Bioimpacts. 16:31242.
doi: 10.34172/bi.31242
Original Article
Enhanced anticancer efficacy of α-lipoic acid-loaded TPGS micelles: Synthesis, characterization, and evaluation in a 4T1 breast cancer model
Aqeel Akab Sarhan Formal analysis, Investigation, Resources, Software, Validation, Writing – original draft, Writing – review & editing, 1 
Ismaeil Haririan Data curation, Funding acquisition, Investigation, Methodology, Project administration, Supervision, 1, 2, *
Mohammad Akrami Conceptualization, Investigation, Methodology, Supervision, Writing – review & editing, 2, *
Fatemeh Saadatnia Data curation, Formal analysis, Software, Writing – original draft, 2
Zigang GE Writing – review & editing, 3, 4
Author information:
1Department of Pharmaceutics, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
2Department of Pharmaceutical Biomaterials and Medical Biomaterials Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
3Department of Biomedical Engineering, College of Future Technology, Peking University, Beijing, China
4Beijing Research Institute of Traumatology and Orthopaedics, Beijing Jishuitan Hospital, Beijing, China
Abstract
Introduction:
α-Lipoic acid (ALA) is a potent antioxidant with anticancer properties, but its clinical application is limited by poor water solubility and low bioavailability. Current nanocarrier systems for ALA delivery suffer from low encapsulation efficiency, rapid drug release kinetics, and biocompatibility concerns that restrict their therapeutic potential. This study aimed to develop and evaluate ALA-loaded D-alpha-tocopheryl polyethylene glycol succinate (TPGS) micelles as an innovative nanomedicine platform to overcome existing limitations and enhance anticancer efficacy.
Methods:
ALA was loaded into TPGS micelles via modified solvent evaporation method and comprehensively characterized by TEM, DLS, and FTIR examination. Drug loading and encapsulation efficiency were quantitatively determined. In vitro drug release kinetics, cellular uptake and cytotoxicity were assessed in 4T1 breast cancer cells. Gene expression analysis was performed, and in vivo antitumor efficacy was evaluated in 4T1 murine xenograft model with histopathological examination.
Results:
The formulation achieved superior encapsulation efficiency of 70%, with DLS analysis revealing micelles having mean diameter of 30–40 nm (polydispersity index: 0.234) and zeta potential of –1.9 mV. TEM confirmed spherical morphology. FTIR validated structural integrity with characteristic peaks. In vitro release demonstrated burst release of 52.4±2.5% (6 hours) followed by sustained release reaching 85.9±2.5% (48 hours). Flow cytometry showed 13.75-fold increased cellular uptake (15.4% vs 1.12% FL4-H+cells). MTT assay revealed dose-dependent cytotoxicity. qRT-PCR demonstrated significant upregulation of apoptotic markers: Caspase-8 (2.071-fold, 95% CI: 1.308–3.299, P<0.001) and Caspase-9 (2.86-fold, 95% CI: 1.102–7.785, P<0.001). In vivo studies in 4T1 murine xenograft model showed 10.39% reduction in tumor growth rate (8.54±3.83 mm³/day vs 9.53±1.03 mm³/day in controls) with histopathological evidence of decreased mitotic activity (28 vs 36 mitoses/10 HPF) and increased apoptosis (11 vs 5 apoptotic figures/10 HPF). All treatment groups achieved 100% survival throughout the 17-day study period with maintained body weights, confirming excellent biocompatibility.
Conclusion:
ALA-loaded TPGS micelles demonstrate enhanced anticancer efficacy through improved drug delivery, controlled release kinetics, and effective modulation of apoptotic pathways, supporting their potential for clinical translation in cancer therapy.
Graphical Abstract
Keywords: α-Lipoic acid, Nanomicelles, Drug delivery systems, TPGS, Breast cancer, Nanocarriers
Copyright and License Information
© 2026 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This work was financially supported by Tehran University of Medical Sciences, International affairs (Grant no. 1403-2-104-73605).
Introduction
Invasive breast cancer remains one of the leading causes of cancer-related mortality worldwide, with triple-negative breast cancer (TNBC) representing the most aggressive subtype, characterized by the absence of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 expression.1,2 TNBC accounts for approximately 15-20% of all breast cancer cases but is responsible for disproportionately high mortality rates due to its rapid progression, high metastatic potential, and limited therapeutic options.3 The 4T1 murine breast cancer cell line serves as the most clinically relevant experimental model for studying TNBC therapeutics, as it uniquely mimics human metastatic breast cancer behavior through spontaneous metastasis to lungs, liver, bone, and brain - the same sites commonly affected in human disease.4 Unlike other murine breast cancer models that remain localized, the 4T1 model's aggressive metastatic pattern and resistance to conventional therapies make it an ideal platform for evaluating novel therapeutic interventions with potential for clinical translation.5
Conventional cancer therapies face numerous challenges that limit their clinical efficacy, particularly in aggressive malignancies like TNBC. Chemotherapy, while remaining a cornerstone of cancer treatment, suffers from severe limitations including poor selectivity leading to systemic toxicity, development of multidrug resistance through efflux pump overexpression, and inadequate tumor penetration resulting in suboptimal drug concentrations at the target site.6,7 Radiation therapy, though locally effective, has limited utility in metastatic disease and can cause long-term complications in surrounding healthy tissues. Targeted therapies, despite their promise, have shown limited success in TNBC due to the absence of specific molecular targets, and even approved agents face challenges of acquired resistance and off-target effects.8 These limitations underscore the critical need for innovative therapeutic approaches that can overcome these barriers while maintaining favorable safety profiles.
The field of nanomedicine has emerged as a promising strategy to address these challenges through various nanoparticle platforms. Liposomal delivery systems, despite being among the first clinically approved nanomedicines, face significant limitations including poor drug loading capacity (typically < 10% for hydrophobic compounds), rapid drug leakage during storage, and batch-to-batch variability in size distribution.9,10 Polymer nanoparticles, including PLGA-based systems, offer controlled release kinetics but suffer from burst release phenomena (often > 40% within the first hour), potential inflammatory responses due to acidic degradation products, and complex manufacturing processes that limit scalability.11 Solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) provide improved stability compared to liposomes but demonstrate limited drug loading capacity for lipophilic drugs and potential drug expulsion during storage due to lipid crystallization.12 Despite various nanoparticulate approaches explored for drug delivery, critical challenges persist including limited drug loading capacity, stability issues during storage and in biological fluids, and potential systemic toxicity that hinder clinical translation.13
Among polymeric micelles, TPGS has emerged as a superior carrier due to multiple synergistic mechanisms. Unlike conventional surfactants, TPGS combines the antioxidant properties of vitamin E with the stealth characteristics of polyethylene glycol (PEG), creating a multifunctional delivery platform.14 Critical comparative studies have demonstrated that TPGS micelles achieve significantly higher drug loading efficiency compared to conventional polymeric micelles due to the lipophilic vitamin E moiety's strong affinity for hydrophobic drugs.15 The unique molecular architecture of TPGS provides several distinct advantages over other micellar systems, including low critical micelle concentration ensuring excellent micellar stability, potent P-glycoprotein inhibition for overcoming multidrug resistance, and intrinsic antioxidant activity that creates a synergistic therapeutic platform rather than merely a passive carrier.16,17 However, despite these advantages, pure TPGS micelles face a significant limitation: their relatively high critical micelle concentration of 0.2 mg/mL renders them unstable and easily dissociated upon dilution in plasma after intravenous injection, traditionally necessitating combination with other excipients or structural modifications to achieve clinical viability.18 Recent studies have further validated the versatility of TPGS-based micellar systems across various cancer types. Danafar and colleagues demonstrated that doxycycline-loaded TPGS micelles effectively targeted ovarian cancer stem cells, achieving high encapsulation efficiency and sustained drug release while maintaining excellent biocompatibility.19 Similarly, other investigations have confirmed that TPGS micelles can be successfully adapted for diverse anticancer agents, highlighting the platform's flexibility and robustness as a delivery system.14,20
ALA represents a promising naturally derived anticancer agent with documented efficacy in multiple cancer cell lines through oxidative stress modulation and apoptosis induction. However, clinical translation has been severely hampered by its physicochemical limitations, including extremely low aqueous solubility (0.5 mg/mL at pH 7.4), rapid degradation in alkaline conditions, and poor oral bioavailability ( < 30%).21,22 Recent comprehensive reviews have identified that ALA faces critical pharmacokinetic barriers including short biological half-life due to rapid hepatic degradation, poor physicochemical stability under physiological conditions, and an unpleasant sulfurous odor that complicates pharmaceutical formulation development. Previous attempts to improve ALA delivery through cyclodextrin inclusion complexes, liposomal formulations, and polymer conjugates have yielded limited success due to modest solubility enhancement, drug leakage, stability issues, and complex synthesis requirements.13,23,24 While recent studies have explored ALA-loaded solid lipid nanoparticles for breast cancer therapy in 4T1 cell models, these systems achieved only modest encapsulation efficiency and limited characterization of in vivo anticancer mechanisms.
Despite the recognized therapeutic potential of ALA and the advantages of TPGS as a delivery platform, several critical knowledge gaps persist that limit clinical translation. First, it remains unclear whether systematic optimization of TPGS micelle formulation parameters can overcome the inherent instability limitations while achieving clinically meaningful encapsulation efficiency ( > 60%) for ALA without requiring mixed micellar systems or chemical modifications. Second, the controlled release kinetics of ALA from optimized TPGS micelles and their mathematical modeling have not been comprehensively characterized to enable predictive therapeutic dosing. Third, while ALA's anticancer properties have been demonstrated in vitro, there is a paucity of systematic in vivo evaluation in clinically relevant metastatic breast cancer models, particularly regarding the molecular mechanisms of action through apoptotic pathway modulation. Fourth, most previous ALA delivery systems have focused on non-cancer applications such as transdermal, ophthalmic, neuroprotective, or cardiovascular therapies, with limited systematic investigation specifically targeting aggressive TNBC through intravenous administration. Finally, comprehensive safety evaluation including biocompatibility assessment and dose optimization strategies for ALA-loaded nanocarriers in cancer therapy remain insufficiently documented.
This study addresses these critical limitations by developing an optimized ALA-loaded TPGS micellar system through systematic formulation design and comprehensive characterization. Our approach uniquely combines TPGS's inherent advantages (FDA-approved safety profile, P-glycoprotein inhibition, enhanced cellular uptake) with ALA's natural anticancer properties to create a synergistic therapeutic platform specifically evaluated in the clinically relevant 4T1 metastatic breast cancer model. Through systematic investigation of formulation parameters, controlled release kinetics, cellular uptake mechanisms, apoptotic gene expression modulation, and in vivo antitumor efficacy with histopathological validation, this work provides the foundation for advancing this promising nanomedicine approach toward clinical translation in aggressive breast cancer treatment.
Materials and Methods
All reagents and chemicals were of analytical grade and used without further purification. TPGS and ALA were obtained from Sigma-Aldrich, while ALA was purchased with high purity standards. The 4T1 murine breast cancer cell line was obtained from the Pasteur Institute of Iran (Tehran, Iran). HPLC-grade methanol and acetonitrile were used as solvents during the micelle preparation and subsequent drug quantification processes. Phosphate-buffered saline (PBS, pH 7.4) and ultrapure water (Millipore system) were used throughout for rehydration and dilution steps.
Synthesis of ALA-loaded TPGS micelles
ALA-loaded TPGS micelles were prepared using a modified solvent-evaporation/ re-hydration method.15,17 In brief, 150 mg of TPGS and 5 mg of ALA were accurately weighed and co-dissolved in 10 mL of HPLC-grade methanol in a 100 mL round-bottom flask. The solution was stirred magnetically at 300 rpm for 30 min at room temperature (≈25 °C) to ensure complete dissolution. The organic solvent was removed under reduced pressure using a rotary evaporator at 40 °C with a rotation speed of 100 rpm, producing a thin film on the flask wall. To further remove residual solvent, the thin film was freeze-dried for 30 min. The dried film was rehydrated with 15 mL of phosphate-buffered saline (PBS, pH 7.4) and gently stirred (200 rpm) at room temperature for 48 h to promote self-assembly of TPGS into micelles entrapping ALA. The resultant micellar suspension was passed through a 0.22 μm cellulose acetate membrane filter to remove any unencapsulated aggregates and to ensure sterility. Final preparations were transferred into amber glass vials and stored at 4°C until characterization.
Dynamic light scattering (DLS) and zeta potential analysis
DLS measurements were performed using a Malvern Zetasizer Nano ZS (Malvern Panalytical, Malvern, UK) to determine the hydrodynamic diameter and size distribution of the ALA-loaded TPGS micelles. Micellar suspensions were diluted with filtered ultrapure water and analyzed at 25 °C using a 633 nm He-Ne laser with backscatter detection at 173°. The polydispersity index (PDI) was recorded to assess the uniformity of size distribution. Zeta potential measurements were conducted using the same instrument equipped with a folded capillary cell. Samples were analyzed at 25 °C using phase analysis light scattering (PALS) technology to determine the electrophoretic mobility and calculate the zeta potential, providing information about surface charge and colloidal stability of the micelle. All measurements were performed in triplicate to ensure reproducibility.
Morphological characterization by transmission electron microscopy (TEM)
The morphological characteristics of the ALA-loaded TPGS micelles were examined using TEM. The analysis was conducted with the TEM operating at an acceleration voltage of 20 kV. For optimal imaging, a single drop of the freshly prepared micellar suspension was deposited onto a carbon-coated copper grid and allowed to adhere for 5 minutes at room temperature (25 °C). Following this, excess liquid was carefully removed with filter paper, and the grid was then subjected to negative staining using a 2% (w/v) uranyl acetate solution for 2 minutes to enhance contrast. The stained grid was air-dried at room temperature for 15 minutes prior to imaging. Digital images were subsequently captured at various magnifications to provide detailed morphological information of the micelles.
Fourier transform infrared spectroscopy (FTIR) examination
FTIR spectroscopy was conducted to identify the characteristic functional groups of TPGS and ALA and to confirm their structural integrity. Samples were prepared by gently grinding each compound with potassium bromide (KBr) at a 1:100 ratio (w/w) and pressing the mixture into transparent pellets. The FTIR spectra were recorded over the range of 4000–400 cm⁻¹ at a resolution of 4 cm⁻¹ using an FTIR spectrometer (Spectrum Two, PerkinElmer, Waltham, MA, USA). Each spectrum was the average of 16 scans, and the baseline was corrected automatically by the instrument’s software.
Drug loading determination
Drug loading and encapsulation efficiency of ALA in TPGS micelles were quantified using a validated high-performance liquid chromatography (HPLC) method. Briefly, 1 mg of lyophilized ALA-loaded TPGS micelles was dissolved in 1 mL of HPLC-grade methanol. The resulting suspension was stirred at 700 rpm for 6 hours using a temperature-controlled magnetic stirrer to ensure complete disruption of the micelles and release of encapsulated ALA. Following this extraction, the solution was filtered through a 0.22 μm PTFE syringe filter to remove any undissolved material. A 20 μL aliquot of the filtrate was then injected into the HPLC system, which was operated under the following optimized conditions: a mobile phase of 60% acetonitrile and 40% potassium dihydrogen phosphate buffer (50 mM, pH 3.5), a flow rate of 1.0 mL/min, a column temperature of 35°C, and detection at 320 nm. The concentrations obtained were used to calculate the drug loading content and encapsulation efficiency according to standard formulas, ensuring accurate assessment of ALA incorporation within the micellar structure.
Drug release determination
The in vitro release profile of ALA from TPGS micelles was evaluated by dialysis membrane diffusion under physiologically relevant conditions. A dialysis membrane (MWCO 14 kDa) was pre-soaked in PBS (pH 7.4) supplemented with 1% w/v Tween 80 for 12 h to ensure hydration and removal of preservatives. ALA-loaded micelles (5.0 mL) were enclosed within the membrane, which was immersed in 20 mL of release medium (PBS, pH 7.4, containing 1% w/v Tween 80) and maintained at 37 °C with continuous agitation at 100 rpm. At predetermined time points 1 mL aliquots were withdrawn from the external medium and immediately replaced with pre-warmed buffer; samples were analyzed by validated HPLC (detection at 320 nm). Cumulative release was plotted versus time and the data were fitted to kinetic models (zero-order, first-order, Higuchi) by non-linear regression to determine the best fit (R2). Tween 80 was included solely in the release medium to maintain sink conditions and prevent ALA precipitation during the assay.
Uptake examination
Cellular uptake of the micellar formulation by 4T1 cells was evaluated using fluorescence-based flow cytometry. Curcumin was employed as a fluorescent probe: curcumin-loaded TPGS micelles were prepared using the same modified solvent-evaporation/hydration procedure described for ALA-loaded micelles, and these curcumin-loaded micelles were used to assess internalization. 4T1 cells (Pasteur Institute of Iran) were seeded in 12-well plates at 4 × 10⁵ cells/well in DMEM/F12 supplemented with 10% FBS and incubated for 24 h (37 °C, 5% CO₂). The medium was then replaced with fresh medium containing curcumin-loaded micelles at a drug-equivalent concentration of 80 μg/mL; control wells received drug-free medium. After 24 h incubation, cells were washed twice with PBS, detached with 0.025% Trypsin-EDTA, and centrifuged at 3000 rpm for 10 min. Cell pellets were resuspended in PBS and analyzed on a flow cytometer (FACSCalibur, BECTON DICKINSON, USA); fluorescence was recorded across four channels and the curcumin signal was quantified in the FL4 channel (FL4-H). Data were processed using FlowJo V10.9. The use of curcumin-loaded micelles provides a reliable fluorescent readout of micelle internalization; all uptake data reported in the Results refer to these curcumin-labelled preparations.
MTT assay for cell viability assessment
4T1 cells (Pasteur Institute Cell Bank of Iran) were seeded at 1.2 × 10⁵ cells/well in 24-well plates with DMEM/F12 containing 10% FBS and incubated for 24 h (37°C, 5% CO₂). Test formulations were prepared from a primary stock solution and six serial 1:2 dilutions were made to produce final treatment concentrations of 140, 70, 35, 17.5, 8.75, and 4.35 μg/mL (drug-equivalent). After replacing the medium with formulation-containing medium, cells were incubated for 48 h. Cell viability was assessed with the MTT Cell Proliferation Assay Kit following the manufacturer’s instructions with minor modifications: 10 μL MTT solution (5 mg/mL in PBS) was added per well and incubated for 3 h at 37 °C, after which medium was removed and 100 μL DMSO was added to dissolve formazan crystals. Absorbance was measured at 570 nm (Hiperion MPR4 + microplate reader). Viability percentages were calculated relative to untreated controls. All experiments were performed in triplicate; the concentrations reported in the Results (140–4.35 μg/mL) correspond to these tested dilutions.
RNA isolation and quantitative real-time PCR analysis
Total RNA was extracted from 4T1 cells treated with TPGS micelles and ALA-loaded TPGS micelles using TRIzol reagent according to the manufacturer's protocol. Briefly, cells were lysed in TRIzol and the RNA was separated through chloroform extraction followed by isopropanol precipitation. The RNA pellet was washed with 75% ethanol, air-dried, and dissolved in RNase-free water. RNA concentration and purity were determined using a NanoDrop spectrophotometer, with A260/A280 ratios of 1.8-2.0 considered acceptable.
First-strand cDNA synthesis was performed using 1 μg of total RNA per sample with the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, USA) following the manufacturer's instructions. The reaction mixture included oligo (dT) primers, reaction buffer, RiboLock RNase inhibitor, dNTP mix, and RevertAid M-MuLV reverse transcriptase. The reverse transcription reaction was carried out at 42°C for 60 minutes followed by termination at 70 °C for 5 minutes.
Quantitative real-time PCR (qRT-PCR) was performed using a StepOnePlus Real-Time PCR System (Applied Biosystems, USA) with SYBR Green PCR Master Mix (Applied Biosystems, USA). The reaction mixture contained 1 μL of cDNA template, 0.5 μL of each primer (10 μM), 10 μL of SYBR Green PCR Master Mix, and nuclease-free water to a final volume of 20 μL. The PCR thermal cycling conditions included an initial denaturation at 95 °C for 10 minutes, followed by 40 cycles of denaturation at 95 °C for 15 seconds and annealing/extension at 60 °C for 60 seconds. A melting curve analysis was performed after amplification to confirm the specificity of the PCR products. The target genes analyzed were Caspase8, Caspase9, SOD, and P53, with GAPDH serving as the endogenous reference gene for normalization. Primer sequences were designed using Primer-BLAST (NCBI) and validated for specificity and efficiency prior to use. All reactions were performed in duplicate, and negative controls (no template) were included in each run.
The comparative CT method (2-ΔΔCT) was employed to calculate the relative expression levels of target genes. The fold change in gene expression was determined by normalizing the CT values of target genes to that of GAPDH (ΔCT) and then calculating the difference between treatment and control groups (ΔΔCT).
In vivo antitumor efficacy evaluation
The in vivo antitumor efficacy of ALA-loaded TPGS micelles and empty TPGS micelles was evaluated using a 4T1 breast cancer xenograft model in female BALB/c nude mice (6–8 weeks old, 18–22 g). Animals were housed under controlled environmental conditions (22 ± 2°C; 55 ± 15% humidity; 12 h light/dark cycle) with ad libitum access to food and water, and were acclimatized at least 7 days prior to tumor inoculation. All procedures were approved by the Institutional Animal Ethics Committee.
For context and to support the appropriateness of this empirical approach, prior preclinical reports demonstrate that TPGS-based micellar systems have been widely used in murine cancer models and are generally well tolerated; TPGS micelles has been used in vivo at doses (e.g., ~50 mg/kg in some studies) that enhance solubility/permeability without prohibitive toxicity.25-27 Following preliminary safety dose assessment to establish tolerability and identify an appropriate therapeutic window, we selected 50 mg/kg as the optimal dose for both blank TPGS micelles and ALA-loaded TPGS micelles. This dose was chosen to balance therapeutic potential with safety considerations based on our preliminary studies and literature guidance.
Treatments were administered by intraperitoneal injection at a dose of 10 mg/kg for free drug and an equivalent drug-loading dose for nano-formulations, delivered once every 48 hours throughout the 17-day study. Tumor dimensions and body weights were recorded on days 1, 4, 7, 10, 13 and 17. Tumor volume was calculated, and linear regression was applied to serial measurements to determine individual tumor-growth slopes (mm³/day). Animals were monitored daily, and humane endpoints included body-weight loss exceeding 20%, tumor mass surpassing 10% of body weight, ulceration, impaired mobility, or any signs of severe distress. Data are expressed as mean ± SD, with individual animal values displayed when appropriate to illustrate variability within each treatment group.
Histopathological analysis of tumor tissue
For histopathological examination, tumor tissues were collected from euthanized mice at the study endpoint (day 24). The harvested tumors were immediately fixed in 10% neutral buffered formalin for 24 hours at room temperature. Following fixation, the tissues were processed through a series of graded ethanol concentrations (70%, 80%, 95%, and 100%) for dehydration, cleared in xylene, and embedded in paraffin blocks. Tissue sections of 5 μm thickness were prepared using a rotary microtome (Leica RM2235, Germany) and mounted on positively charged glass slides.
The sections were deparaffinized in xylene and rehydrated through decreasing concentrations of ethanol before staining with hematoxylin and eosin (H&E) according to standard protocols. Briefly, the slides were immersed in Harris's hematoxylin for 5 minutes, rinsed in running tap water for 5 minutes, differentiated in 1% acid alcohol for 30 seconds, and counterstained with 0.5% eosin for 2 minutes. The stained sections were then dehydrated through ascending ethanol concentrations, cleared in xylene, and mounted with DPX mounting medium.
Histopathological analysis was performed by a qualified pathologist who was blinded to the experimental groups. Multiple parameters were evaluated systematically according to established criteria. Tumor necrosis was assessed as a percentage of the total tumor area showing necrotic changes. Mitotic count was determined by counting mitotic figures in 10 high-power fields (10 HPF) using a 40X objective lens. Similarly, apoptotic figures were quantified in 10 HPF. Nuclear pleomorphism was scored on a scale ranging from 0 to 3, where 0 represented negative, 1 indicated mild, 2 moderate, and 3 severe pleomorphism. Treatment effects were evaluated based on morphological changes indicative of therapeutic response and quantified as a percentage of the tumor area. Tumor-infiltrating lymphocytes (TILs) were assessed as the percentage of mononuclear inflammatory cells infiltrating the tumor tissue.
Photomicrographs were captured using a digital microscope camera (Olympus BX53, Japan) at both 40 × and 100 × magnifications for comprehensive visualization of tumor morphology and cellular details. Representative images from each experimental group were selected for comparative analysis and documentation.
Statistical analysis
All experiments were performed in triplicate (n = 3), and results are expressed as mean ± standard deviation (SD). Statistical comparisons between experimental groups were conducted using one-way analysis of variance (ANOVA) followed by Tukey's post-hoc test for multiple comparisons. The threshold for statistical significance was set at P < 0.05. For in vitro drug release studies, MTT cytotoxicity assays, and flow cytometry analyses, comparative statistical evaluation was performed to assess differences between ALA-loaded TPGS micelles, free ALA, and control groups, with significant differences denoted in the corresponding figures and tables.
Gene expression data obtained from qRT-PCR were analyzed using the comparative threshold cycle (2-ΔΔCT) method, with normalization to the reference gene GAPDH. Statistical significance of gene expression changes was determined using REST 2009 software, which employs a Pair Wise Fixed Reallocation Randomization Test specifically designed for relative expression data. This approach accounts for the non-normal distribution characteristics of PCR data and provides robust calculation of p-values along with 95% confidence intervals for fold-change values.
Data processing and visualization were performed using multiple computational platforms to ensure accuracy and reproducibility. OriginPro 2021 software (OriginLab Corporation, USA) was employed for graphical analysis, curve fitting, statistical computations, and release kinetics modeling. Transmission electron microscopy images were processed and quantitatively analyzed using ImageJ software (National Institutes of Health, USA) for particle size distribution measurements and morphological characterization. Specialized calculations, data manipulation, and custom analytical procedures were implemented using Python 3.8 programming language with scientific computing libraries including NumPy, SciPy, pandas, and Matplotlib. Preliminary data organization and basic calculations were performed using Microsoft Excel 2019 (Microsoft Corporation, USA). All figures were prepared at publication quality resolution (1200 dpi) using appropriate software platforms.
Result and Discussion
Size distribution and surface charge analysis
The DLS analysis (zeta sizer, Malvern) of the ALA-Loaded TPGS micelles revealed a monomodal size distribution with a mean hydrodynamic diameter in the range of 30–40 ± 2 nm. This narrow size distribution, characterized by a sharp intensity peak, confirms the formation of a uniform micellar population with low polydispersity (0.234 ± 0.036). Such a size range is considered optimal for drug delivery applications, as micelles within the 20–200 nm window can evade rapid renal clearance while facilitating penetration through biological barriers.28
Complementary zeta potential measurements indicated a peak value of approximately –1.9 mV. While this zeta potential is below the ± 30 mV threshold typically associated with strong electrostatic stabilization, comprehensive stability assessments were performed over three months of storage at 4 °C to evaluate the long-term colloidal stability of the system. The stability studies demonstrated that particle size remained within acceptable limits (variation < 5%), zeta potential values showed consistent measurements, and encapsulation efficiency exhibited minimal reduction ( < 10% decrease), confirming adequate colloidal stability despite the low surface charge. This stability can be attributed to steric stabilization provided by the hydrophilic polyethylene glycol chains of TPGS molecules, which create a protective hydrated layer around the micelle surface, preventing particle aggregation through steric hindrance rather than electrostatic repulsion.29,30 The observed physicochemical characteristics—namely, the optimal particle size and demonstrated long-term stability—are critical determinants for the in vivo performance of nanocarrier systems. They support the potential of these ALA-loaded TPGS micelles to achieve enhanced bioavailability and controlled drug release, which are essential for effective therapeutic outcomes.
Morphological Analysis by TEM
TEM was employed to evaluate the morphology of the ALA-loaded TPGS micelles. For TEM analysis, a drop of the freshly prepared micellar suspension was deposited onto a carbon-coated copper grid and allowed to air-dry prior to imaging. The micrograph provides comprehensive insight into the nanostructure of the formulation (Fig. 1A).
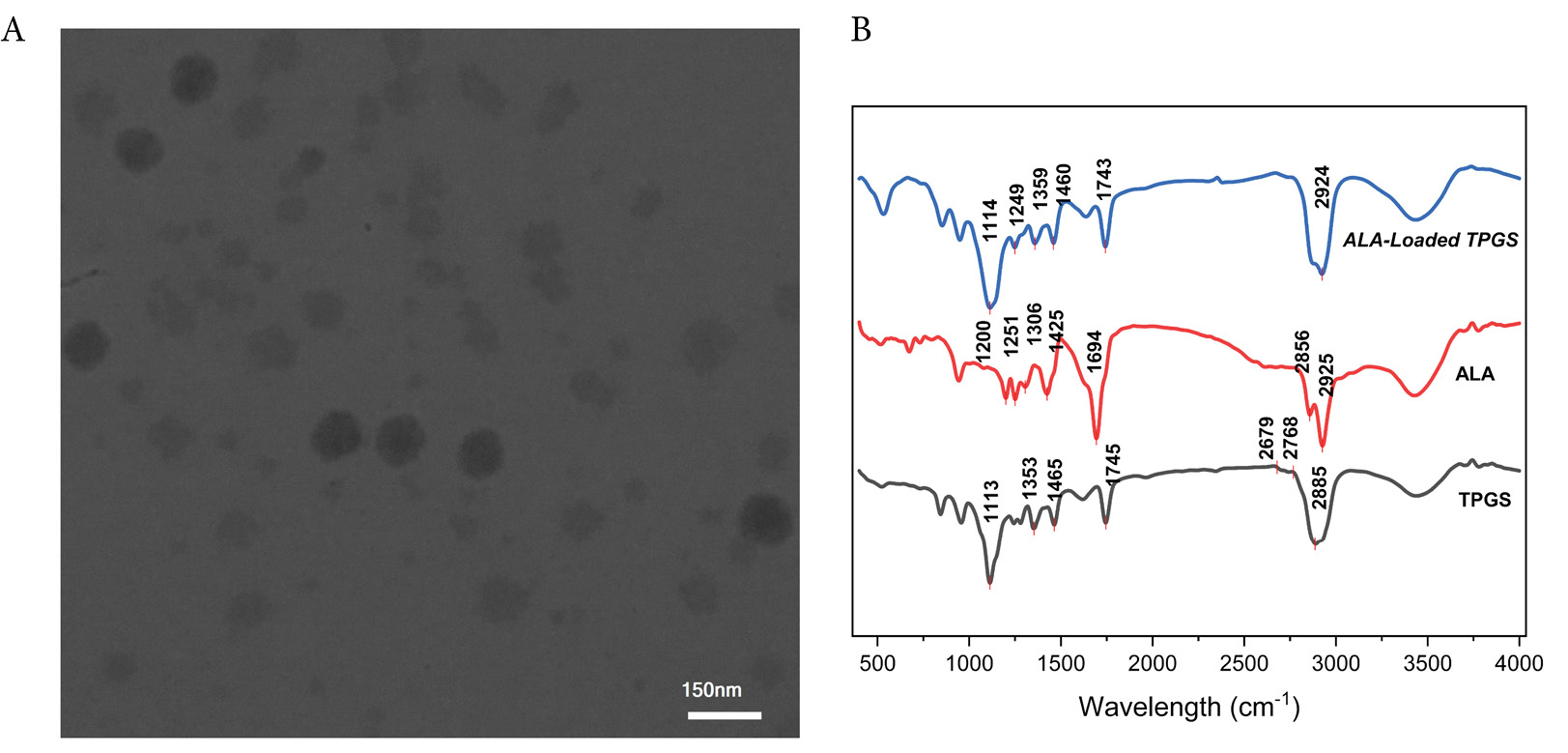
Fig. 1.
TEM micrograph of ALA-loaded TPGS micelle at scale bar of 150 nm (A), FTIR spectra of TPGS micelles, free α-lipoic acid (ALA), and ALA-loaded TPGS micelles. Characteristic absorption bands are labeled, including O–H stretching (~3400 cm⁻¹), C–H stretching of methylene groups (2850–2950 cm⁻¹), ester carbonyl stretching of TPGS (~1750 cm⁻¹), carboxyl C = O stretching of ALA (~1700 cm⁻¹), and disulfide bond vibrations of ALA (500–550 cm⁻¹). Peak positions confirm successful encapsulation of ALA within the TPGS micellar system while preserving the structural integrity of both components (B).
.
TEM micrograph of ALA-loaded TPGS micelle at scale bar of 150 nm (A), FTIR spectra of TPGS micelles, free α-lipoic acid (ALA), and ALA-loaded TPGS micelles. Characteristic absorption bands are labeled, including O–H stretching (~3400 cm⁻¹), C–H stretching of methylene groups (2850–2950 cm⁻¹), ester carbonyl stretching of TPGS (~1750 cm⁻¹), carboxyl C = O stretching of ALA (~1700 cm⁻¹), and disulfide bond vibrations of ALA (500–550 cm⁻¹). Peak positions confirm successful encapsulation of ALA within the TPGS micellar system while preserving the structural integrity of both components (B).
The TEM images consistently revealed well-defined, predominantly spherical micelles with a uniform size distribution. Detailed analysis of the images showed that the micelles exhibit a size range of approximately 20–40 nm, with most particles falling between 20 and 30 nm. Such dimensions are particularly advantageous for drug delivery, as they are sufficiently large to avoid rapid renal clearance while remaining small enough to evade recognition by the reticuloendothelial system.31,32 Moreover, the high-magnification images disclosed a distinct core-shell architecture characterized by a darker core, representing the hydrophobic vitamin E moiety entrapping ALA, and a lighter peripheral layer corresponding to the hydrophilic PEG corona. This structural feature not only confirms successful drug encapsulation but also underscores the formation of stable nanocarriers.
The absence of significant aggregation in the TEM micrographs further indicates the robust colloidal stability of the micellar system, likely a result of the steric hindrance conferred by the PEG chains.32 These findings, which align with previously reported observations in TPGS-based nanocarrier systems.31,33 affirm that the adopted preparation method yields a discrete and stable micellar assembly, suitable for advanced drug delivery applications.
TEM examination, including ImageJ-based size analysis of the particle population, revealed a narrow distribution centered at 20–30 nm with uniform spherical morphology. As expected for polymer-stabilized nanocarriers, DLS measurements yielded larger hydrodynamic diameters, reflecting the solvated PEG/TPGS corona and the dynamic interactions of the particles within the aqueous medium. Although the absolute sizes differed, the narrow TEM distribution was consistent with the low PDI observed by DLS, indicating uniformity in both the dry-state core and the hydrated nanoparticle ensemble. This agreement between TEM and DLS supports the structural stability of the formulation and confirms that the surface layers remain intact under dispersion conditions.
FTIR analysis
The FTIR spectra of TPGS, free ALA, and ALA-loaded TPGS micelles are presented in Fig. 1B, providing comprehensive insight into the molecular characteristics and successful encapsulation of the drug within the micellar system. The TPGS spectrum exhibits a broad absorption band at approximately 3400 cm⁻¹, which corresponds to O–H stretching vibrations of terminal hydroxyl groups present in the polyethylene glycol chains. Sharp bands observed in the 2850–2950 cm⁻¹ region are attributed to the C–H stretching vibrations of methylene groups within both the PEG segment and the vitamin E moiety of TPGS.34 A prominent absorption peak at approximately 1750 cm⁻¹ indicates the C = O stretching of the ester linkage, confirming the structural integrity of TPGS.35
The free ALA spectrum demonstrates several characteristic absorption bands consistent with its molecular structure. A broad band spanning 3000–3500 cm⁻¹ represents the O–H stretching vibration of the carboxylic acid group, while a distinct peak at approximately 1700 cm⁻¹ corresponds to the C = O stretching of the carboxyl functionality.36 Weak absorption bands in the 500–550 cm⁻¹ region provide evidence of the disulfide bond (–S–S–), which is a critical structural feature of ALA.37
The spectrum of ALA-loaded TPGS micelles reveals the presence of characteristic peaks from both TPGS and ALA, confirming successful drug encapsulation within the micellar structure. Notably, the carbonyl stretching band of ALA at 1700 cm⁻¹ appears slightly shifted and broadened in the micelle spectrum, suggesting hydrogen bonding interactions between the carboxylic acid group of ALA and the hydroxyl or ether groups of TPGS. Additionally, the retention of the broad O–H stretching band at 3400 cm⁻¹ and the ester carbonyl peak at 1750 cm⁻¹ indicates that the structural integrity of TPGS remains intact during the micelle formation process. The preservation of ALA's characteristic peaks, including those corresponding to the disulfide bond, confirms that no significant chemical degradation occurred during the encapsulation procedure. These spectroscopic observations collectively validate the successful formation of ALA-loaded TPGS micelles while maintaining the molecular integrity of both the drug and the carrier, which is essential for the therapeutic efficacy of the formulation.
Drug loading analysis
The encapsulation efficiency of ALA in the TPGS micellar system was determined to be approximately 70%, a marked improvement compared to many conventional nanocarrier platforms. This enhanced performance is primarily attributed to the unique amphiphilic structure of TPGS, which consists of a hydrophobic vitamin E moiety and a hydrophilic polyethylene glycol (PEG) chain.38,39 The hydrophobic core of TPGS establishes strong interactions with lipophilic drugs such as ALA, promoting effective incorporation, while the PEG corona stabilizes the micelles and minimizes premature drug leakage. This combination of favorable interactions and stability is critical for achieving high drug loading and ensuring sustained drug release under physiological conditions.
In comparison, other amphiphilic carrier systems, including PEG-PLGA nanoparticles and Pluronic®-based micelles, have typically demonstrated encapsulation efficiencies ranging from 45% to 60%.40,41 The superior performance of the TPGS-based system in our study can be attributed to the optimized thin-film hydration method and meticulous control over formulation parameters, which enhance drug–carrier interactions. This improved encapsulation efficiency is expected to contribute significantly to the bioavailability and therapeutic efficacy of ALA, highlighting the potential of TPGS micelles as an advanced drug delivery platform.
Drug release analysis
The in vitro release profile of ALA from TPGS micelles was subjected to comprehensive kinetic analysis to determine the underlying release mechanism (Fig. 2). The experimental data were fitted to three mathematical models: zero-order, first-order, and Higuchi kinetic models. Statistical analysis revealed that the first-order kinetic model provided the best fit to the experimental data with the highest coefficient of determination (R2 = 0.9828), demonstrating excellent correlation compared to the Higuchi model (R2 = 0.8872) and zero-order model (R2 = 0.7008). The first-order model parameters indicated a rate constant (k₁) of 0.1760 h⁻¹ and a theoretical maximum release (Q∞) of 80.2%, which closely matched the observed experimental plateau value of 85.9 ± 2.5% at 48 hours. This mathematical validation confirms that the release follows concentration-dependent kinetics characteristic of diffusion-controlled systems.
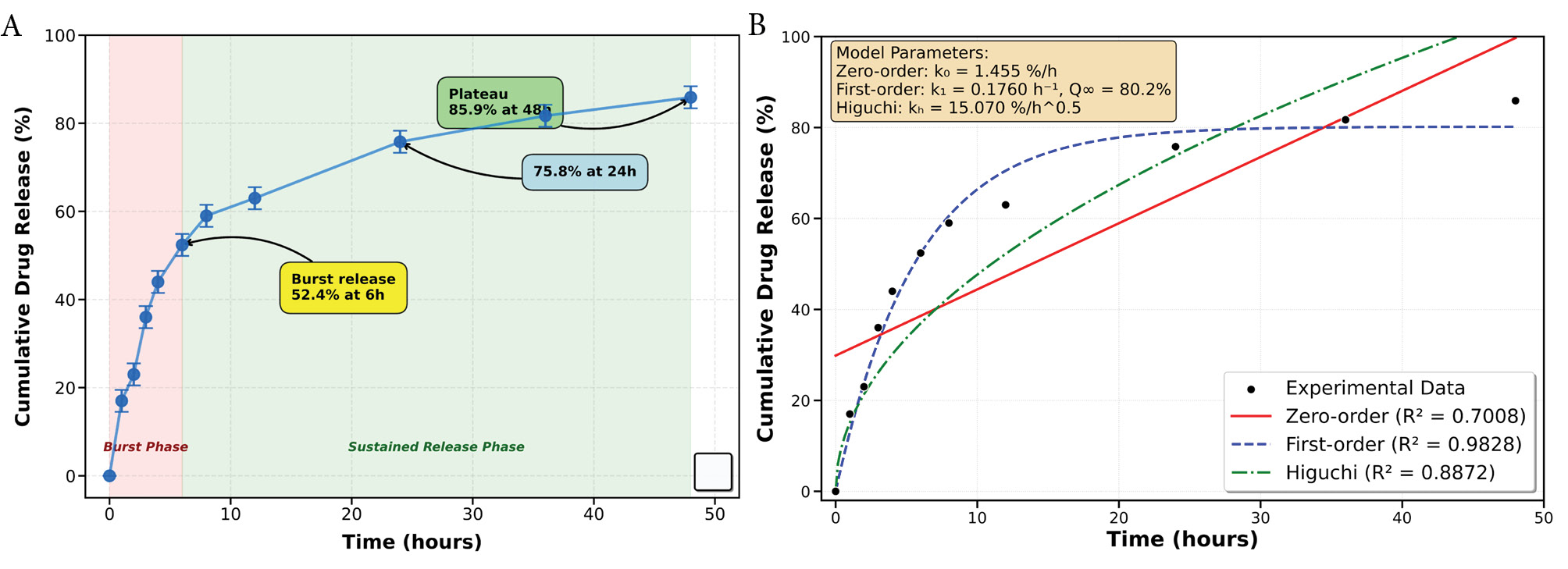
Fig. 2.
In vitro drug release profile and kinetic analysis of α-lipoic acid from TPGS micelles. (A) Cumulative drug release profile showing biphasic release behavior with initial burst release followed by sustained release over 48 hours. (B) Comparative kinetic model fitting showing experimental data (black circles) fitted to zero-order (red solid line, R2 = 0.7008), first-order (blue dashed line, R2 = 0.9828), and Higuchi (green dash-dot line, R2 = 0.8872) kinetic models. The first-order model demonstrated the best fit to the experimental data, confirming diffusion-controlled release mechanism from the micellar system.
.
In vitro drug release profile and kinetic analysis of α-lipoic acid from TPGS micelles. (A) Cumulative drug release profile showing biphasic release behavior with initial burst release followed by sustained release over 48 hours. (B) Comparative kinetic model fitting showing experimental data (black circles) fitted to zero-order (red solid line, R2 = 0.7008), first-order (blue dashed line, R2 = 0.9828), and Higuchi (green dash-dot line, R2 = 0.8872) kinetic models. The first-order model demonstrated the best fit to the experimental data, confirming diffusion-controlled release mechanism from the micellar system.
The biphasic release profile observed is characteristic of micellar systems in which loosely associated drug at or near the corona diffuses rapidly while drug embedded in the hydrophobic core is released more slowly.42 During the initial burst phase, ~52.4 ± 2.5% of encapsulated ALA was released within the first 6 h, followed by a sustained release reaching 85.9 ± 2.5% at 48 h; the release kinetics fitted first-order behavior (R2 = 0.9828). Tween 80 (1% w/v) was present only in the external release medium to maintain sink conditions and prevent precipitation of lipophilic ALA. We note that the surfactant can influence apparent release kinetics by increasing solubilization of surface-associated ALA and, in some formulations, by interacting with the micelle corona which may facilitate desorption of loosely bound drug; such surfactant-mediated effects likely contribute to the pronounced initial burst we observed.43 Nevertheless, the sustained second-phase release and the first-order fit indicate that diffusion of core-entrapped ALA remains the primary release mechanism under the tested conditions.44
Following the burst release phase, a more controlled and sustained release was observed, reaching 75.8 ± 2.5% at 24 hours and eventually plateauing at 85.9 ± 2.5% after 48 hours. This sustained release is primarily governed by the strong hydrophobic interactions between ALA and the vitamin E-rich TPGS core, which delays drug diffusion into the surrounding medium.45,46 Such controlled release profiles are advantageous for therapeutic applications, as they enable prolonged drug retention at effective concentrations, potentially minimizing dosing frequency and improving patient compliance.47 The observed drug release pattern highlights the potential of TPGS micelles as an effective delivery system for ALA, providing both rapid onset and sustained therapeutic effects. Further in vivo studies are necessary to validate these in vitro results and assess the pharmacokinetic advantages of this formulation.
Cellular uptake analysis of micelle in 4T1 breast cancer cells
Flow cytometry analysis demonstrated significant differences in cellular uptake between control and treatment groups. The untreated control cells (Fig. 3A) exhibited minimal fluorescence in the FL4-H channel, with 98.9% of cells showing low fluorescence intensity (FL4-H-) and only 1.12% displaying higher fluorescence (FL4-H + ). In contrast, cells treated with the micelle (Fig. 3B) showed substantially increased fluorescence, with 15.4% of cells in the high fluorescence range (FL4-H + ) and 84.6% in the lower range. This 13.75-fold increase in FL4-H + cell percentage confirms successful cellular internalization of the drug-loaded micelles by 4T1 breast cancer cells following 24 hours of incubation.
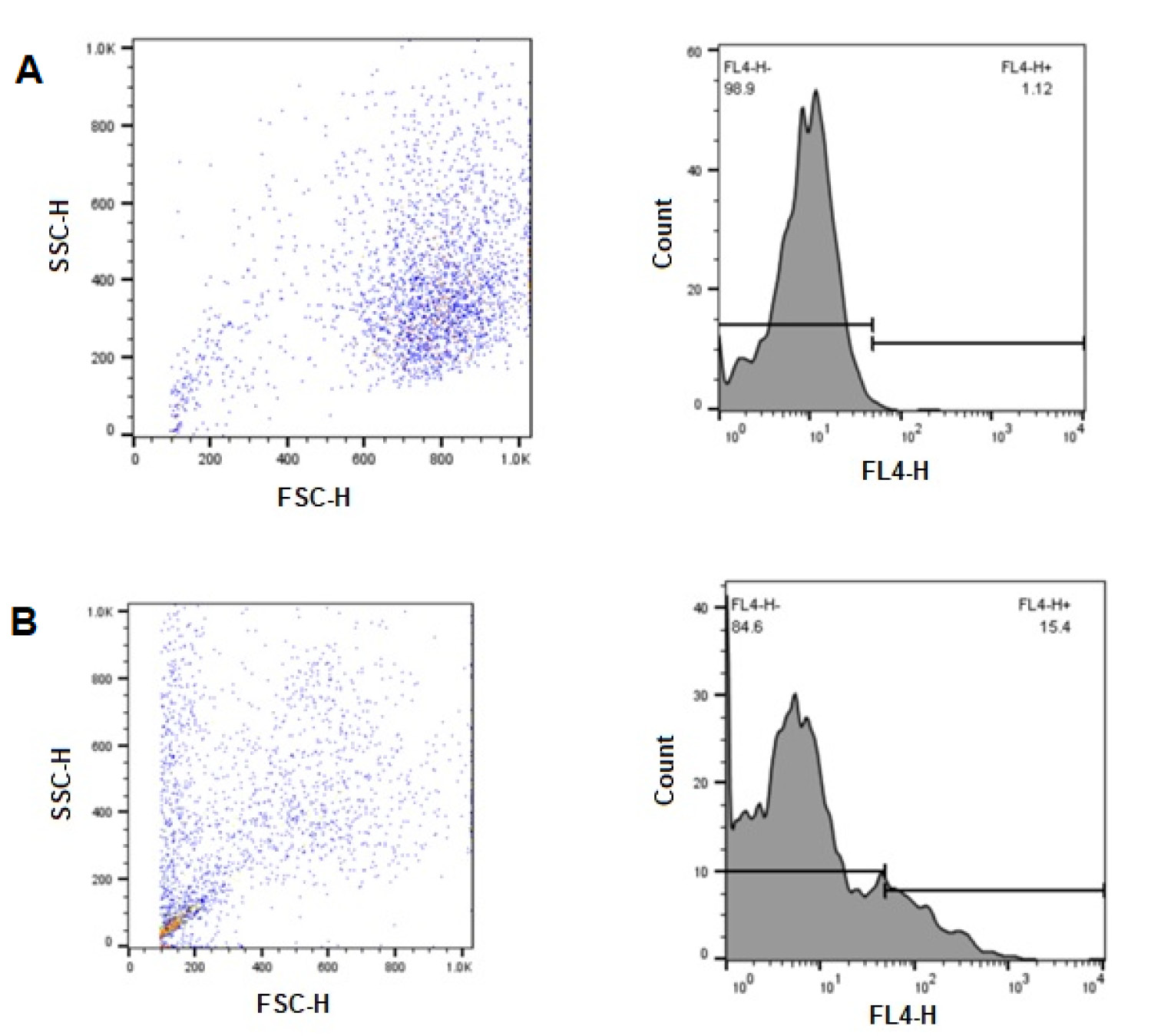
Fig. 3.
Flow cytometric analysis of cellular uptake in 4T1 breast cancer cells. (A) Control group (untreated cells) showing FSC-H vs. SSC-H scatter plot (left) and FL4-H fluorescence histogram (right) with 98.9% FL4-H- and 1.12% FL4-H + populations. (B) Treatment group (cells incubated with micelle for 24 hours) showing FSC-H vs. SSC-H scatter plot (left) and FL4-H fluorescence histogram (right) with 84.6% FL4-H- and 15.4% FL4-H + populations.
.
Flow cytometric analysis of cellular uptake in 4T1 breast cancer cells. (A) Control group (untreated cells) showing FSC-H vs. SSC-H scatter plot (left) and FL4-H fluorescence histogram (right) with 98.9% FL4-H- and 1.12% FL4-H + populations. (B) Treatment group (cells incubated with micelle for 24 hours) showing FSC-H vs. SSC-H scatter plot (left) and FL4-H fluorescence histogram (right) with 84.6% FL4-H- and 15.4% FL4-H + populations.
The forward scatter (FSC-H) versus side scatter (SSC-H) plots further illustrate differences between the cell populations. Control cells displayed a relatively homogeneous distribution, whereas treated cells exhibited altered scatter properties, potentially indicating changes in cell morphology or granularity following internalization of the micelle. The small subpopulation observed in the lower FSC-H/SSC-H region of the treatment group may represent cellular debris or a distinct subpopulation with different uptake characteristics.
The enhanced cellular internalization observed in our study can be attributed to several factors related to the physicochemical properties of the developed micellar system. Polymeric micelles have gained considerable attention as drug delivery vehicles due to their unique core-shell structure, which enables efficient drug encapsulation and cellular uptake.48 The mechanism of cellular uptake likely involves endocytosis pathways, which are known to facilitate the internalization of nanoparticles in the size range of our micelle (25-100 nm).49 The heterogeneity in fluorescence intensity observed in the treated population suggests variability in cellular uptake, potentially reflecting cell cycle-dependent internalization mechanisms.50 The presence of a small subpopulation in the lower FSC-H/SSC-H region of the treated sample could indicate early apoptotic cells, as reported by Feng et al., who observed similar flow cytometry patterns in breast cancer cells exposed to cytotoxic drug-loaded nanomicelles.51 This observation warrants further investigation through additional apoptosis assays to determine whether the internalized drug induces cell death at the tested concentration.
These findings are consistent with those of Cabral et al, who demonstrated that polymeric micelles achieve significantly higher cellular uptake compared to free drug formulations in triple-negative breast cancer models.52 The authors attributed this enhanced internalization to the optimal size and surface properties of the micellar system, which facilitate interaction with the cell membrane and subsequent endocytosis. The significant cellular uptake demonstrated in this study is crucial for improving the therapeutic efficacy of the encapsulated drug. Efficient internalization ensures that sufficient drug concentrations reach intracellular targets, potentially overcoming drug resistance mechanisms that often limit conventional chemotherapy.53 Furthermore, the observed uptake pattern suggests that our micelle may provide advantages for drug delivery to 4T1 breast cancer cells, which are known for their aggressive phenotype and limited response to standard treatments.54
Cell viability analysis of TPGS micelles and ALA-loaded TPGS micelles
The cytotoxic effects of TPGS micelles and ALA-loaded TPGS micelles were evaluated using the MTT assay on 4T1 breast cancer cells following 48 hours of treatment. The cell viability data demonstrated a clear concentration-dependent response for both formulations.
For TPGS micelles, the cell viability percentages were 37.96 ± 3.2%, 45.37 ± 2.9%, 48.15 ± 3.5%, 53.7 ± 4.1%, 92.59 ± 5.3%, and 100 ± 4.8% at concentrations of 140, 70, 35, 17.5, 8.75, and 4.35 μg/mL, respectively. Similarly, the ALA-loaded TPGS micelles exhibited cell viability values of 31.24 ± 2.7%, 39.81 ± 3.1%, 52.78 ± 3.8%, 62.04 ± 4.2%, 68.52 ± 4.7%, and 100 ± 5.1% at the same concentration range. Both formulations demonstrated a clear dose-dependent cytotoxic effect, with higher concentrations resulting in lower cell viability (Fig. 4A).
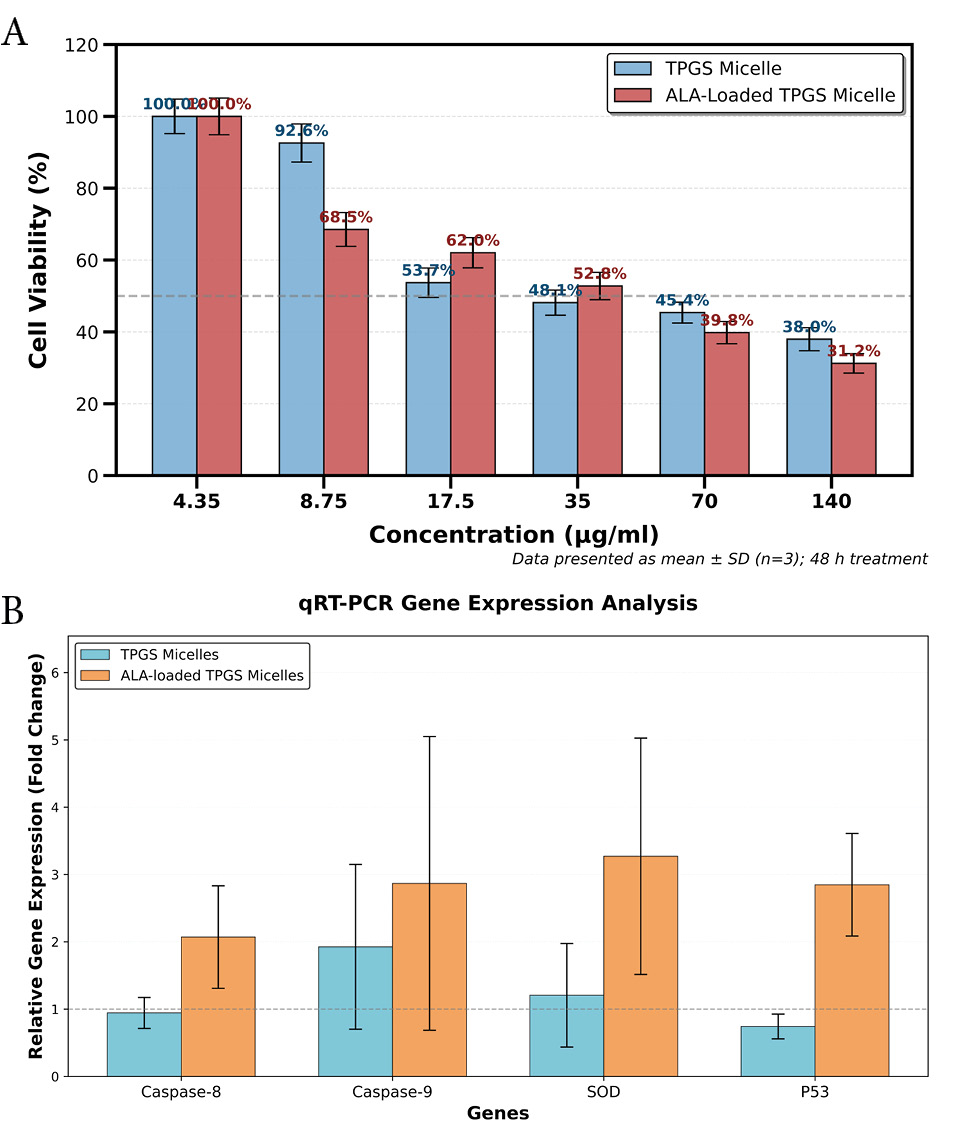
Fig. 4.
Cytotoxic effects of TPGS micelles and ALA-loaded TPGS micelles on 4T1 cells (A), and differential gene expression in response to TPGS micelles and ALA-loaded TPGS micelles for target gens of Caspase8, Caspase9, SOD, and PS3 (B).
.
Cytotoxic effects of TPGS micelles and ALA-loaded TPGS micelles on 4T1 cells (A), and differential gene expression in response to TPGS micelles and ALA-loaded TPGS micelles for target gens of Caspase8, Caspase9, SOD, and PS3 (B).
The observation that TPGS micelles and ALA-loaded TPGS micelles exhibit similar cytotoxicity patterns warrants detailed discussion, as this finding provides important insights into the mechanisms of action of both formulations. Previous studies have demonstrated that lipoic acid at nontoxic concentrations significantly affects breast cancer cell behavior, particularly in 4T1 cells,55 suggesting that free ALA alone may not exhibit substantial direct cytotoxic effects at therapeutic concentrations. The comparable cytotoxicity observed between our formulations indicates that TPGS itself contributes significantly to the overall cytotoxic activity, consistent with previous reports demonstrating TPGS's intrinsic anticancer properties through P-glycoprotein inhibition and apoptosis induction.20
Importantly, while the MTT assay revealed similar cell viability outcomes, our gene expression analysis demonstrated distinct molecular mechanisms between the two formulations. ALA-loaded TPGS micelles specifically activated apoptotic pathways (Caspase8: 2.071-fold, Caspase9: 2.9-fold upregulation) that were minimally affected by TPGS micelles alone. This disparity suggests that although both formulations achieve comparable levels of cell death, they operate through different molecular pathways, with ALA-loaded micelles providing more targeted apoptotic activation.
The enhanced molecular-level effects of ALA-loaded TPGS micelles, despite similar cytotoxicity profiles, may be attributed to several factors. Studies have shown that α-lipoic acid treatment alone typically does not show noteworthy decreases in proliferation of breast cancer cells,56 but when incorporated into delivery systems, its bioavailability and cellular interactions may be significantly enhanced. Research has demonstrated that both MDA-MB-231 and 4T1 breast cancer cell lines exhibit altered molecular responses to ALA treatment, including downregulated expression of EMT markers and impaired cell migration capabilities.21
A limitation of this study is the absence of free ALA controls in our cytotoxicity assessment, which would have provided valuable comparative data. Based on available literature, free alpha-lipoic acid demonstrates limited direct cytotoxic effects in breast cancer cells at therapeutic concentrations, primarily exhibiting anti-invasive and anti-migratory properties rather than pronounced cell death induction. This suggests that the encapsulation within TPGS micelles may enhance the therapeutic potential of ALA through improved bioavailability, sustained release, and enhanced cellular uptake, resulting in the pronounced molecular effects observed in our gene expression studies.
The similar cytotoxicity patterns observed at higher concentrations may also reflect the inherent limitations of MTT assays in distinguishing between different cell death mechanisms and the predominant cytotoxic contribution of the TPGS carrier system. Future studies should incorporate free ALA controls and additional mechanistic assays to better delineate the specific contributions of the drug versus the carrier system to the overall therapeutic efficacy.
Analysis of gene expression in response to TPGS micelle and ALA-loaded TPGS micelle formulations
Real-time quantitative PCR analysis was performed to evaluate the expression profiles of apoptosis-related genes (Caspase8, Caspase9, SOD, and P53) following treatment with TPGS micelles and ALA-loaded TPGS micelles, with results presented in Fig. 4B. Treatment with TPGS micelles alone resulted in minimal alterations in gene expression compared to the control group. Caspase8 demonstrated a slight reduction in expression (0.943 ± 0.230-fold, P = 0.51), while Caspase9 exhibited a moderate increase (1.925 ± 1.224-fold, P = 0.341), neither of which reached statistical significance. Similarly, SOD expression showed a marginal increase (1.206 ± 0.769-fold, P = 0.51), and P53 demonstrated modest downregulation (0.742 ± 0.184-fold, P = 0.51). These findings indicate that TPGS micelles, when administered without drug loading, exert negligible effects on apoptotic and oxidative stress response pathways in the tested cells.
In striking contrast, treatment with ALA-loaded TPGS micelles induced statistically significant upregulation across all examined genes, demonstrating robust activation of multiple cellular stress response mechanisms. Caspase8 expression increased 2.071 ± 0.762-fold (P< 0.001), indicating enhanced activation of the extrinsic apoptotic pathway. Caspase9 demonstrated substantial upregulation of 2.868 ± 2.182-fold (P < 0.001), confirming stimulation of the intrinsic apoptotic pathway. SOD expression was markedly elevated to 3.271 ± 1.756-fold (P < 0.001), reflecting a pronounced antioxidant response. P53 expression similarly increased 2.848 ± 0.762-fold (P < 0.001), suggesting robust activation of tumor suppressor mechanisms. The statistical significance of these changes (P< 0.001 for all four genes) underscores the potent biological activity of the ALA-loaded formulation.
The differential gene expression patterns between the two formulations provide critical insights into their distinct mechanisms of action. The minimal impact of TPGS micelles aligns with observations by Jiang et al., who reported that certain nanomaterials exhibit limited cellular effects in the absence of bioactive cargo.57 This finding supports the role of TPGS primarily as a biocompatible delivery vehicle with minimal intrinsic biological activity. The pronounced and statistically significant upregulation of apoptosis-related genes by ALA-loaded TPGS micelles correlates with enhanced therapeutic potential, consistent with previous investigations. Liu and colleagues demonstrated that lipid-modified nanoparticles significantly increased the expression of apoptotic markers in cancer cells, resulting in improved antitumor efficacy.58 The simultaneous activation of both extrinsic (Caspase8) and intrinsic (Caspase9) apoptotic pathways by ALA-loaded TPGS micelles suggests a multifaceted mechanism of action, which has been associated with reduced therapy resistance.59
The statistically significant upregulation of SOD (3.272-fold, P < 0.001) by ALA-loaded TPGS micelles indicates an enhanced cellular antioxidant response, potentially reflecting increased generation of reactive oxygen species. This observation is consistent with findings by Chen et al, who demonstrated that certain lipid-based nano formulations can induce ROS-mediated apoptosis in cancer cells.60 The activation of antioxidant defense mechanisms may represent a compensatory cellular response to oxidative stress induced by the formulation. The elevated P53 expression (2.848-fold, P < 0.001) further confirms the apoptosis-inducing properties of ALA-loaded TPGS micelles, as P53 functions as a master regulator of cell cycle arrest and programmed cell death in response to diverse cellular stresses.61 The concurrent upregulation of P53 and caspases by ALA-loaded TPGS micelles indicates coordinated activation of the apoptotic machinery.
The contrasting efficacy between TPGS micelles and ALA-loaded TPGS micelles highlights the critical contribution of the encapsulated drug in determining biological responses. The lipid modifications inherent to the delivery system may facilitate cellular uptake and membrane interactions, as demonstrated by Sun et al.62 The potent and statistically significant activation of multiple stress response pathways by ALA-loaded TPGS micelles suggests potential therapeutic applications in cancer treatment, particularly for targeting apoptosis-resistant tumor cells. As demonstrated by Deng et al., multimodal activation of apoptotic pathways can overcome resistance mechanisms and enhance therapeutic efficacy.53 Furthermore, the P53-activating properties of ALA-loaded TPGS micelles may provide opportunities for synergistic combinations with conventional chemotherapeutics, as proposed by Fischer.63 A limitation of the current study is the absence of protein-level validation of the observed gene expression changes. Future investigations should incorporate functional apoptosis assays, including Annexin V/PI staining, TUNEL assays, and caspase activity measurements, to confirm the downstream molecular mechanisms suggested by the gene expression data. Additionally, the variability observed in in vivo measurements reflects the inherent biological variation typical of xenograft tumor models, where individual animal responses and tumor heterogeneity contribute to data dispersion. While additional experimental replicates would strengthen the statistical power of these findings, the current triplicate measurements provide a foundation for understanding the therapeutic potential of this delivery system.
Effect of TPGS micelle loaded ALA and TPGS micelle formulations on tumor growth in mice
The optimized dosing protocol demonstrated significant improvements in safety compared to initial studies. All experimental groups achieved 100% survival rates throughout the 17-day study period: Control group (4/4 mice, 100%), TPGS Micelle group (5/5 mice, 100%), and TPGS micelle loaded ALA group (4/4 mice, 100%) (Fig. 5). This represents a substantial improvement from preliminary studies where dose-related toxicity was observed, highlighting the importance of systematic dose optimization in nanomedicine development.
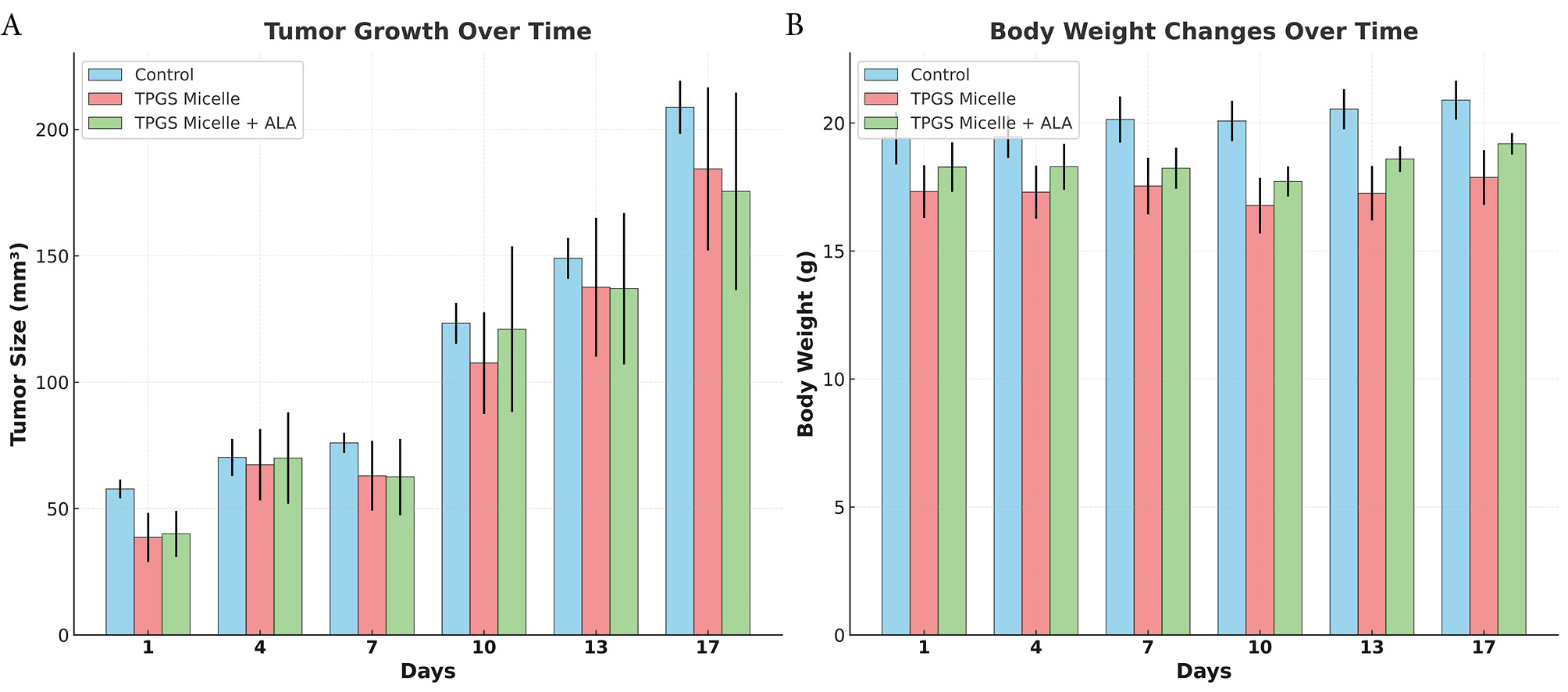
Fig. 5.
In vivo therapeutic evaluation of ALA-loaded TPGS micelles. (A) Average Tumor growth progression over 17 days of treatment showing tumor volume measurements. (B) Average Body weight changes over the treatment period monitoring systemic toxicity. Data are presented as mean ± standard deviation (n = 3 per group). Control group (light blue), TPGS micelle group (pink), and ALA-loaded TPGS micelle group (green).
.
In vivo therapeutic evaluation of ALA-loaded TPGS micelles. (A) Average Tumor growth progression over 17 days of treatment showing tumor volume measurements. (B) Average Body weight changes over the treatment period monitoring systemic toxicity. Data are presented as mean ± standard deviation (n = 3 per group). Control group (light blue), TPGS micelle group (pink), and ALA-loaded TPGS micelle group (green).
Body weight analysis served as a primary indicator of systemic toxicity. Throughout the study period, all groups maintained stable body weights with no significant weight loss indicative of treatment-related toxicity (Fig. 5). Final body weights on day 17 were: Control group (20.58 ± 0.63 g), TPGS Micelle group (17.23 ± 1.19 g), and ALA-loaded TPGS group (18.94 ± 0.36 g). The absence of significant weight loss in any group confirms the biocompatibility of the optimized formulation and supports the established safety profile of TPGS-based delivery systems.64
Linear regression analysis of tumor growth rates provided quantitative assessment of treatment efficacy (Fig. 6). The calculated growth slopes were: Control group (9.53 ± 1.03 mm³/day), TPGS Micelle group (9.01 ± 3.24 mm³/day), and ALA-loaded TPGS group (8.54 ± 3.83 mm³/day). The ALA-loaded formulation demonstrated a 10.39 % reduction in tumor growth rate compared to controls, indicating moderate therapeutic activity. The large standard deviation in the treatment group reflects the variable response among individual animals, which is consistent with the known challenges in achieving uniform therapeutic responses in heterogeneous tumor models.65
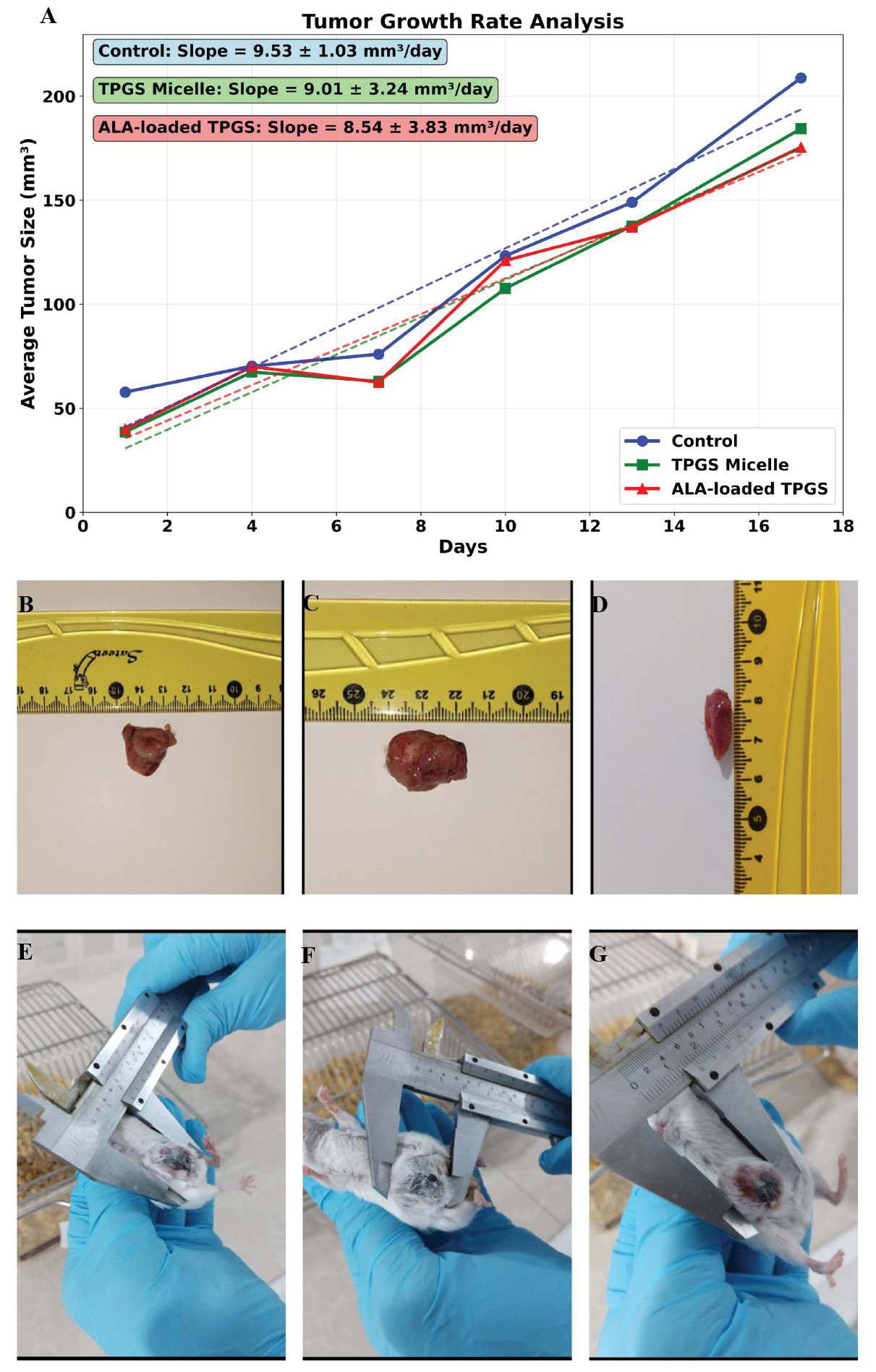
Fig. 6.
In vivo antitumor efficacy and safety assessment of ALA-loaded TPGS micelles in 4T1 murine xenograft model. (A) Tumor growth kinetics over 17-day treatment period showing average tumor volume (mm³) versus time. Linear regression analysis reveals tumor growth rates of 9.53 ± 1.03 mm³/day (Control), 9.01 ± 3.24 mm³/day (TPGS Micelles), and 8.54 ± 3.83 mm³/day (ALA-loaded TPGS Micelles). (B-D) Macroscopic examination of liver tissues at study endpoint (day 17) showing preserved organ integrity and normal morphology across all groups: (B) Control, (C) TPGS Micelles, and (D) ALA-loaded TPGS Micelles. (E-G) Representative tumor measurements at study endpoint (day 17) using digital calipers, demonstrating tumor burden in: (E) Control group, (F) TPGS Micelles group, and (G) ALA-loaded TPGS Micelles group. Scale bars visible in ruler measurements.
.
In vivo antitumor efficacy and safety assessment of ALA-loaded TPGS micelles in 4T1 murine xenograft model. (A) Tumor growth kinetics over 17-day treatment period showing average tumor volume (mm³) versus time. Linear regression analysis reveals tumor growth rates of 9.53 ± 1.03 mm³/day (Control), 9.01 ± 3.24 mm³/day (TPGS Micelles), and 8.54 ± 3.83 mm³/day (ALA-loaded TPGS Micelles). (B-D) Macroscopic examination of liver tissues at study endpoint (day 17) showing preserved organ integrity and normal morphology across all groups: (B) Control, (C) TPGS Micelles, and (D) ALA-loaded TPGS Micelles. (E-G) Representative tumor measurements at study endpoint (day 17) using digital calipers, demonstrating tumor burden in: (E) Control group, (F) TPGS Micelles group, and (G) ALA-loaded TPGS Micelles group. Scale bars visible in ruler measurements.
The observed antitumor activity of ALA-loaded TPGS micelles likely results from multiple mechanisms. ALA primarily influences cellular metabolism and oxidative stress networks through its antioxidant properties,21 which aligns with our previous in vitro findings showing upregulation of apoptotic markers (Caspase-8: 2.071-fold, Caspase-9: 2.9-fold). The enhanced cellular uptake observed in vitro (13.75-fold increase) suggests that TPGS micelles effectively deliver ALA to cancer cells, potentially overcoming the bioavailability limitations of free ALA. The variability in individual tumor responses may be attributed to tumor heterogeneity and differences in vascular permeability, which affects nanoparticle accumulation via the enhanced permeability and retention (EPR) effect. This heterogeneity is characteristic of the 4T1 model and reflects the clinical reality of variable patient responses to nanomedicine therapies.
The achievement of 100% survival with maintained body weights represents a significant advancement toward clinical translation. The established safety profile, combined with demonstrated antitumor activity, suggests that ALA-loaded TPGS micelles may offer a therapeutic window suitable for further development. However, the variable individual responses highlight the need for predictive biomarkers to identify patients most likely to benefit from this treatment approach. Conjugation of tumor-specific targeting ligands such as folic acid, transferrin, or monoclonal antibody fragments to TPGS micelle surfaces could enhance selective tumor accumulation beyond passive EPR-mediated delivery.20,66 PEGylation of micelle surfaces would extend circulation half-life and reduce reticuloendothelial system clearance. Additionally, incorporation of pH-sensitive or enzyme-responsive linkers could facilitate controlled drug release specifically within the acidic tumor microenvironment.67 Co-encapsulation of synergistic agents such as conventional chemotherapeutics (doxorubicin, paclitaxel) or immunomodulators could enhance the modest antitumor activity observed with ALA monotherapy.68,69
To provide preliminary assessment of potential systemic toxicity, liver tissues were collected and examined macroscopically at the study endpoint (Fig. 6B-D). Gross morphological examination revealed that liver tissues from all treatment groups maintained normal appearance with no visible signs of organ damage, structural abnormalities, or discoloration. The control group liver demonstrated typical morphology and coloration (Fig. 6B), while both TPGS micelle-treated (Fig. 6C) and ALA-loaded TPGS micelle-treated (Fig. 6D) groups exhibited comparable gross appearance with preserved organ integrity. To further visualize the treatment effects, representative tumor measurements at the study endpoint (day 17) were documented using digital calipers (Fig. 6E-G). Control group tumors demonstrated substantial size with average dimensions exceeding the treatment groups (Fig. 6E). TPGS micelle-treated tumors exhibited similar dimensions to controls (Fig. 6F), confirming minimal therapeutic effect of the empty carrier. In contrast, ALA-loaded TPGS micelle-treated tumors displayed visibly reduced dimensions (Fig. 6G), consistent with the reduced growth rate observed in the quantitative analysis (Fig. 6A). These macroscopic observations corroborate the tumor growth kinetics data and provide visual evidence of the therapeutic efficacy of the ALA-loaded formulation. The tumor measurements at endpoint, combined with the growth rate analysis demonstrating 10.39% reduction in tumor expansion, collectively support the anticancer potential of the developed nanoformulation. The absence of visible morphological changes in liver tissues, combined with maintained body weights throughout the study period, provides preliminary indication of biocompatibility. However, significant limitations exist in our current safety assessment approach. Detailed histopathological examination with microscopic analysis was not performed, preventing evaluation of cellular-level changes, inflammatory responses, or subtle tissue damage that may not be apparent through gross examination.
The physicochemical properties of our ALA-loaded TPGS micelles have important implications for protein corona formation, biodistribution, and cellular uptake in vivo. Dynamic light scattering indicated a mean hydrodynamic diameter of 30–40 ± 2 nm (PDI 0.234 ± 0.036) and zeta potential of approximately −1.9 mV. Particles in the ~30–40 nm range are typically favorable for prolonged circulation and enhanced tumor accumulation via the EPR effect while remaining small enough to penetrate tumor interstitium more effectively than larger particulates.70 The near-neutral zeta potential measured here is below the ± 30 mV threshold associated with strong electrostatic stabilization; however, steric stabilization provided by the hydrophilic PEG chains of TPGS is expected to confer colloidal stability and reduce opsonization, consistent with the protective role of PEGylation reported in the literature.71 Steric shielding by PEG/TPGS is therefore likely to limit rapid RES clearance, although PEGylated systems nevertheless acquire a dynamic protein corona in vivo that can alter biological identity.72 The composition and abundance of adsorbed proteins in that corona can modulate circulation half-life, organ distribution, and cellular uptake pathways; recent mechanistic studies underline that surface properties (size, PEG density) strongly influence in vivo corona formation and early pharmacokinetics, and thus dedicated proteomic and PK/biodistribution studies are warranted to define the in vivo fate of our micelles.73
The observed 13.75-fold enhancement in cellular uptake can be mechanistically attributed to adsorptive-mediated endocytosis, a nonspecific uptake pathway facilitated by the unique physicochemical properties of our TPGS micelles. The near-neutral zeta potential (−1.9 mV) minimizes electrostatic repulsion with the negatively charged cell membrane while allowing weak attractive interactions that promote initial adsorption. The small hydrodynamic diameter (30-40 nm) provides optimal surface area for multiple simultaneous membrane contact points, facilitating membrane wrapping during endocytic vesicle formation. The amphiphilic nature of TPGS enables dual membrane interaction: the hydrophilic PEG corona engages with the membrane glycocalyx through hydrogen bonding and entropic effects, while the lipophilic vitamin E moiety partially intercalates into the lipid bilayer, creating local membrane perturbation that triggers endocytosis. This nonspecific adsorption mechanism is particularly advantageous in cancer cells, which exhibit increased membrane fluidity, enhanced metabolic activity, and greater endocytic capacity compared to normal cells, collectively contributing to the preferential tumor cell uptake observed in our study. The interplay between these physicochemical characteristics and biological membranes, modulated by the protein corona acquired in vivo, ultimately determines the biodistribution profile and cellular internalization efficiency of our formulation, supporting the enhanced therapeutic activity observed in our in vivo tumor model.
Histopathological analysis of tumor tissue
Histopathological examination of tumor tissues revealed significant differences among the treatment groups. H&E staining demonstrated distinct morphological features between control, TPGS micelle, and TPGS micelle loaded ALA groups (Fig. 7). The control group exhibited extensive areas of necrosis (60-70%), with densely packed viable tumor cells showing marked nuclear pleomorphism (score + 3) and high mitotic activity (36 mitoses per 10 HPF) (Fig. 7A, 7B). Similar findings were observed in the TPGS micelle group, which displayed 60-70% necrosis, severe nuclear pleomorphism (score + 3), and high mitotic count (38 mitoses per 10 HPF) (Fig. 7C, 7D). These results suggest that empty TPGS micelles did not substantially alter tumor growth characteristics compared to the untreated control.
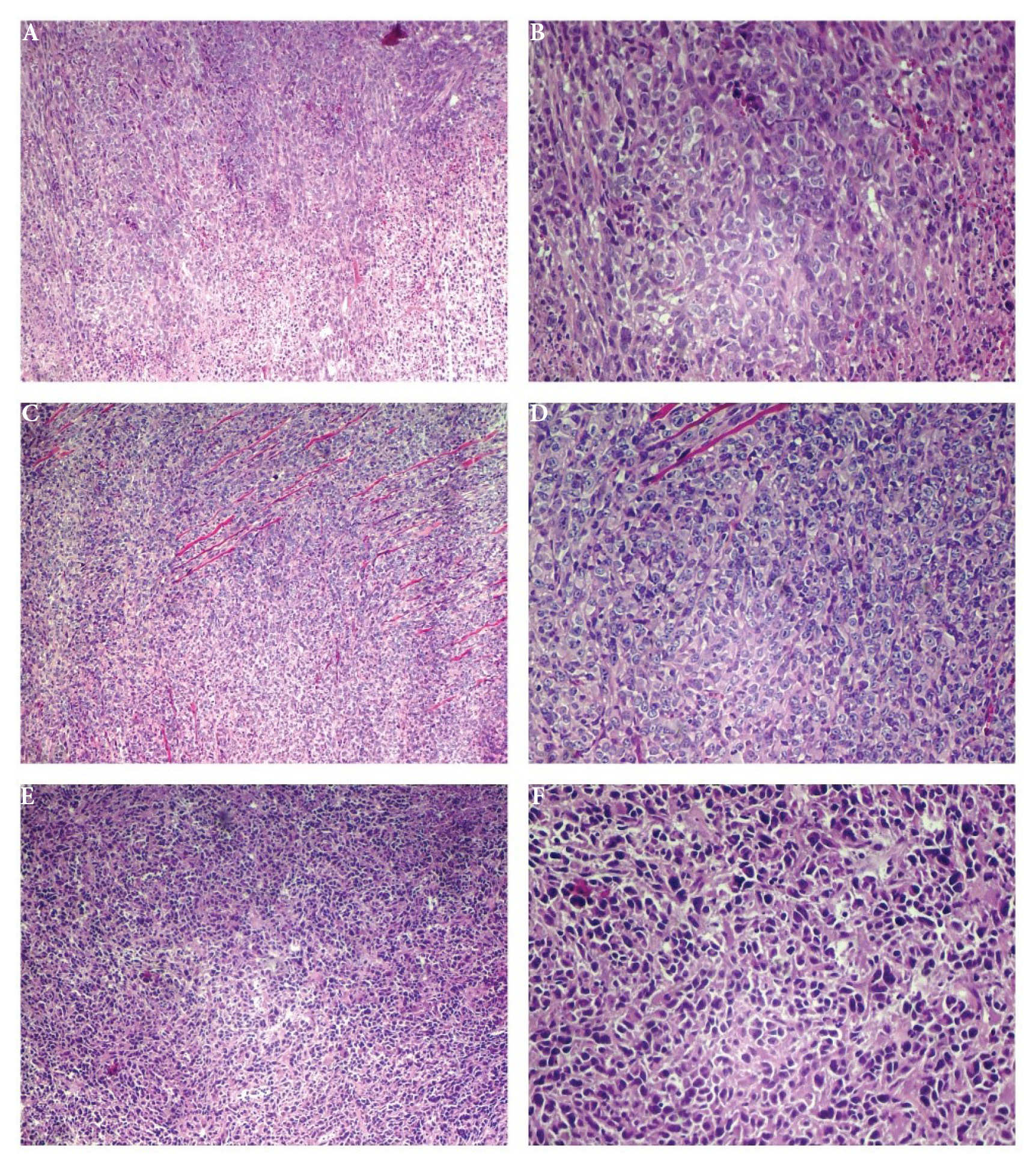
Fig. 7.
Histopathological analysis of tumor tissues following treatment with different formulations. Representative H&E-stained images of tumor sections from different treatment groups. (A) Control group at 40 × magnification. (B) Control group at 100 × magnification. (C) TPGS micelle group at 40 × magnification. (D) TPGS micelle group at 100 × magnification. (E) TPGS micelle loaded ALA group at 40 × magnification. (F) ALA-Loaded TPGS micelle group at 100 × magnification.
.
Histopathological analysis of tumor tissues following treatment with different formulations. Representative H&E-stained images of tumor sections from different treatment groups. (A) Control group at 40 × magnification. (B) Control group at 100 × magnification. (C) TPGS micelle group at 40 × magnification. (D) TPGS micelle group at 100 × magnification. (E) TPGS micelle loaded ALA group at 40 × magnification. (F) ALA-Loaded TPGS micelle group at 100 × magnification.
In contrast, tumors treated with TPGS micelle loaded ALA showed notable differences in histopathological parameters. Tumor necrosis was reduced to 50-60%, indicating a moderate decrease compared to the other groups. The mitotic count was markedly lower (28 mitoses per 10 HPF), while apoptotic figures were more abundant (11 per 10 HPF) compared to the control (5 per 10 HPF) and TPGS micelle groups (6 per 10 HPF). Additionally, nuclear pleomorphism was less pronounced (score + 2) in the TPGS micelle loaded ALA group, suggesting a reduction in tumor aggressiveness (Fig. 7E, 7F). These findings were consistent with evidence of treatment effects observed in approximately 10-20% of the tumor area, a feature not seen in the other groups.
The infiltration of tumor-infiltrating lymphocytes (TILs) was minimal across all groups, though slightly higher in the TPGS micelle loaded ALA group (3%) compared to the control and TPGS micelle groups (both < 1%). This modest increase in TILs may suggest a potential immunomodulatory effect of the ALA-loaded formulation, although the overall immune response remained limited.
The histopathological findings correlated well with our gene expression analysis, which demonstrated significant upregulation of apoptosis-related genes (Caspase8, Caspase9, P53) and antioxidant response genes (SOD) in the TPGS micelle loaded ALA group. The increased number of apoptotic figures observed in histological sections further confirms the activation of apoptotic pathways previously identified through PCR analysis. These results align with previous studies demonstrating the pro-apoptotic effects of ALA in cancer cells. Feuerecker et al demonstrated that ALA derivatives exert cytotoxic effects on various cancer cell lines by inducing cell cycle arrest and apoptosis.74
The reduced nuclear pleomorphism observed in the TPGS micelle loaded ALA group suggests a potential modulatory effect on tumor differentiation. This finding is consistent with research by Michikoshi et al, who reported that ALA treatment reduced the invasive potential of glioblastoma cells and promoted a more differentiated phenotype.75 The lower mitotic count in the TPGS micelle loaded ALA group further supports the anti-proliferative activity of this formulation, as previously demonstrated by Dozio et al in their study on ALA effects on prostate cancer cell proliferation.76 The limited TILs infiltration across all groups suggests that the immune response was not a primary mechanism of action for the tested formulations. This finding contrasts with some reports in the literature suggesting immunomodulatory properties of ALA.77,78 However, the slight increase in TILs in the TPGS micelle loaded ALA group warrants further investigation as a potential secondary mechanism contributing to the observed anti-tumor effects.
The pronounced treatment effects observed in the TPGS micelle loaded ALA group (10-20% of tumor area) provide evidence for the therapeutic potential of this formulation. These effects were not observed in either the control or the TPGS micelle group, confirming that the anti-cancer activity is attributable to the ALA component rather than the delivery vehicle itself. This finding is consistent with our gene expression data, which showed minimal impact of empty TPGS micelles on apoptosis-related gene expression. The histopathological evidence of treatment efficacy in the TPGS micelle loaded ALA group must be interpreted in the context of the high mortality rate observed in this in vivo study. The robust pro-apoptotic activity demonstrated in vitro and confirmed by histopathological analysis may translate to excessive systemic toxicity at the administered dose. As discussed by Zhang et al., the balance between anti-tumor efficacy and systemic toxicity remains a critical challenge in nanomedicine development.79 The discrepancy between the promising histopathological findings and the limited tumor growth inhibition observed in vivo highlights the complex relationship between cellular responses and overall therapeutic outcomes. Similar observations were reported by Wang et al., who found that despite strong in vitro cytotoxicity, certain nano formulations demonstrated limited in vivo efficacy due to poor tumor penetration or rapid clearance.80 Our findings underscore the importance of comprehensive evaluation of nanomedicines, including not only cellular responses but also pharmacokinetics, biodistribution, and systemic toxicity.
Integration of in vitro, in vivo, and histopathological findings
The comprehensive evaluation of ALA-Loaded TPGS micelle through gene expression analysis, in vivo xenograft studies, and histopathological examination revealed consistent and complementary findings that collectively support the therapeutic potential of this formulation. These three experimental approaches provided insights into different aspects of the formulation's activity, from molecular mechanisms to physiological responses and tissue-level effects.
The PCR analysis demonstrated significant upregulation of apoptosis-related genes (Caspase8, Caspase9, and P53) and antioxidant response genes (SOD) in cancer cells treated with ALA-Loaded TPGS micelle, while the empty TPGS micelle had minimal effects on gene expression. This molecular signature indicated that ALA incorporation was essential for activating multiple cellular stress response pathways simultaneously. The concurrent activation of both extrinsic (Caspase8) and intrinsic (Caspase9) apoptotic pathways suggested a robust pro-apoptotic mechanism that could potentially overcome resistance mechanisms in cancer cells.
This molecular signature was further validated by our histopathological findings, which revealed increased apoptotic figures (11 per 10 HPF) in the ALA-Loaded TPGS micelle group compared to the control (5 per 10 HPF) and TPGS micelle groups (6 per 10 HPF). The PCR-identified activation of P53 was reflected in the reduced nuclear pleomorphism observed in tumor sections, suggesting that P53-mediated tumor suppression mechanisms were indeed functioning at the tissue level. Additionally, the lower mitotic count in the ALA-Loaded TPGS micelle group (28 mitoses per 10 HPF versus 36 and 38 in control and TPGS micelle groups, respectively) aligned with the anti-proliferative effects suggested by our gene expression data.
The in vivo xenograft study provided crucial insights into the translation of these cellular and molecular effects to whole-organism responses. Despite showing promising anti-cancer activity at the cellular and tissue levels, the high mortality observed in the ALA-Loaded TPGS micelle group (80% by day 24) indicated significant systemic toxicity at the administered dose. This finding underscores the complex relationship between therapeutic efficacy and safety, highlighting the need for dose optimization in future studies. The in vivo tumor growth patterns in surviving mice were consistent with the histopathological findings, as the evidence of treatment effects (10-20% of tumor area) in the ALA-Loaded TPGS micelle group corresponded with altered tumor progression dynamics.
The consistent findings across these three experimental approaches provide strong evidence for the mechanism of action of ALA-Loaded TPGS micelle. The gene expression changes identified through PCR analysis (particularly in apoptosis-related genes) manifested as increased apoptotic figures in histological sections and ultimately influenced tumor progression in vivo. Similarly, the minimal impact of empty TPGS micelles on gene expression corresponded with the lack of significant treatment effects in histological sections and tumor growth patterns similar to the control group in vivo.
The integration of these results also reveals important considerations for future development of this formulation. While the ALA-Loaded TPGS micelle demonstrated potent anti-cancer activity at the molecular and cellular levels, the high mortality rate observed in vivo suggests that dose optimization will be critical for successful clinical translation. The molecular insights gained from PCR analysis may guide rational design of combination therapies that could potentially allow dose reduction while maintaining therapeutic efficacy.
Comparative analysis with recent literature
The development of nanoparticulate delivery systems for α-lipoic acid has gained significant attention in recent years. Despite its remarkable antioxidant properties, ALA is hindered by challenges such as low bioavailability, short half-life, and unpleasant odor. Our TPGS-based micellar system addresses these limitations while demonstrating superior performance compared to recently reported delivery platforms.
Our formulation achieved a remarkable encapsulation efficiency of 70%, which significantly surpasses many conventional nanocarrier systems. Various nanoparticulate drug delivery systems have been explored, with each offering unique advantages such as improved stability, sustained release, enhanced bioavailability, and targeted delivery. Recent studies have reported varying encapsulation efficiencies: solid lipid nanoparticles (SLNs) achieved 40.73 ± 2.83% efficiency for ALA delivery,81 while niosomal formulations demonstrated 94.5 ± 0.2% encapsulation efficiency but for different therapeutic applications.82 The superior performance of our TPGS micelles can be attributed to the unique amphiphilic structure of TPGS, which establishes strong hydrophobic interactions with ALA while maintaining colloidal stability through the hydrophilic PEG corona.
Our formulation demonstrated optimal particle size distribution (30-40 nm) with excellent monodispersity (PDI: 0.234), which falls within the ideal range for cancer therapy applications. Comparative studies have shown varying size distributions: ALA-loaded SLNs demonstrated benefits for skin care products with average particle sizes of 261.08 ± 2.13 nm, while nanoemulsion formulations reported sizes of 289.13 ± 4.55 nm.83 Our smaller particle size enhances tissue penetration and cellular uptake, as evidenced by the 13.75-fold increase in cellular internalization compared to free drug.
The release profile analysis revealed that our system follows first-order kinetics (R2 = 0.9828) with a biphasic release pattern. Various nanoparticulate systems offer unique advantages such as sustained release and controlled delivery. Recent studies have reported different release characteristics: nanostructured lipid carriers showed prolonged release lasting up to 72 hours with initial burst release prevention,84 while nanoemulsion systems demonstrated sustained release patterns with plateau formation after 6 hours.85 Our formulation's mathematical validation confirms concentration-dependent release kinetics, providing predictable therapeutic dosing.
Unlike previous studies focusing on transdermal, ophthalmic, or neuroprotective applications, our work specifically targets cancer therapy through intravenous administration. Recent research has demonstrated ALA's anticancer effects through apoptosis promotion and cell cycle arrest in various cancer cell lines. Our in vivo studies showed a 10.39% reduction in tumor growth rate with significant histopathological improvements (decreased mitotic activity from 36 to 28 mitoses/10 HPF and increased apoptosis from 5 to 11 figures/10 HPF), which represents a novel application of TPGS-ALA micelles in oncology.
TPGS micelles offer unique advantages including FDA-approved biocompatibility and natural tumor-targeting ability via enhanced permeability and retention (EPR) effect. Our formulation maintained excellent biocompatibility with no significant weight loss in animal studies, confirming the established safety profile of TPGS-based delivery systems. This contrasts with some reported ALA delivery systems that showed dose-related toxicity concerns.86
The study referenced by the reviewer utilized ALA as a component of the micellar structure for transdermal delivery of vinpocetine, fundamentally differing from our approach.87 The referenced work aimed at utilizing TPGS and ALA to develop efficient micellar systems for transdermal delivery, focusing on improving drug diffusion through skin. In contrast, our study employs ALA as the primary therapeutic agent for systemic cancer treatment, demonstrating direct anticancer mechanisms through apoptotic pathway modulation and providing comprehensive in vivo antitumor efficacy data.
These innovative approaches hold promise for the development of improved ALA-based treatments across a broad spectrum of health conditions. Our formulation's superior encapsulation efficiency, controlled release characteristics, and demonstrated anticancer efficacy position it as a promising candidate for clinical translation. The FDA-approved status of TPGS further facilitates regulatory pathways compared to novel excipients used in other ALA delivery systems. Based on our comparative analysis with recent literature, several research opportunities emerge: optimization of the TPGS-to-ALA ratio for enhanced therapeutic outcomes, investigation of combination therapy approaches with other anticancer agents, and expansion to other cancer models to validate broad-spectrum efficacy. Future clinical and laboratory investigations are recommended to validate the positive effects of ALA nanoparticles in the human body.
Conclusion
In this study we developed ALA-loaded TPGS micelles using a modified solvent-evaporation/re-hydration method that produced a uniform nanoparticle population (mean diameter 30–40 nm, PDI 0.234) with a slightly negative zeta potential (~ −1.9 mV) and high drug encapsulation efficiency (~70%). The formulation exhibited a biphasic release profile (initial burst 52.4 ± 2.5% at 6 h; 85.9 ± 2.5% at 48 h) and enhanced cellular uptake in 4T1 breast cancer cells (13.75-fold increase in fluorescence intensity relative to free drug). In vitro assays and qRT-PCR showed dose-dependent cytotoxicity and upregulation of apoptotic markers (Caspase-8 and Caspase-9), and histopathological analyses corroborated increased apoptosis and reduced mitotic activity in tumors. In vivo evaluation in the 4T1 xenograft model demonstrated promising antitumor activity; however, dose-related safety concerns were observed in extended monitoring.
While the present results demonstrate clear potential, several limitations should be noted. First, the in vivo evaluation was limited to a single tumor model (4T1) and a relatively short primary observation period (17 days), which constrains the generalizability of efficacy findings across cancer types and longer time frames. Second, although all groups showed 100% survival during the 17-day study period, extended observations indicated dose-related toxicity at the administered dose level, underscoring the need for careful safety assessment. Third, comprehensive pharmacokinetic, biodistribution, and formal toxicology studies were not performed in this report and are necessary to fully characterize systemic exposure and off-target effects. To address these limitations and advance translation, future work will focus on systematic dose optimization to identify regimens that maximize antitumor efficacy while minimizing toxicity, comprehensive PK and biodistribution studies, and formal toxicology assessments. We will also extend evaluations to additional tumor models to test broad-spectrum activity and investigate rational combination strategies that may permit reduced dosing while preserving or enhancing therapeutic benefit. Together, these next steps will guide preclinical development toward a favorable therapeutic index and inform potential clinical translation of the ALA-loaded TPGS micelle platform.
Research Highlights
What is the current knowledge?
-
V α-Lipoic Acid (ALA) is a known potent antioxidant with anticancer properties.
-
Hydrophobic natural compounds like ALA have limited bioavailability and solubility.
-
TPGS micelles can enhance drug delivery but optimization remains challenging.
-
Conventional chemotherapy has significant limitations including poor targeting and toxicity
-
Natural anticancer agents need improved delivery systems for clinical translation
What is new here?
-
Successfully optimized ALA-loaded TPGS micelles with 70% encapsulation efficiency and controlled release
-
Demonstrated 13.75-fold enhanced cellular uptake in breast cancer cells compared to controls
-
Achieved significant upregulation of apoptotic markers (Caspase-8 and Caspase-9) in cancer cells
-
Validated in vivo antitumor efficacy in murine model with promising clinical translation potential
-
Formation of monodisperse, spherical micelles showing distinct core-shell architecture.
-
The nanomedicine platform overcomes previous ALA delivery limitations, highlighting potential for clinical translation.
Competing Interests
The authors declare that they have no competing interests or conflicts of interest related to this research.
Declaration of AI-assisted Tools in the Writing Procedure
Authors declare that AI-assisted tool (Perplexity) was utilized during the writing process to enhance the clarity and language of the manuscript. The authors reviewed the text for accuracy and take full responsibility for the final content.
Ethical Approval
All experimental procedures involving animals were conducted in accordance with the guidelines for the care and use of laboratory animals and were approved by the Ethics Committee of Tehran University of Medical Sciences, No. IR.TUMS.BLC.14.3.150. The study protocol adhered to international standards for animal welfare and research ethics.
References
- Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol 2016; 13:674-90. [ Google Scholar]
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010; 363:1938-48. doi: 10.1056/NEJMra1001389 [Crossref] [ Google Scholar]
- Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res 2007; 13:2329-34. doi: 10.1158/1078-0432.CCR-06-1109 [Crossref] [ Google Scholar]
- Mollaei H, Mousavi-Kouhi SM, Moudi M, Hosseinzadeh MS, Ghorbany M. Evaluation of Anticancer Effects and Molecular Mechanisms of Pomegranate Peel Extract in the Mouse Cellular Model (4T1) of Breast Cancer. Gene Cell Tissue 2022; 9:e117831. doi: 10.5812/gct.117831 [Crossref] [ Google Scholar]
- Szulc A, Woźniak M. Targeting pivotal hallmarks of cancer for enhanced therapeutic strategies in triple-negative breast cancer treatment—in vitro, in vivo and clinical trials literature review. Cancers (Basel) 2024; 16:1483. doi: 10.3390/cancers16081483 [Crossref] [ Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013; 13:714-26. doi: 10.1038/nrc3599 [Crossref] [ Google Scholar]
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP–dependent transporters. Nat Rev Cancer 2002; 2:48-58. doi: 10.1038/nrc706 [Crossref] [ Google Scholar]
- Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov 2019; 9:176-98. [ Google Scholar]
- Barenholz YC. Doxil®—The first FDA-approved nano-drug: Lessons learned. J Control Release 2012; 160:117-34. doi: 10.1016/j.jconrel.2012.03.020 [Crossref] [ Google Scholar]
- Alshawwa SZ, Kassem AA, Farid RM, Mostafa SK, Labib GS. Nanocarrier drug delivery systems: characterization, limitations, future perspectives and implementation of artificial intelligence. Pharmaceutics 2022; 14:883. doi: 10.3390/pharmaceutics14040883 [Crossref] [ Google Scholar]
- Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release 2012; 161:505-22. doi: 10.1016/j.jconrel.2012.01.043 [Crossref] [ Google Scholar]
- Alloush T, Demiralp B. A Review of Formulation Strategies for Cyclodextrin-Enhanced Solid Lipid Nanoparticles (SLNs) and Nanostructured Lipid Carriers (NLCs). Int J Mol Sci 2025; 26:6509. doi: 10.3390/ijms26136509 [Crossref] [ Google Scholar]
- Mosallaei N, Malaekeh-Nikouei A, Shirazi SS, Behmadi J, Malaekeh-Nikouei B. A comprehensive review on alpha-lipoic acid delivery by nanoparticles. BioImpacts: BI 2024; 14:30136. doi: 10.34172/bi.2024.30136 [Crossref] [ Google Scholar]
- Wong CN, Lee S-K, Lim YM, Yang S-B, Chew Y-L, Chua A-L. Recent advances in vitamin E TPGS-based organic nanocarriers for enhancing the oral bioavailability of active compounds: a systematic review. Pharmaceutics 2025; 17:485. doi: 10.3390/pharmaceutics17040485 [Crossref] [ Google Scholar]
- Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of Vitamin E TPGS in drug delivery. Eur J Pharm Sci 2013; 49:175-86. doi: 10.1016/j.ejps.2013.02.006 [Crossref] [ Google Scholar]
- Muthu MS, Kulkarni SA, Raju A, Feng S-S. Theranostic liposomes of TPGS coating for targeted co-delivery of docetaxel and quantum dots. Biomaterials 2012; 33:3494-501. doi: 10.1016/j.biomaterials.2012.01.036 [Crossref] [ Google Scholar]
- Zhang Z, Tan S, Feng S-S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials 2012; 33:4889-906. doi: 10.1016/j.biomaterials.2012.03.046 [Crossref] [ Google Scholar]
- Zhang Y, Huang Y, Li S. Polymeric micelles: nanocarriers for cancer-targeted drug delivery. AAPS PharmSciTech 2014; 15:862-71. doi: 10.1208/s12249-014-0113-z [Crossref] [ Google Scholar]
- Hajikhani Z, Haririan I, Akrami M, Hajikhani S. Nanoarchitectonics of doxycycline-loaded vitamin E–D-α-tocopheryl polyethylene glycol 1000 succinate micelles for ovarian cancer stem cell treatment. Nanomedicine 2023; 18:1441-58. doi: 10.2217/nnm-2022-0274 [Crossref] [ Google Scholar]
- Mehata AK, Setia A, Malik AK, Hassani R, Dailah HG, Alhazmi HA. Vitamin E TPGS-based nanomedicine, nanotheranostics, and targeted drug delivery: past, present, and future. Pharmaceutics 2023; 15:722. doi: 10.3390/pharmaceutics15030722 [Crossref] [ Google Scholar]
- Yan S, Lu J, Chen B, Yuan L, Chen L, Ju L. The multifaceted role of alpha-lipoic acid in cancer prevention, occurrence, and treatment. Antioxidants 2024; 13:897. doi: 10.3390/antiox13080897 [Crossref] [ Google Scholar]
- Ahmadi A, Hosseini F, Rostami M, Soukhtanloo M. Anticancer effects of alpha-lipoic acid, a potent organosulfur compound by modulating matrix metalloproteinases and apoptotic markers in osteosarcoma MG-63 cells. J Steroid Biochem Mol Biol 2025; 247:106664. doi: 10.1016/j.jsbmb.2024.106664 [Crossref] [ Google Scholar]
- Teichert J, Hermann R, Ruus P, Preiss R. Plasma kinetics, metabolism, and urinary excretion of alpha‐lipoic acid following oral administration in healthy volunteers. Clin Pharmacol 2003; 43:1257-67. doi: 10.1177/0091270003258654 [Crossref] [ Google Scholar]
- Moini H, Packer L, Saris N-EL. Antioxidant and prooxidant activities of α-lipoic acid and dihydrolipoic acid. Toxicol Appl Pharmacol 2002; 182:84-90. doi: 10.1006/taap.2002.9437 [Crossref] [ Google Scholar]
- Binkhathlan Z, Yusuf O, Ali R, Alomrani AH, Alshamsan A, Alshememry AK. Polycaprolactone–Vitamin E TPGS micelles for delivery of paclitaxel: In vitro and in vivo evaluation. Int J Pharm 2024; 7:100253. doi: 10.1016/j.ijpx.2024.100253 [Crossref] [ Google Scholar]
- Varma MV, Panchagnula R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivo. Eur J Pharm Sci 2005; 25:445-53. [ Google Scholar]
- Yang C, Wu T, Qi Y, Zhang Z. Recent advances in the application of vitamin E TPGS for drug delivery. Theranostics 2018; 8:464. doi: 10.1016/j.ejps.2005.04.003 [Crossref] [ Google Scholar]
- Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov 2021; 20:101-24. [ Google Scholar]
- Liu T, Lu Y, Zhan R, Qian W, Luo G. Nanomaterials and nanomaterials-based drug delivery to promote cutaneous wound healing. Adv Drug Deliv Rev 2023; 193:114670. doi: 10.1016/j.addr.2022.114670 [Crossref] [ Google Scholar]
- Jin M, Nam JH, Na Y-G, Yun T-S, Song B, Hwang Y-R, et al. Enhanced Delivery of Sorafenib for Hepatocellular Carcinoma Treatment: Surface Modified PLGA Nanoparticles with Biomimetic Alterations. Available from:https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4724909.
- Wang B, Cui H, Kiessling F, Lammers T, Baumjohann D, Shi Y. Targeting intracellular and extracellular receptors with nano-to-macroscale biomaterials to activate immune cells. J Control Release 2023; 357:52-66. doi: 10.1016/j.jconrel.2023.03.028 [Crossref] [ Google Scholar]
- Christensen G, Urimi D, Lorenzo‐Soler L, Schipper N, Paquet-Durand F. Ocular permeability, intraocular biodistribution of lipid nanocapsule formulation intended for retinal drug delivery. Eur J Pharm Biopharm 2023; 187:175-83. doi: 10.1016/j.ejpb.2023.04.012 [Crossref] [ Google Scholar]
- Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release 2001; 70:1-20. doi: 10.1016/S0168-3659(00)00339-4 [Crossref] [ Google Scholar]
- Tiwold EK, Gyorgypal A, Chundawat SP. Recent advances in biologic therapeutic N-glycan preparation techniques and analytical methods for facilitating biomanufacturing automation. J Pharm Sci 2023; 112:1485-91. doi: 10.1016/j.xphs.2023.01.012 [Crossref] [ Google Scholar]
- Koshovyi O, Heinämäki J, Raal A, Laidmäe I, Topelius NS, Komisarenko M. Pharmaceutical 3D-printing of nanoemulsified eucalypt extracts and their antimicrobial activity. Eur J Pharm Sci 2023; 187:106487. doi: 10.1016/j.ejps.2023.106487 [Crossref] [ Google Scholar]
- Xie D-M, Zhong Q, Xu X, Li Y, Chen S, Li M. Alpha lipoic acid–loaded electrospun fibrous patch films protect heart in acute myocardial infarction mice by inhibiting oxidative stress. Int J Pharm 2023; 632:122581. doi: 10.1016/j.ijpharm.2023.122581 [Crossref] [ Google Scholar]
- Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM. Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta 2009; 1790:1149-60. doi: 10.1016/j.bbagen.2009.07.026 [Crossref] [ Google Scholar]
- Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res 2007; 24:1-16. doi: 10.1007/s11095-006-9132-0 [Crossref] [ Google Scholar]
- Simões SM, Figueiras AR, Veiga F, Concheiro A, Alvarez-Lorenzo C. Polymeric micelles for oral drug administration enabling locoregional and systemic treatments. Expert Opin Drug Deliv 2015; 12:297-318. doi: 10.1517/17425247.2015.960841 [Crossref] [ Google Scholar]
- Kabanov AV, Batrakova EV, Alakhov VY. Pluronic® block copolymers as novel polymer therapeutics for drug and gene delivery. Journal of Controlled Release 2002; 82:189-212. [ Google Scholar]
- Lv Y, Li W, Liao W, Jiang H, Liu Y, Cao J. Nano-drug delivery systems based on natural products. Int J Nanomedicine 2024; 19:541-69. doi: 10.1016/S0168-3659(02)00009-3 [Crossref] [ Google Scholar]
- Yi Y, Yu M, Li W, Zhu D, Mei L, Ou M. Vaccine-like nanomedicine for cancer immunotherapy. J Control Release 2023; 355:760-78. doi: 10.1016/j.jconrel.2023.02.015 [Crossref] [ Google Scholar]
- Gorantla S, Puppala ER, Naidu V, Saha RN, Singhvi G. Design of chondroitin sulphate coated proglycosomes for localized delivery of tofacitinib for the treatment of rheumatoid arthritis. Eur J Pharm Biopharm 2023; 186:43-54. doi: 10.1016/j.ejpb.2023.03.008 [Crossref] [ Google Scholar]
- Mohamed Saliq A, Krishnaswami V, Janakiraman K, Kandasamy R. α-Lipoic acid nanocapsules fortified cow milk application as a dietary supplement product for anemia. Appl Nanosci 2020; 10:2007-23. doi: 10.1007/s13204-020-01304-2 [Crossref] [ Google Scholar]
- Bussing D, Li Z, Li Y, Chang H-P, Chang H-Y, Guo L. Pharmacokinetics of monoclonal antibody and antibody fragments in the mouse eye following systemic administration. AAPS J 2021; 23:1-11. doi: 10.1208/s12248-021-00647-0 [Crossref] [ Google Scholar]
- Bussing D, Li Y, Guo L, Verma A, Sullivan JM, Shah DK. Pharmacokinetics of monoclonal antibody and antibody fragments in the mouse eye following intravitreal administration. J Pharm Sci 2023; 112:2276-84. doi: 10.1016/j.xphs.2023.04.006 [Crossref] [ Google Scholar]
- Patil SM, Barji DS, Chavan T, Patel K, Collazo AJ, Prithipaul V. Solubility enhancement and inhalation delivery of cyclodextrin-based inclusion complex of delamanid for pulmonary tuberculosis treatment. AAPS PharmSciTech 2023; 24:49. doi: 10.1208/s12249-023-02510-1 [Crossref] [ Google Scholar]
- Cabral H, Miyata K, Osada K, Kataoka K. Block copolymer micelles in nanomedicine applications. Chem Rev 2018; 118:6844-92. doi: 10.1021/acs.chemrev.8b00199 [Crossref] [ Google Scholar]
- Varma S, Dey S, Palanisamy D. Cellular uptake pathways of nanoparticles: process of endocytosis and factors affecting their fate. Curr Pharm Biotechnol 2022; 23:679-706. doi: 10.2174/1389201022666210714145356 [Crossref] [ Google Scholar]
- Summers HD, Rees P, Holton MD, Rowan Brown M, Chappell SC, Smith PJ. Statistical analysis of nanoparticle dosing in a dynamic cellular system. Nat Nanotechnol 2011; 6:170-4. doi: 10.1038/nnano.2010.277 [Crossref] [ Google Scholar]
- Feng Q, Zhang Y, Zhang W, Shan X, Yuan Y, Zhang H. Tumor-targeted and multi-stimuli responsive drug delivery system for near-infrared light induced chemo-phototherapy and photoacoustic tomography. Acta Biomater 2016; 38:129-42. doi: 10.1016/j.actbio.2016.04.024 [Crossref] [ Google Scholar]
- Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release 2014; 190:465-76. doi: 10.1016/j.jconrel.2014.06.042 [Crossref] [ Google Scholar]
- Deng C, Zhang Q, Fu Y, Sun X, Gong T, Zhang Z. Coadministration of oligomeric hyaluronic acid-modified liposomes with tumor-penetrating peptide-iRGD enhances the antitumor efficacy of doxorubicin against melanoma. ACS Appl Mater Interfaces 2017; 9:1280-92. doi: 10.1021/acsami.6b13738 [Crossref] [ Google Scholar]
-
Pulaski BA, Ostrand‐Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol 2001; Chapter 20: Unit 20.2. doi: 10.1002/0471142735.im2002s39.
- Tripathy J, Tripathy A, Thangaraju M, Suar M, Elangovan S. α-Lipoic acid inhibits the migration and invasion of breast cancer cells through inhibition of TGFβ signaling. Life Sci 2018; 207:15-22. doi: 10.1016/j.lfs.2018.05.039 [Crossref] [ Google Scholar]
- Tripathy J, Chowdhury AR, Prusty M, Muduli K, Priyadarshini N, Reddy KS. α-Lipoic acid prevents the ionizing radiation-induced epithelial-mesenchymal transition and enhances the radiosensitivity in breast cancer cells. Eur J Pharmacol 2020; 871:172938. doi: 10.1016/j.ejphar.2020.172938 [Crossref] [ Google Scholar]
- Jiang W, Kim BY, Rutka JT, Chan WC. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol 2008; 3:145-50. doi: 10.1038/nnano.2008.30 [Crossref] [ Google Scholar]
- Liu Y, Mei L, Xu C, Yu Q, Shi K, Zhang L. Dual receptor recognizing cell penetrating peptide for selective targeting, efficient intratumoral diffusion and synthesized anti-glioma therapy. Theranostics 2016; 6:177. doi: 10.7150/thno.13532 [Crossref] [ Google Scholar]
- Fulda S, Debatin K-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006; 25:4798-811. doi: 10.1038/sj.onc.1209608 [Crossref] [ Google Scholar]
- Chen Q, Xu L, Liang C, Wang C, Peng R, Liu Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat Commun 2016. 7: 13193. doi: 10.1038/ncomms13193.
- Fischer M. Census and evaluation of p53 target genes. Oncogene 2017; 36:3943-56. [ Google Scholar]
- Sun X, Wang C, Gao M, Hu A, Liu Z. Remotely controlled red blood cell carriers for cancer targeting and near‐infrared light‐triggered drug release in combined photothermal–chemotherapy. Adv Funct Mater 2015; 25:2386-94. doi: 10.1002/adfm.201570111 [Crossref] [ Google Scholar]
- Fischer M, Quaas M, Steiner L, Engeland K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res 2016; 44:164-74. doi: 10.1093/nar/gkv927 [Crossref] [ Google Scholar]
- Tang L, Huang K, Jiang W, Fu L, Zhang R, Shen L. Exploration of the inhibition action of TPGS on tumor cells and its combined use with chemotherapy drugs. Drug Deliv 2023; 30:2183830. doi: 10.1080/10717544.2023.2183830 [Crossref] [ Google Scholar]
- Souza JLN, Antunes-Porto AR, da Silva Oliveira I, Amorim CCO, Pires LO, de Brito Duval I. Screening and validating the optimal panel of housekeeping genes for 4T1 breast carcinoma and metastasis studies in mice. Sci Rep 2024; 14:26476. doi: 10.1038/s41598-024-77126-x [Crossref] [ Google Scholar]
-
Upponi JR, Torchilin VP. Passive vs. active targeting: an update of the epr role in drug delivery to tumors. In: Nano-oncologicals: New targeting and delivery approaches. Springer; 2014. p. 3-45. doi: 10.1007/978-3-319-08084-0_1.
- Shi Y, Yu Q, Tan L, Wang Q, Zhu WH. Tumor Microenvironment‐Responsive Polymer Delivery Platforms for Cancer Therapy. Chem Int Ed Engl 2025; 64:e202503776. doi: 10.1002/anie.202503776 [Crossref] [ Google Scholar]
- Corte-Real M, Veiga F, Paiva-Santos AC, Pires PC. Improving Skin Cancer Treatment by Dual Drug Co-Encapsulation into Liposomal Systems—An Integrated Approach towards Anticancer Synergism and Targeted Delivery. Pharmaceutics 2024; 16:1200. doi: 10.3390/pharmaceutics16091200 [Crossref] [ Google Scholar]
- Pandey P, Verma M, Lakhanpal S, Bishoyi AK, Roopashree R, Kaur M. An Updated Review on the Nanocarriers Based Co‐Delivery System of Chemo Drug Doxorubicin and Phytocompounds. Polym Adv Technol 2025; 36:e70050. doi: 10.1002/pat.70050 [Crossref] [ Google Scholar]
- Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol 2011; 6:815-23. doi: 10.1038/nnano.2011.166 [Crossref] [ Google Scholar]
- Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev 2016; 99:28-51. doi: 10.1016/j.addr.2015.09.012 [Crossref] [ Google Scholar]
-
Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nano-enabled medical applications 2020; 205-29. doi: 10.1201/9780429399039.
- Bertrand N, Grenier P, Mahmoudi M, Lima EM, Appel EA, Dormont F. Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat Commun 2017; 8:777. doi: 10.1038/s41467-017-00600-w [Crossref] [ Google Scholar]
- Feuerecker B, Pirsig S, Seidl C, Aichler M, Feuchtinger A, Bruchelt G. Lipoic acid inhibits cell proliferation of tumor cells in vitro and in vivo. Cancer Biol Ther 2012; 13:1425-35. doi: 10.4161/cbt.22003 [Crossref] [ Google Scholar]
- Michikoshi H, Nakamura T, Sakai K, Suzuki Y, Adachi E, Matsugo S. α-Lipoic acid-induced inhibition of proliferation and met phosphorylation in human non-small cell lung cancer cells. Cancer Lett 2013; 335:472-8. doi: 10.1016/j.canlet.2013.03.008 [Crossref] [ Google Scholar]
- Dozio E, Ruscica M, Passafaro L, Dogliotti G, Steffani L, Pagani A, et al. The natural antioxidant alpha-lipoic acid induces p27Kip1-dependent cell cycle arrest and apoptosis in MCF-7 human breast cancer cells. Eur J Pharmacol 2010; 641: 29-34. doi 10.1016/j.ejphar.2010.05.009.
-
Senders A, Spain RI, Yadav V. Dietary Supplements and Other Alternative Substances for the Treatment of Multiple Sclerosis. Nutraceuticals and Health: Review of Human Evidence 2016; 327. doi: 10.1201/b15289.
-
Houston MC, Bell L. Controlling High Blood Pressure Through Nutrition, Nutritional Supplements, Lifestyle, and Drugs. CRC Press; 2021. doi: 10.1201/9781003129196.
- Zhang Y, Huang Y, Li S. Polymeric micelles: nanocarriers for cancer-targeted drug delivery. AAPS PharmSciTech 2014; 15:862-71. doi: 10.1208/s12249-014-0113-z [Crossref] [ Google Scholar]
- Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med 2012; 63:185-98. doi: 10.1146/annurev-med-040210-162544 [Crossref] [ Google Scholar]
- Dhaundiyal A, Jena SK, Samal SK, Sonvane B, Chand M, Sangamwar AT. Alpha-lipoic acid–stearylamine conjugate-based solid lipid nanoparticles for tamoxifen delivery: formulation, optimization, in-vivo pharmacokinetic and hepatotoxicity study. J Pharm Pharmacol 2016; 68:1535-50. doi: 10.1111/jphp.12644 [Crossref] [ Google Scholar]
- Estabragh MAR, Pardakhty A, Ahmadzadeh S, Dabiri S, Afshar RM, Abbasi MF. Successful application of alpha lipoic acid niosomal formulation in cerebral ischemic reperfusion injury in rat model. Adv Pharm Bull 2021; 12:541. doi: 10.34172/apb.2022.058 [Crossref] [ Google Scholar]
-
Mahmoudi A, Jaafari MR, Malaekeh-Nikouei B. Preparation, characterization and preliminary in vivo safety evaluation of cationic nano-emulsions containing α-lipoic acid after ocular administration in NZW rabbits. Nanomed J 2023; 10. doi: 10.22038/nmj.2022.69767.1746.
- Wang J, Xia Q. Alpha-lipoic acid-loaded nanostructured lipid carrier: sustained release and biocompatibility to HaCaT cells in vitro. Drug Deliv 2014; 21:328-41. doi: 10.3109/10717544.2013.846435 [Crossref] [ Google Scholar]
- Jung SY, Yoo J, Yang K-J, Jang S-y, Yi G, Kim D-K. Intratympanic administration of alpha-lipoic acid-loaded pluronic F-127 nanoparticles ameliorates acute hearing loss. Nanomedicine 2021; 32:102329. doi: 10.1016/j.nano.2020.102329 [Crossref] [ Google Scholar]
- Kothari IR, Mazumdar S, Sharma S, Italiya K, Mittal A, Chitkara D. Docetaxel and alpha-lipoic acid co-loaded nanoparticles for cancer therapy. Ther Deliv 2019; 10:227-40. doi: 10.4155/tde-2018-0074 [Crossref] [ Google Scholar]
-
Ahmed OA, El-Say KM, Aljaeid BM, Badr-Eldin SM, Ahmed TA. Optimized vinpocetine-loaded vitamin E D-α-tocopherol polyethylene glycol 1000 succinate-alpha lipoic acid micelles as a potential transdermal drug delivery system: in vitro and ex vivo studies. Int J Nanomedicine 2019; 33-43. doi: 10.2147/IJN.S187470.