Bioimpacts. 15:30994.
doi: 10.34172/bi.30994
Original Article
Designing of multi-epitope vaccine against Varicella zoster virus (VZV) using immunoinformatics and structural analysis: In silico study
Mohamed J. Saadh Conceptualization, Investigation, Methodology, Writing – original draft, 1 
Mareb Hamed Ahmed Conceptualization, Project administration, Supervision, Writing – original draft, 2, *
Rafid Jihad Albadr Conceptualization, Investigation, Writing – original draft, 3
Gaurav Sanghvi Methodology, Validation, Writing – review & editing, 4
R. Roopashree Data curation, Validation, Writing – review & editing, 5
Aditya Kashyap Formal analysis, Methodology, Visualization, 6
A. Sabarivani Formal analysis, Methodology, Writing – review & editing, 7
Zafar Aminov Data curation, Formal analysis, Visualization, 8
Waam Mohammed Taher Funding acquisition, Project administration, Resources, Validation, 9
Mariem Alwan Investigation, Writing – review & editing, 10
Mahmod Jasem Jawad Data curation, Investigation, 11
Ali M. Ali Al-Nuaimi Funding acquisition, Resources, Supervision, 12
Author information:
1Faculty of Pharmacy, Middle East University, Amman, 11831, Jordan
2College of Dentistry, Alnoor University, Mosul, Iraq
3Ahl al Bayt University, Karbala, Iraq
4Marwadi University Research Center, Department of Microbiology, Faculty of Science, Marwadi University, Rajkot-360003, Gujarat, India
5Department of Chemistry and Biochemistry, School of Sciences, JAIN (Deemed to be University), Bangalore, Karnataka, India
6Centre for Research Impact & Outcome, Chitkara University Institute of Engineering and Technology, Chitkara University, Rajpura, 140401, Punjab, India
7Department of Biomedical, Sathyabama Institute of Science and Technology, Chennai, Tamil Nadu, India
8Department of Public Health and Healthcare management, Samarkand State Medical University, Samarkand, Uzbekistan
9College of Nursing, National University of Science and Technology, Dhi Qar, Iraq
10Pharmacy College, Al-Farahidi University, Baghdad, Iraq
11Department of Pharmacy, Al-Zahrawi University College, Karbala, Iraq
12Gilgamesh Ahliya University, Baghdad, Iraq
Abstract
Introduction:
The Varicella-zoster virus (VZV) causes varicella (chickenpox) and herpes zoster (shingles), posing significant global health challenges. Despite existing vaccines, gaps in coverage and efficacy persist, necessitating novel vaccine designs. This study aimed to develop a multi-epitope vaccine targeting VZV using immunoinformatics and structural bioinformatics approaches.
Methods:
MHC-I and MHC-II binding epitopes from VZV proteins (glycoprotein E, glycoprotein B, tegument protein IE63) were predicted using IEDB tools, prioritizing conserved epitopes with high binding affinity. A chimeric construct was engineered with 18 epitopes, adjuvants (β-defensin 3), and cell-penetrating peptides (HIV TAT), linked with GPGPG/AAY spacers. Antigenicity (VaxiJen), allergenicity (AlgPred), physicochemical properties (ProtParam), and solubility (SOLpro) were assessed. Tertiary structure modeling (GalaxyWEB) and refinement (GalaxyRefine) were performed. Docking (PatchDock) and dynamics simulations (GROMACS, 100 ns) evaluated TLR2-vaccine binding stability. Immune response was simulated (C-ImmSim), and codon optimization (JCAT) ensured E. coli expression compatibility.
Results:
Non-allergenic, antigenic (VaxiJen score: 0.52), stable (instability index: 30.20), and soluble (GRAVY: -0.548). Molecular weight: 34 kDa; pI: 9.65. RMSD (3.8 nm) and RMSF analyses confirmed complex stability. Free energy landscape revealed low-energy binding states (0.3–1.8 kcal/mol). Simulated results showed robust IgG/IgM production, Th1 cytokines (IFN-γ, IL-2), and memory cell activation. Epitopes covered 100% of populations in Europe/North America and > 77% in Africa/South Asia.
Conclusion:
The multi-epitope vaccine demonstrated strong immunogenicity, structural stability, and broad population coverage. Computational validation supports its potential as a candidate for preventing VZV infections, pending experimental verification in the future.
Keywords: Varicella zoster virus (VZV), Multi-epitope vaccine, Immunoinformatics, Molecular dynamic simulation
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This study funded by Ali M. Ali Al-Nuaimi, Waam Mohammed Taher.
Introduction
Varicella zoster virus (VZV), which is classified as a human alphaherpesvirus belonging to the Varicellovirus genus, is the causative agent of both varicella, commonly referred to as chickenpox, and zoster, which is more commonly known as shingles.1 The existing epidemiological data strongly indicates that the initial infection by VZV occurs through its replication within the epithelial cells located in the upper respiratory tract, and this process is followed, after an incubation period that typically ranges from 10 to 21 days, by the emergence of the characteristic widespread vesicular rash associated with varicella.2 This progression likely mirrors the dissemination of the virus to the tonsils and other nearby lymphoid tissues, from which point the infected T cells are capable of transporting the virus through the circulatory system to various skin regions.3 During this primary infection, it is presumed that the virions gain entry into the sensory nerve cell bodies situated within the ganglia through mechanisms such as retrograde axonal transport originating from the sites within the skin where replication occurs, or through T cell-mediated viraemia, thus leading to the establishment of latent infection.4 Upon reactivation of viral replication, the VZV subsequently travels back to the skin using anterograde axonal transport, resulting in the manifestation of zoster, which is distinctly characterized by a vesicular rash appearing in the dermatome that is innervated by the ganglion that has been affected by the virus. It is important to note that both varicella and zoster skin lesions harbor elevated concentrations of infectious virus, thereby serving as significant sources for transmission to individuals who are susceptible to the virus. Before the introduction of a varicella vaccine, which is a live attenuated formulation derived from the VZV Oka strain and was launched in 1995, varicella epidemics occurred every year within the United States; however, it is noteworthy that such epidemics persist among children in nations where no immunization programs are in place 5 VZV reactivation has emerged as an important point of discussion during the COVID-19 pandemic, especially after vaccination.6 The varicella vaccine, commonly referred to as the chickenpox vaccine, serves as a prophylactic measure against varicella-zoster virus infection. A single administration of the vaccine confers an efficacy of 95% against moderate manifestations of the disease and achieves a complete prevention of severe manifestations.7,8 Administration of two doses of the vaccine has been demonstrated to yield superior efficacy in comparison to a singular dose. Furthermore, if the vaccine is administered to individuals who lack immunity within a five-day window following exposure to varicella, it is effective in averting the majority of disease occurrences.7,9 In this study, we introduced a novel multi-epitope vaccine design targeting three specific proteins of the VZV. Using advanced immunoinformatics approaches, we identified and selected highly conserved and immunogenic epitopes capable of eliciting robust cellular and humoral immune responses. The design strategy incorporated multiple layers of computational analysis, including epitope prediction for cytotoxic T lymphocytes (CTL), and helper T lymphocytes (HTL) ensuring the inclusion of epitopes with high antigenicity, non-allergenicity, and non-toxicity. Additionally, structural bioinformatics techniques were employed to refine the vaccine construct, optimize its 3D structure, and assess its stability through molecular docking with immune receptors, such as TLR2, and extensive molecular dynamics simulations. This innovative approach provides a promising framework for the development of effective VZV vaccines, reducing reliance on traditional methodologies while accelerating the design process.
Methods
Detecting protein sequences and locating conserved regions
The amino acid composition of three proteins, namely envelope glycoprotein E (gE) (Accession no P09259), envelope glycoprotein B (gB) (Accession no. P09257), and large tegument protein deneddylase (IE63) (Accession no. P09278), were obtained from the National Center for Biotechnology Information (NCBI) and Universal Protein Resource (UniProt) (http://www.uniprot.org) The data was saved in FASTA format. To predict potential T-cell epitopes, we utilized the IEDB Analysis Resource tools, including NetMHCIIpan and other embedded algorithms. While prioritization was initially based on percentile ranks and binding affinities, we acknowledged inherent limitations and potential biases in these tools, such as restricted training datasets or MHC allele representation. Therefore, we adopted a more integrative approach that also considered additional immunological attributes, including epitope conservancy across multiple VZV clades, cross-reactivity potential, and population coverage. Epitopes with suboptimal percentile rank but high conservation or immunogenicity were not automatically excluded and were re-evaluated for inclusion. A comprehensive multiple sequence alignment was performed using Clustal Omega to assess conservation across representative strains of VZV, retrieved from NCBI Virus. Only epitopes demonstrating > 85% conservation among strains (Oka and wild-type clades 1–5) were prioritized. This transparent and multi-criteria selection strategy enhances the reproducibility and biological relevance of the predicted vaccine candidates. The workflow of this investigation is demonstrated in Fig. 1. To evaluate the presence of transmembrane regions, we employed the TMHMM server, which utilizes a Hidden Markov model to predict transmembrane helices within protein sequences.10 Proteins predicted to possess multiple transmembrane helices were excluded from further consideration due to the practical challenges they present in terms of expression, purification, and cloning factors that significantly hinder their feasibility as vaccine candidates. Instead, we prioritized highly antigenic proteins exhibiting either no transmembrane helices or a single transmembrane segment, ensuring their suitability for downstream vaccine development processes.
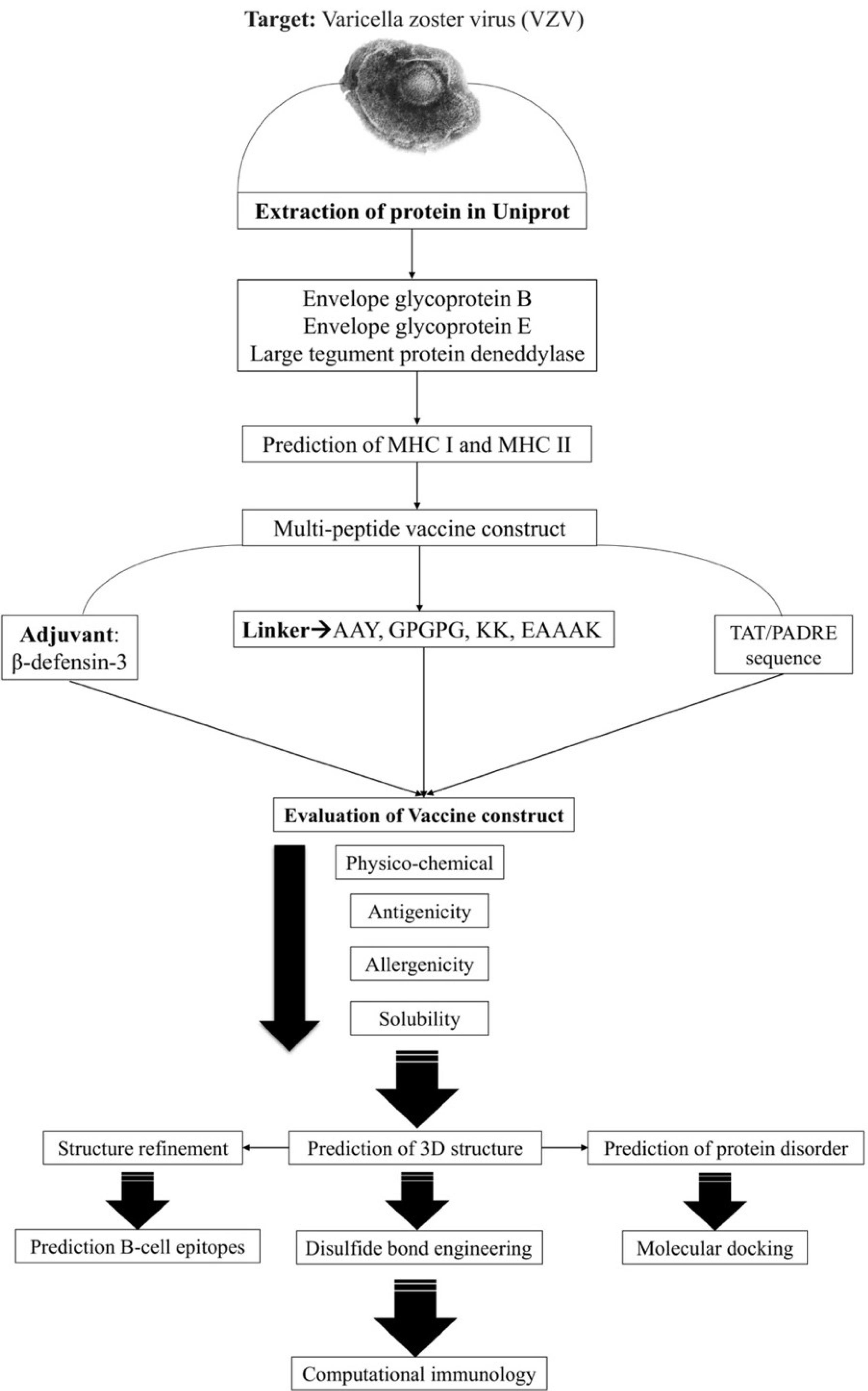
Fig. 1.
The workflow designed for in-silico vaccine design against the VZV.
.
The workflow designed for in-silico vaccine design against the VZV.
MHC-I and MHC-II epitope prediction
The immune system activation relies on MHC-I peptide presentation, essential for binding MHC-I peptides to mouse alleles (H2-Dd, H2-Kb, H2-Ld, H2-Kd, H2-Db, H2-Kk). We used IEDB's default method to predict this binding. MHC-II molecules are vital for the adaptive immune response with CD4 T-lymphocytes. To find MHC-II peptide binders, we used the IEDB tool and various prediction methods like SMM-align, NN-align, Sturniolo, and CombLib. For alleles H2-IAd, H2-IAb, and H2-Ied, we relied on NetMHCIIpan.
Chimeric vaccine construct
The predicted epitopes were fused using innovative linkers (GPGPG, KK, AAY, and EAAAK) to create a chimera. The chimeric protein's N-terminal section included the HIV TAT peptide as cell-penetrating peptides (CPPs) and the adjuvant (β-defensin 3) acting as the TLR2 agonist.11 Python scripts were used to shuffle the epitopes, generating multiple vaccine sequences while keeping the linkers, adjuvant, and other components intact. Another code determined the optimal sequence based on 3D structure and physicochemical properties.
Prediction of antigenicity, allergenicity, solubility, and physicochemical parameters prediction of the chimeric construct
We make use of the ExPASy ProtParam tool (https://web.expasy.org/protparam/) to calculate various parameters, including atomic composition, theoretical pI, amino acid composition, half-life, molecular weight, extinction coefficient, instability index, aliphatic index, and grand average of hydropathicity (GRAVY). One of the potential adverse effects of vaccination is the immune system's hypersensitivity to the vaccine components. To predict allergenicity for the submitted vaccine based on IgE epitope similarity, we rely on the AlgPred server (http://crdd.osdd.net/raghava/algpred/). Additionally, for antigenicity prediction based on the physicochemical properties of proteins in an alignment-free manner, we use VaxiJen v2.0 (http://www.ddgharmfac.net/vaxijen/VaxiJen/VaxiJen.html). Moreover, protein solubility is a crucial characteristic for proper functionality; thus, it needs to be determined for each vaccine. To assess the solubility of a submitted vaccine through overexpression in E. coli, we employ the SOLpro online software (http://scratch.proteomics.ics.uci.edu). This evaluation method relies on two-stage multiple representations of the primary sequence and SVM to determine protein solubility. The evaluation of toxicity was carried out by ToxinPred to prevent toxicity.12 To further prevent potential autoimmune reactions, we conducted a BLASTp analysis to identify and exclude any epitopes with similarity to human proteins.
Prediction of tertiary and secondary structure and refinement of the tertiary structure
We utilized PSIPRED (http://expasy.org/tool/gor4.html), an accessible online server to predict secondary structure, to accurately estimate the proportions of strands (S), helices (H), and coils (C) in the protein structure. Additionally, we employed the GalaxyWEB server (https://galaxy.seoklab.org/) to construct the initial tertiary protein structure.13 To refine and validate the 3D modeled structure, we iteratively utilize the GalaxyRefine server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) to enhance the structural modeling process.14
Forecasting of intrinsic protein disorder
In the field of vaccine development and immunoinformatics, a crucial obstacle is forecasting various aspects of protein behavior, such as disorder/unstructured regions, nucleic acid recognition, gene expression, foldability, and stability. These properties can be influenced by regions with high flexibility and low sequence complexity known as disorder parts. To predict the locations of disorder within the protein sequence, we used DISOPRED3 (http://bioinf.cs.ucl.ac.uk/psipred/) and DisEMBL 1.5 (http://dis.embl.de/).
Detection of conformational and linear B-cell epitopes
B-cell epitopes play a vital role in antigen recognition and their interaction with antibodies, which is essential for activating the immune response. To predict both conformational and linear B-cell epitopes present in the tertiaryvaccine structure, we utilized the ElliPro server (http://tools.iedb.org/ellipro/). The server employs the Protrusion Index (pI) score to identify discontinuous epitopes and cluster them based on a distance threshold R (measured in Ångströms).15
Disulfide bond engineering of chimeric vaccine
The process of disulfide bond engineering is highly beneficial as it helps enhance protein stability and enables the study of protein interactions and dynamics. To quickly identify and evaluate residue pairs that could potentially form disulfide bonds in a given protein model, assuming the residues are converted to cysteines, we rely on the Disulfide by Design 2 (DbD2) tool (http://cptweb.cpt.wayne.edu/DbD2/index.php).
Docking the vaccine with TLR4 and MHC molecules
Toll-like receptor 2 (TLR2) is a critical component of the innate immune system, playing a pivotal role in recognizing pathogen-associated molecular patterns (PAMPs) from a wide range of microorganisms, including bacteria, fungi, viruses, and parasites. It functions as a pattern recognition receptor (PRR) and forms heterodimers with TLR1 or TLR6 to detect specific ligands, such as lipoproteins, lipoteichoic acids, and peptidoglycans. Upon activation, TLR2 triggers downstream signaling cascades through the MyD88-dependent pathway, leading to the activation of nuclear factor kappa B (NF-κB) and the production of pro-inflammatory cytokines and type I interferons. This receptor is also implicated in modulating adaptive immune responses, linking innate recognition to long-term immunity.16 Dysregulation of TLR2 signaling has been associated with inflammatory diseases, autoimmune disorders, and cancer, making it a potential target for therapeutic interventions. The TLR2 PDB format was extracted in RCSB (PDB ID: 6NIG). To investigate how the final vaccine construct interacts with TLR4, we utilized the PatchDock server (http://bioinfo3d.cs.tau.ac.il/PatchDock/php.php), which employs a hybrid docking strategy to predict binding complexes between the vaccine and the receptor. Furthermore, the docking between epitopes and their MHC alleles was carried out by the HDock server (http://hdock.phys.hust.edu.cn/) to study the molecular docking17 and also the Prodigy used to calculate the energy binding18 that the formula of binding affinity demonstrated below.
ΔG = RT lnKd
Normal mode analysis
To thoroughly explore and assess the stability as well as the physical movements associated with the TLR3-vaccine docked complex, a comprehensive molecular dynamics simulation was conducted utilizing the iMODS online server, which can be accessed at the web address (http://imods.Chaconlab.org). This sophisticated online platform employs Normal Mode Analysis (NMA) specifically in internal (dihedral) coordinates, a methodology that is particularly adept at predicting the collective motions exhibited by proteins during their functional processes.19 Furthermore, the iMODS server meticulously computes a variety of important parameters related to the vaccine-receptor complex, including its deformability, B-factor, eigenvalues, variance, covariance map, and the overall elastic network, all of which contribute to a deeper understanding of the structural dynamics at play. Such detailed analyses are essential for elucidating the intricate mechanisms governing protein interactions and their implications for vaccine design and efficacy.
Molecular dynamic simulation
Furthermore, in conjunction with the preceding methodologies, the TLR2-vaccine complex was meticulously simulated utilizing the advanced GROMACS software, which is widely recognized for its capability in molecular dynamics simulations. In the subsequent steps of this intricate procedure, the TLR2-vaccine complex was strategically recentered within a meticulously defined triclinic simulation box, ensuring that the spatial arrangement of the molecular entities was optimal for accurate results. It is imperative to note that, by best practices in computational modeling, the margins of this box were established to maintain a minimum distance of at least 1 nm from the edges, thereby facilitating an environment conducive to realistic molecular interactions. To comprehensively encapsulate the protein molecule and to prevent any potential unfolding phenomena during the rigorous molecular dynamics (MD) simulation, this distance was judiciously increased following a thorough visual inspection utilizing VMD software, which is proficient in molecular visualization tasks. The dimensions of the finalized simulation box, specifically for the Vaccine-TLR2 complex, were precisely measured to be 8.654 × 10.453 × 8.653 nm³, which reflects careful consideration of the spatial requirements for the components involved. Subsequently, to create a realistic three-dimensional environment for the MD simulation, the TIP3P water model was employed to fill the designated simulation box, ensuring that the solvation conditions mimicked physiological relevance. In a further step to ensure the system's neutrality and to achieve an ionic concentration of 150 mM, sodium (Na⁺) and chloride (Cl⁻) ions were systematically introduced into the simulation box, which is a critical aspect of ensuring realistic electrostatic interactions in the molecular dynamics environment.20 To optimize the energy state of the system, the steepest descent method was deployed to minimize the energy of the components within the simulation box until convergence was attained at a predefined maximum force threshold of Fmax 1000 kJ mol⁻¹ nm⁻¹, representing a crucial step towards stabilization. Following this energy minimization phase, a two-phase equilibration process was undertaken, consisting of an initial 100 ps of NVT (constant number of particles, volume, and temperature) followed by an additional 100 ps of NPT (constant number of particles, pressure, and temperature) equilibration, which are essential for achieving a stable simulation environment. During this equilibration process, a base pressure of 1 bar was meticulously maintained at a temperature of 310 K, utilizing the Brendsen pressure coupling method to ensure that the system was equilibrated under desired thermodynamic conditions. Ultimately, a production MD run lasting 100 ns was executed to capture the dynamic behavior of the system over time, allowing for the observation of molecular interactions and conformational changes. To enhance the accuracy of the analysis regarding the evolution of the vaccine-TLR2 complex throughout the simulation, the UCSF Chimera software was employed for the visualization of the MD run, which is renowned for its capabilities in molecular modeling and analysis. In the subsequent analysis of the molecular dynamics trajectory, various metrics such as the root mean square deviation (RMSD), root mean square fluctuation (RMSF), and free energy landscape (FEL) were utilized to quantify and elucidate the structural dynamics and stability of the vaccine-TLR2 system throughout the simulation timeframe.
Calculation binding affinity
The intricate and comprehensive computation of binding free energy was meticulously performed utilizing advanced functional energy methodologies, specifically known as MM-GBSA (Molecular Mechanics-Generalized Born Surface Area), which were adeptly executed through the sophisticated Hawkdock web tool.21 To ascertain the binding free energy, it is essential to consider a multitude of energy components, which collectively include van der Waals energy, electrostatic energy, polar solvation-free energy, solvation-free energy, and the aggregate total energy across the molecular system under investigation. To accurately calculate the binding free energy, we systematically retrieved a molecular snapshot at regular intervals of every 5 nanoseconds throughout the MD simulation, which allowed for a thorough estimation of the binding free energy based on the temporal evolution of the system. It is noteworthy that lower values of binding energy, particularly those that are negative, are indicative of a more favorable binding interaction occurring at the residue level, which suggests a stronger affinity between the interacting molecular entities involved. The significance of these calculated binding energies is paramount, as they provide critical insights into the thermodynamic stability and interactions that govern the molecular behavior within the biological context. Ultimately, the application of such methodologies not only enhances our understanding of molecular binding interactions but also contributes to the broader field of computational biochemistry and drug design.
Immune simulation
To assess the vaccine immunogenicity and predict the immune response, it is necessary to identify B cell and T cell epitopes. To achieve this goal, we employed C-ImmSim (http://kraken.iac.rm.cnr.it/C-IMMSIM/index.php?page=0), a specialized Immunoinformatics simulation tool. This tool was utilized to simulate specific immune processes in three regions of the body: the bone marrow, thymus, and lymphatic organs.22
Codon optimization and cloning
Ultimately, the effectiveness and overall efficacy of the vaccine model employed for cloning and subsequent expression is of paramount importance when considering the in vitro production of the vaccine itself. To achieve an optimal construction of the vaccine, the initial step involved the utilization of the Java Codon Adaptation Tool (JCat), which can be accessed at the website http://www.JCat.de/, to ascertain the most favorable conditions for protein expression specifically within the E. coli strain K12.23 The quantification of protein expression levels is meticulously calculated based on the codon adaptation index (CAI) values, which must exceed the threshold of 0.8, along with the GC content that should ideally range between 30 to 70 percent. Subsequently, to provide empirical confirmation of the vaccine’s expression, the optimized sequence corresponding to the vaccine was meticulously cloned into the E. coli plasmid vector pET-30a ( + ), ensuring that this was achieved within the designated restriction sites of HindIII and BamHI, utilizing the sophisticated functionalities of SnapGene 8.0.2 software to facilitate this process.
Population coverage
The global applicability of the selected epitopes was evaluated through comprehensive population coverage analysis using the IEDB Population Coverage Tool (http://tools.iedb.org/population/). All predicted MHC-I and MHC-II epitopes, along with their frequent human corresponding binding alleles, were analyzed using default parameters for epitope length and integrated allele frequency databases.
Results
Selected protein analysis
We performed transmembrane topology prediction using TMHMM to evaluate the extracellular accessibility of the selected proteins. Our analysis confirmed that key regions of glycoprotein E (gE) and glycoprotein B (gB) are exposed on the viral surface, making them accessible to the host immune system and suitable as vaccine targets. Although the tegument protein IE63 is predominantly intracellular, its inclusion aims to stimulate cellular immunity by targeting latent infection phases.
Initial assessment of the acquired sequences and MHC I/MHC II binding epitopes prediction
We collected FASTA sequences for nine proteins, namely envelope glycoprotein B, envelope glycoprotein E, and large tegument protein deneddylase, from the UniProt database. Through bioinformatics assessments, we identified MHC I and MHC II binding epitopes with the most favorable percentile ranks. Furthermore, using the IEDB server, we discovered MHC-I epitopes that bind to specific mouse alleles, including H2-Dd, H2-Kb, H2-Ld, H2-Kd, H2-Db, and H2-Kk. For mice MHC-II alleles, we used the IEDB MHC-II prediction platform to predict MHC II binding epitopes based on percentile rank and IC50. Out of the predicted MHC I and MHC II binding epitopes, we selected eighteen epitopes. It is important to note that higher percentile ranks indicate better binding potential, as shown in Table 1.
Table 1.
The predicted CTL and HTL epitopes
|
Antigen
|
MHC I epitope
|
Position
|
Allele
|
Percentile rank
|
Polarity
|
| Envelope glycoprotein E |
RYDDFHTDEDKL |
34-45 |
H-2-Kd |
0.55 |
Polar |
| Envelope glycoprotein E |
RYTETWSFL |
15-23 |
H-2-Kd |
0.01 |
Polar |
| Envelope glycoprotein E |
HEAPFDLLL |
8-16 |
H-2-Kk |
0.07 |
Non-polar |
| Glycoprotein B |
SSVEFAML |
30-37 |
H-2-Kb |
0.01 |
Non-polar |
| Glycoprotein B |
EEVEARSI |
40-47 |
H-2-Kk |
0.02 |
Polar |
| Glycoprotein B |
NEFNLNQI |
45-52 |
H-2-Kk |
0.03 |
Polar |
| Tegument protein |
ISILNSPII |
23-31 |
H-2-Db |
0.01 |
Non-polar |
| Tegument protein |
SEGLLIRAI |
37-45 |
H-2-Kk |
0.04 |
Non-polar |
| Tegument protein |
RYMREHTAL |
5-13 |
H-2-Kd |
0.01 |
Polar |
| Envelope glycoprotein E |
AVTPQPRGA |
329-338 |
H2-IAb |
0.21 |
Non- polar |
| Envelope glycoprotein E |
YHSDHAESS |
52-61 |
H2-IAb |
0.24 |
Polar |
| Envelope glycoprotein E |
ENHPFTLRA |
176-185 |
H2-IAb |
0.7 |
Polar |
| Glycoprotein B |
YNSSHVRTG |
368-377 |
H2-IAb |
0.12 |
Polar |
| Glycoprotein B |
YESLQVEPT |
13-22 |
H2-IAd |
0.62 |
Polar |
| Glycoprotein B |
YSRPLISIV |
544-553 |
H2-IAb |
1.1 |
Non-polar |
| Tegument protein |
ESAINAVVA |
583-592 |
H2-IAd |
0.01 |
Non-polar |
| Tegument protein |
VDSIKALQA |
1177-1186 |
H2-IAd |
0.05 |
Non-polar |
| Tegument protein |
TRALIAEQS |
233-243 |
H2-IAd |
0.06 |
Polar |
Creating the chimeric vaccine
The vaccine design comprises eighteen epitopes, strategically separated by linkers to avoid any conformational clashes. Furthermore, the inclusion of the TAT peptide aids in improving vaccine delivery. To identify a promising candidate for further in silico experiments, all sequences generated by our vaccine sequence generator were thoroughly analyzed. Polarity calculations revealed a favorable proportion of polar amino acids in the selected candidate Fig. 2.
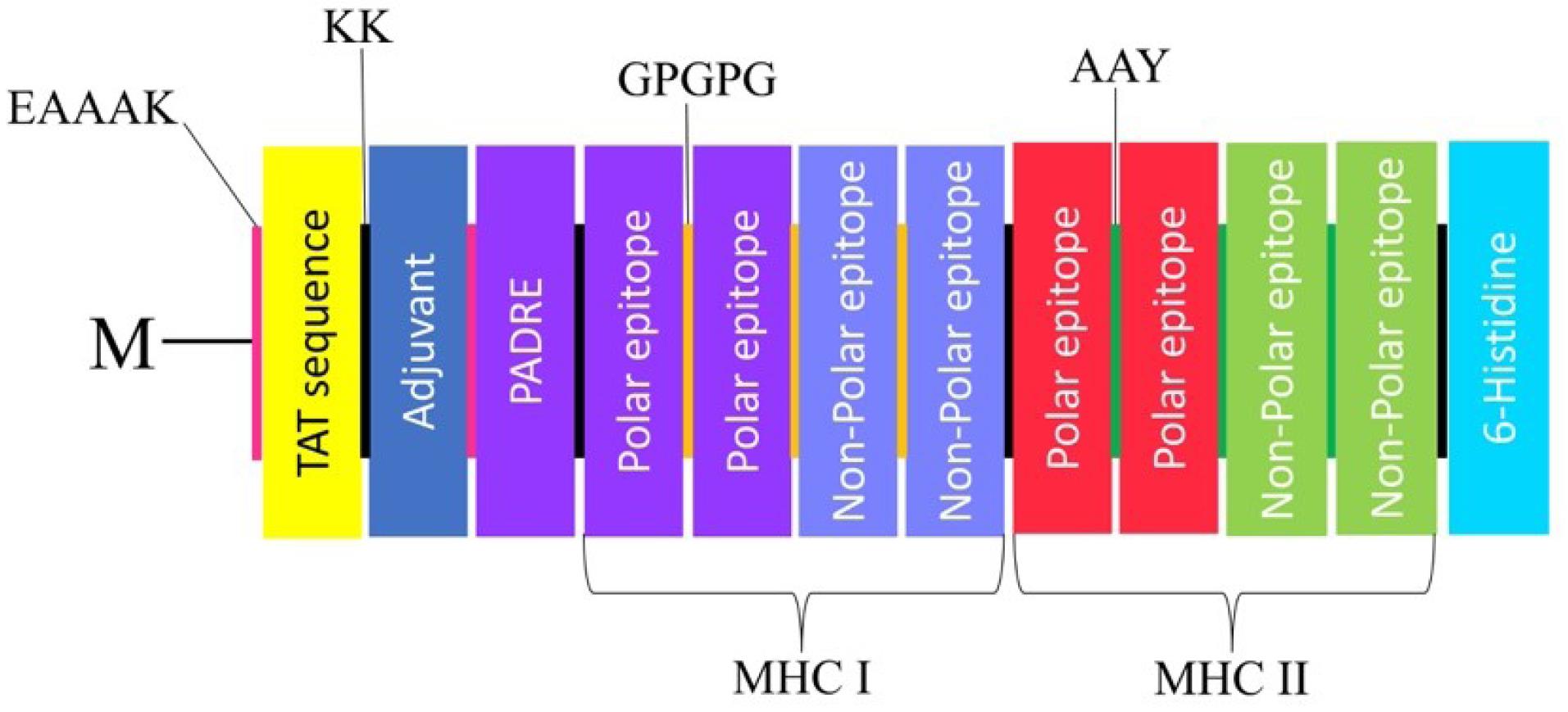
Fig. 2.
The construction of a vaccine consists of adjuvant, linkers, TAT and PADRE sequence, HTL and CTL epitopes, and 6-histidine. At the N-terminal of the vaccine construct the methionine linked with EAAAK to TAT sequence and also adjuvant attached with KK then the PADRE sequences bonded with EAAAK. In addition, the polar and non-polar epitopes are attached separately with GPGPG and AAY linkers (MHC I epitopes connected by GPGPG and the MHC II epitopes connected by AAY). At the C-terminal the 6-histidine linked with KK linkers.
.
The construction of a vaccine consists of adjuvant, linkers, TAT and PADRE sequence, HTL and CTL epitopes, and 6-histidine. At the N-terminal of the vaccine construct the methionine linked with EAAAK to TAT sequence and also adjuvant attached with KK then the PADRE sequences bonded with EAAAK. In addition, the polar and non-polar epitopes are attached separately with GPGPG and AAY linkers (MHC I epitopes connected by GPGPG and the MHC II epitopes connected by AAY). At the C-terminal the 6-histidine linked with KK linkers.
Assessment of antigenicity, allergenicity, physiochemical properties, and solubility of the vaccine candidate
The AlgPred server was used to predict the probability of vaccine allergenicity with highly specific methods, and the results confirmed that the final vaccine construct is non-allergenic. Additionally, no experimentally proven IgE epitopes were found in the vaccine. To assess antigenicity, the VaxiJen v2.0 platform yielded a probability of approximately 0.52 at the cut-off level of 0.4, confirming the vaccine's antigenicity. The theoretical isoelectric point (pI) of the vaccine structure was calculated to be 9.65, and its molecular weight (Mw) was found to be 34 kDa. Furthermore, the half-life of the vaccine was estimated to be 30 hours in mammalian reticulocytes (in vitro), more than 20 hours in yeast (in vivo), and more than 10 hours in E. coli (in vivo). The Grand Average of Hydropathy (GRAVY) value for the vaccine construct was determined to be -0.548, indicating a slightly hydrophobic nature. Furthermore, the aliphatic index was computed and yielded a value of 67.80, indicating that the engineered chimera possesses favorable thermodynamic stability across different temperatures. The selected construct also exhibited an instability index of 30.20, providing additional evidence of its stability. Moreover, the estimated protein solubility after overexpression in E. coli was determined to be 0.90% (Table 2). The vaccine construct was analyzed for potential homology with human proteins, and the results confirmed no significant similarity, indicating a lower risk of autoimmunity.
Table 2.
The characterization of vaccine
|
Property
|
Value
|
| Number of residues |
322 |
| Allergenicity |
Non-allergenic (AlgPred prediction) |
| IgE epitopes |
None experimentally proven |
| Antigenicity |
0.52 (VaxiJen v2.0, cut-off 0.4) |
| Toxicity |
Non-toxicity |
| Isoelectric point (pI) |
9.65 |
| Molecular weight (Mw) |
34 kDa |
| Half-Life |
30 hours (mammalian reticulocytes, in
vitro) > 20 hours (yeast, in vivo) > 10 hours (E. coli, in vivo) |
| GRAVY |
-0.548 (slightly hydrophobic) |
| Aliphatic index |
67.80 |
| Instability index |
30.20 (stable) |
| Solubility in E. coli |
0.90% |
Tertiary and secondary structure and Refinement of the tertiary structure
The PSIPRED computational server analyzed the vaccine and identified its alpha-helix, beta-strand, and coil secondary structural components (Fig. 3). These predicted secondary structures are crucial for improving the 3D structure of the protein. The final vaccine construct was then modeled using the GalaxyWEB server. To further enhance the structural modeling, we applied the GalaxyRefine server iteratively (Fig. S1).

Fig. 3.
2D structure of vaccine construct that used the PSIPRED server
.
2D structure of vaccine construct that used the PSIPRED server
Prediction of intrinsic protein disorder
The results from DisEMBL revealed the presence of disorder segments at positions 9-23, 33-62, 87-200, 212-225, 267-278, and 315-322, based on the Loops/coils definition. Similarly, disorder intervals were identified at positions 1-31, 40-61, and 84-104 using the Hot-loops definition, and at positions 1-19, 53-73, 211-218, and 313-322 based on the Remark-465 definition. Additionally, potentially disordered regions were identified using a cut-off value of 0.5 and visualized through PSIPRED.
Predicted B-cell epitopes
The Ellipro online tool was utilized to conduct a comprehensive analysis for the detection of both linear and discontinuous B cell epitopes. Following that, the results of this study were arranged and shown in Table 3 and Table S1 for linear and discontinuous epitopes, respectively. These data provide a concise summary of the identified B cell epitopes for further evaluation and consideration in the vaccine development process.
Table 3.
The linear B-cell epitopes
|
Number
|
Start
|
End
|
Peptide
|
Number of residues
|
Score
|
| 1 |
1 |
45 |
MEAAAKGRKKRRQRRRPPQKKGIINTLQKYYCRVRGGRCAVLSCL |
45 |
0.813 |
| 2 |
120 |
148 |
VEARSIGPGPGNEFNLNQIGPGPGRYMRE |
29 |
0.696 |
| 3 |
234 |
254 |
YNSSHVRTGAAYYESLQVEPT |
21 |
0.674 |
| 4 |
50 |
61 |
QIGKCSTRGRKC |
12 |
0.713 |
| 5 |
190 |
198 |
SPIIGPGPG |
9 |
0.516 |
| 6 |
104 |
111 |
RYTETWSF |
8 |
0.609 |
| 7 |
271 |
277 |
VTPQPRG |
7 |
0.629 |
| 8 |
96 |
102 |
DKLGPGP |
7 |
0.569 |
Vaccine disulfide bond engineering
Disulfide engineering involves modifying residues located in highly flexible regions, and introducing cysteines to enhance vaccine stability. We examined the residue pairs based on parameters such as energy on chi3 value and B-factor, and the results are depicted in Fig. S2.
Molecular docking of the vaccine construct with TLR4
The vaccine was docked with TLR2 using PatchDock, resulting in a docking score of -280.81 and a ligand RMSD of 145.05 Å for the top-ranked model. The 10 most favorable docked complexes were acquired with PatchDock, and visualizations of the docked complex and interacting residues were provided using UCSF Chimera (Fig. 4). The analysis of protein-protein interaction is carried out via RING. The result showcases the analysis of the protein-protein interaction between TLR2 and the vaccine candidate, highlighting various edge types involved in their inter-chain interactions. Here is the integrated interpretation: The interaction profile between TLR2 and the vaccine reveals the formation of six inter-chain hydrogen bonds, which are critical for establishing and maintaining the stability of the complex. Hydrogen bonds serve as primary contributors to the specificity and binding affinity between the two molecules. Additionally, two π-π stacking interactions were observed, indicating the involvement of aromatic residues in stabilizing the protein-protein interface through their planar stacking. One ionic interaction was identified, signifying an electrostatic contribution to the binding interface, which further enhances the strength of the complex. Van der Waals forces accounted for the majority of interactions, with 34 inter-chain contacts contributing to the stabilization of the complex through non-covalent dispersive forces. These interactions highlight the close packing and complementary surface geometry between TLR2 and the vaccine candidate. No π-cation, π-H bond, disulfide, metal coordination, halogen, or interatomic contacts were detected in this analysis, suggesting the absence of these interaction types within the binding interface. The lack of intra-chain interactions also emphasizes that the observed interactions are purely inter-chain, reinforcing the specificity of the TLR2-vaccine interaction. Overall, the interaction profile underscores a stable binding complex facilitated predominantly by hydrogen bonds, van der Waals forces, and π-π stacking, with ionic interactions providing additional strength. This stability is essential for effective vaccine recognition and immune activation via TLR2. Further computational and experimental validation would confirm the robustness of this interaction and its implications for immune response induction.
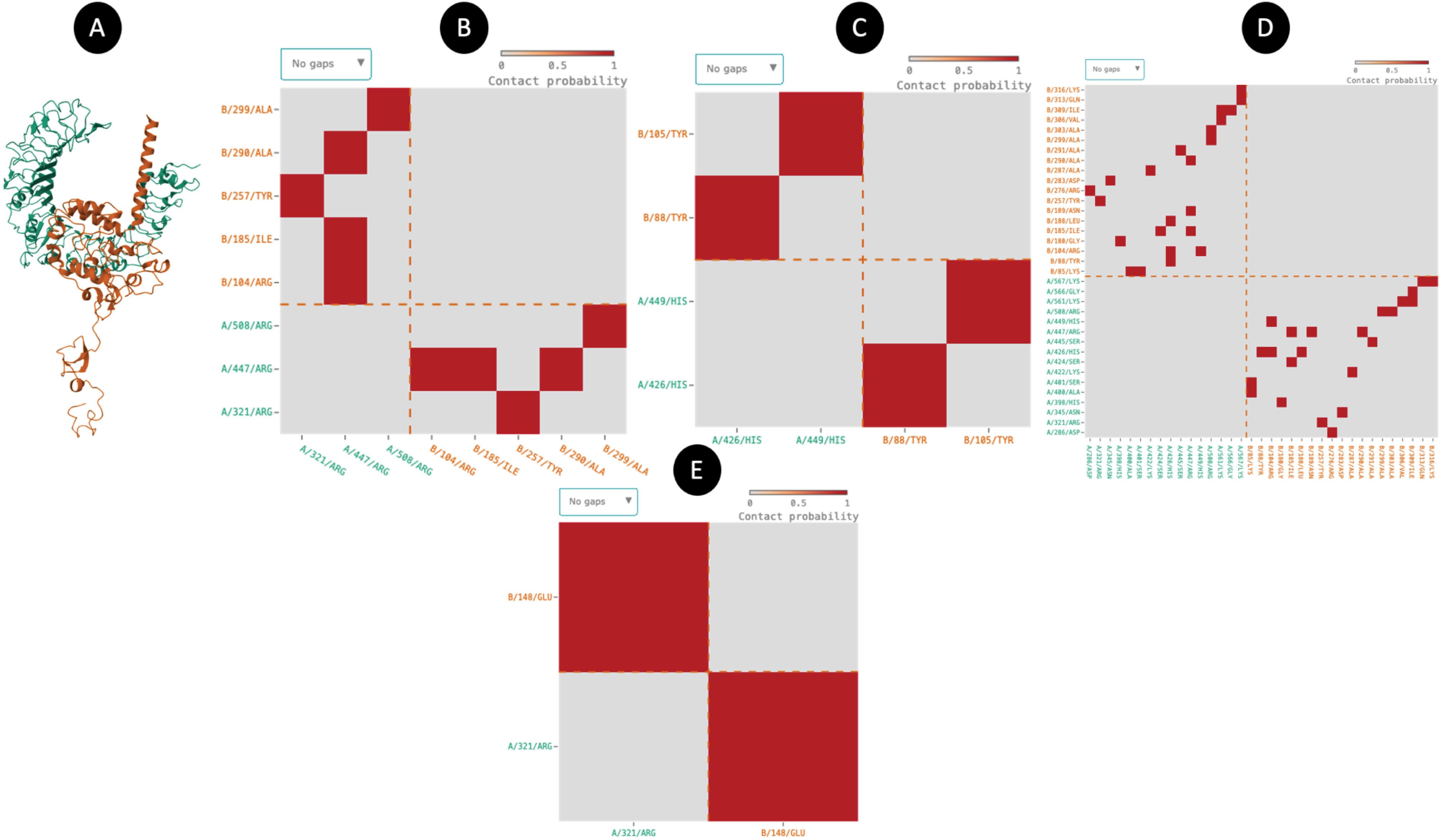
Fig. 4.
Molecular docking result and residue-residue interactions. (A) The TLR2 and vaccine docked with the Patchdock server. (B) H-bond (C) pi-pi stacking (D) Van der Walls.
.
Molecular docking result and residue-residue interactions. (A) The TLR2 and vaccine docked with the Patchdock server. (B) H-bond (C) pi-pi stacking (D) Van der Walls.
The molecular docking analysis was conducted to evaluate the binding affinity and stability of selected epitopes to MHC I and MHC II molecules. The docking scores and binding free energy (ΔG binding) values highlight the immunological relevance of these epitopes as vaccine candidates. The results for MHC I alleles show strong binding interactions, with HEAPFDLLL (H-2-Kk, PDB ID: 1ZT7) demonstrating the highest docking score (-181.05) and ΔG binding value (-9.0 kcal/mol). This high binding energy suggests a stable interaction, likely due to the peptide's hydrophobic nature and compatibility with the H-2-Kk binding groove. Similarly, SSVEFAML (H-2-Kb, PDB ID: 1LK2) and RYDDFHTDEDKL (H-2-Kd, PDB ID: 2FWO) exhibited docking scores of -178.55 and -169.62, respectively, with corresponding ΔG values of -8.4 and -8.8 kcal/mol. These results emphasize the potential of these peptides to elicit CTL responses by stably binding to MHC I molecules, essential for antigen presentation. For MHC II alleles, AVTPQPRGA (H2-IAb, PDB ID: 1MUJ) showed the most negative ΔG binding value (-26.1 kcal/mol), indicating exceptional binding stability. The docking score (-150.65) and strong molecular interactions suggest that this peptide is highly immunogenic and capable of activating CD4 + T-helper cells effectively. YESLQVEPT (H2-IAd, PDB ID: 7RDV) followed closely, with a ΔG value of -25.3 kcal/mol and a docking score of -152.65. The interaction of this peptide with the H2-IAd molecule reflects its structural compatibility and immunogenic potential. VDSIKALQA (H2-IAb, PDB ID: 1MUJ) displayed the least negative docking score (-100.56) but maintained a strong ΔG binding value of -24.7 kcal/mol, suggesting a stable yet slightly weaker interaction compared to the other MHC II epitopes.
The comparison of ΔG binding values between MHC I and MHC II alleles highlights notable differences. MHC II epitopes showed significantly more negative ΔG values, indicating stronger binding stability. This aligns with the broader and more flexible peptide-binding grooves of MHC II molecules, allowing for diverse peptide interactions. On the other hand, MHC I alleles, characterized by compact binding grooves, demonstrated slightly lower binding energies but still within a range indicative of strong and stable interactions. The immunological implications of these findings are substantial. The robust binding affinities observed, particularly for HEAPFDLLL (H-2-Kk) among MHC I epitopes and AVTPQPRGA (H2-IAb) among MHC II epitopes, underscore their potential as key components in vaccine formulations. These epitopes can trigger both CTL and helper T-cell responses, crucial for inducing a comprehensive immune response. Additionally, the inclusion of multiple MHC alleles in the analysis ensures broad population coverage, enhancing the potential efficacy of the designed vaccine. Overall, ΔG binding values provide critical insights into the stability and immunogenic potential of the selected epitopes, supporting their further evaluation and potential inclusion in vaccine development efforts. Further experimental validation, including binding assays and immunogenicity testing, is required to confirm these computational findings and establish their applicability in real-world settings. The docking scores for MHC-I/MHC-II and epitopes were demonstrated in Table S2.
Normal mode analysis
The structural dynamics and flexibility of the TLR2-vaccine complex were analyzed using normal mode analysis (NMA). The covariance matrix (Fig. 5A) shows the correlated and anti-correlated motions between residues, with significant clustering observed, indicating functional and structural domains within the complex. The stiffness matrix (Fig. 5B) illustrates the elastic network model, with denser regions signifying strong inter-atomic interactions. The deformability plot (Fig. 5C) highlights the regions of the complex with high flexibility, particularly in loop regions and termini, which are more susceptible to conformational changes. The variance explained by individual modes (Fig. 5D) demonstrates that the first few modes contribute significantly to the overall motion of the complex, suggesting a dominance of low-frequency motions. Comparison of the B-factor (Fig. 5E) derived from NMA and experimental data (PDB) shows consistency in flexibility patterns, validating the computational results. Finally, the eigenvalue distribution (Fig. 5F) confirms the dominance of the first mode, with subsequent modes exhibiting decreasing contributions, indicative of the structural stability and the constrained nature of the complex. These results provide a detailed understanding of the TLR2-vaccine complex's structural dynamics, offering insights into its stability and interaction potential.
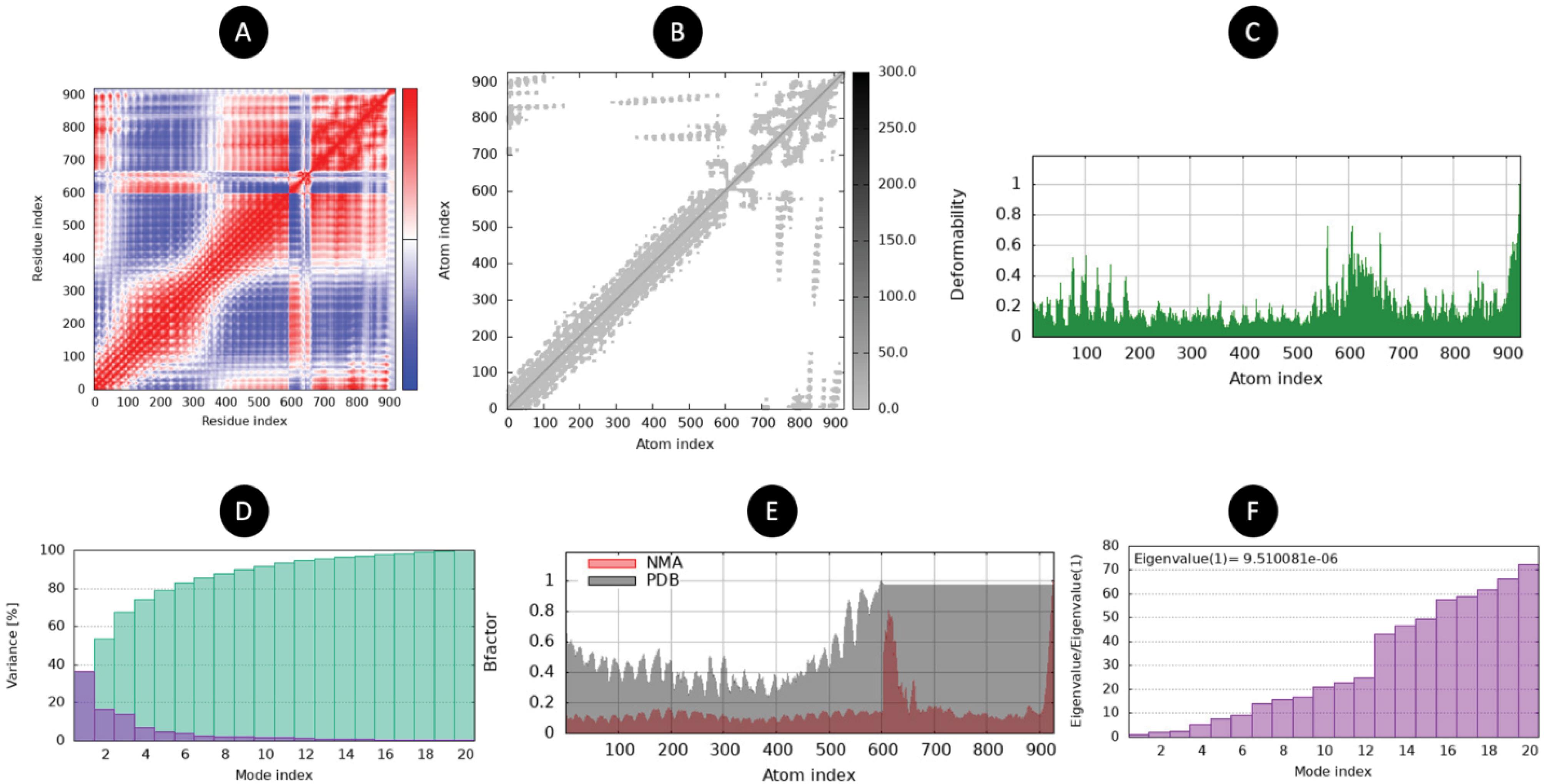
Fig. 5.
The normal mode analysis. (A) Covariance map (red: correlated, white: uncorrelated, blue: anti-correlated). (B) Elastic network (darker regions mean stiffer regions) (C) Deformability (D) Variance (red color: individual variances and green color: cumulative variances). (E) B-factor (F) Eigenvalues (lower value means easier deformation).
.
The normal mode analysis. (A) Covariance map (red: correlated, white: uncorrelated, blue: anti-correlated). (B) Elastic network (darker regions mean stiffer regions) (C) Deformability (D) Variance (red color: individual variances and green color: cumulative variances). (E) B-factor (F) Eigenvalues (lower value means easier deformation).
Molecular dynamic simulation
The RMSD plot for the TLR2-vaccine complex over a 100 ns molecular dynamics simulation provides valuable insights into the system's structural stability and conformational changes. Initially, the RMSD shows a rapid increase, reaching approximately 3.5 nm within the first 20 ns, indicating significant structural rearrangements as the system stabilizes. This phase reflects the relaxation of the complex from its starting conformation to an energetically favorable state. After the initial equilibration, the RMSD stabilizes around 4.0 nm, with minor fluctuations, indicating that the system reaches a steady state. This consistent RMSD value throughout the remainder of the simulation suggests that the TLR2-vaccine complex maintains a stable conformation and does not experience significant structural deviations (Fig. 6A). The observed stability is critical for effective receptor-ligand binding, implying that the vaccine construct forms a robust interaction with TLR2. The high initial RMSD can be attributed to the flexible regions of the complex or adjustments at the binding interface, while the subsequent plateau demonstrates that these movements lead to a stable and energetically favorable binding conformation. This result supports the structural integrity of the complex, reinforcing the potential of the vaccine candidate for efficient immune activation through TLR2 engagement.
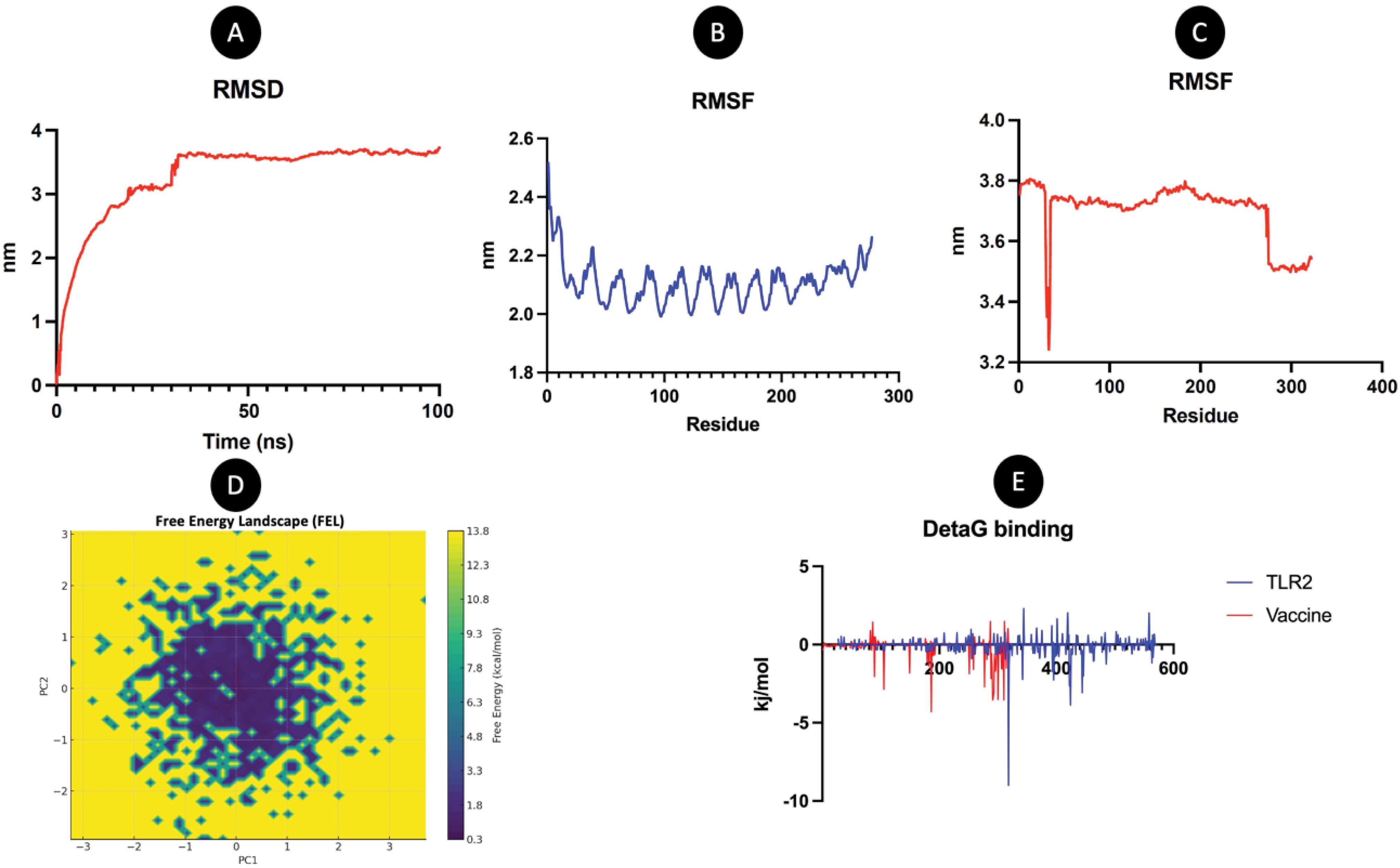
Fig. 6.
Molecular dynamic simulation. (A) RMSD (B) RMSF of TLR2 (C) RMSF of vaccine (D) Free energy landscape (E) Delta G (Binding energy).
.
Molecular dynamic simulation. (A) RMSD (B) RMSF of TLR2 (C) RMSF of vaccine (D) Free energy landscape (E) Delta G (Binding energy).
The RMSF analysis compares the flexibility of residues in TLR2 (blue) (Fig. 6B) and the vaccine (red) (Fig. 6C). The graph for TLR2 shows fluctuations ranging from 1.8 to 2.6 Å, with peaks around specific residues, indicating regions of higher flexibility. In contrast, the vaccine exhibits higher overall fluctuations, ranging from 3.2 to 3.8 Å, suggesting greater structural flexibility across its residues. TLR2 displays moderate flexibility, with relatively stable regions (lower RMSF values) and some flexible peaks. The vaccine shows consistently higher flexibility, which may reflect its dynamic nature or adaptive properties. This comparison highlights distinct dynamic behaviors: TLR2 maintains relatively stable regions, while the vaccine is more flexible, potentially influencing their interaction mechanisms or functional roles. Further analysis could correlate these fluctuations with specific structural or functional features of both molecules. The FEL of the TLR2-vaccine complex reveals insights into the thermodynamic stability and conformational dynamics of their interaction (Fig. 6D). The landscape shows a broad range of free energy values, spanning from 0.3 to 13.8 kcal/mol, with multiple minima and transitions, indicating distinct stable states and intermediate conformations during binding. Key observations: The presence of low-energy basins (e.g., near 0.3–1.8 kcal/mol) suggests highly stable conformations, likely representing tightly bound states of the TLR2-vaccine complex. Higher-energy regions (e.g., 10.8–13.8 kcal/mol) correspond to less favorable or transient states, possibly reflecting dissociation intermediates or flexible, unbound configurations. The smooth transitions between energy minima imply a dynamic but cooperative binding process, where the system samples multiple conformations before settling into stable complexes. This landscape underscores the complexity of TLR2-vaccine interactions, with both stable binding modes and flexible regions that may facilitate adaptive immune recognition. Further analysis could link specific energy minima to functional structural features. The Comparative Evaluation of Different Linkers in Multi-Epitope Vaccine Design was demonstrated in Table S3.
MMGBSA
The energy decomposition analysis during a 100 ns molecular dynamics simulation of the TLR2-vaccine complex reveals key insights into the energetic contributions to the binding interaction (Table S4). The van der Waals (VDW) interactions, with an average energy of -153.81 kcal/mol, contribute significantly to the stability of the complex, indicating effective packing and complementary surface interactions. Electrostatic energy (ELE) further enhances the binding, showing a substantial contribution of -907.61 kcal/mol, reflecting the strong ionic and polar interactions at the binding interface. The Generalized Born (GB) solvation energy, with a value of 1007.18 kcal/mol, offsets some of these stabilizing forces, as expected for polar and charged interfaces interacting in an aqueous environment. The surface area energy (SA) is recorded as -20.83 kcal/mol, representing hydrophobic contributions that aid in stabilizing the interface by minimizing solvent exposure of hydrophobic residues. Overall, the total binding free energy is calculated at -75.07 kcal/mol, demonstrating a thermodynamically favorable interaction between TLR2 and the vaccine (Fig. 6E). This energy profile highlights the critical balance of van der Waals and electrostatic forces in maintaining the integrity of the complex, with solvation effects providing essential context for the interaction within a biological system. The results suggest a robust and stable interaction, supporting the vaccine's potential for TLR2-mediated immune activation. Further experimental validation could solidify these findings and assess their implications for vaccine efficacy.
Immune simulation
The immune response simulation of the vaccine revealed significant dynamics of antigen presentation and adaptive immune activation over time. The antigen count rapidly declined (Fig. 7A) following its initial spike, corresponding to antigen processing and immune system engagement. Immunoglobulin levels, particularly IgG1 and IgG2, increased steadily, indicating a robust humoral response, with IgM peaking earlier as part of the primary immune response. Cytokine profiling (Fig. 7B) demonstrated a sharp rise in IFN-γ, IL-2, and IL-12, highlighting a Th1-dominated response critical for cellular immunity. IL-4 and IL-10 levels also increased, suggesting a balanced regulatory environment, while TGF-β remained relatively stable. The inset indicates the dynamic interplay of cytokines over time. The cytotoxic T-cell (TC) population analysis (Fig. 7C) showed an increase in both total and memory TC cells, reflecting the vaccine’s potential to elicit long-term cellular immunity. Memory cell formation was prominent, and critical for durable protection. Similarly, macrophage (MA) populations (Fig. 7D) exhibited dynamic transitions between resting, active, and antigen-presenting states, with active MAs stabilizing after initial fluctuations, indicating sustained antigen presentation. TC cell states (Fig. 7E) revealed a decline in resting cells and an increase in active and duplicating cells, confirming effective T-cell activation and proliferation. Anergic cells remained minimal, suggesting the absence of significant immune tolerance or exhaustion. Lastly, B-cell populations (Fig. 7F) showed a substantial increase in total B cells and memory B cells, with isotype switching favoring IgG1 and IgG2 production, consistent with a robust and targeted humoral response. Overall, the simulation indicates a strong and balanced immune response with efficient antigen clearance, cytokine activation, T- and B-cell proliferation, and memory cell generation, demonstrating the vaccine's potential efficacy in eliciting protective immunity.
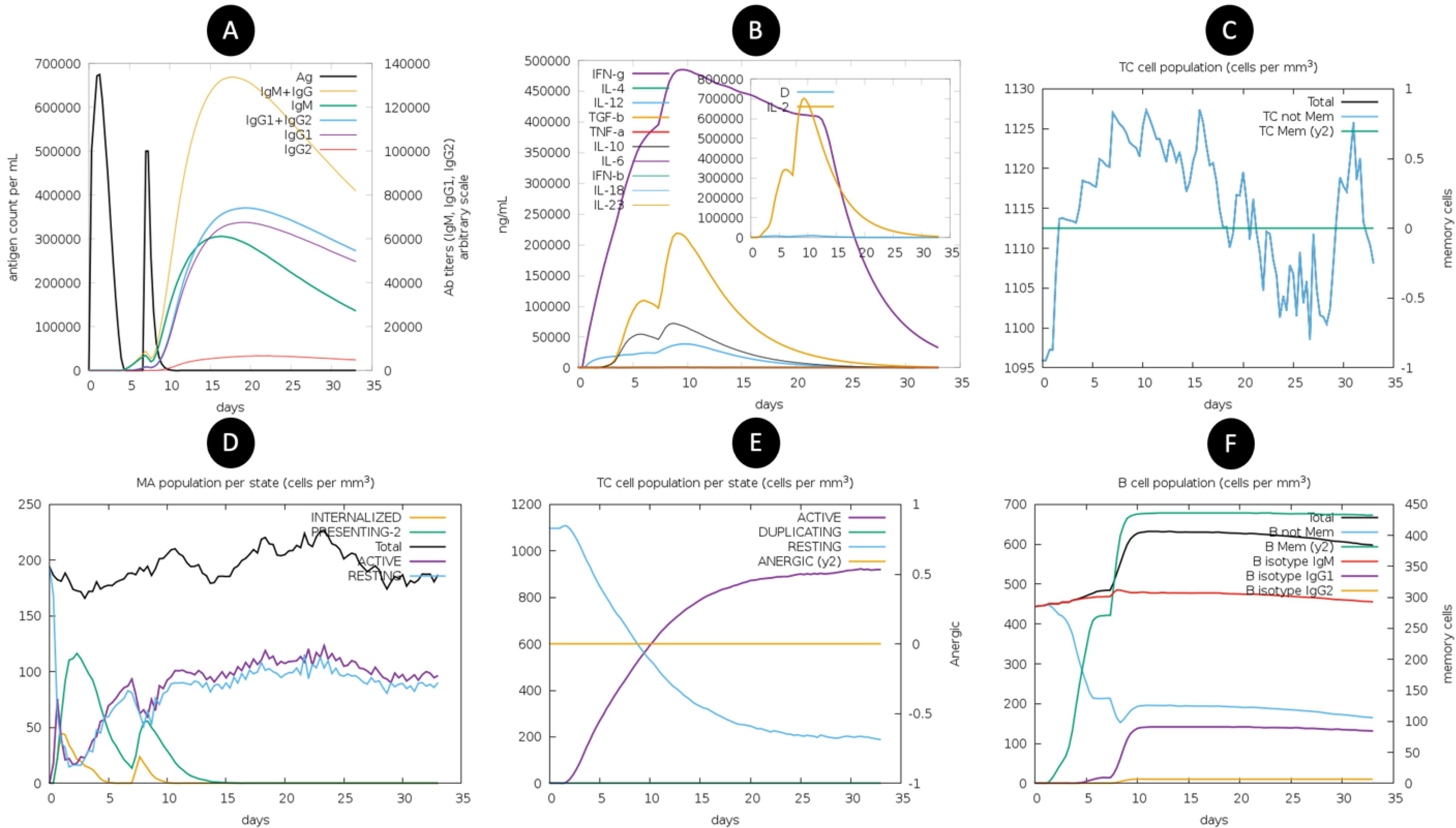
Fig. 7.
Immune simulation.
.
Immune simulation.
In silico cloning
After codon optimization using the JCAT tool, the vaccine construct resulted in a sequence of 978 nucleotides. The optimized sequence achieved a Codon Adaptation Index (CAI) of 1.0, reflecting ideal codon usage for high-level expression in the E. coli K12 host system. Additionally, the GC content of 54.14% was within the optimal range for stability and efficient transcription in E. coli. While this is appropriate for initial cloning and expression trials, it is important to note that E. coli lacks the machinery for certain post-translational modifications (e.g., glycosylation, disulfide bond isomerization) that may be essential for the proper folding and immunogenic function of eukaryotic epitopes. Additionally, codon usage preferences differ across species, and optimization for mammalian systems may be necessary in future studies aimed at therapeutic development and clinical application. These considerations will guide the subsequent phases of vaccine development. These attributes strongly suggest that the vaccine sequence is well-suited for robust expression in the selected bacterial host (Fig. 8). To further validate the construct, the optimized sequence was successfully inserted into the pET30a ( + ) expression vector. The recombinant plasmid design ensures compatibility with the bacterial system for downstream production and includes regulatory elements such as a strong T7 promoter for enhanced expression. The final vaccine sequence integrated into the plasmid is depicted in the accompanying figure, providing a visual representation of the cloning strategy and construct organization. This step marks a critical milestone in the development pipeline, enabling efficient production and further characterization of the vaccine candidate.
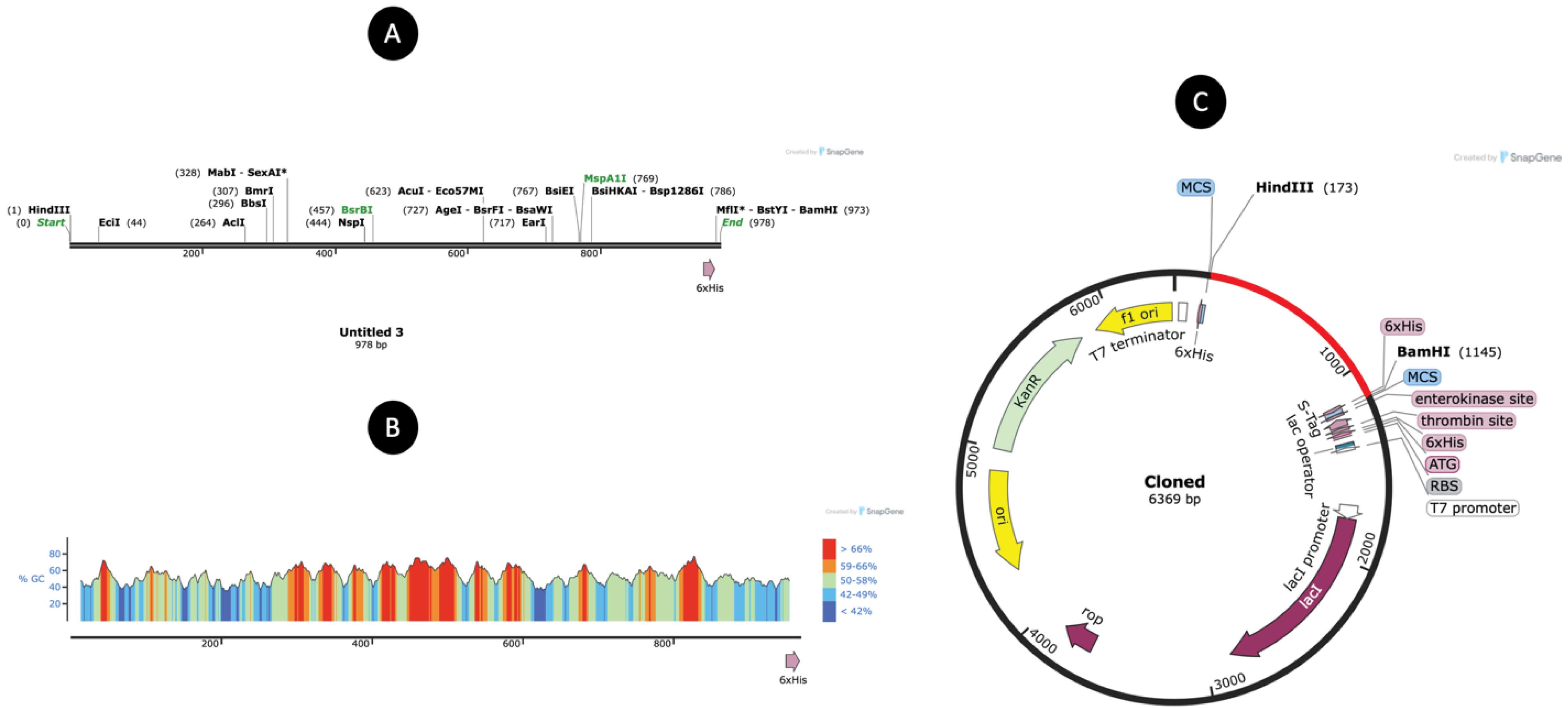
Fig. 8.
(A) Diagram ofvaccine sequence and enzyme retractions. (B) GC content plot (C) The cloned vaccine map (Red line).
.
(A) Diagram ofvaccine sequence and enzyme retractions. (B) GC content plot (C) The cloned vaccine map (Red line).
Population coverage
Population coverage analysis of the selected epitopes demonstrated a high level of immunogenic breadth, with global population coverage reaching 100%, supported by an average of 33.99 epitope-HLA hits per individual and a pc90 of 27.48 (Table S5). Regional analysis revealed robust coverage in developed regions, particularly in Europe and North America (100% coverage, average hits > 36), suggesting strong compatibility of the epitope set with prevalent HLA alleles in these populations. Notably, South Asia (91.25%), Oceania (88.77%), South America (82.04%), and Central/East Africa (78.74% and 77.37%, respectively) also exhibited favorable coverage, supporting the vaccine's global utility. However, lower coverage was observed in Central America (2.19%), Southeast Asia (46.1%), and Southwest Asia (40.63%), indicating potential limitations in these populations that may warrant the inclusion of additional region-specific epitopes to ensure equitable immunoprotection. The standard deviation across regions ( ± 24.64%) underscores the heterogeneity of HLA allele distributions globally, which remains a critical consideration in multi-epitope vaccine design. This result confirms that the constructed vaccine has the potential to provide broad immunogenic coverage, especially in regions with high HLA allele diversity, and highlights target populations for further epitope refinement in future work. The Predicted MHC Class I and II Epitopes with Population Coverage is showed in Table S6.
Discussion
VZV is a globally prevalent pathogen responsible for two distinct clinical conditions: varicella (chickenpox) during primary infection and herpes zoster (shingles) upon reactivation. The World Health Organization estimates that varicella affects approximately 140 million individuals annually, leading to 4.2 million severe complications requiring hospitalization and around 4,200 deaths worldwide. In temperate regions, over 90% of individuals acquire VZV by adolescence in the absence of vaccination programs.24 Herpes zoster incidence has been increasing globally, particularly among older adults. Studies indicate that individuals over 50 years of age have a lifetime risk of approximately 50% for developing herpes zoster. This trend is attributed to factors such as aging populations and increased use of immunosuppressive therapies. Complications from herpes zoster, including postherpetic neuralgia, vision, and hearing loss, and increased risk of cardiovascular events, underscore the need for effective preventive measures.25
Current vaccines, such as the live attenuated Zostavax and the recombinant subunit vaccine Shingrix, have significantly reduced the incidence of herpes zoster. However, limitations exist, including reduced efficacy in immunocompromised individuals and the need for cold-chain storage. The development of a multi-epitope vaccine against the VZV using immunoinformatics and structural analysis represents a significant advancement in the field of vaccine design. This study leveraged computational tools to predict, design, and evaluate a chimeric vaccine construct targeting three key VZV proteins such as envelope glycoprotein E, envelope glycoprotein B, and large tegument protein deneddylase.26-28 Additionally, in silico design allows for rapid development and optimization, potentially overcoming the limitations of current vaccines. Implementing a novel multi-epitope vaccine could enhance global VZV control efforts, reduce the burden of disease, and address the needs of populations inadequately protected by existing vaccines. Such advancements are crucial for improving public health outcomes related to VZV infections.
The selection of Glycoprotein E (gE), Glycoprotein B (gB), and a tegument protein as the primary targets for epitope prediction and vaccine construction was based on their biological significance in the VZV life cycle and their immunogenic profiles.29 Glycoprotein E is the most abundant surface protein expressed on the viral envelope and plays a vital role in viral cell-to-cell spread and immune evasion. Importantly, gE is the primary antigen in the licensed recombinant subunit vaccine Shingrix, underscoring its proven immunogenicity and protective potential.30
Glycoprotein B, another conserved viral envelope protein, facilitates membrane fusion and viral entry into host cells. It has been identified as a major target for neutralizing antibodies and CTL responses in herpesviruses, including VZV. The selection of gB aims to elicit strong cellular and humoral immune responses and provide cross-protection across different viral strains due to its conserved nature.31 The inclusion of a tegument protein adds a strategic dimension to the vaccine design. Tegument proteins are structural components situated between the capsid and envelope and are released early during viral entry, modulating host cell signaling and immune responses. While they have historically received less attention as vaccine targets, recent studies have highlighted their antigenic potential and role in priming innate and adaptive immune responses. Collectively, these three proteins represent distinct viral compartments surface, entry machinery, and internal structures providing comprehensive immune coverage. Their combined inclusion is intended to induce a balanced and broad-spectrum immune response, enhancing the overall protective efficacy of the vaccine candidate.32 While other VZV proteins, such as glycoprotein H (gH), glycoprotein I (gI), and ORF9, have been explored as vaccine targets, they present certain limitations. Glycoprotein H, although involved in viral entry, is less immunogenic compared to gE and gB, with limited evidence supporting its efficacy as a standalone vaccine antigen. Glycoprotein I forms a complex with gE but is not as abundantly expressed, making it a less favorable target. The tegument protein ORF9 interacts with IE62, a major transactivator, yet its role in eliciting protective immune responses remains poorly characterized. By focusing on gE, gB, and the immediate early protein IE63, our vaccine design aims to elicit a comprehensive immune response encompassing both humoral and cellular immunity. This combination targets distinct stages of the viral life cycle entry, replication, and latency thereby potentially offering broader and more effective protection against VZV infection and reactivation. We have updated the Discussion section to include this comparative analysis to provide a clearer and stronger rationale for the selection of these proteins in our multi-epitope vaccine design.33
The integration of bioinformatics approaches allowed for the identification of highly conserved and immunogenic epitopes, which were subsequently fused using innovative linkers to create a stable and effective vaccine candidate. The inclusion of adjuvants, such as β-defensin 3, and cell-penetrating peptides, like the HIV TAT peptide, further enhanced the vaccine's potential to elicit robust immune responses. One of the key strengths of this study lies in the comprehensive computational analysis employed to ensure the vaccine's efficacy and safety. The predicted epitopes were rigorously evaluated for their antigenicity, non-allergenicity, and non-toxicity, ensuring that the final construct would be both immunogenic and safe for potential clinical use. The use of tools such as VaxiJen, AlgPred, and SOLpro provided critical insights into the vaccine's physicochemical properties, solubility, and stability, which are essential for its successful expression and functionality in a host system. The vaccine's theoretical isoelectric point (pI) of 9.65 and molecular weight of 34 kDa, along with its favorable solubility and stability indices, suggest that it is well-suited for further experimental validation. The recent study, the designed vaccine construct, TgVax452, comprising 452 residues with a molecular weight of 44.07 kDa and pI of 6.7, demonstrated favorable physicochemical properties, including stability (instability index: 33.20), solubility (0.498), and strong antigenicity (0.9639), without allergenic potential. Molecular dynamics simulation confirmed a stable interaction (average potential energy: 3.38 × 10⁶ kJ/mol) between the RS09 peptide of TgVax452 and human TLR4, suggesting effective TLR4-mediated innate immune activation. Immune stimulation further predicted elevated IgM, IgG, IFN-γ, and lymphocyte responses.34-36
The structural refinement of the vaccine construct using GalaxyWEB and GalaxyRefine servers further underscores the robustness of this approach. The iterative refinement process ensured that the 3D structure of the vaccine was optimized for stability and interaction with immune receptors, such as TLR2. Molecular docking studies revealed strong binding interactions between the vaccine and TLR2, with a docking score of -280.81, indicating a stable and energetically favorable complex. This interaction is crucial for the activation of innate immune responses, which are essential for the vaccine's efficacy. Additionally, the molecular dynamics simulations demonstrated the stability of the vaccine-TLR2 complex over a 100 ns period, further supporting the vaccine's potential for inducing long-lasting immune responses. The immune simulation results provided valuable insights into the vaccine's ability to elicit both humoral and cellular immune responses. The simulation predicted a robust increase in immunoglobulin levels, particularly IgG1 and IgG2, indicating a strong humoral response. Furthermore, the cytokine profiling revealed a Th1-dominated response, characterized by elevated levels of IFN-γ, IL-2, and IL-12, which are critical for cellular immunity. The increase in TC and memory B-cell populations further highlights the vaccine's potential to provide durable protection against VZV. These findings align to develop a vaccine that can effectively prevent both varicella and zoster, particularly in populations where immunization programs are lacking.37
The codon optimization and in silico cloning of the vaccine construct into the pET-30a ( + ) expression vector represent a critical step toward its experimental production. The optimized sequence achieved a Codon Adaptation Index (CAI) of 1.0 and a GC content of 54.14%, ensuring high-level expression in E. coli K12. This step is essential for the large-scale production of the vaccine, which will be necessary for further preclinical and clinical testing. Despite the promising results, it is important to acknowledge the limitations of this study. While the computational predictions provide a strong foundation for vaccine design, experimental validation is necessary to confirm the vaccine's efficacy and safety in vivo.38,39
The population coverage analysis of the selected epitopes confirms the high immunogenic breadth of the designed multi-epitope vaccine, with global coverage reaching 100%. This is supported by an average of 33.99 epitope-HLA hits per individual and a pc90 value of 27.48, indicating substantial immune system activation potential across diverse populations. Regional breakdown highlights exceptional coverage in Europe and North America, both achieving 100% coverage with more than 36 average hits per individual, suggesting strong compatibility of the selected epitope set with prevalent HLA alleles in these regions. Additionally, high coverage was observed in South Asia (91.25%), Oceania (88.77%), South America (82.04%), Central Africa (78.74%), and East Africa (77.37%), further supporting the broad global applicability of the vaccine. Nevertheless, lower coverage rates in Central America (2.19%), Southeast Asia (46.1%), and Southwest Asia (40.63%) indicate potential gaps in immunoprotection, likely due to regional HLA polymorphisms. These discrepancies highlight the need for further epitope refinement or inclusion of region-specific epitopes to enhance vaccine efficacy in underrepresented populations. The observed standard deviation across regions ( ± 24.64%) underscores the considerable variability in global HLA allele distribution, which remains a key challenge in universal vaccine design. Despite these regional disparities, the current vaccine construct demonstrates substantial potential for eliciting broad-based immune responses and serves as a strong candidate for further preclinical evaluation and population-specific optimization.
In future work on the development of a multi-epitope vaccine against VZV, we intend to incorporate strategies demonstrated in other successful multi-antigenic vaccine models. For example, a recombinant vaccine targeting the dog-sheep transmission cycle of cystic echinococcosis (CE), composed of EG95, Eg14-3-3, and EgEnolase antigens, demonstrated promising immunogenicity and protective efficacy in a pilot trial. In this study, significant increases in serum IgG, IgE, and IL-4 particularly in dogs were observed following two subcutaneous doses, with an 85.43% reduction in hydatid cysts, indicating strong immune protection. Similarly, we aim to design a VZV vaccine capable of inducing robust humoral and cellular responses, validated through immune simulation and in vivo models. The immune simulation results, while encouraging, are based on computational models and may not fully capture the complexity of the human immune system. Additionally, the vaccine's stability and immunogenicity in human subjects remain to be tested, and potential side effects or adverse reactions must be carefully evaluated in clinical trials.40,41 In conclusion, this study presents a novel and comprehensive approach to the design of a multi-epitope vaccine against VZV. The integration of immunoinformatics, structural analysis, and molecular dynamics simulations has resulted in a promising vaccine candidate with the potential to elicit robust immune responses. While further experimental validation is required, the findings from this study provide a strong foundation for the development of effective vaccines against VZV, particularly in regions where immunization programs are not yet established. The success of this approach could also pave the way for the application of similar strategies in the design of vaccines against other infectious diseases.
Limitation
Multi-epitope vaccines, developed through immunoinformatics approaches, offer a promising strategy for combating pathogens like the VZV. By integrating computational tools, researchers can design vaccine constructs that include multiple epitopes, aiming to elicit robust immune responses. However, despite their potential, these vaccines face certain limitations that need to be addressed. One significant challenge is the inherently low immunogenicity of peptide-based vaccines. Peptides are susceptible to enzymatic degradation in the serum, which can reduce their stability and effectiveness. To mitigate this, strategies such as incorporating suitable adjuvants and employing specific linkers (e.g., AAY, GPGPG) are utilized to enhance immunogenicity and ensure proper epitope presentation.42 Another concern is the variability in HLA alleles among different populations. This polymorphism can affect the binding affinity of epitopes to MHC molecules, leading to inconsistent immune responses across individuals. Immunoinformatics tools address this by analyzing population coverage and selecting epitopes with broad HLA binding profiles. Additionally, the lack of post-translational modifications in synthetic peptides may impact the vaccine's ability to mimic natural antigens, potentially affecting the immune response. Computational methods can predict and model these modifications to some extent, but experimental validation remains crucial. Despite these challenges, immunoinformatics provides a powerful platform for vaccine design. It enables the rapid identification of immunodominant epitopes, assessment of antigenicity, allergenicity, and toxicity, and facilitates structural modeling and molecular docking studies. These approaches streamline the vaccine development process, making it more efficient and targeted. In conclusion, while multi-epitope vaccines present certain limitations, the application of immunoinformatics approaches offers solutions to overcome these hurdles, paving the way for the development of effective and safe vaccines against VZV and other pathogens.
Conclusion
In conclusion, this study successfully designed a novel multi-epitope vaccine against the VZV using advanced immunoinformatics and structural bioinformatics approaches. By targeting three key VZV proteins, the vaccine was engineered to include highly conserved and immunogenic epitopes, enhanced with adjuvants and cell-penetrating peptides to boost immune activation and delivery. Comprehensive computational analyses confirmed the vaccine's antigenicity, safety, and stability, with favorable physicochemical properties and strong binding interactions with TLR2. Immune simulations predicted robust humoral and cellular immune responses, including memory cell generation, essential for long-term protection. Codon optimization and in silico cloning confirmed its suitability for expression in E. coli K12. While experimental validation is needed, this study represents a significant step forward in VZV vaccine development, offering a promising tool for preventing varicella and zoster, especially in regions lacking immunization programs, and setting a precedent for future vaccine design against other infectious diseases.
Research Highlights
-
A novel multi-epitope vaccine against VZV was designed using immunoinformatics and structural bioinformatics.
-
The vaccine construct demonstrated strong binding interactions with TLR2, as confirmed through molecular docking and a 100 ns molecular dynamics simulation.
-
Immune simulations indicated a strong humoral and cellular immune response, with high levels of IgG1 and IgG2 antibodies, and increased cytokine activity (IFN-γ, IL-2, IL-12.
Competing Interests
The authors confirm that they do not have a conflict of interest.
Ethical Approval
Not applicable.
Supplementary files
Supplementarily file 1 contains Figs. S1-S2 and Tables S1-S6.
(pdf)
Acknowledgements
We thanked Biorender for supporting us.
References
- Zerboni L, Sen N, Oliver SL, Arvin AM. Molecular mechanisms of varicella zoster virus pathogenesis. Nat Rev Microbiol 2014; 12:197-210. doi: 10.1038/nrmicro3215 [Crossref] [ Google Scholar]
- Ku CC, Zerboni L, Ito H, Graham BS, Wallace M, Arvin AM. Varicella-zoster virus transfer to skin by T cells and modulation of viral replication by epidermal cell interferon-alpha. J Exp Med 2004; 200:917-25. doi: 10.1084/jem.20040634 [Crossref] [ Google Scholar]
- Gilden DH, Vafai A, Shtram Y, Becker Y, Devlin M, Wellish M. Varicella-zoster virus DNA in human sensory ganglia. Nature 1983; 306:478-80. doi: 10.1038/306478a0 [Crossref] [ Google Scholar]
- Gershon AA, Gershon MD. Perspectives on vaccines against varicella-zoster virus infections. Curr Top Microbiol Immunol 2010; 342:359-72. doi: 10.1007/82_2010_12 [Crossref] [ Google Scholar]
- Weibel RE, Neff BJ, Kuter BJ, Guess HA, Rothenberger CA, Fitzgerald AJ. Live attenuated varicella virus vaccine Efficacy trial in healthy children. N Engl J Med 1984; 310:1409-15. doi: 10.1056/nejm198405313102201 [Crossref] [ Google Scholar]
- Patil A, Goldust M, Wollina U. Herpes zoster: a review of clinical manifestations and management. Viruses 2022; 14:192. doi: 10.3390/v14020192 [Crossref] [ Google Scholar]
- Varicella and herpes zoster vaccines: WHO position paper, June 2014. Wkly Epidemiol Rec 2014; 89: 265-87.
- Zhao S, Sun W, Sun J, Peng L, Wang C. Clinical features, treatment, and outcomes of nivolumab induced psoriasis. Invest New Drugs 2025; 43:42-9. doi: 10.1007/s10637-024-01494-4 [Crossref] [ Google Scholar]
- Alishvandi A, Barancheshemeh M, Firuzpour F, Aram C, Kamali MJ, Keikha M. Decoding virulence and resistance in Klebsiella pneumoniae: pharmacological insights, immunological dynamics, and in silico therapeutic strategies. Microb Pathog 2025. 205: 107691. doi: 10.1016/j.micpath.2025.107691.
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001; 305:567-80. doi: 10.1006/jmbi.2000.4315 [Crossref] [ Google Scholar]
- Funderburg N, Lederman MM, Feng Z, Drage MG, Jadlowsky J, Harding CV. Human β-defensin-3 activates professional antigen-presenting cells via toll-like receptors 1 and 2. Proc Natl Acad Sci U S A 2007; 104:18631-5. doi: 10.1073/pnas.0702130104 [Crossref] [ Google Scholar]
- Rathore AS, Arora A, Choudhury S, Tijare P, Raghava GP. ToxinPred 3.0: an improved method for predicting the toxicity of peptides. bioRxiv [Preprint]. August 14, 2023. Available from: https://www.biorxiv.org/content/10.1101/2023.08.11.552911v1.full.
- Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res 2012; 40:W294-7. doi: 10.1093/nar/gks493 [Crossref] [ Google Scholar]
- Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res 2013; 41:W384-8. doi: 10.1093/nar/gkt458 [Crossref] [ Google Scholar]
- Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 2008; 9:514. doi: 10.1186/1471-2105-9-514 [Crossref] [ Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol 2010; 11:373-84. doi: 10.1038/ni.1863 [Crossref] [ Google Scholar]
- Yan Y, Zhang D, Zhou P, Li B, Huang SY. HDOCK: a web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res 2017; 45:W365-73. doi: 10.1093/nar/gkx407 [Crossref] [ Google Scholar]
- Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A. PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016; 32:3676-8. doi: 10.1093/bioinformatics/btw514 [Crossref] [ Google Scholar]
- López-Blanco JR, Aliaga JI, Quintana-Ortí ES, Chacón P. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res 2014; 42:W271-6. doi: 10.1093/nar/gku339 [Crossref] [ Google Scholar]
- Atefi A, Ghanaatpisheh A, Ghasemi A, Haghshenas H, Eyvani K, Bakhshi A. Meningitis after COVID-19 vaccination, a systematic review of case reports and case series. BMC Infect Dis 2024; 24:1138. doi: 10.1186/s12879-024-10043-6 [Crossref] [ Google Scholar]
- Weng G, Wang E, Wang Z, Liu H, Zhu F, Li D. HawkDock: a web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res 2019; 47:W322-30. doi: 10.1093/nar/gkz397 [Crossref] [ Google Scholar]
- Rapin N, Lund O, Castiglione F. Immune system simulation online. Bioinformatics 2011; 27:2013-4. doi: 10.1093/bioinformatics/btr335 [Crossref] [ Google Scholar]
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 2005; 33:W526-31. doi: 10.1093/nar/gki376 [Crossref] [ Google Scholar]
- Shah HA, Meiwald A, Perera C, Casabona G, Richmond P, Jamet N. Global prevalence of varicella-associated complications: a systematic review and meta-analysis. Infect Dis Ther 2024; 13:79-103. doi: 10.1007/s40121-023-00899-7 [Crossref] [ Google Scholar]
- Pan CX, Lee MS, Nambudiri VE. Global herpes zoster incidence, burden of disease, and vaccine availability: a narrative review. Ther Adv Vaccines Immunother 2022; 10:25151355221084535. doi: 10.1177/25151355221084535 [Crossref] [ Google Scholar]
- Pourseif MM, Masoudi-Sobhanzadeh Y, Azari E, Parvizpour S, Barar J, Ansari R. Self-amplifying mRNA vaccines: mode of action, design, development and optimization. Drug Discov Today 2022; 27:103341. doi: 10.1016/j.drudis.2022.103341 [Crossref] [ Google Scholar]
- Omidi Y, Pourseif MM, Ansari RA, Barar J. Design and development of mRNA and self-amplifying mRNA vaccine nanoformulations. Nanomedicine (Lond) 2024; 19:2699-725. doi: 10.1080/17435889.2024.2419815 [Crossref] [ Google Scholar]
- Salemi A, Pourseif MM, Omidi Y. Next-generation vaccines and the impacts of state-of-the-art in-silico technologies. Biologicals 2021; 69:83-5. doi: 10.1016/j.biologicals.2020.10.002 [Crossref] [ Google Scholar]
- Oliver SL. The structures and functions of VZV glycoproteins. Curr Top Microbiol Immunol 2023; 438:25-58. doi: 10.1007/82_2021_243 [Crossref] [ Google Scholar]
- Singh G, Song S, Choi E, Lee PB, Nahm FS. Recombinant zoster vaccine (Shingrix®): a new option for the prevention of herpes zoster and postherpetic neuralgia. Korean J Pain 2020; 33:201-7. doi: 10.3344/kjp.2020.33.3.201 [Crossref] [ Google Scholar]
- Mo C, Schneeberger EE, Arvin AM. Glycoprotein E of varicella-zoster virus enhances cell-cell contact in polarized epithelial cells. J Virol 2000; 74:11377-87. doi: 10.1128/jvi.74.23.11377-11387.2000 [Crossref] [ Google Scholar]
- Mo C, Lee J, Sommer M, Grose C, Arvin AM. The requirement of varicella zoster virus glycoprotein E (gE) for viral replication and effects of glycoprotein I on gE in melanoma cells. Virology 2002; 304:176-86. doi: 10.1006/viro.2002.1556 [Crossref] [ Google Scholar]
- Wu Z, Li X, Huang R, He B, Wang C. Clinical features, treatment, and prognosis of pembrolizumab -induced Stevens-Johnson syndrome/toxic epidermal necrolysis. Invest New Drugs 2025; 43:74-80. doi: 10.1007/s10637-024-01499-z [Crossref] [ Google Scholar]
- Majidiani H, Pourseif MM, Kordi B, Sadeghi MR, Najafi A. TgVax452, an epitope-based candidate vaccine targeting Toxoplasma gondii tachyzoite-specific SAG1-related sequence (SRS) proteins: immunoinformatics, structural simulations and experimental evidence-based approaches. BMC Infect Dis 2024; 24:886. doi: 10.1186/s12879-024-09807-x [Crossref] [ Google Scholar]
- Ma Z, Chen Z, Zheng X, Wang T, You Y, Zou S. A biological immunity-based neuro prototype for few-shot anomaly detection with character embedding. Cyborg Bionic Syst 2024; 5:0086. doi: 10.34133/cbsystems.0086 [Crossref] [ Google Scholar]
- Yang X, Pan Y, Zhang Y, Meng Y, Tong T, Zhao M. Association of systemic immune-inflammation index (SII) with risk of psoriasis: a cross-sectional analysis of National Health and Nutrition Examination Survey 2011-2014. Eur J Med Res 2025; 30:58. doi: 10.1186/s40001-025-02304-0 [Crossref] [ Google Scholar]
- Firuzpour F, Saleki K, Aram C, Rezaei N. Nanocarriers in glioblastoma treatment: a neuroimmunological perspective. Rev Neurosci 2025; 36:431-53. doi: 10.1515/revneuro-2024-0097 [Crossref] [ Google Scholar]
- Aram C, Alijanizadeh P, Saleki K, Karami L. Development of an ancestral DC and TLR4-inducing multi-epitope peptide vaccine against the spike protein of SARS-CoV and SARS-CoV-2 using the advanced immunoinformatics approaches. Biochem Biophys Rep 2024; 39:101745. doi: 10.1016/j.bbrep.2024.101745 [Crossref] [ Google Scholar]
- Saleki K, Aram C, Alijanizadeh P, Khanmirzaei MH, Vaziri Z, Ramzankhah M. Matrix metalloproteinase/Fas ligand (MMP/FasL) interaction dynamics in COVID-19: an in-silico study and neuroimmune perspective. Heliyon 2024; 10:e30898. doi: 10.1016/j.heliyon.2024.e30898 [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Nematollahi A, Khordadmehr M, Naghili B, Dehghani J. Vaccination with rEGVac elicits immunoprotection against different stages of Echinococcus granulosus life cycle: a pilot study. Acta Trop 2021; 218:105883. doi: 10.1016/j.actatropica.2021.105883 [Crossref] [ Google Scholar]
- Pourseif MM, Moghaddam G, Daghighkia H, Nematollahi A, Omidi Y. A novel B- and helper T-cell epitopes-based prophylactic vaccine against Echinococcus granulosus. Bioimpacts 2018; 8:39-52. doi: 10.15171/bi.2018.06 [Crossref] [ Google Scholar]
- Kolla HB, Tirumalasetty C, Sreerama K, Ayyagari VS. An immunoinformatics approach for the design of a multi-epitope vaccine targeting super antigen TSST-1 of Staphylococcus aureus. J Genet Eng Biotechnol 2021; 19:69. doi: 10.1186/s43141-021-00160-z [Crossref] [ Google Scholar]