Bioimpacts. 15:30815.
doi: 10.34172/bi.30815
Original Article
Marine fungal metabolites as antiviral agents: Computer-aided drug screening for selective inhibition of African swine fever virus dUTPase
Mark Andrian B. Macalalad Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing, 1, 2 
Fredmoore L. Orosco Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review & editing, 1, 3, 4, *
Author information:
1Virology and Vaccine Research Program, Department of Science and Technology - Industrial Technology Development Institute, Taguig, Metro Manila, 1631, Philippines
2Career Incentive Program, Department of Science and Technology – Science Education Institute, Taguig, Metro Manila, 1631, Philippines
3S&T Fellows Program, Department of Science and Technology, Taguig, Metro Manila, 1631, Philippines
4The UPLB Graduate School, University of the Philippines Los Baños, Los Baños, Laguna, 4031, Philippines
Abstract
Introduction:
African swine fever (ASF) continues to be a significant threat to the global livestock industry due to its severe impact on pig populations. Currently, there are no approved therapeutic agents for the virus, and biosecurity measures such as culling have led to substantial economic losses. In light of its effects on food security and the economy, our study aims to identify potential antiviral compounds from marine fungal metabolites that target the dUTPase enzyme of the African swine fever virus (ASFV).
Methods:
We screened 4,683 marine fungal metabolites using a series of virtual screening techniques. These included ADMET profiling to assess drug-likeness, consensus molecular docking to predict preferred docking poses and rank the docking scores, 300 ns molecular dynamics (MD) simulations to determine stability, principal component analysis (PCA) to verify simulation convergence, and MMPB(GB)SA analysis to estimate binding affinity.
Results:
Of the 4,683 compounds, 328 passed our ADMET filter, and the 10 highest-scoring ligands from molecular docking were evaluated for stability and binding affinity against both swine and ASFV dUTPase. Among the candidates, tricycloalternarene C (M1421), derived from Alternaria sp., emerged as a promising candidate. It exhibited excellent drug-likeness, stability, and binding affinity comparable to the three control compounds, while showing less affinity towards the swine dUTPase.
Conclusion:
Tricycloalternarene C holds potential as a selective inhibitor of ASFV dUTPase. We recommend further experimental validation to confirm its efficacy as an antiviral agent against African swine fever.
Keywords: ASFV, Docking, Molecular dynamics, Marine fungi, Tricycloalternarene C, In silico
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
The authors are also thankful to the Philippine Council for Agriculture, Aquatic and Natural Resources Research and Development (PCAARRD), which funded the project under the Virology and Vaccine Research and Development Program.
Introduction
African swine fever (ASF) poses a serious global concern to the livestock industry owing to its devastating impact on swine population. The disease is highly contagious, and without effective treatment, it remains highly fatal to pigs. It can lead to the disruption of the entire pork supply chain, threatening food security, and resulting in substantial economic losses.1-3 In 2018, China culled over 1.2 million pigs due to a major ASF outbreak that infected over 150 million pigs and led to economic losses estimated between US$89.5 billion and US$196.2 billion.4,5 As the world's largest pork producer, this outbreak not only harmed China’s economy but also had significant repercussions on global pork markets.6 As of May 2024, there have been 19,172 reported ASF outbreaks across four continents since January 2022.7 Biosecurity measures have been undertaken to prevent and mitigate the ongoing transmission of the disease; however, the lack of a commercially available cure or vaccine remains a major obstacle to achieve complete disease eradication.5,8 Thus, ASF continues to be a significant threat that demands urgent attention.
ASF is caused by African swine fever virus (ASFV), a large enveloped double-stranded DNA virus of the Asfarviridae family. The virus encodes 150–165 structural and non-structural proteins, each playing varying roles in viral entry, attachment, and replication.9 Additionally, ASFV has its own repair proteins to rectify any damage caused by mutagenic reactions. Among these, the ASFV enzyme deoxyuridine 5’-triphosphate nucleotidohydrolase (dUTPase) (Supplementary file 1, Fig. S1) plays an important role in maintaining DNA integrity by preventing the misincorporation of uracil into the ASFV DNA.10,11 The enzyme dUTPase catalyzes the hydrolysis of deoxyuridine triphosphate (dUTP) into deoxyuridine monophosphate (dUMP) and pyrophosphate, thereby reducing the cellular concentration of dUTP. If dUTPase function is disrupted, there is a higher chance that uracil will be mistakenly added to the viral DNA. The ASFV repair proteins then excise the integrated uracil in the DNA, creating an abasic site. Without the conversion of dUTP to dUMP, the dUTP/deoxythymine triphosphate (dTTP) ratio increases, hindering the proper and complete repair of the abasic site with the correct thymine base. The repeated cycle of excision and repair involving newly incorporated uracil triggers numerous DNA strand breaks, ultimately leading to thymine-free cell death.10-12 Given its essential role in maintaining the fidelity of DNA replication and genome integrity, dUTPase is considered a promising target for ASFV antiviral therapies.9
Marine fungal metabolites are a rich source of bioactive compounds known for their diverse biological activities. Of over 4000 identified metabolites, 46% show anticancer activity, 13% are antibacterial, and 14% possess antifungal, antiviral, or pesticide resistance properties.13-15 Notable examples include cephalosporins, the first marine fungal antibiotic, isolated from Acremonium chrysogenum, and gliotoxin, a new type of diketopiperazine antibiotic discovered from Aspergillus sp. Plinabulin, also sourced from Aspergillus sp., is currently in late-stage clinical trials for anticancer therapies.14-16 Furthermore, certain compounds derived from the strains of Aspergillus, Penicillium, Cladosporium, and Fusarium, have demonstrated potent antiviral properties against a range of viruses, such as enterovirus, HSV, HIV, influenza, PRRSV, MCV, and RSV.17 Despite this progress, many marine fungal metabolites remain underexplored. Their pharmacological diversity highlights their potential as novel antiviral inhibitors targeting ASFV dUTPase.
Recent advancements in computational methods have renewed research interest in exploring natural compounds as potential sources for new antiviral drugs.18-20 Through computer-aided drug discovery (CADD) methods, various pharmacological factors such as bioavailability, toxicity, protein-ligand stability, and binding strength can be accurately predicted, enabling researchers to filter out less promising compounds that are likely to fail, early in the process. Furthermore, faster computers and advanced screening techniques have significantly enhanced the efficiency of CADD, reducing the time, cost, and effort associated with the traditional experimental methods.21,22
In this study, we used a range of computer-aided drug screening tools to identify selective inhibitors of ASFV dUTPase from a pool of 4,683 fungal metabolites. The most promising inhibitors were selected based on their predicted safety, efficacy, and strong structural stability with ASFV dUTPase, while ensuring they exhibited less favorable affinity for swine dUTPase. Additionally, we used two different docking software programs and applied consensus docking to validate our results. Principal component analysis (PCA) was employed to verify molecular dynamics simulation convergence. We also analyzed the binding site residues with the most significant energy contribution and compared them to the known ASFV dUTPase active site residues.
Materials and Methods
Protein and ligand preparation
The 3D representations of the ASFV dUTPase (PDB ID: 6LJ3) and swine dUTPase (PDB ID: 6LJJ)11 structures were obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) (https://www.rcsb.org/). Before the molecular docking experiments, the structures were adjusted using Modeller23 to incorporate any missing residues and refine the newly generated loops. Subsequently, a 5000-step relaxation utilizing the steepest descent algorithm was conducted to eliminate any steric clashes and inappropriate geometries.
For the candidate compounds, the Comprehensive Marine Natural Products Database (CMNPD)24 provided the structures of 4,683 marine fungal metabolites in Structure Data Format (SDF). These structures were then optimized using the OPLS 2005 force field in Schrödinger Maestro to attain the configuration with the lowest intramolecular potential energy.
ADMET profiling
The absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of the marine fungal metabolites were assessed using ADMETLab 2.0 (https://admetmesh.scbdd.com/).25 A total of 31 descriptors, each with their own range of accepted values, were used to evaluate the efficacy, safety, and drug-likeness of the metabolites. These descriptors were selected based on their relevance to pharmacokinetic and pharmacodynamic properties in mammals, particularly pigs, to ensure the potential applicability of the compounds as antiviral agents in swine.26-29 These included four descriptors for absorption, three for distribution, five for metabolism, two for excretion, ten for toxicity, and seven drug-likeness and bioavailability scoring tools, including Lipinski's Rule of Five (Ro5) and Quantitative Estimate of Drug-likeness (QED) (Table 1). According to Lipinski's Rule of Five (Ro5), a compound is more likely to have good permeation and absorption when administered orally if it meets the following criteria: a molecular weight of less than 500 g/mol, fewer than five hydrogen bond donors, fewer than ten hydrogen bond acceptors, and an octanol-water partition coefficient of less than five. QED provides a more flexible classification than conventional ADMET scoring tools based on the molecular properties of 771 existing oral drugs.30 Compounds with a QED score greater than 0.67 are considered to exhibit favorable drug-likeness, whereas those with scores below 0.67 are classified as non-drugs.25 In selecting compounds with promising ADMET properties, two screening stages were implemented. The first screening was based on the seven drug-likeness and bioavailability tools, allowing only those that passed all seven to proceed. In the second stage, to avoid prematurely excluding potentially promising compounds, ligands with minor ADMET violations were allowed to proceed, while those with multiple violations were excluded. Specifically, only metabolites that met at least 20 out of the 24 ADMET criteria were selected for further evaluation. This approach ensured a balance between maintaining reasonable pharmacokinetic and toxicity profiles and retaining chemical diversity for the docking experiments.
Table 1.
List of ADMET parameters used in this study
|
Property
|
Parameter
|
Accepted values
|
| Drug-likeness |
Quantitative estimate of drug-likeness (QED) |
> 0.67 |
| Number of sp3 hybridized carbons |
≥ 0.42 |
| Pan assay interference compounds (PAIN) |
0 |
| Lipinski’s rule of five |
Accepted |
| Pfizer rule |
Accepted |
| GSK rule |
Accepted |
| Golden triangle rule |
Accepted |
| Absorption |
Intestinal absorption |
0 – 0.3 |
| Oral bioavailability |
0 – 0.3 |
| Caco-2 permeability |
> -5.15 |
| MDCK permeability |
> 2 × 10-6 cm/s |
| Distribution |
Plasma protein binding |
≤ 90 |
| Volume distribution |
0.04 – 20 |
| Fraction unbound in plasma |
≥ 5% |
| Metabolism |
CYP 1A2 / 2C19 / 2C9 / 2D6 / 3A4 inhibitor |
0 – 0.3 |
| Excretion |
Clearance of drug |
≥ 5 |
| Half-life |
0 – 0.3 |
| Toxicity |
Hepatoxicity |
0 – 0.3 |
| Drug-induced liver injury |
0 – 0.3 |
| Ames toxicity |
0 – 0.3 |
| Maximum recommended daily dose |
0 – 0.3 |
| Carcinogenicity |
0 – 0.3 |
| Nongenotoxic carcinogenicity rule |
0 |
| Genotoxic carcinogenicity rule |
0 |
| Acute Toxicity rule |
0 |
| SureChEMBL rule |
0 |
| FAF-Drugs4 rule |
0 |
Consensus docking
Two docking software, AutoDock 4.231 and AutoDock Vina 1.1,32 were used to predict the preferred binding conformation and orientation of candidate ligands against ASFV and swine dUTPase. Both programs were developed by the Scripps Research Institute and utilized similar empirical scoring functions but differed in their conformational search algorithms. AutoDock 4.2 uses a stochastic approach through the Lamarckian Genetic Algorithm (LGA), while AutoDock Vina 1.1 employs a gradient-based local search genetic algorithm, which is generally faster and can provide more accurate predictions than AutoDock 4.2.33-35
For the docking parameters, the amino acid residues were kept rigid, and only the rotatable bonds of the ligands were allowed to change their conformation. Both software packages used a 15 Å × 15 Å × 15 Å cubic grid box to define the boundary of the docking experiment. In AutoDock 4.2, the grid point spacing was set to 0.375 Å, with 40 grid points used to create a 15 Å box length. Employing smaller grid point spacing enhances both the efficiency and accuracy of docking.36 In AutoDock Vina, the default grid spacing is always set to 1 Å; therefore, 15 grid points were used to achieve similar box dimensions. The binding site coordinates in Table S2 were determined based on the locations of the active site residues. The grid box was centered on these residues to localize the docking process.
The protein was prepared in AutoDock Tools36 by adding polar hydrogens and converting it to the PDBQT format. The candidate compounds were also saved in the PDBQT format using the Python scripts prepare_gp4.py and prepare_dp4.py The grid parameter files (GPF) and docking parameter files (DPF) were then used as input files for AutoDock 4.2, where docking experiments were conducted using the Lamarckian genetic algorithm with 1,000 iterations. For AutoDock Vina, the exhaustiveness was set to 20, the energy range to 1, and the number of binding modes to 1000, with other parameters maintained at their default values.
Since two docking software programs were used, consensus docking approach was implemented to consolidate the results. Studies have shown that validation using consensus docking improved hit rates and increased the likelihood of identifying the correct binding pose of the ligand.37 The top binding pose of the ligand with the most negative docking score in AutoDock 4.2 was compared to its corresponding docking pose from AutoDock Vina 1.1. If the root mean square deviation (RMSD) between the two poses was less than 2 Å, the ligand was accepted for further analysis. However, if the RMSD was greater than 2 Å, the ligand was excluded from the list of potential candidate inhibitors. The final docking score of each ligand was calculated as the average score from AutoDock 4.2 and AutoDock Vina 1.1. Only the ligands with more negative docking scores than all three control compounds were selected for molecular dynamics (MD) simulations. The three controls include deoxyuridine phosphate (dUTP), the main substrate of dUTPase, deoxyuridine monophosphate (dUMP), the product of dUTP hydrolysis, and 2′-deoxyuridine 5′-(α,β-imido)triphosphate (DUPNPP), a potent inhibitor of dUTPase.10 A decoy ligand was also generated using LIDEB's Useful Decoys (LUDe) server38 to assess the docking protocol’s ability to identify true binders. The server filters compounds from the CHEMBL database, selecting those with similar physicochemical properties to a known active compound but with distinct topological features.
MD simulation
To assess stability and study the dynamics between the protein and docked ligands, all-atom 300 ns MD simulations were conducted using GROMACS (Groningen Machine for Chemical Simulations) 2023.3. The force fields for the protein-ligand complex were assigned separately. The GROMACS built-in Chemistry at Harvard Macromolecular Mechanics (CHARMM) 36 force field was used to parameterize the atoms and define the potential energy of the proteins, while force field parameters for the candidate ligands were assigned using the CHARMM General Force Field (CGenFF) server (cgenff.silcsbio.com).39,40 The PDB files of both the protein and ligands were converted and combined into a single GROMACS-compatible GRO file to generate the molecular structure of the entire protein-ligand system. The system was then enclosed in a cubic box with at a distance of 10 Å from the protein edge to prevent interactions with periodic images from adjacent unit cells. The box was subsequently filled with TIP3P water molecules and neutralized with sodium and chlorine ions. The TIP3P model was specifically used because CHARMM is parameterized in conjunction with this water model. TIP3P is essential when employing the CHARMM 36 force field to ensure an accurate representation of solvent–solute interactions.41,42 Additional ions were added to achieve a salt concentration of 0.15 M. The complete system, now containing solvent and ions, underwent 50,000 relaxation steps using steepest descent minimization to resolve any steric clashes present. The water molecules and ions were then equilibrated around the protein-ligand complex using isochoric-isothermal (NVT) and isothermal-isobaric (NPT) canonical ensembles to reach the desired temperature and pressure of 312 K and 1 bar, respectively. NVT equilibration was regulated using the modified Berendsen thermostat, while NPT equilibration was controlled using a Berendsen barostat. Finally, a 300 ns MD simulation under NPT ensemble was conducted with 150,000,000 integration steps, saving snapshots every 100 ps, resulting in 3,000 trajectory frames.
The MD trajectories were analyzed using GROMACS tools to evaluate the stability and dynamics of the protein–ligand complexes. RMSD (gmx rms) was used to assess structural stability, RMSF (gmx rmsf) to examine residue flexibility, and gmx distance to calculate average distances between the ligand and the binding pocket. Hydrogen bonds formed during the simulation were identified using gmx hbond. PCA was also performed to verify convergence and identify the dominant motions of the protein–ligand complexes. The GROMACS tool gmx covar was used to calculate the covariance matrix, which describes how atomic positions vary together over time. This matrix was then diagonalized to obtain eigenvectors and eigenvalues, where the eigenvectors represent directions of motion and the eigenvalues indicate the magnitude of movement along each direction. The eigenvector and eigenvalue files were subsequently used as input for gmx anaeig to project the simulation data onto the top principal components, which capture the most significant motions in the system. A 2D projection plot between the first and second principal components (PC1 and PC2) was then generated to provide insight into large-scale motions and conformational changes that occurred during the simulation.
Molecular mechanics Poisson Boltzmann (generalized born) surface area estimation
The complexes with stable behaviors were further analyzed for their binding affinity using the molecular mechanics Poisson-Boltzmann surface area (MMPBSA) and molecular mechanics generalized born surface area (MMGBSA) approaches. Both methods combine molecular mechanics and continuum solvation models through equation 1, and differ only in the calculation of the polar solvation energy: the Poisson-Boltzmann equation for MMPBSA and the generalized Born approximation for MMGBSA. The molecular mechanics energies were derived from the sum of the van der Waals and electrostatic forces, obtained using the Lennard-Jones and Coulomb potential functions, respectively. The nonpolar solvation energy is assumed to be linearly proportional to the solvent accessible surface area (SASA).43,44 The entropic contribution was estimated using the interaction entropy method, which approximates entropy based on the fluctuations in the interaction energy between the ligand and the receptor. This method offers a less computationally expensive alternative to traditional entropy estimation techniques such as normal mode analysis.45
(1)
Binding free energy calculations were conducted using the GROMACS-compatible package gmx_MMPBSA45 under the same set of conditions as the molecular dynamics simulation. To ensure that the analysis reflected the stabilized phase of the system, frames were extracted from the last 100 ns of the 300 ns trajectory. Sampling was performed every 10 frames, resulting in 100 representative snapshots for MMPBSA and MMGBSA computations.
Results and Discussion
ADMET profiling
The initial screening process utilized seven drug-likeness scoring tools, narrowing the pool of candidate compounds from 4,683 to 507 that met the specified criteria. These tools evaluated the similarity of the marine fungal metabolites to existing drugs, which reflects their potential success rate in clinical trials. All 507 compounds passed Lipinski’s Rule of Five (Ro5), indicating good absorption and permeability when administered orally, and their favorable QED scores further suggested potential drug efficacy. In the second screening phase, the remaining metabolites were evaluated against 24 ADMET parameters to refine the selection, with only those passing at least 20 of these tests being considered. Fig. S2 of the Supplementary file 1 summarizes the results of the entire ADMET screening process.
Of the 507 metabolites that passed the initial screening, 328 showed favorable results for at least 20 ADMET properties, suggesting good safety and efficacy. These compounds were selected as the final candidates for the molecular docking experiments. Notably, 27 metabolites passed all 24 ADMET tests. Coupled with excellent drug-likeness scores, the 328 compounds demonstrate a higher likelihood of success in downstream drug development stages. Although some compounds did not meet the required criteria for certain ADMET parameters, their excellent overall ADMET profile justifies further investigation. Additionally, since this is only a preliminary screening, their structures can be modified or used as templates to create semi-synthetic compounds with improved ADMET properties. The complete ADMET screening is presented in Table S1 (see Supplementary file 2).
Consensus docking
Following ADMET profiling, the 328 metabolites were docked against the known active region of ASFV dUTPase to predict their preferred binding conformations and associated docking scores. To ensure selectivity and minimize off-target effects, these metabolites were also docked against swine dUTPase.
Due to differences in the scoring algorithms used, AutoDock 4.2 and AutoDock Vina generated different docking scores and overall rankings (Table S3 of Supplementary file 1). AutoDock 4.2 can better discriminate the poses compared to Vina, which yielded similar docking scores. Nevertheless, a consistent pattern was observed when considering the ranking of all 328 metabolites. The top-ranked metabolites from AutoDock 4.2 also appeared among the highest scoring metabolites identified by AutoDock Vina.
Using the LUDe server, the compound CHEMBL55407 (Fig. S3) was identified as a decoy ligand to validate the performance of the two docking programs. As shown in Table 2, all three control compounds exhibited stronger binding affinities toward both ASFV and swine dUTPase, whereas the decoy ligand showed noticeably weaker binding. This supports the ability of the docking protocol to reliably distinguish true binders from weak or non-binders.Out of the 328 metabolites subjected to molecular docking, only nine compounds exhibited better or more negative scores (-7.64 kcal/mol to -8.19 kcal/mol) than all three control compounds (-7.32 kcal/mol to -7.59 kcal/mol). Among these, tricycloalternarene K obtained the best docking score, followed by tricycloalternarene C and corynechromone H. Interestingly, the highest-scoring ligands included three isomers: tricycloalternarene K, tricycloalternarene C, and tricycloalternarene L, all of which fall under the chemical class, sesquiterpenoid. In addition to the three isomers, thomimarine A, which ranked ninth, was also classified the same chemical classification. The final candidate nine ligands also registered less negative docking scores (-6.58 kcal/mol to -8.78 kcal/mol) against the swine dUTPase compared to all the three controls (-9.88 kcal/mol to -11.90 kcal/mol). These compounds demonstrated favorable affinity against the ASFV dUTPase while presenting weaker binding associations with swine dUTPase. This suggests that they are likely to have minimal interference with the swine dUTPase, ensuring that their normal physiological functions remain unaffected and thereby reducing potential side effects. Fig. 1 illustrates the binding poses of the ligands against the active site of ASFV dUTPase, while Fig. 2 shows the interaction diagrams of the nine ligands and the three controls with the protein.
Table 2.
Docking scores (kcal/mol) of the top nine highest-scoring ligands against ASFV dUTPase and swine dUTPase
|
Ligand
|
Compound name
|
ASFV dUTPase
|
Swine dUTPase
|
|
Vina
|
AD4
|
RMSD
|
Average
|
Vina
|
AD4
|
RMSD
|
Average
|
| (+)dUMP |
Deoxyuridine monophosphate |
-7.5 |
-7.68 |
0.732 |
-7.59 |
-9.1 |
-10.66 |
1.590 |
-9.88 |
| (+)dUTP |
Deoxyuridine triphosphate |
-7.4 |
-7.54 |
1.310 |
-7.47 |
-10.2 |
-13.60 |
1.468 |
-11.90 |
| (+)dUPNPP |
α,β-imido-dUTP |
-7.3 |
-7.34 |
1.604 |
-7.32 |
-9.5 |
-13.32 |
1.386 |
-11.41 |
| (-)Decoy |
CHEMBL554071 |
-5.9 |
-6.94 |
1.981 |
-6.77 |
-6.6 |
-9.04 |
1.179 |
-7.82 |
| M3120 |
Tricycloalternarene K |
-7.6 |
-8.78 |
1.489 |
-8.19 |
-4.0 |
-11.12 |
1.870 |
-7.56 |
| M1421 |
Tricycloalternarene C |
-6.9 |
-9.19 |
1.972 |
-8.05 |
-3.3 |
-9.86 |
0.881 |
-6.58 |
| M4555 |
Corynechromone H |
-7.3 |
-8.31 |
0.892 |
-7.81 |
-6.1 |
-9.45 |
0.926 |
-7.78 |
| M1231 |
Nigrospin C |
-6.9 |
-8.70 |
1.235 |
-7.80 |
-6.4 |
-11.16 |
1.768 |
-8.78 |
| M0549 |
Aurantiomide B |
-7.6 |
-8.00 |
1.268 |
-7.80 |
-4.0 |
-7.77 |
0.703 |
-5.89 |
| M0288 |
Fusaquinon A |
-7.7 |
-7.84 |
1.241 |
-7.77 |
-6.6 |
-10.07 |
1.190 |
-8.34 |
| M1628 |
5′-Hydroxyasperentin |
-7.7 |
-7.72 |
1.159 |
-7.71 |
-6.7 |
-10.83 |
1.662 |
-8.77 |
| M2440 |
Tricycloalternarene L |
-6.9 |
-8.42 |
1.730 |
-7.66 |
-5.9 |
-10.10 |
1.893 |
-8.00 |
| M2989 |
Thomimarine A |
-7.4 |
-7.88 |
1.694 |
-7.64 |
-4.2 |
-9.33 |
1.624 |
-6.77 |
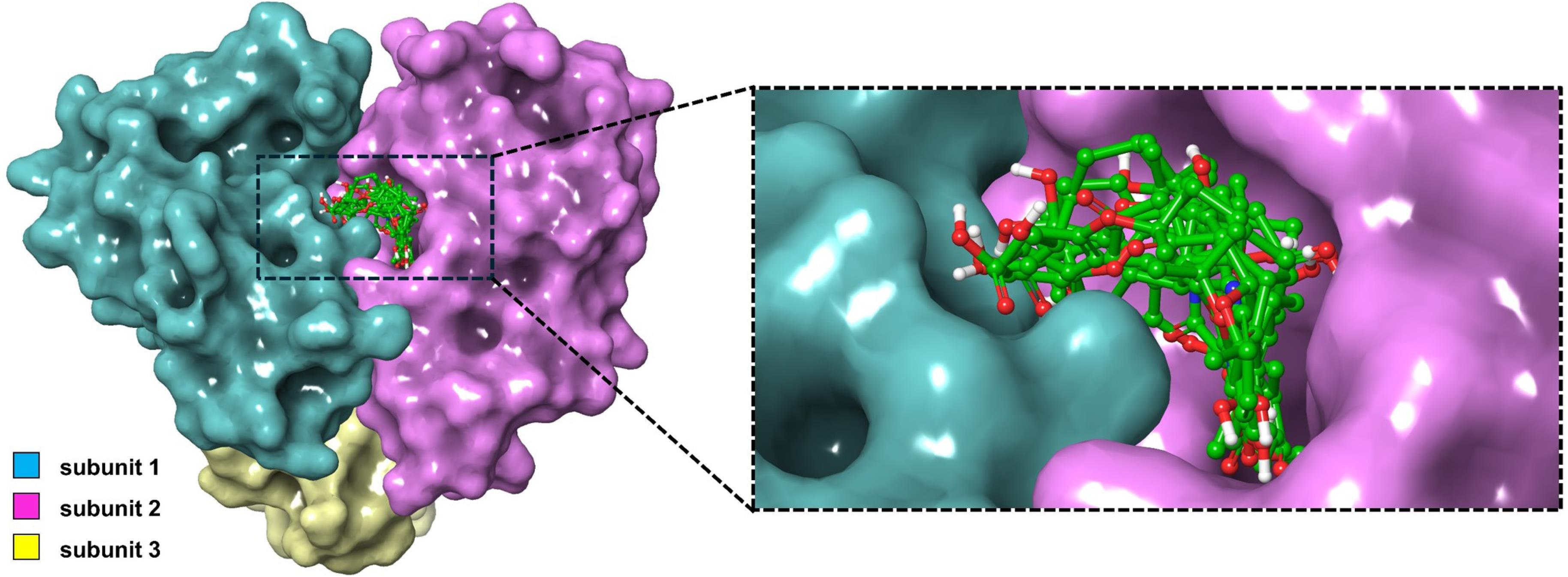
Fig. 1.
Binding poses of the nine highest-scoring ligands against the dual-subunit active site of the ASFV dUTPase.
.
Binding poses of the nine highest-scoring ligands against the dual-subunit active site of the ASFV dUTPase.
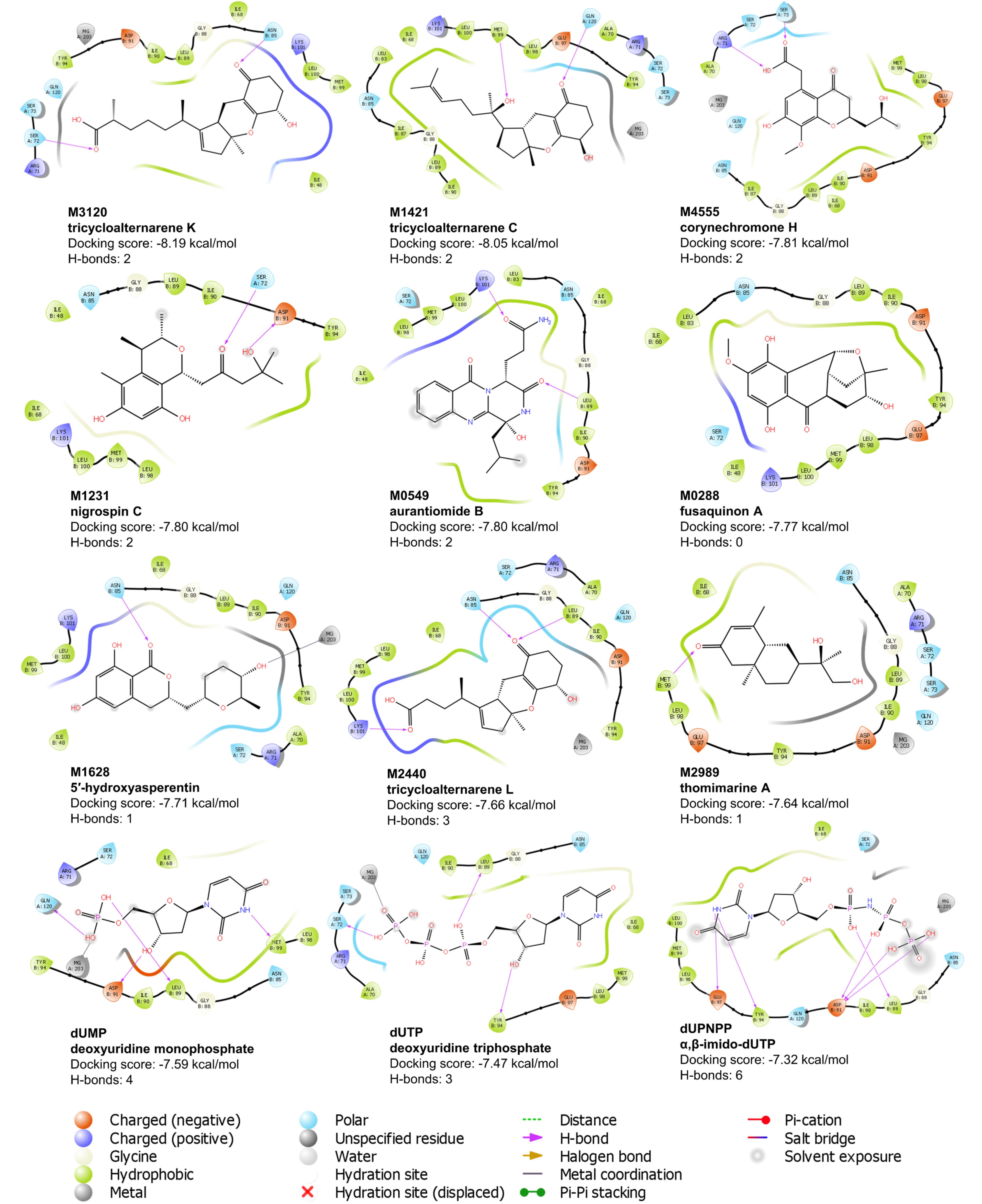
Fig. 2.
Ligand interaction diagram of the top nine ligands and the three control compounds against the binding pocket of ASFV dUTPase. Residues shown are within 4 Å from the ligand.
.
Ligand interaction diagram of the top nine ligands and the three control compounds against the binding pocket of ASFV dUTPase. Residues shown are within 4 Å from the ligand.
The final nine candidate ligands adopted highly similar binding poses against the two adjacent protomers within the binding pocket of the ASFV dUTPase enzyme (Fig. 1). A significant portion of the ligands was embedded in subunit 2, with some moieties extending into the first subunit. As shown in Fig. 2, most amino acids surrounding the ligands were hydrophobic with a few polar residues. Additionally, charged residues such as Glu 97, Asp 91, Arg 71, and Lys 101 were also observed. The majority of the hydrogen bonds are formed with polar residues, including Asn 86 and Asn 72. For some ligands including M4555 and M0549, hydrogen bonds were paired with the charged residues Arg 71 and Lys 101.
Seven residues interacted with all nine ligands and the three control compounds: Asn 85, Gly 88, Leu 89, Ile 90, Tyr 94, and Met 99 from subunit 2, and Ser 72 from subunit 1 (Fig. 2). Among these, Tyr 94 plays a crucial role in the catalytic process. Mutation of this residue, along with Asp 91, Arg 71, and Arg 149, led to a reduced or complete loss of enzymatic activity. Additionally, mutation of Leu 89 resulted in a 4 to 7-fold increase in the Michaelis-Menten constant (Km), indicating decreased substrate binding efficiency. A similar case was also observed with the mutation of residue Ile 90, highlighting its critical role in the catalytic process.11 Given the well-established catalytic importance of these residues, ligand interactions at these sites are likely to interfere with substrate binding or turnover. This could potentially impair enzymatic activity and may contribute to potential antiviral effects. Apart from the protein residues, the magnesium ion is also an integral component of the catalytic mechanism, as it coordinates and stabilizes intermediates formed during substrate binding and hydrolysis.10 From Fig. 2, five candidate ligands—M1421, M1628, M2440, M3120, and M4555—maintained close contact with the magnesium ion after docking. Establishing a stable connection with the ion can also potentially enhance the efficacy of the inhibitor.
MD simulation
After obtaining the structure of the protein with the bound ligands, the nine final complexes and three control complexes were subjected MD simulations to determine their stability and assess their dynamic interactions, using RMSD, RMSF, and mean distance analysis.
Before production runs, the systems underwent energy minimization and equilibration to correct geometries, equilibrate solvent and ions, and reach target temperature and pressure. As shown in Fig. S4, potential energy decreased steadily during equilibration, stabilizing around -850,000 kJ/mol and remaining consistent during production, indicating well-relaxed systems.46 All systems reached the target temperature of 312 K with a minimal SEM of ~0.250. Pressure stabilized between 1.356 and 1.976 bar (SEM: 2.6), with expected fluctuations typical of MD simulations, while average pressure stayed near 1 bar. Density profiles also stabilized around 1035 kg/m³, confirming system equilibration.46,47
Fig. 3A illustrates the RMSD trajectories over the 300 ns simulation period. Eight of the nine top-scoring ligands exhibited unstable behavior, as indicated by rising and erratic RMSD fluctuations. In contrast, only M1421 displayed a stable trajectory throughout the simulation. Ligands M0288, M1231, M1628, M2440, and M3120 initially displayed stable behaviors at the start of the simulation. However, before reaching 100 ns, a sharp increase in the RMSD was observed, indicating that the ligands had detached from the binding pocket. Moreover, ligands M0549, M2989, and M4555 also presented plateaued trajectories within the first 100 ns but eventually became unstable towards 150 ns.
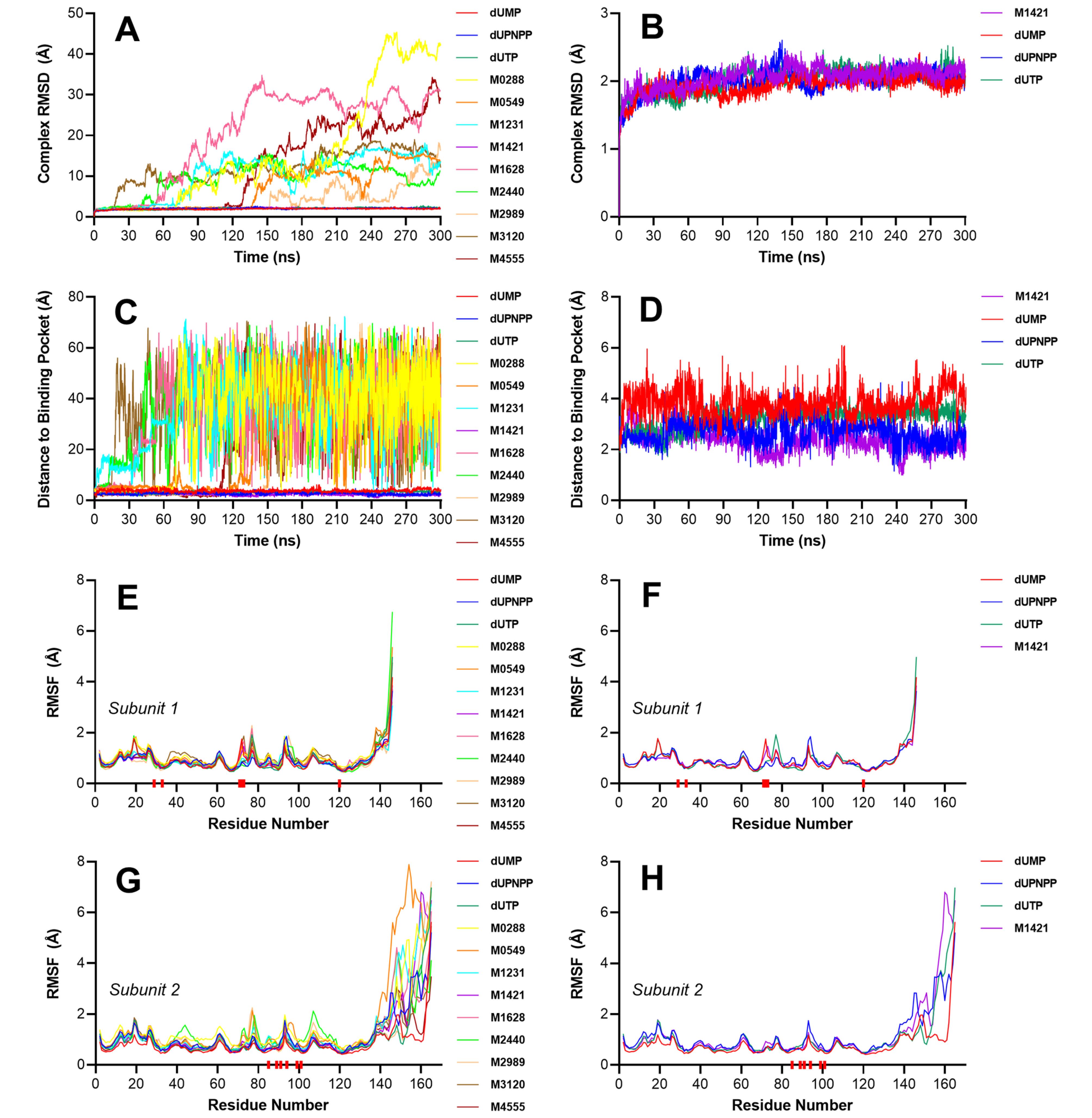
Fig. 3.
Stability analysis of the protein–ligand complexes using RMSD and distance-based metrics. (A) Complex RMSD of all nine ligands and the three control compounds against dUTPase. (B) Complex RMSD of the stable complexes. (C) Time-resolved distance between the center of mass of each ligand and the center of mass of the binding pocket. (D) Time-resolved distance between the center of mass of the stable ligands and the center of mass of the binding pocket. (E) RMSF of subunit 1 for all simulated complexes, including nine candidate ligands and three control compounds. (F) RMSF of subunit 1 for the stable complexes only. (G) RMSF of subunit 2 for the same set of complexes. (H) RMSF of subunit 2 for the stable complexes. Red blocks below the x-axis indicate the positions of the active site residues.
.
Stability analysis of the protein–ligand complexes using RMSD and distance-based metrics. (A) Complex RMSD of all nine ligands and the three control compounds against dUTPase. (B) Complex RMSD of the stable complexes. (C) Time-resolved distance between the center of mass of each ligand and the center of mass of the binding pocket. (D) Time-resolved distance between the center of mass of the stable ligands and the center of mass of the binding pocket. (E) RMSF of subunit 1 for all simulated complexes, including nine candidate ligands and three control compounds. (F) RMSF of subunit 1 for the stable complexes only. (G) RMSF of subunit 2 for the same set of complexes. (H) RMSF of subunit 2 for the stable complexes. Red blocks below the x-axis indicate the positions of the active site residues.
Most of the eight unstable ligands had over five oxygen- and nitrogen-containing groups, which extensively interacted with water during MD simulations. To maintain stable binding within the active site, an optimal number of internal interactions are required to anchor these groups in place. As shown in Fig. 2, aside from ion–dipole and dipole–dipole interactions, only hydrogen bonds contributed significantly to the binding stability. Ligands with multiple polar groups, including M0549, M0288, and M1628, depended on forming several hydrogen bonds, similar to the control compounds, to remain stably bound in the binding pocket. However, in these cases, the number of hydrogen bonds formed may have been insufficient to fully stabilize the ligands against the protein, leading to their observed instability. M2989 possessed only three polar functional groups with one hydrogen bond; however, two of these were oriented toward a glycine residue, which has a side chain with zero net charge. As a result, its interaction with M2989 was relatively weak due to the lack of strong electrostatic attraction. The binding poses of the eight unstable ligands may have also been suboptimal due to the spatial positioning of their oxygen and nitrogen atoms, with polar groups near nonpolar residues and vice versa, leading to weak interactions. In contrast, the polar groups of M1421 are clustered at one end, forming favorable interactions with the oppositely charged polar residues like Gln 120 and Glu 97, while its nonpolar tail aligned with the hydrophobic pocket which enhanced its binding stability.
Among the nine final candidate ligands, only M1421 exhibited a stable behavior comparable to that of the three reference compounds (Fig. 3B). During the first few nanoseconds, the increasing RMSD plot indicated that the system was undergoing initial conformational adjustments to adapt to the simulated physiological conditions, including the presence of water molecules and ions that were absent in the original docked structure. This phase involved ligand repositioning and movement of flexible loops and side chains toward a more stable, energetically favorable state. Once the system stabilized and the ligand settled into its final pose, the RMSD plateaued around 2 Å, similar to the control compounds. Distance plots (Fig. 3C and 3D) further support these observations. The eight unstable ligands exhibited erratic and highly fluctuating distance profiles, with some drifting as far as 70 Å from the binding site. In contrast, M1421 maintained a relatively stable distance throughout the simulation, closely matching the profiles of the control compounds, reinforcing its stable binding within the pocket.
These observations suggest that M1421 successfully adopted a low-energy conformation and remained stably bound within the active site over the entire 300 ns simulation, highlighting its structural stability.
The ligand binding poses at different time points, as shown in Fig. 4, further confirmed its stability, with the initial conformation largely preserved throughout the simulation. The ligand consistently adopted similar orientations across various time frames and maintained close contact with key active site residues, indicating strong and persistent interactions. This conformational consistency underscores the robustness of the ligand's binding within the pocket. Moreover, the molecular dynamics simulation video (Supplementary file 3, Video S1) visually demonstrated that the ligand remained confined within the binding site for the duration of the simulation. While minor conformational adjustments were observed, likely due to interactions with surrounding amino acid residues and solvent molecules, no significant translational displacement of the ligand occurred.
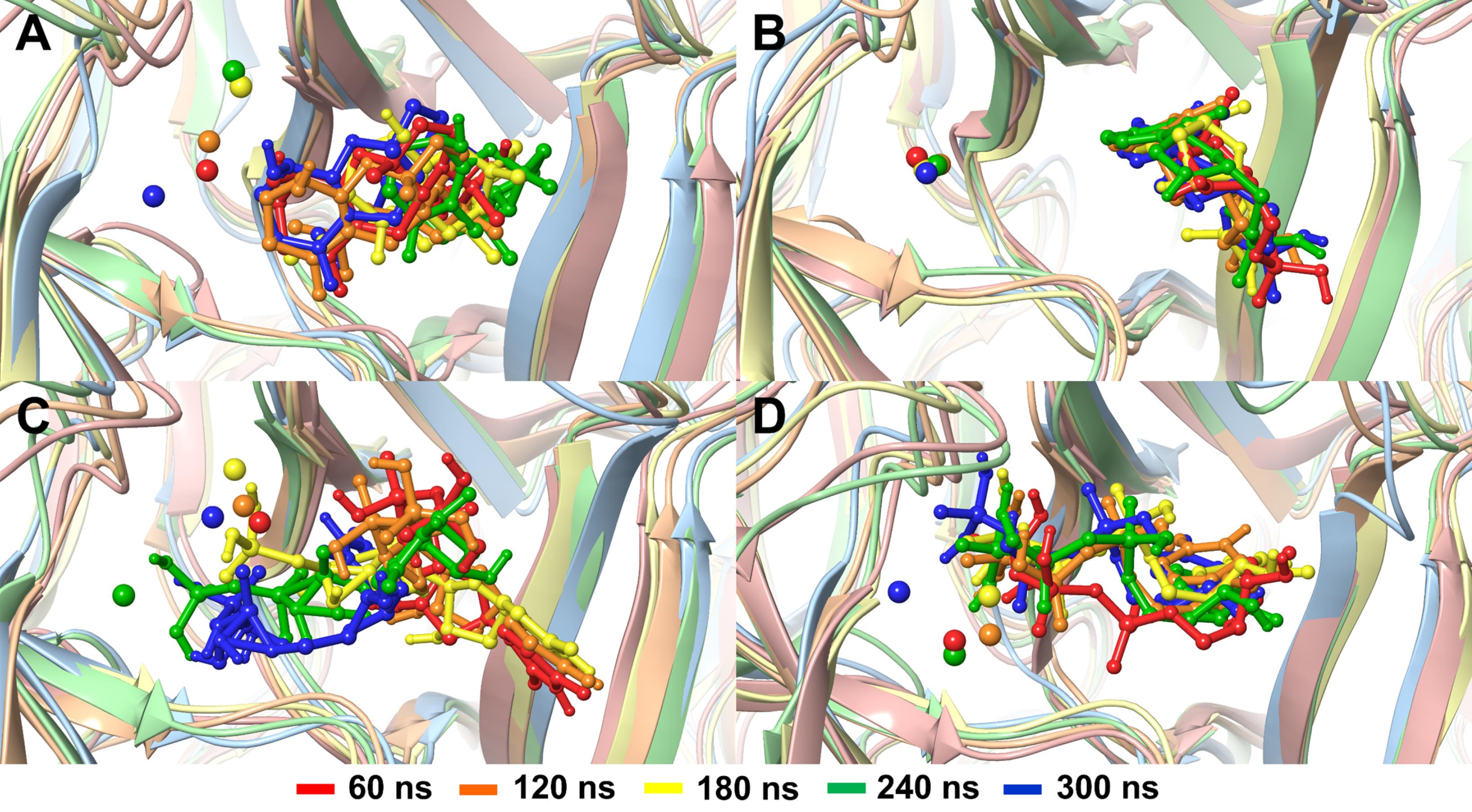
Fig. 4.
Binding poses of (A) M1421, (B) dUMP, (C) dUPNPP, and (D) dUTP at different trajectory frames, captured every 60 ns. Magnesium ions, represented by a small sphere, are also shown.
.
Binding poses of (A) M1421, (B) dUMP, (C) dUPNPP, and (D) dUTP at different trajectory frames, captured every 60 ns. Magnesium ions, represented by a small sphere, are also shown.
The RMSF plots in Fig. 3E to 3F illustrate the rotational movements of the ASFV dUTPase residues. The known critical residues, highlighted by the red bars on the axis, consistently exhibited minimal movements, suggesting that the binding pocket remained unchanged and that the stable ligands maintained a strong connection with the active site residues. Additionally, ligands that exhibited instability, such as M0288, M0549, M2440, and M2989, showed higher RMSF values, particularly in subunit 2 of the protein, where most of the ligand interactions occurred. Since the unstable ligands dissociated from the binding pocket, the active site residues gained more freedom to interact with other residues and water molecules, leading to increased residue movements and higher RMSF values. High RMSF values were also observed in residues at the ends of the protein sequence, particularly in the second subunit. These regions, located external to the protein trimer, exhibited increased flexibility due to direct interactions with water molecules. Additionally, subunit 2 has a longer end chain that moves freely with the solvent, leading to elevated RMSF values at residues 140 to 160. Since these residues are distant from the active site and do not directly interact with the ligand, they do not influence the binding of the compounds.
To confirm the convergence of the simulations and further validate the stability of the complexes, PCA was performed. PCA provides a qualitative and descriptive overview of the dominant conformational motions sampled by the protein–ligand complexes during the MD simulations. While RMSD and distance-based metrics capture structural deviations and stability over time, PCA complements these by visualizing large-scale, collective motions. This allows for assessment of whether the system remains confined to a stable conformational space or continues to explore alternative structural states. Tightly clustered data points in the PCA plot, representing a smaller conformational space, suggest limited atomic motion and minimal structural fluctuations. Accordingly, the more stable complexes occupy compact regions in the principal component space, supporting the convergence of the simulations. In contrast, more scattered data points reflect increased atomic mobility and may indicate significant shifts in ligand binding positions.
The scree plot in Fig. 5A highlights the top two principal components (PC1 and PC2) as the dominant contributors to the total atomic motion in the simulated systems. For the complexes of compounds M1421, dUMP, and dUPNPP, PC1 and PC2 together accounted for 71.03% to 80.85% of the total variance, indicating that the majority of the conformational fluctuations can be described along just two principal axes. In contrast, for the control compound dUTP, PC1 and PC2 accounted for only 48.88% of the total variance. While this is lower than the values observed for the other complexes, it remains sufficient for qualitative analysis. The reduced variance in the first two components suggests that the dUTP complex undergoes more distributed or multidimensional motions, potentially involving higher-order principal components. Importantly, a lower percentage of variance captured by PC1 and PC2 does not necessarily indicate poor stability convergence. This is further supported by the stability metrics, including RMSD and mean distance analyses, which confirm the overall structural stability of the dUTP complex.
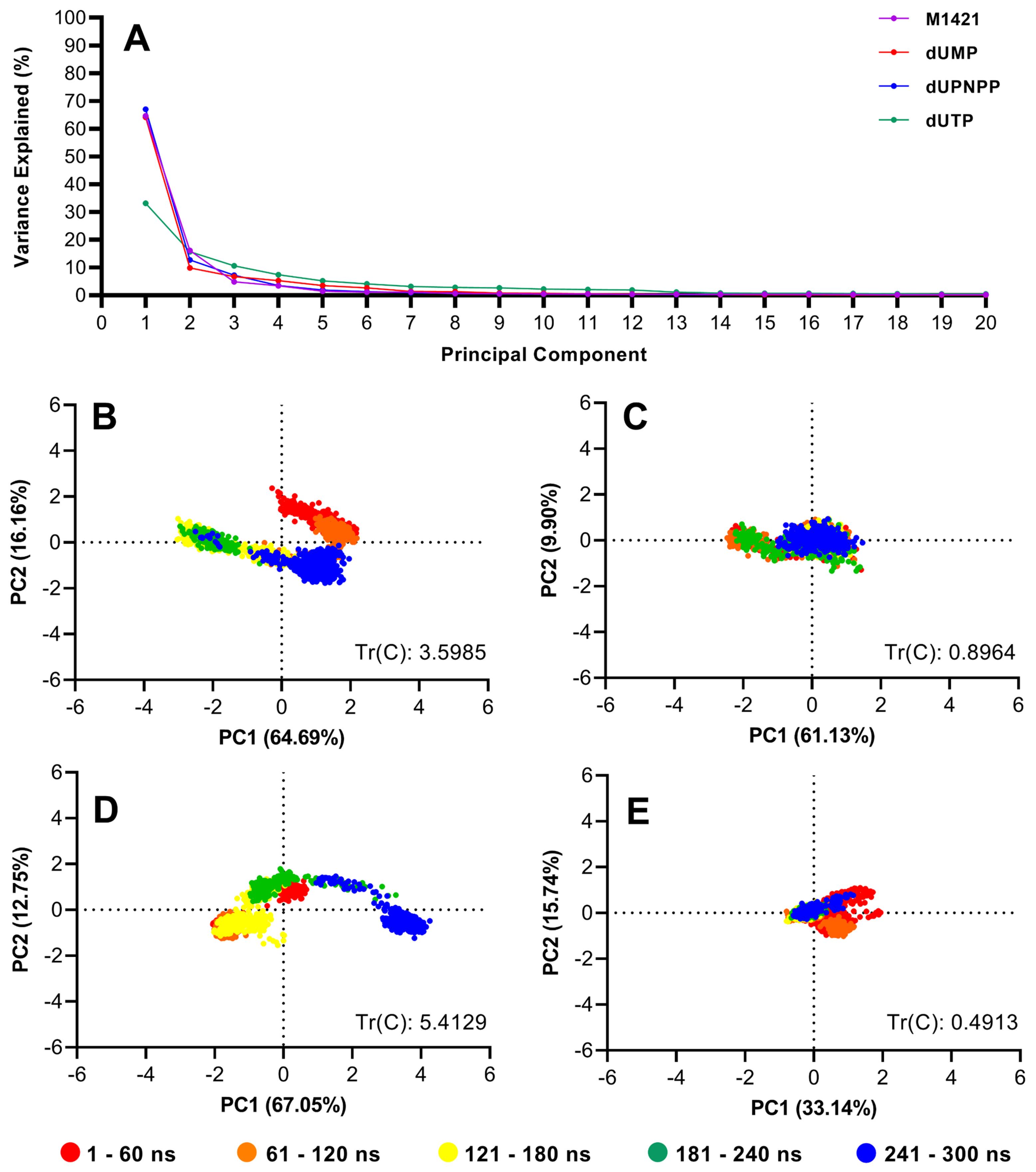
Fig. 5.
Principal component analysis of M1421 and three control compounds complexed with swine dUTPase. (A) Scree plot of the first 20 eigenvectors against the explained variance. Also shown are projections of ligand motions along PC1 and PC2: (B) M1421, (C) dUMP, (D) dUPNPP, and (E) dUTP, and the corresponding trace of the covariance matrix.
.
Principal component analysis of M1421 and three control compounds complexed with swine dUTPase. (A) Scree plot of the first 20 eigenvectors against the explained variance. Also shown are projections of ligand motions along PC1 and PC2: (B) M1421, (C) dUMP, (D) dUPNPP, and (E) dUTP, and the corresponding trace of the covariance matrix.
The PCA plot for M1421 revealed tight clusters over different time durations (Fig. 5B). During the first 60 ns, the data points occupied a relatively larger area as the complex equilibrated to a lower energy state. This binding pose was maintained until 120 ns, at which point the ligand transitioned to a different conformation and held this new pose until approximately 240 ns (Fig. 4A). Toward the end of the simulation, the cluster of blue points moved closer to the red and orange clusters, indicating that the ligand's pose had shifted back to its original conformation. From the visual representation in Fig. 4A, the ligand's pose at 60, 120, and 300 ns were highly superimposed, consistent with the PCA plot and confirming the stability of the ligand over time. Furthermore, the tight blue cluster near the end of the 300 ns run signified convergence of the simulation. These results are also consistent with RMSD and mean distance analyses, collectively reinforcing the observed stability of the protein–ligand interactions
In contrast, the control compounds, dUMP and dUTP, formed tighter clusters throughout the simulation and displayed uniform binding poses (Fig. 5C to 5E), reflecting a higher degree of stability compared to dUPNPP and M1421. Additionally, the trace values of the control compounds dUMP and dUTP, were lower than those of dUPNPP and M1421. The trace of the covariance matrix corresponds to the total variance present in the dataset.48 It provides a quantitative measure of the total atomic motion throughout the simulation. Lower trace values are typically associated with stable and well-converged complexes. Although dUTP exhibited a lower percentage of variance in its first two principal components, it recorded the smallest trace value among all the complexes. This low trace (0.398) indicates reduced overall atomic motion, suggesting that the dUTP-bound complex remained structurally stable throughout the entire 300 ns. In contrast, the control compound dUPNPP, which has a larger structure and more functional groups, engaged in more extensive interactions with surrounding residues and water molecules. These interactions likely contributed to increased atomic fluctuations, resulting in the highest trace value observed (5.4129), and reflecting greater overall motion compared to the other ligands. As shown in Fig. 5D, the blue clusters of dUPNPP shifted away from the other clusters as the ligand settled into its final binding conformation. This shift was consistent with Fig. 4C, where the binding pose at 300 ns differed from the initial configurations. Despite this minimal shift, dUPNPP remained stable within the binding pocket.
M1421 consistently formed an average of two to three hydrogen bonds throughout the simulation (Fig. 6). Notably, subunit 2 exhibited more hydrogen bonding interactions compared to subunit 1, suggesting that its residues provide a more favorable environment for ligand stabilization. This is consistent with the structural composition of the binding pocket, which is primarily formed by residues from subunit 2. Among the three controls, dUTP formed the most hydrogen bonds due to its structure having more polar functional groups. The control dUPNPP also formed a substantial number of hydrogen bonds toward the end of the simulation. Despite having fewer moieties capable of hydrogen bonding compared to the controls, M1421 generated nearly the same number of hydrogen bonds as dUMP. The consistent hydrogen bonding of M1421 likely contributed to its observed stability, underscoring its potential as an antiviral drug.
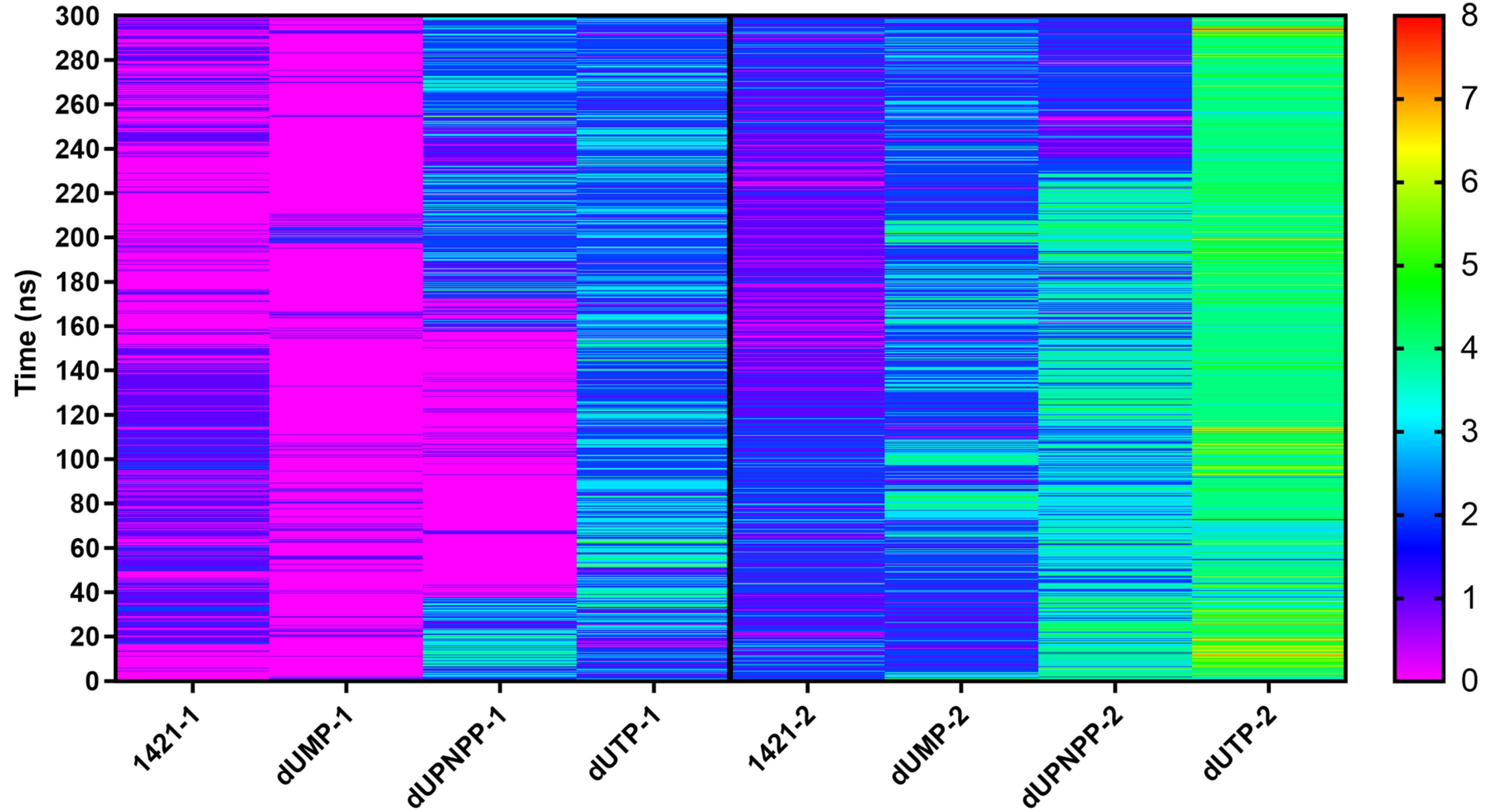
Fig. 6.
Number of hydrogen bonds between the ASFV dUTPase (Subunit 1 and 2) and M1421. Also shown are the number of hydrogen bonds of three control compounds.
.
Number of hydrogen bonds between the ASFV dUTPase (Subunit 1 and 2) and M1421. Also shown are the number of hydrogen bonds of three control compounds.
Molecular mechanics Poisson Boltzmann (generalized born) surface area estimation
The binding energies of M1421 and the three control compounds were predicted to estimate their relative binding strengths. Although the MMGBSA and MMPBSA methods yielded different magnitudes for the polar and non-polar solvation energies, they showed similar rankings across all energy terms (Table S4). M1421 exhibited stronger binding energies (MMGBSA: -28.05 kcal/mol, MMPBSA: -23.70 kcal/mol) than the two control compounds, dUMP (MMGBSA: -13.61 kcal/mol, MMPBSA: -12.21 kcal/mol) and dUPNPP (MMGBSA: -5.98 kcal/mol, MMPBSA: -4.4 kcal/mol), but had weaker binding affinity compared to dUTP (MMGBSA: -32.19 kcal/mol, MMPBSA: -32.19 kcal/mol) (Fig. 7A and 7B). M1421, dUMP, and dUTP all exhibited stronger binding affinities toward subunit 2, whereas dUPNPP showed a clear preference for subunit 1 and even displayed repulsive behavior toward subunit 2 (Fig. 7C–7F). This is further supported by the hydrogen bond analysis in Fig. 6, which shows that dUPNPP formed an increasing number of hydrogen bonds with subunit 1 over time, while interactions with subunit 2 remained minimal. In contrast, M1421, dUMP, and dUTP consistently favored hydrogen bond formation with subunit 2 throughout the simulation.
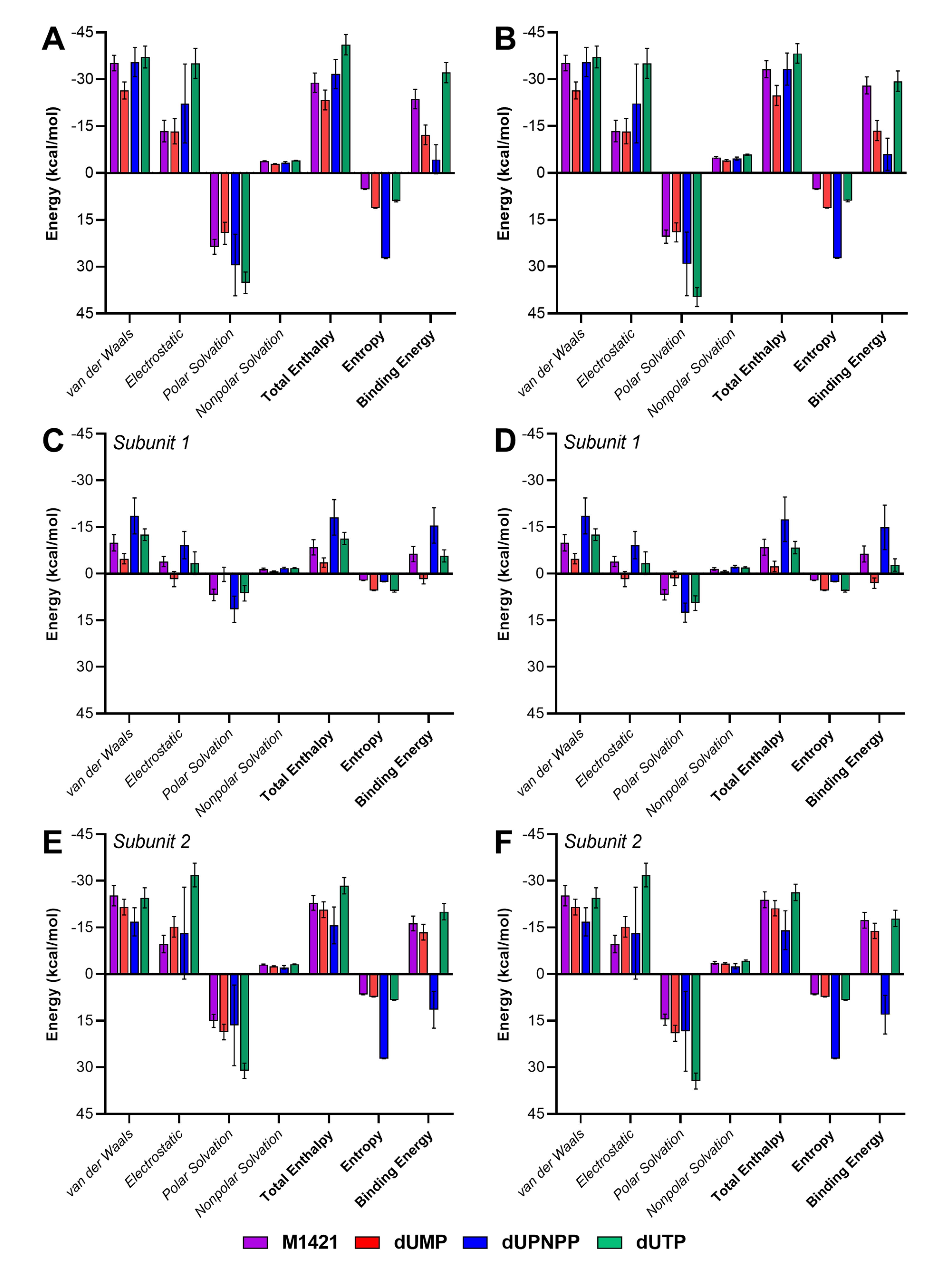
Fig. 7.
Binding free energy decomposition of compound M1421 and the control ligands dUMP, dUPNPP, and dUTP. (A) MMPBSA and (B) MMGBSA total binding energy components for the entire protein over the last 100 nanoseconds of simulation. (C) MMPBSA and (D) MMGBSA decomposition by subunit 1. (E) MMPBSA and (F) MMGBSA decomposition by subunit 2. Total enthalpy is the sum of van der Waals, electrostatic, polar and non-polar solvation energies.
.
Binding free energy decomposition of compound M1421 and the control ligands dUMP, dUPNPP, and dUTP. (A) MMPBSA and (B) MMGBSA total binding energy components for the entire protein over the last 100 nanoseconds of simulation. (C) MMPBSA and (D) MMGBSA decomposition by subunit 1. (E) MMPBSA and (F) MMGBSA decomposition by subunit 2. Total enthalpy is the sum of van der Waals, electrostatic, polar and non-polar solvation energies.
Statistically, the enthalpic binding energy (excluding entropy) of compound M1421 is significantly more favorable than that of the control compound dUMP, as indicated by the very low P value ( < 0.0001) and a large effect size (R2 = 0.4303) (Table 3). In contrast, M1421 shows weaker binding than both dUPNPP (P < 0.0001, R2 = 0.1132) and dUTP (P < 0.0001, R2 = 0.7859). The relatively low effect size in the M1421 vs dUPNPP comparison suggests that, although the difference is statistically significant, the actual magnitude is small. Specifically, only about 11% of the variance in binding energy can be attributed to differences between the two ligands. This is supported by the significant F-test result (P < 0.0001), indicating the inconsistent binding behavior of dUPNPP across simulation frames. Interestingly, this variability also corresponds to its stronger binding affinity toward subunit 1 (Fig. 7C and 7D), signifying that dUPNPP exhibits a distinct binding preference for subunit 1 compared to the other ligands, which show stronger binding affinity toward subunit 2 (Fig. 7E and 7F). In the MMGBSA results, M1421 binds significantly more strongly than dUMP (p < 0.0001, R2 = 0.6658), while dUTP again exhibits more favorable binding than M1421 (P < 0.0001, R2 = 0.4295). When comparing M1421 to dUPNPP, it reveals a negligible and statistically insignificant mean difference (–0.0502 kcal/mol, P = 0.9317), with a small effect size (R2 = 3.68 × 10⁻⁵), suggesting nearly identical enthalpic binding energies under this method.
Table 3.
Pairwise statistical comparison of the MMPBSA and MMGBSA binding energies between the candidate compound 1421 and the control compounds dUMP, dUPN, and dUTP, based on 100 data points per group
|
Comparison
|
Mean Difference
|
95% CI of Difference
|
t-test
P
-value
|
Effect Size (R2)
|
F-test
P
-value
|
| MMPBSA |
| 1421 vs dUMP |
5.462 ± 0.4443 |
4.585 to 6.338 |
< 0.0001 |
0.4303 |
0.8205 |
| 1421 vs dUPNPP |
-2.816 ± 0.5574 |
-3.915 to -1.717 |
< 0.0001 |
0.1132 |
< 0.0001 |
| 1421 vs dUTP |
-12.21 ± 0.4508 |
13.10 to -11.32 |
< 0.0001 |
0.7859 |
0.6130 |
| MMGBSA |
| 1421 vs dUMP |
8.413 ± 0.4215 |
7.582 to 9.244 |
< 0.0001 |
0.6658 |
0.1185 |
| 1421 vs dUPNPP |
-0.0502 ± 0.585 |
-1.204 to 1.103 |
0.9317 |
3.682e-005 |
< 0.0001 |
| 1421 vs dUTP |
-5.096 ± 0.415 |
-5.915 to -4.277 |
< 0.0001 |
0.4295 |
0.1924 |
Note: The values presented are binding energy estimates derived from molecular mechanics and polar solvation energies, excluding entropic contributions*.
*The gmx_MMPBSA package does not provide entropic values per frame, but only a single final entropic estimate per system.
In both binding energy estimation approaches, the inclusion of entropic contributions reveals that M1421 exhibits significantly stronger total binding free energy than both dUPNPP. The reduced affinity of dUPNPP is attributed to its larger molecular size and greater flexibility, which result in a higher entropic penalty. This highlights the importance of including entropy in binding free energy calculations, especially for larger ligands with a higher number of rotatable bonds, whose entropic costs can substantially diminish their overall binding affinity.
For all systems, van der Waals interactions provided the strongest energy contributions, effectively counteracting the positive polar solvation and entropic energies. The control compounds dUTP and dUPNPP, which have the most polar groups, exhibited the strongest electrostatic energies but also the most positive polar solvation energies. Again, the larger molecular size and greater conformational flexibility of dUPNPP resulted in a significantly higher entropic cost, ultimately leading to the weakest overall binding affinity among all tested complexes. Although M1421 showed weaker affinity than dUTP, one of the main substrates of ASFV dUTPase, its negative binding energy still indicates spontaneity. Moreover, since it had a stronger affinity than the other two controls, the core structure of M1421 could be modified or used as a scaffold for creating new compounds with enhanced binding strength and increased potency as an antiviral drug.
Since all ligands interacted more with subunit 2, the residues in this subunit provided more attractive interactions compared to those in subunit 1. As shown in Fig. 8A and 8B, the residues that consistently contributed the most to binding in both the MMGBSA and MMPBSA methods were Asp 29, Leu 30, Gly 31, Arg 71, Ser 72, and Gln 120 from subunit 1, and Ile 48, Ile 68, Gly 88, Leu 89, Ile 90, Asp 91, Tyr 94, Leu 98, Met 99, Leu 100, and Lys 101 from subunit 2. Among these, Asp 29, Arg 71, Ser 72, Gly 89, Ile 90, Tyr 94, and Gln 120 have been identified through kinetic studies as crucial for binding. Mutations or deletions of any of these residues led to decreased binding efficiency, weakening or disrupting the interaction of dUTPase with its natural substrate and severely impacting enzymatic activity.11 The strong binding of M1421 to these key residues underscores its potential as an antiviral compound for inhibiting ASFV dUTPase and highlights the importance of these residues as specific targets in designing more effective inhibitor compounds. Similar observations for the three control compounds further validated the results of our binding energy experiments.
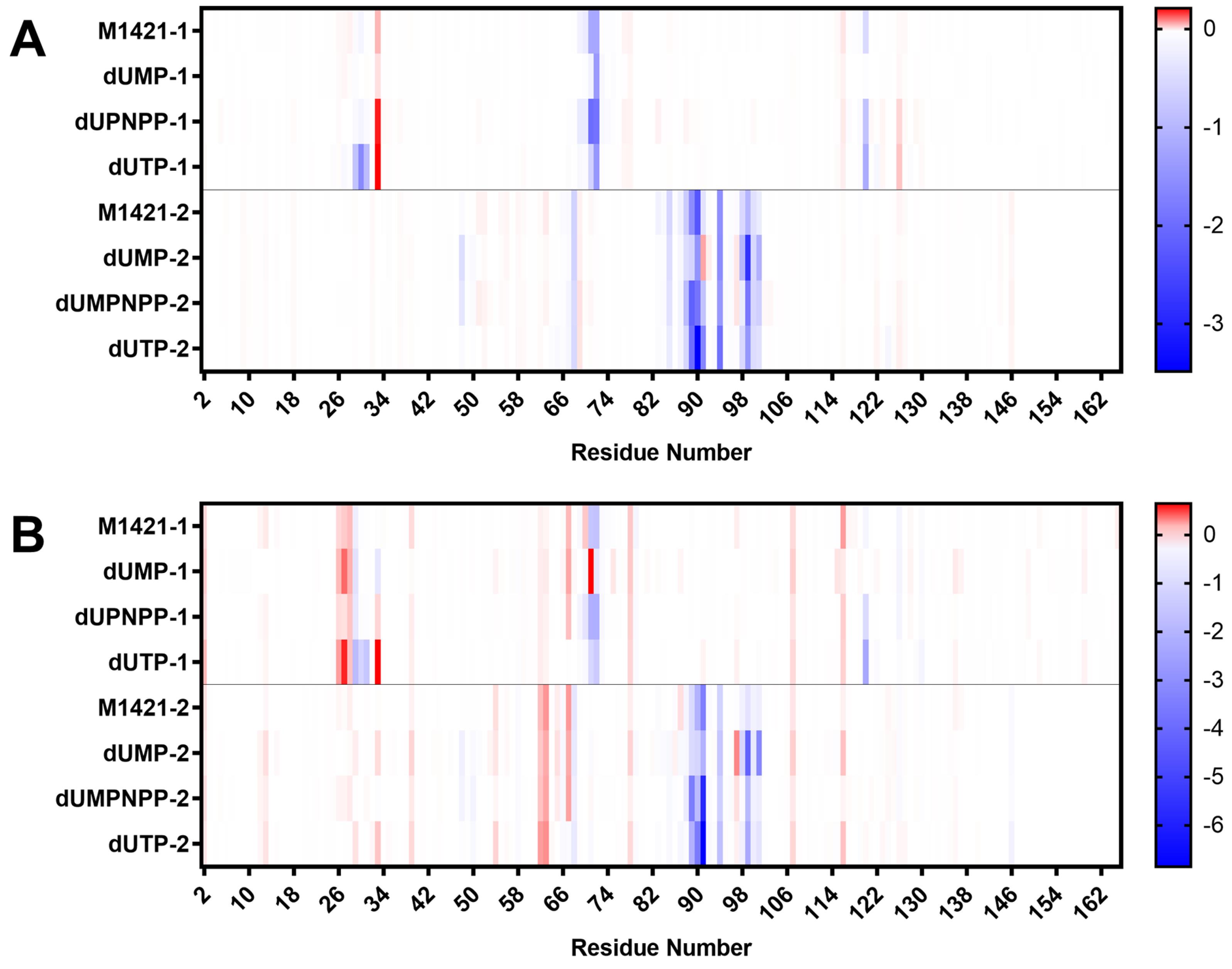
Fig. 8.
Binding energy contribution of the residues from subunit 1 and 2 of ASFV dUTPase, calculated using (A) MMGBSA and (B) MMPBSA. Blue bars represent attractive forces, while red bars represent repulsive forces.
.
Binding energy contribution of the residues from subunit 1 and 2 of ASFV dUTPase, calculated using (A) MMGBSA and (B) MMPBSA. Blue bars represent attractive forces, while red bars represent repulsive forces.
Bioactivity of tricycloalternarene C (M1421)
M1421, identified as tricycloalternarene C, is a sesquiterpenoid fungal metabolite derived from the sponge-associated fungus Alternaria sp.49 It is known for its inhibitory activity against the NF-κB protein complex, which is linked to autoimmune diseases, inflammatory conditions, and cancer.49,50 However, to the best of our knowledge, its antiviral activity against any pathogenic swine viruses has not been explored. Going back to the ADMET profiling, tricycloalternarene C performed favorably in 23 of the 24 ADMET tests. It scored 92.3% in the Protein Plasma Binding (PPB) test under Distribution, which exceeds the recommended threshold of less than 90%. Despite this, the compound met the criteria for other Distribution descriptors, such as Volume Distribution (VD) and Fraction Unbound in Plasma (Fu). Additionally, the fungus Alternaria sp., from which it was derived, is known for its wide range of bioactivities, including antitumor, antibacterial, and antitoxic effects. Apart from its observed antiviral activity against SARS-CoV-2 with Alternaria sp. HJT-Y7, no other antiviral activities against other viruses have been reported.51,52
While there are currently no approved antiviral drugs specifically targeting ASFV, several naturally occurring small molecules have shown promising inhibitory effects against key viral proteins. Flavonoids like kaempferol, apigenin, and dihydromyricetin have demonstrated antiviral activity,53-56 but most findings are limited to in vitro studies, and their specific viral targets remain unconfirmed. Despite favorable ADMET profiles, their potential interaction with native swine proteins raises safety concerns. In contrast, tricycloalternarene C specifically targets the ASFV A179L protein, showing strong binding and stability, suggesting better selectivity and safety.
Other strategies include nucleoside analogues (NAs) and siRNAs.55,57 NAs, such as genistein and HPMPC, inhibit viral replication but suffer from poor solubility, bioavailability, and dose-limiting toxicity.58 Their activation also depends on phosphorylation, a rate-limiting step that can significantly reduce their overall antiviral efficacy.59 Similarly, siRNAs can suppress viral replication but face challenges like off-target effects, poor stability, and limited delivery efficiency.57,60,61 In contrast, small-molecule inhibitors are generally more stable and can directly bind to viral target proteins with high specificity.62,63 These properties make tricycloalternarene C a more practical and potentially promising ASFV antiviral candidate.
Conclusion
A series of CADD techniques were used to screen and identify potential antiviral inhibitors against dUTPase from a pool of 4,683 marine fungal metabolites. The compounds were initially assessed for safety and efficacy through ADMET profiling, resulting in 328 metabolites with acceptable drug-likeness properties. These metabolites were then subjected to consensus molecular docking to predict their binding conformations with the lowest potential energy against the ASFV dUTPase active site. Ligands were further tested for their binding affinity to ensure they did not interfere with the normal function of swine dUTPase. Subsequently, the stability of the ten best-scoring complexes was examined through 300 ns molecular dynamics simulations. Among these, tricycloalternarene C (M1421) was the only metabolite that demonstrated a stable behavior with ASFV dUTPase, exhibiting RMSD trajectories similar to the three control compounds. Principal component analysis of the 300 ns MD run confirmed simulation convergence. The binding strength of tricycloalternarene C against the protein was also predicted using MMGBSA and MMPBSA methods and showed comparable binding energy with the control compounds. Van der Waals and electrostatic forces, along with hydrogen bonding, contributed to the stability and observed favorable binding affinity. Critical residues such as Asp 29, Arg 71, Ser 72, Gly 89, Ile 90, Tyr 94, and Gln 120 were also identified as significant for binding. Tricycloalternarene C has demonstrated activity in inhibiting the NF-κB protein, which is associated with autoimmune diseases and cancer. However, its antiviral properties against pathogenic viruses have not yet been explored. Further in vivo and in vitro studies are needed to confirm its potential as an antiviral inhibitor of ASFV dUTPase.
Research Highlights
What is the current knowledge?
-
ASFV continues to be a persistent threat to the livestock industry
-
There are no commercially available antiviral drugs for ASFV
-
Marine fungal metabolites are valuable sources of antiviral compounds against pathogenic swine viruses.
What is new here?
-
Computer-aided drug screening techniques and PCA were used to identify theoretical leads from marine fungal metabolites to inhibit the dUTPase enzyme of ASFV.
-
Tricycloalternarene C holds potential as a theoretical lead candidate for selectively inhibiting ASFV dUTPase.
Competing Interests
Authors declare no conflict of interests.
Ethical Approval
Not applicable.
Supplementary files
Supplementary file 1 contains Figs. S1-S4 and Tables S2-S4.
(pdf)
Supplementary file 2 contains Table S1.
(pdf)
Supplementary file 3 contains Video S1.
(mp4)
Acknowledgements
The authors would also like to thank the Department of Science and Technology - S&T Fellows Program, the Department of Science and Technology - Science Education Institute (DOST-SEI) Career Incentive Program, the Department of Science and Technology - Industrial Technology Development Institute (DOST-ITDI), and the Department of Science and Technology – Advanced Science and Technology Institute (DOST-ASTI) Computing and Archiving Research Environment (COARE) for their support to this research.
References
- Galindo I, Alonso C. African swine fever virus: a review. Viruses 2017; 9:103. doi: 10.3390/v9050103 [Crossref] [ Google Scholar]
- Kedkovid R, Sirisereewan C, Thanawongnuwech R. Major swine viral diseases: an Asian perspective after the African swine fever introduction. Porcine Health Manag 2020; 6:20. doi: 10.1186/s40813-020-00159-x [Crossref] [ Google Scholar]
- Orosco FL. Current progress in diagnostics, therapeutics, and vaccines for African swine fever virus. Vet Integr Sci 2023; 21:751-81. doi: 10.12982/vis.2023.054 [Crossref] [ Google Scholar]
- Wang T, Sun Y, Qiu HJ. African swine fever: an unprecedented disaster and challenge to China. Infect Dis Poverty 2018; 7:111. doi: 10.1186/s40249-018-0495-3 [Crossref] [ Google Scholar]
- You S, Liu T, Zhang M, Zhao X, Dong Y, Wu B. African swine fever outbreaks in China led to gross domestic product and economic losses. Nat Food 2021; 2:802-8. doi: 10.1038/s43016-021-00362-1 [Crossref] [ Google Scholar]
- Mason-D'Croz D, Bogard JR, Herrero M, Robinson S, Sulser TB, Wiebe K. Modelling the global economic consequences of a major African swine fever outbreak in China. Nat Food 2020; 1:221-8. doi: 10.1038/s43016-020-0057-2 [Crossref] [ Google Scholar]
- World Organisation for Animal Health (WOAH). African Swine Fever (ASF) – Situation Report 50. WOAH; 2024. Available from: https://www.woah.org/en/document/african-swine-fever-asf-situation-report-50/. Accessed May 20, 2024.
- Orosco FL. Host immune responses against African swine fever virus: insights and challenges for vaccine development. Open Vet J 2023; 13:1517-35. doi: 10.5455/OVJ.2023.v13.i12.2 [Crossref] [ Google Scholar]
- Wang G, Xie M, Wu W, Chen Z. Structures and functional diversities of ASFV proteins. Viruses 2021; 13:2124. doi: 10.3390/v13112124 [Crossref] [ Google Scholar]
- Li C, Chai Y, Song H, Weng C, Qi J, Sun Y. Crystal structure of African swine fever virus dUTPase reveals a potential drug target. mBio 2019; 10:e02483-19. doi: 10.1128/mBio.02483-19 [Crossref] [ Google Scholar]
- Liang R, Wang G, Zhang D, Ye G, Li M, Shi Y. Structural comparisons of host and African swine fever virus dUTPases reveal new clues for inhibitor development. J Biol Chem 2021; 296:100015. doi: 10.1074/jbc.RA120.014005 [Crossref] [ Google Scholar]
- Oliveros M, García-Escudero R, Alejo A, Viñuela E, Salas ML, Salas J. African swine fever virus dUTPase is a highly specific enzyme required for efficient replication in swine macrophages. J Virol 1999. 73: 8934-43. doi: 10.1128/jvi.73.11.8934-8943.1999.
- Durães F, Szemerédi N, Kumla D, Pinto M, Kijjoa A, Spengler G. Metabolites from marine-derived fungi as potential antimicrobial adjuvants. Mar Drugs 2021; 19:475. doi: 10.3390/md19090475 [Crossref] [ Google Scholar]
- Gonçalves MF, Esteves AC, Alves A. Marine fungi: opportunities and challenges. Encyclopedia 2022; 2:559-77. doi: 10.3390/encyclopedia2010037 [Crossref] [ Google Scholar]
- Hasan S, Ansari MI, Ahmad A, Mishra M. Major bioactive metabolites from marine fungi: a review. Bioinformation 2015; 11:176-81. doi: 10.6026/97320630011176 [Crossref] [ Google Scholar]
- Silber J, Kramer A, Labes A, Tasdemir D. From discovery to production: biotechnology of marine fungi for the production of new antibiotics. Mar Drugs 2016; 14:137. doi: 10.3390/md14070137 [Crossref] [ Google Scholar]
- Zorofchian Moghadamtousi S, Nikzad S, Abdul Kadir H, Abubakar S, Zandi K. Potential antiviral agents from marine fungi: an overview. Mar Drugs 2015; 13:4520-38. doi: 10.3390/md13074520 [Crossref] [ Google Scholar]
- Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov 2015; 14:111-29. doi: 10.1038/nrd4510 [Crossref] [ Google Scholar]
- Kikiowo B, Ogunleye AJ, Inyang OK, Adelakun NS, Omotuyi OI, Metibemu DS. Flavones scaffold of Chromolaena odorata as a potential xanthine oxidase inhibitor: induced fit docking and ADME studies. Bioimpacts 2020; 10:227-34. doi: 10.34172/bi.2020.29 [Crossref] [ Google Scholar]
- Divyashri G, Krishna Murthy TP, Sundareshan S, Kamath P, Murahari M, Saraswathy GR. In silico approach towards the identification of potential inhibitors from Curcuma amada Roxb against H pylori: ADMET screening and molecular docking studies. Bioimpacts 2021; 11:119-27. doi: 10.34172/bi.2021.19 [Crossref] [ Google Scholar]
- Maia EH, Assis LC, de Oliveira TA, da Silva AM, Taranto AG. Structure-based virtual screening: from classical to artificial intelligence. Front Chem 2020; 8:343. doi: 10.3389/fchem.2020.00343 [Crossref] [ Google Scholar]
- Singh E, Matada GS, Dhiwar PS, Patil RB, Pal R. In-silico based discovery of potential Keap1 inhibitors using the strategies of pharmacophore screening, molecular docking, and MD simulation studies. Bioimpacts 2025; 15:30335. doi: 10.34172/bi.30335 [Crossref] [ Google Scholar]
- Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol 2008; 426:145-59. doi: 10.1007/978-1-60327-058-8_8 [Crossref] [ Google Scholar]
- Lyu C, Chen T, Qiang B, Liu N, Wang H, Zhang L. CMNPD: a comprehensive marine natural products database towards facilitating drug discovery from the ocean. Nucleic Acids Res 2021; 49:D509-15. doi: 10.1093/nar/gkaa763 [Crossref] [ Google Scholar]
- Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C. ADMETlab 20: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res 2021; 49:W5-14. doi: 10.1093/nar/gkab255 [Crossref] [ Google Scholar]
- Zhao R, Hou L, Tesfagaber W, Song L, Zhang Z, Li F. Virtual screening and molecular dynamics simulation targeting the ATP domain of African swine fever virus type II DNA topoisomerase. Viruses 2025; 17:681. doi: 10.3390/v17050681 [Crossref] [ Google Scholar]
- Tang H, Mayersohn M. Porcine prediction of pharmacokinetic parameters in people: a pig in a poke?. Drug Metab Dispos 2018; 46:1712-24. doi: 10.1124/dmd.118.083311 [Crossref] [ Google Scholar]
- Gonzalez LM, Moeser AJ, Blikslager AT. Porcine models of digestive disease: the future of large animal translational research. Transl Res 2015; 166:12-27. doi: 10.1016/j.trsl.2015.01.004 [Crossref] [ Google Scholar]
- Rose EC, Blikslager AT, Ziegler AL. Porcine models of the intestinal microbiota: the translational key to understanding how gut commensals contribute to gastrointestinal disease. Front Vet Sci 2022; 9:834598. doi: 10.3389/fvets.2022.834598 [Crossref] [ Google Scholar]
- Guan L, Yang H, Cai Y, Sun L, Di P, Li W. ADMET-score - a comprehensive scoring function for evaluation of chemical drug-likeness. Medchemcomm 2019; 10:148-57. doi: 10.1039/c8md00472b [Crossref] [ Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009; 30:2785-91. doi: 10.1002/jcc.21256 [Crossref] [ Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010; 31:455-61. doi: 10.1002/jcc.21334 [Crossref] [ Google Scholar]
- Chang MW, Ayeni C, Breuer S, Torbett BE. Virtual screening for HIV protease inhibitors: a comparison of AutoDock 4 and Vina. PLoS One 2010; 5:e11955. doi: 10.1371/journal.pone.0011955 [Crossref] [ Google Scholar]
- Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules 2015; 20:13384-421. doi: 10.3390/molecules200713384 [Crossref] [ Google Scholar]
- Sarkar A, Concilio S, Sessa L, Marrafino F, Piotto S. Advancements and novel approaches in modified AutoDock Vina algorithms for enhanced molecular docking. Results Chem 2024; 7:101319. doi: 10.1016/j.rechem.2024.101319 [Crossref] [ Google Scholar]
- Huey R, Morris GM, Forli S. Using AutoDock 4 and Vina with AutoDockTools: A Tutorial. California, USA: Scripps Research Institute; 2011. Available from: http://www.bch.cuhk.edu.hk/croucher11/tutorials/day1_autodock_tutorial.pdf. Accessed June 5, 2024.
- Houston DR, Walkinshaw MD. Consensus docking: improving the reliability of docking in a virtual screening context. J Chem Inf Model 2013; 53:384-90. doi: 10.1021/ci300399w [Crossref] [ Google Scholar]
- Alberca LN, Prada Gori DN, Fallico MJ, Fassio AV, Talevi A, Bellera CL. LIDEB's Useful Decoys (LUDe): a freely available decoy-generation tool Benchmarking and scope. Artif Intell Life Sci 2025; 7:100129. doi: 10.1016/j.ailsci.2025.100129 [Crossref] [ Google Scholar]
- Vanommeslaeghe K, MacKerell AD Jr. Automation of the CHARMM general force field (CGenFF) I: bond perception and atom typing. J Chem Inf Model 2012; 52:3144-54. doi: 10.1021/ci300363c [Crossref] [ Google Scholar]
- Vanommeslaeghe K, Raman EP, MacKerell AD Jr. Automation of the CHARMM general force field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model 2012; 52:3155-68. doi: 10.1021/ci3003649 [Crossref] [ Google Scholar]
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 2010; 31:671-90. doi: 10.1002/jcc.21367 [Crossref] [ Google Scholar]
- Gil Pineda LI, Milko LN, He Y. Performance of CHARMM36m with modified water model in simulating intrinsically disordered proteins: a case study. Biophys Rep 2020; 6:80-7. doi: 10.1007/s41048-020-00107-w [Crossref] [ Google Scholar]
- Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 2015; 10:449-61. doi: 10.1517/17460441.2015.1032936 [Crossref] [ Google Scholar]
- Wang E, Sun H, Wang J, Wang Z, Liu H, Zhang JZH. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev 2019; 119:9478-508. doi: 10.1021/acs.chemrev.9b00055 [Crossref] [ Google Scholar]
- Valdés-Tresanco MS, Valdés-Tresanco ME, Valiente PA, Moreno E. gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J Chem Theory Comput 2021; 17:6281-91. doi: 10.1021/acs.jctc.1c00645 [Crossref] [ Google Scholar]
- Majidiani H, Pourseif MM, Kordi B, Sadeghi MR, Najafi A. TgVax452, an epitope-based candidate vaccine targeting Toxoplasma gondii tachyzoite-specific SAG1-related sequence (SRS) proteins: immunoinformatics, structural simulations and experimental evidence-based approaches. BMC Infect Dis 2024; 24:886. doi: 10.1186/s12879-024-09807-x [Crossref] [ Google Scholar]
- Lemkul JA. Introductory tutorials for simulating protein dynamics with GROMACS. J Phys Chem B 2024; 128:9418-35. doi: 10.1021/acs.jpcb.4c04901 [Crossref] [ Google Scholar]
- El-Demerdash A, Al-Karmalawy AA, Abdel-Aziz TM, Elhady SS, Darwish KM, Hassan AHE. Investigating the structure-activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv 2021; 11:31339-63. doi: 10.1039/d1ra05817g [Crossref] [ Google Scholar]
- Zhang G, Wu G, Zhu T, Kurtán T, Mándi A, Jiao J. Meroterpenoids with diverse ring systems from the sponge-associated fungus Alternaria sp JJY-32. J Nat Prod 2013; 76:1946-57. doi: 10.1021/np4005757 [Crossref] [ Google Scholar]
- Dąbek J, Kułach A, Gąsior Z. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB): a new potential therapeutic target in atherosclerosis?. Pharmacol Rep 2010; 62:778-83. doi: 10.1016/s1734-1140(10)70338-8 [Crossref] [ Google Scholar]
- Lu X, Tang XY, Wang HX, Huang WJ, Feng WX, Feng BM. Polyketone metabolites isolated from Rhodiola tibetica endohytic fungus Alternaria sp HJT-Y7 and their SARS-CoV-2 virus inhibitory activitives. Bioorg Chem 2021; 116:105309. doi: 10.1016/j.bioorg.2021.105309 [Crossref] [ Google Scholar]
- Zhao S, Li J, Liu J, Xiao S, Yang S, Mei J. Secondary metabolites of Alternaria: a comprehensive review of chemical diversity and pharmacological properties. Front Microbiol 2022; 13:1085666. doi: 10.3389/fmicb.2022.1085666 [Crossref] [ Google Scholar]
- da Fonseca RN, Maggioli MF, de Oliveira Hübner S, Bauermann FV. Antiviral effects of flavonoids on animal viruses. Virology 2025; 610:110596. doi: 10.1016/j.virol.2025.110596 [Crossref] [ Google Scholar]
- Juszkiewicz M, Walczak M, Woźniakowski G, Szczotka-Bochniarz A. Virucidal activity of plant extracts against African swine fever virus. Pathogens 2021; 10:1357. doi: 10.3390/pathogens10111357 [Crossref] [ Google Scholar]
- Orosco FL. Recent progress in plant-derived antiviral compounds against African swine fever virus. Vet Integr Sci 2024; 22:969-91. doi: 10.12982/vis.2024.066 [Crossref] [ Google Scholar]
- Zhang X, Chen S, Li X, Zhang L, Ren L. Flavonoids as potential antiviral agents for porcine viruses. Pharmaceutics 2022; 14:1793. doi: 10.3390/pharmaceutics14091793 [Crossref] [ Google Scholar]
- Keita D, Heath L, Albina E. Control of African swine fever virus replication by small interfering RNA targeting the A151R and VP72 genes. Antivir Ther 2010; 15:727-36. doi: 10.3851/imp1593 [Crossref] [ Google Scholar]
- Goulding LV, Kiss E, Goatley L, Vrancken R, Goris NE, Dixon L. In vitro and in vivo antiviral activity of nucleoside analogue cHPMPC against African swine fever virus replication. Antiviral Res 2022; 208:105433. doi: 10.1016/j.antiviral.2022.105433 [Crossref] [ Google Scholar]
- Kamzeeva PN, Aralov AV, Alferova VA, Korshun VA. Recent advances in molecular mechanisms of nucleoside antivirals. Curr Issues Mol Biol 2023; 45:6851-79. doi: 10.3390/cimb45080433 [Crossref] [ Google Scholar]
- Paul A, Muralidharan A, Biswas A, Kamath BV, Joseph A, Alex AT. siRNA therapeutics and its challenges: recent advances in effective delivery for cancer therapy. OpenNano 2022; 7:100063. doi: 10.1016/j.onano.2022.100063 [Crossref] [ Google Scholar]
- Zhang J, Chen B, Gan C, Sun H, Zhang J, Feng L. A comprehensive review of small interfering RNAs (siRNAs): mechanism, therapeutic targets, and delivery strategies for cancer therapy. Int J Nanomedicine 2023; 18:7605-35. doi: 10.2147/ijn.S436038 [Crossref] [ Google Scholar]
- Ngo HX, Garneau-Tsodikova S. What are the drugs of the future?. Medchemcomm 2018; 9:757-8. doi: 10.1039/c8md90019a [Crossref] [ Google Scholar]
- Wu K, Karapetyan E, Schloss J, Vadgama J, Wu Y. Advancements in small molecule drug design: a structural perspective. Drug Discov Today 2023; 28:103730. doi: 10.1016/j.drudis.2023.103730 [Crossref] [ Google Scholar]