Bioimpacts. 15:30443.
doi: 10.34172/bi.30443
Original Article
A subspace learning aided matrix factorization for drug repurposing
Amir Mahdi Zhalefar Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing, 
Zahra Narimani Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing, , *
Author information:
Department of Computer Science and Information Technology, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan 45137-66731, Iran
Abstract
Introduction:
Design and development of new drugs needs a huge amount of investment of time and money. The advent of machine learning and computational biology has led to sophisticated techniques for drug repositioning, i.e., recommending available drugs for new diseases or, more specifically, protein targets. However, there remains a critical need for improved synergy between these techniques to enhance their predictive accuracy and practical application in clinical settings.
Methods:
This study presents a novel approach that integrates two methodologies: SLSDR, a sparse and low-redundant subspace learning-based dual-graph regularized robust feature selection technique, and the iDrug method for drug repurposing which integrates different domains. SLSDR is a subspace learning algorithm based on matrix factorization, and iDrug is a matrix factorization-based drug repositioning method that integrates data from two different domains (drug-disease and drug-target domains). By leveraging SLSDR's ability to extract essential features from drug-disease and drug-target spaces, we enhance the iDrug objective function. Our approach includes constructing a drug-drug similarity matrix using a feature space derived from SLSDR, and target-target and disease-disease similarity matrices. This ensures a comprehensive representation of drug-disease and drug-target associations. We introduce a novel objective function that captures the nuanced interactions between drugs and diseases, considering the complex interrelationships among features within all the datasets.
Results:
By integrating these components, our strategy offers a holistic solution for drug repositioning, optimizing the prediction process. In terms of prediction accuracy, AUC, AUPR and computing efficiency, the results indicate notable gains over the state of the art drug repurposing methods. Fig. 1, represents the comparison of the performance of the proposed method with existing approaches across various metrics.
Conclusion:
The proposed matrix factorization based method for drug repurposing, benefits from integrating knowledge from two domains, drug-disease and drug-target domains, and also is capable of preserve the geometry of the data in both feature space, and s ample space. Comparing to existing state of the art methods, this shows accuracy improvement in drug repurposing.
Keywords: Drug repurposing, Subspace learning, Matrix factorization, Feature selection
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This research is conducted in the Bioinformatics lab, CS and IT department, Institute for Advanced Studies in Basic Sciences, Zanjan, Iran. No specific funding is provided for this study.
Introduction
Traditionally, the drug design process has been an exceedingly costly and time-consuming task. However, the emergence of high-throughput technologies and machine learning techniques has significantly made effect on this field, enabling researchers to harness these tools in both drug discovery and drug repurposing (repositioning). The primary objective of drug repurposing is to identify new therapeutic applications for existing drugs, targeting diseases or proteins beyond their originally intended use. This not only extends the utility of known drugs but also deepens our understanding of their mechanisms within the human body. Computational methods are often employed as an initial step before conducting wet-lab experiments, substantially reducing the search space, and thereby minimizing the time and costs associated with drug development. Over the past decade, a wealth of research has focused on drug repurposing, with various computational approaches being explored. This section provides a concise review of these approaches.
Graph-based methods for drug repositioning typically model the relationships between entities such as drugs, targets (proteins), side effects, and diseases in a graph structure. Techniques like random walks, and community detection, are then applied to analyze these graphs.1-3 Another widely used approach is matrix factorization, which decomposes the sparse drug-target matrix to reconstruct missing elements, enabling the extraction of linear or non-linear latent features from the drug space and facilitating drug similarity predictions. For instance, Zhang et al., utilized matrix factorization to detect drug-disease similarities through learning a latent cluster space (over similar drugs and similar diseases) and multiple similarity measures, while Chen et al., refined this approach using different kernels and data sources.4,5 Deep learning has also become increasingly popular for predicting drug-disease relationships. DeepDR, for example, employs a deep learning model to capture non-linear drug features from a heterogeneous network, using a random walk method to represent the network.6 Chen et al introduced a matrix factorization-based method called iDrug, which simultaneously considers drug-disease and drug-target interactions in a unified model, forming the basis for our proposed approach.7
Recent advancements in deep learning have significantly enhanced the field of drug repurposing. The Integrated Deep Drug-Disease Neural Network (IDDI-DNN), and DeepAVP, leverages diverse datasets to accurately predict new therapeutic applications for existing drugs.8,9 Chen et al., introduced an innovative framework that evaluates the therapeutic potential for individual medicine by analyzing their feature space through a combination of Long Short-Term Memory (LSTM) networks and attention mechanisms.10 This approach effectively accounts for confounding factors and disease progression, demonstrating notable success in identifying drugs with promising therapeutic properties.
In parallel, deep learning has been transformative in the discovery of novel antimicrobial agents. For instance, Halicin, identified via the ZINC database, emerged as a potent antibiotic capable of combating resistant bacterial strains.11 Furthermore, Timmons and Hewage developed ENNAVIA, an advanced deep learning model combined with chemoinformatics, designed to identify peptides with low toxicity and high biological activity. This innovative method holds significant potential in the development of antiviral drugs.12
In the current study, we aim to enhance the iDrug model by incorporating matrices derived from a subspace learning technique known as SLSDR into its objective function. The following sections provide an overview of the iDrug and SLSDR methods, followed by a detailed discussion of the objective function and optimization process. Our results demonstrate significant improvements over existing methods in drug repurposing tasks. The general idea of the proposed methods is introduced in the following.
The iDrug model is a matrix-factorization based methods for predicting drug-target interactions and therapeutic repositioning. Drug-disease networks and drug-target networks are two interrelated domains that it makes use of. Effective knowledge transfer across domains is made possible by the iDrug model, which contains partially shared drug nodes and anchor linkages that connect these networks. This skill aids in more precise identification of the molecular targets of current medications and promotes the development of new therapeutic uses for them.
Integrating SLSDR technique into the iDrug and including both within-network and cross-network connections, the final model considers drug repositioning and target prediction as a cross-network embedding problem and also considers both data and feature manifolds preservation in the process of feature selection.13
This paper's structure is set as follows:
We describe the iDrug technique, the objective function and thorough presentation of its iterative updating rules. In the next section the SLSDR approach is introduced, including its iterative updating rules. Finally, the combination of the SLSDR method with the iDrug method, leading to the development of our new methodology. The new objective function and update rules are derived. Finally, the last section displays the experimental results, displaying the efficacy and robustness of our suggested method through rigorous evaluations and comparison analysis (Fig. 1).
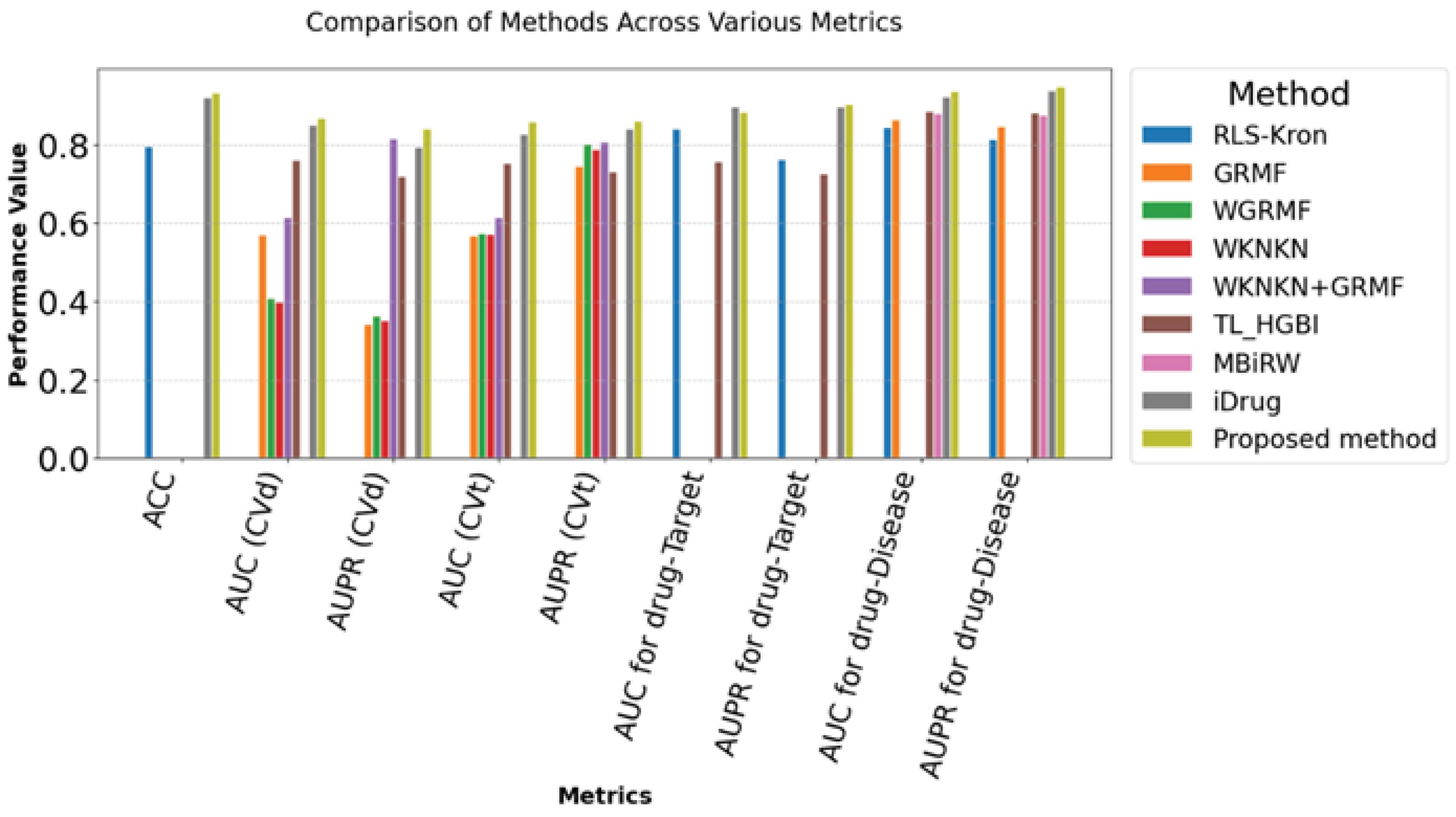
Fig. 1.
Different evaluation criteria reported on CTD dataset, comparing our method (the proposed method), iDrug, MBiRW, TL_HGBI, WKNKN + GRMF, WKNKN, WGRMF, GRMF, RLS_Kron.
.
Different evaluation criteria reported on CTD dataset, comparing our method (the proposed method), iDrug, MBiRW, TL_HGBI, WKNKN + GRMF, WKNKN, WGRMF, GRMF, RLS_Kron.
iDrug
In this section we explain the cost iDrug cost function, which is proposed to find the best factorization leading to estimating new drug-target relationships. The proposed objective function by Chen et al. in iDrug consists of within network and cross-network based components. In the following these terms are explained.
Within-network factorization
Within-network factorization focuses on solving single-domain challenges, such as drug-disease prediction, by leveraging graph-regularized non-negative matrix factorization.13-15 This technique decomposes the drug-disease interaction matrix X(1) ϵ ℝ(n1× m1) into two latent feature matrices: U(1) ϵ ℝ(n1× r1), which encapsulates the feature space of drugs, and V(1) ∈ ℝ(m1× r1), which represents the feature space of diseases. The term α1 Tr(U(1)TLu(1)TU(1)), is used in order to guarantee that the similarity of the features in feature space is preserved in the new feature space. The decomposition process is governed by minimizing the following objective function:
Eq. (1)
Cross-network consistency
The iDrug framework offers an advanced approach to understanding cross-network relationships by hypothesizing that drugs appearing in multiple domains represent identical entities and must exhibit consistent properties. To implement this concept, a drug mapping matrix, denoted as S(1,2)ϵ ℝ(n2× n1), is introduced. This matrix encodes the anchor links bridging domains D1 and D2 . Specifically, S(1,2)(i,j) = 1 if the i-th row of U(2) corresponds to the j-th row of U(1), signifying that they represent the same drug; otherwise, S(1,2)(i,j) = 0.
To maintain the integrity of these anchor links, a one-to-one mapping constraint is enforced. This constraint ensures that each row of S(1,2) contains no more than one non-zero element, thus guaranteeing that a drug in one domain maps to at most one corresponding drug in the other domain. This formulation not only strengthens the theoretical underpinnings of the iDrug framework but also enhances its ability to accurately model and predict cross-domain drug relationships.
Leveraging S(1,2) in conjunction with U(1), the shared drug feature space from domain D1 is seamlessly projected onto domain D2. Moreover, the model ensures that if two drugs exhibit similarity (correlation) within domain D1, this similarity is retained upon projection to domain D2. To guarantee consistency across networks, the following discrepancy measure is minimized14:
Eq. (2)
Cross-domain integration in iDrug
The iDrug framework unifies the objectives derived from domain-specific internal networks, including drug-target interactions and drug-disease associations represented in (Eq. 1), with the cross-network alignment strategy outlined in (Eq. 2). This synthesis results in a cohesive optimization function, encapsulating both aspects into a single formulation, as demonstrated below:
Eq. (3)
The symbols used in the objective function (Eq. 3) are defined in Table 1, which provides a detailed description of each matrix and parameter involved, such as the data matrices X(i), i ∈ {1,2}, the low-dimensional representations U(1), V(1), U(2), V(2), the mapping matrix S(1,2) and the, etc details.
Table 1.
The symbols used in the objective function (
Eq. 3) and their descriptions
|
Symbol
|
Definition and Description
|
| X(1), W(1) |
Matrices representing the structural information and interaction weights within the drug-disease network. |
| X(2), W(2) |
Matrices capturing the structural information and interaction weights within the drug-target network. |
| U(1), V(1) |
Low-dimensional representations of drugs and diseases derived from the drug-disease interaction network. |
| U(2), V(2) |
Low-dimensional representations of drugs and targets derived from the drug-target interaction network. |
| S(1,2) |
Mapping matrix that establishes correspondences between the drug-disease and drug-target domains, indicating cross-domain alignments. |
| Au(1), Du(1) |
Drug-drug similarity matrix and its corresponding degree matrix within the drug-disease network. |
| Au(2), Du(2) |
Drug-drug similarity matrix and its corresponding degree matrix within the drug-target network. |
| Av(1), Dv(1) |
Similarity matrix and degree matrix for diseases in the drug-disease network. |
| Av(2), Dv(2) |
Similarity matrix and degree matrix for targets in the drug-target network. |
| n1,m1 |
Total number of drugs and diseases analyzed within the drug-disease network. |
| N2,m2 |
Total number of drugs and targets analyzed within the drug-target network. |
| r1,r1 |
Ranks of the matrices {U(1), V(1)} and {U(2), V(2)}, representing the dimensions of their latent feature spaces. |
The first summation term,
represents the "domain-specific factorization" of the two data matrices X(i), i ∈ {1,2}, corresponds to drug-target and drug-disease domains, respectively. Here, U(i) and V(i) are the low-rank matrices that their product approximates the original data,X(i). For example for i = 1, X(i) represents the drug-disease interaction matrix, which the goal is to decompose it to U(1) and V(1). U(1) represents drugs in latent space and V(1) represent diseases in latent space (the dimension of this latent space is optimized). The operator ∘ denotes the Hadamard (element-wise) product, and W(i) is a weight matrix to emphasize the significance of certain interactions in the data.
The second term,
is responsible for ensuring "cross-network consistency". The parameter β controls the trade-off between factorizing individual networks and ensuring consistency between them. The idea is that similar (correlated) drugs in the disease space, should also be correlated in the target space. Therefore, UUT in the disease space, should be similar to the UUT in the target space. S(1,2) is a selector matrix that identifies the common drugs between disease and target space. As a conclusion, this term encourages the feature representations of drugs in the drug-target network to align with those in the drug-disease network, reinforcing consistency across domains.
The third term,
promotes within network smoothness. Here, Au(i) and Av(i) are adjacency matrices for the drug and target (or disease) networks, while Du(i) and Dv(i) are their corresponding degree matrices. This term encourages similar nodes (e.g., drugs or targets) in the network to have similar feature representations, effectively smoothing the learned representations. The parameter α controls the strength of this smoothness constraint.
The final term,
is a regularization term enforcing sparsity in the learned matrices U(i) and V(i). The L1-norm (‖⋅‖1) encourages many entries in these matrices to be zero, leading to simpler and more interpretable representations. The regularization parameter γ controls the sparsity level, preventing overfitting by ensuring that only the most significant features are captured in the model.
The regularization parameters α, β, and γ allow us to control the relative importance of smoothness, consistency, and sparsity in the model. These parameters can be tuned to achieve the optimal balance for the given data.
The objective function presented in (Eq. 3) is inherently non-convex when all variables are considered simultaneously. To address this challenge, the authors adopt a multiplicative update minimization strategy, as detailed in.16 This approach alternates the minimization process by optimizing one variable at a time while keeping the others fixed. The procedure is repeated iteratively until convergence is achieved, defined as ‖J(t + 1) - J(t)‖ ≤ δ, where δ represents a small predefined constant. Further details regarding the optimization process can be found in the original iDrug paper.7
The objective function described in equation (3) is fundamentally non-convex when all variables are considered simultaneously. To address this complexity, the authors utilize a specialized optimization framework grounded in the multiplicative update minimization technique, as elaborated.16 This method strategically alternates the minimization process by optimizing one variable at a time while holding the remaining variables constant. By leveraging specialized computational tools and techniques, this approach ensures both precision and computational efficiency in navigating the challenging optimization landscape.
SLSDR
The SLSDR method is a subspace learning-based graph regularized feature selection framework that is integrated in this research with iDrug model for drug discovery. This method enhances the iDrug model by considering both the feature and data manifolds, ensuring sparsity and low redundancy in feature selection, and maintaining robustness to outlier samples. As explained in the previous section, iDrug is a matrix factorization method designed to preserve cross-domain consistency between drug-disease and drug-target domains. SLSDR, is a feature selection methods that reconstruct the original matrix by estimating it using only a subset of important features. In addition to preserving the structure of features (similar to iDrug), SLSDR, preserves the topological structure underlying data samples.
Subspace learning has emerged as a powerful technique for effectively reducing data dimensionality and deriving low-dimensional representations from high-dimensional spaces. Utilizing matrix decomposition methodologies, subspace learning broadens its applications from merely feature extraction to the realm of feature selection. In groundbreaking research, Wang et al., introduced an advanced method for unsupervised feature selection based on matrix factorization, known as Matrix Factorization for Feature Selection (MFFS).17,18 This approach reframes unsupervised feature selection as a problem of matrix decomposition. Subsequently, Wang et al. proposed the Maximum Projection and Minimum Redundancy (MPMR) framework, which quantifies the relevance of selected feature subsets by analyzing the entire feature set and incorporates a redundancy minimization term to ensure minimal overlap among selected features.17
Moreover, Shang et al., made significant advancements with their Subspace Learning-Based Graph Regularized Feature Selection (SGFS), which constructs a feature graph to preserve the intrinsic geometric structure of the feature manifold.19 Despite the effectiveness of these algorithms in feature selection, certain limitations remain. Specifically, MFFS and MPMR do not consider the local geometric characteristics of both data and feature manifolds. On the other hand, SGFS, while accounting for the geometric structure of the feature manifold, neglects the structural properties of the data manifold. To address these shortcomings, the proposed Subspace Learning for Simultaneous Dimensionality Reduction (SLSDR) framework integrates the local geometric information of both data and feature manifolds, thereby achieving superior performance in feature selection tasks.
The SLSDR framework is composed of three principal components: sparse and low-redundancy subspacℝℝe learning, manifold structure preservation, and feature evaluation. Formally, let X = [x1,x2,…,xn] ϵ ℝ(m × n) denote the data matrix, where m represents the number of features per sample, and n is the total number of samples in the dataset. Each column xi = ℝm corresponds to the ith sample within X.
The similarity between features, in the feature manifold, and similarity between samples, in the sample manifold, is computed in SLSDR. The final objective function of SLSDR is provided in (Eq. 4)
Eq. (4)
in which, S represents the feature selection matrix, which assigns significance to individual features. The matrix V contains the reconstruction coefficients, while L denotes the graph Laplacian matrix corresponding to the feature manifold. Additionally, Ω(S) refers to the inner product regularization term, whose details are elaborated in the subsequent subsection discussing the update rules for SLSDR. It's shown in the SLSDR paper that this objective function is able to select a subset of features while preserving distances between instances of data in the sample manifold and distances between features in the feature manifold.20 The term α1 Tr(VLVVT), is used in order to guarantee that the similarity of the features in feature space is preserved in the new feature space. This term is used commonly in NMF techniques. Similarly, the term Tr(STX LSXTS) is used for ensuring that the similarity between samples in sample manifold is preserved in the transformed data.
More details about the objective function and objective function in provided in Shang et al.20
Update rules for SLSDR
This section provides a comprehensive overview of the Sparse Linear Square Dimension Reduction (SLSDR) algorithm; we have to mention that the original update rules are used in our research also. The SLSDR approach addresses non-convex optimization problems by utilizing an alternating iterative update method to optimize the objective function, which is particularly designed for feature selection and dimensionality reduction.
Objective function and update rules
The objective function of SLSDR is defined as:
Eq. (5)
where S and V are the matrices to be optimized, and α, β, and λ are balancing parameters.
Updating S
Given fixed U and V, the update rule for S can be derived by setting the gradient of with respect to S to zero, which results in:
Eq. (6)
Eq. (7)
Updating V
With S and U held fixed, the update rule for V is obtained in a similar fashion:
Eq. (9)
SLSDR Algorithm Workflow
The detailed workflow of the SLSDR algorithm is outlined below:
-
Develop the k-nearest neighbor graphs, denoted as G0 and G1, to effectively capture the intrinsic structures of the feature space and the data space, respectively.
-
Calculate the similarity matrices Wv and WS for representing pairwise relationships and derive their corresponding graph Laplacian matrices, LV and LS, to encode the geometric and topological properties of the data.
-
Initialize the matrices U, S, and V as initial estimations to facilitate the iterative optimization process.
-
Iteratively update the matrices U, S, and V by applying the designated optimization rules until either convergence criteria are satisfied or the maximum number of iterations is reached.
-
Quantify the significance of each feature by analyzing their respective contributions, rank them accordingly, and select the top l features to construct the refined data representation, Xnew.
Materials and Methods
As discussed, iDrug unifies drug-target and drug-disease domains by introducing a matrix factorizing technique for drug-target and drug-disease prediction. A feature selection method called SLSD preserves the geometric structure of the feature manifolds as well as the data. Here we offer a subspace learning method based on SLSDR, meaning that it retains the feature and data manifolds, and also uses the domain integration technique introduced in iDrug; i.e., the loss function of SLSDR is applied to the two drug-target and drug-disease domains.
Our objective is to enhance and refine the iDrug objective function that is expressed in Equation (Eq. 3), by transitioning from the conventional drug and target feature spaces to the broader feature space derived from the SLSDR objective function. In order to define our loss function, first we consider a part of loss function inspired from the SLSDR loss function; we name this Termslsdr (Eq. 10).
In order to integrate the iDrug domain integration, we need to adopt the objective function in iDrug to be considered along with Termslsdr with Termslsdr as as the final objective function. In order to do this adaptation, instead of using the traditional decomposition of X(i) into U(i) and V(i) matrices (i.e., X(i) ≈U(i)V(i)T), we adopt the SLSDR approach and decompose X(i) into X(i)S(i)V(i), where S(i) is a sparse subspace learning matrix that captures low-redundant information from the data. This method leverages both sparse representation and graph-based regularization to enhance feature selection and cross-domain consistency. With this approach instead of "extracting latent feature space" on drug in disease and target domains, the algorithm will "select a subset of features" in disease and target space, which can be representative of drugs such that the geometry of data manifold and feature manifold is preserved (in both domains).
For SLSDR integration, the term TermSLSDR is:
Eq. (10)
in which h is an index which refers to domains; h = 1 represent drug-target domain and h = 2represent drug disease domain (it has similar purpose to using i in iDrug objective function in equation 3). Finally, within-network smoothness is incorporated through term1 - Eq. 11 (this is achieved by replacing Uh in iDrug with X(h)T S(h) in the within-network smoothness term in Eq. 3):
Eq. (11)
term1 ensures that the similarity between samples is preserved in the new feature space. term2 is the Laplacian matrix for (X(h)T S(h)). As explained before, this term is common in NMF techniques.
Eq. (12)
We unify the domain-specific objective function across drug-target and drug-disease networks by incorporating the SLSDR components into the iDrug framework. The unified objective can be expressed as:
Eq. (13)
Update rules for the proposed method
To enhance iDrug's performance by incorporating SLSDR features into the objective function, we must adapt the parameter updates accordingly. This entails aligning the modifications with the intended alterations brought about by SLSDR for both h = 1 and h = 2. By carefully tweaking the update mechanism, we ensure compatibility with the newly introduced features, thereby boosting the system's overall effectiveness.
Update rules for Sij(h)
A Degree (diagonal) matrix Q(h) ∈ ℝ(n(h) × n(h)) is first introduced, and its ith element is defined as follows:
Where E(h) = X(h)T - X(h)TS(h)V(h), and ei(h) is the i th row of the matrix E(h). To avoid overflow, a small constant ε is introduced into (Eq. 14), and the obtained formula is as follows:
Eq. (15)
Eq. (16)
Eq. (17)
Update rules for Vij(h)
Eq. (18)
Eq. (19)
Eq. (20)
The workflow of the proposed method is summarized in Table 2, and also with more detail (pseudocode) in Supplementary file 1.
Table 2.
Elaborated steps of the proposed methodology
|
Step
|
Description
|
| 0 |
Input: Input matrices X(h) ∈ ℝ(m(h) × n(h)) for h = 1,2; neighborhood size parameters K(h) for h = 1,2; weighting factors α(h), β(h), and λ(h) for h = 1,2; maximum permissible iterations NIter; Gaussian scale parameters σ(h) for h = 1,2; and the desired number of features l(h) for h = 1,2. |
|
Output: Selected feature indices Index for h = 1,2 along with processed data matrices optimized for iDrug model implemkentation. |
| 1 |
Formulate K-nearest neighbor graphs G0(h) = (V0(h), E0(h)) and G1(h) = (V1(h), E1(h)) for h = 1,2, representing feature and data manifolds, respectively. |
| 2 |
Derive similarity matrices Wv(h) and WS(h) along with graph Laplacian matrices LV(h) and LS(h) for h = 1,2. |
| 3 |
Initialize matrices Q(h), S(h), and V(h) for h = 1,2. |
| 4 |
Iteratively refine Q(h), S(h), and V(h) for h = 1,2, using the specified update rules until reaching the maximum iteration threshold NIter. |
| 5 |
Evaluate the relevance of each feature i for h = 1,2 by computing ‖ Si(h) ‖2. Rank features in descending order of importance, select the top l(h) features, and identify their indices Index. Construct refined data matrices Xnew(h) ∈ ℝ(l(h) × n(h)) for h = 1,2. |
Results
Dataset
This study presents an in-depth assessment of a range of computational methodologies applied to a meticulously curated dataset originally compiled by Gottlieb et al. This dataset, extensively referenced in prior research, comprises 1,933 confirmed drug-disease associations, encompassing 593 drugs and 313 diseases.2,21,22
To enhance the utility of this dataset, Chen et al. expanded it by incorporating 1,011 known molecular targets associated with the 593 drugs, sourced from the DrugBank database, resulting in 3,427 documented drug-target interactions, and the final dataset is used for evaluation.7 The performance of various predictive models was systematically evaluated for both drug-disease and drug-target interaction prediction tasks under a "pair prediction" paradigm.
Comparison and Parameter tuning
A range of state-of-the-art computational approaches were employed to predict drug-target and drug-disease interactions, leveraging techniques such as kernel-based classifiers, matrix factorization, and random-walk algorithms. These methods are already also used by iDrug paper for comparison purposes and the parameter selection is similar to what reported in iDrug comparisons for preserving consistency. The source code and data is available at: https://github.com/amirmahdizhalefar/matrix-factorization-for-drug-repurposing.
-
RLS-Kron: This approach combines chemical and genomic similarity matrices to improve predictions of drug-target interactions. In our implementation, the regularization parameter was set to σ = 1, while the kernel bandwidth was fixed at γ = 1.23
-
TL_HGBI: The methodology employs a refined random-walk algorithm specifically adapted to operate on a tri-layer network encompassing drugs, their molecular targets, and associated diseases.24 This algorithm is structured to identify novel, previously uncharacterized interactions within the drug-disease and drug-target interaction spaces. All threshold parameters were meticulously fine-tuned to their most effective values, as thoroughly detailed in the original foundational research.
-
MBiRW: This bi-random walk algorithm works on bipartite networks and integrates clustering information for drug-disease associations. Parameter initialization was conducted based on the guidelines provided in the original publication.2
-
GRMF (Graph Regularized Matrix Factorization): This method incorporates graph regularization to derive low-rank representations of drugs and targets. Regularization parameters were fine-tuned using grid search, resulting in λ1 = 0.5 and λd = λt = 10-3.22
-
iDrug: our proposed method was configured with rank parameters r1 = 90 and r2 = 70, a weight factor w =0.3, and regularization parameters α = β = λ = 0.01. A sensitivity analysis was conducted to evaluate the effects of these regularization parameters on performance.7
Cross-validation scenarios
In addition to two main test scenarios for drug-disease and drug-target interaction prediction, we also considered two cross-validation scenarios as following:
CVd (Cross-validation on drug profiles): This evaluation scenario omits entire drug interaction profiles throughout the training phase, reserving them exclusively for testing. It tests the model's potential to predict interactions for wholly new medications absent in the training data. Performance under CVd is tested using metrics such as AUC and AUPR, reflecting the model's efficacy in generalizing to unknown medicines.
CVt (Cross-validation on target profiles): In this situation, whole target interaction profiles are omitted from the training dataset and utilized simply for testing. This configuration examines the model's ability to foresee interactions with novel targets. Typically, models obtain superior performance under CVt since the sequence similarity of targets often provides more predictive power compared to the chemical similarity of medications.
The experimental outcomes, as summarized in Table 3, shows that the proposed strategy consistently outperforms all alternative methods across the all scenarios. In drug-disease prediction, the proposed framework achieved an AUROC of 0.936 and an AUPR of 0.947 in drug-disease prediction tasks. In contrast, iDrug attained an AUROC of 0.9213 and an AUPR of 0.938. Other methods TL_HGBI (AUROC: 0.886, AUPR: 0.881), MBiRW (AUROC: 0.879, AUPR: 0.876), GRMF (AUROC: 0.863, AUPR: 0.847), and RLS-Kron (AUROC: 0.844, AUPR: 0.813).
Table 3.
Performance comparison of drug-target interaction prediction methods under different scenarios
|
Method
|
ACC
|
AUC (CVd)
|
AUPR (CVd)
|
AUC (CVt)
|
AUPR (CVt)
|
AUC for drug Target prediction
|
AUPR for drug Target prediction
|
AUC for drug Disease prediction
|
AUPR for drug Disease prediction
|
| RLS-Kron |
0.796 |
- |
- |
- |
- |
0.841 |
0.763 |
0.844 |
0.813 |
| GRMF |
- |
0.569 |
0.341 |
0.567 |
0.745 |
- |
- |
0.863 |
0.847 |
| WGRMF |
- |
0.408 |
0.364 |
0.574 |
0.801 |
- |
- |
- |
- |
| WKNKN |
- |
0.399 |
0.352 |
0.572 |
0.787 |
- |
- |
- |
- |
| WKNKN + GRMF |
- |
0.615 |
0.815 |
0.615 |
0.807 |
- |
- |
- |
- |
| TL_HGBI |
- |
0.761 |
0.720 |
0.753 |
0.732 |
0.757 |
0.726 |
0.886 |
0.881 |
| MBiRW |
- |
- |
- |
- |
- |
- |
- |
0.879 |
0.876 |
| iDrug |
0.921 |
0.851 |
0.793 |
0.826 |
0.841 |
0.897 |
0.897 |
0.9213 |
0.938 |
| Proposed method |
0.932 |
0.867 |
0.841 |
0.857 |
0.859 |
0.884 |
0.902 |
0.936 |
0.947 |
The proposed method regularly outperforms existing approaches in both CVd and CVt evaluations. It exhibits near-perfect performance across numerous parameters, including AUC, AUPR, and F1 scores. This highlights the durability and adaptability of the technique in successfully anticipating drug-target interactions, even when facing previously encountered medicines or targets.
Several key insights can be drawn from these results:
The proposed method, iDrug and TL_HGBI, which incorporate drug-target interactions, significantly outperform other models. This highlights the importance of leveraging target information for drug-disease prediction. Consistent with prior studies, removing drug-target links from the network degrades performance.4,7
Unlike TL_HGBI, which suffers from data sparsity, our method addresses the cold-start issue by jointly learning from both drug-disease and drug-target networks. This allows our method to perform better, particularly in cases involving new drugs or diseases. Larger networks provide richer information, mitigating sparsity issues.
While each of three MBiRW, iDrug, and our method employ drug community/cluster concepts, our method and iDrug applies consistency constraints across domains, yielding more reliable drug communities. MBiRW’s reliance on known drug-disease associations may introduce bias, whereas our method and iDrug benefit from cross-domain knowledge transfer.
The higher AUPR score of our method compared to GRMF is likely due to our method’s incorporation of multi-domain knowledge. GRMF, while effective for single-domain predictions, lacks the cross-domain learning capabilities of our method, resulting in lower prediction accuracy.
Kron, which relies on kernel-based methods, demonstrated the lowest performance. The selection of an appropriate kernel function is challenging and often requires domain-specific expertise, limiting the model's flexibility.
In conclusion, the proposed method achieves significant improvements by aided matrix factorization for drug repurposing and integrating multi-domain knowledge, overcoming the challenges of data sparsity and cold-start problems, and providing a robust framework for drug-disease prediction.
Discussion
Development of new drugs is a highly costly and time-consuming process. One of strategies of drug development companies is to find possible protein targets for already developed drugs, and trying to control diseases other than known target diseases with an available drug. There are different computational approaches to address this problem, such as deep and non-deep machine learning methods. One of the approaches that is common for solving such problems, is matrix factorization. Matrix factorization methods decompose a matrix into factors, leading to discovery of latent features of the data matrix. These latent features are useful in order to identify a feature space in which similarity of drugs and targets is better understood, while in the primary feature space which is sparse it’s not possible. Different matrix factorization based methods have been suggested in existing literature. In this study, we proposed a sparse subspace learning, which is based on matrix factorization. As a result, drugs can be represented in target space. The base subspace learning method we use is SLSDR, a matrix factorization based subspace learning which preserves the data and feature manifold geometric properties. It means that drugs which are similar in the original space, will remain similar in the new feature space. We modified the objective function of SLSDR, so that it considers the subspace learning with respect to two different domains, the drug-disease and drug-target. As a result, subspace learning considers the drug space in both the disease and target spaces; the loss function is defined so that the correlation of disease with respect to diseases and also with respect to drugs is preserved in the new subspace. The results show that the proposed method superiors other state of the art matrix factorization based methods in drug repositioning problem.
Conclusion
Prediction of interaction between existing drugs and potential new targets is a major area of research in drug development. Different machine learning methods are hired by researchers to address this problem. Matrix factorization is a mathematical framework which has been widely used for extracting hidden patterns from sparse datasets. While methods such as deep learning based ones, need very large datasets to extract patterns, matrix factorization based methods do not suffer from this limitation. Patterns inferred using matrix factorization methods are interpretable, and helps to understand the underlying mechanism of the observed pattern. One of the advantages of matrix factorization methods is that different domain knowledge can be integrated to their loss function. At the same time, a finely defined objective function can integrate other objectives such as what used in this paper, preserving the geometry of the data and features in the latent space. This research, confirms these statements, by defining an objective function for integrating different knowledge domains and also paying attention to preserving the data/feature geometry in the latent space. Our results, confirmed that the new objective function which benefits from these criteria, outperforms state of the art matrix factorization methods for drug repurposing. In addition, matrix factorization methods can be combined with other machine learning methods, such as deep learning, and therefore benefit from both computational power and interpretability, and this can be considered in future work.
Research Highlights
What is the current knowledge?
-
In computational drug repurposing, matrix factorization is an established technique for identifying latent drug-target associations. However, existing methodologies often do not fully integrate diverse domain knowledge, such as drug-disease relationships, nor do they consistently preserve the inherent topological structures within drug and disease spaces. This can sometimes limit the biological interpretability and predictive power of their outputs, potentially affecting the discovery of meaningful drug repurposing candidates.
What is new here?
-
We propose a framework that seeks to enhance matrix factorization for drug repurposing by addressing these considerations. Our framework aims to provide a more comprehensive view of drug action by jointly integrating drug-target and drug-disease relational spaces. The method incorporates a feature selection strategy designed to include domain-specific knowledge and maintain the topological structure of both feature and data spaces. This approach is intended to yield decompositions that are both robust and more interpretable. Experimental evaluations suggest that this approach shows improved performance compared to some existing matrix factorization models. Furthermore, its biological relevance is suggested through alignment with known biological pathways and examples of drug repositioning for conditions such as Alzheimer's and various cancers. We believe this framework offers a promising avenue to support drug discovery efforts.
Competing Interests
The authors declare no competing interests.
Ethical Approval
Not applicable.
Supplementary files
Supplementary file 1. Detailed pseudocode implementation of the proposed method.
(pdf)
Acknowledgements
We would like to thank Dr. Saeed Karami Zarandi, Assistant Professor in Applied Mathematics, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan, Iran, for his precious comments on our method. We would like to acknowledge anonymous reviewers for their precious comments on our manuscript.
References
- Chen H, Zhang H, Zhang Z, Cao Y, Tang W. Network-based inference methods for drug repositioning. Comput Math Methods Med 2015; 2015:130620. doi: 10.1155/2015/130620 [Crossref] [ Google Scholar]
- Luo H, Wang J, Li M, Luo J, Peng X, Wu FX. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics 2016; 32:2664-71. doi: 10.1093/bioinformatics/btw228 [Crossref] [ Google Scholar]
- Wu C, Gudivada RC, Aronow BJ, Jegga AG. Computational drug repositioning through heterogeneous network clustering. BMC Syst Biol 2013; 7 Suppl 5:S6. doi: 10.1186/1752-0509-7-s5-s6 [Crossref] [ Google Scholar]
- Zhang P, Wang F, Hu J. Towards drug repositioning: a unified computational framework for integrating multiple aspects of drug similarity and disease similarity. AMIA Annu Symp Proc 2014; 2014:1258-67. [ Google Scholar]
- Chen H, Li J. A flexible and robust multi-source learning algorithm for drug repositioning. In: Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics. Association for Computing Machinery. 2017. p. 510-15. doi: 10.1145/3107411.3107473.
- Zeng X, Zhu S, Liu X, Zhou Y, Nussinov R, Cheng F. deepDR: a network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019; 35:5191-8. doi: 10.1093/bioinformatics/btz418 [Crossref] [ Google Scholar]
- Chen H, Cheng F, Li J. iDrug: integration of drug repositioning and drug-target prediction via cross-network embedding. PLoS Comput Biol 2020; 16:e1008040. doi: 10.1371/journal.pcbi.1008040 [Crossref] [ Google Scholar]
- Amiri R, Razmara J, Parvizpour S, Izadkhah H. A novel efficient drug repurposing framework through drug-disease association data integration using convolutional neural networks. BMC Bioinformatics 2023; 24:442. doi: 10.1186/s12859-023-05572-x [Crossref] [ Google Scholar]
- Li J, Pu Y, Tang J, Zou Q, Guo F. DeepAVP: a dual-channel deep neural network for identifying variable-length antiviral peptides. IEEE J Biomed Health Inform 2020; 24:3012-9. doi: 10.1109/jbhi.2020.2977091 [Crossref] [ Google Scholar]
- Chen Z, Liu X, Hogan W, Shenkman E, Bian J. Applications of artificial intelligence in drug development using real-world data. Drug Discov Today 2021; 26:1256-64. doi: 10.1016/j.drudis.2020.12.013 [Crossref] [ Google Scholar]
- Jukič M, Bren U. Machine learning in antibacterial drug design. Front Pharmacol 2022; 13:864412. doi: 10.3389/fphar.2022.864412 [Crossref] [ Google Scholar]
- Timmons PB, Hewage CM. ENNAVIA is a novel method which employs neural networks for antiviral and anti-coronavirus activity prediction for therapeutic peptides. Brief Bioinform 2021; 22:bbab258. doi: 10.1093/bib/bbab258 [Crossref] [ Google Scholar]
- Chen C, Tong H, Xie L, Ying L, He Q. FASCINATE: fast cross-layer dependency inference on multi-layered networks. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. Association for Computing Machinery. 2016. p. 765-74. doi: 10.1145/2939672.2939784.
- Cheng W, Zhang X, Guo Z, Wu Y, Sullivan PF, Wang W. Flexible and robust co-regularized multi-domain graph clustering. In: Proceedings of the 19th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. Association for Computing Machinery. 2013. p. 320-8. doi: 10.1145/2487575.2487582.
- Pan R, Zhou Y, Cao B, Liu NN, Lukose R, Scholz M, et al. One-class collaborative filtering. In: 2008 Eighth IEEE International Conference on Data Mining. Pisa, Italy: IEEE. 2008. p. 502-11. doi: 10.1109/icdm.2008.16.
- Lee D, Seung HS. Algorithms for non-negative matrix factorization. In: Proceedings of the 14th International Conference on Neural Information Processing Systems. Denver, CO: MIT Press. 2000. p. 535-41. doi: 10.5555/3008751.3008829.
- Wang S, Pedrycz W, Zhu Q, Zhu W. Unsupervised feature selection via maximum projection and minimum redundancy. Knowl Based Syst 2015; 75:19-29. doi: 10.1016/j.knosys.2014.11.008 [Crossref] [ Google Scholar]
- Wang S, Pedrycz W, Zhu Q, Zhu W. Subspace learning for unsupervised feature selection via matrix factorization. Pattern Recognit 2015; 48:10-9. doi: 10.1016/j.patcog.2014.08.004 [Crossref] [ Google Scholar]
- Shang R, Wang W, Stolkin R, Jiao L. Subspace learning-based graph regularized feature selection. Knowl Based Syst 2016; 112:152-65. doi: 10.1016/j.knosys.2016.09.006 [Crossref] [ Google Scholar]
- Shang R, Xu K, Shang F, Jiao L. Sparse and low-redundant subspace learning-based dual-graph regularized robust feature selection. Knowl Based Syst 2020; 187:104830. doi: 10.1016/j.knosys.2019.07.001 [Crossref] [ Google Scholar]
- Gottlieb A, Stein GY, Ruppin E, Sharan R. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol Syst Biol 2011; 7:496. doi: 10.1038/msb.2011.26 [Crossref] [ Google Scholar]
- Ezzat A, Zhao P, Wu M, Li XL, Kwoh CK. Drug-target interaction prediction with graph regularized matrix factorization. IEEE/ACM Trans Comput Biol Bioinform 2017. 14: 646-56. doi: 10.1109/tcbb.2016.2530062.
- van Laarhoven T, Nabuurs SB, Marchiori E. Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 2011; 27:3036-43. doi: 10.1093/bioinformatics/btr500 [Crossref] [ Google Scholar]
- Wang W, Yang S, Zhang X, Li J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 2014; 30:2923-30. doi: 10.1093/bioinformatics/btu403 [Crossref] [ Google Scholar]