Bioimpacts. 16:30943.
doi: 10.34172/bi.30943
Original Article
Designing a novel therapeutic candidate vaccine for HPV16 using immunoinformatics approach: Targeting E6 and E7 epitopes with TLR4 agonist integration
Zahra Samadi Moghaddam Investigation, Methodology, Writing – original draft, 1 
Maryam Mashhadi Abolghasem Shirazi Conceptualization, Investigation, Methodology, Visualization, Writing – original draft, 2
Rosa Jahangiri Writing – review & editing, 1
Babak Negahdari Supervision, Writing – review & editing, 3, *
Nasser Hashemi Goradel Conceptualization, Supervision, Visualization, 1, 4, *
Author information:
1Department of Medical Biotechnology, Maragheh University of Medical Sciences, Maragheh, Iran
2Department of Molecular Virology, Pasteur Institute of Iran, Tehran, Iran
3Department of Medical Biotechnology, School of Advanced Technologies in Medicine, Tehran University of Medical Sciences, Tehran, Iran
4Cancer Gene Therapy Research Center, Zanjan University of Medical Sciences, Zanjan, Iran
Abstract
Introduction:
Although prophylactic vaccinations for human papillomaviruses (HPVs) have been approved, these vaccines lack therapeutic efficacy and cannot eradicate pre-existing infections. Although epitope-based vaccines represent a promising therapeutic vaccine platform, their anti-tumor efficacy has been limited due to low immunogenicity. This study aimed to apply bioinformatics tools to design a built-in adjuvant therapeutic candidate vaccine targeting HPV16 infections and associated cancers.
Methods:
The designed vaccine consists of HPV16 E6 and E7 epitopes conjugated to the domain 4 of pneumolysin (Ply4) from Streptococcus pneumonia, which serves as a potential toll-like receptor 4 (TLR4) agonist. In silico analyses were performed to evaluate the vaccine's physicochemical properties, antigenicity, immunogenicity, and binding interactions with the TLR4 receptor. The designed vaccine was expressed in E. coli and its expression was confirmed by SDS-PAGE and Western blot analysis.
Results:
In silico analysis predicted that the designed vaccine could have desirable qualities, including non-toxicity, non-allergenicity, antigenicity, immunogenicity, hydrophilicity, and stability. Docking analysis between the vaccine and the TLR4 proteins predicted a high binding capacity and efficient binding. Furthermore, immunoinformatics tools showed that the vaccine could induce robust immune responses, specifically helper and cytotoxic T-cell responses, and promote the production of IFN-γ. The vaccine was successfully expressed in the E. coli system after being cloned into the pET28a vector. SDS-PAGE and Western blotting assays confirmed the purification of the target protein.
Conclusion:
The novel built-in adjuvant therapeutic candidate vaccine is a rationally designed construct for eradicating pre-existing HPV infections and HPV-induced cervical cancers that warrants further preclinical evaluation.
Graphical Abstract
Keywords: Human papillomavirus, Epitope, Bioinformatics, Adjuvant, Toll-like receptor 4, Pneumolysin
Copyright and License Information
© 2026 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This experiment was financially supported via the fund from Maragheh University of Medical Sciences, Maragheh, Iran (Grant No. 402000079).
Introduction
The most common and fatal cancer associated with HPV infection is cervical cancer, which is responsible for approximately 350,000 deaths in 2022, resulting from persistent infection with high-risk types of the virus, including HPV16 and HPV18.1,2 The genome of HPV is a circular, double-stranded DNA molecule with approximately 8,000 base pairs in length. It contains eight open reading frames (ORFs), which are categorized into two groups, including early and late genes. The early proteins produced by HPV, involving E1, E2, E4, E5, E6, and E7, are essential for regulating cell division, cellular signaling, viral DNA replication, and gene expression. In contrast, L1 and L2, which are the late ones, are responsible for constructing the viral coat.3 Although five vaccines, including Gardasil®, Gardasil 9®, Cervarix®, Cecolin®, and WalrinVax®, have been licensed against HPV,4 however, none of them have therapeutic efficacy. Existing prophylactic vaccines mechanistically prevent virus entrance into the cells by inducing anti-L1 antibodies; therefore, they do not work on already existing infections.5 Consequently, the development of potent and robust therapeutic candidate vaccines is of paramount importance and could yield substantial benefits in addressing this critical public health concern.
Epitope-based vaccines have received growing interest within the field of vaccine research due to several advantages over traditional vaccine approaches. These advantages include high precision, enhanced stability, excellent safety profiles, and ease of production and storage, making epitope-based vaccines a promising avenue for further exploration and development.6 Nonetheless, the inherent challenge of low immunogenicity associated with epitope-based vaccines has hitherto constrained their broader application; this limitation may be potentially overcome through the strategic incorporation of adjuvants.7 Built-in adjuvants, which means covalently linking adjuvants and antigens, offer a promising approach to elicit robust immune responses against specific targets without compromising safety.8 This strategy leverages the advantages of an ideal candidate vaccine, resulting in vaccines with desirable composition, consistent quality, and controllable properties. Furthermore, by delivering both antigen and adjuvant components simultaneously to antigen-presenting cells (APCs), these integrated adjuvants enhance immune activation and efficacy.9 Toll-like receptor (TLR) agonists based on proteins have become attractive adjuvant candidates in recent decades. These molecules have indicated stimulatory effects on the TLR signaling pathway, increase APC maturation, and trigger the synthesis of pro-inflammatory cytokines. The inherent peptide structure of these adjuvants offers flexibility in molecular design to optimize immunogenicity while minimizing toxicity. Furthermore, the fusion of an adjuvant with peptide antigens facilitates their co-delivery to target cells, thereby potentially inducing immune activation.10 Bacterial proteins that activate TLRs have been explored as potential vaccine adjuvants, such as pneumolysin (Ply) and flagellin.11,12 In particular, the C-terminal domain 4 of Ply (Ply4) from Streptococcus pneumoniae has been identified as a potent TLR4 agonist. Native Ply (pneumolysin) was found by Chiu et al to induce TLR4 activation and have hemolytic activity, but Ply4 was still able to provide TLR4 agonism with less cytotoxicity. They also identified specific mutations that decreased the cell-killing properties of Ply4.13 The decision to use Ply4 as an adjuvant has many advantages: as a protein, it is ideal for the built-in adjuvant approach, which allows direct genetic fusion of the adjuvant with target antigens. The requirement for co-delivery is important for the improved antigen presentation and T-cell activation, which are advantages of built-in adjuvants compared to non-covalently linked adjuvants.8,14 Moreover, TLR4 is particularly relevant in the context of HPV-associated cancers, as TLR4 agonism can help overcome the immune evasion properties of HPV-mediated activity and support activation of anti-tumor immunity.15,16 Using Ply4 to elicit TLR4 activity as part of an HPV candidate vaccine is novel because the strategy relates to established processes with TLR4 termed type of TLR4 agonism as part of a rationally designed multi-epitope protein assembly; strategies that are much less explored than other TLR agonists like MPLA or flagellin for HPV. In the present study, we employed the built-in adjuvant strategy for designing a therapeutic candidate vaccine against HPV16, using immunoinformatics tools. E6 and E7 epitopes of HPV16 were conjugated to Ply4 as an adjuvant. The designed therapeutic vaccine was expressed in Escherichia coli and finally, the recombinant protein was purified and confirmed.
Materials and Methods
Study design
The E6 and E7 proteins of HPV16, as well as pneumolysin (ABJ53672.1) from Streptococcus pneumoniae D39, were used for the vaccine design. Prediction of cytotoxic T lymphocyte epitopes and proteasome cleavage sites, along with population coverage, was performed. Subsequently, the physicochemical characteristics of the designed construct were assessed and then the secondary and tertiary structures were predicted. Following validation and refinement of the vaccine's 3D model, a molecular docking investigation of the construct's refined 3D model with TLR4 was carried out. Molecular dynamics (MD) modeling was used to assess the vaccine's stability and its ability to induce immune responses was simulated. Lastly, after expression in E. coli, the vaccine construct was separated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE), purified using column chromatography, and identified by Western blot. The workflow followed in this study is depicted in Fig. 1.
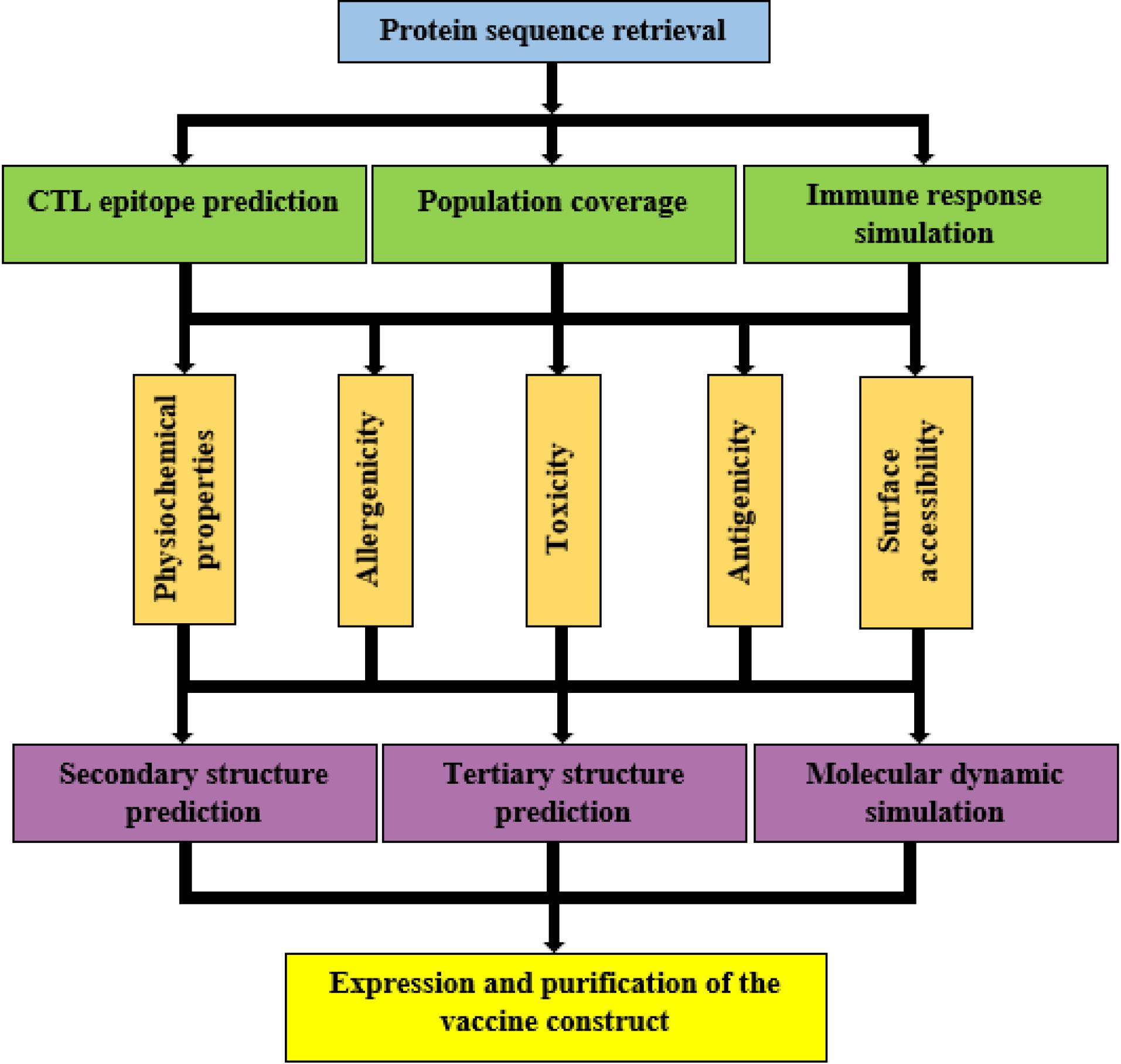
Fig. 1.
The schematicillustration of the methodology in the study.
.
The schematicillustration of the methodology in the study.
Retrieving protein sequences and developing a potential vaccine
Protein sequences for E6 (NP_041325.1) and E7 (NP_041326.1) from HPV16, along with a mutated form of Ply4 (ABJ53672.1) from Streptococcus pneumoniae D39 (a TLR4 agonist with reduced hemolytic activity, specifically the triple mutant Asp385Asn, Cys428Gly, and Trp433Phe as described by Chiu et al13), were acquired in FASTA format from the NCBI database (https://www.ncbi.nlm.nih.gov/).
The 6xHis-tag and the rigid linkers LLSVGG and LRMK were obtained from validated data. The mouse TLR4/MD-2/LPS complex (PDB ID: 3VQ2) sequence was obtained from the Protein Data Bank (PDB) for use in molecular docking studies (http://www.rcsb.org). The vaccine construct was designed to begin with methionine at the N-terminus. The TLR4 agonist, Ply4, was then connected to E6 and E7 epitopes using an LLSVGG linker. Lastly, an LRMK linker was used to join a His-tag encoding sequence to the C-terminus. Fig. 2 represents the graphical map of the designed candidate vaccine.
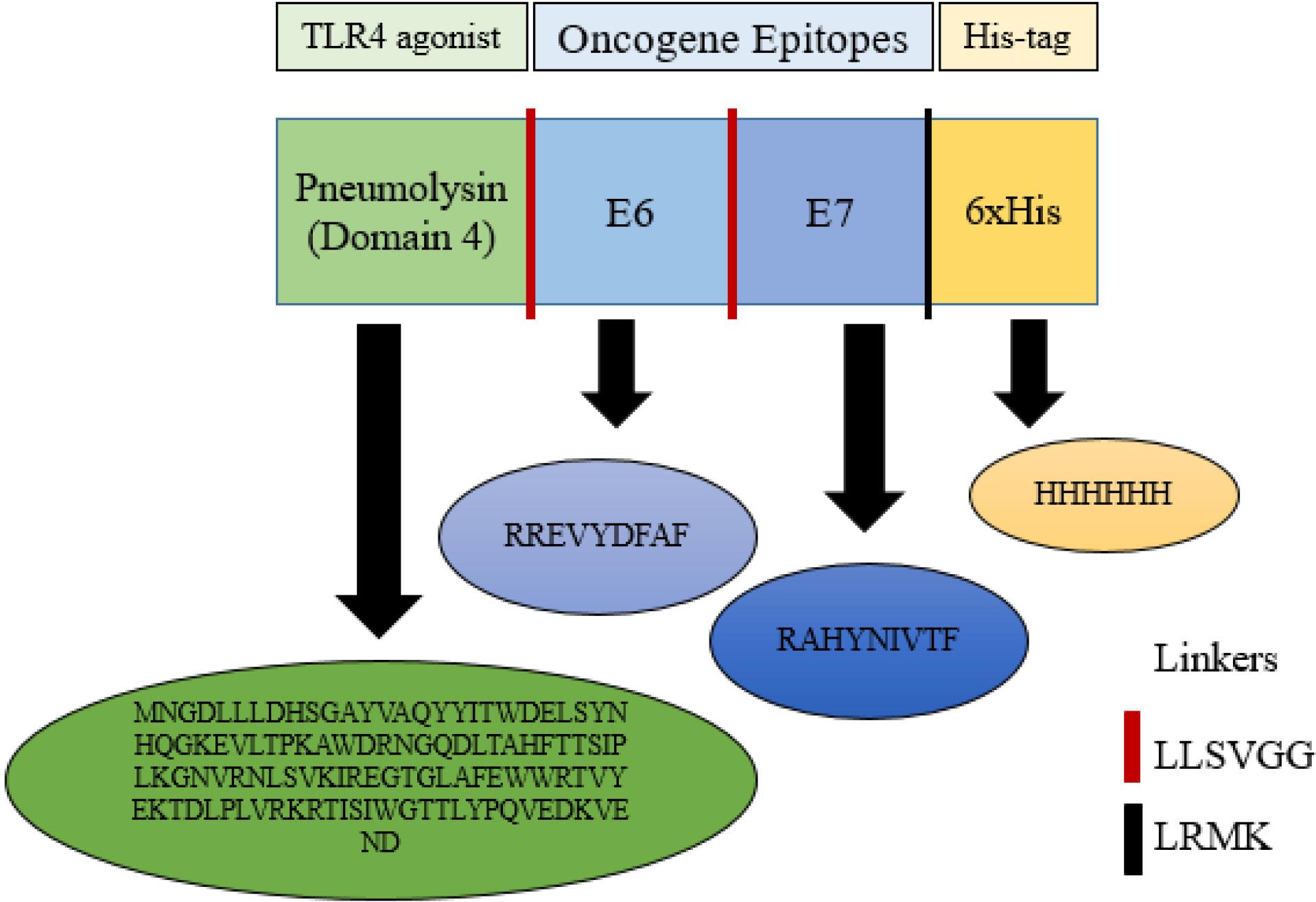
Fig. 2.
Schematic view of the vaccine construct. The Immunogenic platform contained the pneumolysin (the TLR4 agonist) tandem-linked by LLSVGG, displayed as the red dashes. Therefore, the oncogene E6 and E7 epitopes, tandem-linked by short rigid linkers of LLSVGG, are displayed as the red dashes. The C-terminal was His-tag (6xHis) for facilitating purification of the antigen linked by the LRMK linker, which is shown as the black dashes. The presented sequences indicate the amino acid sequences of each fragment.
.
Schematic view of the vaccine construct. The Immunogenic platform contained the pneumolysin (the TLR4 agonist) tandem-linked by LLSVGG, displayed as the red dashes. Therefore, the oncogene E6 and E7 epitopes, tandem-linked by short rigid linkers of LLSVGG, are displayed as the red dashes. The C-terminal was His-tag (6xHis) for facilitating purification of the antigen linked by the LRMK linker, which is shown as the black dashes. The presented sequences indicate the amino acid sequences of each fragment.
Prediction of cytotoxic T lymphocyte epitopes
The IEDB (http://tools.iedb.org/mhci/) is a resource that provides tools for the prediction of T-cell epitopes. An amino acid sequence's capacity to attach to a particular MHC molecule can be assessed using the IEDB MHC-I binding prediction tool. This information can be helpful in the design of candidate vaccines and other immunotherapies. Epitope lengths were set to 9 amino acids for both humans and mice. The inclusion of murine MHC alleles (H-2Db, H-2Dd, H-2Dq, H-2Kb, H-2Kd, H-2Kk, H-2Kq, H-2Ld, H-2Lq, H-2Qa1, and H-2Qa2) alongside human alleles (HLA-A and HLA-B) is based on the common practice in preclinical vaccine development, where mouse models are frequently used for initial in vivo validation studies. This approach helps identify epitopes that are likely to be effective in both human target populations and relevant animal models. Epitopes were selected based on the high binding affinity to MHC I alleles (percentile rank), percentile score, and antigenicity.
Prediction of proteasome cleavage sites
PCPS (http://tools.iedb.org/netchop/) utilizes n-gram models trained on naturally limited CD8 + T-cell epitopes (immunoproteasome models) and peptide fragments obtained from human MHC I molecules (proteasome models).
Prediction of population coverage
The IEDB server population coverage tool can be used to assess the population coverage of candidate vaccine peptides against MHC class I alleles from different populations. This tool allows users to upload a list of peptide sequences and select MHC class I alleles of interest. The tool then calculates the population coverage of the peptides for Iran and the total world population.
Prediction of antigenicity, allergenicity, and toxicity of the construct
Finding potential vaccines that can elicit an immune response is an essential step in the development process of a candidate vaccine. It is worth noting that the safety predictions were human-based in silico analyses, with murine studies planned for experimental validation. The VaxiJen v2.0 online tool (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was used to evaluate the antigenicity of the candidate vaccine platform. VaxiJen v2.0 analyzes the protein's physical and chemical characteristics to predict if it's an antigen. A computer program, called Kolaskar and Tongaonkar antigenicity server, was used to analyze the sequence of epitopes to identify if it might be able to trigger an immune response. The AllergenFP server also was used to assess the potential allergenicity of the proteins. It uses information about the proteins' amino acid sequence, including size, hydrophobicity, relative abundance, alpha-helix content, and beta-sheet formation, to distinguish between allergens and non-allergens (http://ddgpharmfac.net/AllergenFP/). The potential toxicity of the platform was investigated using the ToxinPred server.This server is a computational tool that analyzes protein sequences to predict whether they are toxic or not.
Prediction of physicochemical properties of the construct
A web-based tool called ProtParam (available at http://web.expasy.org/protparam/) was used to analyze the physicochemical properties of the chosen protein.
Surface accessible regions and hydrophilicity prediction
Ply4, a substance that activates the TLR4 immune receptor, must be exposed on the surface of a cell to interact with its receptor. Emini surface accessibility prediction and Parker hydrophilicity prediction of IEDB (http://tools.iedb.org/bcell/result/) tools were used to identify parts of the Ply4 molecule that are likely to be accessible on the surface and hydrophilic, respectively.
Secondary structure prediction
The SOPMA (http://npsa-pbil.ibcp.fr) server, a web-based application for predicting protein secondary structure, was used to analyze the secondary structure of the platform.
Tertiary structure and refinement prediction
The tertiary or 3D structure of the platform was modeled using the I-TASSER (http://zhanglab.ccmb.med.umich.edu/ITASSER/) server. I-TASSER is a computational tool that utilizes threading and assembly techniques to predict protein structure. The quality of the 3D models generated by I-TASSER was assessed using a C-score. The C-score is a numerical value that ranges from -5 to 2, with higher scores indicating more reliable models. This metric provides a quantitative measure of the confidence level associated with the predicted protein structures. To enhance the accuracy of the I-TASSER-generated protein structure model, the 3Drefine server was employed. 3Drefine (https://3drefine.mu.hekademeia.org/) is a computational tool that utilizes deep learning techniques to refine protein structures with high precision.
Validation of tertiary structure
A crucial phase in the model-building process is the validation of the tertiary structure, which aids in identifying any possible errors in the anticipated 3D models. For 3D model validation analyses, three web servers were employed. One of these servers was ProSA (https://prosa.services.came.sbg.ac.at/prosa.php), which estimates a total quality score (Z-score) based on the exact input structure. A higher Z-score indicates a better-quality model, while a lower Z-score suggests potential errors or inconsistencies in the predicted structure. If the Z-score calculated by ProSA falls outside the range of features observed in native proteins, it suggests that the predicted structure is likely to contain errors. This could be due to various factors, such as incorrect amino acid conformations, steric clashes, or violations of physical and chemical principles. Ramachandran plot (https://saves.mbi.ucla.edu/) illustrates the quality of the 3D modeled structure by displaying the percentage of amino acid residues that fall within the allowed and generously allowed regions of the Ramachandran plot. A high percentage of residues within these regions indicates a more accurate structure. ERRAT (http://services.mbi.ucla.edu/ERRAT/) server predicts non-bonded interactions between various atom types, providing insights into potential steric clashes or other structural irregularities.
Molecular docking of the construct with the immune receptor
An antigenic molecule interacts with a particular immune receptor to initiate the immunological response. TLR4/MD-2 is one such receptor that may identify lipopolysaccharide (LPS). The structure of TLR4/MD-2 in complex with LPS was obtained from the Protein Data Bank (PDB) (https://www.rcsb.org). HawkDock server (http://cadd.zju.edu.cn/hawkdock/) was a computational tool used to simulate how two molecules interact with each other at the molecular level. To establish a baseline for comparison, the interaction of unincorporated Ply4 with TLR4/MD-2 (PDB ID: 3VQ2, after computationally removing LPS and docking native Ply4 alone) was also docked. This control experiment allowed for the evaluation of the binding affinity and specificity of Ply4 to TLR4/MD-2 in the absence of any additional factors, providing a benchmark for the construct's interaction. It should be noted that the murine TLR4/MD-2 complex (PDB ID: 3VQ2) was employed for molecular docking because this structure represents one of the most complete and high-resolution crystallographic templates available for the TLR4 receptor. The murine and human TLR4 proteins share a high degree of sequence and structural similarity (approximately 75–80% identity), particularly in the ligand-binding domain, which allows reliable modeling of ligand–receptor interactions across species. Moreover, murine models are routinely used in preclinical immunization and vaccine validation studies; therefore, assessing the construct’s interaction with murine TLR4 provides translationally relevant insights for subsequent in vivo experiments.
Molecular dynamics simulation
The iMODS (http://imods.Chaconlab.org/) server, a freely accessible online tool, was utilized for this simulation. iMODS is a fast and efficient server designed to analyze protein flexibility and dynamics.
Immune simulation against the construct
The C-ImmSim (https://150.146.2.1/C-IMMSIM/index.Php) agent-based modeling server was employed to simulate the interactions between the candidate vaccine and the human immune system. The vaccine construct profile is assumed to be administered three injections at different intervals of 4 weeks. This regimen aimed to induce a robust and sustained immune response against the target pathogen. Time points were selected at 0, 28, and 35 days to match the vaccine delivery schedule, and all simulation parameters were left at their default settings.
Expression and purification of recombinant protein
For expression in E. coli BL21 (DE3), the platform's DNA sequence was synthesized and subcloned into the pET28a vector between the HindIII and XhoI restriction sites (Biomatik, Canada). At 37 °C, 1 mM IPTG (Isopropyl-β-1-thiogalactopyranoside) (Sigma, UK) was added to stimulate protein expression. After 4 h of incubation, the cell pellet was prepared for the purification step. The protein expression was confirmed by SDS-PAGE and Western blot using anti-6x His tag antibody (Abcam, UK) according to the standard protocol. According to the manufacturer's instructions (Qiagen, Germany), the recombinant protein was purified under native circumstances using a Ni-NTA agarose column, and the Bradford technique was used to measure the protein's concentration. The endotoxin level of the purified recombinant protein was evaluated by the chromogenic Limulus amoebocyte lysate (LAL) test (BioWhittaker, UK) according to the manufacturer's instructions.
Results
Prediction of CTL epitopes
The IEDB tool was used to identify potential CTL epitopes within the fusion protein. These epitopes were nine amino acids long and were selected based on their predicted effectiveness, which was determined using a percentile rank threshold. The identified CTL epitopes were found to have a strong binding affinity to MHC-I molecules. This was determined using the vaxigen v.2 Kolaskar and Tongaonkar tool, which calculates antigenicity scores. Human alleles predicted epitopes are HLA-B such as HLA-B*07:02, HLA-B*08:01, HLA-B*51:01, HLA-B*53:01, HLA-B*40:01, and HLA-A such as HLA-A*23:01, HLA-B*24:02, HLA-B*30:01, and HLA-B*30:02. The E6 and E7 epitopes 10 peptides in mice (Table 1) and 9 peptides in human (Table 2) were predicted to binding to MHC-I molecules.
Table 1.
Mice MHC-I binding epitope prediction
|
No.
|
Protein
|
Peptide sequence
|
Allele
|
Start
|
End
|
Percentile score
|
Percentile rank
|
Antigenicity
|
| 1 |
E6 |
RREVYDFAF |
H-2-Kb |
120 |
128 |
0.569376 |
0.08 |
|
| 2 |
|
|
H-2-Lq |
|
|
0.067309 |
0.92 |
0.8597 ( + ) |
| 3 |
|
|
H-2-Qa1 |
|
|
0.701394 |
0.04 |
|
| 4 |
|
|
H-2-Dd |
|
|
0.134196 |
0.09 |
|
| 5 |
|
|
H-2-Ld |
|
|
0.203934 |
0.38 |
|
| 6 |
|
|
|
|
|
0.138867 |
0.38 |
|
| 7 |
E7 |
RAHYNIVTF |
H-2-Kb |
135 |
143 |
0.17384 |
0.6 |
0.7355 ( + ) |
| 8 |
|
|
H-2-Qa2 |
|
|
0.146852 |
0.67 |
|
| 9 |
|
|
H-2-Dq |
|
|
0.292791 |
0.8 |
|
| 10 |
|
|
H-2-Kd |
|
|
0.075327 |
0.97 |
|
Table 2.
Human MHC-I binding epitope prediction
|
No.
|
Protein
|
Peptide sequence
|
Allele
|
Start
|
End
|
Percentile score
|
Percentile rank
|
Antigenicity
|
| 1 |
E6 |
RREVYDFAF |
HLA-B*40:01 |
120 |
128 |
0.193592 |
0.65 |
0.8597 ( + ) |
| 2 |
|
|
HLA-B*53:01 |
|
|
0.201439 |
0.42 |
|
| 3 |
|
|
HLA-A*23:01 |
|
|
0.180955 |
0.47 |
|
| 4 |
|
|
HLA-A*24:02 |
|
|
0.185655 |
0.54 |
|
| 5 |
E7 |
RAHYNIVTF |
HLA-A*30:01 |
135 |
143 |
0.235038 |
0.61 |
0.7355 ( + ) |
| 6 |
|
|
HLA-A*30:02 |
|
|
0.191695 |
0.74 |
|
| 7 |
|
|
HLA-B*07:02 |
|
|
0.150617 |
0.84 |
|
| 8 |
|
|
HLA-B*51:01 |
|
|
0.156262 |
0.89 |
|
| 9 |
|
|
HLA-B*08:01 |
|
|
0.116888 |
0.99 |
|
Prediction of proteasome cleavage sites
The software predicted the potential of the peptide to be a CTL epitope by analyzing its proteasomal processing, TAP transport, and MHC-I binding properties. Peptides with a score above 0.5 were considered potential epitopes. The green regions represent proteasomal cleavage sequences, which include the E6, and E7 epitopes and linker regions (Supplementary file 1, Fig. S1).
Prediction of population coverage
The identified MHC-I binders within the platform were analyzed to determine their population coverage. This analysis predicted the percentage of people in various regions, particularly the Persian population, who would likely be responsive to these epitopes. The goal was to select epitopes that could cover the majority of individuals infected with HPV. The population coverage analysis revealed that the epitopes had the highest coverage (69.53%) in the West Indies. Following the West Indies, the epitopes had the highest coverage in East Asia (66.30%), Oceania (65.43%), North America (64.87%), Europe (62.52%), and Iran (55.51%) (Fig. S2). The overall population coverage for each region is summarized in Table S1. These results demonstrate the variability of coverage in different populations, indicating that while the candidate vaccine is reasonably accessible at a global level, optimizing it for heterogenous and particularly high-incidence populations (e.g., Sub-Saharan African or Central/South American populations) would require further consideration of epitopes and specific HLA profiles in future iterations of design.
Prediction of antigenicity, allergenicity, and safety of the construct
The platform was analyzed using VaxiJen v2.0 and Kolaskar and Tongaonkar tools to determine its potential as an antigen. Both tools predicted that the protein could be a good antigen. The VaxiJen v2.0 tool used a standard threshold of 0.4 to assess antigenicity. The antigen platform showed an antigenicity score of 0.5757. The Kolaskar and Tongaonkar tool identified the E6 and E7 regions of the platform as potential antigens (Fig. S3). According to the AllergenFP server's documentation, an allergenicity score above 0.75 is generally considered non-allergic. The platform's score of 0.77 suggests it might not be allergenic. Additionally, toxicity tests predicted that the platform is non-toxic (Table 3).
Table 3.
Antigenicity, allergenicity, and safety of the platform predicated by bioinformatics analysis
|
No.
|
Evaluated immunogenicity
|
Results
|
| 1 |
Antigenicity (Vaxijen v.2) |
0.5757 antigen |
| 2 |
Allergenicity (Allergen FP) |
0.77 non-allergen |
| 3 |
Toxicity (Toxinpred) |
Non-toxic |
Prediction of physicochemical property
The molecular weight of the protein was calculated at about 17,777.11 kilodaltons (kDa), which consist of 153 amino acids. The instability index below 40 indicates a stable protein. In the present experiment, an index of 16.50 was found which indicates this protein is stable. The Gravy tool assessed the protein's hydrophobicity and hydrophilicity. A more negative Gravy score indicates greater hydrophilicity. The platform's Gravy score of -0.483 suggests that it is hydrophilic. An aliphatic index score of 85.95 suggests that the protein lacks surface electric charge at a pH of 8.04, which is the theoretical isoelectric point, or pI. The protein has 18 positively charged residues and 17 negatively charged residues. Based on these bioinformatics analyses, the platform is polar, hydrophilic, positively charged, and stable (Table 4).
Table 4.
Physicochemical characteristics of the platform predicated by bioinformatics analysis
|
No.
|
Evaluated characteristics
|
Results
|
| 1 |
No. of amino acids |
153 aa |
| 2 |
Molecular weight |
17777.11 kDa |
| 3 |
Instability index |
16.50 |
| 4 |
Gravy |
-0.483 |
| 5 |
Aliphatic index |
85.95 |
| 6 |
Theoretical pI |
8.04 |
| 7 |
Total No. of positively charged residues (Arg + Lys) |
18 |
| 8 |
Total No. of negatively charged residues (Asp + Glu) |
17 |
Prediction of surface-accessible regions
The hydrophilic regions of Ply4, which acts as a TLR4 agonist, are important because they need to be exposed on the surface to allow drugs to enter cells and interact with the receptor. Using the Emini and Parker tools, the surface accessibility and hydrophilicity of Ply4, which acts as a TLR4 agonist were analyzed. We set a threshold of 1.000 for surface accessibility and 1.046 for hydrophilicity. Ply4 scored above both thresholds, indicating high surface accessibility and hydrophilicity (Fig. S4A and Fig. S4B, respectively).
Prediction of secondary structure
We analyzed the overall structure of the platform sequence using the SOPMA tool. The results showed that it has predominantly random coil/extended strand, but has a significant amount of extended strands (35.95%) and random coils (60.78%) (Fig. S5).
Tertiary structure modeling and refinement
Five 3D models of the platform were predicted using the I-TASSER service. These models were based on ten threading templates and had confidence scores (C-score) ranging from -3.43 to -1.77. The C score ranges from -5 to 2, with high scores indicating high confidence. The best 3D model, which had a confidence score of -1.77, was chosen for further analysis (Fig. 3A). Although this C-score is acceptable for protein structure prediction in de novo, it implies that the model is likely a plausible structural hypothesis, rather than a true, experimentally solved structure, which needs to be answered through experiment. The 3D model had a TM-score of 0.50 with a standard deviation of 0.15. The expected RMSD score was 8.6 with a standard deviation of 4.5 Å. The TM-score is a better way to measure how similar a designed structure is to a natural one compared to the RMSD. RMSD is easily affected by errors in the natural structure. The 3Drefine server was used to enhance the stability and energy minimization of the modeled protein. We used the 3Drefine server to refine the platform model and got five new model structures. Based on factors like the RW-plus score (-28291.419953) and 3Drefine score (9414.99), model 5 was the best quality. A lower 3Drefine score and RW-plus score indicate a higher quality model. Because model 5 had the lowest scores, we chose it for further validation (Fig. 3B).
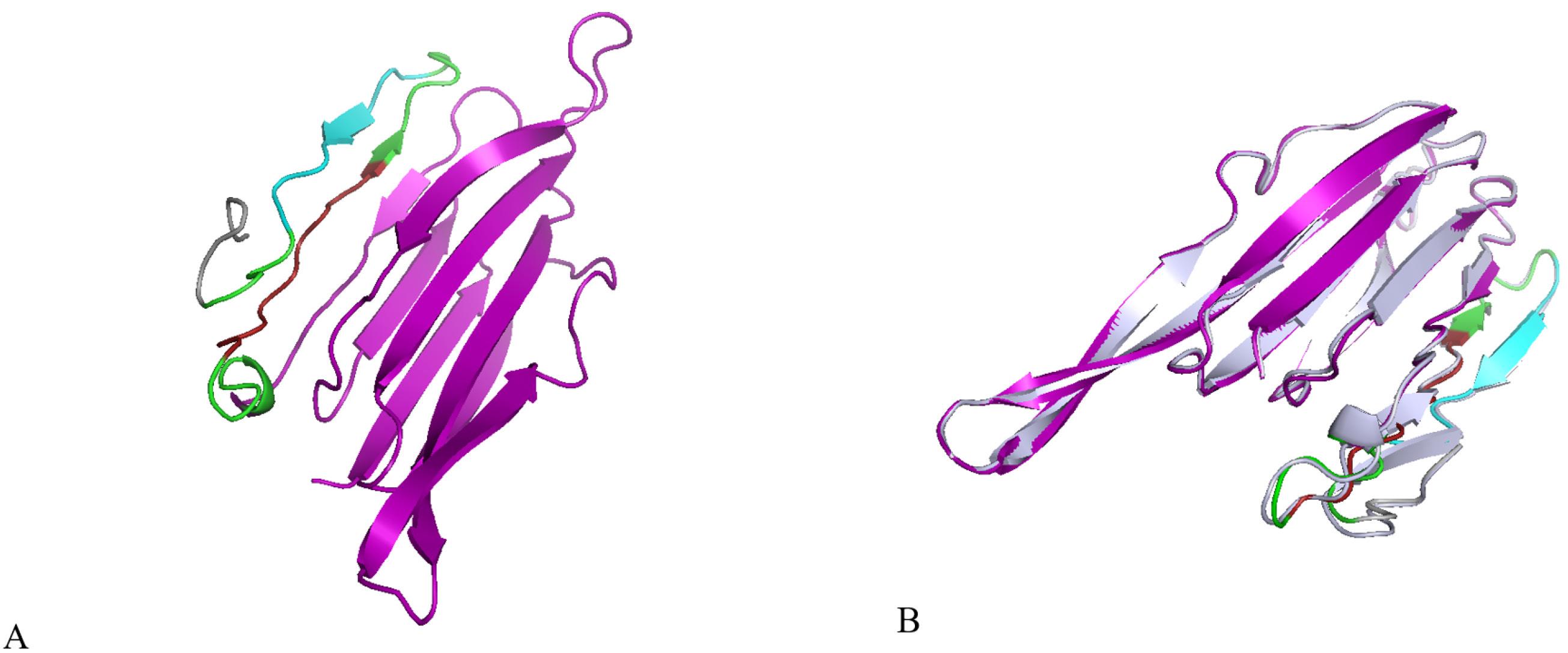
Fig. 3.
Protein 3D structural modelling and refinement. (A) The 3D model of the platform was obtained on the I-TASSER server. (B) Alignment by the 3Drefine server of a refined 3D structure (colored) on a crude model (blue-white). Ply4 is shown in purple, E6 epitope in red, E7 epitope in cyan, linkers in green, and His-tag in gray
.
Protein 3D structural modelling and refinement. (A) The 3D model of the platform was obtained on the I-TASSER server. (B) Alignment by the 3Drefine server of a refined 3D structure (colored) on a crude model (blue-white). Ply4 is shown in purple, E6 epitope in red, E7 epitope in cyan, linkers in green, and His-tag in gray
Tertiary structure validation
The ProSA and ERRAT servers were used to check the quality and find potential errors in the 3D model. ProSA estimated the Z-score of the input platform to be -3.27 (Fig. 4A). The selected model after refinement had an overall quality factor of 86.207% by the ERRAT server (Fig. 4C). The Ramachandran plot of the chosen model was analyzed using the Ramachandran plot server. The results showed that 71.9% of the amino acids were in the favored regions, 20.7% were in the allowed regions, 5.9% were in the generously allowed regions, and 1.5% were in the disallowed regions (Fig. 4B). Although the percentages indicate a generally reliable structure, the presence of a small percentage of residues in disallowed areas implies areas that may need additional refinement or possible inherent flexibility in the predicted structure. Overall, the results from ProSA, ERRAT, and the Ramachandran plot confirmed the good quality of the 3D protein model and provided a solid in silico basis for further experimental studies.
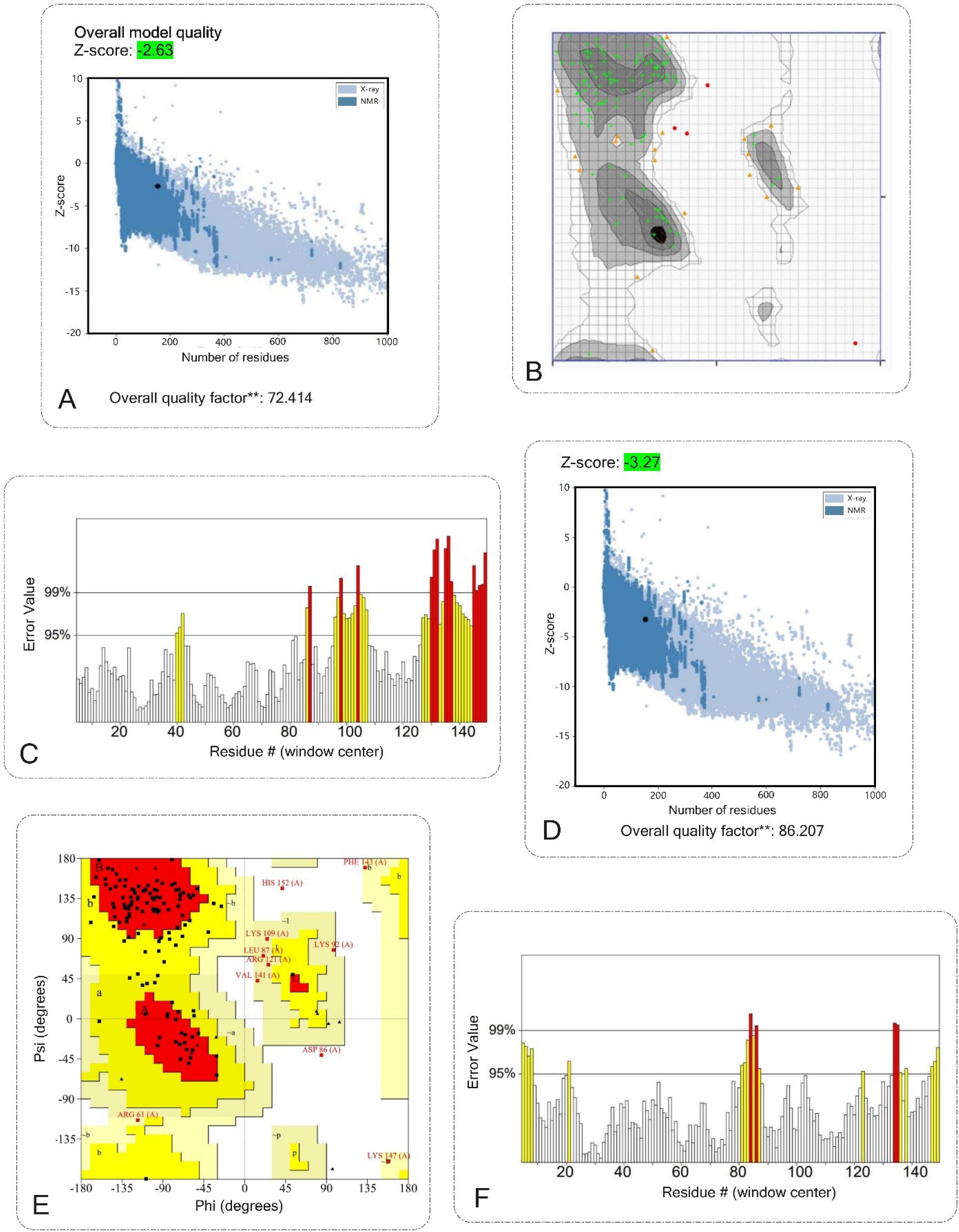
Fig. 4.
Validation of the 3D structure of the vaccine construct. A, B and C validation shows the vaccine structure before modification. D, E and F validation shows the vaccine structure after modification.A, D) ProSA server with a Z-score of before modification was -2.63 and after modification was -3.27. B, E) Ramachandran plot analyses before and after modification show 71.9%, 83.70% favored regions respectively, 20.7%, 13.33% allowed regions respectively, 5.9%, 2.96% generously allowed regions respectively, and 1.5% generously allowed protein residues. C, F) The ERRAT plot analysis showed that the overall quality factor of the before modification was 72.414%** and after refined construct is as high as 86.207%**. * In the error axis, two lines were drawn to ensure that areas that are higher than the error value can be passed. **Expressed as the percentage of the protein. The calculated error value falls below the 95% rejection threshold. High-resolution structures generally produce values of about 95% or higher. For low resolution (2.5 to 3 A), the average overall quality factor is about 91%. All three servers confirmed the quality and possible errors in the 3D model.
.
Validation of the 3D structure of the vaccine construct. A, B and C validation shows the vaccine structure before modification. D, E and F validation shows the vaccine structure after modification.A, D) ProSA server with a Z-score of before modification was -2.63 and after modification was -3.27. B, E) Ramachandran plot analyses before and after modification show 71.9%, 83.70% favored regions respectively, 20.7%, 13.33% allowed regions respectively, 5.9%, 2.96% generously allowed regions respectively, and 1.5% generously allowed protein residues. C, F) The ERRAT plot analysis showed that the overall quality factor of the before modification was 72.414%** and after refined construct is as high as 86.207%**. * In the error axis, two lines were drawn to ensure that areas that are higher than the error value can be passed. **Expressed as the percentage of the protein. The calculated error value falls below the 95% rejection threshold. High-resolution structures generally produce values of about 95% or higher. For low resolution (2.5 to 3 A), the average overall quality factor is about 91%. All three servers confirmed the quality and possible errors in the 3D model.
Protein-protein molecular docking
Researchers investigating TLR4 modulators often focus on the LPS binding site. However, because many potential ligands are based on the LPS structure, molecular docking was needed to verify if Ply4 could be a suitable therapeutic target, given its distinct characteristics. Docking analysis revealed a predicted interaction between the ligand and receptor, with a high HowkDock score for Ply4. This suggests a potential binding affinity between Ply4 and the MD-2/TLR4 receptor. Molecular docking simulations demonstrated that Ply4 from the platform construct could effectively bind to the MD2/TLR4 receptor (Fig. 5A). The alignment of the control docking complex with the platform docking complex was similar to the natural interaction. This was confirmed by a comparable E-total value of -6375.730140 kcal/mol for Ply4's interaction with the MD2/TLR4 complex (Fig. 5B). The visual alignment in Fig. 5B illustrates that the Ply4 epitope within the platform construct is predicted to engage with the MD2/TLR4 binding site in a manner consistent with that of unincorporated Ply4, suggesting a preserved functional interaction. The results indicated that the MD2/TLR4 receptor interacted with Ply4 in the platform in a way similar to its interaction with a natural ligand. The choice of this binding pose for modeling in subsequent MD simulations is also supported by research showing that the C-terminal domain of pneumolysin, specifically Ply4, is an important region for mediating TLR4 activation, and that there are residues within the C-terminal domain that are important for this interaction. The test docking procedure revealed that the Ply4 domain contained in the construct we used corresponds to the native TLR4 binding mechanism that has been characterized with Ply4, further supporting this simulated scenario. In summary, it must be emphasized that the in silico modeling assessment of binding affinity and binding interaction requires experimental confirmation by Surface Plasmon Resonance (SPR) or Isothermal Titration Calorimetry (ITC) for functional binding.
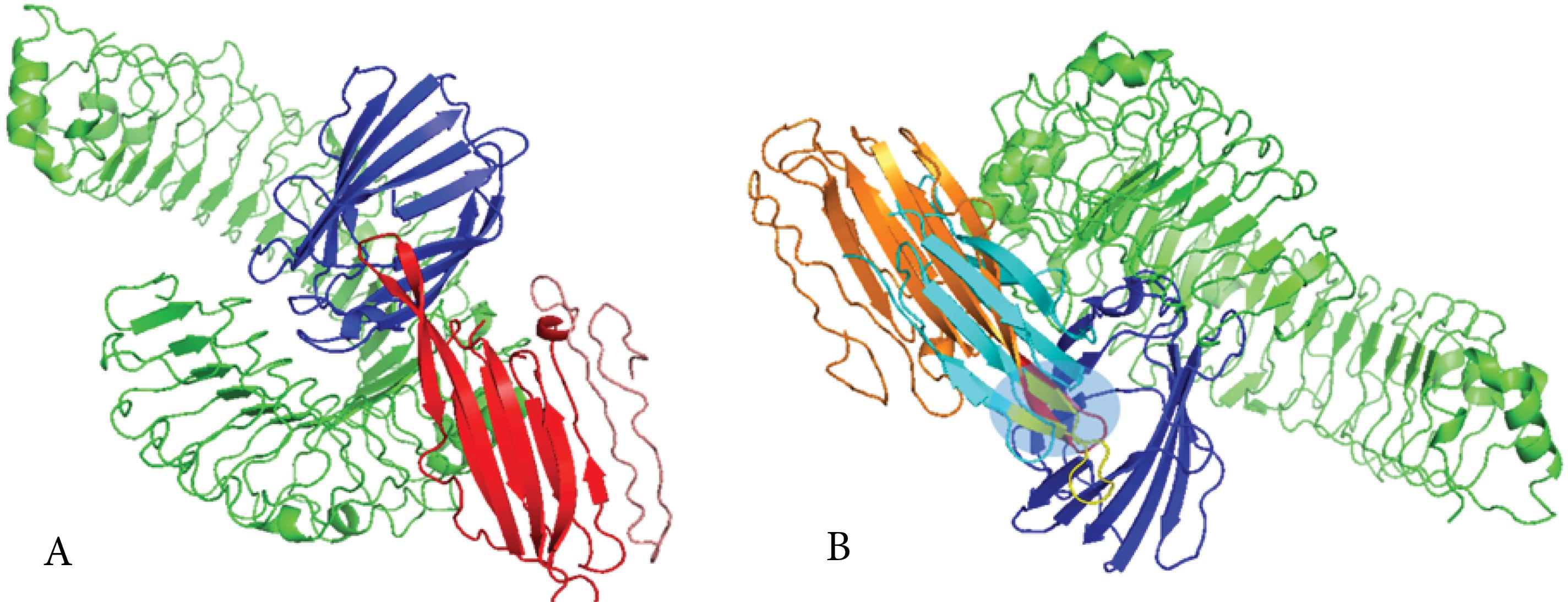
Fig. 5.
Analyses of the molecular docked complex between the platform and MD-2/TLR4. (A) 3D structure of molecular docking simulation between MD-2/TLR4 and the ply epitope in the platform construct. The salmon-red chain is the platform construct, the red ribbon is the Ply4 epitope in the construct, the blue chain is MD-2, and the green chain is TLR4. Results of the interaction analyses of the platform with MD-2/TLR4 by HawkDock server showed good affinities between the co-receptor TLR4 (MD-2) and the Ply4 (TLR4 agonists) modelled platform. (B) Alignment of molecular docking of natural ply with MD2/TLR4 as a control complex and Ply4 of platform construct with MD2/TLR4. The blue chain is MD-2, the green chain is TLR4, the cyan chain is natural ply, the yellow chain is the natural ply, the orange chain is the platform, and the red chain is the ply of the platform. The light blue box shows the binding site of the ligand to the receptor alignment between the natural Ply4 and Ply4 of the platform.
.
Analyses of the molecular docked complex between the platform and MD-2/TLR4. (A) 3D structure of molecular docking simulation between MD-2/TLR4 and the ply epitope in the platform construct. The salmon-red chain is the platform construct, the red ribbon is the Ply4 epitope in the construct, the blue chain is MD-2, and the green chain is TLR4. Results of the interaction analyses of the platform with MD-2/TLR4 by HawkDock server showed good affinities between the co-receptor TLR4 (MD-2) and the Ply4 (TLR4 agonists) modelled platform. (B) Alignment of molecular docking of natural ply with MD2/TLR4 as a control complex and Ply4 of platform construct with MD2/TLR4. The blue chain is MD-2, the green chain is TLR4, the cyan chain is natural ply, the yellow chain is the natural ply, the orange chain is the platform, and the red chain is the ply of the platform. The light blue box shows the binding site of the ligand to the receptor alignment between the natural Ply4 and Ply4 of the platform.
Molecular dynamics simulation
The present study analyzed the molecular dynamics simulation and normal mode analysis of the docked complex between the platform construct and TLR4. The B-factor/mobility simulation was applied to assess the predicted flexibility of atoms and molecules within the platform construct. The mobility map and deformability plot demonstrate areas of the Ply4–TLR4/MD-2 complex that show high flexibility (peaks) and low flexibility (troughs). The majority of the peaks align with loops at the edges of the interface, and the binding interface itself revealed low flexibility, indicative of stable formation of contact (Fig. 6A and Fig. 6B). The B-factor profile is consistent with the deformability assessment and shows that the highest mobility regions are mostly defined by solvent-exposed loops that move more compared to the interface residues that are rigid and therefore have low mobility. This is also consistent with the expected stability of the binding mode as defined over the course of the simulation. (Fig. 6C). The calculated eigenvalue (1.813 × 10-5) represents the amount of energy needed to deform the model in the slowest mode of motion. The eigenvalue’s relatively low value suggests that the complex is not too stiff and would allow a necessary amount of conformational change for a receptor–ligand interaction (Fig. 6D). The variability of each normal mode is inversely proportional to its eigenvalue. Green indicates the cumulative variance, while red represents the individual variances (Fig. 6E). The co-variance diagram of the docked complex illustrates the predicted correlated, uncorrelated, and anti-correlated movements between pairs of residues using red, white, and blue colors, respectively (Fig. 6F). The elastic network plot shows dense gray connectivity in the core binding interface, indicating strong intramolecular connections that maintain structural integrity during simulation (Fig. 6G). Generally, the MD simulations indicate the docked complex remained in a structurally stable conformation over the simulated period, providing some in silico evidence that the construct may associate with TLR4 and motivating further in vitro and in vivo evaluation of the vaccine–TLR4 interaction.
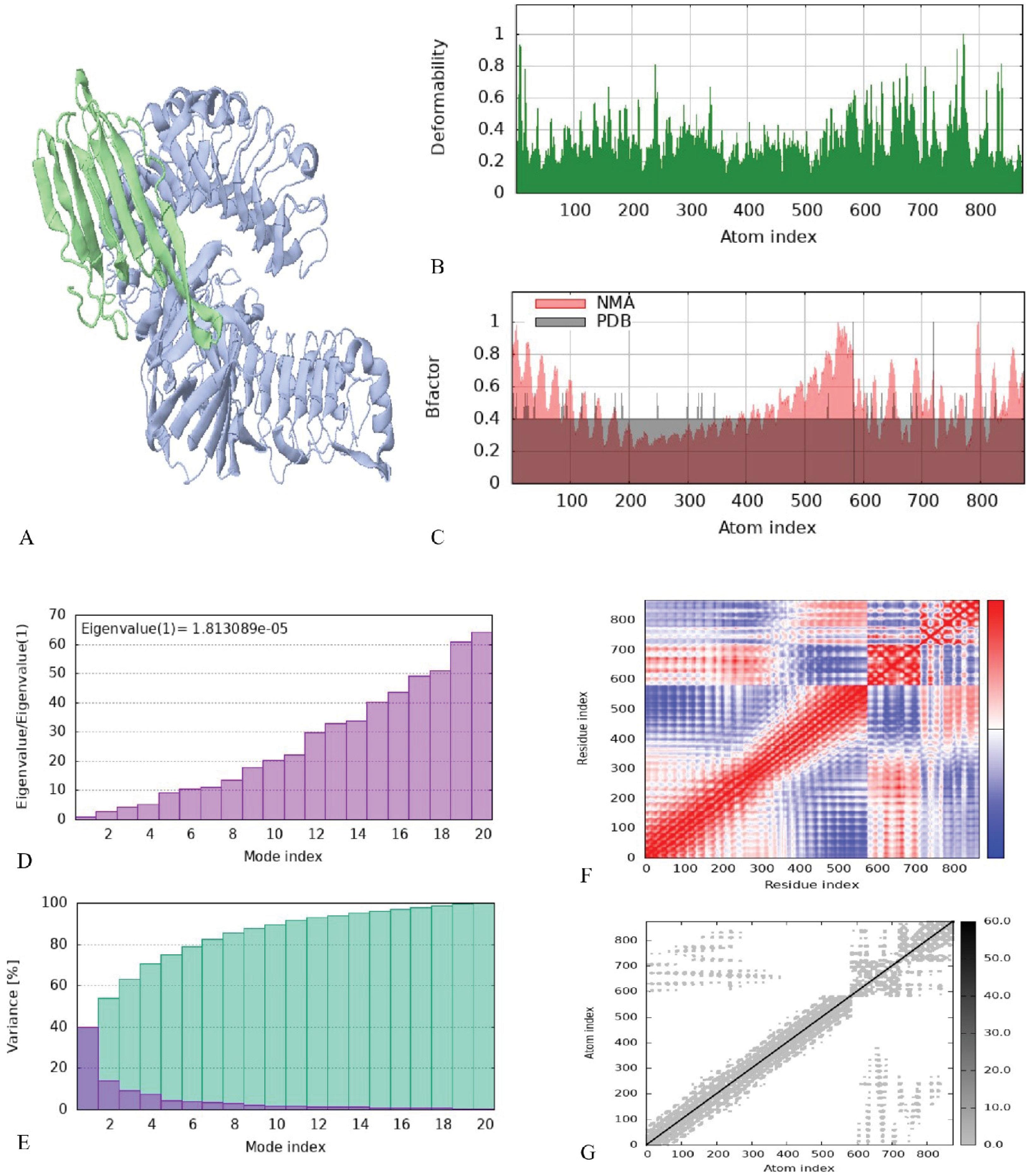
Fig. 6.
Analyses of the molecular dynamics simulation of the platform and TLR4 docked complex. (A) 3D structure of NMA mobility. The purple chain is MD2/TLR4, and the green chain is the platform. (B) Deformability plot, the peaks in the graph are deformability regions of the protein. (C) B-factor diagram. (D) Eigenvalues graph. (E) Variance graph, red color is individual and green color is cumulative variances. (F) Co-variance diagram, red, white, and blue colors are correlated, uncorrelated, and anti-correlated, respectively. (G) Elastic network plot, the darker gray color area is stiffer region
.
Analyses of the molecular dynamics simulation of the platform and TLR4 docked complex. (A) 3D structure of NMA mobility. The purple chain is MD2/TLR4, and the green chain is the platform. (B) Deformability plot, the peaks in the graph are deformability regions of the protein. (C) B-factor diagram. (D) Eigenvalues graph. (E) Variance graph, red color is individual and green color is cumulative variances. (F) Co-variance diagram, red, white, and blue colors are correlated, uncorrelated, and anti-correlated, respectively. (G) Elastic network plot, the darker gray color area is stiffer region
Immune response simulation
C-ImmSim models were used for the sequential and effective interactions between the innate and adaptive immune systems, as well as cell states and immune cell memory. It is critical to interpret these in silico simulation results as predictive hypotheses rather than decisive outcomes, as they represent computational models of complex biological processes. The initial immune response was characterized by high levels of IgM and IgG antibodies, followed by increased IgG1 and IgG2 levels. Additionally, the population of B cell isotypes expanded, indicating a rise in immunoglobulin production after the antigen (candidate vaccine platform) injection. The specific antibody titers were measured as IgM + IgG at approximately 230,000, IgG1 + IgG2 at 140,000, and IgM at 90,000. These levels decreased as the antigen concentration declined (Fig. 7A and Fig. 7B). The injection also led to an increase in the population of T-helper and T-cytotoxic cells, along with the development of memory cells (Fig. 7C and Fig. 7D). The results of the induction of Th responses showed that Th1 was induced about 100% compared to other Th types (Fig. 7C). The results of the T-cell population increased significantly and were in an active state (Fig. 7D). Production of high titers of interferon gamma (IFN-γ) and transforming growth factor β (TGF-b) showed that antigen could trigger a stable and strong response (Fig. 7E).
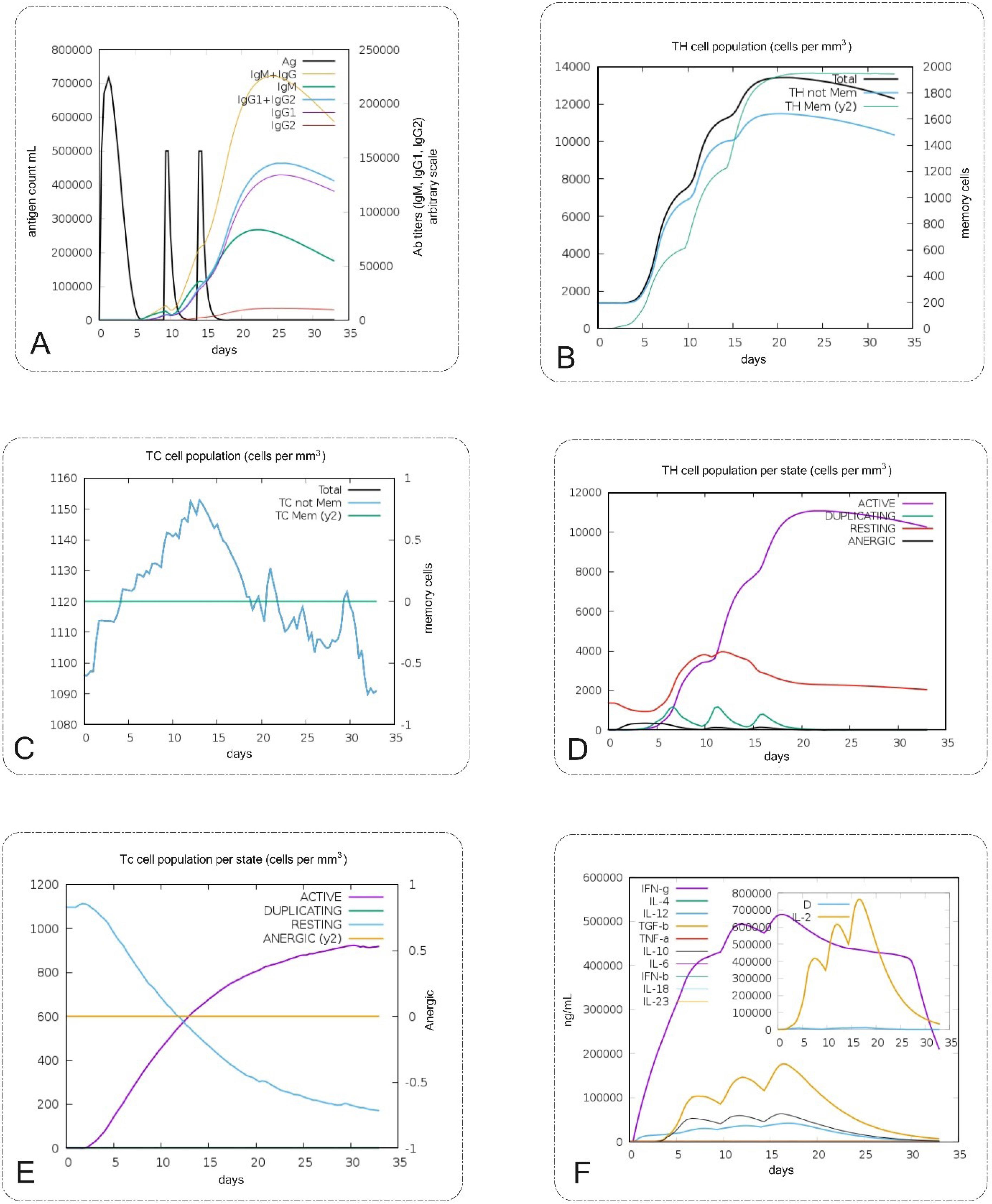
Fig. 7.
In silico immune simulation analyses of the vaccine design (platform) by C-ImmSim. (A) Production of immunoglobulins titers after injection and response to platform (black vertical lines). (B, D) The Th cell population per state levels and percent after the injection. (C, E) The T cytotoxic population per state after the injections that show TC is active. (F) The plot shows interleukins and cytokines levels after the injections. Another plot shows the IL-2 level with the Simpson index and D is shown by the blue line. An increase in the D line over time shows the emergence of epitope-dominant clones of T cells and the smaller the value of D, the lower the variety. High titers of IFN-γ and TGF-b were induced after injections.
.
In silico immune simulation analyses of the vaccine design (platform) by C-ImmSim. (A) Production of immunoglobulins titers after injection and response to platform (black vertical lines). (B, D) The Th cell population per state levels and percent after the injection. (C, E) The T cytotoxic population per state after the injections that show TC is active. (F) The plot shows interleukins and cytokines levels after the injections. Another plot shows the IL-2 level with the Simpson index and D is shown by the blue line. An increase in the D line over time shows the emergence of epitope-dominant clones of T cells and the smaller the value of D, the lower the variety. High titers of IFN-γ and TGF-b were induced after injections.
Expression and purification of the recombinant protein
The pET28a vector, containing the 474-base pair platform DNA sequence, was verified by restriction enzyme analysis using HindIII and XhoI (Fig. 8A). Induction of E. coli BL21 (DE3) cells containing the pET28a-platform vector with IPTG led to the production of a recombinant protein with a molecular weight of approximately 17 kDa (Fig. 8B). Western blot results confirmed the induction of the recombinant protein (Fig. 8C). Purification of the recombinant protein using Ni-NTA chromatography under native conditions yielded a pure protein band with a molecular weight of approximately 17 kDa (shown in Fig. 8D). The endotoxin level of the purified platform was determined to be less than 0.01 EU/mL, which was suitable for immunization purposes.
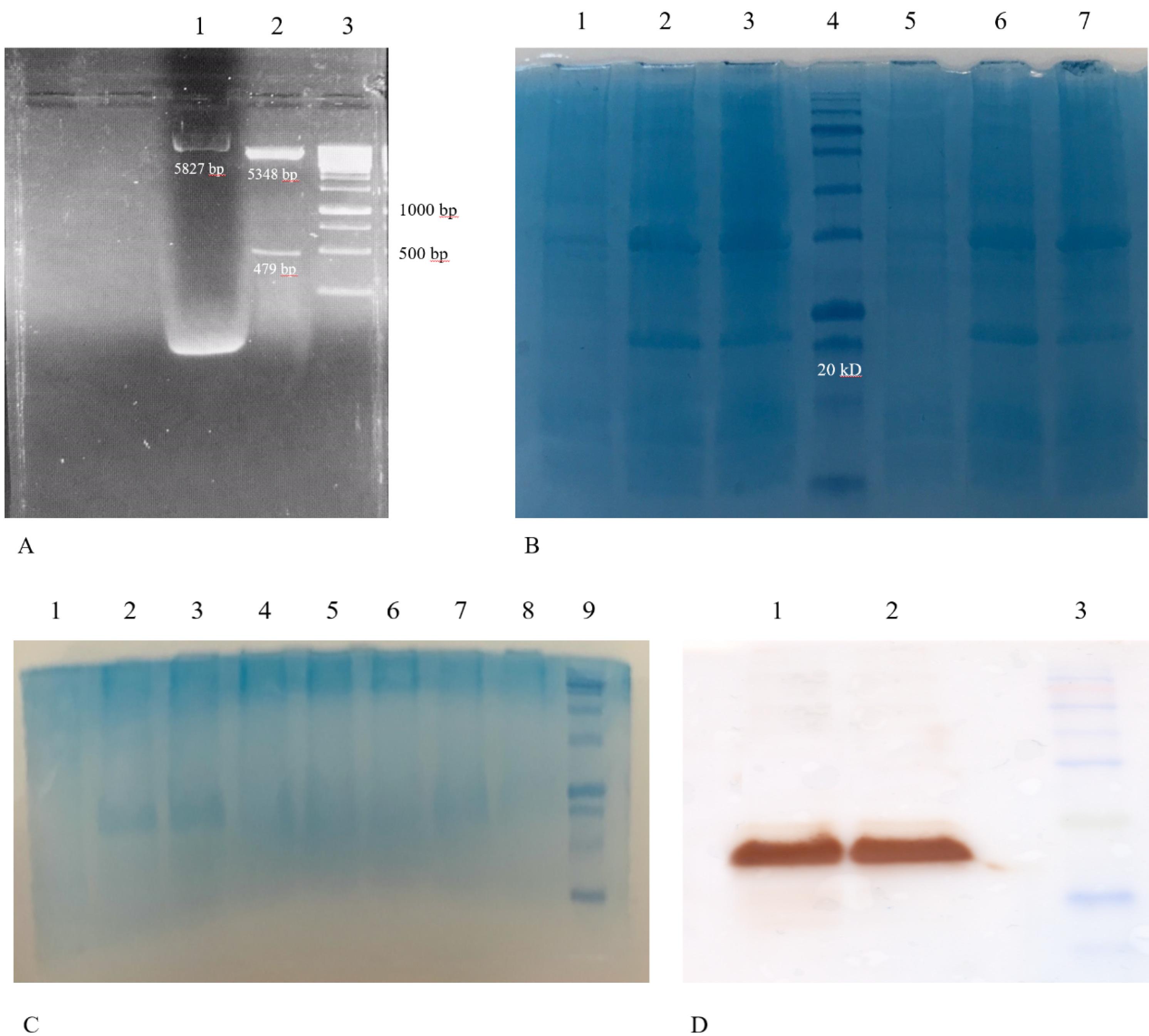
Fig. 8.
Restriction analysis of the pET28a vector-encoded DNA and characterization of the E. coli-derived recombinant protein. A) Lane 1: Undigested pET28a vector harboring 749 bp encoded DNA (the bright, higher molecular weight band may represent supercoiled plasmid DNA, common in undigested preparations). Lane 2: Digestion with HindIII and Xho I enzymes resulted in two fragments of 5348 bp and 749 bp. Lane 3: DNA marker DM3100 (SMOBIO 1kb, Taiwan). B) SDS-PAGE analysis before and after induction of pET28a-RP-harboring E. coli with IPTG revealed a protein band of approximately 17 kDa corresponding to the expressed RP. Lane 1: Un-induced cell lysates of colony 1 of the E. coli BL-21(DE3) cells harboring pET28a-RP. Lane 2: Cell lysates of colony 1 of E. coli cells harboring pET28a-recombinant protein after 3 h induction with IPTG. Lane 3: Cell lysates of colony 1 of E. coli cells harboring pET28a-recombinant protein after 4 h induction with IPTG. Lane 4: Molecular weight marker (sizes: 180, 140, 100, 72, 60, 45, 35, 25, 20, 15, 10 kDa) (SMOBIO PM1600, Taiwan). Lane 5: Un-induced cell lysates of colony 2 of the E. coli BL-21(DE3) cells harboring pET28a-recombinant protein. Lane 6: Cell lysates of colony 2 of E. coli cells harboring pET28a-recombinant protein after 3 h induction with IPTG. Lane 7: Cell lysates of colony 2 of E. coli cells harboring pET28a-recombinant protein after 4 h induction with IPTG. C) SDS-PAGE analysis native purification of pET28a-RP-harboring E. coli. Lane 1: Un-induced cell lysates of colony 2 of the E. coli BL-21(DE3) cells harboring pET28a- recombinant protein. Lane 2: Crude lysates of E. coli cells harboring pET28a-recombinant protein before centrifuge. Lane 3: Crude lysates of E. coli cells harboring pET28a-recombinant protein after centrifuge. Lane 4: Flow throw. Lane 5: Washed 1. Lane 6: Washed 2. Lane 7: Elution 1 of recombinant protein. Lane 8: Elution 2 of recombinant protein. Lane 9: Molecular weight marker. D) Western blotting results for the expressed recombinant protein. Lane 1, 2: Purified recombinant protein (approximately 17 kDa). Lane 3: Molecular weight marker (SinaClon, Iran).
.
Restriction analysis of the pET28a vector-encoded DNA and characterization of the E. coli-derived recombinant protein. A) Lane 1: Undigested pET28a vector harboring 749 bp encoded DNA (the bright, higher molecular weight band may represent supercoiled plasmid DNA, common in undigested preparations). Lane 2: Digestion with HindIII and Xho I enzymes resulted in two fragments of 5348 bp and 749 bp. Lane 3: DNA marker DM3100 (SMOBIO 1kb, Taiwan). B) SDS-PAGE analysis before and after induction of pET28a-RP-harboring E. coli with IPTG revealed a protein band of approximately 17 kDa corresponding to the expressed RP. Lane 1: Un-induced cell lysates of colony 1 of the E. coli BL-21(DE3) cells harboring pET28a-RP. Lane 2: Cell lysates of colony 1 of E. coli cells harboring pET28a-recombinant protein after 3 h induction with IPTG. Lane 3: Cell lysates of colony 1 of E. coli cells harboring pET28a-recombinant protein after 4 h induction with IPTG. Lane 4: Molecular weight marker (sizes: 180, 140, 100, 72, 60, 45, 35, 25, 20, 15, 10 kDa) (SMOBIO PM1600, Taiwan). Lane 5: Un-induced cell lysates of colony 2 of the E. coli BL-21(DE3) cells harboring pET28a-recombinant protein. Lane 6: Cell lysates of colony 2 of E. coli cells harboring pET28a-recombinant protein after 3 h induction with IPTG. Lane 7: Cell lysates of colony 2 of E. coli cells harboring pET28a-recombinant protein after 4 h induction with IPTG. C) SDS-PAGE analysis native purification of pET28a-RP-harboring E. coli. Lane 1: Un-induced cell lysates of colony 2 of the E. coli BL-21(DE3) cells harboring pET28a- recombinant protein. Lane 2: Crude lysates of E. coli cells harboring pET28a-recombinant protein before centrifuge. Lane 3: Crude lysates of E. coli cells harboring pET28a-recombinant protein after centrifuge. Lane 4: Flow throw. Lane 5: Washed 1. Lane 6: Washed 2. Lane 7: Elution 1 of recombinant protein. Lane 8: Elution 2 of recombinant protein. Lane 9: Molecular weight marker. D) Western blotting results for the expressed recombinant protein. Lane 1, 2: Purified recombinant protein (approximately 17 kDa). Lane 3: Molecular weight marker (SinaClon, Iran).
Discussion
Despite the availability of prophylactic vaccines, HPV-induced cervical cancer remains a significant global health problem and current management strategies are inadequate.17 The absence of therapeutic HPV vaccines necessitates the development of a candidate vaccine capable of treating established infections and tumors. Such a candidate vaccine would represent a substantial advancement in the fight against cervical cancer.
As a branch of bioinformatics, immunoinformatics has emerged as a valuable tool for the rational design of vaccines. By employing computational methods, researchers can identify and characterize epitopes and specific regions of antigens that elicit immune responses.18,19 These epitopes serve as promising targets for the development of vaccines against a variety of viral and pathogenic infections. Our results, particularly our reports of predicted antigenicity and immunogenicity, were in agreement with previous immunoinformatics analyses that used similar computational techniques to predict candidate vaccination against viral diseases and cancers, indicating confidence in the validity of these computational approaches.20,21 Recently, Pratiwi et al also used a similar computational product development workflow to characterize a multi-epitope vaccine against HPV and demonstrated similarly high predicted antigenicity scores and predicted immune responses.20 The consistency in our studies strengthens our results as well as the credibility of our analysis approaches overall. Immunoinformatics facilitates the selection of epitopes capable of stimulating both cellular and humoral immune responses, thereby enhancing vaccine efficacy.22 Bahmani et al predicted six CD4+ and six CD8+ T-cell epitopes from the E7 protein of HPV16 using the IEDB server with > 90% population coverage, in which E749-57, RAHYNIVTF, was one of the final predicted epitopes.23 Other studies also predicted the E749-57 epitope for their vaccine construct based on an in silico approach,24,25 and other ones indicated that its adjuvanted form could elicit strong CTL-mediated responses and reduce tumor growth.26,27
The judicious selection of adjuvants in candidate vaccine formulations can significantly augment the immunogenicity, potency, and stability of target antigens, particularly in epitope-based vaccines exhibiting suboptimal responses.28 Adjuvants, when incorporated into vaccine structures, elicit robust and sustained immune responses.29 In recent years, TLR agonists have emerged as promising adjuvant candidates,30 with TLR4 being identified as a key receptor expressed in cervical tumors.31 The interaction between TLR4 and the related DAMPs and PAMPs results in the activation of intracellular signaling pathways, such as Toll-IL-1 receptor domain-containing adapter inducing interferon b (TRIF) and Myeloid differentiation primary response 88 (MYD88), hypothesizing that TLR4 can recognize HPV-related PAMPs and induce immune responses.32 On the other hand, there is evidence that HPVs could downregulate the expression of the TLR4 gene and negatively affect its function in promoting the virus' escape from immune responses.33 Moreover, the activation of TLR4 prevents the integration of HPV DNA into the host genome and its downregulation promotes genomic integration of HPV DNA.34,35 Thus, TLR4 not only plays a protective role against HPV infection, but also induces immune responses to eradicate established viral infection, suggesting that TLR4 agonists could be reliable adjuvants in the development of HPV-induced cervical cancer. In an in silico study, Negahdaripour et al showed that a short synthetic TLR4 agonist could protect against HPV infection by inducing both humoral and cellular immune responses.36 In another study, Gableh et al used monophosphoryl lipid A (MPL), as a TLR4 agonist and adjuvant, in combination with a DNA vaccine directed against HPV to enhance the efficacy of DNA vaccine.37 In addition to MPL, other agonists of TLR4 have been used in vaccine platforms against HPV, such as heat shock protein X (HSPX), lipopolysaccharide (LPS), carrageenan, and heparin-binding hemagglutinin (HBHA).38-41 Our use of a protein-based TLR4 agonist, Ply4, as an integrated adjuvant leverages these considerations with the additional value of co-delivery and local immune stimulation. This approach is similar to other efforts in the literature where built-in adjuvant approaches were studied to increase vaccine immunogenicity.42 For example, Shey et al showed that a fusion protein containing an adjuvant and an antigen was effective and produced immune responses similar to what our in silico results predicted for immune cell activation and cytokine production.43 Due to the TLR4 capability of stimulation of the domain 4 of pneumolysin (Ply4) from Streptococcus pneumonia,13 it emerges as a promising candidate for TLR4 agonist adjuvantation. Besides the stimulatory activity on TLR4, Ply4 has a hemolytic activity in which a triple mutant, including Asp385Asn, Cys428Gly, and Trp433Phe, has been reported to diminish Ply4's cytolytic potential significantly.44 Here, we used the triple-mutant form of Ply4 (Asp385Asn, Cys428Gly, and Trp433Phe), which is known to maintain TLR4 agonistic activity while considerably diminishing hemolytic potential,13 as a built-in adjuvant in the structure of the therapeutic candidate vaccine platform to interact with TLR4 and significantly increase immune responses to fused HPV16 E6 and E7 epitopes. The key criterion for selecting Ply4, and in particular its mutated and truncated form, over other TLR agonists such as MPLA or flagellin, is that it is a protein. As a protein, we can genetically fuse it into our multi-epitope construct such that it becomes a 'built-in' adjuvant. The covalent linkage between Ply4 and the rest of our multi-epitope construct would ensure co-delivery to APCs, which is generally a more effective mechanism than co-administration. S. pneumoniae is a common pathogen, and in the context of a highly-attenuated and specific domain (Ply4) multi-epitope construct, we expect that the likelihood of induction of an overwhelming anti-Ply4 immune response will be reduced, as this immune response is necessary to counter S. pneumoniae and could diminish from the HPV-specific response to the multi-epitope construct. Also, it's designed to reduce toxicity limits potentially cross-reactive immune responses against host components. The TLR4 binding region in Ply4 has been well studied for its sense of action, and the preserved TLR4 binding region in Ply4 contributes to the strength of our computational docking and subsequent molecular dynamics simulations, even without currently available experimental site-directed mutagenesis data for our construct.
As shown in our results, the physicochemical and immunological properties of the vaccine platform were evaluated using different computational references. The MW of the protein was 17 kDa, which was suitable for purification and vaccine development as well as SDS-PAGE electrophoresis and Western blot analysis.45,46 The candidate vaccine construct was classified as stable with an instability index of 16.50 since structures with an instability index < 40 are assumed stable.47 The GRAVY index of the recombinant protein was negative (-0.483), indicating the hydrophilic nature of the candidate vaccine.48 The aliphatic index of the designed candidate vaccine was 85.95, which suggests the candidate vaccine construct is thermostable.49 The theoretical pI is about 8.04, indicating that the protein is basic in nature. Additionally, the candidate vaccine construct was predicted to be non-toxic, non-allergen, and antigenic using Toxinpred, Allergen FP, and Vaxijen v.2 servers, respectively.
Current best practice for multi-epitope vaccines is to include both MHC I and MHC II epitopes to stimulate helper T cells (and often B-cell epitopes), enhancing immunogenicity. For example, Negahdaripour et al et al explicitly incorporated universal T-helper sequences to ensure robust Th responses.36 As a limitation of our study, we did not specifically select or present MHC II-restricted peptides in the current construct, however, our in silico immune simulation experiment resulted in activation of Th1-type helper T cells, which we attribute to the inclusion of natural MHC II, presentable sequences in the fused E6/E7-Ply4 construct as well as the significant TLR4 agonist activity of Ply4 that enhanced antigen presentation. Another important issue for future studies is the population coverage of the designed vaccine. The results of the vaccine populations coverage analysis illustrated certainly positive trends in the West Indies and East Asia, but also distinct differences in coverage within these regions' sub-groups as well. This highlights the issue of developing a truly universal vaccine for a global virus such as HPV. Also, our focus on the "Persian" population was based on where the research team was located, and that the vaccine is intended to be regionally universal. Future studies should focus on more targeted epitope selection based on regional considerations of HLA distributions, particularly in areas with a high incidence of HPV infection (i.e., the region of Sub-Saharan Africa and certain areas of Latin America). Our current design serves as a strong starting point, providing a platform with generalizability for future optimization to achieve a more universal efficacy profile globally. It is worth noting that the in silico immune simulation experiment provides encouraging evidence of strong humoral and cellular responses with a robust Th1 polarization, it indicates a computational model, and therefore these data should be viewed as strong supportive hypotheses supporting the vaccine's potential immunogenicity, confirming these predicted immune outcomes will require careful in vitro and in vivo experimental validation. Functional immunological assays, such as T-cell proliferation, activation markers, and cytokine secretion profiling (e.g., IFN-γ ELISPOT and CD107a), are next crucial steps for in vitro validation.
It should be noted that the MD analysis performed in this study was based on the iMODS platform, which applies Normal Mode Analysis (NMA) to predict the flexibility, stability, and dynamic behavior of protein complexes. NMA-based MD simulations provide a computationally efficient and widely accepted approach for assessing the stability of docked complexes, particularly in early-stage vaccine design. While this approach captures the essential dynamic motions of macromolecules, it does not account for solvent effects or atomic-level fluctuations as in all-atom MD simulations. Therefore, future studies will incorporate longer, atomistic MD simulations (e.g., GROMACS-based) to further validate the dynamic stability and binding energetics of the designed vaccine–TLR4 complex. This multi-tiered approach will enhance the robustness and translational relevance of the computational findings.
Conclusion
In this study, using immunoinformatics tools, we designed a novel therapeutic candidate vaccine based on the built-in adjuvant strategy using HPV 16 E6 and E7 epitopes and the domain 4 of pneumolysin (Ply4) from S. pneumonia, as an agonist for TLR4. The physicochemical properties, antigenicity, allergenicity, toxicity, binding interactions with the TLR4 receptor, and immune response simulation revealed that the designed candidate vaccine is appropriate for therapeutic purposes against HPV-related infections and cancers, but further examinations, both in vitro and in vivo, are required for efficacy and safety confirmation. It should be emphasized that the present study represents a computational and preliminary experimental effort. The designed vaccine construct will require comprehensive in vitro immunological assessments and in vivo preclinical validations before any consideration for clinical translation.
Research Highlights
What is the current knowledge?
What is new here?
-
Computational vaccine design assists in evaluating the designed vaccine's efficacy and safety.
-
Built-in adjuvants can be used in vaccine design to improve the efficacy of therapeutic vaccines.
-
Incorporating the domain 4 of pneumolysin from Streptococcus pneumonia, as a TLR4 agonist, in the epitope-based vaccine is a reliable strategy for designing therapeutic vaccines against HPV infections.
Competing Interests
The authors have declared no conflict of interest related to this paper.
Declaration of AI-assisted Tools in the Writing Procedure
The authors declare that no AI or AI-assisted technologies were used in the writing of this paper.
Ethical Approval
No animal studies were involved in this research.
Supplementary files
Supplementary file 1 contains Figs. S1-S5 and Table S1.
(pdf)
References
- Kombe AJK, Zoa-Assoumou S, Bounda GA, Nsole-Biteghe FA, Jin T, Zouré AA. Advances in Etiopathological Role and Control of HPV in Cervical Cancer Oncogenesis. Front Biosci (Landmark Ed) 2023; 28:245. doi: 10.31083/j.fbl2810245 [Crossref] [ Google Scholar]
- Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2024; 74:229-63. doi: 10.3322/caac.21834 [Crossref] [ Google Scholar]
- Namvar A, Panahi HA, Agi E, Bolhassani A. Development of HPV(16,18,31,45) E5 and E7 peptides-based vaccines predicted by immunoinformatics tools. Biotechnol Lett 2020; 42:403-18. doi: 10.1007/s10529-020-02792-6 [Crossref] [ Google Scholar]
- Wang R, Huang H, Yu C, Li X, Wang Y, Xie L. Current status and future directions for the development of human papillomavirus vaccines. Front Immunol 2024; 15:1362770. doi: 10.3389/fimmu.2024.1362770 [Crossref] [ Google Scholar]
-
Mlynarczyk-Bonikowska B, Rudnicka L. HPV Infections-Classification, Pathogenesis, and Potential New Therapies. Int J Mol Sci 2024; 25. doi: 10.3390/ijms25147616.
-
Forner M, Cañas-Arranz R, Defaus S, de León P, Rodríguez-Pulido M, Ganges L, et al. Peptide-Based Vaccines: Foot-and-Mouth Disease Virus, a Paradigm in Animal Health. Vaccines (Basel) 2021; 9. doi: 10.3390/vaccines9050477.
- Lim HX, Lim J, Jazayeri SD, Poppema S, Poh CL. Development of multi-epitope peptide-based vaccines against SARS-CoV-2. Biomed J 2021; 44:18-30. doi: 10.1016/j.bj.2020.09.005 [Crossref] [ Google Scholar]
- Lei Y, Zhao F, Shao J, Li Y, Li S, Chang H. Application of built-in adjuvants for epitope-based vaccines. PeerJ 2019; 6:e6185. doi: 10.7717/peerj.6185 [Crossref] [ Google Scholar]
- Li Q, Li Z, Deng N, Ding F, Li Y, Cai H. Built-in adjuvants for use in vaccines. Eur J Med Chem 2022; 227:113917. doi: 10.1016/j.ejmech.2021.113917 [Crossref] [ Google Scholar]
- Kumar S, Sunagar R, Gosselin E. Bacterial Protein Toll-Like-Receptor Agonists: A Novel Perspective on Vaccine Adjuvants. Front Immunol 2019; 10:1144. doi: 10.3389/fimmu.2019.01144 [Crossref] [ Google Scholar]
- Shafaghi M, Bahadori Z, Madanchi H, Ranjbar MM, Shabani AA, Mousavi SF. Immunoinformatics-aided design of a new multi-epitope vaccine adjuvanted with domain 4 of pneumolysin against Streptococcus pneumoniae strains. BMC Bioinformatics 2023; 24:67. doi: 10.1186/s12859-023-05175-6 [Crossref] [ Google Scholar]
- Mashhadi Abolghasem Shirazi M, Sadat SM, Haghighat S, Roohvand F, Arashkia A. Alum and a TLR7 agonist combined with built-in TLR4 and 5 agonists synergistically enhance immune responses against HPV RG1 epitope. Sci Rep 2023; 13:16801. doi: 10.1038/s41598-023-43965-3 [Crossref] [ Google Scholar]
- Chiu FF, Leng CH, Ding YJ, Chang JC, Chang LS, Lien SP. Domain 4 of pneumolysin from Streptococcus pneumoniae is a multifunctional domain contributing TLR4 activating and hemolytic activity. BiochemBiophys Res Commun 2019; 517:596-602. doi: 10.1016/j.bbrc.2019.07.063 [Crossref] [ Google Scholar]
- Zhang RY, Zhou SH, He CB, Wang J, Wen Y, Feng RR. Adjuvant-Protein Conjugate Vaccine with Built-In TLR7 Agonist on S1 Induces Potent Immunity against SARS-CoV-2 and Variants of Concern. ACS Infect Dis 2022; 8:1367-75. doi: 10.1021/acsinfecdis.2c00259 [Crossref] [ Google Scholar]
- Zhang H, Zhang S. The expression of Foxp3 and TLR4 in cervical cancer: association with immune escape and clinical pathology. Arch GynecolObstet 2017; 295:705-12. doi: 10.1007/s00404-016-4277-5 [Crossref] [ Google Scholar]
- Yang X, Cheng Y, Li C. The role of TLRs in cervical cancer with HPV infection: a review. Signal Transduct Target Ther 2017; 2:17055. doi: 10.1038/sigtrans.2017.55 [Crossref] [ Google Scholar]
- Ashique S, Hussain A, Fatima N, Altamimi MA. HPV pathogenesis, various types of vaccines, safety concern, prophylactic and therapeutic applications to control cervical cancer, and future perspective. Virusdisease 2023; 34:1-19. doi: 10.1007/s13337-023-00824-z [Crossref] [ Google Scholar]
- Oli AN, Obialor WO, Ifeanyichukwu MO, Odimegwu DC, Okoyeh JN, Emechebe GO. Immunoinformatics and Vaccine Development: An Overview. Immunotargets Ther 2020; 9:13-30. doi: 10.2147/itt.S241064 [Crossref] [ Google Scholar]
-
Rawat SS, Keshri AK, Kaur R, Prasad A. Immunoinformatics Approaches for Vaccine Design: A Fast and Secure Strategy for Successful Vaccine Development. Vaccines (Basel) 2023; 11. doi: 10.3390/vaccines11020221.
- Pratiwi SE, Ysrafil Y, Mardhia M, Mahyarudin M, Ilmiawan MI, Trianto HF. A novel therapeutic multiepitope vaccine based on oncoprotein E6 and E7 of HPV 16 and 18: An in silico approach. Bioimpacts 2024; 14:27846. doi: 10.34172/bi.2024.27846 [Crossref] [ Google Scholar]
- Parvizpour S, Razmara J, Pourseif MM, Omidi Y. In silico design of a triple-negative breast cancer vaccine by targeting cancer testis antigens. Bioimpacts 2019; 9:45-56. doi: 10.15171/bi.2019.06 [Crossref] [ Google Scholar]
- Shawan M, Sharma AR, Halder SK, Arian TA, Shuvo MN, Sarker SR. Advances in Computational and Bioinformatics Tools and Databases for Designing and Developing a Multi-Epitope-Based Peptide Vaccine. Int J Pept Res Ther 2023; 29:60. doi: 10.1007/s10989-023-10535-0 [Crossref] [ Google Scholar]
- Bahmani B, Amini-Bayat Z, Ranjbar MM, Bakhtiari N, Zarnani AH. HPV16-E7 Protein T Cell Epitope Prediction and Global Therapeutic Peptide Vaccine Design Based on Human Leukocyte Antigen Frequency: An In-Silico Study. Int J Pept Res Ther 2021; 27:365-78. doi: 10.1007/s10989-020-10089-5 [Crossref] [ Google Scholar]
- Guo N, Niu Z, Yan Z, Liu W, Shi L, Li C, et al. Immunoinformatics Design and In Vivo Immunogenicity Evaluation of a Conserved CTL Multi-Epitope Vaccine Targeting HPV16 E5, E6, and E7 Proteins. Vaccines (Basel) 2024; 12. doi: 10.3390/vaccines12040392.
-
Tîrziu A, Avram S, Madă L, Crișan-Vida M, Popovici C, Popovici D, et al. Design of a Synthetic Long Peptide Vaccine Targeting HPV-16 and -18 Using Immunoinformatic Methods. Pharmaceutics 2023; 15. doi: 10.3390/pharmaceutics15071798.
- Gendron KB, Rodriguez A, Sewell DA. Vaccination with human papillomavirus type 16 E7 peptide with CpG oligonucleotides for prevention of tumor growth in mice. Arch Otolaryngol Head Neck Surg 2006; 132:327-32. doi: 10.1001/archotol.132.3.327 [Crossref] [ Google Scholar]
- Goradel NH, Negahdari B, Mohajel N, Malekshahi ZV, Shirazi MMA, Arashkia A. Heterologous administration of HPV16 E7 epitope-loaded nanocomplexes inhibits tumor growth in mouse model. Int Immunopharmacol 2021; 101:108298. doi: 10.1016/j.intimp.2021.108298 [Crossref] [ Google Scholar]
- Montomoli E, Piccirella S, Khadang B, Mennitto E, Camerini R, De Rosa A. Current adjuvants and new perspectives in vaccine formulation. Expert Rev Vaccines 2011; 10:1053-61. doi: 10.1586/erv.11.48 [Crossref] [ Google Scholar]
- Parvizpour S, Pourseif MM, Razmara J, Rafi MA, Omidi Y. Epitope-based vaccine design: a comprehensive overview of bioinformatics approaches. Drug Discov Today 2020; 25:1034-42. doi: 10.1016/j.drudis.2020.03.006 [Crossref] [ Google Scholar]
- Reed SG, Hsu FC, Carter D, Orr MT. The science of vaccine adjuvants: advances in TLR4 ligand adjuvants. CurrOpin Immunol 2016; 41:85-90. doi: 10.1016/j.coi.2016.06.007 [Crossref] [ Google Scholar]
- Jiang N, Xie F, Chen L, Chen F, Sui L. The effect of TLR4 on the growth and local inflammatory microenvironment of HPV-related cervical cancer in vivo. Infect Agent Cancer 2020; 15:12. doi: 10.1186/s13027-020-0279-9 [Crossref] [ Google Scholar]
- Bahramabadi R, Dabiri S, Iranpour M, Kazemi Arababadi M. TLR4: An Important Molecule Participating in Either Anti-Human Papillomavirus Immune Responses or Development of Its Related Cancers. Viral Immunol 2019; 32:417-23. doi: 10.1089/vim.2019.0061 [Crossref] [ Google Scholar]
- Tobouti PL, Bolt R, Radhakrishnan R, de Sousa S, Hunter KD. Altered Toll-like receptor expression and function in HPV-associated oropharyngeal carcinoma. Oncotarget 2018; 9:236-48. doi: 10.18632/oncotarget.18959 [Crossref] [ Google Scholar]
- Damasdi M, Kovacs K, Farkas N, Jakab F, Kovacs G. Down-regulation of Toll-like Receptor TLR4 Is Associated with HPV DNA Integration in Penile Carcinoma. Anticancer Res 2017; 37:5515-9. doi: 10.21873/anticanres.11982 [Crossref] [ Google Scholar]
- Pannone G, Bufo P, Pace M, Lepore S, Russo GM, Rubini C. TLR4 down-regulation identifies high risk HPV infection and integration in head and neck squamous cell carcinomas. Front Biosci (Elite Ed) 2016; 8:15-28. doi: 10.2741/e747 [Crossref] [ Google Scholar]
- Negahdaripour M, Eslami M, Nezafat N, Hajighahramani N, Ghoshoon MB, Shoolian E. A novel HPV prophylactic peptide vaccine, designed by immunoinformatics and structural vaccinology approaches. Infect Genet Evol 2017; 54:402-16. doi: 10.1016/j.meegid.2017.08.002 [Crossref] [ Google Scholar]
- Gableh F, Saeidi M, Hemati S, Hamdi K, Soleimanjahi H, Gorji A. Combination of the toll like receptor agonist and α-Galactosylceramide as an efficient adjuvant for cancer vaccine. J Biomed Sci 2016; 23:16. doi: 10.1186/s12929-016-0238-3 [Crossref] [ Google Scholar]
- Jung ID, Shin SJ, Lee MG, Kang TH, Han HD, Lee SJ. Enhancement of tumor-specific T cell-mediated immunity in dendritic cell-based vaccines by Mycobacterium tuberculosis heat shock protein X. J Immunol 2014; 193:1233-45. doi: 10.4049/jimmunol.1400656 [Crossref] [ Google Scholar]
- Chen XZ, Mao XH, Zhu KJ, Jin N, Ye J, Cen JP. Toll like receptor agonists augment HPV 11 E7-specific T cell responses by modulating monocyte-derived dendritic cells. Arch Dermatol Res 2010; 302:57-65. doi: 10.1007/s00403-009-0976-0 [Crossref] [ Google Scholar]
- Zhang YQ, Tsai YC, Monie A, Hung CF, Wu TC. Carrageenan as an adjuvant to enhance peptide-based vaccine potency. Vaccine 2010; 28:5212-9. doi: 10.1016/j.vaccine.2010.05.068 [Crossref] [ Google Scholar]
- Nezafat N, Ghasemi Y, Javadi G, Khoshnoud MJ, Omidinia E. A novel multi-epitope peptide vaccine against cancer: an in silico approach. J Theor Biol 2014; 349:121-34. doi: 10.1016/j.jtbi.2014.01.018 [Crossref] [ Google Scholar]
- Rezaei M, Habibi M, Ehsani P, Asadi Karam MR, Bouzari S. Design and computational analysis of an effective multi-epitope vaccine candidate using subunit B of cholera toxin as a build-in adjuvant against urinary tract infections. Bioimpacts 2024; 14:27513. doi: 10.34172/bi.2023.27513 [Crossref] [ Google Scholar]
- Shey RA, Ghogomu SM, Nebangwa DN, Shintouo CM, Yaah NE, Yengo BN. Rational design of a novel multi-epitope peptide-based vaccine against Onchocerca volvulus using transmembrane proteins. Front Trop Dis 2022; 3:1046522. doi: 10.3389/fitd.2022.1046522 [Crossref] [ Google Scholar]
- Berry AM, Alexander JE, Mitchell TJ, Andrew PW, Hansman D, Paton JC. Effect of defined point mutations in the pneumolysin gene on the virulence of Streptococcus pneumoniae. Infect Immun 1995; 63:1969-74. doi: 10.1128/iai.63.5.1969-1974.1995 [Crossref] [ Google Scholar]
- Sanami S, Zandi M, Pourhossein B, Mobini GR, Safaei M, Abed A. Design of a multi-epitope vaccine against SARS-CoV-2 using immunoinformatics approach. Int J Biol Macromol 2020; 164:871-83. doi: 10.1016/j.ijbiomac.2020.07.117 [Crossref] [ Google Scholar]
- Zhao X, Zhang F, Li Z, Wang H, An M, Li Y. Bioinformatics analysis of EgA31 and EgG1Y162 proteins for designing a multi-epitope vaccine against Echinococcus granulosus. Infect Genet Evol 2019; 73:98-108. doi: 10.1016/j.meegid.2019.04.017 [Crossref] [ Google Scholar]
-
Gasteiger E, Hoogland C, Gattiker A, Duvaud Se, Wilkins MR, Appel RD, et al. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005. p. 571–607. doi: 10.1385/1-59259-890-0:571.
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982; 157:105-32. doi: 10.1016/0022-2836(82)90515-0 [Crossref] [ Google Scholar]
- Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem 1980; 88:1895-8. [ Google Scholar]