in interaction with the receptor, thus was selected as the best-docked complex (Fig. 6A, B). The intramolecular interactions including hydrophobic and hydrogen bonds are represented in Fig. 6C. The DIMPLOT analysis exhibited that nineteen residues of the vaccine construct were involved in hydrogen bond (H-bond) with TLR-2 and the length of H-bonds was ranged from 2.55 to 3.04 Å (Fig. 6).
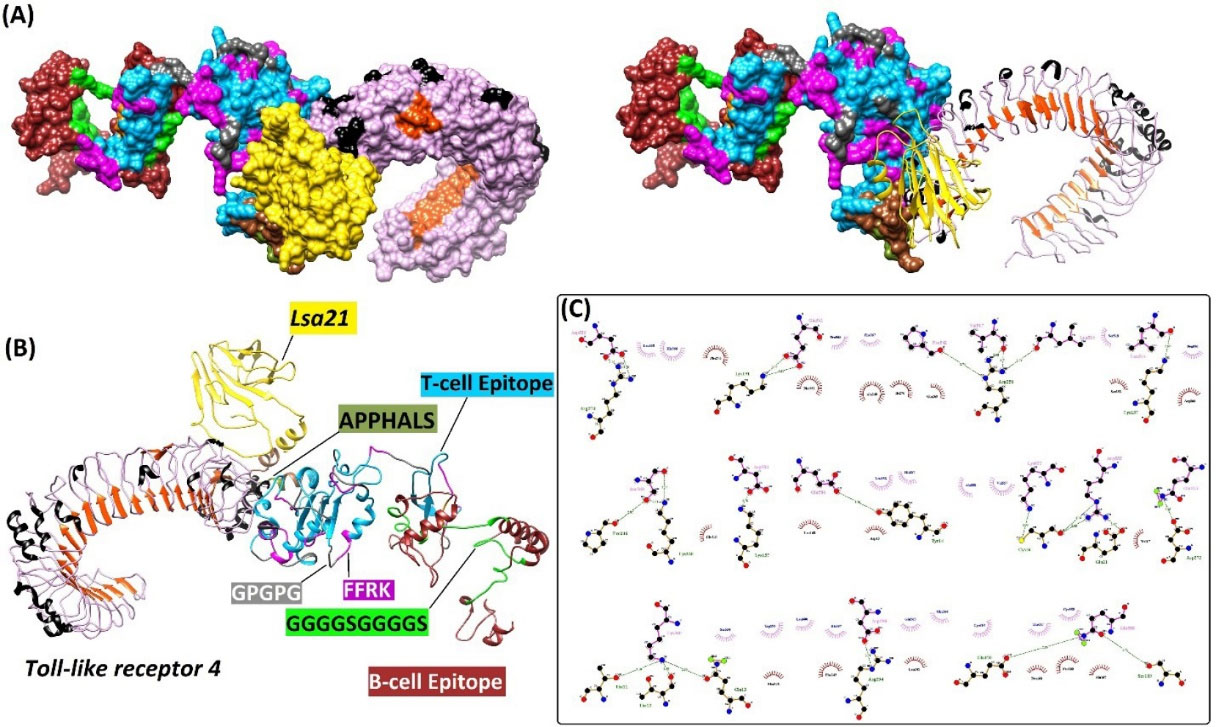
Fig. 6.
The interaction between the minigene vaccine and TLR-4 receptor as three- and two-dimensional representations. (A) The docked complex of vaccine construct and its receptor TLR-4. (B) Various components of the vaccine construct, including intramolecular adjuvants (e.g., Leptospira surface adhesion (Lsa21), and APPHALS), linkers (e.g., EAAAK, FFRK, GPGPG, GPSL, and GGGGSGGGGS), and B- and T-cell epitopes are shown as a docked complex with TLR-4. (C) The hydrophobic and hydrogen bonds between the vaccine construct and TLR-4.
.
The interaction between the minigene vaccine and TLR-4 receptor as three- and two-dimensional representations. (A) The docked complex of vaccine construct and its receptor TLR-4. (B) Various components of the vaccine construct, including intramolecular adjuvants (e.g., Leptospira surface adhesion (Lsa21), and APPHALS), linkers (e.g., EAAAK, FFRK, GPGPG, GPSL, and GGGGSGGGGS), and B- and T-cell epitopes are shown as a docked complex with TLR-4. (C) The hydrophobic and hydrogen bonds between the vaccine construct and TLR-4.
Codon optimization and analysis of mRNA stability
To achieve the maximal protein expression in the E. coli expression system, the protein sequence of the vaccine construct was backtranslated to the DNA sequence and then codon usage of the sequence was adapted based on E. coli K12 codon frequency table and using the visual gene developer v1.7 software. In this sense, the value of codon adaptation index (CAI), the GC-content, effective number of codons (Nc), and tRNA adaptation index (tAI) before and after codon optimization are represented in Table 4.
Table 4.
The parameters of codon usage bias pre- and post-codon adaptation of the vaccine construct
|
Parameter
|
Before
|
After
|
| CAI |
1.0 (0.558) |
0.9 |
| Nc |
13.8 |
15.0 |
| tAI |
0.4 |
0.4 |
| Overall G/C content (%) |
57.5 |
57.3 |
|
G/C content at 1st place (%)
|
57.4 |
57.4 |
|
G/C content at 2nd place (%)
|
47.8 |
47.8 |
|
G/C content at 3rd place (%)
|
66.9 |
65.7 |
CAI: Codon adaptation index; Nc: Effective number of codons; tAI: tRNA adaptation index.
The overall Gibbs free energy (ΔG) of mRNA secondary structure pre- and post-optimization were -832.90 and -815.80 kcal/mol, respectively. The ΔG shows the favorability of the mRNA folding process.
Discussion
Vaccination of animals might be one of the important treatment modalities for efficient, rapid and affordable controlling of the zoonotic infectious diseases among the human populations. The current study is focused on the rational vaccinology (e.g., epitope-based vaccines) in comparison with the traditional vaccinology (e.g., killed, attenuated or live vaccines) because the rationally designed vaccines contain specific immunogenic machinery of the organisms responsible for the disease rather than the whole-cell vaccines.
75
Over the past decades, the genome sequencing projects lead to accessibility to the genomic and proteomic data of E. granulosus and the other pathogenic microorganisms in databases. This information from the pathogens aid vaccinologists in rational antigen discovery using the computational methods.
76
The excretory/secretory (ES) products of adult E. granulosus have a critical role in physical interaction with the host proteins (likely plasminogen) during the host-pathogen interactions. Among various ES products, enolase is the most abundant one, which is known as a multifunctional tegumental enzyme. The EgEnolase in various parasites shows antigenic properties and in E. granulosus is also involved in the invasion and evasion mechanisms through the immunomodulation of the host immune system.
77,78
An epitope-based vaccine administrated to animal or human host body should ideally (i) be immunogenic sufficiently, (ii) has a high ability to stimulate significant humoral and cellular immune response, and finally (iii) activate the memory cells against the pathogenic epitopes. In our study, seven E. granulosus enolase protein sequences were checked in terms of antigenicity and the best ones were selected for further immunoinformatics analysis (Table S1, Supplementary file 1). Surface accessible clusters of residues in the folded antigenic proteins predominantly consist of hydrophilic amino acids, which can be mainly defined as BCE. The surface regions of antigens contribute to the direct interaction with the humoral immune cells especially immunoglobulins (e.g., IgG). Therefore, identification of the solvent surface accessible residues is the necessary step for more accurate recognition of the conformational BCEs.
79
Therefore, prior to the BCE mapping, the primary sequence was scanned for the presence of non-accessible residues such as a hydrophobic, transmembrane and signal peptide. B lymphocyte cells are the main arm of the humoral immune system. In vaccine designing against extracellular pathogens, the main immunological function is the detection of the epitope by the B-cell receptor.
80
In the current study, BCEs were identified based on the multi-method process that consists of different physicochemical, structural, and machine learning based methods (Fig. 2). CD4+ T-lymphocytes are the main player for coordination of both humoral and cellular immune responses.
81
Therefore, the helper T-cell receptor specific epitopes are the main components for vaccine designing against infectious diseases
13,61
as well as cancers.
73
The estimated physicochemical parameters of the final vaccine construct showed the vaccine protein contains aliphatic side chains and is hydrophobic, thermos-stable, and basic in nature.
82
The 3D structure of the EgEnolase, TLR-2, TLR-4, and DLA-DRB1*01101 was modeled with the minimum structural error (Fig. 1). The structural validation scores and tools including GA341, DOPE, ERRAT, Verify3D, and ProSA showed that the overall quality of the modeled vaccine construct was satisfactory. The GA341 score is ranged from 0.0 (worst model) to 1.0 (native-like model) and the model with more negative DOPE score counts as a stable model. The zero and negative ProSA Z-score values are related to the stabilize models.
37
Ramachandran plot indicated that a high percentage of residues (~96%) were clustered tightly into the favored and allowed regions of the map (Fig. 4 and Table1). The codon optimization parameters especially GC-content and CAI value of the improved sequence showed the nucleotide sequence can be effectively expressed in the E. coli system. Some disapproved hairpin loops in initial nucleotides of the mRNA were optimized to improve the mRNA stability especially in the ribosome binding site.
9
The formation of mRNA secondary structure is a key step in translation initiation, elongation, and generally mRNA biogenesis.
83
In this sense, the mRNA sequence should be optimized in terms of the codon usage bias of expression system (E. coli K12). In our research, this step was accomplished by analysis of guanine-cytosine (GC) content, codon adaptation index (CAI), an effective number of codons (Nc), and tRNA adaptation index (tAI) of the nucleotide sequence. The hydrophilicity and thermos-stability nature of the overexpressed vaccine construct in E. coli K12 are emergence factors for the investigation of the protein function. Leptospirosis surface adhesion 21 (Lsa21) is the main virulence factor in Leptospira pathogenesis and it is identified that has strong TLR-2 and TLR-4 activity leading to the control of Leptospira infection. Thus, Lsa21 suggested has a potent immunostimulatory capacity (or adjuvanticity) to induce strong innate responses.
58
Our designed vaccine showed an acceptable extent of the solubility in an overexpressed state. Moreover, we observed a high binding affinity between the selected HTEs and DRB1*01101 allele (Table 3). The overall vaccine construct displayed has a high binding affinity to the TLR-2 and TLR-4 receptors (Figs. 5 and 6). In this study, we aimed to integrate the comprehensive immunoinformatics tools to the known immunological data of dog-echinococcus interactions to design a non-allergen, safe, and immunogenic minigene vaccine. The immunological potency of the predicted epitopes for animal trial against E. granulosus is under the investigation by histopathological and immunological bioassays.
Conclusion
E. granulosus is an asymptomatic infection that can lead to the high-frequent cyst development to the various internal organs, such as liver, lung, kidney, and central nervous system. At any one time, more than one million people are affected with echinococcosis. Lack of an effective vaccine and high fiscal burden of the controlling programs especially in the developing countries resulted in the erosion condition among the people living in the endemic areas. On the other side, vaccination in animals is a cost-effective strategy to develop animal health and have an important impact on public health by controlling the zoonotic diseases. Therefore, it is of great necessity to develop the novel strategies to combat against the zoonotic pathogens such as E. granulosus. The previous studies revealed that the secretory proteins of E. granulosus (e.g., enolase) not only respond to the pathogenesis but also are the vital immunogenic proteins in the host-pathogen interactions. Therefore, this study was designed to establish a step ahead in the vaccinology. We used EgEnolase protein sequence to design an epitope-based vaccine construct. The minigene vaccine has T-helper and B-cell determinants that were selected based on the multiple methods. The minigene vaccine showed stable physicochemical properties as well as a proper binding affinity for the TLR-2 and TLR-4 receptors. Generally, this study was performed based on the step-by-step immunoinformatics analysis to design a rational vaccine that may fight prophylactically against the E. granulosus infection. In our on-going project, we are going to revalidate through in vitro and in vivo bioassays to prove the vaccine safety and immunogenicity.
Funding sources
This work is part of the military soldier service project, financially supported by the AJA University of Medical Sciences (grant No.: 997889).
Ethical Statement
There is none to be declared.
Acknowledgments
The authors thank Professor Yadollah Omidi for providing some facilities in terms of the computational methods and his helpful comments in improving the manuscript quality.
Competing interests
No potential conflict of interests was reported by the authors.
Author contributions
MMP contributed to the bioinformatics analysis and wrote the manuscript. MY, and MA supervised the study. GM and AN did result interpretation and discussion.
Supplementary Materials
Supplementary file 1 contains Figs. S1-S6 and Tables S1-S3.
(pdf)
Research Highlights
What is the current knowledge?
simple
-
√ The multi-method and structure-based methodology was served for designing a host-specific epitope-based vaccine against E. granulosus.
What is new here?
simple
-
√ Echinococcus granulosus enolase indicate an antigenic nature and can be used for the vaccine designing and development.
-
√ High accuracy structure modeling represents some comprehensive immunological properties about the identified B-cell epitopes.
-
√ The vaccine construct has helper T- and B-cell epitopes of varying length which are identified in a high accuracy sequential and structural manner.
-
√ The minigene vaccine showed a high binding affinity to the TLR-2 and TLR-4 receptors.
References
- Bingham GM, Budke CM, Larrieu E, Del Carpio M, Mujica G, Slater MR. A community-based study to examine the epidemiology of human cystic echinococcosis in Rio Negro Province, Argentina. Acta Trop 2014; 136:81-8. doi: 10.1016/j.actatropica.2014.04.005 [Crossref] [ Google Scholar]
- Carmena D, Cardona GA. Echinococcosis in wild carnivorous species: epidemiology, genotypic diversity, and implications for veterinary public health. Vet Parasitol 2014; 202:69-94. doi: 10.1016/j.vetpar.2014.03.009 [Crossref] [ Google Scholar]
-
WHO. Echinococcosis. WHO; 2017. Available from: https://www.who.int/echinococcosis/en/.
- Ansar W, Ghosh S. C-reactive protein and the biology of disease. Immunol Res 2013; 56:131-42. doi: 10.1007/s12026-013-8384-0 [Crossref] [ Google Scholar]
- Rivas-Santiago B, Cervantes-Villagrana AR. Novel approaches to tuberculosis prevention: DNA vaccines. Scand J Infect Dis 2014; 46:161-8. doi: 10.3109/00365548.2013.871645 [Crossref] [ Google Scholar]
- Borriello F, van Haren SD, Levy O. First International Precision Vaccines Conference: Multidisciplinary Approaches to Next-Generation Vaccines. mSphere 2018; 3:e00214-18. doi: 10.1128/mSphere.00214-18 [Crossref] [ Google Scholar]
- Bethony JM, Cole RN, Guo X, Kamhawi S, Lightowlers MW, Loukas A. Vaccines to combat the neglected tropical diseases. Immunol Rev 2011; 239:237-70. doi: 10.1111/j.1600-065X.2010.00976.x [Crossref] [ Google Scholar]
- Louise R, Skjot V, Agger EM, Andersen P. Antigen discovery and tuberculosis vaccine development in the post-genomic era. Scand J Infect Dis 2001; 33:643-7. doi: 10.1080/00365540110026971 [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Saeedi N, Barzegari A, Dehghani J, Omidi Y. Current status and future prospective of vaccine development against Echinococcus granulosus. Biologicals 2018; 51:1-11. doi: 10.1016/j.biologicals.2017.10.003 [Crossref] [ Google Scholar]
- Jahangiri A, Rasooli I, Gargari SL, Owlia P, Rahbar MR, Amani J. An in silico DNA vaccine against Listeria monocytogenes. Vaccine 2011; 29:6948-58. doi: 10.1016/j.vaccine.2011.07.040 [Crossref] [ Google Scholar]
- Li YJ, Yang J, Zhao H, Jia HY, Zhang LN, Liu XX. Bioinformatics prediction of egA31 recombinant antigen epitopes of Echinococcus granulosus. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 2012; 30:78-80. [ Google Scholar]
- Lu G, Yu XB, Huang C, Xu J, Wu ZD, Chen SY. Bioinformatics analysis for the structure and function of lactate dehydrogenase from Schistosoma japonicum. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 2007; 25:202-5. [ Google Scholar]
- Pourseif MM, Moghaddam G, Daghighkia H, Nematollahi A, Omidi Y. A novel B- and helper T-cell epitopes-based prophylactic vaccine against Echinococcus granulosus. Bioimpacts 2018; 8:39-52. doi: 10.15171/bi.2018.06 [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Naghili B, Saeedi N, Parvizpour S, Nematollahi A. A novel in silico minigene vaccine based on CD4(+) T-helper and B-cell epitopes of EG95 isolates for vaccination against cystic echinococcosis. Comput Biol Chem 2018; 72:150-63. doi: 10.1016/j.compbiolchem.2017.11.008 [Crossref] [ Google Scholar]
- Riffkin M, Seow HF, Jackson D, Brown L, Wood P. Defence against the immune barrage: helminth survival strategies. Immunol Cell Biol 1996; 74:564-74. doi: 10.1038/icb.1996.90 [Crossref] [ Google Scholar]
- Janeway CA, Jr Jr. How the immune system protects the host from infection. Microbes Infect 2001; 3:1167-71. [ Google Scholar]
- Lorenzatto KR, Monteiro KM, Paredes R, Paludo GP, da Fonseca MM, Galanti N. Fructose-bisphosphate aldolase and enolase from Echinococcus granulosus: genes, expression patterns and protein interactions of two potential moonlighting proteins. Gene 2012; 506:76-84. doi: 10.1016/j.gene.2012.06.046 [Crossref] [ Google Scholar]
- Labbe M, Peroval M, Bourdieu C, Girard-Misguich F, Pery P. Eimeria tenella enolase and pyruvate kinase: a likely role in glycolysis and in others functions. Int J Parasitol 2006; 36:1443-52. doi: 10.1016/j.ijpara.2006.08.011 [Crossref] [ Google Scholar]
- Pal-Bhowmick I, Vora HK, Jarori GK. Sub-cellular localization and post-translational modifications of the Plasmodium yoelii enolase suggest moonlighting functions. Malar J 2007; 6:45. doi: 10.1186/1475-2875-6-45 [Crossref] [ Google Scholar]
- Pomel S, Luk FC, Beckers CJ. Host cell egress and invasion induce marked relocations of glycolytic enzymes in Toxoplasma gondii tachyzoites. PLoS Pathog 2008; 4:e1000188. doi: 10.1371/journal.ppat.1000188 [Crossref] [ Google Scholar]
- Dehghani J, Adibkia K, Movafeghi A, Barzegari A, Pourseif MM, Maleki Kakelar H. Stable transformation of Spirulina (Arthrospira) platensis: a promising microalga for production of edible vaccines. Appl Microbiol Biotechnol 2018. doi: 10.1007/s00253-018-9296-7 [Crossref]
- Gan W, Zhao G, Xu H, Wu W, Du W, Huang J. Reverse vaccinology approach identify an Echinococcus granulosus tegumental membrane protein enolase as vaccine candidate. Parasitol Res 2010; 106:873-82. doi: 10.1007/s00436-010-1729-x [Crossref] [ Google Scholar]
- Cheng J, Randall AZ, Sweredoski MJ, Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res 2005; 33:W72-6. doi: 10.1093/nar/gki396 [Crossref] [ Google Scholar]
- Tsirigos KD, Peters C, Shu N, Kall L, Elofsson A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res 2015; 43:W401-7. doi: 10.1093/nar/gkv485 [Crossref] [ Google Scholar]
- Kall L, Krogh A, Sonnhammer EL. Advantages of combined transmembrane topology and signal peptide prediction--the Phobius web server. Nucleic Acids Res 2007; 35:W429-32. doi: 10.1093/nar/gkm256 [Crossref] [ Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001; 305:567-80. doi: 10.1006/jmbi.2000.4315 [Crossref] [ Google Scholar]
- Zhang YZ, Shen HB. Signal-3L 20: A Hierarchical Mixture Model for Enhancing Protein Signal Peptide Prediction by Incorporating Residue-Domain Cross-Level Features. J Chem Inf Model 2017; 57:988-99. doi: 10.1021/acs.jcim.6b00484 [Crossref] [ Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 40: discriminating signal peptides from transmembrane regions. Nat Methods 2011; 8:785-6. doi: 10.1038/nmeth.1701 [Crossref] [ Google Scholar]
- Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 1999; 294:1351-62. doi: 10.1006/jmbi.1999.3310 [Crossref] [ Google Scholar]
-
Gupta R, Jung E, Brubak S. NetNGlyc: Prediction of N-glycosylation sites in human proteins. 2005. Available from: http://www.cbs.dtu.dk/services/NetNGlyc/.
- Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J 2013; 32:1478-88. doi: 10.1038/emboj.2013.79 [Crossref] [ Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982; 157:105-32. doi: 10.1016/0022-2836(82)90515-0 [Crossref] [ Google Scholar]
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 1999; 112:531-52. doi: 10.1385/1-59259-890-0:571 [Crossref] [ Google Scholar]
- Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Protein Sci 2016; 86:2 9 1-2 9 37. doi: 10.1002/cpps.20 [Crossref] [ Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605-12. doi: 10.1002/jcc.20084 [Crossref] [ Google Scholar]
- Case DA, Cheatham TE, 3rd 3rd, Darden T, Gohlke H, Luo R, Merz KM. The Amber biomolecular simulation programs. J Comput Chem 2005; 26:1668-88. doi: 10.1002/jcc.20290 [Crossref] [ Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 2007; 35:W407-10. doi: 10.1093/nar/gkm290 [Crossref] [ Google Scholar]
- Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 1997; 277:396-404. doi: 10.1016/S0076-6879(97)77022-8 [Crossref] [ Google Scholar]
- Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 1993; 2:1511-9. doi: 10.1002/pro.5560020916 [Crossref] [ Google Scholar]
- Lovell SC, Davis IW, Arendall WB, 3rd 3rd, de Bakker PI, Word JM, Prisant MG. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 2003; 50:437-50. doi: 10.1002/prot.10286 [Crossref] [ Google Scholar]
- Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol 1978; 47:45-148. doi: 10.1002/9780470122921.ch2 [Crossref] [ Google Scholar]
- Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol 1985; 55:836-9. [ Google Scholar]
- Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 1990; 276:172-4. [ Google Scholar]
- Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 1986; 25:5425-32. doi: 10.1021/bi00367a013 [Crossref] [ Google Scholar]
- Singh H, Ansari HR, Raghava GP. Improved method for linear B-cell epitope prediction using antigen's primary sequence. PLoS One 2013; 8:e62216. doi: 10.1371/journal.pone.0062216 [Crossref] [ Google Scholar]
- Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res 2006; 2:2. doi: 10.1186/1745-7580-2-2 [Crossref] [ Google Scholar]
-
Saha S, Raghava G, editors. BcePred: prediction of continuous
B-cell epitopes in antigenic sequences using physico-chemical
properties. International Conference on Artificial Immune Systems;
2004: Springer. doi:10.1007/978-3-540-30220-9_16
- Saha S, Raghava GP. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006; 65:40-8. doi: 10.1002/prot.21078 [Crossref] [ Google Scholar]
- Yao B, Zhang L, Liang S, Zhang C. SVMTriP: a method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PLoS One 2012; 7:e45152. doi: 10.1371/journal.pone.0045152 [Crossref] [ Google Scholar]
- Qi T, Qiu T, Zhang Q, Tang K, Fan Y, Qiu J. SEPPA 20--more refined server to predict spatial epitope considering species of immune host and subcellular localization of protein antigen. Nucleic Acids Res 2014; 42:W59-63. doi: 10.1093/nar/gku395 [Crossref] [ Google Scholar]
- Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 2008; 9:514. doi: 10.1186/1471-2105-9-514 [Crossref] [ Google Scholar]
-
Hall TA, editor. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic acids symposium series; 1999.
- Pandey RK, Sundar S, Prajapati VK. Differential Expression of miRNA Regulates T Cell Differentiation and Plasticity During Visceral Leishmaniasis Infection. Front Microbiol 2016; 7:206. doi: 10.3389/fmicb.2016.00206 [Crossref] [ Google Scholar]
- Scully C, Georgakopoulou EA, Hassona Y. The Immune System: Basis of so much Health and Disease: 3 Adaptive Immunity. Dent Update 2017; 44:322-4, 7. doi: 10.12968/denu.2017.44.4.322 [Crossref] [ Google Scholar]
- Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C. The ClusPro web server for protein-protein docking. Nat Protoc 2017; 12:255-78. doi: 10.1038/nprot.2016.169 [Crossref] [ Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 2011; 51:2778-86. doi: 10.1021/ci200227u [Crossref] [ Google Scholar]
- Shan JY, Ji WZ, Li HT, Tuxun T, Lin RY, Wen H. TLR2 and TLR4 expression in peripheral blood mononuclear cells of patients with chronic cystic echinococcosis and its relationship with IL-10. Parasite Immunol 2011; 33:692-6. doi: 10.1111/j.1365-3024.2011.01335.x [Crossref] [ Google Scholar]
- Faisal SM, Varma VP, Subathra M, Azam S, Sunkara AK, Akif M. Leptospira surface adhesin (Lsa21) induces Toll like receptor 2 and 4 mediated inflammatory responses in macrophages. Sci Rep 2016; 6:39530. doi: 10.1038/srep39530 [Crossref] [ Google Scholar]
- Shanmugam A, Rajoria S, George AL, Mittelman A, Suriano R, Tiwari RK. Synthetic Toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PLoS One 2012; 7:e30839. doi: 10.1371/journal.pone.0030839 [Crossref] [ Google Scholar]
- Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev 2013; 65:1357-69. doi: 10.1016/j.addr.2012.09.039 [Crossref] [ Google Scholar]
- Pandey RK, Bhatt TK, Prajapati VK. Novel Immunoinformatics Approaches to Design Multi-epitope Subunit Vaccine for Malaria by Investigating Anopheles Salivary Protein. Sci Rep 2018; 8:1125. doi: 10.1038/s41598-018-19456-1 [Crossref] [ Google Scholar]
- Ivanciuc O, Schein CH, Braun W. SDAP: database and computational tools for allergenic proteins. Nucleic Acids Res 2003; 31:359-62. doi: 10.1093/nar/gkg010 [Crossref] [ Google Scholar]
- Saha S, Raghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res 2006; 34:W202-9. doi: 10.1093/nar/gkl343 [Crossref] [ Google Scholar]
- Pandey RK, Kumbhar BV, Srivastava S, Malik R, Sundar S, Kunwar A. Febrifugine analogues as Leishmania donovani trypanothione reductase inhibitors: binding energy analysis assisted by molecular docking, ADMET and molecular dynamics simulation. J Biomol Struct Dyn 2017; 35:141-58. doi: 10.1080/07391102.2015.1135298 [Crossref] [ Google Scholar]
- Kallberg M, Margaryan G, Wang S, Ma J, Xu J. RaptorX server: a resource for template-based protein structure modeling. Methods Mol Biol 2014; 1137:17-27. doi: 10.1007/978-1-4939-0366-5_2 [Crossref] [ Google Scholar]
- Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol 2014; 1137:1-15. doi: 10.1007/978-1-4939-0366-5_1 [Crossref] [ Google Scholar]
- Blundell TL, Hendrickson WA. What is 'current opinion' in structural biology?. Curr Opin Struct Biol 2011; 21:447-9. doi: 10.1016/j.sbi.2011.06.005 [Crossref] [ Google Scholar]
- Jung SK, McDonald K. Visual gene developer: a fully programmable bioinformatics software for synthetic gene optimization. BMC Bioinformatics 2011; 12:340. doi: 10.1186/1471-2105-12-340 [Crossref] [ Google Scholar]
- Kahali B, Basak S, Ghosh TC. Delving deeper into the unexpected correlation between gene expressivity and codon usage bias of Escherichia coli genome. J Biomol Struct Dyn 2008; 25:655-61. doi: 10.1080/07391102.2008.10507212 [Crossref] [ Google Scholar]
- Heo L, Park H, Seok C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res 2013; 41:W384-8. doi: 10.1093/nar/gkt458 [Crossref] [ Google Scholar]
- Yang Z, Lasker K, Schneidman-Duhovny D, Webb B, Huang CC, Pettersen EF. UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J Struct Biol 2012; 179:269-78. doi: 10.1016/j.jsb.2011.09.006 [Crossref] [ Google Scholar]
- Yang Y, Sun W, Guo J, Zhao G, Sun S, Yu H. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum Vaccin Immunother 2015; 11:795-805. doi: 10.1080/21645515.2015.1012017 [Crossref] [ Google Scholar]
- Sepideh Parvizpour JR, Mohammad Mostafa Pourseif, Yadollah Omidi. In silico design of a triple-negative breast cancer vaccine by targeting cancer testis antigens. Bioimpacts 2019; 9:55-64. doi: 10.15171/bi.2019.06 [Crossref] [ Google Scholar]
- Peng J, Xu J. A multiple-template approach to protein threading. Proteins 2011; 79:1930-9. doi: 10.1002/prot.23016 [Crossref] [ Google Scholar]
- Phythian CJ, Jackson B, Bell R, Citer L, Barwell R, Windsor PA. Abattoir surveillance of Sarcocystis spp, Cysticercosis ovis and Echinococcus granulosus in Tasmanian slaughter sheep, 2007-2013. Aust Vet J 2018; 96:62-8. doi: 10.1111/avj.12670 [Crossref] [ Google Scholar]
- Rueckert C, Guzman CA. Vaccines: from empirical development to rational design. PLoS Pathog 2012; 8:e1003001. doi: 10.1371/journal.ppat.1003001 [Crossref] [ Google Scholar]
- Wang Y, Xiao D, Shen Y, Han X, Zhao F, Li X. Proteomic analysis of the excretory/secretory products and antigenic proteins of Echinococcus granulosus adult worms from infected dogs. BMC Vet Res 2015; 11:119. doi: 10.1186/s12917-015-0423-8 [Crossref] [ Google Scholar]
- Bernal D, de la Rubia JE, Carrasco-Abad AM, Toledo R, Mas-Coma S, Marcilla A. Identification of enolase as a plasminogen-binding protein in excretory-secretory products of Fasciola hepatica. FEBS Lett 2004; 563:203-6. doi: 10.1016/S0014-5793(04)00306-0 [Crossref] [ Google Scholar]
- Getzoff ED, Tainer JA, Lerner RA, Geysen HM. The chemistry and mechanism of antibody binding to protein antigens. Adv Immunol 1988; 43:1-98. doi: 10.1016/S0065-2776(08)60363-6 [Crossref] [ Google Scholar]
- Hoft DF, Brusic V, Sakala IG. Optimizing vaccine development. Cell Microbiol 2011; 13:934-42. doi: 10.1111/j.1462-5822.2011.01609.x [Crossref] [ Google Scholar]
- Okoye IS, Wilson MS. CD4+ T helper 2 cells--microbial triggers, differentiation requirements and effector functions. Immunology 2011; 134:368-77. doi: 10.1111/j.1365-2567.2011.03497.x [Crossref] [ Google Scholar]
- Khatoon N, Pandey RK, Prajapati VK. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep 2017; 7:8285. doi: 10.1038/s41598-017-08842-w [Crossref] [ Google Scholar]
- Marin A, Gallardo M, Kato Y, Shirahige K, Gutierrez G, Ohta K. Relationship between G+C content, ORF-length and mRNA concentration in Saccharomyces cerevisiae. Yeast 2003; 20:703-11. doi: 10.1002/yea.992 [Crossref] [ Google Scholar]