Bioimpacts. 11(1):65-84.
doi: 10.34172/bi.2021.11
Original Research
A domain-based vaccine construct against SARS-CoV-2, the causative agent of COVID-19 pandemic: development of self-amplifying mRNA and peptide vaccines
Mohammad Mostafa Pourseif 1  , Sepideh Parvizpour 1, Behzad Jafari 2, 1, Jaber Dehghani 1 , Behrouz Naghili 3, Yadollah Omidi 4, *
, Sepideh Parvizpour 1, Behzad Jafari 2, 1, Jaber Dehghani 1 , Behrouz Naghili 3, Yadollah Omidi 4, *
Author information:
1Research Center for Pharmaceutical Nanotechnology, Biomedicine Institute, Tabriz University of Medical Sciences, Tabriz, Iran
2Department of Medicinal Chemistry, Faculty of Pharmacy, Urmia University of Medical Sciences, Urmia, Iran
3Research Center for Infectious and Tropical Diseases, Tabriz University of Medical Sciences, Tabriz, Iran
4Nova Southeastern University, College of Pharmacy, Florida, USA
*Corresponding author: Yadollah Omidi, Tel: +1 954 262 1350, E-mail:
yomidi@nova.edu
Abstract
Introduction:
Coronavirus disease 2019 (COVID-19) is undoubtedly the most challenging pandemic in the current century with more than 293,241 deaths worldwide since its emergence in late 2019 (updated May 13, 2020). COVID-19 is caused by a novel emerged coronavirus named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Today, the world needs crucially to develop a prophylactic vaccine scheme for such emerged and emerging infectious pathogens.
Methods:
In this study, we have targeted spike (S) glycoprotein, as an important surface antigen to identify its B- and T-cell immunodominant regions. We have conducted a multi-method B-cell epitope (BCE) prediction approach using different predictor algorithms to discover the most potential BCEs. Besides, we sought among a pool of MHC class I and II-associated peptide binders provided by the IEDB server through the strict cut-off values. To design a broad-coverage vaccine, we carried out a population coverage analysis for a set of candidate T-cell epitopes and based on the HLA allele frequency in the top most-affected countries by COVID-19 (update 02 April 2020).
Results:
The final determined B- and T-cell epitopes were mapped on the S glycoprotein sequence, and three potential hub regions covering the largest number of overlapping epitopes were identified for the vaccine designing (I531–N711; T717–C877; and V883–E973). Here, we have designed two domain-based constructs to be produced and delivered through the recombinant protein- and gene-based approaches, including (i) an adjuvanted domain-based protein vaccine construct (DPVC), and (ii) a self-amplifying mRNA vaccine (SAMV) construct. The safety, stability, and immunogenicity of the DPVC were validated using the integrated sequential (i.e. allergenicity, autoimmunity, and physicochemical features) and structural (i.e. molecular docking between the vaccine and human Toll-like receptors (TLRs) 4 and 5) analysis. The stability of the docked complexes was evaluated using the molecular dynamics (MD) simulations.
Conclusion:
These rigorous in silico validations supported the potential of the DPVC and SAMV to promote both innate and specific immune responses in preclinical studies.
Keywords: COVID-19, Emerging virus, Epitope, SARS-CoV-2, Self-amplifying mRNA vaccine, Spike glycoprotein, Structural modeling, Pandemic
Copyright and License Information
© 2021 The Author(s)
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by-nc/4.0/
). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Introduction
Despite notable progress in medical sciences during the 20th century, still, infectious diseases have significant consequences on the public health systems worldwide. Of these, emerging infectious diseases (EIDs) and re-emerging infectious diseases (RIDs) are always considered as striking threats to humans all around the world.
1
The majority of such infectious diseases are zoonotic and mostly originated from animals, including severe acute respiratory syndrome coronavirus (SARS-CoV), influenza A virus subtype H1N1, Middle East respiratory syndrome coronavirus (MERS-CoV), Ebola, and Zika virus.
Today, the world is confronting a novel coronavirus so officially named SARS-CoV-2, and World Health Organization (WHO) has named its relevant disease as “Coronavirus disease 2019 (COVID-19)”. The first known SARS-CoV-2 was discovered in late December 2019 in Wuhan, Hubei province, China. Since then, it has become a global pandemic, in large part due to its rapid rate of human-to-human transmission, lack of vaccine, and delay in global functional protocols.
2
The infection of SARS-CoV-2 can lead to some severe respiratory damages with a different range of symptoms and complications – ranging from mild symptoms (e.g., fever, cough, myalgia or fatigue, and shortness of breath) to severe illness and death.
3
The SARS-CoV-2 belongs to the family Coronaviridae and the Betacoronavirus genus.
4
Coronaviruses (CoVs) are a large group of zoonotic viruses with unique features, including the crown-like surface projections with club-shaped spike proteins, and the enveloped positive-sense single-stranded RNA viruses with helical nucleocapsids. The structure of SARS-CoV-2 and its genome data is schematically illustrated in Fig. 1.

Fig. 1.
Schematic illustration of the structure of the novel coronavirus 2019 and its annotated genome. A) Different parts of the full-length genome of SARS-CoV-2. B) The genomic RNA of SARS-CoV-2 encodes four structural proteins with key roles in the structure of the virus: (i) Surface spike protein (S), (ii) Nucleocapsid protein (N), (iii) Membrane protein (M), and (iv) envelope protein (E). R1a: Replicase polyprotein 1a; R1ab: Replicase polyprotein 1ab; 3a: protein 3a; 6: non-structural protein 6; 7a: protein 7a (NS7A); 7b: protein 7b (NS7B); 8: Non-structural protein 8 (NS8); 9b: protein 9b; 14: uncharacterized protein 14; ORF10: hypothetical ORF10 protein. Source: ViralZone: www.expasy.org/viralzone,SIB Swiss Institute of Bioinformatics.
.
Schematic illustration of the structure of the novel coronavirus 2019 and its annotated genome. A) Different parts of the full-length genome of SARS-CoV-2. B) The genomic RNA of SARS-CoV-2 encodes four structural proteins with key roles in the structure of the virus: (i) Surface spike protein (S), (ii) Nucleocapsid protein (N), (iii) Membrane protein (M), and (iv) envelope protein (E). R1a: Replicase polyprotein 1a; R1ab: Replicase polyprotein 1ab; 3a: protein 3a; 6: non-structural protein 6; 7a: protein 7a (NS7A); 7b: protein 7b (NS7B); 8: Non-structural protein 8 (NS8); 9b: protein 9b; 14: uncharacterized protein 14; ORF10: hypothetical ORF10 protein. Source: ViralZone: www.expasy.org/viralzone,SIB Swiss Institute of Bioinformatics.
Presently, along with the basic predictive measures and therapeutic modalities, the development of effective vaccine(s) is extremely vital for the controlling of the SARS-CoV-2. The empirical vaccinology against emerging and re-emerging infectious (EREI) pathogens such as SARS-CoV-2 might contend with several critical challenges, in large measure because of the paucity of the basic knowledge about their pathogenic mechanisms and behavior.
5
In contrast, the rational vaccinology through the bioinformatics, statistical meta-analyses (or mining) among the pathogen's genome/proteome, and comparative pathogenomic analyses might provide key detailed estimates for the vaccine design.
6,7
Recent progress in the next-generation sequencing technology and the relevant computational approaches have offered vaccinologists to take a holistic and deep analysis of the whole genomes, and proteomes of the EREI pathogens like SARS-CoV-2.
8,9
The vaccine design and delivery strategies can be optimized based on a "vaccine on-demand" approach. The target-pathogen (i.e., pathogens causing chronic infectious or emerging ones) and its outbreak rate are vital factors to apply the best vaccine design, production, formulation, and delivery strategy. The production of injectable recombinant protein vaccines needs some additional cost- and time-consuming in vitro steps (e.g., upstream and downstream processing) compared to the edible and nucleic acid-based vaccine delivery systems.
10-12
These novel platforms can be used for rapid (or emergency) response applications like the COVID-19 pandemic.
In the current study, we focused on the SARS-CoV-2 glycoprotein S due to its ability to trigger the most dominant and long-lasting neutralizing immune cells against SARS-CoV.
13,14
Our main objective was to identify the immunodominant regions of the target antigen through the robust immunoinformatics approaches to accelerate the development process rationally. The regions of spike glycoprotein that cover the largest number of overlapping predicted B- and T-cell epitopes were used to logically design two different immunogenic constructs, including (i) an adjuvanted domain-based protein vaccine construct (DPVC), and (ii) a self-amplifying mRNA vaccine (SAMV). The immunizing efficiency of DPVC was validated through, (i) the analysis of the vaccine sequence and its three-dimensional (3D) structure, (ii) molecular docking between the vaccine structure and the human toll-like receptors (TLRs) 4 and 5, and (iii) the molecular dynamics (MD) simulations.
Materials and Methods
Spike protein sequence retrieval, and phylogenetic analysis
The whole-genome reference sequence of SARS-CoV-2 was retrieved from the National Center for Biotechnology Information (NCBI) genome database (accession no. NC_045512). The reference protein sequence of spike protein (accession no. YP_009724390.1) in FASTA format was used for BLAST against non-redundant protein sequences (nr) database through the blastp (protein-protein BLAST) algorithm. The FASTA sequence of 100 spike protein of different countries and different dates of isolation with significant alignments (identity ≥ 75.80% and E-value 0.0) were taken and multiple-sequence-alignment was carried out using the MUSCLE program of MEGA v10.0 software.
15,16
The aligned sequences were then analyzed to find the best substitution model of amino acid evolution using MEGA 10 software. The phylogenetic tree of the protein S dataset was inferred by using the Maximum Likelihood (ML) method and JTT matrix-based model
17
and via bootstraps replications of 1000.
18
The putative spike protein isolated from Zaria Bat coronavirus (GenBank: ADY17911.1) was served as an outgroup.
Preliminary features of S protein sequence
Signal peptide and sub-cellular localization
In domain-based vaccine design, one important criterion is selecting epitopes that have an extracellular localization and are more accessible for the epitope-paratope interactions. In this regard, the spike protein was analyzed for the possible presence of signal peptide, transmembrane helices, and also intracellular regions. These structural features were predicted using the online web-servers, including TOPCONS,
19
CCTOP v2.0,
20
and TMHMM.
21
Annotation of conserved domains and regions
The NCBI's Conserved Domain Database (CDD) v3.16 tool with default E-value threshold was used to annotate the conserved domain(s) of SARS-CoV-2 S glycoprotein.
22
Besides, the aligned sequences of the protein S were imported to the BioEdit v7.2.5 to determine conserved regions of the S protein sequence by use of Shannon's entropy (Hx) plot.
23
This measure was also carried out to compare mutated regions of SARS-CoV-2 to SARS-CoV (Reference sequence accession no. NP_828851) using BioEdit v7.2.5 software and via Shannon entropy (Hx) analysis.
Secondary and tertiary structure prediction of S glycoprotein
The secondary structure of S protein was predicted employing the PSIPRED web-server.
24
The 3D structure of S protein was homology modeled using the SWISS-MODEL online tool
25
and the newly reported crystal structures in Protein Data Bank (6LVN, 6LXT, 6VSB, 6VXX, and 6VYB).
Structure refinement, molecular dynamics simulation, and validation
To refine the 3D model for the hydrogen bonds and overall structural relaxation, it was subjected to the GalaxyRefine server processing.
26
To optimize the model's free energy, the refined model was subjected to an MD simulation recruiting GROMACS 5.0.7 software together with the GROMOS 96 force field.
27
The MD simulation procedure was carried out at 310 K by placing the model into a cubic box that had a suitable size and two Na+ ions to neutralize the environment. Subsequently, the RMSD graph was drawn for the analysis of the dynamic behavior of the constructed model.
28
The local and overall quality of the improved 3D model was checked using online web-servers, including PROCHECK,
29
verify3D,
30
ERRAT.
31
In silico B-cell epitope mapping: a multi-method approach
The potential B-cell epitopes (BCEs) were predicted by using the sequence- and structure-based tools. To predict linear and conformational BCEs with high accuracy, we implemented a multi-method approach based on the different currently available online BCE prediction web-servers.
32
We exploited the physicochemical and machine learning methods such as all the predictor tools of the Immune Epitope Database and Analysis Resource (IEDB) as a repository of curated epitope related information (http://tools.iedb.org/main/bcell/), BepiPred v2.0,
33
LBtope,
34
IgPred,
35
CBTOPE,
36
BEPITOPE v2.0,
37
ABCPred,
38
SEPPA v3.0,
39
DiscoTope v2.0,
40
ElliPro,
41
BcePred (https://webs.iiitd.edu.in/raghava/bcepred/index.html). The energy minimized 3D structure of protein S was utilized to predict and map the potential discontinuous BCEs. The FASTA sequence of the protein was imported into the Excel program and any single amino acid was separated in a single cell as a set of consecutive cells using a user-defined function named "AddSpace" (the Excel VBA code is shown in Table S1, see supplementary material). The scores of each of the twenty-one prediction algorithms were normalized to have values between 0 and 1. Then, an average of all normalized scores for each residue was represented as a plot, in which the immunodominant regions of the S protein sequence were highlighted based on a strict threshold value of ≥ 0.6. For the residue-based comparison analysis of the final predicted BCEs, the pairwise sequence alignment was implemented employing Clustal Omega web-server
42
between the reference sequences of the spike proteins of SARS-CoV (accession ID: NP_828851.1) and SARS-CoV-2 (accession ID: YP_009724390.1). All the experimentally-determined spike glycoprotein SARS-CoV-derived BCEs were obtained from the NIAID Virus Pathogen Database and Analysis Resource (ViPR) (accessed on April 1st, 2020) and IEDB web-server to have a comparative evaluation with SARS-CoV-2 dominant predicted BCEs (Table S2).
43
T-cell epitope prediction
SARS coronavirus-associated T-cell epitopes are almost all correlated to the HLA complex antigen recognition. However, the HLA alleles are highly polymorphic among populations and there is no entire screening system to clarify the possible association between the occurrence of SARS-CoV-2 and the susceptibility/resistance of various HLA alleles. Therefore, in such diseases, it is logical to use the reference sets of HLA alleles with the maximal population coverage. The T-cell epitope prediction was performed using the reference isolate of SARS-CoV-2, i.e., spike protein sequence (NCBI: YP_009724390.1). Due to utilizing a vast number of the human leukocyte antigen (HLA) alleles during the calculation of peptide-MHC binding, the predicted output table might be quite substantial. Therefore, the prediction of peptide binders for class I and II MHC molecules was carried out based on the strict cut-offs to give more accurate and reliable peptide binders. To have a final set of the epitope for vaccine designing, those candidate epitopes that displayed overlap for multiple alleles were selected.
CD8+ T-cell epitope prediction
The cytotoxic T-lymphocyte (CTL) epitopes were predicted by utilizing the IEDB recommended v2.22 algorithm,
44
which was performed against the HLA allele reference set covering > 97% of the global population.
45
Of note, the HLA allele reference set is a library of 16 alleles for class A (01:01, 02:01, 02:03, 02:06, 03:01, 11:01, 23:01, 24:02, 26:01, 30:01, 30:02, 31:01, 32:01, 33:01, 68:01, 68:02), and 11 alleles for HLA class B (07:02, 08:01 15:01, 35:01, 40:01, 44:02, 44:03, 51:01, 53:01, 57:01, 58:01). To find the best consensus epitopes among a pool of peptide binders, we first sorted the IEDB's output table based on the rank of any binder in the three binding prediction methods (i.e., percentile rank, artificial neural network (ANN) IC50, and stabilized matrix method (SMM) IC50. Then, the sorted binders were filtered based on an MHC binding affinity (IC50) value of ≤ 50 nM, and the percentile rank of ≤ 1.0, as strict thresholds. In the end, we selected the best candidate peptide binders via defining a ranking score, the so-called "consensus rank" (CR). This CR score was calculated by the following equation [i.e., CR = average rank of a mapped peptide binder/n], where, "n" refers to the total number of alleles covered by a peptide binder. Therefore, it provides a small list of candidate peptide binders that not only possess the highest prediction rank but also can bind to a wide range of MHC alleles.
CD4+ T-cell epitope prediction
To predict the most potential CD4+ helper T-cell epitopes, we used the IEDB recommended algorithm v2.22 (consensus approach)
46
based on the full HLA reference set that can cover > 99% of the global population.
47
The epitope length was specified on a variable-length option 12-18 that can cover 82.89% of epitope frequency. To generate a consensus list of CD4+ T cell epitopes, we selected the best peptides based on the adjusted percentile rank ≤ 1.0 (as a strict cut-off) and the number of MHC-II alleles covered by the candidate predicted peptide binders.
Population coverage for selection consensus T-cell epitopes
HLA molecules are extremely polymorphic, thus using multiple peptides with various HLA binding specificities will give more coverage of the population targeted by domain-based vaccines. Accordingly, in this study, we computed population coverage of the final T cell epitopes using the allele frequency net database
48
and the tool provided by the IEDB server.
49
The measured population coverage indicates the percentage of individuals within the population that are likely to stimulate an immune response to at least one T cell epitope from the set. We estimated the population coverage of T-cell epitopes for the top most-affected countries by the COVID-19 pandemic (updated data on April 2nd, 2020).
Designing the candidate vaccine constructs
In this study, we designed two different vaccine constructs optimized based on the two different vaccine platforms and using the identified immunodominant B- and T-cell regions of SARS-CoV-2 spike glycoprotein.
i. A DPVC for in vitro expression and purification as an injectable recombinant vaccine.
ii. A self-amplifying mRNA vaccine (SAMV) construct for in vitro transcription and purification, and in vivo expression.
The DPVC was designed based on the immunodominant B- and T-cell epitopes, intramolecular adjuvants, and different peptide linkers. The residues of the spike protein covering the largest number of overlapping predicted epitopes were used to design the DVC. Currently, it is known that the TLRs 4 and 5 are effectively contributed to the recognition and induction of immune responses against respiratory coronavirus infectious.
50,51
Therefore, to potentially enhance the vaccine immunogenicity, we capitalized on two TLR agonist sequences as intramolecular adjuvants, including (i) a synthetic TLR4 agonist 7-mer peptide, named RS09 (APPHALS),
52
and (ii) Salmonella typhimurium Flagellin C (UniProtKB: P06179) as a bacterial ligand for binding to TLR5.
53
To improve the CD4+ T-cell immune responses, an invariant Pan HLA-DR reactive epitope (PADRE) was exploited in the vaccine construct. The intramolecular adjuvants (Flagellin C, and RS09) were linked to the PADRE sequence at the N-terminal site of the construct and joined each other using an in vivo cleavable linker (sequence: PPGVS). This peptide appears as the optimal cleavage site of matrix metalloproteinase-9 (MMP-9), which is a member of the metalloendopeptidase distributed in the human skin.
54,55
The PADRE sequence was linked to the main domain of the vaccine construct using the Cathepsin S cleavable linker (PMGLP). In the human skin, the protease activity of cathepsin S has the main role in the antigen presentation pathways mediated by MHC class II molecules.
56-58
It is discussed before that signal peptides not only can improve vaccine immunogenicity but also have an intrinsic nature to direct the protein to the desired cellular compartment (e.g. secretion out of the cell or into cell membrane).
59
Here, according to the goal of vaccination, the final localization of the cytosolic expressed SAM vaccine can be engineered by antigen-specific signal sequences to be secreted extracellular or translocated into the host’s cell membrane.
The second vaccine construct was designed as a self-amplifying mRNA (SAM) replicon vaccine. In this construct, we used the identified immunodominant regions of the glycoprotein S as a vaccine sequence. Further, to have a SAM construct we used the genes encoding non-structural proteins (nsp) of the Semliki Forest virus (NCBI reference sequence: NC_003215.1) as a genomic (+) single-strand RNA alphavirus.
60
The nsp1-4 region can improve properly the mRNA capping, stability, translational efficiency, and can form properly the RNA-dependent RNA polymerase (RdRp) complex.
12
The SAMV construct was flanked between the newly designed 5' and 3' untranslated regions (UTRs) named as NASAR.
61
NCA-7d, as the 5' untranslated region (UTR), and S27a+R3U, as the 3' UTR. We propose a newly developed CleanCapTM method (by TriLink BioTechnologies, US) with base analogs Adenosine and Uridine for the mRNA capping process (cap residue: m7G(5')ppp(5')(2'OMeA)pU). This 5'-capping, as a co-transcriptional capping technology, is specialized for the high efficient production of the SAMVs with naturally creating Cap 1 structure.
Prediction of vaccine antigenicity, safety, and stability
The antigenicity analysis was varied out using the VaxiJen v2.0 server.
62
The potential allergenicity of the vaccine construct was evaluated in the AlgPred (using the hybrid method)
63
and AllerTOP v2.0
64
web-servers and based on the FAO/WHO allergenicity rules. To prevent possible autoimmunity of the designed vaccine, the vaccine amino acid sequence was blasted against non-redundant protein sequences of Homo sapiens using the blastp algorithm of the NCBI. The physicochemical properties of the designed vaccine such as molecular weight, theoretical isoelectric point (pI), half-life in vitro and in vivo, stability, aliphatic index, extinction coefficient, and grand average of hydropathicity (GRAVY) were predicted using the ProtParam tool of ExPASy web-server.
65
Structural simulation of the vaccine binding affinity
The tertiary and secondary structure of the vaccine construct was predicted using the I-TASSER and the Garnier Osguthorpe and Robson (GOR) version IV online servers.
66,67
The highest quality 3D model was refined through the GalaxyRefine server
26
and then was executed for the energy minimization by the GROMACS 5.0.7 software package.
27
The structural quality of the optimized 3D model was validated using PROCHECK
29
web-server. The molecular docking was performed via ClusPro v2.0 online server
68
to assess the binding affinity between the DVC and extracellular regions of the human TLR4 (PDB ID: 4G8A), and TLR5 (PDB ID: 3J0A) molecules. The output of docking simulations was visualized and analyzed using the Chimera v1.14
69
and DIMPLOT schematic diagram of LigPlot+ v2.2,
70
respectively.
Results
Evolutionary analysis of SARS-CoV-2 spike protein
Different features of the SARS-CoV-2 genome are categorized and presented in Table S3 (Supplementary file 1). To further assay the phylogenetic relationship between the SARS-CoV-2 genome and all other strains of CoVs, as shown in Fig. 2A, we built an evolutionary tree with the highest log likelihood (-11665.31). According to the phylogenetic analysis, among all known CoVs, the bat coronavirus RaTG13 (Accession no. QHR63300.2) showed the closest relation to the recent emergent human coronavirus (HCoV).
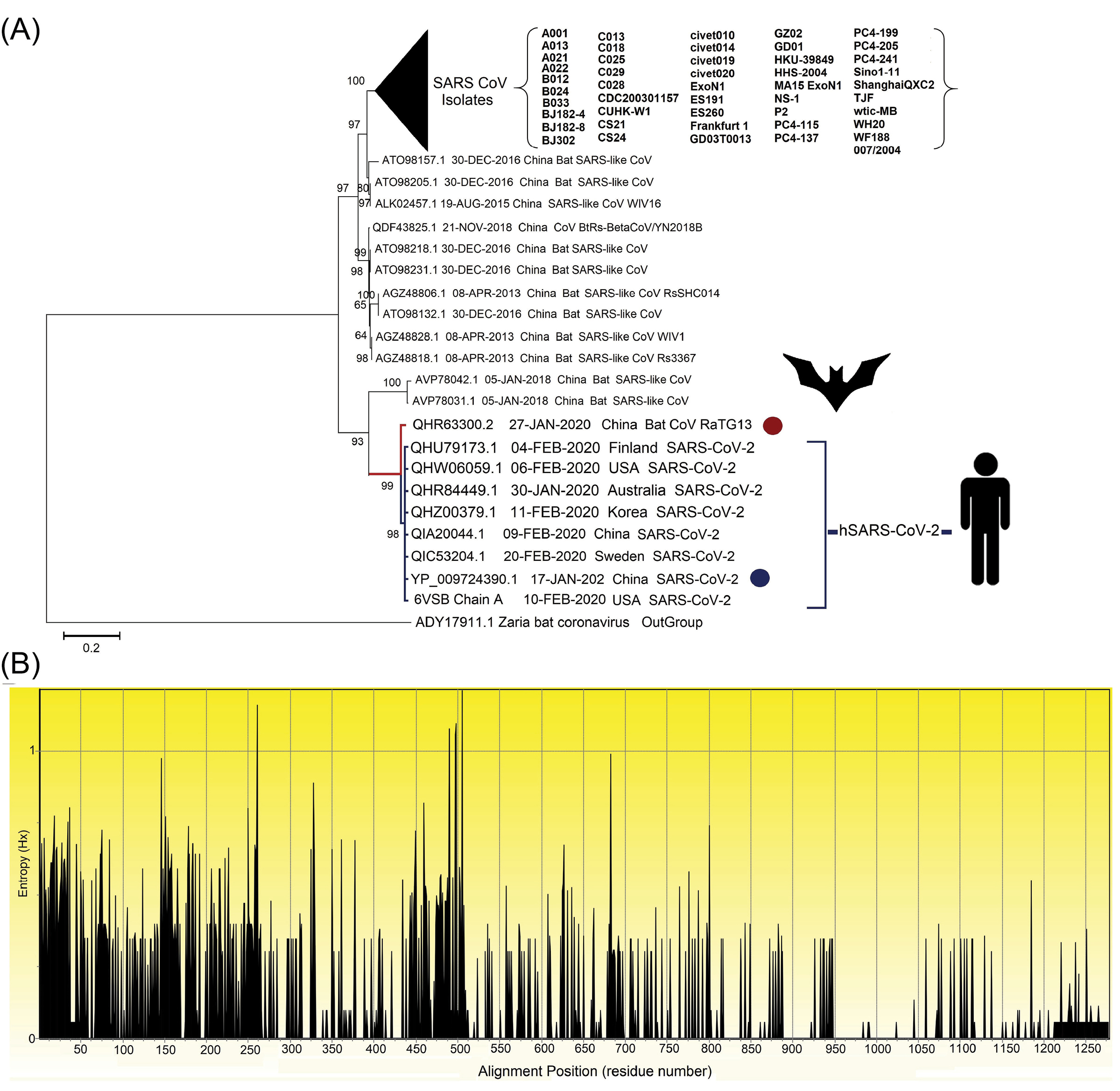
Fig. 2.
The evolutionary analysis of multiple strains of SARS-related coronavirus spike glycoprotein. (A) The phylogenetic tree built based on 101 amino acid sequences with high identity percentage and the reference protein sequence of glycoprotein S (accession no. YP_009724390.1; specified using the dark blue circle) using MEGA v1015 and rooted with the outgroup putative spike protein of Zaria Bat coronavirus (GenBank: ADY17911.1; specified using the dark red circle). There were a total of 1334 positions in the final dataset. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The evolutionary tree was created through the Jones-Taylor-Thornton (JTT) model by a discrete gamma distribution (+G) of 0.66 and assuming that a certain fraction of sites might be evolutionary invariable ([+I], 16.94% sites). Initial tree(s) for the statistical heuristic search algorithm were obtained automatically by applying the Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then, the selecting of the topology with the superior log-likelihood value. The clades correspond to the different isolates of SARS-CoVs that are collapsed facing the triangle for better presentation. (B) The Shannon entropy plot of different isolates of the protein S. The entropy (Hx) values ranged between 0.0 and 1.0, where the values more than 1.0 are related to the diverse residues.
.
The evolutionary analysis of multiple strains of SARS-related coronavirus spike glycoprotein. (A) The phylogenetic tree built based on 101 amino acid sequences with high identity percentage and the reference protein sequence of glycoprotein S (accession no. YP_009724390.1; specified using the dark blue circle) using MEGA v1015 and rooted with the outgroup putative spike protein of Zaria Bat coronavirus (GenBank: ADY17911.1; specified using the dark red circle). There were a total of 1334 positions in the final dataset. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The evolutionary tree was created through the Jones-Taylor-Thornton (JTT) model by a discrete gamma distribution (+G) of 0.66 and assuming that a certain fraction of sites might be evolutionary invariable ([+I], 16.94% sites). Initial tree(s) for the statistical heuristic search algorithm were obtained automatically by applying the Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then, the selecting of the topology with the superior log-likelihood value. The clades correspond to the different isolates of SARS-CoVs that are collapsed facing the triangle for better presentation. (B) The Shannon entropy plot of different isolates of the protein S. The entropy (Hx) values ranged between 0.0 and 1.0, where the values more than 1.0 are related to the diverse residues.
Identification of spike glycoprotein conserved domain(s) and region(s)
The conserved and variable regions of the spike glycoprotein among the hundred CoV strains are shown based on the Shannon entropy plot (Fig. 2B). The most variable residues have entropy (Hx) values more than 1.0. According to the NCBI-CDD's output, there are two domain hits in the glycoprotein S sequence, including (i) a large polypeptide (CoV S2 protein, residues from 662 to 1270), and (ii) spike receptor-binding domain (residues from 331 to 583) that mediates the affinity binding of the virus to angiotensin-converting enzyme 2 (ACE2) (Fig. 3B). The conserved regions have a higher probability to be as a part of functional domains of the protein, however, epitope escape mutations may be also a potential consequence to the emergence of such zoonotic EREI viruses.
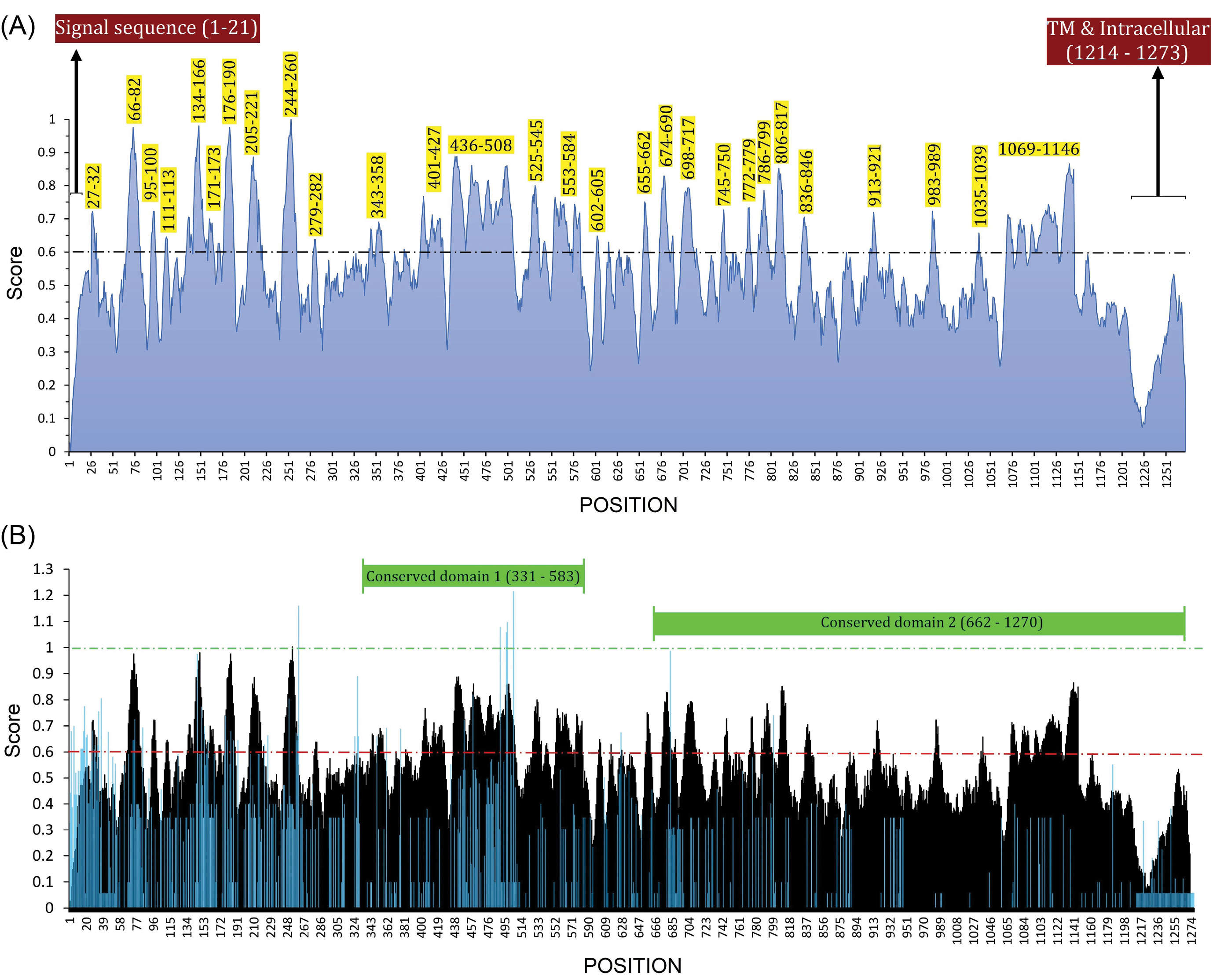
Fig. 3.
B-cell immunodominant regions of SAS-CoV-2 spike glycoprotein and its superimposed format with the sequence entropy-variability plot. (A) The plot was created based on the consensus result of the twenty-one B-cell epitope prediction algorithms. The normalized average scores ≥ 0.60 are marked as potential B-cell immunodominant regions. The residues which are in the signal sequence (residues 1–21), transmembrane (TM), and intracellular regions (residues 1214–1273) cannot be considered as B-cell immunodominant regions. (B) The B-cell immunodominant plot (shown as black) is superimposed with a sequence entropy-variability plot (shown as blue). The most variable residues have entropy (Hx) values ≥ 1.0. The two predicted conserved domain hits of S glycoprotein (331–583 and 662–1270) are exhibited top of the plot.
.
B-cell immunodominant regions of SAS-CoV-2 spike glycoprotein and its superimposed format with the sequence entropy-variability plot. (A) The plot was created based on the consensus result of the twenty-one B-cell epitope prediction algorithms. The normalized average scores ≥ 0.60 are marked as potential B-cell immunodominant regions. The residues which are in the signal sequence (residues 1–21), transmembrane (TM), and intracellular regions (residues 1214–1273) cannot be considered as B-cell immunodominant regions. (B) The B-cell immunodominant plot (shown as black) is superimposed with a sequence entropy-variability plot (shown as blue). The most variable residues have entropy (Hx) values ≥ 1.0. The two predicted conserved domain hits of S glycoprotein (331–583 and 662–1270) are exhibited top of the plot.
In total, 28 immunodominant B-cell peptides were predicted. All the predicted peptides are located on the accessible surface of the S glycoprotein (Fig. 4C). Therefore, those peptides, which have the highest prediction score, were selected for the vaccine design (Table 1). Besides, the reference sequences of the S glycoproteins of SARS-CoV (accession ID: NP_828851.1) and SARS-CoV-2 (accession ID: YP_009724390.1) were used for pairwise sequence alignment, and the final predicted BCEs were marked for comparison with the experimentally-determined SARS-CoV-derived BCEs
71
(Fig. S4; Table S4).
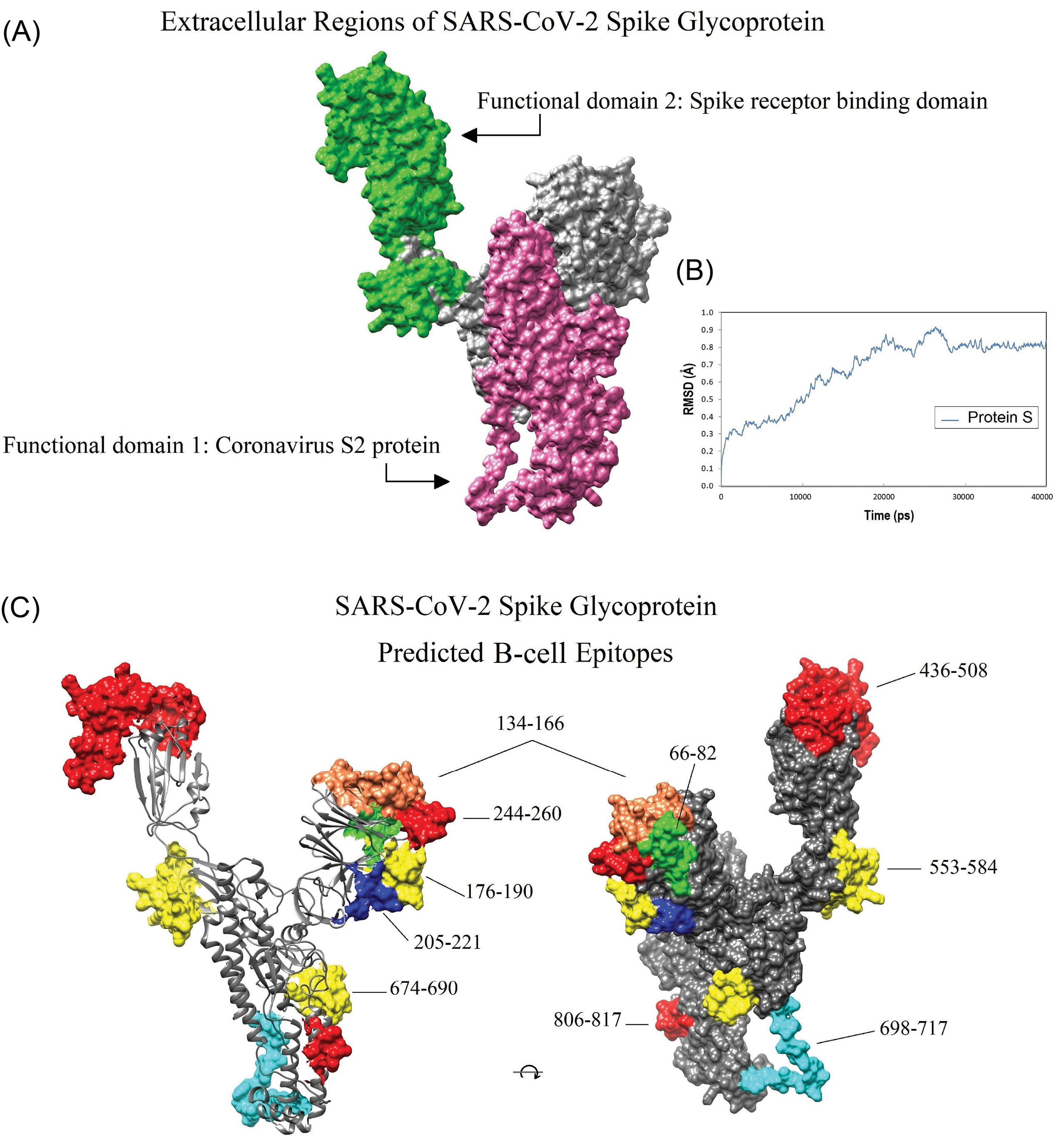
Fig. 4.
Location of the conserved domains and the immunodominant B-cell epitopes of SARS-CoV-2 spike glycoprotein on the homology modeled structure. A) The receptor-binding domain (green), and S2 subunit (pink) of SARS-CoV-2 S glycoprotein. B) The dynamic root-mean-square deviation (RMSD) graph corresponding to all atoms of the modeled spike glycoprotein shows that the simulation time (40000 ps) was long enough to achieve convergence (or stability) for the protein. C) The position of the ten dominant predicted B-cell epitopes. Owing to the lack of crystallized template protein, some residues in the beginning and end of the 3D model (1-27, and 1021-1273, respectively) are missed. The 3D structures were visualized by UCSF Chimera v1.14 software.
72
.
Location of the conserved domains and the immunodominant B-cell epitopes of SARS-CoV-2 spike glycoprotein on the homology modeled structure. A) The receptor-binding domain (green), and S2 subunit (pink) of SARS-CoV-2 S glycoprotein. B) The dynamic root-mean-square deviation (RMSD) graph corresponding to all atoms of the modeled spike glycoprotein shows that the simulation time (40000 ps) was long enough to achieve convergence (or stability) for the protein. C) The position of the ten dominant predicted B-cell epitopes. Owing to the lack of crystallized template protein, some residues in the beginning and end of the 3D model (1-27, and 1021-1273, respectively) are missed. The 3D structures were visualized by UCSF Chimera v1.14 software.
72
Table 1.
Predicted B-cell epitopes from SARS-CoV-2 S protein
|
Sequence
|
Position
|
Entropy score
a
|
Entropy score
b
|
CBPS*
|
| AYTNSF |
27–32 |
0.545 |
0.346 |
0.67 |
|
HAIHVSGTNGTKRFDNP*
|
66–82 |
0.381 |
0.489 |
0.79 |
| TEKSNI |
95–100 |
0.050 |
0.115 |
0.66 |
| DSK |
111–113 |
0.115 |
0.231 |
0.64 |
|
QFCNDPFLGVYYHKNNKSWMESEFRVYSSANNC*
|
134–166 |
0.400 |
0.567 |
0.71 |
| VSQ |
171–173 |
0.0 |
0.0 |
0.62 |
|
LMDLEGKQGNFKNLR*
|
176–190 |
0.320 |
0.370 |
0.78 |
|
SKHTPINLVRDLPQGFS*
|
205–221 |
0.299 |
0.367 |
0.74 |
|
LHRSYLTPGDSSSGWTA*
|
244–260 |
0.435 |
0.652 |
0.81 |
| YNEN |
279–282 |
0.075 |
0.173 |
0.62 |
| NATRFASVYAWNRKRI |
343–358 |
0.091 |
0.130 |
0.63 |
| VIRGDEVRQIAPGQTGKIADYNYKLPD |
401–427 |
0.071 |
0.102 |
0.66 |
WNSNNLDSKVGGNYNYLYRLFRKSNLKPFERDISTEI
YQAGSTPCNGVEGFNCYFPLQSYGFQPTNGVGYQPY*
|
436–508 |
0.319 |
0.351 |
0.76 |
| CGPKKSTNLVKNKCVNFNFNG |
525–545 |
0.064 |
0.132 |
0.65 |
|
TESNKKFLPFQQFGRDIADTTDAVRDPQTLEI*
|
553–584 |
0.107 |
0.195 |
0.68 |
| TNTS |
602–605 |
0.0 |
0.0 |
0.62 |
| HVNNSYEC |
655–662 |
0.116 |
0.173 |
0.66 |
|
YQTQTNSPRRARSVASQ*
|
674–690 |
0.210 |
0.367 |
0.68 |
|
SLGAENSVAYSNNSIAIPTN*
|
698–717 |
0.080 |
0.138 |
0.69 |
| DSTECS |
745–750 |
0.0 |
0.0 |
0.65 |
| VEQDKNTQ |
772–779 |
0.110 |
0.173 |
0.64 |
| KQIYKTPPIKDFGG |
786–799 |
0.114 |
0.148 |
0.67 |
|
LPDPSKPSKRSF*
|
806–817 |
0.074 |
0.115 |
0.73 |
| QYGDCLGDIAA |
836–846 |
0.068 |
0.126 |
0.63 |
| QNVLYENQK |
913–921 |
0.0 |
0.0 |
0.64 |
| RLDKVEA |
983–989 |
0.008 |
0.0 |
0.64 |
| GQSKR |
1035–1039 |
0.0 |
0.0 |
0.60 |
PAQEKNFTTAPAICHDGKAHFPREGVFVSNGTHWFVTQR
NFYEPQIITTDNTFVSGNCDVVIGIVNNTVYDPLQPELD*
|
1069–1146 |
0.053 |
0.089 |
0.68 |
Abbreviation:CBPS, Consensus B-cell epitope prediction score.
aThe average Shannon entropy score calculated using the multiple sequence alignment of the reference sequence of SARS-CoV-2 S protein (accession no. YP_009724390.1) and the hundred different isolates of spike glycoprotein of CoVs. b The average Shannon entropy score calculated using pairwise sequence alignment of the reference sequence of the S glycoproteins of SARS-CoV (accession no. NP_828851.1) and SARS-CoV-2. Final selected B-cell epitopes are indicated by *.
Prediction of SARS-CoV-2 T-cell epitopes
Cytotoxic T-cell epitope
The IEDB server predicted a list of 2529 unique peptides of S glycoprotein binding to the 27 alleles of HLA class A and B as raw data (Table S5). Of these, the consensus rank (CR) score of the peptide binders which had percentile rank ≤ 1.0, and ANN– and SMM–based IC50 ≤ 50.0 were calculated. In this approach, peptide binders were sorted and then selected based on (i) their rank in terms of percentile rank, ANN–IC50, and SMM–IC50 measures (e.g., consensus rank or CR), and (ii) the number of the HLA alleles that are covered by these binders. As a result of the CR score-based screening, we plotted the most dominant peptide binders for both HLA-A and B alleles in Fig. 5A. The CR scores allow screening a subset of binder hits covering a large range of the human population. The most potent peptide binders of the SARS-CoV-2 spike glycoprotein sequence corresponding to the HLA-A and B alleles are shown in Fig. 5A.

Fig. 5.
The mapped peptide binders of the SARS-CoV-2 derived from spike glycoprotein and their respective HLA class I and II restrictions. (A) The column chart of 16 most dominant peptide binders with the most binding affinity and maximum population coverage. A lower CR score shows peptides with a higher binding affinity and greater HLA coverage. (B) The epitope mapping plot showing the predicted HLA-restriction T-helper epitope hits. The calculation was based on the average adjusted ranks for all the multiple corresponding HLA-II restrictions for each region. Of all the analyzed HLA class II alleles, only the peptide binders with adjusted rank ≤ 1.0 were considered for CD4+ T-cell epitope prediction. A small numbered percentile rank indicates a high binding affinity. The names of alleles that are covered by each peptide binder are written at the top of each column.
.
The mapped peptide binders of the SARS-CoV-2 derived from spike glycoprotein and their respective HLA class I and II restrictions. (A) The column chart of 16 most dominant peptide binders with the most binding affinity and maximum population coverage. A lower CR score shows peptides with a higher binding affinity and greater HLA coverage. (B) The epitope mapping plot showing the predicted HLA-restriction T-helper epitope hits. The calculation was based on the average adjusted ranks for all the multiple corresponding HLA-II restrictions for each region. Of all the analyzed HLA class II alleles, only the peptide binders with adjusted rank ≤ 1.0 were considered for CD4+ T-cell epitope prediction. A small numbered percentile rank indicates a high binding affinity. The names of alleles that are covered by each peptide binder are written at the top of each column.
The raw output table of IEDB's was contained 132195 peptides of different length binding to HLA-DRB alleles (Table S6). The predicted HLA-II peptide binders were filtered using a strict threshold (adjusted rank ≤ 1.0) to choose all the top-scoring peptides for each specific HLA-II allele. Of these, 24 most immunodominant peptides were chosen for more analysis (Fig. 5B). The potentially effective CD4+ T-cell epitopes were selected based on the population coverage of each peptide and also the number of covered HLA-II alleles.
Population coverage of T-cell epitopes
According to the announcement of the WHO on March 12th, 2020, the COVID-19 outbreak was characterized as a pandemic, indicating that vaccinologists may confront with the broad-spectrum immunophenotypes that can complicate the vaccine design and development.
73
Therefore, in this study, we provided a list of most potent peptide binders associated with most frequent MHC alleles to design a broad coverage vaccine construct. The population coverage of the most potent T-cell epitopes in the countries that are impacted the most by SARS-CoV-2 is reported in Table S7.
Selection of the most dominant CD8+ T-cell epitopes
Among the pool of CD8+ T-cell peptide binders (Table S5) we sought to found the most potent regions of S glycoprotein as the CTL epitope. Generally, we found 16 epitope sequences with the highest binding affinity to a maximum number of the most frequent HLA-I alleles. The most dominant predicted CD8+ T-cell epitopes were selected based on their CR score, MHC allele coverage, and percentage of population coverage (Table 2). As presented in Table 2, the average population coverage for the eleven of the best CD8+T-cell epitopes and their corresponding HLA-alleles were observed between 36.48% for the "SGWTAGAAAYYV" and 79.05% for the "GYLQPRTFLLKY" peptides. For details of the results of population coverage analysis of each of 16 predicted CD8+ T-cell epitopes in the most-affected countries by COVID-19, readers are directed to see Table S8.
Table 2.
List of dominant SARS-CoV-2-derived cytotoxic T-cell peptides, their consensus rank (CR) scores, and the population coverage results in the most-affected countries with COVID-19
|
Sequence
|
Position
|
CR score
|
Coverage of MHC class I allele
|
Coverage
a
(%)
|
Average hit
b
|
Pc90
c
|
|
IKWPWYIWLGFI#
|
1210–1221 |
5.94 |
A*02:06, A*23:01, A*24:02, A*32:01, B*35:01, B*51:01, B*53:01 |
41.88±14.94 |
0.50±0.20 |
0.18±0.04 |
|
LQIPFAMQMAYRF#
|
894–906 |
6.55 |
A*02:06, A*23:01, A*24:02, A*26:01, A*33:01, A*68:01, B*08:01, B*15:01, B*35:01, B*53:01, B*58:01 |
54.66±17.07 |
0.72±0.25 |
0.24±0.06 |
|
CEFQFCNDPFL#
|
131–141 |
6.61 |
A*02:06, A*02:01, A*23:01, B*44:03, B*40:01, B*44:02 |
55.88±10.14 |
0.68±0.16 |
0.24±0.05 |
|
GVFVSNGTHWFV#
|
1093–1104 |
7.48 |
A*02:01, A*02:03, A*02:06, A*23:01, A*24:02, A*26:01, A*68:02, B*35:01, B*58:01 |
66.93±9.42 |
0.84±0.18 |
0.33±0.1 |
|
FPNITNLCPF#
|
329–338 |
8.64 |
B*07:02, B*35:01, B*51:01, B*53:01 |
35.56±11.58 |
0.38±0.13 |
0.16±0.03 |
|
GFIAGLIAIVM#
|
1219–1229 |
9.01 |
A*02:01, A*02:03, A*02:0, A*26:01, A*68:02, B*15:01 |
51.59±9.07 |
0.58±0.14 |
0.22±0.05 |
|
EVFNATRFASVYAW#
|
340–353 |
9.18 |
A*30:01, A*68:02, B*08:01, B*15:01, B*35:01, B*57:01, B*58:01 |
39.95±10.45 |
0.45±0.14 |
0.17±0.03 |
|
SGWTAGAAAYYV#
|
256–267 |
9.42 |
A*01:01, A*02:06, A*30:02, A*26:01, A*68:01, A*68:02, B*15:01 |
36.48±15.59 |
0.42±0.19 |
0.17±0.04 |
|
LYNSASFSTFKCY#
|
368–380 |
9.99 |
A*03:01, A*11:01, A*23:01, A*24:02, A*68:01, B*15:01, B*58:01 |
58.69±14.05 |
0.73±0.21 |
0.27±0.09 |
|
NFTISVTTEILPV#
|
717–729 |
10.27 |
A*02:01, A*02:03, A*02:06, A*26:01, A*68:02, B*51:01, B*58:01 |
54.44±8.9 |
0.64±0.14 |
0.23±0.05 |
|
GYLQPRTFLLKY#
|
268–279 |
10.5 |
A*02:01, A*02:03, A*02:06, A*03:01, A*11:01, A*23:01, A*24:02, B*08:01, B*15:01 |
79.05±12.03 |
1.14±0.29 |
0.59±0.26 |
| YTNSFTRGVYY |
28–38 |
11.8 |
A*01:01, A*02:03, A*26:01, A*30:02, A*68:02, B*15:01 |
31.69±14.91 |
0.35±0.18 |
0.15±0.03 |
| FLPFFSNVTWF |
55–65 |
12.19 |
B*35:01, B*51:01, B*53:01, B*57:01 |
25.09±11.79 |
0.27±0.13 |
0.14±0.02 |
| EQYIKWPWYIW |
1207–1217 |
14.3 |
A*23:01, A*24:02, B*44:02, B*44:03 |
35.34±9.59 |
0.4±0.12 |
0.16±0.02 |
| VYSSANNCTFEY |
159–170 |
15.8 |
A*30:02, A*23:01, A*24:02, B*58:01, B*35:01, B*15:01 |
40.5±12.11 |
0.48±0.16 |
0.18±0.04 |
| CTLKSFTVEKGIY |
301–313 |
16.6 |
A*03:01, A*11:01, A*30:02, A*68:01, B*57:01, B*58:01 |
42.77±8.86 |
0.48±0.1 |
0.18±0.03 |
a Average (±SD) projected population coverage. b Average number of epitope hits/HLA combinations recognized by the population. c Minimum number of epitope hits/HLA combinations recognized by 90% of the population. Final selected CD8+ T-cell epitopes are indicated by #.
Selection of final CD4+ T-cell epitopes
The selected HLA class II binders contain the most frequently occurring amino acids that have the highest capacity to attach different MHC class II alleles (Table 3). Thereupon, they might have good potential to elicit effective cellular immunity in most human populations. The detailed results of population coverage analysis for all 16 predicted CD4+ T-cell epitopes in the most-affected countries by COVID-19 are presented in Table S9.
Table 3.
List of top-scoring SARS-CoV-2-derived helper T-cell epitopes, their average adjusted ranks, and population coverage results in the most-affected countries with COVID-19
|
Sequence
|
Position
|
APR
*
|
Coverage of MHC class I allele
|
Coverage
a
(%)
|
Average hit
b
|
Pc90
c
|
|
TLDSKTQSLLIVNNATNVVIKVCEFQF#
|
109–135 |
0.19 |
DRB1*04:01, DRB1*13:02, DRB3*02:02 |
14.24±9.16 |
0.15±0.1 |
0.12±0.01 |
|
YRVVVLSFELLHAPATVCGPKKS#
|
508–530 |
0.25 |
DRB1*01:01 |
10.74±6.4 |
0.11±0.07 |
0.11±0.01 |
|
FKNLREFVFKNIDGYFKIYSKHTPI#
|
186–210 |
0.39 |
DRB5*01:01 |
NA |
NA |
NA |
|
IGINITRFQTLLALHRSYLTP#
|
231–251 |
0.50 |
DRB1*01:01, DRB1*15:01, DRB5*01:01 |
23.81±12.87 |
0.25±0.14 |
0.14±0.02 |
| MFVFLVLLPLVSSQCVNLT |
1–19 |
0.52 |
DRB1*01:01, DRB1*11:01 |
20.44±10.42 |
0.21±0.11 |
0.13±0.02 |
|
KVGGNYNYLYRLFRKSNLKPFER#
|
444–466 |
0.57 |
DRB1*11:01 |
11.09±4.95 |
0.11±0.05 |
0.11±0.01 |
|
IAIPTNFTISVTTEILPVSMT#
|
712–732 |
0.58 |
DRB1*07:01 |
17.35±7.49 |
0.17±0.07 |
0.12±0.01 |
|
TITSGWTFGAGAALQIPFAMQ#
|
881–901 |
0.58 |
DRB1*01:01, DRB1*09:01 |
15.2±9.3 |
0.15±0.1 |
0.12±0.02 |
|
HFPREGVFVSNGTHWFVTQRNF#
|
1088–1109 |
0.59 |
DRB1*13:02, DRB3*01:01, DRB3*02:02 |
6.98±4.07 |
0.07±0.04 |
0.11±0.01 |
| VYADSFVIRGDEVRQIAPGQTGK |
395–417 |
0.64 |
DRB3*01:01 |
NA |
NA |
NA |
|
SKHTPINLVRDLPQGFSALEP#
|
205–225 |
0.64 |
DRB1*03:01, DRB3*01:01 |
19.12±12.55 |
0.19±0.13 |
0.13±0.03 |
| KCVNFNFNGLTGTGVLTES |
537–555 |
0.69 |
DRB1*09:01 |
6.21±7.34 |
0.06±0.07 |
0.11±0.01 |
| ADYSVLYNSASFSTFKC |
363–379 |
0.70 |
DRB3*02:02 |
NA |
NA |
NA |
| NATRFASVYAWNRKRISN |
343–360 |
0.71 |
DRB5*01:01 |
NA |
NA |
NA |
| ECSNLLLQYGSFCTQLNR |
748–765 |
0.71 |
DRB1*15:01 |
15.01±8.07 |
0.15±0.08 |
0.12±0.01 |
| ENQKLIANQFNSAIGKI |
918–934 |
0.72 |
DRB3*02:02 |
NA |
NA |
NA |
| GNCDVVIGIVNNTVYDPL |
1124–1141 |
0.72 |
DRB1*13:02 |
6.98±4.07 |
0.07±0.04 |
0.11±0.01 |
| AALQIPFAMQMAYRFNGI |
892–909 |
0.74 |
DRB4*01:01 |
NA |
NA |
NA |
| VQPTESIVRFPNITNLCPFG |
320–339 |
0.78 |
DRB1*04:05, DRB1*15:01 |
18.42±8.21 |
0.19±0.08 |
0.12±0.01 |
| FGGFNFSQILPDPSK |
797–811 |
0.81 |
DRB1*04:05 |
4.81±6.13 |
0.05±0.06 |
0.1±0.01 |
| ALNTLVKQLSSNFGAIS |
958–974 |
0.81 |
DRB1*04:01 |
8.0±7.17 |
0.08±0.07 |
0.11±0.01 |
| DLFLPFFSNVTWFHAI |
53–68 |
0.91 |
DRB1*04:01, DRB3*02:02 |
8.0±7.17 |
0.08±0.07 |
0.11±0.01 |
| RAAEIRASANLAATKM |
1014–1029 |
0.93 |
DRB3*02:02 |
NA |
NA |
NA |
| LTDEMIAQYTSALLAGT |
865–881 |
0.94 |
DRB1*15:01 |
15.01±8.07 |
0.15±0.08 |
0.12±0.01 |
Abbreviation: APR,Average percentile rank.
a Average (±SD) projected population coverage. b Average number of epitope hits/HLA combinations recognized by the population. c Minimum number of epitope hits/HLA combinations recognized by 90% of the population. Four HLA-II alleles (DRB5*01:01, DRB3*01:01, DRB4*01:01, and DRB3*02:02) were not available in population coverage calculation. NA: not available. Final selected CD4+ T-cell epitopes are indicated by #.
The scaffold of vaccine constructs and their features
For the rational design of the DPVC, we rendered the position of all final chosen B- and T-cell epitopes in the SARS-CoV-2 spike protein sequence (Fig. S5). Consequently, we found three peptide fragments (100–280, 430–590, and 1060–1150) containing the largest number of the overlapping immunodominant B- and T- cell epitopes. These fragments can cover 7 BCEs, 7 CD4+ T-cell epitopes, and 4 CD8+ T-cell epitopes (Fig. S5). Here, we designed two vaccine constructs based on the two different platforms:
(i) An adjuvanted DPVC, which needs to be produced, expressed, and purified in vitro, and injected subcutaneously.
(ii) A self-adjuvanted SAMV construct, which needs to be synthesized, produced as in vitro transcription process, delivered by employing a designated non-viral delivery system such as liposomal nanoformulation, administrated intramuscularly, and expressed in vivo.
The recombinant DPVC
In this platform, we designed an adjuvanted vaccine construct with a full-length of 984 amino acid residues. The different components of the vaccine are schematically represented in Fig. 6A. The result of PSIPRED web-server showed among 984 amino acids, 257 (26.12%), 204 (20.73%), and 523 (53.15%) amino acids are involved in α-helix, extended strand, and random coil, respectively. The map of the predicted secondary structure is shown in Fig. S6. The 3D structure of the MD-refined vaccine model is represented in Fig. 6B.
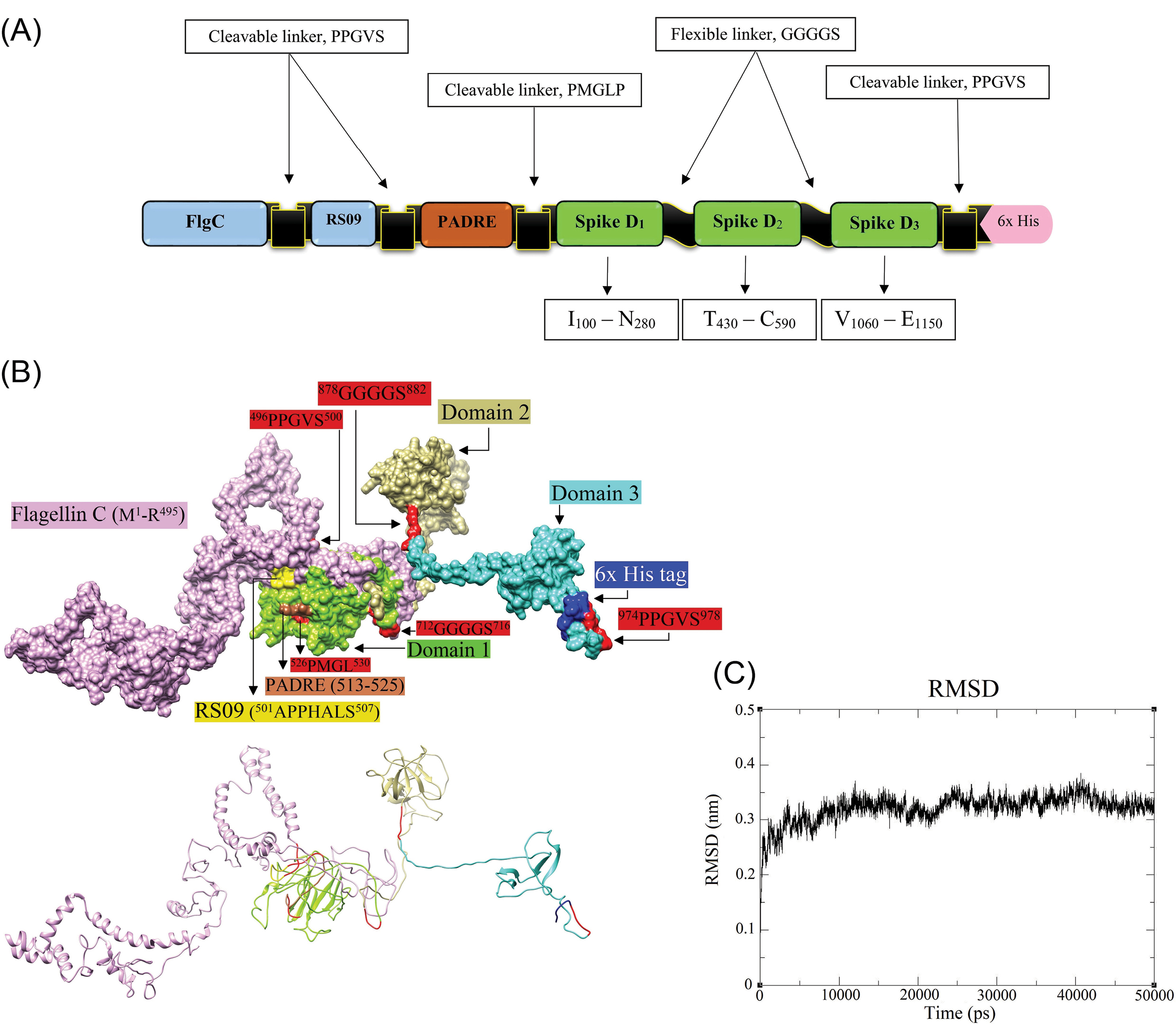
Fig. 6.
The molecular modeling of the designed DPVC. A) schematic diagram of the DPVC, including Flagellin C (1–495), and RS09 (101–107), two in vivo cleavable linkers (PPGVS and PPGVS), one PADRE sequence (AKFVAAWTLKAAA), and three peptide fragments covering the immunodominant B- and T-cell epitopes of spike glycoprotein. B) The structure of the molecular dynamics (MD)-refined vaccine model and its various constituent parts are visualized in surface (upper panel) and ribbon (lower panel) styles. The 3D models are represented by UCSF-Chimera software.
72
C) The root-mean-square deviation (RMSD) trajectory of the DVC, showing the structural stability of the optimized vaccine model during a course of MD simulations (50000 ps). DPVC: domain-based protein vaccine construct. D: domain.
.
The molecular modeling of the designed DPVC. A) schematic diagram of the DPVC, including Flagellin C (1–495), and RS09 (101–107), two in vivo cleavable linkers (PPGVS and PPGVS), one PADRE sequence (AKFVAAWTLKAAA), and three peptide fragments covering the immunodominant B- and T-cell epitopes of spike glycoprotein. B) The structure of the molecular dynamics (MD)-refined vaccine model and its various constituent parts are visualized in surface (upper panel) and ribbon (lower panel) styles. The 3D models are represented by UCSF-Chimera software.
72
C) The root-mean-square deviation (RMSD) trajectory of the DVC, showing the structural stability of the optimized vaccine model during a course of MD simulations (50000 ps). DPVC: domain-based protein vaccine construct. D: domain.
The C- and TM-scores, and RMSD of the initially modeled vaccine by the I-TASSER were calculated as -2.63, 0.41±0.14, and 13.6±3.1Å, respectively. The C-score is usually ranged from -5 to 2, where the C-score of higher values implies a model with higher confidence.
74
The TM-score and RMSD, as the standard metrics, are measured based on the C-score following the correlation observed between these qualities.
75
The TM-score threshold is independent of the size of proteins and values more than 0.5 are relevant to the correct model topology.
The energy level of the homology 3D modeled vaccine was minimized through the MD simulations for 50 ns to improve structural stability. The RMSD trajectory graph of the MD optimized vaccine model is shown in Fig. 6C. The RMSD of the structure reached 3.2Å after 5ns and remained approximately stable until the end of the simulations. This observation indicated the model expansion during the simulation and that the simulation duration was long enough to obtain an equilibrium structure for the constructed vaccine. Consequently, the extracted equilibrium structure at 310K was used for the subsequent evaluation of the vaccine-receptor binding affinity and interactions.
The backbone torsion angles (psi/phi) of the vaccine model and its overall quality before (i.e., initially modeled vaccine) and after MD simulation were analyzed based on the validation plots obtained from the PROCHECK (Fig. S7). The energy minimized vaccine model showed that 710 of all residues (82.8%) were in the most favored regions of the Ramachandran plot. Whereas in the initial DPVC model only 399 of residues (46.4%) were in these regions (Fig. S7). The comparison assessments showed that the MD-minimized vaccine model can be reliable to predict the binding affinity between the vaccine and TLRs 4 and 5.
Vaccine safety, antigenicity, stability, and solubility
Based on the result of both AlgPred and AllerTOP web-servers, the DPVC have no allergenic nature. The NCBI protein-protein BLAST against Homo sapiens showed the DPVC has no sequence similarity with the human proteome. This implies that the candidate vaccine should not trigger the autoimmune responses in the human body but activate the desired specific immunogenic reactions. The VaxiJen antigenicity score for the DPVC was 0.5097 indicating it as a probable antigen.
The molecular weight of the vaccine obtained from the ProtParam tool was about 105 kDa. The theoretical isoelectric point (pI) was calculated to be 5.95 showing the vaccine is slightly neural. The total numbers of positively and negatively charged residues were computed to be 81 and 91, respectively. The extinction-coefficient was 83660 M-1 cm-1 at 280 nm measured in water, which means all Cys residues are reduced. The half-life of the vaccine construct in mammalian reticulocytes was estimated at 30 hours(in vitro), more than 20 hours in yeast (in vivo), and more than 10 hours in Escherichia coli (in vivo) obtained by ProtParam tool. The computed instability index (II) classified the vaccine construct as a stable protein with a score of 28.47. The aliphatic index and GRAVY were calculated to be 80.50, and -0.296, respectively. These measures indicate that the vaccine construct is highly thermostable and also hydrophilic. The safe, immunogenic, and stable nature of the designed vaccine makes it a good candidate for more structural analysis.
Vaccine adjuvanticity and molecular docking simulations
The protein-protein molecular docking between the MD-optimized DPVC and the immune receptors (TLR4 and TLR5) was performed using the ClusPro v2.0 tool (Fig. 7). The best docked-complexes with the lowest energy scores were -1350.3 kcal/mol, and -1369.5 kcal/mol, for vaccine-TLR4, and vaccine-TLR5 complexes, respectively. The binding energies of the docked complexes were measured in the form of coefficient wattage using the formula E=0.40Erep+-0.40Eatt+600Eelec+1.00EDARS in the Balanced model.
68
The complexes with the highest binding affinities were subjected to the MD simulations by the GROMCAS software to survey their conformational stability (Fig. 7). The simulations were carried out in a 10 Å cubic box containing water molecules at 310K. The protein solvation was done using the spc216 template. The charges on the proteins were neutralized based on the Varlet cut-off scheme. Then, the system was subjected to energy minimization using the 1500 steps of steepest descent. The geometrical quality of the Cα backbone conformation was investigated using the root mean square deviation (RMSD) that is produced during MD simulation. According to the RMSD plots (Fig. 7), both docked complexes are stable mostly during the simulation. Based on the RMSD plot of the vaccine-TLR4 complex (Fig. 7A), the system reaches equilibrium at 15 ns (≈3.8 Å), whereas the RMSD values narrowly fluctuate between 3.5–4 Å. Nonetheless, the analysis of simulations for the vaccine-TLR5 reveals that it equilibrates much faster at 5 ns (≈3.8 Å) without significant fluctuations (Fig. 7B). As represented in Figs. 7 and 8, the DPVC functional parts (spike glycoprotein domains 1, 2, and 3; TLR4 agonistic motif RS09; and TLR5 agonistic domain flagellin C (FlgC) have a high binding affinity to the extracellular domains of the TLR4 and TLR5. Of these, the vaccine domains 2, and 3 (Figs. 7 and 8) indicated a more binding affinity to the TLRs. Here, we observed that the domains of SARS-CoV-2 spike glycoprotein can interact with the TLRs 4 and TLR5 on the cell surface, possibly triggering the intracellular NF-κB pathway and subsequent production of cytokine. Wang et al demonstrated that the interaction between the SARS-CoV spike glycoprotein and the murine macrophages could elicit the NF-κB activation pathway and then up-regulation of cytokines IL-6 and tumor necrosis factor alpha (TNF-α).
76
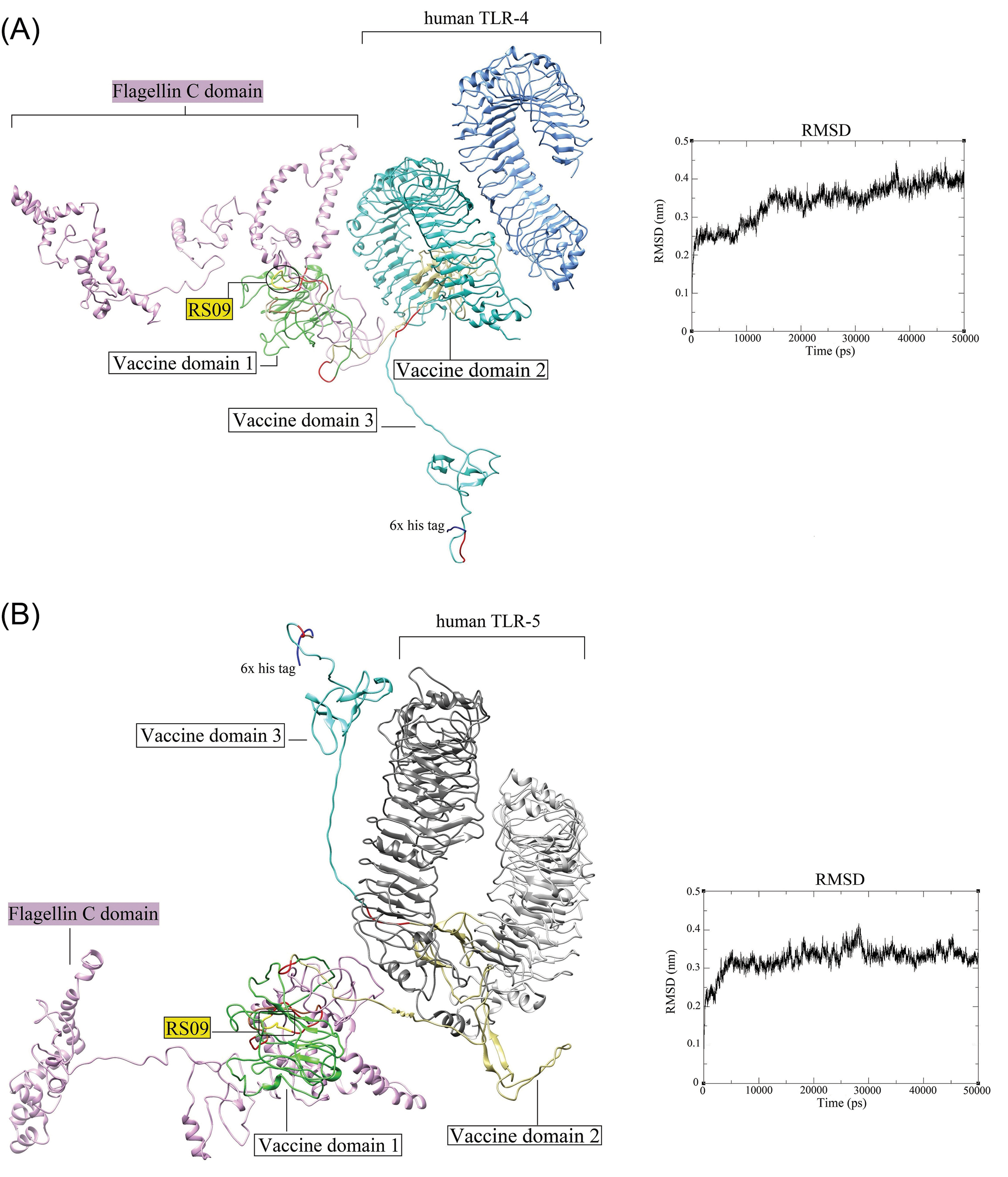
Fig. 7.
The docked complexes between the DPVC and the TLR4 and TLR5. (A) The interaction between the DPVC 3D model and the human TLR4 (PDB: 4G8A). (B) The docked complex of the DPVC and human TLR5 (PDB: 3J0A). The RMSD plots corresponding to the docked complexes are indicated on the right side of each panel. The 3D structures are visualized by the UCSF Chimera v1.14 software.
72
TLR: toll-like receptor. DPVC: domain-based polypeptide vaccine construct.
.
The docked complexes between the DPVC and the TLR4 and TLR5. (A) The interaction between the DPVC 3D model and the human TLR4 (PDB: 4G8A). (B) The docked complex of the DPVC and human TLR5 (PDB: 3J0A). The RMSD plots corresponding to the docked complexes are indicated on the right side of each panel. The 3D structures are visualized by the UCSF Chimera v1.14 software.
72
TLR: toll-like receptor. DPVC: domain-based polypeptide vaccine construct.
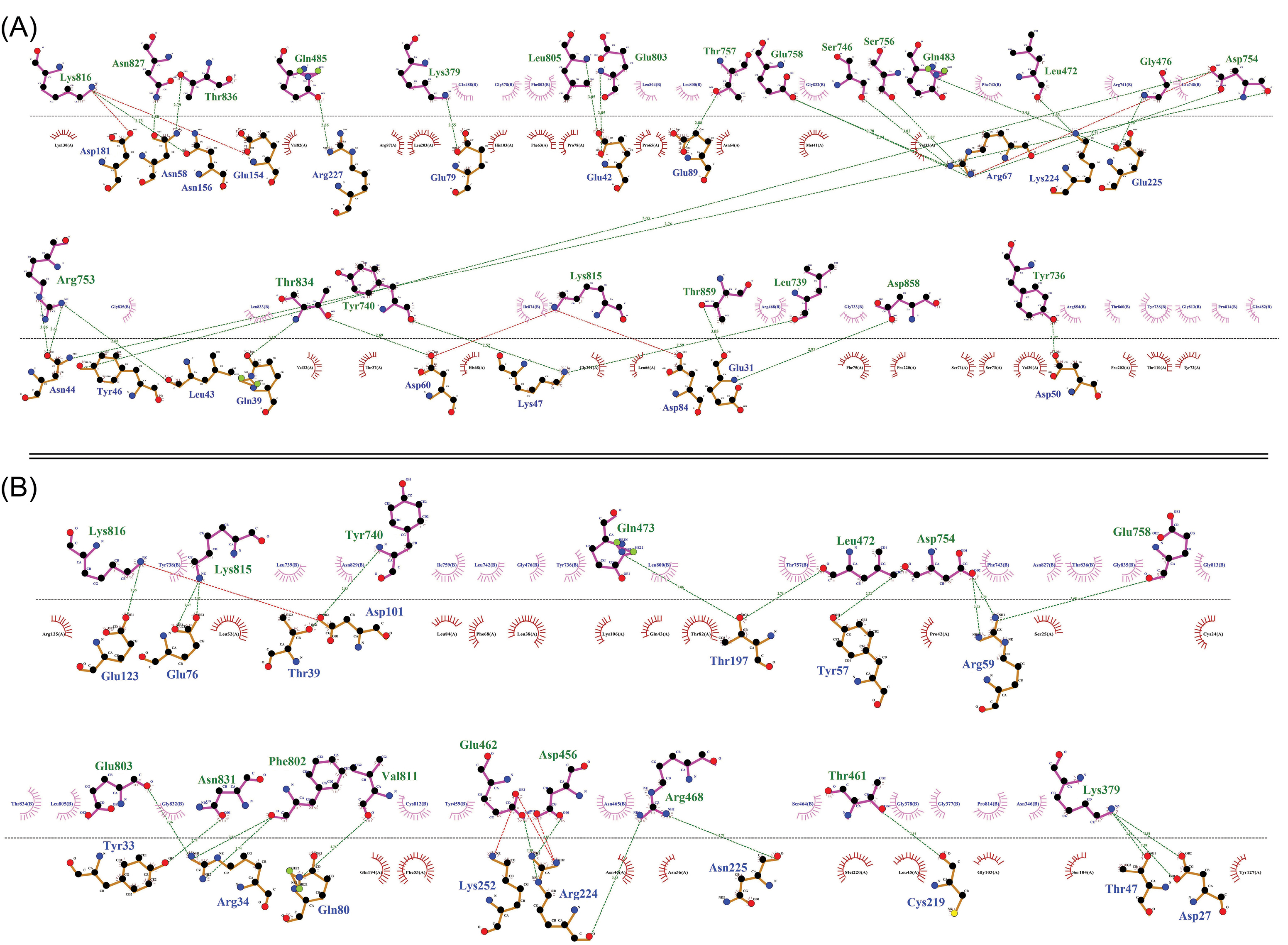
Fig. 8.
The two-dimensional diagram of the vaccine-receptor docked complexes. Intramolecular interactions between the vaccine-TLR4 (A) and vaccine-TLR5 (B). The hydrogen bonds and hydrophobic interactions are shown as a green dashed line, and a red spline curve, respectively. The intermolecular bonds of the vaccine and TLRs are shown as purple and brown lines, respectively. The plots provided by the DIMPLOT tool of LigPlot+ v2.2 program.
70
.
The two-dimensional diagram of the vaccine-receptor docked complexes. Intramolecular interactions between the vaccine-TLR4 (A) and vaccine-TLR5 (B). The hydrogen bonds and hydrophobic interactions are shown as a green dashed line, and a red spline curve, respectively. The intermolecular bonds of the vaccine and TLRs are shown as purple and brown lines, respectively. The plots provided by the DIMPLOT tool of LigPlot+ v2.2 program.
70
The H-bonds and hydrophobic interactions between the immune receptors (i.e., TLR4 and TLR5) and the DPVC are represented as a two-dimensional graph in Fig. 8.
Having capitalized on the in vivo cleavable linker (PPGVS) between the PADRE sequence and intramolecular adjuvants, it is expected to have a high level of either TLR-dependent innate immunity by the in vivo cleaved intramolecular adjuvants (FlgC and RS09) and S glycoprotein domains, and also the adaptive immune responses by PADRE sequence and SARS-CoV-2 S glycoprotein domains.
The self-amplifying mRNA (replicon) vaccine construct
In this approach, we designed a SAMV construct using the genes encoding the non-structural proteins (nsp1-4) of the positive-sense single-stranded RNA of Semliki Forest virus which are linked to the codon-optimized genes encoding the three identified immunodominant regions of the spike glycoprotein (I531–N711; T717–C877; V883–E973) to support the translation machinery in human cells. The different compounds of the designed SAMV and its cap structure are represented in Fig. 9.
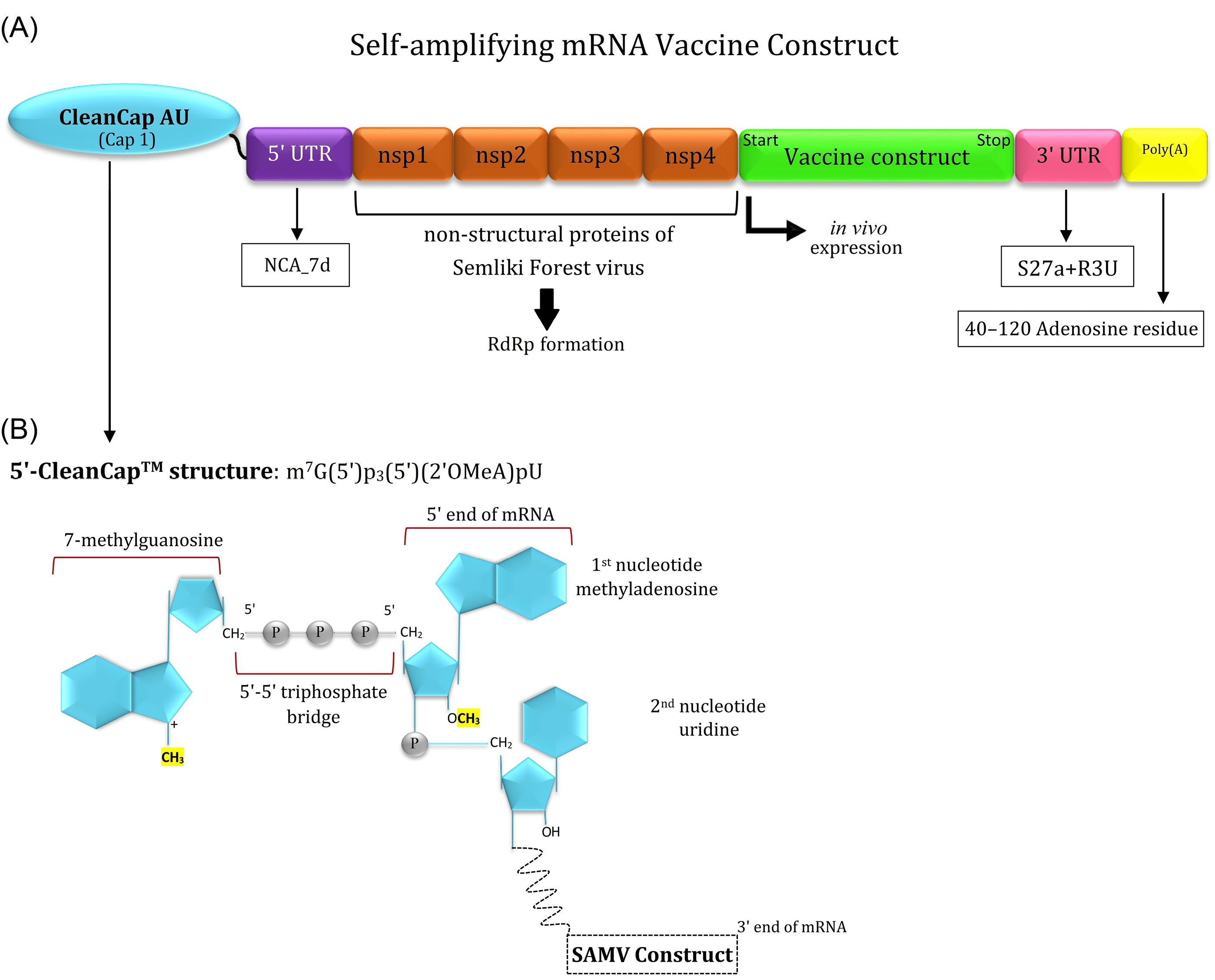
Fig. 9.
Schematic representation of the different parts of the designed self-amplifying mRNA vaccine (SAMV). (A) The designed SAMV consists of the genes encoding non-structural proteins (nsp1-4) of the Semliki Forest virus (NCBI reference sequence: NC_003215.1). The identified immunodominant regions of the glycoprotein S were used as vaccine sequences of interest. The nsp1-4 regions can form the RNA-dependent RNA polymerase (RdRp) complex. The SAMV construct was flanked between the 5' and 3' untranslated regions (NCA-7d, and S27a+R3U, respectively). A tail of 40–120 adenosine residues (Poly(A) tail) is inserted in the 3' end of the construct to improve the SAMV stability and functionality. (B) The 5' end of the SAMV construct contains a cap 1 structure with base analogs AU for the mRNA capping process.
.
Schematic representation of the different parts of the designed self-amplifying mRNA vaccine (SAMV). (A) The designed SAMV consists of the genes encoding non-structural proteins (nsp1-4) of the Semliki Forest virus (NCBI reference sequence: NC_003215.1). The identified immunodominant regions of the glycoprotein S were used as vaccine sequences of interest. The nsp1-4 regions can form the RNA-dependent RNA polymerase (RdRp) complex. The SAMV construct was flanked between the 5' and 3' untranslated regions (NCA-7d, and S27a+R3U, respectively). A tail of 40–120 adenosine residues (Poly(A) tail) is inserted in the 3' end of the construct to improve the SAMV stability and functionality. (B) The 5' end of the SAMV construct contains a cap 1 structure with base analogs AU for the mRNA capping process.
The designed SAMV consisted of the replication machinery of the Semliki Forest virus, therefore it might result in the injection-site intrinsic adjuvant reactions by the induction of pattern recognition receptors (PRRs), chemokines, cytokines (e.g., IL-12), and TNF.
77
These innate immune responses are critical for the maturation of dendritic cells (DCs) to boost up the subsequent direct adaptive immune responses. The mechanism of SAMV cellular uptake, activation of innate immunity, vaccine antigen's cellular processing, and the MHC presentation machinery in the injection site is projected in Fig. 10.
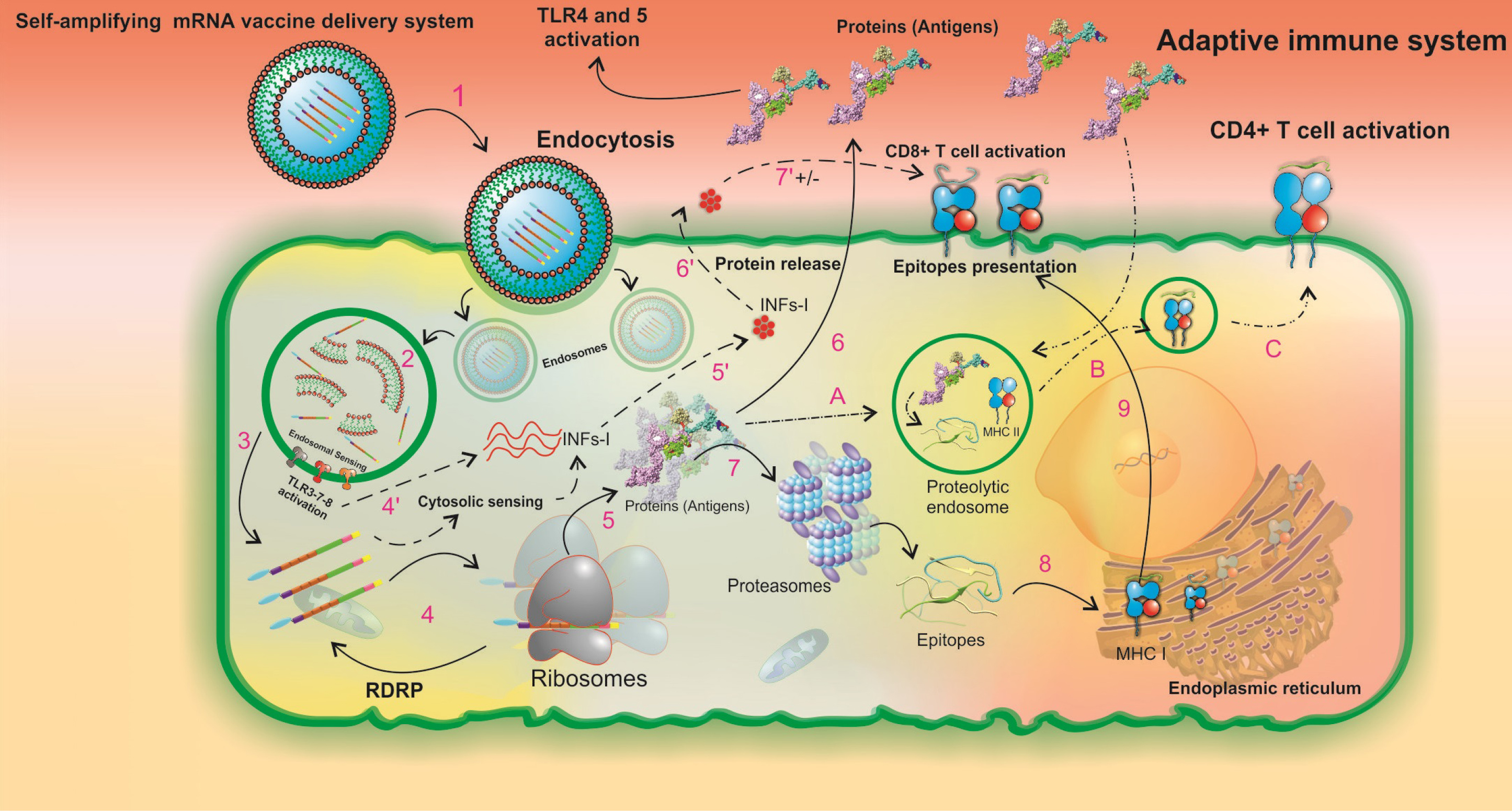
Fig. 10.
A schematic illustration of the intracellular processing of LNPs formulated the SAM vaccine and the subsequent innate and pathogen-specific immune responses. The in vitro transcribed SAM vaccine is formulated as a targeted vaccine delivery system (VDS), which is internalized by the antigen-presenting cells through receptor-mediated endocytosis (1). The targeted VDS is escaped from the endosomal compartment, and the initial endosomal RNA sensing by TLRs (mainly TLRs 3, 7, and 8) is activated (2). Upon SAMV endosomal escape, two main pathways of innate and adaptive immune responses can be activated (3). In the innate immune responses, steps 4' to 7' can occur. Both the SAM vaccine construct and the initial endosomal RNA sensing system activate the secondary RNA sensing system which is induced by cytosolic pathogen recognition receptors and then results in the production of type I interferons (INFα/β) (4', 5'). INFs are secreted (6'). The regulatory impacts of INFs are imposed on T-cell activity pathways (7'). In the Adaptive immune responses,steps 4-9 can occur. The in vivo translation of SAMV construct, the formation of RNA-dependent RNA polymerase (RdRp) complex, and the beginning a self-replication machinery for enhancement of the protein yield occur (4). The newly produced recombinant proteins have three possible destinies (5). First, protein is released to the extracellular space and its TLR agonists (i.e. RS09, and FlgC) can activate both TLRs 4 and 5, respectively (6). Second, protein is degraded by proteasomes to the small peptide fragments (7). The peptide fragments are processed by the endoplasmic reticulum (8). The MHC class I-epitope complexes are presented on the cell surface (9). Third, the peptides enter the proteolytic endosomes (A) to form the MHC class II-epitope complexes (B) and to be presented on the cell surface (C). LNP: lipid-nanoparticle. SAMV: self-amplifying mRNA vaccine.
.
A schematic illustration of the intracellular processing of LNPs formulated the SAM vaccine and the subsequent innate and pathogen-specific immune responses. The in vitro transcribed SAM vaccine is formulated as a targeted vaccine delivery system (VDS), which is internalized by the antigen-presenting cells through receptor-mediated endocytosis (1). The targeted VDS is escaped from the endosomal compartment, and the initial endosomal RNA sensing by TLRs (mainly TLRs 3, 7, and 8) is activated (2). Upon SAMV endosomal escape, two main pathways of innate and adaptive immune responses can be activated (3). In the innate immune responses, steps 4' to 7' can occur. Both the SAM vaccine construct and the initial endosomal RNA sensing system activate the secondary RNA sensing system which is induced by cytosolic pathogen recognition receptors and then results in the production of type I interferons (INFα/β) (4', 5'). INFs are secreted (6'). The regulatory impacts of INFs are imposed on T-cell activity pathways (7'). In the Adaptive immune responses,steps 4-9 can occur. The in vivo translation of SAMV construct, the formation of RNA-dependent RNA polymerase (RdRp) complex, and the beginning a self-replication machinery for enhancement of the protein yield occur (4). The newly produced recombinant proteins have three possible destinies (5). First, protein is released to the extracellular space and its TLR agonists (i.e. RS09, and FlgC) can activate both TLRs 4 and 5, respectively (6). Second, protein is degraded by proteasomes to the small peptide fragments (7). The peptide fragments are processed by the endoplasmic reticulum (8). The MHC class I-epitope complexes are presented on the cell surface (9). Third, the peptides enter the proteolytic endosomes (A) to form the MHC class II-epitope complexes (B) and to be presented on the cell surface (C). LNP: lipid-nanoparticle. SAMV: self-amplifying mRNA vaccine.
Discussion
Today, the sudden emergence with the quick spread of the novel zoonotic infectious agent, SARS-CoV-2 (Fig. 1), has led to a serious pandemic. Currently, several vaccine research teams in several countries are working to design, develop, and formulate an efficient prophylactic vaccine/adjuvant.
2,78-80
However, the conventional vaccine platforms against such a high transmissible and less-known infectious agent is an extremely time-consuming and risky task. Accordingly, among different vaccine platforms, self-amplifying mRNA vaccines as the next generation of mRNA vaccines provide a cost-effective and time-efficient strategy for the development of vaccines compared to the traditional methods.
81
Conducting a rapid vaccine engineering approach during such a viral pandemic may need three important preliminary research steps, including (i) viral genome sequencing, (ii) bioinformatics and data analysis, and (iii) designing a gene-based vaccine construct. Under these circumstances, computational modeling and simulation methods can assist the vaccinologists to extrapolate close to real biological evidence for designing a promising recombinant vaccine with high accuracy, least cost, and minimal time.
32,82
The in silico vaccinology, as a synergistic strategy is mainly based on (i) discovering of candidate vaccine antigens through the computer-aided data analysis approaches (e.g., reverse vaccinology),
83,84
and (ii) identification of immunodominant epitopes by applying an immunoinformatics pipeline.
85-87
In this context, along with releasing multiple whole-genome sequences of SARS-CoV-2 together with our previous experience in designing and developing an epitope-based recombinant vaccine against Echinococcus granulosus through comprehensive field trials (National Patent number: 100538; IPC: C12R 32/1;A61P 00/33;C12N 00/15), we designed two domain-based vaccine constructs based on the two different vaccine production and delivery platforms (i.e. recombinant protein vaccine, and self-replicating mRNA vaccine) as candidate prophylactic treatment against COVID-19. In this line, we used the reference sequence of SARS-CoV-2 spike glycoprotein (accession ID: YP_009724390.1) to rationally design the vaccines. First, to find out the virus origin and its conserved/variable regions, we carried out a multiple sequence alignment and also phylogenetic analysis based on all the sequenced spike glycoprotein of SARS-related CoVs. According to our phylogenetic analysis, SARS-CoV-2 has a close genetic similarity to the bat-derived CoVs (Fig. 2). A previous analysis using the haplotype network analysis announced that SARS-CoV-2 has emerged (or maybe emerging) due to the high frequently recurrent genetic recombination especially in the receptor-binding domain (RBD) of spike glycoprotein.
88
Theoretically, this natural occurrence has been likely affected in the virus transmissibility and pathogenicity through multiple amino acid alterations than SARS-CoV.
89
Based on the sequence variability analysis presented in the Shannon entropy plot (Fig. 2B), the RBD was found to be highly variable among different SARS-related CoVs. Tai et al represented a residue fragment (N331–V524) in the RBD domain of spike protein which can significantly bound to human and bat ACE2 receptors with higher affinity than SARS-CoV.
80
They suggested this region as a candidate for the development of a prophylactic domain-based vaccine against SARS-CoV-2. Amino acid insertion or deletion can disrupt or make significant changes in the physiological function of an antigen. Ting et al observed the single amino acid substitutions in protein L1 of human papillomavirus 16 (HPV16) can change its susceptibility to neutralization by monoclonal antibodies or vaccinated sera.
90
It is newly reported that SARS-CoV and SARS-CoV-2 have either high binding capability to the ACE2 receptor but probably with different affinities. Walls et al and Zhang et al found a furin cleavage site (P681–V687) of SARS-CoV-2 spike protein that is missed in the spike protein of all other SARS-related CoVs, and this insertion mutation has improved the mechanism of virus entry into the host cells.
91,92
Existing knowledge about the SARS-CoV-2 is mainly based on the prediction and simulation algorithms derived from the experimental data of other SARS-related CoVs. Grifoni et al used SARS-CoV surface proteins (S, M, Orf 3a, Orf 1ab, and N) as a homolog model for SARS-CoV-2 to predict candidate B- and T- cell epitopes of SARS-CoV-2.
78
In a recent study, Ahmed et al utilized immunological data of SARS-CoV to predict the potential epitopes of SARS-CoV-2 spike and nucleocapsid proteins.
79
In another study, peptide binders to HLA-DR types of the Asia-pacific region were predicted based on the four surface proteins (S, E, M, and N) and five accessory proteins (ORF3a, ORF6, ORF7a, ORF8, and ORF10) of SARS-CoV-2.
93
Despite these homology-based methodologies for epitope mapping, we believe that an emerged virus may develop sparse peculiar epitopes. Especially, in the variable residues of the spike antigen, emerging probable neo-epitopes may render different physicochemical features to form a stable complex with paratope site of antibodies and also binding groove of specific HLA molecules.
94,95
At this stage, the prediction of SARS-CoV-2 epitopes by monitoring its homolog viruses (i.e. SARS-related CoVs) seems to be a reliable method for conserved epitopes. By the same token, we computed the S glycoprotein sequence based on a multi-method BCE prediction approach through various machine learning and physicochemical algorithms to find out the hub regions (not exact epitope sequence) with high potential for B-cell immune responses (Fig. 3A). Then, through a stringent cut-off value (≥ 0.6) we identified a list of n=11 most immunodominant BCEs (Table 1), which are almost compatible with the predicted BCEs by Bhattacharya et al.
96
As showed in the 3D structure of the spike protein, these immunodominant BCEs are in the surface accessible areas of the protein (Fig. 4).
The currently developed methods for the T-cell epitope prediction are as a shortcut in epitope discovery; however, antigen processing and presentation in antigen-presenting cells (APCs) are followed through several complicated pathways. The T-cell epitope prediction servers specialized to provide widely dispersed dominant peptide binders with different lengths in a queried protein. Moreover, It is known that many of the cleaved peptides that are translocated into the endoplasmic reticulum (ER) have lengths of more than 8-10 amino acids, and some residues will be removed during processing by ER aminopeptidases.
97,98
The structural studies verified that there are many different mechanisms whereby a long peptide binder originated from either structural and nonstructural antigens can proceed into the APCs, attached, and presented by MHC class I and II molecules.
99-102
Currently, there is a lack of knowledge about the binding configuration/mechanism of SARS-CoV-2 epitopes and that how they make stable MHC-peptide complexes. In this regard, we used of online predictor tool IEDB to map potential high-rank T-cell peptide binders based on the reference set of HLA alleles covering > 97% (HLA-I) and > 99% (HLA-II) of the global population. To select candidate CD8+ T-cell epitopes, we defined a consensus ranking (CR) score to find out peptide binders with the lowest CR score and the highest HLA allele coverage (Fig. 5A). To predict the most potential CD4+ T-cell binders, we selected peptide fragments with the lowest adjusted percentile rank (Fig. 5B). The final T-cell epitopes were chosen based on the population coverage result of each predicted peptide fragment (Tables 2 and 3).
Having considered the scaled map indicated in Fig. S6, three hub domains of the spike glycoprotein covering the largest number of the best overlapping B- and T-cell epitopes were selected for the designing of the DVC (Fig. 6A). Despite the high consistency between our predicted epitopes and the recently reported epitopes,
78,79
we decided to target immunodominant domains of spike glycoprotein for vaccine designing, in large part due to the uncertainty about the exact sequence of B- and T-cell epitopes in different studies. This strategy allowed to have the optimal B- and T-cell epitopes through the natural humoral and cellular adaptive immune trafficking and APC-based proteolytic processing systems in the human body. We have joined the RS09 and S. typhimurium FlgC fragments at the N-terminal of the vaccine construct using an in vivo cleavable linker (Fig. 6A). The RS09 and FlgC are agonists for TLR4 and TLR5, respectively. RS09 is an LPS peptide mimicking entity that can bind to TLR4 and stimulate it, resulting in the subsequent activation of NF-κB signaling pathways and secretion of chemokines.
103
FlgC is the structural unit of the bacterial flagellum, which can interact with TLR5-expressing cells (e.g., monocytes, neutrophils, DCs, lymphocytes, and macrophages) as an agonist of TLR5.
104,105
Some studies reported the synergistic effects of the TLR4 and 5 signaling pathways; therefore, the use of FlgC might modulate initial innate and then the subsequent adaptive immune responses.
104,106
We have validated the interaction of vaccine construct with the TLR4 and TLR5 using molecular docking and then MD simulations (Fig. 7). Of note, as a strength, the self-amplifying mRNA vaccines have a high self-adjuvanted nature and both the endosomal and cytosolic RNA sensors (e.g., TLRs 3, 7, 8 and retinoic acid-inducible gene I (RIG-I) receptors, respectively) can recognize the viral derived agents and then trigger the innate immune signaling cascades (Fig. 10).
107
The Pan-DR epitope (i.e. PADRE sequence), a 13-mer synthetic T helper epitope, was also used to elicit more efficient adaptive immune responses (Fig. 6). It is demonstrated that the linear PADRE epitope in conjugation with the carbohydrate BCE can stimulate specific IgG antibodies.
108
The PADRE sequence was added between the RS09 and spike glycoprotein's domains using the intracellular cleavable linker to facilitate its independent processing and presentation by APCs (Fig. 6).
To produce the designed recombinant protein vaccine in a lab setting, a suitable expression host such as microalgae can be used to express the recombinant vaccine with the optimal post-translational modifications.
109,110
In the case of SAMV construct, although both the non-viral delivery systems (e.g., lipid nanoparticles,
111
polymeric nanoparticles,
112
and cell-penetrating peptides
113
), and in vivo transfection systems (e.g., injection, electroporation, and gene gun) can improve the stability and cellular uptake efficacy, however, the naked SAM vaccine can be taken up as well by significantly antigen-presenting cells without any additional required formulation.
114
Conclusion
Having capitalized on bioinformatics tools in the current study, for the first time, we designed two domain-based vaccine constructs against SARS-CoV-2 based on the two different vaccine production and delivery platforms including, (i) a recombinant protein vaccine, and (ii) a self-amplifying mRNA vaccine. We believe that the results of this study can be a step ahead in the vaccine development campaign against SARS-CoV-2. The methods used for the identification of the hub residue fragments of S glycoprotein were conducted based on the rational data filtering and also the precise multi-method analyses of various immunological datasets. The sequential and structural analysis of the DPVC showed that the vaccine is stable, safe, and immunogenic. In this context, these constructs are our urgent ongoing project to monitor the vaccine's potential to trigger properly both innate and specific B- and T-cell immune responses in animal models. Altogether, we have considered comprehensive key factors in the prediction of epitopes and the designing of both the DPVC and SAMV to ensure the proposed vaccines can induce both innate and pathogen-specific immune responses. As a result, we proposed the designed vaccines are promising vaccines against SARS-CoV-2 after being further examined through accelerated animal studies and clinical trials.
Acknowledgments
The authors are very thankful to all the nurses, physicians, and every one of the workers in hospitals who have been being exposed to the SARS-CoV-2 infectious agent worldwide. Further, the authors are grateful to the Research Center for Pharmaceutical Nanotechnology (RCPN) at the Tabriz University of Medical Sciences (TUOMS) for the financial and technical support. This work has synchronically been applied to be patented.
Funding sources
This study was supported by the Research Center for Pharmaceutical Nanotechnology, Tabriz University of Medical Sciences (#65207).
Ethical statement
This study was approved by the Research Ethics Committee of Tabriz University of Medical Sciences (Ethics No. IR.TBZMED.REC.1399.858).
Competing interests
It should be stated that the corresponding author of this study, YO, acts as the EIC of the journal. The peer-review process and acceptance of this study was performed according to the rules and regulations of the journal based on the ICMJE and COPE guidelines.
Authors’ contribution
The study protocol and research concept were designed by YO and MMP; The original draft, and the data analyses were performed by MMP; The molecular dynamics simulations were carried out by SP; The manuscript wrote by MMP; The manuscript was reviewed and edited by YO, BN, BJ and JD; The project was supervised by YO.
Supplementary Materials
Supplementary file 1 contains Figs. S1-S7 and Tables S1-S3.
(pdf)
Supplementary file 2 contains Table S4.
(xlsx)
Supplementary file 3 contains Table S5.
(xlsx)
Supplementary file 4 contains Table S6.
(xlsx)
Supplementary file 5 contains Tables S7-S9.
(pdf)
Research Highlights
What is the current knowledge?
simple
-
√ The B- and T-cell multi-epitope mapping provided versatile results for the immunodominant regions of SARS-CoV-2 spike protein.
-
√ Using the consensus rank (CR) score and the approach used for T-cell epitope mapping, one can design a potentially immunogenic candidate vaccine with high population coverage.
-
√ The self-amplifying mRNA (SAM) vaccine can be used as a nanoparticle-based vaccine (so-called nanovaccine) with an intrinsic adjuvanticity feature.
What is new here?
simple
-
√ The multi-method approach for the prediction of spike protein B-cell epitopes improved the accuracy of the in silicoepitope mapping.
-
√ The CR score as a precise method could promote selection of best T-cell epitopes with highest binding affinity and population coverage.
-
√ The designed SAM vaccine is a nanovaccine that offer both B-cell and T-cell immunity with an intrinsic adjuvanticity feature.
References
- Zumla A, Hui DSC. Emerging and Reemerging Infectious Diseases: Global Overview. Infect Dis Clin North Am 2019; 33:xiii-xix. doi: 10.1016/j.idc.2019.09.001 [Crossref] [ Google Scholar]
- Lucchese G. Epitopes for a 2019-nCoV vaccine. Cell Mol Immunol 2020; 17:539-540. doi: 10.1038/s41423-020-0377-z [Crossref] [ Google Scholar]
- Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020; 323:1061-1069. doi: 10.1001/jama.2020.1585 [Crossref] [ Google Scholar]
- The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 2020; 5:536-544. doi: 10.1038/s41564-020-0695-z [Crossref] [ Google Scholar]
- Maslow JN. The cost and challenge of vaccine development for emerging and emergent infectious diseases. Lancet Glob Health 2018; 6:e1266-e7. doi: 10.1016/S2214-109X(18)30418-2 [Crossref] [ Google Scholar]
- Oh SJ, Choi YK, Shin OS. Systems Biology-Based Platforms to Accelerate Research of Emerging Infectious Diseases. Yonsei Med J 2018; 59:176-86. doi: 10.3349/ymj.2018.59.2.176 [Crossref] [ Google Scholar]
- Shahid F, Ashfaq UA, Javaid A, Khalid H. Immunoinformatics guided rational design of a next generation multi epitope based peptide (MEBP) vaccine by exploring Zika virus proteome. Infect Genet Evol 2020; 80:104199. doi: 10.1016/j.meegid.2020.104199 [Crossref] [ Google Scholar]
- Raeven RHM, van Riet E, Meiring HD, Metz B, Kersten GFA. Systems vaccinology and big data in the vaccine development chain. Immunology 2019; 156:33-46. doi: 10.1111/imm.13012 [Crossref] [ Google Scholar]
- Parvizpour S, Pourseif MM, Razmara J, Rafi MA, Omidi Y. Epitope-based vaccine design: a comprehensive overview of bioinformatics approaches. Drug Discovery Today 2020; 25:1034-1042. doi: 10.1016/j.drudis.2020.03.006 [Crossref] [ Google Scholar]
- Barzegari A, Saeedi N, Zarredar H, Barar J, Omidi Y. The search for a promising cell factory system for production of edible vaccine. Hum VaccinImmunother 2014; 10:2497-502. doi: 10.4161/hv.29032 [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Saeedi N, Barzegari A, Dehghani J, Omidi Y. Current status and future prospective of vaccine development against Echinococcus granulosus. Biologicals 2018; 51:1-11. doi: 10.1016/j.biologicals.2017.10.003 [Crossref] [ Google Scholar]
- Maruggi G, Zhang C, Li J, Ulmer JB, Yu D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol Ther 2019; 27:757-72. doi: 10.1016/j.ymthe.2019.01.020 [Crossref] [ Google Scholar]
- McPherson C, Chubet R, Holtz K, Honda-Okubo Y, Barnard D, Cox M. Development of a SARS Coronavirus Vaccine from Recombinant Spike Protein Plus Delta Inulin Adjuvant. Methods Mol Biol 2016; 1403:269-84. doi: 10.1007/978-1-4939-3387-7_14 [Crossref] [ Google Scholar]
- Ng OW, Tan YJ. Understanding bat SARS-like coronaviruses for the preparation of future coronavirus outbreaks - Implications for coronavirus vaccine development. Hum VaccinImmunother 2017; 13:186-9. doi: 10.1080/21645515.2016.1228500 [Crossref] [ Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 2018; 35:1547-9. doi: 10.1093/molbev/msy096 [Crossref] [ Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004; 32:1792-7. doi: 10.1093/nar/gkh340 [Crossref] [ Google Scholar]
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 1992; 8:275-82. doi: 10.1093/bioinformatics/8.3.275 [Crossref] [ Google Scholar]
- Dopazo J. Estimating errors and confidence intervals for branch lengths in phylogenetic trees by a bootstrap approach. J Mol Evol 1994; 38:300-4. doi: 10.1007/bf00176092 [Crossref] [ Google Scholar]
- Tsirigos KD, Peters C, Shu N, Kall L, Elofsson A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res 2015; 43:W401-7. doi: 10.1093/nar/gkv485 [Crossref] [ Google Scholar]
- Dobson L, Remenyi I, Tusnady GE. CCTOP: a Consensus Constrained TOPology prediction web server. Nucleic Acids Res 2015; 43:W408-12. doi: 10.1093/nar/gkv451 [Crossref] [ Google Scholar]
- Chen Y, Yu P, Luo J, Jiang Y. Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm Genome 2003; 14:859-65. doi: 10.1007/s00335-003-2296-6 [Crossref] [ Google Scholar]
- Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 2017; 45:D200-D3. doi: 10.1093/nar/gkw1129 [Crossref] [ Google Scholar]
- Hall TA. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symposium Series 1999; 41:95-8. [ Google Scholar]
- Buchan DWA, Jones DT. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res 2019; 47:W402-W7. doi: 10.1093/nar/gkz297 [Crossref] [ Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018; 46:W296-W303. doi: 10.1093/nar/gky427 [Crossref] [ Google Scholar]
- Heo L, Park H, Seok C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res 2013; 41:W384-8. doi: 10.1093/nar/gkt458 [Crossref] [ Google Scholar]
- Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R. GROMACS 45: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013; 29:845-54. doi: 10.1093/bioinformatics/btt055 [Crossref] [ Google Scholar]
- Parvizpour S, Razmara J, Pourseif MM, Omidi Y. In silico design of a triple-negative breast cancer vaccine by targeting cancer testis antigens. Bioimpacts 2019; 9:45-56. doi: 10.15171/bi.2019.06 [Crossref] [ Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 1996; 8:477-86. doi: 10.1007/bf00228148 [Crossref] [ Google Scholar]
- Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 1997; 277:396-404. doi: 10.1016/s0076-6879(97)77022-8 [Crossref] [ Google Scholar]
- Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 1993; 2:1511-9. doi: 10.1002/pro.5560020916 [Crossref] [ Google Scholar]
- Pourseif MM, Yousefpour M, Aminianfar M, Moghaddam G, Nematollahi A. A multi-method and structure-based in silico vaccine designing against Echinococcus granulosus through investigating enolase protein. Bioimpacts 2019; 9:131-44. doi: 10.15171/bi.2019.18 [Crossref] [ Google Scholar]
- Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-20: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res 2017; 45:W24-W9. doi: 10.1093/nar/gkx346 [Crossref] [ Google Scholar]
- Singh H, Ansari HR, Raghava GP. Improved method for linear B-cell epitope prediction using antigen's primary sequence. PLoS One 2013; 8:e62216. doi: 10.1371/journal.pone.0062216 [Crossref] [ Google Scholar]
- Gupta S, Ansari HR, Gautam A, Raghava GP. Identification of B-cell epitopes in an antigen for inducing specific class of antibodies. Biol Direct 2013; 8:27. doi: 10.1186/1745-6150-8-27 [Crossref] [ Google Scholar]
- Ansari HR, Raghava GP. Identification of conformational B-cell Epitopes in an antigen from its primary sequence. Immunome Res 2010; 6:6. doi: 10.1186/1745-7580-6-6 [Crossref] [ Google Scholar]
- Odorico M, Pellequer JL. BEPITOPE: predicting the location of continuous epitopes and patterns in proteins. J Mol Recognit 2003; 16:20-2. doi: 10.1002/jmr.602 [Crossref] [ Google Scholar]
- Saha S, Raghava GP. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006; 65:40-8. doi: 10.1002/prot.21078 [Crossref] [ Google Scholar]
- Zhou C, Chen Z, Zhang L, Yan D, Mao T, Tang K. SEPPA 30-enhanced spatial epitope prediction enabling glycoprotein antigens. Nucleic Acids Res 2019; 47:W388-W94. doi: 10.1093/nar/gkz413 [Crossref] [ Google Scholar]
- Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoSComput Biol 2012; 8:e1002829. doi: 10.1371/journal.pcbi.1002829 [Crossref] [ Google Scholar]
- Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 2008; 9:514. doi: 10.1186/1471-2105-9-514 [Crossref] [ Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 2011; 7:539. doi: 10.1038/msb.2011.75 [Crossref] [ Google Scholar]
- Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res 2012; 40:D593-8. doi: 10.1093/nar/gkr859 [Crossref] [ Google Scholar]
- Moutaftsi M, Peters B, Pasquetto V, Tscharke DC, Sidney J, Bui HH. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat Biotechnol 2006; 24:817-9. doi: 10.1038/nbt1215 [Crossref] [ Google Scholar]
- Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 2003; 12:1007-17. doi: 10.1110/ps.0239403 [Crossref] [ Google Scholar]
- Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoSComput Biol 2008; 4:e1000048. doi: 10.1371/journal.pcbi.1000048 [Crossref] [ Google Scholar]
- Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics 2011; 63:325-35. doi: 10.1007/s00251-011-0513-0 [Crossref] [ Google Scholar]
- Gonzalez-Galarza FF, Takeshita LY, Santos EJ, Kempson F, Maia MH, da Silva AL. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res 2015; 43:D784-8. doi: 10.1093/nar/gku1166 [Crossref] [ Google Scholar]
- Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 2019; 47:D339-D43. doi: 10.1093/nar/gky1006 [Crossref] [ Google Scholar]
- Patel MC, Shirey KA, Pletneva LM, Boukhvalova MS, Garzino-Demo A, Vogel SN. Novel drugs targeting Toll-like receptors for antiviral therapy. Future Virol 2014; 9:811-29. doi: 10.2217/fvl.14.70 [Crossref] [ Google Scholar]
- Shah M, Anwar MA, Kim JH, Choi S. Advances in Antiviral Therapies Targeting Toll-like Receptors. Expert OpinInvestig Drugs 2016; 25:437-53. doi: 10.1517/13543784.2016.1154040 [Crossref] [ Google Scholar]
- Shanmugam A, Rajoria S, George AL, Mittelman A, Suriano R, Tiwari RK. Synthetic Toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PLoS One 2012; 7:e30839. doi: 10.1371/journal.pone.0030839 [Crossref] [ Google Scholar]
- Skountzou I, Martin Mdel P, Wang B, Ye L, Koutsonanos D, Weldon W. Salmonella flagellins are potent adjuvants for intranasally administered whole inactivated influenza vaccine. Vaccine 2010; 28:4103-12. doi: 10.1016/j.vaccine.2009.07.058 [Crossref] [ Google Scholar]
- Kridel SJ, Chen E, Kotra LP, Howard EW, Mobashery S, Smith JW. Substrate hydrolysis by matrix metalloproteinase-9. J Biol Chem 2001; 276:20572-8. doi: 10.1074/jbc.M100900200 [Crossref] [ Google Scholar]
- Varani J, Hattori Y, Chi Y, Schmidt T, Perone P, Zeigler ME. Collagenolytic and gelatinolytic matrix metalloproteinases and their inhibitors in basal cell carcinoma of skin: comparison with normal skin. Br J Cancer 2000; 82:657-65. doi: 10.1054/bjoc.1999.0978 [Crossref] [ Google Scholar]
- Schonefuss A, Wendt W, Schattling B, Schulten R, Hoffmann K, Stuecker M. Upregulation of cathepsin S in psoriatic keratinocytes. Exp Dermatol 2010; 19:e80-8. doi: 10.1111/j.1600-0625.2009.00990.x [Crossref] [ Google Scholar]
- Riese RJ, Mitchell RN, Villadangos JA, Shi GP, Palmer JT, Karp ER. Cathepsin S activity regulates antigen presentation and immunity. J Clin Invest 1998; 101:2351-63. doi: 10.1172/JCI1158 [Crossref] [ Google Scholar]
- Lucke M, Mottas I, Herbst T, Hotz C, Romer L, Schierling M. Engineered hybrid spider silk particles as delivery system for peptide vaccines. Biomaterials 2018; 172:105-15. doi: 10.1016/j.biomaterials.2018.04.008 [Crossref] [ Google Scholar]
- Kovjazin R, Carmon L. The use of signal peptide domains as vaccine candidates. Hum VaccinImmunother 2014; 10:2733-40. doi: 10.4161/21645515.2014.970916 [Crossref] [ Google Scholar]
- Tews BA, Meyers G. Self-Replicating RNA. Methods Mol Biol 2017; 1499:15-35. doi: 10.1007/978-1-4939-6481-9_2 [Crossref] [ Google Scholar]
- Zeng C, Hou X, Yan J, Zhang C, Li W, Zhao W. Leveraging mRNAs sequences to express SARS-CoV-2 antigens in vivo. bioRxiv 2020:2020.04.01.019877. doi: 10.1101/2020.04.01.019877 [Crossref]
- Zaharieva N, Dimitrov I, Flower DR, Doytchinova I. VaxiJen Dataset of Bacterial Immunogens: An Update. CurrComput Aided Drug Des 2019; 15:398-400. doi: 10.2174/1573409915666190318121838 [Crossref] [ Google Scholar]
- Saha S, Raghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res 2006; 34:W202-9. doi: 10.1093/nar/gkl343 [Crossref] [ Google Scholar]
- Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP v2--a server for in silico prediction of allergens. J Mol Model 2014; 20:2278. doi: 10.1007/s00894-014-2278-5 [Crossref] [ Google Scholar]
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 1999; 112:531-52. doi: 10.1385/1-59259-584-7:531 [Crossref] [ Google Scholar]
- Garnier J, Gibrat JF, Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol 1996; 266:540-53. doi: 10.1016/s0076-6879(96)66034-0 [Crossref] [ Google Scholar]
- Yang J, Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 2015; 43:W174-81. doi: 10.1093/nar/gkv342 [Crossref] [ Google Scholar]
- Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C. The ClusPro web server for protein-protein docking. Nat Protoc 2017; 12:255-78. doi: 10.1038/nprot.2016.169 [Crossref] [ Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605-12. doi: 10.1002/jcc.20084 [Crossref] [ Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 2011; 51:2778-86. doi: 10.1021/ci200227u [Crossref] [ Google Scholar]
- Guo JP, Petric M, Campbell W, McGeer PL. SARS corona virus peptides recognized by antibodies in the sera of convalescent cases. Virology 2004; 324:251-6. doi: 10.1016/j.virol.2004.04.017 [Crossref] [ Google Scholar]
- Yang Z, Lasker K, Schneidman-Duhovny D, Webb B, Huang CC, Pettersen EF. UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J Struct Biol 2012; 179:269-78. doi: 10.1016/j.jsb.2011.09.006 [Crossref] [ Google Scholar]
- Peters B, Bui HH, Frankild S, Nielson M, Lundegaard C, Kostem E. A community resource benchmarking predictions of peptide binding to MHC-I molecules. PLoSComput Biol 2006; 2:e65. doi: 10.1371/journal.pcbi.0020065 [Crossref] [ Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008; 9:40. doi: 10.1186/1471-2105-9-40 [Crossref] [ Google Scholar]
- Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins 2004; 57:702-10. doi: 10.1002/prot.20264 [Crossref] [ Google Scholar]
- Wang W, Ye L, Ye L, Li B, Gao B, Zeng Y. Up-regulation of IL-6 and TNF-alpha induced by SARS-coronavirus spike protein in murine macrophages via NF-kappaB pathway. Virus Res 2007; 128:1-8. doi: 10.1016/j.virusres.2007.02.007 [Crossref] [ Google Scholar]
- Chen N, Xia P, Li S, Zhang T, Wang TT, Zhu J. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 2017; 69:297-304. doi: 10.1002/iub.1625 [Crossref] [ Google Scholar]
- Grifoni A, Sidney J, Zhang Y, Scheuermann RH, Peters B, Sette A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 2020; 27,:671-680. doi: 10.1016/j.chom.2020.03.002 [Crossref] [ Google Scholar]
- Ahmed SF, Quadeer AA, McKay MR. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020; 12:254. doi: 10.3390/v12030254 [Crossref] [ Google Scholar]
- Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol 2020; 17:613-620. doi: 10.1038/s41423-020-0400-4 [Crossref] [ Google Scholar]
- Salemi A, Pourseif MM, Omidi Y. Next-generation vaccines and the impacts of state-of-the-art in-silico technologies. Biologicals 2020. doi: 10.1016/j.biologicals.2020.10.002 [Crossref]
- Robson B. Robson BComputers and viral diseasesPreliminary bioinformatics studies on the design of a synthetic vaccine and a preventative peptidomimetic antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) coronavirus. Comput Biol Med 2020; 119:103670. doi: 10.1016/j.compbiomed.2020.103670 [Crossref] [ Google Scholar]
- Moxon R, Reche PA, Rappuoli R. Editorial: Reverse Vaccinology. Front Immunol 2019; 10:2776. doi: 10.3389/fimmu.2019.02776 [Crossref] [ Google Scholar]
- Sanchez-Trincado JL, Gomez-Perosanz M, Reche PA. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J Immunol Res 2017; 2017:2680160. doi: 10.1155/2017/2680160 [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Daghighkia H, Nematollahi A, Omidi Y. A novel B- and helper T-cell epitopes-based prophylactic vaccine against Echinococcus granulosus. Bioimpacts 2018; 8:39-52. doi: 10.15171/bi.2018.06 [Crossref] [ Google Scholar]
- Shey RA, Ghogomu SM, Esoh KK, Nebangwa ND, Shintouo CM, Nongley NF. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci Rep 2019; 9:4409. doi: 10.1038/s41598-019-40833-x [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Naghili B, Saeedi N, Parvizpour S, Nematollahi A. A novel in silico minigene vaccine based on CD4(+) T-helper and B-cell epitopes of EG95 isolates for vaccination against cystic echinococcosis. Comput Biol Chem 2018; 72:150-63. doi: 10.1016/j.compbiolchem.2017.11.008 [Crossref] [ Google Scholar]
- Yi H. 2019 novel coronavirus is undergoing active recombination. Clin Infect Dis 2020; 71:884-887. doi: 10.1093/cid/ciaa219 [Crossref] [ Google Scholar]
- Kristian G. Andersen AR, W Ian Lipkin, Edward C Holmes and Robert F Garry The proximal origin of SARS-CoV-2. Nature Medicine 2020; 26:450-452. doi: 10.1038/s41591-020-0820-9 [Crossref] [ Google Scholar]
- Ning T, Wolfe A, Nie J, Huang W, Chen XS, Wang Y. Naturally Occurring Single Amino Acid Substitution in the L1 Major Capsid Protein of Human Papillomavirus Type 16: Alteration of Susceptibility to Antibody-Mediated Neutralization. J Infect Dis 2017; 216:867-76. doi: 10.1093/infdis/jix274 [Crossref] [ Google Scholar]
- Zhang T, Wu Q, Zhang Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr Biol 2020; 30:1346-51 e2. doi: 10.1016/j.cub.2020.03.022 [Crossref] [ Google Scholar]
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020; 181:281-292. doi: 10.1016/j.cell.2020.02.058 [Crossref] [ Google Scholar]
- Arunachalam Ramaiah VA. Insights into Cross-species Evolution of Novel Human Coronavirus 2019-nCoV and Defining Immune Determinants for Vaccine Development. bioRxiv 2020:925867. doi: 10.1101/2020.01.29.925867 [Crossref]
- Besser H, Yunger S, Merhavi-Shoham E, Cohen CJ, Louzoun Y. Level of neo-epitope predecessor and mutation type determine T cell activation of MHC binding peptides. J Immunother Cancer 2019; 7:135. doi: 10.1186/s40425-019-0595-z [Crossref] [ Google Scholar]
- Moradi A, Pourseif MM, Jafari B, Parvizpour S, Omidi Y. Nanobody-based therapeutics against colorectal cancer: Precision therapies based on the personal mutanome profile and tumor neoantigens. Pharmacol Res 2020; 156:104790. doi: 10.1016/j.phrs.2020.104790 [Crossref] [ Google Scholar]
- Bhattacharya M, Sharma AR, Patra P, Ghosh P, Sharma G, Patra BC. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J Med Virol 2020; 92:618-31. doi: 10.1002/jmv.25736 [Crossref] [ Google Scholar]
- Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol 2013; 31:443-73. doi: 10.1146/annurev-immunol-032712-095910 [Crossref] [ Google Scholar]
- Trolle T, McMurtrey CP, Sidney J, Bardet W, Osborn SC, Kaever T. The Length Distribution of Class I-Restricted T Cell Epitopes Is Determined by Both Peptide Supply and MHC Allele-Specific Binding Preference. J Immunol 2016; 196:1480-7. doi: 10.4049/jimmunol.1501721 [Crossref] [ Google Scholar]
- Tian Y, Grifoni A, Sette A, Weiskopf D. Human T Cell Response to Dengue Virus Infection. Front Immunol 2019; 10:2125. doi: 10.3389/fimmu.2019.02125 [Crossref] [ Google Scholar]
- Sette A, Moutaftsi M, Moyron-Quiroz J, McCausland MM, Davies DH, Johnston RJ. Selective CD4+ T cell help for antibody responses to a large viral pathogen: deterministic linkage of specificities. Immunity 2008; 28:847-58. doi: 10.1016/j.immuni.2008.04.018 [Crossref] [ Google Scholar]
- Burrows SR, Rossjohn J, McCluskey J. Have we cut ourselves too short in mapping CTL epitopes?. Trends Immunol 2006; 27:11-6. doi: 10.1016/j.it.2005.11.001 [Crossref] [ Google Scholar]
- Samino Y, Lopez D, Guil S, Saveanu L, van Endert PM, Del Val M. A long N-terminal-extended nested set of abundant and antigenic major histocompatibility complex class I natural ligands from HIV envelope protein. J Biol Chem 2006; 281:6358-65. doi: 10.1074/jbc.M512263200 [Crossref] [ Google Scholar]
- Li M, Jiang Y, Gong T, Zhang Z, Sun X. Intranasal Vaccination against HIV-1 with Adenoviral Vector-Based Nanocomplex Using Synthetic TLR-4 Agonist Peptide as Adjuvant. Mol Pharm 2016; 13:885-94. doi: 10.1021/acs.molpharmaceut.5b00802 [Crossref] [ Google Scholar]
- Hajam IA, Dar PA, Shahnawaz I, Jaume JC, Lee JH. Bacterial flagellin-a potent immunomodulatory agent. Exp Mol Med 2017; 49:e373. doi: 10.1038/emm.2017.172 [Crossref] [ Google Scholar]
- Kim E, Erdos G, Huang S, Kenniston TW, Balmert SC, Carey CD. Microneedle array delivered recombinant coronavirus vaccines: Immunogenicity and rapid translational development. EBioMedicine 2020; 55:102743. doi: 10.1016/j.ebiom.2020.102743 [Crossref] [ Google Scholar]
- Hussain S, Johnson CG, Sciurba J, Meng X, Stober VP, Liu C. TLR5 participates in the TLR4 receptor complex and promotes MyD88-dependent signaling in environmental lung injury. Elife 2020; 9:e50458. doi: 10.7554/eLife.50458 [Crossref] [ Google Scholar]
- Versteeg L, Almutairi MM, Hotez PJ, Pollet J. Enlisting the mRNA Vaccine Platform to Combat Parasitic Infections. Vaccines (Basel) 2019; 7:122. doi: 10.3390/vaccines7040122 [Crossref] [ Google Scholar]
- Alexander J, del Guercio MF, Maewal A, Qiao L, Fikes J, Chesnut RW. Linear PADRE T helper epitope and carbohydrate B cell epitope conjugates induce specific high titer IgG antibody responses. J Immunol 2000; 164:1625-33. doi: 10.4049/jimmunol.164.3.1625 [Crossref] [ Google Scholar]
- Dehghani J, Adibkia K, Movafeghi A, Barzegari A, Pourseif MM, Maleki Kakelar H. Stable transformation of Spirulina (Arthrospira) platensis: a promising microalga for production of edible vaccines. Appl Microbiol Biotechnol 2018; 102:9267-78. doi: 10.1007/s00253-018-9296-7 [Crossref] [ Google Scholar]
- Dehghani J, Adibkia K, Movafeghi A, Maleki-Kakelar H, Saeedi N, Omidi Y. Towards a new avenue for producing therapeutic proteins: Microalgae as a tempting green biofactory. Biotechnol Adv 2020; 40:107499. doi: 10.1016/j.biotechadv.2019.107499 [Crossref] [ Google Scholar]
- Zhang C, Maruggi G, Shan H, Li J. Advances in mRNA Vaccines for Infectious Diseases. Front Immunol 2019; 10:594. doi: 10.3389/fimmu.2019.00594 [Crossref] [ Google Scholar]
- Daryabari SS, Fathi M, Mahdavi M, Moaddab Y, Hosseinpour Feizi MA, Shokoohi B. Overexpression of CFL1 in gastric cancer and the effects of its silencing by siRNA with a nanoparticle delivery system in the gastric cancer cell line. J Cell Physiol 2020; 235:6660-6672. doi: 10.1002/jcp.29562 [Crossref] [ Google Scholar]
- Jafari B, Pourseif MM, Barar J, Rafi MA, Omidi Y. Peptide-mediated drug delivery across the blood-brain barrier for targeting brain tumors. Expert Opin Drug Deliv 2019; 16:583-605. doi: 10.1080/17425247.2019.1614911 [Crossref] [ Google Scholar]
- Lundstrom K. Self-Replicating RNA Viruses for RNA Therapeutics. Molecules 2018; 23:3310. doi: 10.3390/molecules23123310 [Crossref] [ Google Scholar]