Bioimpacts. 11(2):119-127.
doi: 10.34172/bi.2021.19
Original Research
In silico approach towards the identification of potential inhibitors from Curcuma amada Roxb against H. pylori: ADMET screening and molecular docking studies
G Divyashri 1, *  , T P Krishna Murthy 1, Subramaniam Sundareshan 1, Pavan Kamath 1, Manikanta Murahari 2, 3, *, G R Saraswathy 2, 4, Bindu Sadanandan 1
, T P Krishna Murthy 1, Subramaniam Sundareshan 1, Pavan Kamath 1, Manikanta Murahari 2, 3, *, G R Saraswathy 2, 4, Bindu Sadanandan 1
Author information:
1Department of Biotechnology, M S Ramaiah Institute of Technology, Bangalore, Karnataka, India
2Pharmacological Modelling and Simulation Centre, M.S. Ramaiah University of Applied Sciences, Bangalore, Karnataka, India
3Department of Pharmaceutical Chemistry, Faculty of Pharmacy, M.S. Ramaiah University of Applied Sciences, Bangalore, Karnataka, India
4Department of Pharmacy Practice, Faculty of Pharmacy, M.S. Ramaiah University of Applied Sciences, Bangalore, Karnataka, India
Abstract
Introduction:
The present study attempts to identify potential targets of H. pylori for novel inhibitors from therapeutic herb, mango ginger (Curcuma amada Roxb.).
Methods:
Crystal structure of all the selected drug targets obtained from Protein Data Bank (PDB) were subjected to molecular docking against a total of 130 compounds (found to have biological activity against H. pylori ) were retrieved from public databases. Compounds with good binding affinity were selected for Prime MM-GBSA rescoring and molecular dynamics (MD) simulation. Final list of compounds were taken for ADMET predictions.
Results:
Based on binding affinity denoted by glide score and ligand efficiency, mango ginger compounds were found selective to shikimate kinase and type II dehydroquinase through hydrogen bonding and salt bridge interactions. Stability of the interactions and free energy calculations by Prime MM-GBSA results confirmed the affinity of mango ginger compounds towards both shikimate kinase and type II dehydroquinase. From the above results, 15 compounds were calculated for ADMET parameters, Lipinski’s rule of five, and the results were found promising without any limitations. MD simulations identified gentisic acid as hit compound for shikimate kinase of H. pylori.
Conclusion:
Current study could identify the in silico potential of mango ginger compounds against shikimate kinase and type II dehydroquinase targets for H. pylori infections and are suitable for in vitro and in vivo evaluation.
Keywords: Mango ginger, H. pylori, Gentisic acid, Molecular dynamics, Docking, Lipinski’s rule of five
Copyright and License Information
© 2021 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Helicobacter pylori infection is reported to be the most common chronic bacterial infections in humans, affecting approximately “4.4 billion individuals worldwide.”
1
Infection with H pylori is associated with the development of chronic gastritis, peptic ulcer, gastric adenocarcinoma, mucosa-associated lymphoid tissue lymphoma, idiopathic thrombocytopenic purpura, vitamin B12, and iron deficiency.
2,3
Management of H. pylori-associated gastrointestinal disorders necessitates the early treatment of H. pylori infection.
4
However, the current commercially available therapeutic regime (standard triple therapy consisting of proton pump inhibitor and antibiotics viz., amoxicillin and clarithromycin) are regularly been adversely indicated due to their side effects and related toxicity.
5
Emergence and the increasing prevalence of multi-drug resistant strains of H. pylori has led to reduced success rate in various treatment regimens.
6
In line with this, it is required to choose new alternatives with more efficiency and less toxicity.
7
In this regard, therapeutic herbs can be attractive and desirable options because of their positive outcomes in the treatment of H pylori-associated gastric ulcers.
8
Furthermore, in recent years’ number of studies have suggested that H. pylori infection can be suppressed through the use of medicinal plants accounting for their antimicrobial activity.
9
Mango ginger (Curcuma amada Roxb.) is a unique spice having morphological similarity with ginger but imparts a raw mango flavor. This plant is significantly recognized in Ayurveda and Unani medicinal systems because of its antipyretic, diuretic, expectorant, and laxative health attributes.
10
The numerous biological activities of mango ginger such as antioxidant, antibacterial, anti-inflammatory, antiallergic, antifungal, platelet aggregation inhibition activity, and analgesic activity is recognized for the presence of major constituents in its rhizomes namely curcuminoids (curcumin, demethoxycurcumin, bis-demethoxycurcumin), phenolic compounds (caffeic acid, gentisic acid, ferulic acid, gallic acid, cinnamic acid), terpenoids (difurocumenol, amadannulen, amadaldehyde) and essential oils (β-myrcene and α-asarone).
11
Enzymes involved in shikimate pathway (biosynthesis of folates and aromatic amino acids) namely type II dehydroquinase, and shikimate kinase inhibition might show promising activity in therapy of H. pylori infections.Type II dehydroquinase and shikimate kinase are the third and fifth enzymes in the shikimate pathway and are widely targeted by many research groups resulting in inhibitors with a low micromolar affinity, including a compound with a non-competitive inhibition mechanism.
12
Furthermore, due to the major difference between the enzymes involved in the type II fatty acid (FAS II) synthetic pathway in H. pylori and mammals, the enzymes involved in FAS II can also be treated as potential drug targets. In this line, hydroxyacyl-acyl carrier proteins dehydratase has been used as drug target by few researchers.
13
Glutamate racemase, involved in the production and maintenance of d-glutamate levels is highly required for H pylori growth. This enzyme is selected in the discovery of both narrow and broad-spectrum inhibitors.
14
It is also being suggested for many decades that vital ubiquitous enzyme, fructose 1,6-bisphosphate aldolase (FBA) could be a good drug target against bacteria. Because these organisms possess a metal-dependent (Class II) FBA, in contrast to higher organisms which possess a Schiff-base forming and metal-independent (Class I) FBA.
15
The present study was aimed to screen 130 compounds (reported in the literature for their biological activity against H. pylori) from mango ginger to investigate the in silico binding affinity by glide docking, prime energy calculations, and molecular dynamics (MD) for selected targets of H. pylori and predict their pharmacokinetic properties (Fig. 1). Selected drug targets were the enzymes viz., type II dehydroquinase, fructose 1,6-biphosphate aldolase, hydroxyacyl-acyl carrier proteins dehydratase, shikimate kinase, and glutamate racemase found in H. pylori.
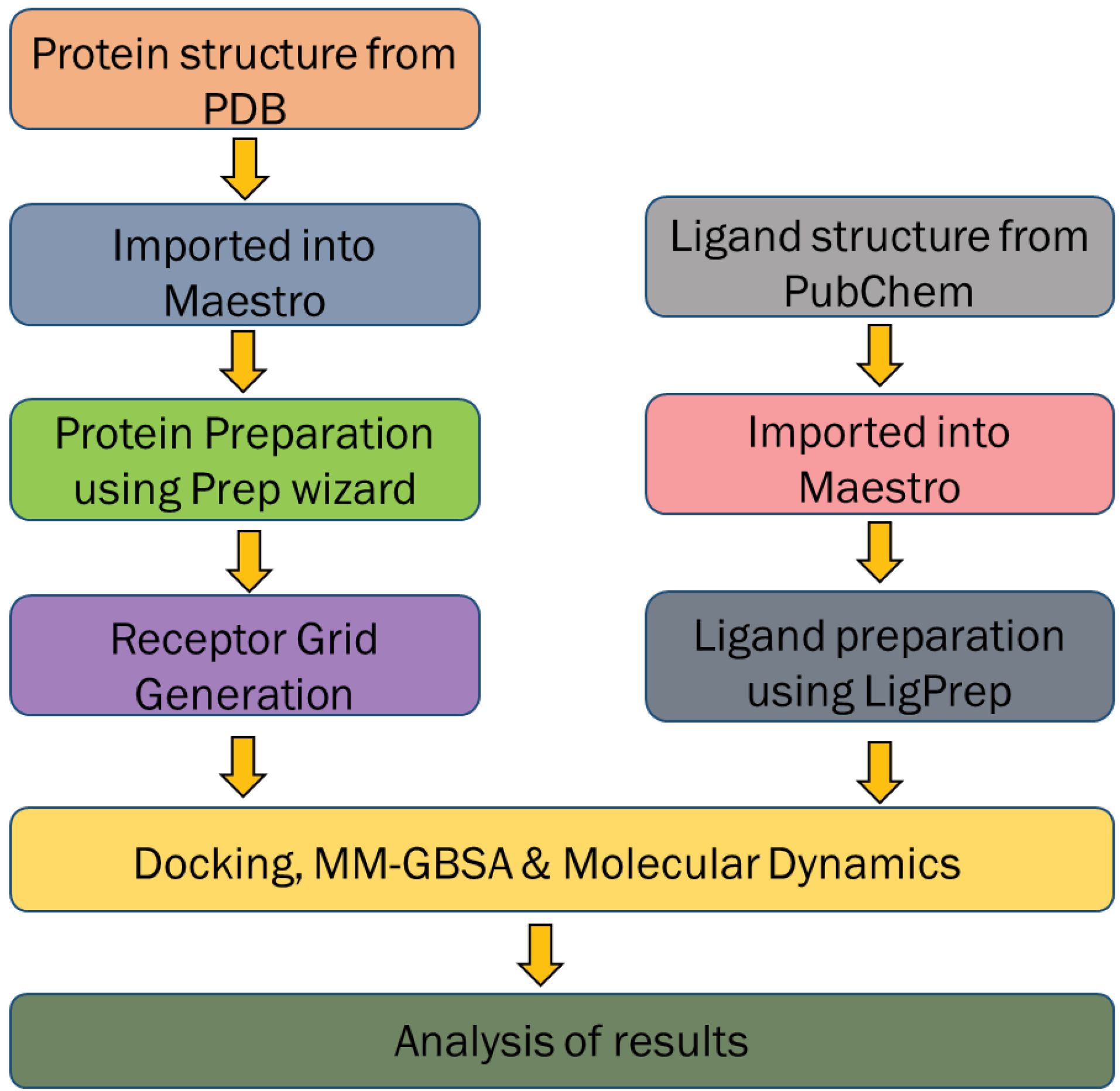
Fig. 1.
Overall scheme for the screening of compounds from mango ginger against the selected drug target.
.
Overall scheme for the screening of compounds from mango ginger against the selected drug target.
Materials and Methods
Retrieval of H. pylori drug targets and preparation for docking
Retrieval of H. pylori drug targets: All the crystal structures of H. pylori drug targets viz., type II dehydroquinase, fructose 1,6-biphosphate aldolase, hydroxyacyl-acyl carrier proteins dehydratase, shikimate kinase, and glutamate racemase were retrieved from Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) (http://www.rcsb.org/pdb/home/home.do). To the retrieved structures, necessary changes (removal of heteroatoms, addition of missing hydrogen atoms, assigning proper bond orders, and removal of water molecules) were made using the optimized potentials for liquid simulations (OPLS) force field.
16
Grid was generated by selecting the co-crystalized ligand for respective target proteins as per the instructions of Glide tool.
17
Ligand preparation
The chemical structures of 130 compounds from mango ginger which are known for their biological activity against H. pylori were retrieved from PubChem and ChEMBL databases. Few structures were drawn using Maestro utility of the Schrodinger software.
18
Ligand preparation process involved mainly estimating the partial atomic charge using the OPLS-3 force field, preserving the chiralities and generating maximum of 64 low-energy stereoisomers per ligand, followed by geometry and energy minimization. The resulting ligands were subjected to docking using the Glide module of Schrodinger suite.
Molecular docking studies
Molecular docking was performed with grid-based ligand docking with energetics (Glide) module of Schrodinger suite to determine the binding affinity and protein-ligand interactions.
19
All the structures of target proteins were retrieved from PDB and subjected to protein preparation wizard of Maestro. During the process of structure refinement, water molecules were removed, hydrogens were added and missing bond orders were adjusted. Missing residues were verified as notified by the Maestro, subjected for structure optimization and minimization, and proceeded to receptor grid generation. Grid was generated at the centroid of workspace co-crystallized ligands for type II dehydroquinase (PDB ID: 2XDA, 1.85 Å), fructose 1,6-biphosphate aldolase (PDB ID:3C56, 2.3 Å), and glutamate racemase (PDB ID: 2JFZ; 1.86 Å). For hydroxyacyl-acyl carrier proteins dehydratase (PDB ID: 3B7J, 2.4 Å) and shikimate kinase (PDB ID: 1ZUI; 2.3 Å), grid dimensions were modified manually to (30 x 30 x 30 Å) as the co-crystallized ligands were small. Only fructose 1,6-biphosphate aldolase has a zinc metal and it was added to the constraints tab. Selected Hydroxyl groups of amino acids as rotatable groups. Once the receptor grid was generated, ligands were docked to the protein using Glide docking protocol. The ligands were docked by using a three tire docking process viz., "High throughput Virtual Screening" (HTVS) followed by "Standard Precision" (SP) and then by "Extra Precision" mode (XP). Docking was selected for flexibility, haven’t opted for ring conformations, for amide bonds- penalize nonpolar conformation. Selected 5000 poses for the initial phase of docking and to save 500 poses per ligand. Ligand parameters were kept on default. Force field version utilized for calculations is OPLS_2005. The docked conformers were evaluated using Glide (G) Score.
20,21
Overall scheme of our approach is shown in Fig. 1.
MM-GBSA rescoring
Here, Prime MM-GBSA approach was applied to predict the binding free energies (ΔGbind) of top 5 compounds identified from Glide XP docking with shikimate kinase and type II dehydroquinase targets using OPLS_2005 force field.
22
In addition to the binding free energy (ΔGbinding), individual components like hydrogen bond correction (ΔGH-bond), van der Waals energy (ΔGvdw), lipophilic energy (ΔGlipo), and Coulomb energy (ΔGcoulomb) contributing to free energy were analyzed.
23
Molecular Dynamics simulations
MD simulations were carried out using Desmond tool of Schrodinger Drug Design Suite for gentisic acid with shikimate kinase and (E)-Labda-8(17),13-diene-15,16-olide with type II dehydroquinase for 100 ns.
24
Methodology involved was respective complex is placed in a box of TI3P type water model. Overall charge of the complex was calculated and neutralized by adding counter ions, the entire system was minimized using Desmond minimization tools and subjected to dynamics simulations. The whole simulation was run at 300°K temperature and atmospheric pressure. Trajectory was recorded and stability of respective complex was analyzed for RMSD fluctuations, Protein-Ligand interactions, and plots using Simulation Event Analysis tool of Desmond.
25
ADME prediction
Compounds with good docking score and ligand affinity, lowest free binding energy obtained from Glide docking and Prime MM-GBSA calculations, were further subjected to absorption, distribution, metabolism, excretion (ADME) predictions using QikProp tool of Schrodinger suite.
26
Lipinski filter was applied before virtual screening to avoid false-positive lead molecule using OSIRIS Property explorer. Lipinski filter rejected ligands not following Lipinski rule of five and reactive filter rejected ligands with reactive functional groups.
Results and Discussion
Screening and identification of potential inhibitors from mango ginger plant against H. pylori: Molecular docking studies
A list of 130 isolated compounds from mango ginger were selected and docked into the active site of the drug targets with the generated receptor grid by Extra Precision (XP) mode of Glide. Further, compounds were ranked based on docking scores and the score above -7.0 were categorized as in silico potential compounds and below -7.0 as less active.
27
Using this threshold, a total of 110, 124, 127, 103, and 117 compounds with docking score over -7.0 were identified as potential inhibitory compounds against hydroxyacyl-acyl carrier proteins dehydratase, glutamate racemase, shikimate kinase, type II dehydroquinase and fructose 1,6-biphosphate aldolase, respectively. The result of the docking studies against each drug target is listed in Table 1.
Table 1.
Molecular docking results of compounds from mango ginger (only top 5 compounds shown) against each drug target
|
Compound Name
|
Glide Score
|
Ligand efficiency
|
Glide
Emodel
|
Type of interactions
|
Residue Information
|
|
Hydroxyacyl-acyl carrier proteins dehydratase
|
| Gentisic acid |
-7.999 |
-0.726 |
-48.399 |
H |
GLY67; PHE101; PHE65; VAL113 |
| Ledol |
-7.939 |
-0.496 |
-38.304 |
H |
PHE101 |
| Spathulenol |
-7.807 |
-0.488 |
-41.704 |
H |
GLY67 |
| E-Sabinol |
-7.782 |
-0.707 |
-36.132 |
H |
VAL113; PHE65 |
| Amadannulen |
-7.588 |
-0.281 |
-55.519 |
H |
PHE101; PHE109 |
|
Glutamate racemase
|
| Torreyol |
-7.685 |
-0.452 |
-35.846 |
H |
ILE149 |
| Alpha-Muurolene |
-7.430 |
-0.495 |
-34.675 |
No |
|
| Zerumin A |
-7.420 |
-0.323 |
-47.660 |
H; Salt bridge |
LYS17; LYS17 |
| Spathulenol |
-7.365 |
-0.460 |
-36.281 |
H |
ILE149 |
| Demethoxycurcumin |
-7.358 |
-0.273 |
-58.984 |
H |
GLY11; GLN248; TRP252 |
|
Shikimate kinase
|
| Protocatechuic acid |
-8.458 |
-0.769 |
-79.921 |
H; Salt bridge |
ARG57; ASP33; GLY80; GLU114; ARG132; RG116 |
| Gentisic acid |
-8.427 |
-0.766 |
-77.690 |
H |
ASP33; ARG132; ARG116; ARG57 |
| Gallic acid |
-8.061 |
-0.672 |
-74.465 |
H |
ASP33; ARG132; ARG116; ARG57 |
| Syringic acid |
-7.362 |
-0.526 |
-45.894 |
H; Salt bridge |
ASP33; ARG132; ARG116, ARG57 |
| 3-Exo-hydroxy-1,8-cineole |
-5.893 |
-0.491 |
-21.211 |
H |
ARG116; GLU114; GLY81; ARG57 |
|
Type II dehydroquinase
|
| (E)-Labda-8(17),13-diene-15,16-olide |
-8.435 |
-0.444 |
-53.605 |
H |
LEU103; THR104; HIP102 |
| Zerumin A |
-8.338 |
-0.363 |
-55.321 |
H |
THR104, HIP102 |
| Gentisic acid |
-8.316 |
-0.756 |
-64.652 |
H; Salt bridge |
THR104; ARG113; HIS82; LEU103; ASN76; HIP102 |
| Gallic acid |
-7.99 |
-0.666 |
-66.844 |
H; Salt bridge |
ASN76; LEU103; THR104; HIS82; HIP102 |
| Protocatechuic acid |
-7.911 |
-0.719 |
-62.483 |
H; Salt bridge |
ASN76; LEU103; THR104; ARG113; HIS82; HIP102 |
|
Fructose 1,6-biphosphate aldolase
|
| Gentisic acid |
-7.785 |
-0.707 |
-64.122 |
H; Salt bridge |
THR256; LYS184; ASP255; HIE210; LYS184 |
| Gallic acid |
-7.741 |
-0.645 |
-68.456 |
H |
LYS184; THR256; ASP255; HIP83; ASP82; ASN253; GLY211; HIE210 |
| Protocatechuic acid |
-7.255 |
-0.651 |
-65.181 |
H; Salt bridge |
THR256; LYS184; SER213; ASP255; HIP83; ASP82; ASN253; LYS184 |
| Zerumin A |
-7.206 |
-0.314 |
-66.036 |
H; Salt bridge |
GLY211; LYS184; THR256; LYS184 |
| Caffeic acid |
-7.046 |
-0.542 |
-66.278 |
H; Salt bridge |
ASP255; THR256; LYS184; SER213; ASN253; ASP82; LYS184 |
H : Hydrogen bond; No: no molecular interactions found.
Based on the ligand affinity and docking scores (Tables 1 and 2), mango ginger compounds were found more selective to type II dehydroquinase and shikimate kinase. For type II dehydroquinase, docking score for the top five compounds was observed in the range of -8.435 to -7.911 kcal/mol and three compounds have exhibited scores more than -8.0 kcal/mol. Interestingly, all the five compounds formed hydrogen bonding with THR104 and four compounds with LEU103. Similarly, three compounds have formed salt bridge interactions with HIP102. Following hydrogen bonding and salt bridge interactions with amino acid residues of target protein might further strengthen the complex stability or binding affinity. Further experimental screening might help in claiming significant interactions for the target. Coming to shikimate kinase, three compounds demonstrated docking score more than -8.0 kcal/mol and protocatechuic acid has exhibited highest docking score -8.458 compared to all mango ginger compounds with all the five targets. Concerning interactions, all the five compounds have formed hydrogen bonding with ARG57 and only two compounds have formed salt bridge interactions with arginine residue but of different numbers (Table 1). Interestingly, gentisic acid and gallic acid have formed hydrogen bonding interactions exactly with the same residues. But syringic acid also displayed similar interactions, but salt bridge interaction with ARG57 might have reduced the docking score. Similarly, mango ginger compounds have exhibited good docking scores and interactions with other three selected targets.
Table 2.
Common targets identified for mango ginger compounds by Glide docking
|
Compound Name
|
Target Names
|
Docking score
|
Ligand efficiency
|
| Gentisic acid |
Hydroxyacyl-acyl carrier proteins dehydratase |
-7.999 |
-0.726 |
|
Shikimate kinase
|
-8.427
|
-0.766
|
| Type II dehydroquinase |
-8.316 |
-0.756 |
| Fructose 1,6-biphosphate aldolase |
-7.785 |
-0.707 |
| Zerumin A |
Glutamate racemase |
-7.420 |
-0.323 |
|
Type II dehydroquinase
|
-8.338
|
-0.363
|
| Fructose 1,6-biphosphate aldolase |
-7.206 |
-0.314 |
| Protocatechuic acid |
Shikimate kinase
|
-8.458
|
-0.769
|
| Type II dehydroquinase |
-7.911 |
-0.719 |
| Fructose 1,6-biphosphate aldolase |
-7.255 |
-0.651 |
| Gallic acid |
Shikimate kinase
|
-8.061
|
-0.672
|
| Type II dehydroquinase |
-7.990 |
-0.666 |
| Fructose 1,6-biphosphate aldolase |
-7.741 |
-0.645 |
Comparison of the molecular docking results of mango ginger compounds identified that five compounds have common targets (Table 2). Gentisic acid is sharing with four targets and other three compounds zerumin A, protocatechuic acid, and gallic acid with three targets. In terms of docking score and ligand efficiency, compounds exhibited affinity towards shikimate kinase and Type II dehydroquinase. Out of which, three compounds gentisic acid, protocatechuic acid and gallic acid with shikimate kinase, and zerumin A with type II dehydroquinase. Based on the docking score and ligand efficiency, top five compounds identified with shikimate kinase and type II dehydroquinase (Table 1) were taken for free binding energy calculations using Prime MM-GBSA approach.
Prime MM-GBSA rescoring
From the docking results with Glide of Schrodinger Suite, mango ginger compounds have demonstrated better affinity with molecular targets of H. pylori particularly shikimate kinase and type II dehydroquinase. With these findings, top five identified from docking were further taken for free binding energy calculations using Prime module. The free binding energy of respective complex and individual contributions for the total energy values were shown in Table 3. Energy values help us in understanding the stability factor of individual complex and in turn, suggest modifying the chemical structure of ligand to bring down the free binding energy. As the Prime MM-GBSA calculations were done for natural compounds, following results might help in the design of synthetic compounds by taking lead molecules based on experimental results. Though prime energy calculations have more significance for synthetic compounds, we have conducted to investigate the energy contributions of individual ligand-protein complex to further refine the list of in silico potential compounds.
Table 3.
Prime MM-GBSA rescoring of in silico potential compounds
|
Compound name
|
Target name
|
ΔG
binding
|
ΔG
H-bond
|
ΔG
lipo
|
ΔG
vdw
|
ΔG
coulomb
|
| Protocatechuic acid |
Shikimate kinase |
-37.74 |
-6.95 |
-9.78 |
-20.46 |
-72.68 |
| Gallic acid |
-35.82 |
-7.04 |
-9.26 |
-21.81 |
-72.52 |
| Gentisic acid |
-44.65 |
-6.39 |
-9.20 |
-22.01 |
-84.74 |
| Syringic acid |
-22.15 |
-3.58 |
-5.23 |
-24.80 |
-38.15 |
| 3-Exo-hydroxy-1,8-cineole |
0.97 |
-1.22 |
-13.03 |
-1.60 |
-17.54 |
| (E)-Labda-8(17),13-diene-15,16-olide |
Type II dehydroquinase |
-48.45 |
-2.47 |
-21.71 |
-33.93 |
-18.62 |
| Gallic acid |
-37.08 |
-5.15 |
-6.89 |
-25.28 |
-41.36 |
| Protocatechuic acid |
-36.92 |
-5.08 |
-6.94 |
-23.50 |
-40.49 |
| Gentisic acid |
-39.96 |
-5.02 |
-6.83 |
-24.07 |
-49.22 |
| Zerumin A |
1.89 |
-4.25 |
-22.29 |
-0.42 |
-32.47 |
*ΔGbinding: MM-GBSA free energy of binding; ΔGH-bond: hydrogen-bonding correction; ΔGlipo: lipophilic energy of the complex; ΔGvdw: van der Waals energy of the complex; ΔGcoulomb: Coulomb energy of the complex.
Based on the free binding energy calculations, gentisic acid with shikimate kinase and (E)-Labda-8(17),13-diene-15,16-olide with type II dehydroquinase were found with lowest free energy values, and complexes were stabilized with Coulomb and Van der Waals energies. But the complex of (E)-Labda-8(17),13-diene-15,16-olide with type II dehydroquinase was found stabilized more with Van der Waals and lipophilic energies. Concerning free energy calculations, both the above-identified complexes were taken for MD simulations to further investigate the stability. 3-Exo-hydroxy-1,8-cineole with shikimate kinase and zerumin A with type II dehydroquinase were found with highest free binding energy among the five compounds selected for each target. The other four compounds for both the targets have demonstrated better energy values. For shikimate kinase, ligand-protein complexes were more stabilized with coulomb energy. Whereas for type II dehydroquinase, compound with lowest binding energy was stabilized with van der Waals, lipophilic, and Coulomb energies, and other three compounds with van der Waals and Coulomb energies.
Molecular dynamics simulations
To further investigate the stability of complex and binding interactions at the active pocket, MD simulations were carried out to gentisic acid and shikimate kinase complex for 100 ns. In the ligand RMSD plot, large drift was observed for the first 15 ns then it got stabilized. Later a deep fluctuation was observed around 70 ns. Except for the fluctuation around 70 ns, complex observed stable with acceptable RMSD fluctuations in the range of 1 Å. The compound has exhibited hydrogen bonding interactions with ARG116 (94%), ASP33 (67%), ARG57 (48%), and few more interactions with ARG132, GLU53 and GLU60 mediated through water molecules (Fig. 2). Similarly, MD simulation was conducted to (E)-Labda-8(17),13-diene-15,16-olide, and type II dehydroquinase complex for 100 ns (Fig. 3). The first 20 ns of simulation can be omitted for the stabilization of complex. But after 20 ns also, ligand has undergone RMSD fluctuations in the range of 2 Å. Overall, stability of ligand was not observed in the active pocket of target protein. Also, protein-ligand contacts were found less with poor interaction fraction. With these findings, we suggest gentisic acid can be further studied experimentally for shikimate kinase inhibition.
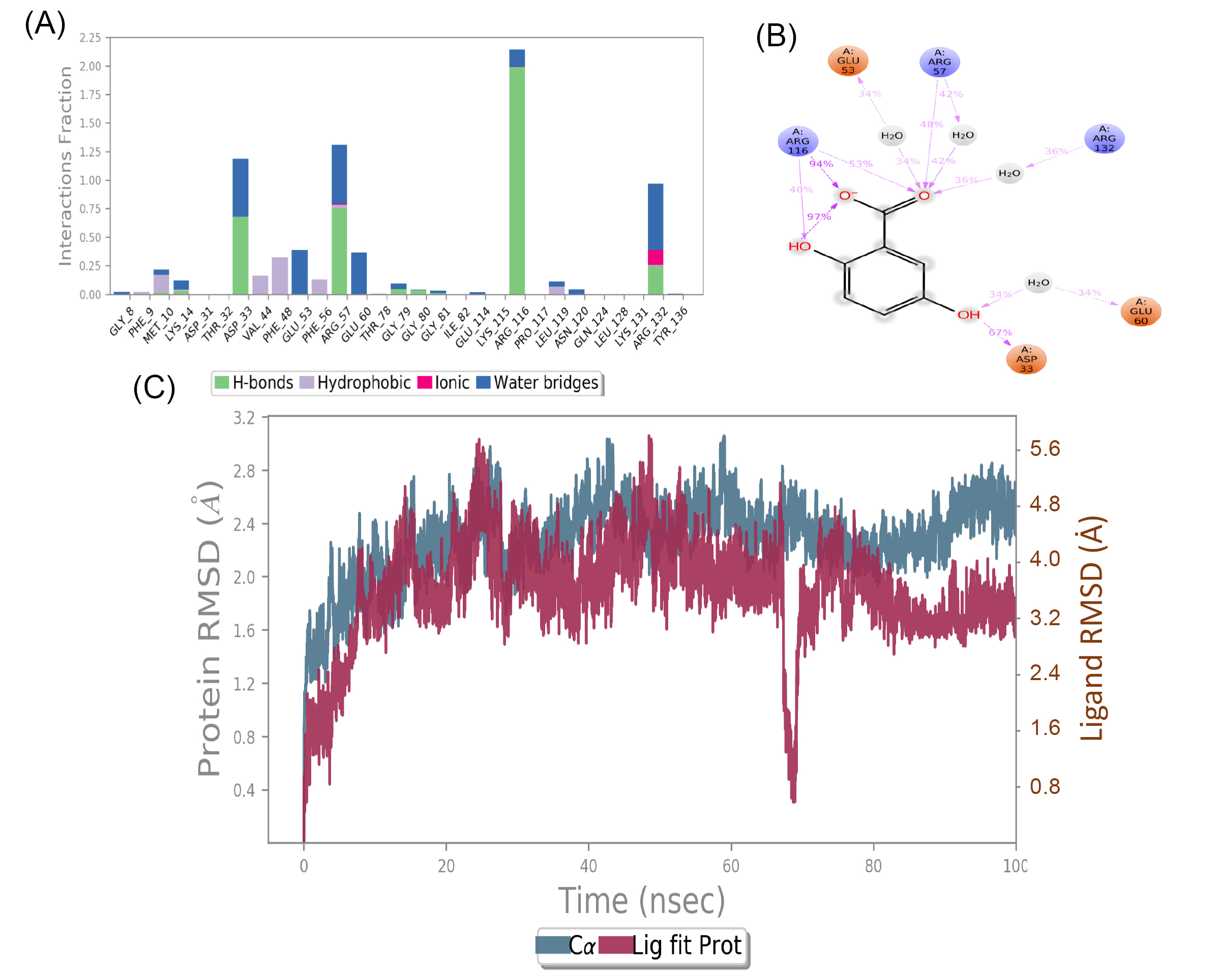
Fig. 2.
Molecular Dynamics (MD) simulations for gentisic acid and shikimate kinase complex. (A) Protein-ligand contacts; (B) 2D pose of ligand interactions; (C) RMSD of protein-ligand complex Schematic representation of ligand interactions with target protein for a period of 100 ns was conducted. Figs. 2A and 2B indicate that the complex is more stabilized by hydrogen bonds and to some extent with water bridges. Fig. 2C indicates the conformations changes in both protein and ligand complex during the simulation process. Overall results indicate that the protein-ligand complex is stabilized after 30 ns and large conformational change took place in ligand at 70 ns and later got equilibrated.
.
Molecular Dynamics (MD) simulations for gentisic acid and shikimate kinase complex. (A) Protein-ligand contacts; (B) 2D pose of ligand interactions; (C) RMSD of protein-ligand complex Schematic representation of ligand interactions with target protein for a period of 100 ns was conducted. Figs. 2A and 2B indicate that the complex is more stabilized by hydrogen bonds and to some extent with water bridges. Fig. 2C indicates the conformations changes in both protein and ligand complex during the simulation process. Overall results indicate that the protein-ligand complex is stabilized after 30 ns and large conformational change took place in ligand at 70 ns and later got equilibrated.
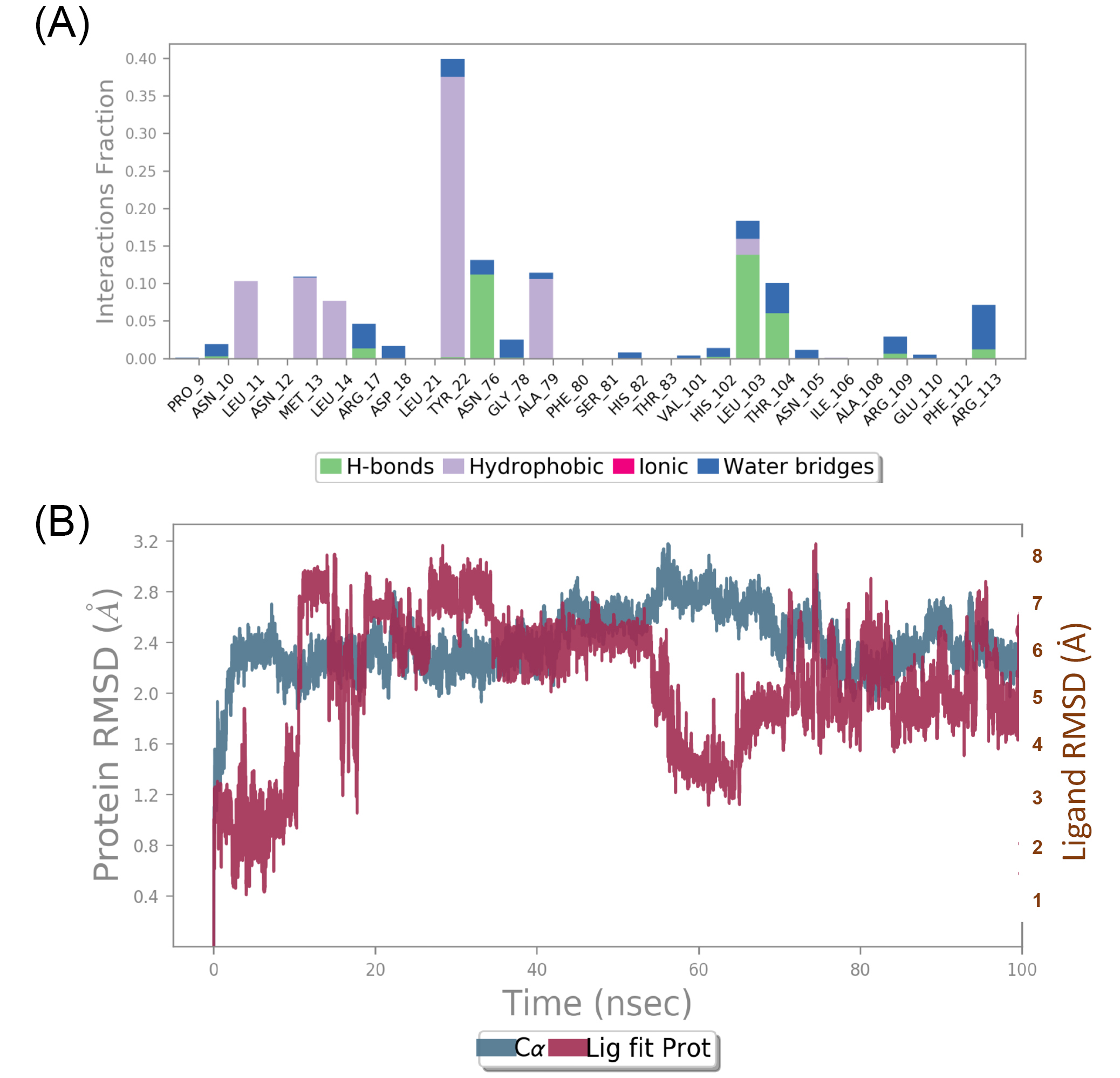
Fig. 3.
Molecular Dynamics (MD) simulations for (E)-Labda-8(17),13-diene-15,16-olide and type II dehydroquinase complex (A) Protein-ligand contacts; (B) RMSD of protein-ligand complex Schematic representation of ligand interactions with target protein for a period of 100 ns was conducted. Fig. 3a indicates that complex is more connected by hydrophobic interactions and to some extent with hydrogen bonds. Fig 3c indicates the conformations changes in both protein and ligand complex during the simulation process. Overall results indicate that the protein-ligand complex is not stabilized for 100 ns simulation particularly ligand has undergone more conformational changes during the process.
.
Molecular Dynamics (MD) simulations for (E)-Labda-8(17),13-diene-15,16-olide and type II dehydroquinase complex (A) Protein-ligand contacts; (B) RMSD of protein-ligand complex Schematic representation of ligand interactions with target protein for a period of 100 ns was conducted. Fig. 3a indicates that complex is more connected by hydrophobic interactions and to some extent with hydrogen bonds. Fig 3c indicates the conformations changes in both protein and ligand complex during the simulation process. Overall results indicate that the protein-ligand complex is not stabilized for 100 ns simulation particularly ligand has undergone more conformational changes during the process.
In silico prediction of pharmacokinetic parameters
ADME modeling has gained considerable attention in the field of pharmaceutical research for drug discovery as they are high-throughput in nature and cost-effective.
28
The results of Lipinski’s rule of five and ADME prediction obtained from Qikprop of the 15 compounds are shown in Tables 4 and 5. Analyzing Lipinski’s rule of five for 15 compounds exhibited no violations for molecular weight, hydrogen bond donors, and acceptors. Two compounds alpha-Muurolene and amadannulen have violated for predicted logP values of 5.531 and 5.396, respectively. Further experimental determination of log P values might give a better understanding.
Table 4.
Lipinski’s rule of five for in silico potential compounds
|
Compound Name
|
Molecular Weight
|
HB Donor
|
Hydrogen Bond Donor Acceptor
|
QPlogPo/w
|
Rule of five
|
| (E)-Labda-8(17),13-diene-15,16-olide |
260.3 |
0 |
3 |
3.431 |
0 |
| 3-Exo-hydroxy-1,8-cineole |
170.2 |
1 |
2.45 |
1.973 |
0 |
| Alpha-muurolene |
204.3 |
1 |
0.75 |
5.531 |
1 |
| Amadannulen |
376.5 |
1 |
3.7 |
5.396 |
1 |
| Caffeic acid |
180.1 |
3 |
3.5 |
0.559 |
0 |
| Demethocycurcumin |
366.4 |
2 |
4 |
3.292 |
0 |
| E-Sabinol |
152.2 |
1 |
1.70 |
2.237 |
0 |
| Gallic acid |
170.1 |
4 |
4.25 |
-0.568 |
0 |
| Gentisic acid |
154.1 |
2 |
2.5 |
0.792 |
0 |
| Ledol |
222.3 |
1 |
0.75 |
3.936 |
0 |
| Protocatechuic acid |
154.1 |
3 |
3.5 |
0.034 |
0 |
| Spathulenol |
220.3 |
1 |
0.75 |
3.891 |
0 |
| Syringic acid |
198.1 |
2 |
4.25 |
1.074 |
0 |
| Torreyol |
236.3 |
1 |
0.75 |
4.203 |
0 |
| Zerumin A |
318.4 |
1 |
4 |
3.842 |
0 |
Table 5.
Predicted ADME parameters of in silico potential compounds
|
Compound Name
|
SASA
|
FOSA
|
FISA
|
PISA
|
Volume
|
QPPCaco
|
QPlogBB
|
HOA (%)
|
| (E)-Labda-8(17),13-diene-15,16-olide |
525.25 |
395.58 |
75.753 |
53.914 |
928.53 |
1894.67 |
-0.275 |
100 |
| 3-Exo-hydroxy-1,8-cineole |
382.61 |
343.34 |
39.27 |
0.0 |
637.50 |
4202.46 |
0.205 |
100 |
| Alpha-muurolene |
478.22 |
429.64 |
0.0 |
48.583 |
826.81 |
9906.03 |
1.052 |
100 |
| Amadannulen |
713.43 |
614.49 |
95.413 |
3.527 |
1322.56 |
1233.38 |
-0.571 |
100 |
| Caffeic acid |
391.8 |
29.482 |
216.32 |
146.01 |
611.87 |
22.29 |
-1.56 |
54.34 |
| Demethocycurcumin |
697.2 |
256.71 |
173.75 |
266.74 |
1221.37 |
222.97 |
-1.943 |
88.25 |
| E-Sabinol |
386.7 |
312.2 |
41.675 |
32.82 |
632.24 |
3987.49 |
0.116 |
100 |
| Gallic acid |
342.01 |
0.0 |
252.8 |
89.208 |
526.2 |
10.05 |
-1.662 |
41.55 |
| Gentisic acid |
328.83 |
0.0 |
197.02 |
131.81 |
502.34 |
33.97 |
-1.138 |
58.98 |
| Ledol |
459.97 |
437.18 |
22.786 |
0.0 |
826.45 |
6023.07 |
0.326 |
100 |
| Protocatechuic acid |
331.4 |
0.0 |
206.29 |
125.11 |
505.35 |
27.75 |
-1.222 |
52.97 |
| Spathulenol |
462.32 |
399.34 |
32.035 |
30.943 |
823.88 |
4921.66 |
0.251 |
100 |
| Syringic acid |
391.64 |
181.42 |
138.19 |
72.025 |
632.08 |
122.74 |
-0.791 |
70.62 |
| Torreyol |
485.32 |
439.88 |
33.328 |
12.114 |
876.98 |
4784.67 |
0.176 |
100 |
| Zerumin A |
590.75 |
403.85 |
162 |
24.898 |
1083.73 |
72.98 |
-1.186 |
82.78 |
SASA. Solvent Accessible Surface Area; FOSA, Hydrophobic Component of SASA; FISA, Hydrophilic Component of SASA; PISA, Pi Component of SASA; QPPCaco, Predicted apparent Caco-2 cell permeability in nm/sec; QPlogBB, Predicted brain/blood partition coefficient; HOA (%), Percent Humal Oral Absorption.
From the surface area components, SASA (total solvent accessible surface area), FOSA (a hydrophobic component of SASA), FISA (a hydrophilic component of SASA), PISA (Π component of SASA), and volume (total solvent-accessible volume) were predicted. Most of the values were found in acceptable range as mentioned by Qikprop manual of Schrodinger. Permeability of compounds was predicted by QPPCaco (model for gut-blood barrier) and QPlogBB (brain-blood partition coefficient) values. QPPCaco values greater than 500 will have good permeability. Alpha-Muurolene has shown the highest predicted value of 9906.03. Compounds except caffeic acid, gallic acid, demethoxycurcumin, gentisic acid, protocatechuic acid, syringic acid, and zerumin A have exhibited values greater than 1000. All acid derivatives have shown poor gut membrane permeability prediction values. Coming to QPlogBB, negatives values indicate that compounds are polar and poor permeability. Alpha-Muurolene has displayed highest value of 1.052 indicating that compound crosses blood-brain barrier. Five compounds have values closer to 0.3 and remaining all other compounds have negative values. The percentage of human oral absorption was observed 100% for eight compounds. Absorption values of acid derivatives were little poor and observed in the range of 40%-60%. Overall, compounds have exhibited good values of Lipinski’s rule of five and predicted pharmacokinetic parameters without too many violations with drug-likeness features. The study suggests that in vitro potential compounds can be further taken for preclinical evaluation.
Conclusion
The search for herbal therapeutic compounds as inhibitors for treating diseases is one of the approaches for drug discovery and development. Herbal plants have been analyzed and examined for centuries in the development of novel drugs.
28,29
Different studies have inspected variety of herbal plants and have confirmed that they contain compounds with anti-Helicobacter activities. In our present study, a total of 130 compounds from mango ginger that are known to have biological activity against H. pylori
30
were screened and ranked based on the docking and Prime MM-GBSA prediction results. Out of the five drug targets, mango ginger compounds were found selective to shikimate kinase and type II dehydroquinase. MD simulations revealed that gentisic acid can be further studied for in vitro shikimate kinase inhibition and in vivo anti-H. pylori activity to validate the mechanism. Furthermore, mango ginger compounds can be considered safe and effective (Lipsinki’s RO5 and predicted ADME properties) to treat infections associated with H. pylori and also for development into a commercial formulation. The overall study suggests that mango ginger compounds can be studied for experimental evaluation of shikimate kinase and type II dehydroquinase inhibition to compare and validate the computational and experimental results and for identification of potential inhibitors against H. pylori.
Acknowledgment
All the authors are thankful to Department of Biotechnology, M S Ramaiah Institute of Technology and Pharmacological Modelling and Simulation Centre (PMSC), M.S. Ramaiah University of Applied Sciences, Bangalore for the support and providing necessary facilities to carry out this research work.
Funding sources
None to be declared.
Ethical statement
There is none to be stated.
Competing interests
The authors report no conflicts of interest. All the authors alone are responsible for the content and writing of this article.
Authors’ contribution
KMTP and MM wrote the concept of the study and designed the experiments. SS and PK performed the experiments. DG, SGR, and BS analyzed and interpreted the data. All authors drafted and revised the manuscript.
Research Highlights
What is the current knowledge?
simple
-
√ H. pylori infection is the common chronic bacterial infections affecting humans worldwide.
-
√ Mango ginger (Curcuma amada Roxb.) is a unique spice known for its antipyretic, diuretic, expectorant, and laxative health attributes.
What is new here?
simple
-
√ Novel inhibitors from the therapeutic herb, Mango ginger (Curcuma amada Roxb.) were identified against selected five drug targets of H. pylori.
-
√ Molecular dynamics simulations identified the in silico potential of Mango ginger compound, gentisic acid with the drug target, shikimate kinase.
-
√ The results from ADME parameters and Lipinski’s rule of five were found promising without any limitations.
References
- Hooi JK, Lai WY, Ng WK, Suen MM, Underwood FE, Tanyingoh D. Global prevalence of Helicobacter pylori infection: systematic review and meta-analysis. Gastroenterology 2017; 153(2):420-9. doi: 10.1053/j.gastro.2017.04.022 [Crossref] [ Google Scholar]
- Kuipers EJ. Helicobacter pylori and the risk and management of associated diseases: gastritis, ulcer disease, atrophic gastritis and gastric cancer. Aliment Pharmacol Ther 1997; 11:71-88. doi: 10.1046/j.1365-2036.11.s1.5.x [Crossref] [ Google Scholar]
- Amgalanbaatar A, Shimomura H, Sugano K, Bira TO, Okamoto H, Hirai Y. Correlation between Helicobacter pylori infection and gastric atrophy examined in the sera of mongolian people. Gastrointest Disord 2019; 1:241-52. doi: 10.3390/gidisord1020019 [Crossref] [ Google Scholar]
- Goderska K, Pena SA, Alarcon T. Helicobacter pylori treatment: antibiotics or probiotics. Appl Microbiol Biotechnol 2018; 102:1-7. doi: 10.1007/s00253-017-8535-7 [Crossref] [ Google Scholar]
- Salehi B, Sharopov F, Martorell M, Rajkovic J, Ademiluyi AO, Sharifi-Rad M. Phytochemicals in Helicobacter pylori infections: What are we doing now?. Int J Mol Sci 2018; 19(8):2361. doi: 10.3390/ijms19082361 [Crossref] [ Google Scholar]
- Alebouyeh M, Yadegar A, Farzi N, Miri M, Zojaji H, Gharibi S. Impacts of H pylori mixed-infection and heteroresistance on clinical outcomes. Gastroenterol Hepatol Bed Bench 2015; 8:S1. [ Google Scholar]
- Zamani M, Zamani V. Helicobacter pylori antibiotic resistance: Can herbal medicine be an alternative for the treatment?. J Res Med Sci 2016; 21:97. doi: 10.4103/1735-1995.193169 [Crossref] [ Google Scholar]
- Zaidi SF, Muhammad JS, Usmanghani K, Sugiyama T. Pharmacological ins and outs of medicinal plants against Helicobacter pylori: A review. Pak J Pharm Sci 2015; 28:1171-6. [ Google Scholar]
- Rezaeimanesh N, Farzi N, Pirmanesh S, Emami S, Yadegar A. Management of multi-drug resistant Helicobacter pylori infection by supplementary, complementary and alternative medicine; a review. Gastroenterol Hepatol Bed Bench 2017; 10:S8. [ Google Scholar]
- Policegoudra RS, Aradhya SM, Singh L. Mango ginger (Curcuma amada Roxb)–A promising spice for phytochemicals and biological activities. J Biosci 2011; 36(4):739-48. doi: 10.1007/s12038-011-9106-1 [Crossref] [ Google Scholar]
- Gupta RK, Gaur H, Pati JK, Pandey A. Evaluation of antioxidant potential of different extracts of mango ginger (Curcuma amada Roxb) Rhizome. Int J Pharm Sci Res 2015; 6:3986. doi: 10.13040/IJPSR.0975-8232.6(9).3986-89 [Crossref] [ Google Scholar]
- Debraekeleer A, Remaut H. Future perspective for potential Helicobacter pylori eradication therapies. Future Microbiol 2018; 13:671-687. doi: 10.2217/fmb-2017-0115 [Crossref] [ Google Scholar]
- Chen J, Zhang L, Zhang Y, Zhang H, Du J, Ding J. Emodin targets the β-hydroxyacyl-acyl carrier protein dehydratase from Helicobacter pylori: enzymatic inhibition assay with crystal structural and thermodynamic characterization. BMC Microbiol 2009; 9(1):91. doi: 10.1186/1471-2180-9-91 [Crossref] [ Google Scholar]
- Fisher SL. Glutamate racemase as a target for drug discovery. Microb Biotechnol 2008; 1(5):345-360. doi: 10.1111/j.1751-7915.2008.00031.x [Crossref] [ Google Scholar]
- Labbé G, Krismanich AP, de Groot S, Rasmusson T, Shang M, Brown MD, Dmitrienko GI, Guillemette JG. Development of metal-chelating inhibitors for the Class II fructose 1, 6-bisphosphate (FBP) aldolase. J Inorg Biochem 2012; 112:49-58. doi: 10.1016/j.jinorgbio.2012.02.032 [Crossref] [ Google Scholar]
-
Schrödinger Release 2018-3: Schrödinger Suite 2018-3 Protein Preparation Wizard; Epik. New York, NY: Schrödinger, LLC; 2018.
- Murahari M, Prakash KV, Peters GJ, Mayur YC. Acridone-pyrimidine hybrids-design, synthesis, cytotoxicity studies in resistant and sensitive cancer cells and molecular docking studies. Eur J Med Chem 2017; 139:961-981. doi: 10.1016/j.ejmech.2017.08.023 [Crossref] [ Google Scholar]
-
Schrödinger Release 2018-3: LigPrep. New York, NY: Schrödinger, LLC; 2018.
-
Schrödinger Release 2018-3: Glide. New York, NY: Schrödinger, LLC; 2018.
- Murahari M, Kharkar PS, Lonikar N, Mayur YC. Design, synthesis, biological evaluation, molecular docking and QSAR studies of 2, 4-dimethylacridones as anticancer agents. Eur J Med Chem 2017; 130:154-170. doi: 10.1016/j.ejmech.2017.02.022 [Crossref] [ Google Scholar]
- Gudipati S, Muttineni R, Mankad AU, Pandya HA, Jasrai YT. Molecular docking based screening of Noggin inhibitors. Bioinformation 2018; 14:15-20. doi: 10.6026/97320630014015 [Crossref] [ Google Scholar]
- Cutinho PF, Roy J, Anand A, Cheluvaraj R, Murahari M, Chimatapu HV. Design of metronidazole derivatives and flavonoids as potential non-nucleoside reverse transcriptase inhibitors using combined ligand-and structure-based approaches. J Biomol Struct Dyn 2019; 27:1-23. doi: 10.1080/07391102.2019.1614094 [Crossref] [ Google Scholar]
-
Schrödinger Release 2018-3: Prime. New York, NY: Schrödinger, LLC; 2018.
-
Schrödinger Release 2018-3: Desmond Molecular Dynamics System. New York, NY: D. E. Shaw Research; 2018.
- Cutinho PF, Shankar RC, Anand A, Roy J, Mehta CH, Nayak UY, Murahari M. Hit identification and drug repositioning of potential non-nucleoside reverse transcriptase inhibitors by structure-based approach using computational tools (part II). J Biomol Struct Dyn 2019; 23:1-8. doi: 10.1080/07391102.2019.1663263 [Crossref] [ Google Scholar]
-
Schrödinger Release 2018-3: Qikprop. New York, NY: Schrödinger, LLC; 2018.
- Veeramachaneni GK, Raj KK, Chalasani LM, Bondili JS, Talluri VR. High-throughput virtual screening with e-pharmacophore and molecular simulations study in the designing of pancreatic lipase inhibitors. Drug Des Devel Ther 2015; 9:4397. doi: 10.2147/DDDT.S84052 [Crossref] [ Google Scholar]
- Sharma D, Kumar S, Narasimhan B, Ramasamy K, Lim SM, Shah SA, Mani V. 4-(4-Bromophenyl)-thiazol-2-amine derivatives: synthesis, biological activity and molecular docking study with ADME profile. BMC Chem 2019; 13:60. doi: 10.1186/s13065-019-0575-x [Crossref] [ Google Scholar]
- Ekor M. The growing use of herbal medicines: issues relating to adverse reactions and challenges in monitoring safety. Front Pharmacol 2014; 4:177. doi: 10.3389/fphar.2013.00177 [Crossref] [ Google Scholar]
- Murthy TP, Manohar B. Modelling solubility of phenolics of mango ginger extract in supercritical carbon dioxide using equation of state and empirical models. J Food Sci Technol 2015; 52:5557-67. doi: 10.1007/s13197-014-1667-1 [Crossref] [ Google Scholar]